 Nicolas Bachmann
Nicolas Bachmann Dominic Leiser1
Dominic Leiser1 Barbara Bachtiary
Barbara Bachtiary Damien C. Weber
Damien C. Weber
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 27 June 2022
Sec. Radiation Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.881665
Objective: Peripheral nerve sheath tumors (PNSTs) commonly arise from peripheral nerve roots and grow locally invasive. Malignant PNSTs (mPNSTs) represent aggressive sarcomas of neural origin that can originate from PNSTs. Radiation therapy is commonly used as part of the required multimodal treatment. However, both entities tend to occur early in life and are associated with the genetic disorder neurofibromatosis type 1 (NF-1), which is known to cause increased radiosensitivity. Pencil beam scanning proton therapy (PBSPT) allows for a minimization of the dose delivered to organs at risk and the integral dose and, thus, potentially also a reduction of radiation-induced adverse events. We report the clinical outcome and toxicity rates of patients with (m)PNSTs treated with PBSPT.
Methods: We retrospectively reviewed 36 patients who received PBSPT (median dose, 64 GyRBE) with curative intent for (m)PNSTs between 1999 and 2020 at our institute. Twenty-eight (78%) and 8 (22%) patients were treated at diagnosis and for tumor recurrence/progression, respectively. The median age was 32 years (range, 3–75), and 25 (69%) patients were male. mPNST and PNST were diagnosed in 31 (86%) and 5 (14%) patients, respectively. Underlying NF-1 disease was found in 8 (22%) patients. Acute and late toxicities were recorded according to Common Terminology Criteria for Adverse Events, version 4.1 (CTCAE v4.1). Overall survival (OS), local control (LC), and distant control (DC) were estimated using the Kaplan–Meier method.
Results: With a median follow-up time of 31 months (range, 4–194), 13 (36%) patients died from a progressive disease, 8 (22%) experienced local failure, and 14 (39%) experienced distant failure after PBSPT. Estimated 2-year OS, LC, and DC were 75.5%, 73.5%, and 61.2%, respectively. Acute grade 3 toxicity (dermatitis, mucositis, and pain) was observed in 5 (14%) patients. Late grade 3 cataract and osteonecrosis were both observed in 1 (3%) patient at 34 and 194 months after PBSPT, respectively. There was no late grade >3 toxicity or radiation-induced secondary cancer.
Conclusion: To our knowledge, this is the first study to analyze the outcome of (m)PNSTs treated with proton therapy using a PBS delivery paradigm. In our cohort, consisting mainly of patients with mPNSTs, we report reasonable oncological outcomes and low toxicity rates after PBSPT.
Peripheral nerve sheath tumors (PNSTs) and malignant PNSTs (mPNSTs) are neoplasms arising from peripheral nerves and are frequently also described as neurofibroma or schwannoma (PNST), neurofibrosarcoma, neurogenic sarcomas, or malignant schwannomas (mPNST). Both entities are often associated with the genetic disorder neurofibromatosis type 1 (NF-1) (1). PNSTs usually arise from Schwann cells (2) and are typically of benign character but can cause progressive and uncontrolled pain, neurologic deficits, compression/destruction of vital structures, and severe disfigurement (1). mPNST is a rare and highly aggressive soft tissue sarcoma of neural origin with an incidence of 1.46 per 1,000,000 person-years (3). mPNSTs account for approximately 10% of all soft tissue sarcomas and can originate from precursor PNSTs, particularly from plexiform neurofibromas (2, 4–10). An NF-1 disorder is found in approximately 50% of mPNST cases, 10% of all mPNSTs are radiation-induced, and the remaining 40% occur sporadically (6). Tumors of the peripheral nerves tend to present earlier in life (1, 4, 11, 12) than most other sarcomas, and mPNSTs represent one of the most frequent non-rhabdomyosarcomatous soft tissue sarcomas in the pediatric population (4, 13). A multimodal treatment approach including surgery and radiotherapy, with or without chemotherapy, is often administered due to the local aggressiveness and high potential to metastasize (14). Despite aggressive treatment regimens, the outcome for most patients remains poor, with median 5-year survival rates ranging from 15% to 50% (1). Studies have shown an increased radiosensitivity in NF-1 patients, which can lead to increased side effects from irradiation and a higher risk of secondary tumor induction (15, 16). With proton therapy (PT), the dose to organs at risk (OARs) can be reduced, and hence a lower integral dose is achievable. Considering that PNSTs/mPNSTs are often in a critical location (i.e., head or spine) and the young age of patients, PT seems an appropriate treatment strategy for this patient group.
The aim of this study is to report the oncological outcome and toxicity after pencil beam scanning PT (PBSPT) for PNSTs and mPNSTs and to assess the major prognostic factors for these challenging tumors.
Medical records of all non-metastatic patients who were treated with PBSPT for a PNST or an mPNST with curative attempt between 1999 and 2020 at our institution were retrospectively reviewed. Peripheral schwannoma, benign neurilemmoma, neurofibroma, and benign PNST were considered PNST. Malignant schwannoma, malignant neurilemmoma, triton tumor, malignant perineurioma, neurogenic sarcoma, neurofibrosarcoma, and mPNST were categorized as mPNST.
Patients with any age (pediatric patients <18 years, adolescents and young adults (AYA) 18–39 years, and adults >39 years) and any Karnofsky performance status (KPS for adults and AYA) or Lansky score (for pediatric patients) were included. Previous photon irradiations and combined treatments with protons and photons were allowed. Out of 164 patients screened in our institutional database, 1 patient with no follow-up data, 121 patients with inappropriate histology, and 6 patients with suspected PNSTs of cranial nerves diagnosed only radiologically were excluded. In total, 36 patients were included in the analysis. Approval from the competent ethics committee was obtained for this study (Ethikkommission Nordwest- und Zentralschweiz 2021-00369).
Patients treated in 1999 and early 2000 received gantry-delivered PBSPT with a beam from the main 590-MeV cyclotron. From mid-2000 onward, PBSPT was delivered with an energy-degraded beam from a 250-MeV cyclotron. PBSPT was delivered as neoadjuvant, adjuvant, or definitive treatment for primary or recurrent tumors.
Treatment planning was performed either on the in-house planning system PSIplan or Eclipse® (Varian Medical Systems, Palo Alto, CA, USA). Multi-field optimization (MFO) and single-field optimization (SFO) techniques were used. For patients who underwent macroscopic complete tumor resection, the initial gross tumor was contoured in pre-surgery images (PET-CT, CT, and MRI). The pre-surgery images were fused with the PT planning-CT, and the tumor bed was delineated. In the case of persisting gross tumor after surgery and for the non-operated patients, a gross tumor volume (GTV) was defined by the tumor visualized on the planning CT and planning MRI. The clinical target volume (CTV) was defined as the GTV or the tumor bed with an additional margin (median 20 mm) dependent on histology and pathological features. In most cases, a boost dose to the high-risk area was delivered sequentially or as a simultaneous integrated boost (SIB). For the CTV boost, an additional margin (median 10 mm) was added to the GTV or the tumor bed, again dependent on the histology and pathological characteristics. The planning target volume (PTV) was defined as the CTV plus a 4–10-mm safety margin, depending on tumor location and patient immobilization. For PBSPT planning, a relative biological effectiveness (RBE) value of 1.1 was used.
Acute toxicity was recorded weekly during PT and assessed within the first 3 months after PBSPT. All subsequent institutional and external clinical notes were collected by our study and research office and reviewed during our weekly follow-up meeting to determine disease status and toxicity. All observed adverse events were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 4.1 (CTCAE v4.1).
Local failure (LF) was either proven histologically or defined radiologically as residual tumor progression (an increase of ≥25% in size visible in MRI, CT, or PET-CT) or as the development of new nodular contrast enhancement and/or FDG uptake in the surgical bed compared to the baseline images. LFs occurring within the 95%, 50%–95%, and <50% isodose were classified as “in-field,” “marginal,” or “out-of-field,” respectively (17). Distant failure (DF) was defined as the development of new distant lesions in MRI, CT, or PET-CT follow-up or histologically proven by biopsy or resection.
Time to event data were calculated from the first day of PBSPT to the date of death or censored at the last follow-up using the Kaplan–Meier method. Death from any cause, LF, and DF were the defined events for the calculation of overall survival (OS), local control (LC), and distant control (DC), respectively. Group differences were analyzed with the log-rank test. Univariate Cox regression was used to investigate prognostic factors for LF, DF, and OS. Due to the cohort size and the limited number of events, a multivariate analysis was not deemed reasonable. Fisher’s exact test was used to compare toxicity rates in NF-1 and non-NF-1 patients. A p-value ≤0.05 was considered statistically significant. All statistical analyses were computed using SPSS version 26 (IBM, Armonk, NY, USA).
Thirty-six patients with a median age of 32 years (range, 3–75) were included in this study, comprising 11 (31%) women and 25 (69%) men. Baseline characteristics are summarized in Table 1. Nine (25%) patients were younger than 18 years, and 15 (42%) patients were adolescents and young adults at the time of PBSPT. Histological confirmation was obtained for all patients. mPNST and PNST were diagnosed in 31 (86%) and 5 (14%) patients, respectively. Eight (22%) tumors were associated with underlying NF-1 disorder and 5 (14%) tumors (initial diagnosis: nasopharyngeal squamous cell carcinoma, n = 1; seminoma, n = 2; leukemia [total body irradiation], n = 1; and medulloblastoma [craniospinal irradiation], n = 1) were believed to be radiation induced because of the close proximity of the tumor with the previous radiation fields. About half of the tumors (n = 20, 56%) were located in the trunk, and two-thirds (n = 24, 67%) showed an initial local tumor extension of >5 cm. At the start date of PBSPT, all patients were assessed as non-metastatic. Histological workup of mPNSTs after complete resection (R0, n = 11, 31%) or partial resection (R1/R2 or biopsy, n = 15, 42%) showed FNCLCC (Fédération Nationale des Centres de Lutte Contre Le Cancer) Grades 1, 2, and 3 in 2 (6%), 14 (39%), and 10 (28%) cases, respectively. For 5 (14%) mPNST cases, there was insufficient information to assess an FNCLCC grade.
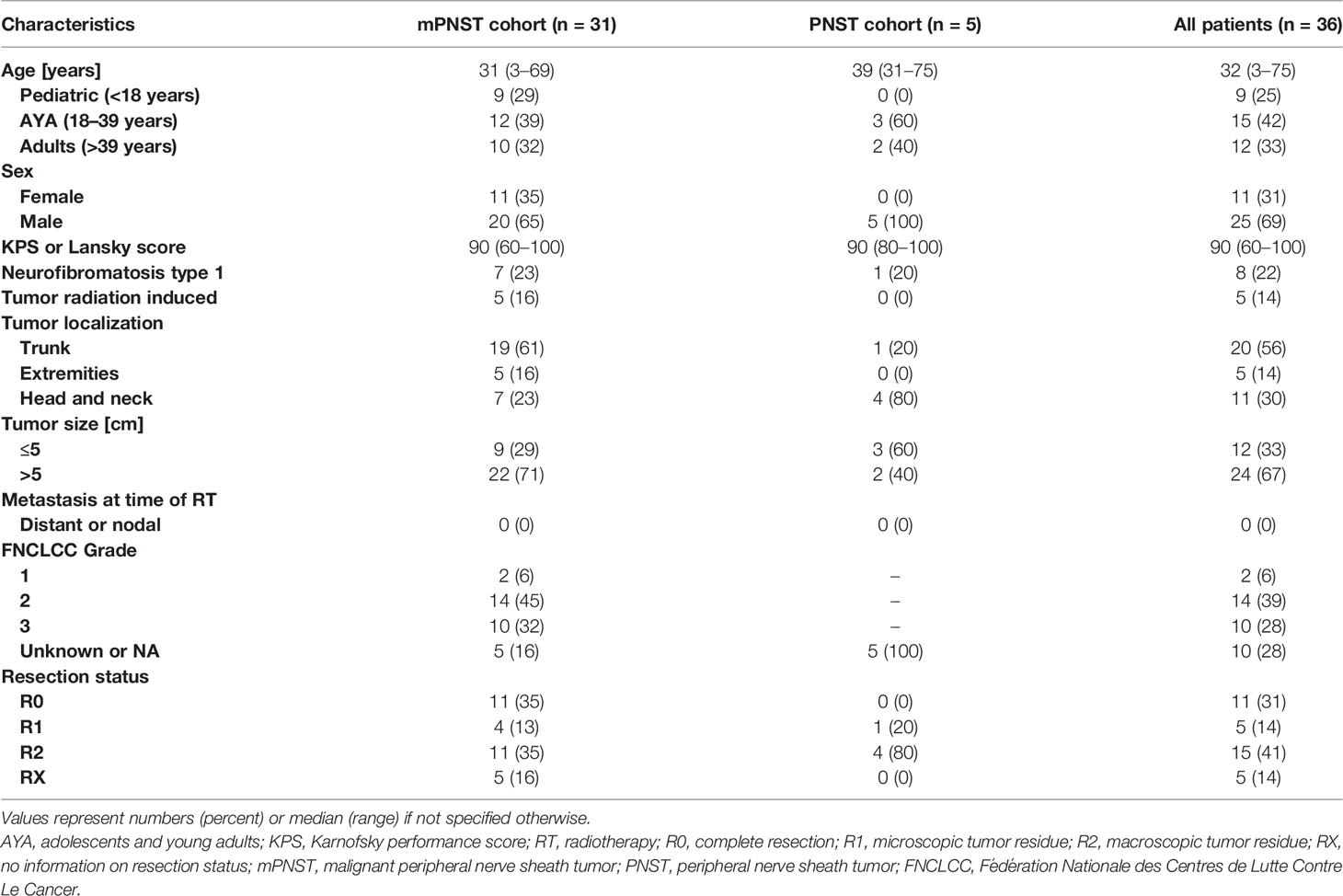
Table 1 Patient characteristics.
A summary of the treatment characteristics is detailed in Table 2. Eight (22%) patients received PBSPT for a recurrent tumor, whereas the majority of patients (n = 28, 78%) underwent PBSPT at diagnosis. Of the former group, 1 patient (3%) was treated previously with photon radiotherapy 42 months before PBSPT. PBSPT was delivered as adjuvant, neoadjuvant, and definitive treatments in 28 (78%), 5 (14%), and 3 (8%) cases, respectively. In total, 10 (28%) patients received chemotherapy as part of their treatment, as specified in Table 2. The median prescribed total dose was 64 GyRBE (range, 50–74) in 32 fractions (range, 17–39). Only 3 (8%) patients underwent combined irradiation with protons and photons: 2 patients (6%) received a boost with photons after completion of PT, and another patient (3%) started irradiation with photons and was boosted with protons. Most patients (n = 22, 61%) received a boost dose sequentially or as a SIB.
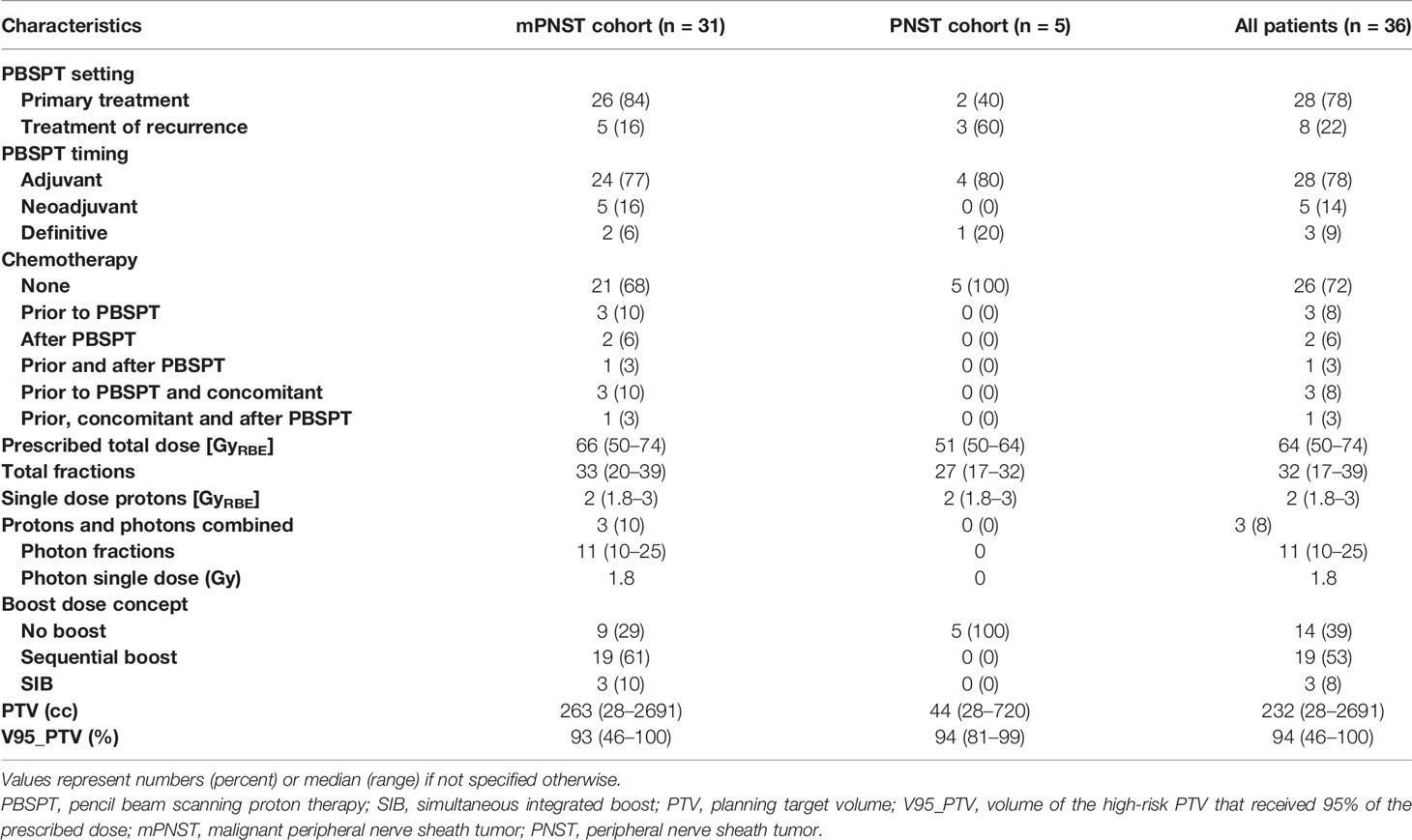
Table 2 Treatment characteristics.
LF was observed in a total of 8 (22%; 1 PNST and 7 mPNST) patients, with 6 failures being classified as “in-field” and 2 others as “marginal” failures. Estimated 2-year LC rate was 73.5% (95% CI: 57.6%–89.4%). DF after PBSPT was observed in 14 (39%) patients, which resulted in an estimated overall 2-year DC rate of 61.2% (95% CI: 44.7%–77.7%). Sites of DF were the central nervous system (n = 5, 36%), lungs (n = 3, 21%), and soft tissue (n = 2, 14%). Four (29%) patients failed distantly at multiple sites. None of the PNST patients failed distantly. Analyzing only the mPNST cohort revealed an estimated 2-year DC rate of 56.7% (95% CI: 39.1%–74.3%) for this group. With a median follow-up time of 31 months (range, 4–194), 13 (36%) patients died from progressive mPNST disease. None of the patients with PNST had died. Overall, the estimated 2-year survival rates were 75.5% (95% CI: 60.6%–90.4%) and 72.8% (95% CI: 56.7%–88.9%) in the mPNST cohort.
On univariate analysis, no prognostic factor for LF was identifiable. However, univariate analysis showed a significant negative association between DF and higher FNCLCC grade (hazard ratio (HR) 3.79, 95% CI: 1.32–10.9, p = 0.013) and R2/RX resection status (HR 3.97, 95% CI: 1.1–14.3, p = 0.035, Table 3). These two factors demonstrate a similar impact on survival in univariate and log-rank analyses: the 2-year survival rates for patients with FNCLCC grade 3 tumors and R2/RX resection status were 67.5% (95% CI: 37.1%–97.8%) and 59.8% (95% CI: 36.7%–82.9%), respectively, while FNCLCC grade ≤2 tumors and R0/R1 resection status had 2-year survival rates of 78.7% (95% CI: 62%–95.4%) and 93.3% (95% CI: 80.8%–100%), respectively (Figure 1 and Table 3).
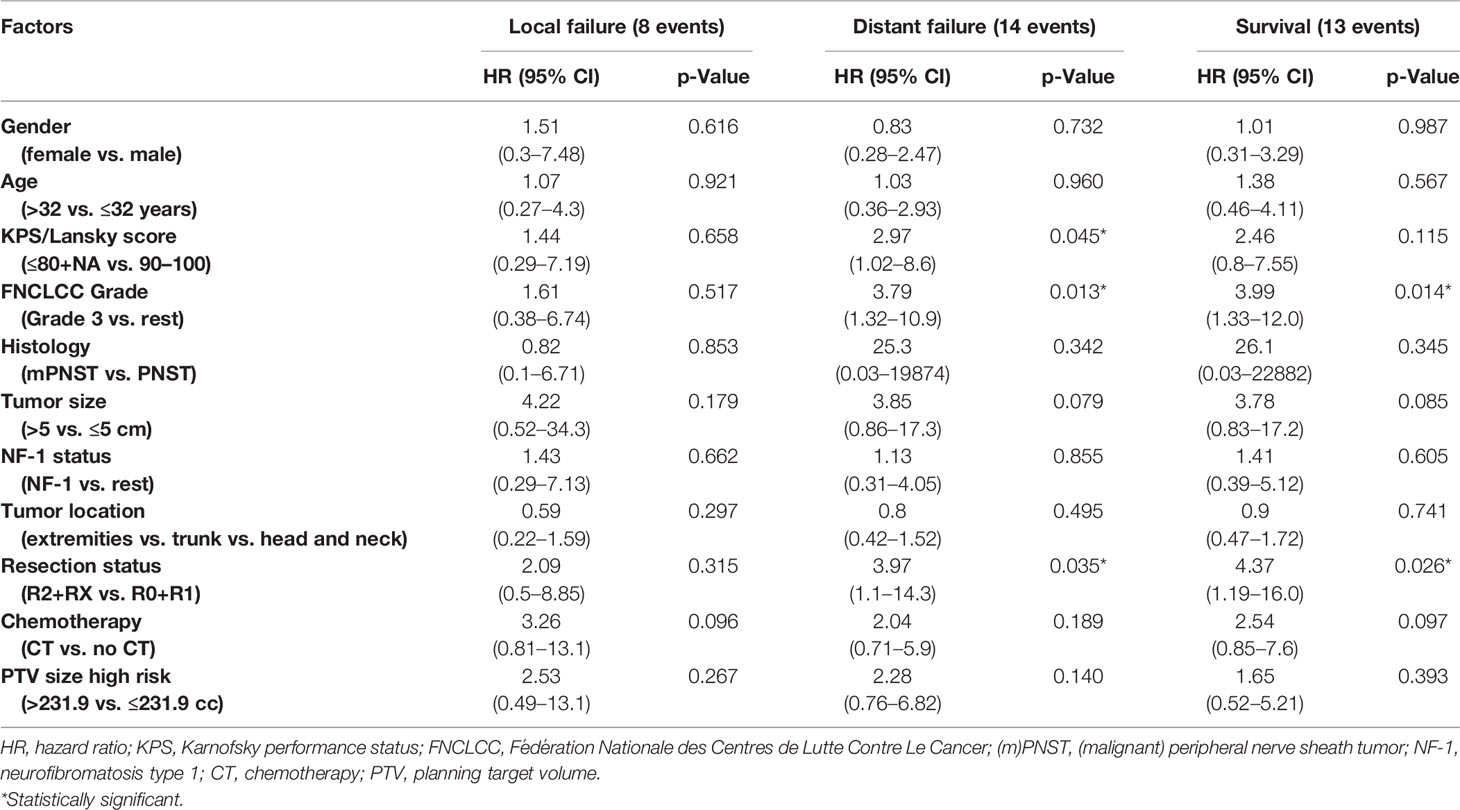
Table 3 Univariate analysis.
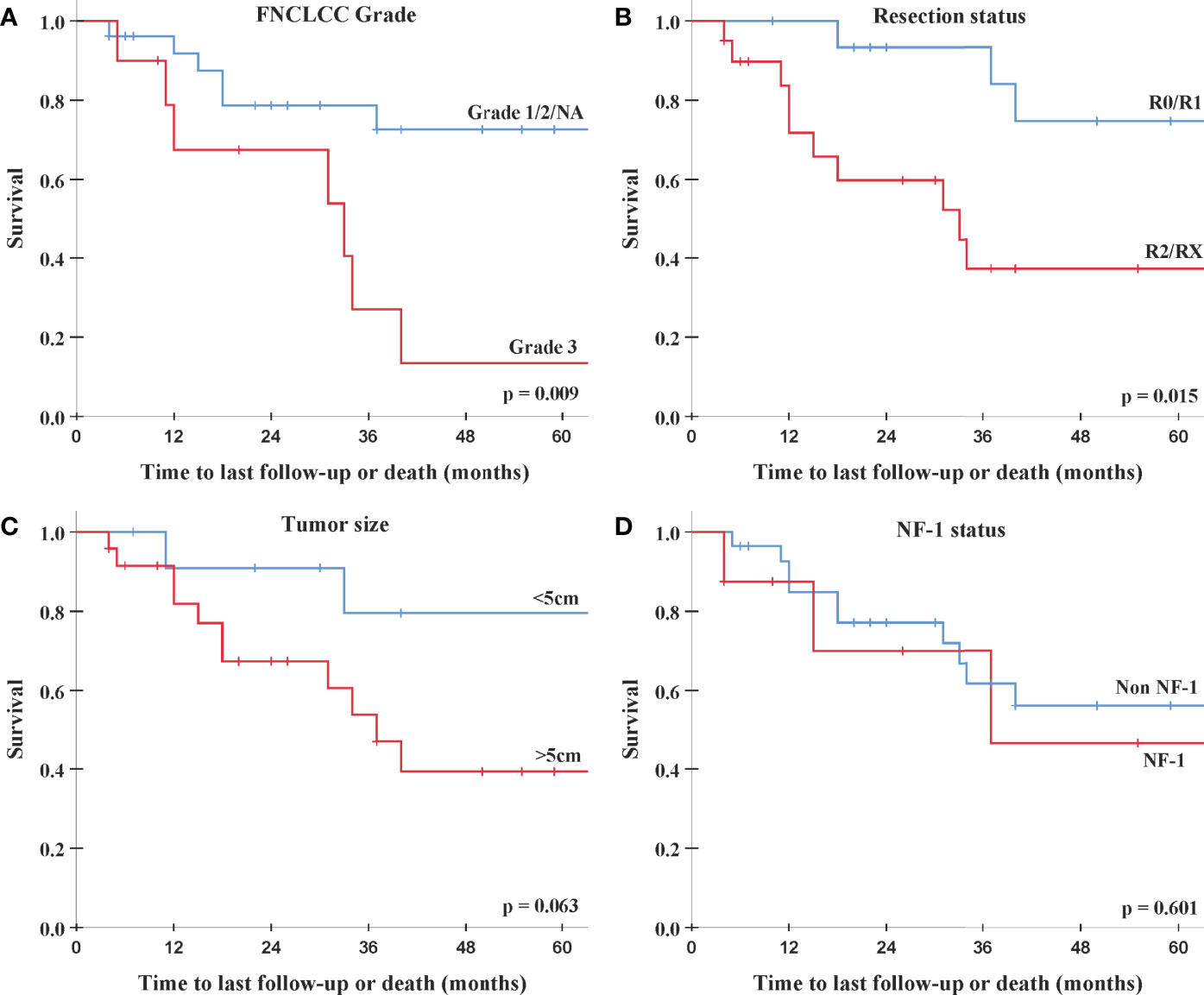
Figure 1 Log-rank analysis for overall survival. (A) FNCLCC Grade 1/2/NA (blue) vs. Grade 3 (red). (B) Resection status R0/R1 (blue) vs. R2/RX (red). (C) Tumor size <5 cm (blue) vs. >5 cm (red). (D) Non-NF-1 (blue) vs. NF-1 (red). FNCLCC, Fédération Nationale des Centres de Lutte Contre Le Cancer; NF-1, neurofibromatosis type 1.
Additionally, on univariate analysis lower performance score (KPS/Lansky ≤ 80) was significantly associated with increased DF, and patients with larger tumors (>5 cm) showed a trend toward an increased risk for DF and worse survival. NF-1 patients had a similar failure and survival rates as non-NF-1 patients (Table 3 and Figure 1).
Acute grade 1 dermatitis was the most common acute toxicity (n = 20, 56%). Acute grade 2 dermatitis, conjunctivitis, and mucositis were seen in 8 (22%), 1 (3%), and 1 (3%) patients, respectively. In 4 (11%) patients, acute grade 3 dermatitis was observed, while 1 (3%) patient presented with acute grade 3 mucositis, and 1 (3%) patient developed grade 3 pain in the irradiated extremity.
All observed late toxicities are detailed in Table 4. Overall, late toxicity was noted in 16 (44%) patients, mostly as grade 1 hyperpigmentation (n = 6, 17%). Out of these 16 patients, 5 (14%) presented with multiple late toxicities. Late grade 2 toxicity was observed in 4 (11%) patients as otitis media (n = 1, 3%), nasal crusting (n = 1, 3%), musculoskeletal deformity (n = 1, 3%), and hyperpigmentation (n = 1, 3%). Of these 4 patients, later on, one (3%) patient developed two late grade 3 toxicities (cataract and osteoradionecrosis) 34 and 194 months after PBSPT. Of note, this non-NF-1 patient was treated 11 years before PBSPT for a nasopharyngeal tumor with 70 Gy. Two-year late grade >3 toxicity rate was 0%. There was no observed grade 4 or 5 toxicity or radiation-induced secondary cancer.
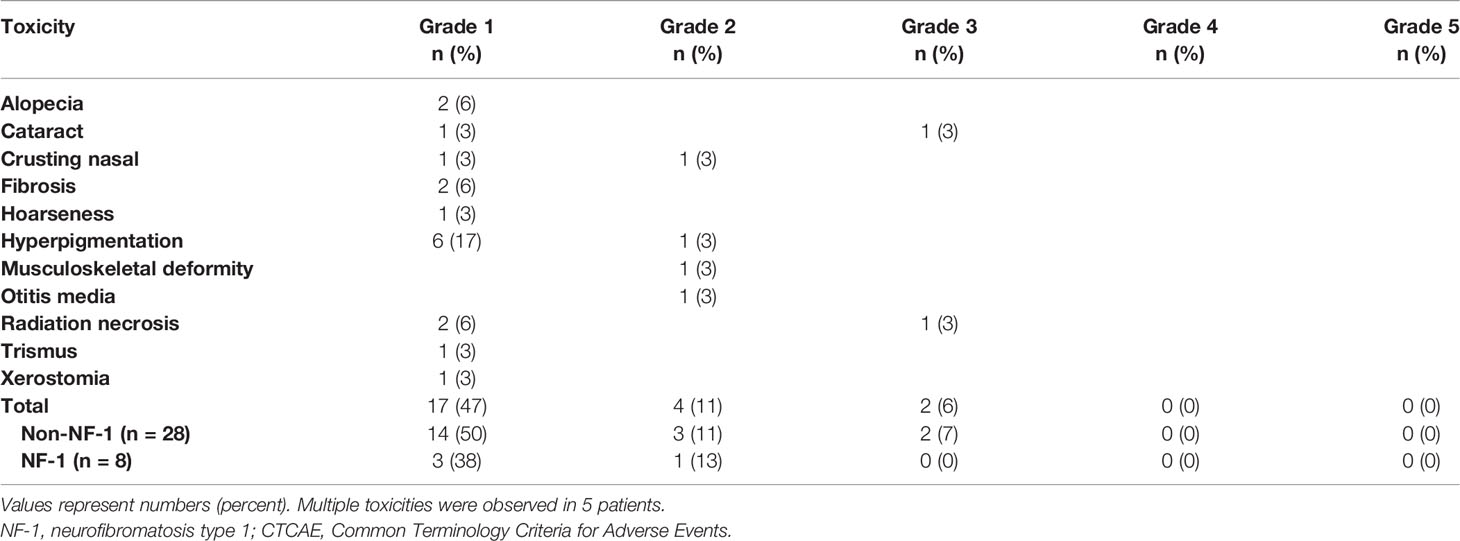
Table 4 Summary of late toxicities according to CTCAE v4.1 in alphabetical order.
The rate of any grade ≥3 acute toxicity and any late toxicity in NF-1 patients was 25% and 50% and for non-NF-1 patients 11% and 43%, respectively. These differences in toxicity rates in NF-1 and non-NF-1 patients did not translate into statistical significance using Fisher’s exact test (p = 0.305 for any grade ≥3 acute toxicity and p = 0.514 for any late toxicity).
In this study, we analyzed the clinical outcome of non-metastatic patients with PNSTs and mPNSTs treated with PBSPT. To the best of our knowledge, this is the first report of the oncological outcome and toxicity rates of mPNST/PNST patients treated with protons using a PBS delivery paradigm. Of note, two retrospective studies and two case reports analyzing mPNSTs treated with carbon ion (C12) particles were published (18–20). Jensen et al., 2015 (18) reported on the outcome and toxicity of 11 patients with unresected or incompletely resected mPNSTs treated with C12 irradiation. Patients with combined photon irradiation received 50 Gy photon intensity-modulated radiation therapy (IMRT) in 25 fractions and a 24 GyRBE boost with C12 irradiation in 8 fractions, while patients treated solely with C12 irradiation received 60–66 GyRBE in 20–22 fractions. With a median follow-up of 17 months, the authors reported a 2-year LC, progression-free survival, and survival rates of 65%, 56%, and 75%, respectively. Similar results were observed by Vitolo et al., 2019 (19) in their retrospective series of 13 patients with unresected mPNSTs treated with C12 irradiation (median dose of 73.6 GyRBE in 16 fractions) and a median follow-up of 24.6 months: reported 2-year LC and survival rates were 63% and 60%, respectively. Compatible with the C12 literature, we report in our cohort consisting mainly of patients with mPNSTs 2-year LC, DC, and OS rates of 73.5%, 61.2%, and 75.5%, respectively (Figure 1). The superior LC rate in our study is possibly due to the fact that in both C12 studies, mainly patients with gross residual tumors were included. While prospective studies examining the impact of irradiation for PNST and mPNSTs are still missing, some larger retrospective series are available. The outcome of the selected studies and the two aforementioned C12 studies is summarized in Table 5. One of the biggest series was published by Bishop et al., 2018 (22). These authors analyzed 71 mPNSTs treated with external beam radiation therapy (EBRT) either preoperatively with a median dose of 50 Gy or postoperatively with a median dose of 64 Gy. They reported excellent 5-year LC and survival rates of 84% and 66%, respectively (Table 5). However, patients with recurrent and radiation-induced tumors, which are known to have the worse outcome (1, 25–28), were excluded from their analysis. Furthermore, they found positive/uncertain surgical margin status to be adversely associated with local recurrence at 5 years (28% vs. 5% for negative margins, p = 0.02).

Table 5 Summary of studies detailing the outcome after irradiation of mPNSTs.
In our cohort, univariate analysis revealed no factor to be significantly associated with LF. Patients with FNCLCC Grade 3 mPNSTs were however significantly more likely to develop distant metastases (HR 3.79, 95% CI: 1.32–10.9, p = 0.013; Table 3) and to die (HR 3.99, 95% CI: 1.33–12.0, p = 0.014; Table 3). For patients with macroscopic or uncertain resection status (R2/RX), a similar circa 4-fold risk increase to develop DFs (HR 3.97, 95% CI: 1.1–14.3, p = 0.035; Table 3) and decrease (HR 4.37, 95% CI: 1.19–16, p = 0.026; Table 3) was observed. Furthermore, on univariate analysis, a performance status score of ≤80 was negatively associated with DF (p = 0.045), and a trend toward increased DF and worse survival was seen for tumors larger than 5 cm (see Table 3). Tumor location seemed to have no impact on outcome in our cohort. These findings are largely in line with the existing literature where higher tumor grade (21, 23, 27, 29–33), incomplete resection (11–13, 23, 26, 27, 29, 31, 34), larger tumor size (11, 12, 21, 25, 27, 30–35), truncal tumors (12, 23, 25, 26, 30) and decreased performance status (36) were shown to be associated with worse prognosis.
There is some conflicting information in the literature concerning the impact of NF-1 disorder in mPNST patients. The majority of studies report decreased survival for mPNSTs occurring in NF-1-patients (11, 13, 26, 27, 29, 30), while several other studies report similar outcomes for NF-1 associated mPNSTs (12, 25, 31, 34). More recent studies have shown that genetically NF-1 and non-NF-1 mPNSTs seem to be indistinguishable (37, 38), and so far, no determining molecular differences have been identified (31). Most authors, therefore, attribute the observed poorer outcome of NF-1-associated mPNSTs to several accompanying factors known to be associated with worse prognosis, namely, 1) mPNSTs in NF-1 patients tend to be larger in size at the time of diagnosis (13, 26, 30), 2) the tumors are often in a non-extremity location and are therefore less amenable to surgery (11, 39), 3) NF-1 patients are more likely to develop other malignancies that might impair survival (13), and finally, 4) NF-1-associated tumors seem to respond less to chemotherapy (13). In 2013, Kolberg et al. (31) published survival meta-analyses comprising >1,800 mPNSTs comparing the survival of NF-1 and non-NF-1 patients. Indeed, they found that non-NF-1 patients had significantly increased OS (HR 1.38, 95% CI: 1.1–1.72, p = 0.004) and disease-specific survival rates (HR 1.4, 95% CI: 1.13–1.75, p = 0.002). Interestingly, this observed difference in survival is diminishing when only analyzing the patients treated after the year 2000. While disease-specific survival rates remain borderline significantly increased for non-NF-1 patients (HR 1.32, 95% CI: 1.0–1.74, p = 0.05), OS for NF-1 patients treated in the last 2 decades did not differ significantly compared to non-NF-1 patients (HR 1.19, 95% CI: 0.85–1.66, p = 0.3). A similar survival improvement for more recently treated NF-1-associated mPNSTs has been observed by Ingham et al., 2011 (40). Kolberg et al. (2013) considered that increased awareness among NF-1 patients and improved monitoring routines in the last years have led to earlier detection of mPNSTs and, thus, increased survival in NF-1 patients. One might also speculate that in the past, NF-1-associated mPNSTs were treated differently and that nowadays, due to improved and safer treatment modalities, NF-1 patients benefit from the same anticancer treatment as non-NF-1 patients.
In our cohort, NF-1 disorder was not associated with worse oncological outcomes (Table 3 and Figure 1). On the one hand, only 8 (22%) patients had proven NF-1 disorder, which rendered finding a statistical difference for this sub-cohort difficult. On the other hand, 35 (97%) of our patients were treated after the year 2000 and 28 (78%) after the year 2010.
Cells from patients harboring a mutated NF-1 gene are considered to be more radiosensitive. No significant rate of relevant acute or late toxicity was observed in our NF-1 patients. In general, we report very low toxicity rates for our cohort after PBSPT. Late grade ≥2 toxicity rate was observed in 4 (11%) patients. There was no observed late grade 4 or 5 toxicity. These rates are in line with the reported toxicity in the literature, particularly when comparing PBSPT with C12 irradiation (Table 5). Jensen et al. (2015) (18) reported late grade 3 toxicity in 2 (18%) patients, and Vitolo et al. (2019) (19) observed late grade ≥2 toxicity in 2 (15%) patients. No secondary tumor after PBSPT was observed in our cohort, although the follow-up period of our series is short. Kahn et al. (2014) (23) and Sloan et al. (2018) (24) reported in their series secondary tumor rates of 15% and 13%, respectively. However, their cohorts consisted of 50% and 100% NF-1 patients, which highlights the concern of secondary tumors after irradiation for this selected patient group.
Undoubtedly, our study has certain limitations, mainly being retrospective in nature with its inherent biases. A larger sample size and a longer follow-up period would have further increased the clinical validity of this study. The small sample size of 36 patients limited the statistical power to detect associations between outcome and some of the clinical factors examined. Our inability to perform a multivariate analysis also prohibits definitive conclusions regarding the outcome and prognostic factors. Additionally, this study consists of a rather heterogeneous cohort, including patients of any age, any resection status, and any histologic grade as well as five PNST cases. However, PNSTs are known to be a precursor of mPNSTs and are sometimes difficult to distinguish from mPNSTs (2, 10, 41, 42). As such, PNSTs usually require the same multimodal treatment including surgery and irradiation due to their destructive growth.
In our cohort, consisting mainly of patients with mPNSTs, we report 2-year LC and OS rates of 73.5% and 75.5%, respectively. The majority (n = 14, 39%) of patients failed distantly, and FNCLCC grade and resection type were significantly associated with DF. Only 1 (3%) patient presented with high-grade toxicity. No difference in toxicity rates was observed between NF-1 and non-NF-1 patients. Additional PT series are needed to legitimate the administration of protons in these young challenging patients.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethikkommission Nordwest- und Zentralschweiz (2021-00369). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
NB established the concept and design of the study; acquired, analyzed, and interpreted the data; and drafted the article. DL supported the statistical analysis and critically reviewed the submitted version of the manuscript. AP critically reviewed the submitted version of the manuscript. BB critically reviewed the submitted version of the manuscript. DW established the concept and design of the study, supervised the study, interpreted the data, and critically reviewed the submitted version of the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Open access funding provided by PSI - Paul Scherrer Institute.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Widemann BC. Current Status of Sporadic and Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Curr Oncol Rep (2009) 11(4):322–8. doi: 10.1007/s11912-009-0045-z
2. Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of Peripheral Nerve Sheath Tumors: Diagnostic Overview and Update on Selected Diagnostic Problems. Acta Neuropathol (2012) 123(3):295–319. doi: 10.1007/s00401-012-0954-z
3. Bates JE, Peterson CR, Dhakal S, Giampoli EJ, Constine LS. Malignant Peripheral Nerve Sheath Tumors (MPNST): A SEER Analysis of Incidence Across the Age Spectrum and Therapeutic Interventions in the Pediatric Population. Pediatr Blood Cancer (2014) 61(11):1955–60. doi: 10.1002/pbc.25149
4. Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, et al. Malignant Peripheral Nerve Sheath Tumors. Oncologist (2014) 19(2):193–201. doi: 10.1634/theoncologist.2013-0328
5. Ng VY, Scharschmidt TJ, Mayerson JL, Fisher JL. Incidence and Survival in Sarcoma in the United States: A Focus on Musculoskeletal Lesions. Anticancer Res (2013) 33(6):2597–604.
6. Doorn PF, Molenaar WM, Buter J, Hoekstra HJ. Malignant Peripheral Nerve Sheath Tumors in Patients With and Without Neurofibromatosis. Eur J Surg Oncol J Eur Soc Surg Oncol Br Assoc Surg Oncol (1995) 21(1):78–82. doi: 10.1016/S0748-7983(05)80073-3
7. Bradford D, Kim A. Current Treatment Options for Malignant Peripheral Nerve Sheath Tumors. Curr Treat Options Oncol (2015) 16(3):328. doi: 10.1007/s11864-015-0328-6
8. Meany H, Widemann BC, Ratner N. Malignant Peripheral Nerve Sheath Tumors: Prognostic and Diagnostic Markers and Therapeutic Targets. In: Upadhyaya M, Cooper DN, Editors. Neurofibromatosis Type 1: Molecular and Cellular Biology. Berlin, Heidelberg: Springer Berlin Heidelberg (2012). p. 445–67. doi: 10.1007/978-3-642-32864-0_29
9. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (Neurilemoma) With Malignant Transformation. A Rare, Distinctive Peripheral Nerve Tumor. Am J Surg Pathol (1994) 18(9):882–95. doi: 10.1097/00000478-199409000-00003
10. Beert E, Brems H, Daniëls B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical Neurofibromas in Neurofibromatosis Type 1 Are Premalignant Tumors. Genes Chromosomes Cancer (2011) 50(12):1021–32. doi: 10.1002/gcc.20921
11. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant Peripheral Nerve Sheath Tumors. A Clinicopathologic Study of 120 Cases. Cancer (1986) 57(10):2006–21. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6
12. Wanebo JE, Malik JM, Vandenberg SR, Wanebo HJ, Driesen N, Persing JA. Malignant Peripheral Nerve Sheath Tumors. A Clinicopathologic Study of 28 Cases. Cancer (1993) 71(4):1247–53. doi: 10.1002/1097-0142(19930215)71:4<1247::aid-cncr2820710413>3.0.co;2-s
13. Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, et al. Pediatric Malignant Peripheral Nerve Sheath Tumor: The Italian and German Soft Tissue Sarcoma Cooperative Group. J Clin Oncol (2005) 23(33):8422–30. doi: 10.1200/JCO.2005.01.4886
14. Kattan MW, Leung DHY, Brennan MF. Postoperative Nomogram for 12-Year Sarcoma-Specific Death. J Clin Oncol Off J Am Soc Clin Oncol (2002) 20(3):791–6. doi: 10.1200/JCO.2002.20.3.791
15. Hannan MA, Sackey K, Sigut D. Cellular Radiosensitivity of Patients With Different Types of Neurofibromatosis. Cancer Genet Cytogenet (1993) 66(2):120–5. doi: 10.1016/0165-4608(93)90240-M
16. Ducatman BS, Scheithauer BW. Postirradiation Neurofibrosarcoma. Cancer (1983) 51(6):1028–33. doi: 10.1002/1097-0142(19830315)51:6<1028::AID-CNCR2820510610>3.0.CO;2-3
17. Kamran SC, Dworkin M, Niemierko A, Bussiere M, Oh KS, Loeffler JS, et al. Patterns of Failure Among Patients With Low-Grade Glioma Treated With Proton Radiation Therapy. Pract Radiat Oncol (2019) 9(4):e356–61. doi: 10.1016/j.prro.2019.02.002
18. Jensen AD, Uhl M, Chaudhri N, Herfarth KK, Debus J, Roeder F. Carbon Ion Irradiation in the Treatment of Grossly Incomplete or Unresectable Malignant Peripheral Nerve Sheaths Tumors: Acute Toxicity and Preliminary Outcome. Radiat Oncol (2015) 10:109. doi: 10.1186/s13014-015-0414-8
19. Vitolo V, Fiore MR, Barcellini A, Vischioni B, Iannalfi A, Facoetti A, et al. Carbon Ion Radiotherapy in the Management of the Tumors of the Peripheral Nervous System. Anticancer Res (2019) 39(2):909–13. doi: 10.21873/anticanres.13193
20. Honda A, Iizuka Y, Okamoto M, Shiba S, Koshi H, Mieda T, et al. Malignant Peripheral Nerve Sheath Tumor of the Cervical Spine Treated With Surgical Resection Followed by X-Ray Radiotherapy or Carbon Ion Radiotherapy: A Report of Three Cases. Spine Surg Relat Res (2020) 4(3):269–73. doi: 10.22603/ssrr.2019-0100
21. Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant Peripheral Nerve Sheath Tumor: Analysis of Treatment Outcome. Int J Radiat Oncol Biol Phys (1998) 42(2):351–60. doi: 10.1016/S0360-3016(98)00223-5
22. Bishop AJ, Zagars GK, Torres KE, Bird JE, Feig BW, Guadagnolo BA. Malignant Peripheral Nerve Sheath Tumors: A Single Institution’s Experience Using Combined Surgery and Radiation Therapy. Am J Clin Oncol (2018) 41(5):465–70. doi: 10.1097/COC.0000000000000303
23. Kahn J, Gillespie A, Tsokos M, Ondos J, Dombi E, Camphausen K, et al. Radiation Therapy in Management of Sporadic and Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Front Oncol (2014) 4. doi: 10.3389/fonc.2014.00324
24. Sloan L, Terezakis SA, Blakeley JO, Slobogean B, Kleinberg LR. Long-Term Outcomes of Radiation Therapy (RT) in the Management of Malignant Peripheral Nerve Sheath Tumors (MPNST) in Patients With Neurofibromatosis Type 1 (Nf1). Int J Radiat Oncol (2018) 102(3):e474–5. doi: 10.1016/j.ijrobp.2018.07.1357
25. Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, et al. Malignant Peripheral Nerve Sheath Tumors: Prognostic Factors and Survival in a Series of Patients Treated at a Single Institution. Cancer (2006) 107(5):1065–74. doi: 10.1002/cncr.22098
26. Watson KL, Al Sannaa GA, Kivlin CM, Ingram DR, Landers SM, Roland CL, et al. Patterns of Recurrence and Survival in Sporadic, Neurofibromatosis Type 1-Associated, and Radiation-Associated Malignant Peripheral Nerve Sheath Tumors. J Neurosurg (2017) 126(1):319–29. doi: 10.3171/2015.12.JNS152443
27. Miao R, Wang H, Jacobson A, Lietz AP, Choy E, Raskin KA, et al. Radiation-Induced and Neurofibromatosis-Associated Malignant Peripheral Nerve Sheath Tumors (MPNST) Have Worse Outcomes Than Sporadic MPNST. Radiother Oncol J Eur Soc Ther Radiol Oncol (2019) 137:61–70. doi: 10.1016/j.radonc.2019.03.015
28. Yamanaka R, Hayano A. Radiation-Induced Malignant Peripheral Nerve Sheath Tumors: A Systematic Review. World Neurosurg (2017), 105:961–970.e8. doi: 10.1016/j.wneu.2017.06.010
29. Loree TR, North JHJ, Werness BA, Nangia R, Mullins AP, Hicks WLJ. Malignant Peripheral Nerve Sheath Tumors of the Head and Neck: Analysis of Prognostic Factors. Otolaryngol Neck Surg Off J Am Acad Otolaryngol Neck Surg (2000) 122(5):667–72. doi: 10.1016/S0194-5998(00)70193-8
30. Stucky CCH, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, et al. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann Surg Oncol (2012) 19(3):878–85. doi: 10.1245/s10434-011-1978-7
31. Kolberg M, Høland M, Agesen TH, Brekke HR, Liestøl K, Hall KS, et al. Survival Meta-Analyses for >1800 Malignant Peripheral Nerve Sheath Tumor Patients With and Without Neurofibromatosis Type 1. Neuro Oncol (2013) 15(2):135–47. doi: 10.1093/neuonc/nos287
32. Lucas C-HG, Vasudevan HN, Chen WC, Magill ST, Braunstein SE, Jacques L, et al. Histopathologic Findings in Malignant Peripheral Nerve Sheath Tumor Predict Response To Radiotherapy and Overall Survival. Neuro-oncology Adv (2020) 2(1):vdaa131. doi: 10.1093/noajnl/vdaa131
33. Okada K, Hasegawa T, Tajino T, Hotta T, Yanagisawa M, Osanai T, et al. Clinical Relevance of Pathological Grades of Malignant Peripheral Nerve Sheath Tumor: A Multi-Institution TMTS Study of 56 Cases in Northern Japan. Ann Surg Oncol (2007) 14(2):597–604. doi: 10.1245/s10434-006-9053-5
34. LaFemina J, Qin L-X, Moraco NH, Antonescu CR, Fields RC, Crago AM, et al. Oncologic Outcomes of Sporadic, Neurofibromatosis-Associated, and Radiation-Induced Malignant Peripheral Nerve Sheath Tumors. Ann Surg Oncol (2013) 20(1):66–72. doi: 10.1245/s10434-012-2573-2
35. Mowery A, Clayburgh D. Malignant Peripheral Nerve Sheath Tumors: Analysis of the National Cancer Database. Oral Oncol (2019) 98:13–9. doi: 10.1016/j.oraloncology.2019.09.010
36. Kroep JR, Ouali M, Gelderblom H, Le Cesne A, Dekker TJA, Van Glabbeke M, et al. First-Line Chemotherapy for Malignant Peripheral Nerve Sheath Tumor (MPNST) Versus Other Histological Soft Tissue Sarcoma Subtypes and as a Prognostic Factor for MPNST: An EORTC Soft Tissue and Bone Sarcoma Group Study. Ann Oncol Off J Eur Soc Med Oncol (2011) 22(1):207–14. doi: 10.1093/annonc/mdq338
37. Watson MA, Perry A, Tihan T, Prayson RA, Guha A, Bridge J, et al. Gene Expression Profiling Reveals Unique Molecular Subtypes of Neurofibromatosis Type I-Associated and Sporadic Malignant Peripheral Nerve Sheath Tumors. Brain Pathol (2004) 14(3):297–303. doi: 10.1111/j.1750-3639.2004.tb00067.x
38. Brekke HR, Ribeiro FR, Kolberg M, Agesen TH, Lind GE, Eknaes M, et al. Genomic Changes in Chromosomes 10, 16, and X in Malignant Peripheral Nerve Sheath Tumors Identify a High-Risk Patient Group. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28(9):1573–82. doi: 10.1200/JCO.2009.24.8989
39. Sordillo PP, Helson L, Hajdu SI, Magill GB, Kosloff C, Golbey RB, et al. Malignant Schwannoma–Clinical Characteristics, Survival, and Response to Therapy. Cancer (1981) 47(10):2503–9. doi: 10.1002/1097-0142(19810515)47:10<2503::AID-CNCR2820471033>3.0.CO;2-3
40. Ingham S, Huson SM, Moran A, Wylie J, Leahy M, Evans DGR. Malignant Peripheral Nerve Sheath Tumours in NF1: Improved Survival in Women and in Recent Years. Eur J Cancer (2011) 47(18):2723–8. doi: 10.1016/j.ejca.2011.05.031
41. Benz MR, Czernin J, Dry SM, Tap WD, Allen-Auerbach MS, Elashoff D, et al. Quantitative F18-Fluorodeoxyglucose Positron Emission Tomography Accurately Characterizes Peripheral Nerve Sheath Tumors as Malignant or Benign. Cancer (2010) 116(2):451–8. doi: 10.1002/cncr.24755
42. Meany H, Dombi E, Reynolds J, Whatley M, Kurwa A, Tsokos M, et al. 18-Fluorodeoxyglucose-Positron Emission Tomography (FDG-PET) Evaluation of Nodular Lesions in Patients With Neurofibromatosis Type 1 and Plexiform Neurofibromas (PN) or Malignant Peripheral Nerve Sheath Tumors (MPNST). Pediatr Blood Cancer (2013) 60(1):59–64. doi: 10.1002/pbc.24212
Keywords: malignant peripheral nerve sheath tumors, proton therapy, pencil beam scanning, adolescents and young adults, neurofibromatosis type 1, benign peripheral nerve sheath tumors
Citation: Bachmann N, Leiser D, Pica A, Bachtiary B and Weber DC (2022) Clinical Outcome After Pencil Beam Scanning Proton Therapy of Patients With Non-Metastatic Malignant and Benign Peripheral Nerve Sheath Tumors. Front. Oncol. 12:881665. doi: 10.3389/fonc.2022.881665
Received: 22 February 2022; Accepted: 20 May 2022;
Published: 27 June 2022.
Edited by:
Susanne Rogers, Aarau Cantonal Hospital, SwitzerlandReviewed by:
Rishan Thimma Sudarsan, Apollo Proton Cancer Centre, IndiaCopyright © 2022 Bachmann, Leiser, Pica, Bachtiary and Weber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Damien C. Weber, ZGFtaWVuLndlYmVyQHBzaS5jaA==; ZGFtaWVuY2hhcmxlcy53ZWJlckB1emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.