 Qiuyu Wu1,2†
Qiuyu Wu1,2† Zhichao Zheng3†
Zhichao Zheng3† Junwei Zhang1†Zhengguo Piao1Mengyu Xin3Xi Xiang3Antong Wu3Tianyu Zhao1Songkai Huang1Yu Qiao1Jiayu Zhou1
Junwei Zhang1†Zhengguo Piao1Mengyu Xin3Xi Xiang3Antong Wu3Tianyu Zhao1Songkai Huang1Yu Qiao1Jiayu Zhou1 Shaofen Xu3
Shaofen Xu3 Haoyu Cheng3
Haoyu Cheng3 Lihong Wu3*
Lihong Wu3* Kexiong Ouyang1*
Kexiong Ouyang1*- 1Department of Oral and Maxillofacial Surgery, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China
- 2Jiangmen Central Hospital, Affiliated Jiangmen Hospital of Sun Yat-sen University, Jiangmen, China
- 3Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China
Background: Accumulating evidence suggests that dysregulation of Chordin-like 1 (CHRDL1) is associated with malignant biological behaviors in multiple cancers. However, the exact function and molecular mechanism of CHRDL1 in oral squamous cell carcinoma (OSCC) remain unclear.
Methods: The expression levels of CHRDL1 in OSCC tissues and CAL27 cells were determined by RT-qPCR. Immunohistochemical staining was applied to detect CHRDL1 protein expression in sample tissues from OSCC patients. Gain of function and knockdown by lentivirus were further used to examine the effects of CHRDL1 on cell proliferation, migration, invasion, and adhesion in OSCC. Tail vein injection of CAL27 cells with dysregulated CHRDL1 expression was further used to examine the effect of CHRDL1 on lung colonization. RNA sequencing was performed to explore the molecular mechanisms of CHRDL1 that underlie the progression of OSCC.
Results: CHRDL1 was significantly downregulated in OSCC tissues and CAL27 cells compared to controls. CHRDL1 knockdown enhanced migration, invasion, adhesion, and EMT, but not proliferation, in CAL27 cells. Overexpression of CHRDL1 had the opposite effects. Moreover, CHRDL1 was proven to inhibit tumor metastasis in vivo. Mechanistically, MAPK signaling pathway components, including ERK1/2, p38, and JNK, were found to regulate the malignant biological behaviors of CAL27 cells.
Conclusions: Our results suggest that CHRDL1 has an inhibitory effect on OSCC metastasis via the MAPK signaling pathway, which provides a new possible potential therapeutic target against OSCC.
Introduction
Oral squamous cell carcinoma (OSCC) is one of the most common lethal malignancies worldwide, with poor prognosis (1). It is reported that approximately 377,713 cases of oral cancer are newly diagnosed each year, accounting for approximately 2% of all malignancies reported worldwide. The latest data indicate that the overall number of cases will even rise to 510,948 in 2035 (2). The traditional treatments for OSCC are surgical resection, radiotherapy, or chemotherapy or their combination. Although reconstructive surgery of the lesion is always applied to restore appearance and function, the side effects of surgical resection, radiotherapy, or chemotherapy persistently and irreversibly reduce patient quality of life. Recent research has shown an improvement in the survival rates of OCSS patients from 55% in 1986 to 60% in 2003 (3). However, the improvement in the survival rate of OSCC has been much smaller than that for other cancer types, such as breast cancers and colon cancers. It is widely accepted that the accumulation of genetic alterations in oncogenes and tumor suppressors leads to the emergence of OSCC (4, 5). The potential pathogenesis, including the progression mechanism, of OSCC is not fully understood.
Bone morphogenetic proteins (BMPs), belonging to the TGF-β family (6), play an important role in early embryonic development and homeostasis, including bone formation and regeneration (7). Recently, several studies have indicated that several BMPs, such as BMP-2, BMP-4, BMP-6, and BMP-7, are also involved in tumor development and progression (8, 9). Studies have shown that BMP-2 has a positive effect on the invasion ability of oral, gastric, breast, colon, bladder, and pancreatic cancers (10–12). BMP-2 induced the invasion of OSCC cells, possibly through CCL5 release, in coculture models (11). BMP-9 was reported to stimulate the proliferation of ovarian cancer cells (13). Rothhammer et al. demonstrated that BMP-4 promotes melanoma cell invasion and migration (14). In OSCC, BMP-7 was found to be the key to the acquisition of cetuximab resistance (15).
Compared with the fully elucidated role of BMPs in tumors, research on their antagonists in tumors is relatively limited. Chordin like-1 (CHRDL1), a BMP antagonist, was originally reported to be expressed in the developing retina (16). Mutation of CHRDL1 is related to X-linked megalocornea, an ocular anterior segment disorder (17–20). Recent studies have associated CHRDL1 with various cancers, including breast cancer (21–24), lung cancer (25–28), gastric cancer (29, 30), thyroid cancer (31–34), T-cell acute lymphoblastic leukemia (T-ALL) (35), melanoma (36), and OSCC (37). CHRDL1 was proven to block BMP-induced increases in breast cancer cell migration and invasion. High CHRDL1 expression is associated with better clinical outcomes in patients with breast cancer (21). Furthermore, CHRDL1 expression is significantly downregulated in gastric cancer tissues and associated with poor survival, and CHRDL1 knockdown promotes tumor cell proliferation and migration through BMPR II by activating Akt, Erk, and β-catenin (29). In melanoma, CHRDL1 showed growth-suppressing properties in melanoma-derived cell lines with DNA methylation and genomic deletion (36). However, most of these studies provided only correlation analyses of CHRDL1 with different tumors. The specific role of CHRDL1 in cancers should be further explored. Moreover, the function and regulatory mechanism of CHRDL1 in OSCC have not been studied.
Here, we aimed to identify the role of CHRDL1 in OSCC and to explore the relevant mechanisms, which may provide a new potential target for OSCC therapy.
Methods and Materials
Clinical Specimen Collection and Immunohistochemistry Staining for CHRDL1
The studies involving human participants were approved by the Ethics Committee of the Stomatology Hospital of Guangzhou Medical University (Approval No. KY2019026). Thirty primary OSCC specimens and adjacent tissue specimens were obtained from the Department of Oral and Maxillofacial Surgery, Stomatology Hospital of Guangzhou Medical University. All OSCC specimens were pathologically diagnosed as OSCC, and the adjacent tissue specimens were obtained ≥2 cm from the resection margin, which were pathologically diagnosed to have no precancerous or reactive changes histologically. All specimens were frozen immediately in liquid nitrogen after resection.
Immunohistochemical staining (IHC) was performed on 5 human OSCC tumor tissues and paired adjacent specimens. Paraffin-embedded tissues were cut into 4-µm sections. The samples were dewaxed, dehydrated, and then treated with 3% H2O2 solution to inhibit the endogenous peroxidase activity. Antigen retrieval was carried out by microwave heating in sodium citrate buffer (pH 7.8). Slides were then blocked with 5% BSA solution. Tissue sections were further incubated with primary antibody against CHRDL1 (1:150 dilution, Zen-Bio) at 4°C for 12 h and then washed and incubated with secondary antibody (1:100 dilution, ZSJB-Bio) at 37°C for 1 h. Finally, slides were incubated with diaminobenzidine (DAB, ZSJB-Bio) and counterstained with hematoxylin.
IHC slices were scanned by a digital tissue slice scanner (3D HISTECH, Magyarország). The Servicebio image analysis system was used to score the slides for positivity. Positivity grading was first performed (I) as follows: negative without staining, 0 points; weak positive/light yellow, 1 point; medium positive/brownish yellow, 2 points; strong positive/brown, 3 points; then, the cumulative weak, medium, and strong positive area, tissue area, and integrated optical density (IOD) value of positive in the measurement area were calculated. The histochemistry score (H-score) is the output of a histological scoring method for immunohistochemistry. The number of positive cells in each section and their staining intensity were converted into corresponding values for semiquantification of the tissue staining, as follows: H-Score = ∑(pi×i) = (percentage of weak intensity area ×1) + (percentage of moderate intensity area ×2) + (percentage of strong intensity area ×3), where pi represents the percentage of the pixel area of positive signal, and i represents positive grade (38, 39).
Cell Line Acquisition and Culture
The human tongue squamous cell carcinoma cell line CAL27 (ATCC, ATCC® CRL-2095™) was purchased from ATCC, while the normal oral epithelial keratinocyte line HOK was purchased from AULU (Guangdong, China). CAL27 and HOK cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Waltham, MA, USA). The complete cell culture medium contained 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Gibco). The serum-free cell culture medium for carcinoma cells was prepared as DMEM containing 1% penicillin/streptomycin for a number of subsequent experiments (e.g., wound-healing assay, cell migration, and invasion assays). The cells were cultured in a humidified incubator at 37°C and 5% CO2 incubator.
Overexpression/Knockdown of CHRDL1 in CAL27 by Lentiviral Transfection
Commercial lentivirus (lenti-CHRDL1 and shRNA-CHRDL1; OBIO, Shanghai, China) was utilized to overexpress or silence CHRDL1. An empty carrier lentivirus (lenti-NC/shRNA-NC) was used as a negative control. CAL27 cells were infected with the lentivirus (MOI = 60) and screened with puromycin (2 µg/ml) for 15 days. Quantitative reverse transcription-polymerase chain reaction (RT-qPCR) and Western blotting (WB) were used to validate the overexpression/knockdown level of CHRDL1.
RNA Isolation and Quantitative Real-Time PCR
Total RNA from the clinical specimens was extracted using TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions. Total RNA from the cells was extracted using an RNA extraction kit (19221ES50, Yeasen, China). The quantity and quality of RNAs were detected by A260/A280 with a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, Waltham, MA, USA). Two micrograms of total RNA was used to synthesize cDNA (11141ES60, Yeasen, China). RT-qPCR was performed using a SYBR Green qPCR kit (11199ES08, Yeasen, China). Relative mRNA expression was normalized to that of the internal GAPDH control. The primer sequences are listed in Supplementary Table S1. The relative expression of targeted genes was calculated by the 2−ΔΔCt method. Each test was repeated at least three times.
Western Blot Analysis
Total proteins were isolated from cell samples through precooled cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) with protease inhibitor and phosphatase inhibitors (RayBiotech, Guangzhou, China) and quantified by a BCA protein assay kit (SE248351, Thermo Fisher, USA). Equal protein extracts (30 µg) were separated by SDS–PAGE (PG112, Epizyme, China) and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA). After incubation with the primary antibody and the secondary antibody, the target protein was visualized by chemiluminescence using an ECL kit (P001AS, Beyotime, China). The antibodies used in the Western blot assay are listed as follows: CHRDL1 (1:500, Thermo Fisher, USA), Fibronectin (1:1,000, Proteintech, China), E-cadherin (1:1,000, Proteintech, China), E-cadherin (1:1,000, Proteintech, China), Cytokeratin 18 (1:1,000, Abcam, UK), β-actin (1:1,0000, Cell Signaling Technology, USA), JNK (1:1,000, Cell Signaling Technology, USA), p-JNK (1:1,000, Cell Signaling Technology, USA), ERK1/2 (1:1,000, Cell Signaling Technology, USA), p-ERK1/2 (1:1,000, Cell Signaling Technology, USA), p38 (1:1,000, Cell Signaling Technology, USA), p-p38 (1:1,000, Cell Signaling*nbsp;Technology, USA), and goat anti-rabbit IgG H&L (HRP) antibody (1:10,000, Abcam, UK). β-actin served as the internal control to calculate the relative expression of the targeted proteins.
Wound-Healing Assay
For the wound-healing assay, transfected CAL27 cells were first incubated and cultured with complete medium. A scratch was made after a confluent monolayer of cells was formed. Afterward, the cells were washed with phosphate-buffered saline (PBS) and cultured with serum-free medium. Images were taken at 0 and 24 h after wound generation. The wound-healing areas were assessed by ImageJ to calculate the wound-healing rate. Wound-healing rate% = [Area at 0 – Area at 1]/Area at 0 × 100% (Area at 0 is the area of the wound measured immediately after scratching, and Area at 1 is the area of the wound measured at 1 h after scratching). The wound area was measured at 0 and 24 h. Each test was repeated at least three times.
Cell Migration Assay
CAL27 cells were seeded at a density of 1 × 105 in serum-free DMEM in the upper wells of Transwell chambers (8 μm pore size, Corning, New York, NY, USA), while the lower wells were filled with complete medium containing 20% FBS. After 48 h, cells in the upper layer were removed with a swab. The cells on the bottom membrane were fixed with 4% paraformaldehyde and stained with crystal violet. Five random visual fields were photographed for each group, and the experiments were repeated three times.
Cell Invasion Assay
CAL27 cells were seeded at a density of 2 × 105 in serum-free DMEM in the upper wells of chambers that were coated with 60 µl of Matrigel (200 μg/µl, Corning) and incubated for 2 h at 37°C, while the lower wells were filled with complete medium containing 20% FBS. Cells in the upper layer were removed with a swab after 36 h of culture, and cells on the bottom membrane were fixed with 4% paraformaldehyde and stained with crystal violet. Each group was photographed in five randomized visual fields, and the experiments were repeated three times.
Cell Adhesion Assay
A Cell Adhesion Detection kit (BB-48120, BestBio Science, China) was used according to the manufacturer’s instructions to analyze the adhesion ability of CAL27 cells. Briefly, the coating buffer (100 µl) was transferred to each well in a 96-well plate and incubated overnight at 4°C. Cells (5×104) were inoculated into each well of the coated 96-well plate and allowed to adhere for 1.5 h at 37°C. After the cells were washed three times with PBS, cell imaging was performed. The optical density value at 450 nm was measured to calculate the cell adhesion rate. Each test was repeated at least three times.
Rhodamine-Conjugated Phalloidin Staining and Confocal Microscopy
CAL27 cells were seeded at a density of 1,000 in a confocal dish. After 24 h, cell monolayers were fixed with 4% paraformaldehyde and stained with rhodamine-conjugated phalloidin (40737ES75, Yeasen, China) according to the manufacturer’s instructions. Confocal images were captured using a confocal laser scanning microscope (LEIKATCSSP8, Leica, Germany).
mRNA Sequencing
Total RNA was extracted from transfected CAL27 cells (OV-CHRDL1/OV-NC and sh1-CHRDL1/sh-NC) using TRIzol reagent (Invitrogen Life Technologies, USA) following the manufacturer’s protocol. RNA concentration and purity were checked with a Nanodrop 2000 instrument (Thermo Fisher Scientific, USA). After RNA quality control was performed, the libraries for next-generation sequencing were prepared using the TruSeqTM RNA Sample Prep Kit (Illumina, USA) according to the manufacturer’s instructions. Sequencing was performed by Shanghai Origingene Biopharm Technology Co., Ltd. (Shanghai, China). After quality control of the original data, the high-quality sequencing data were compared with the designated reference genome. The expression values were calculated by the StringTie tool, and the tDESeq algorithm was applied to filter the differentially expressed genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to reveal the involved pathways.
Lung Metastasis in Nude Mice
The animal experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Guangzhou Medical University of China (Approval number was 2017-086).
Four-week-old male BALB/c-nude mice were purchased from GemPharmatech (Jiangsu, China). All animal experiments were carried out following the guidelines set by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Guangzhou Medical University of China. Nude mice were randomly divided into four groups. CAL27 cells (1×106) were suspended in 0.1 ml PBS and injected via the tail vein into animals that had been anesthetized with 3% isoflurane. After 9 weeks, the nude mice were anesthetized and euthanized by cervical dislocation. Lungs were collected, weighed, rinsed, fixed, and embedded in paraffin blocks and sectioned for H&E staining to confirm metastasis. Three slices were acquired from each lung sample. The number of pulmonary tumor nodules were counted under the dissecting microscope. The averaged lung colonies from 3 slices were calculated for each lung sample.
Cell Proliferation Assay
Cells (4,000) were cultured in 96-well plates, and cell proliferation was detected using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) at 1, 3, and 5 days of culture according to the manufacturer’s instructions. EdU staining was performed to assess cell proliferation ability by the Cell-Light EdU imaging kit (C0071S, Beyotime, China) following the manufacturer’s instructions. A colony formation assay was performed. One thousand transfected CAL27 cells were seeded in six-well plates and cultured for 8 days in complete medium. The colonies were further fixed with 4% paraformaldehyde and stained with crystal violet. Colonies that contained more than 50 cells were counted. Each test was repeated at least three times.
Statistical Analysis
The data were analyzed using statistical software (SPSS Statistics 16.0). The data are presented as the means and standard deviations. Student’s t-test was used for comparisons of normally distributed data. The Mann–Whitney U test was used for nonnormally distributed data. A p-value <0.05 was considered statistically significant.
Results
CHRDL1 Is Downregulated in OSCC Both In Vivo and In Vitro
The data from the Gene Expression Profiling Interactive Analysis database indicated that the expression of CHRDL1 was downregulated in HNSC tissues compared to normal tissues (Figure 1A). We further collected samples from 30 OSCC patients, and qRT-PCR indicated that the expression of CHRDL1 was significantly decreased in OSCC tissues compared to adjacent tissues (Figure 1B). A lower expression level of CHRDL1 was also detected in the common OSCC cell line CAL27 than in the normal cell line HOK (Figure 1C). IHC analysis of tissue samples showed that CHRDL1 was downregulated in OSCC tissues. Representative IHC images revealed that CHRDL1 protein was primarily distributed in the epithelial structure of OSCC tissues and adjacent tissues. However, there was little staining of CHRDL1 in either the tumor stroma in OSCC or subepithelial tissues in the adjacent normal tissues (Figures 1D, E). These data demonstrated that CHRDL1 was typically negatively correlated with OSCC development.
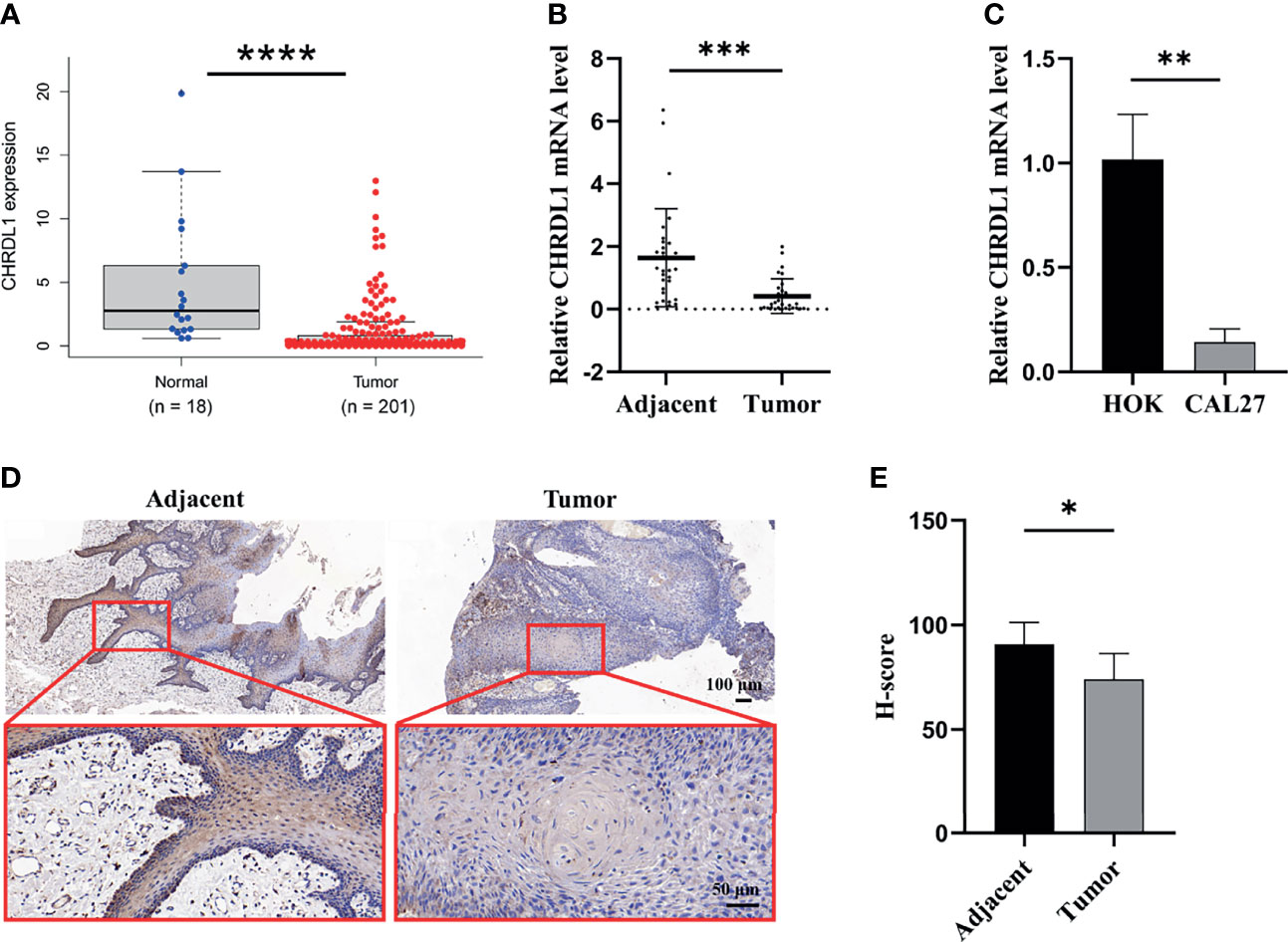
Figure 1 The expression of CHRDL1 was downregulated in OSCC tissue and cell lines. (A) Relative mRNA expression of CHRDL1 in OSCC tissues (n = 201) and normal tissues (n = 18) from the TCGA database. (B) The relative mRNA expression of CHRDL1 in 30 specimens of OSCC and adjacent tissues was determined by qRT-PCR. (C) The relative mRNA expression of CHRDL1 in normal human oral keratinocytes (HOK) and the OSCC cell line CAL27 was determined by RT-qPCR. CHRDL1 mRNA expression was normalized to GAPDH. (D, E) IHC analysis of CHRDL1 in tumor and adjacent tissues (n = 5). Error bars represent the standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Overexpression of CHRDL1 Inhibits Malignant Biological Behaviors of CAL27 Cells
The role of CHRDL1 in the progression of OSCC was explored in the CAL27 cell line. CHRDL1 was overexpressed in the CAL27 cell line by lentivirus infection (Figures 2A–C). Overexpression of CHRDL1 markedly suppressed the wound-healing and migration abilities of CAL27 cells (Figures 2D–G). Furthermore, CHRDL1 overexpression significantly decreased the number of invading CAL27 cells (Figures 2F, G). The cell adhesion assay showed a lower adhesion rate in the CHRDL1 overexpression (OV-CHRDL1) group than in the control group (NC-CHRDL1) (Figures 2H, I). However, the EdU assay, colony formation assay, and CCK-8 assay showed no significant difference between the OV-CHRDL1 group and the NC-CHRDL1 group, indicating that overexpression of CHRDL1 did not affect the proliferation of CAL27 cells (Supplementary Figure 1). These results suggested that upregulation of CHRDL1 inhibited metastasis rather than proliferation in the CAL27 cell line.
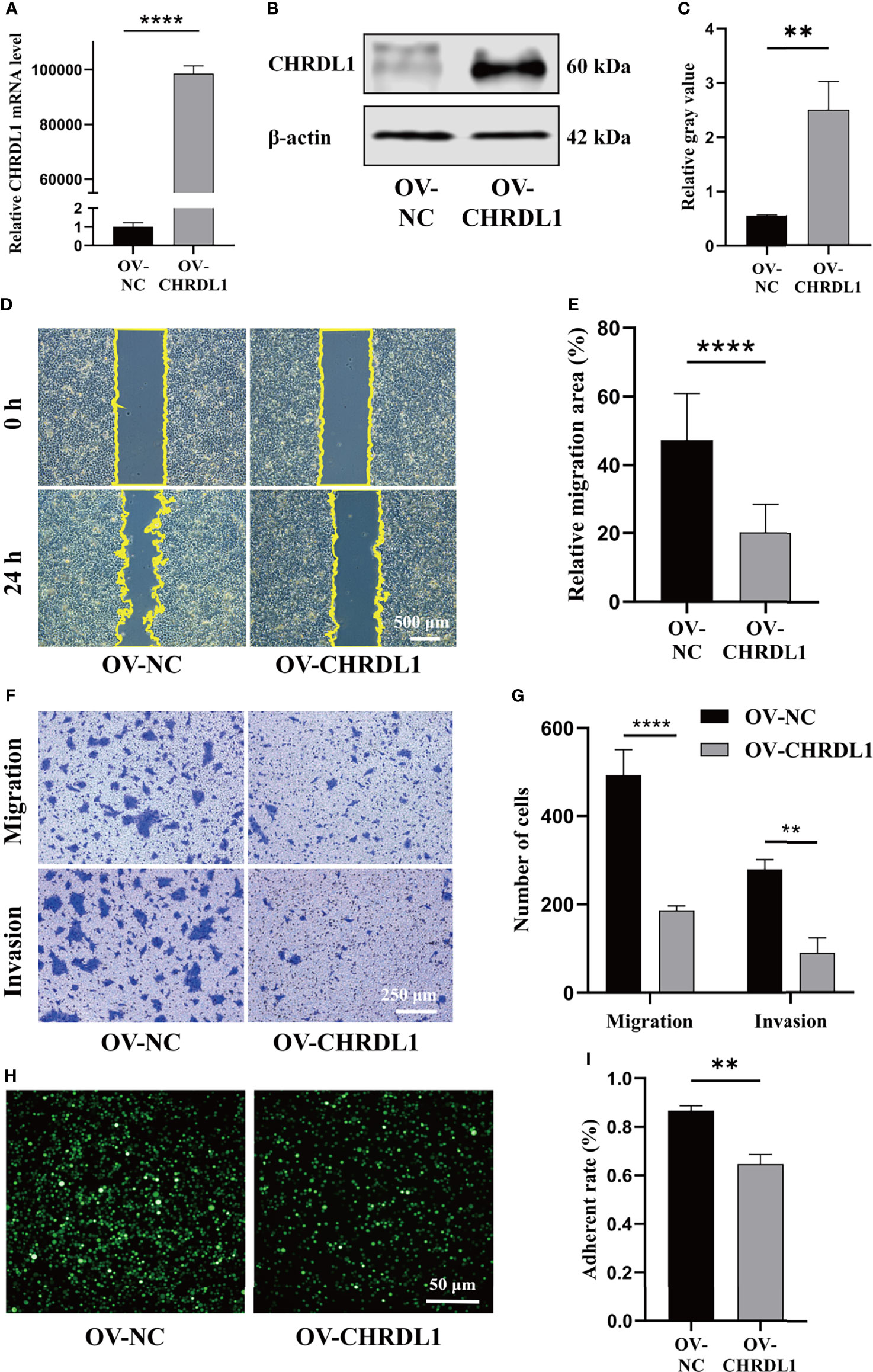
Figure 2 Overexpression of CHRDL1 inhibits CAL27 cell migration, invasion, and adhesion. (A) Relative mRNA expression of CHRDL1 in CAL27 cells after lentivirus transfection. (B, C) CHRDL1 protein expression in CAL27 cells after lentivirus transfection was identified by Western blot and quantitatively analyzed. (D, E) Representative images of wound-healing assays of transfected CAL27 cells at 0 and 24 h and statistical results from each group. (F, G) Migration and invasion assays of transfected CAL27 cells. Representative images of cell migration and invasion in each group are shown in (F). Statistical analyses of cell migration and invasion are shown in (G). (H, I) Cell adhesion assay of transfected CAL27 cells. Representative fluorescence images from each group (H) and statistical analyses of the adherence rate (I). Error bars represent the standard deviation. **p < 0.01, ***p < 0.001.
CHRDL1 Knockdown Promotes Malignant Biological Behaviors in CAL27 Cells
To determine whether silencing CHRDL1 promoted malignant biological behaviors in the CAL27 cell line, two stable CHRDL1-knockdown CAL27 cell lines (sh1-CHRDL1 and sh2-CHRDL1) and their control group (sh-NC) were established. Although CHRDL1 mRNA levels were significantly decreased in both stable CHRDL1-knockdown cell lines (Figure 3A), the protein level was reduced only in the sh1-CHRDL1 cell line and was obviously increased in the sh2-CHRDL1 cell line (Figure 3B, C). Thus, the sh1-CHRDL1 CAL27 cell line was used for the following experiments. We demonstrated that CHRDL1 silencing markedly promoted the wound-healing and migration abilities of CAL27 cells (Figures 3D–G). The sh1-CHRDL1 CAL27 cells also exhibited enhanced invasion (Figures 3D–G) and adhesion ability (Figures 3H, I) compared to sh-NC CAL27 cells. Intriguingly, proliferation assays also showed no significant difference between the sh1-CHRDL1 group and the sh-NC group (Supplementary Figure 1). These findings confirmed that CHRDL1 silencing promoted the progression of CAL27 cells.
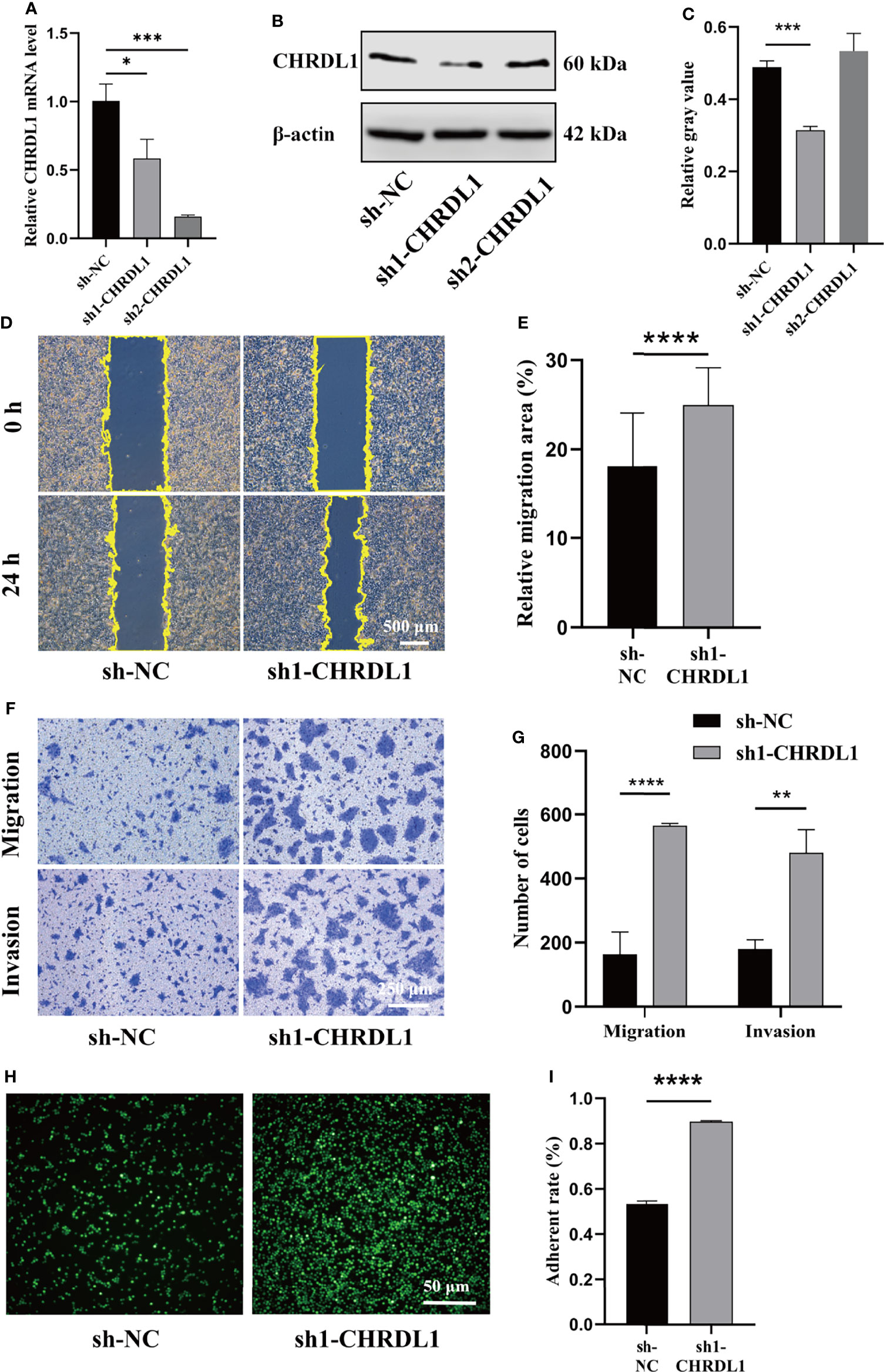
Figure 3 CHRDL1 knockdown promotes CAL27 cell migration, invasion, and adhesion. (A) Relative mRNA expression of CHRDL1 in CAL27 cells after lentivirus transfection. (B, C) CHRDL1 protein expression in CAL27 cells after lentivirus transfection was evaluated by Western blot and quantitatively analyzed. (D, E) Representative images of wound-healing assays of transfected CAL27 cells at 0 and 24 h and statistical analyses of each group. (F, G) Migration and invasion assays of transfected CAL27 cells. Representative images of cell migration and invasion in each group are shown in (F). Statistical analyses of cell migration and invasion are shown in (G). (H, I) Cell adhesion assay of transfected CAL27 cells. Representative fluorescence images of each group (H) and statistical analyses of the adherence rate (I). Error bars represent the standard deviation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CHRDL1 Knockdown Induces the Epithelial–Mesenchymal Transition Phenotype in CAL27 Cells
EMT has been widely reported to increase OSCC metastasis (40, 41). Compared to sh-NC cells, sh1-CHRDL1 cells became fusiform and fibroblast-like in shape and lost their polygonal epithelial morphology (Figure 4A a). The sh1-CHRDL1 CAL27 cells also became more dispersed and showed loose cell-to-cell contact compared to sh-NC CAL27 cells under high cell density conditions (Figure 4A b). Rhodamine phalloidin staining was further performed to detect F-actin. Actin-rich membrane protrusions called filopodia (white arrow) were clearly seen at the periphery of sh1-CHRDL1 CAL27 cells but not sh-NC CAL27 cells (Figure 4A c). To further examine the EMT-like phenotype of sh1-CHRDL1 CAL27 cells, the expression of epithelial and mesenchymal markers, including cytokeratin 18, E-cadherin, N-cadherin, and fibronectin, was examined by WB (Figures 4B, C). The protein expression levels of E-cadherin and cytokeratin 18, epithelial differentiation markers, were significantly upregulated in OV-CHRDL1 CAL27 cells compared to OV-NC CAL27 cells. The protein expression levels of the mesenchymal differentiation markers N-cadherin and fibronectin were significantly downregulated in OV-CHRDL1 CAL27 cells compared to OV-NC CAL27 cells. In contrast, the protein expression levels of E-cadherin and cytokeratin 18 were 1.21- and 2.17-fold downregulated, respectively, in sh1-CHRDL1 CAL27 cells compared to sh-NC CAL27 cells, while the protein expression levels of N-cadherin and fibronectin were 0.64- and 0.53-fold upregulated, respectively, in sh1-CHRDL1 CAL27 cells compared to sh-NC CAL27 cells. These results suggested that CHRDL1 silencing in CAL27 cells led to the EMT phenotype, while overexpression of CHRDL1 in CAL27 cells inhibited the process. Thus, CHRDL1 knockdown may induce metastasis in CAL27 cells by activating EMT.
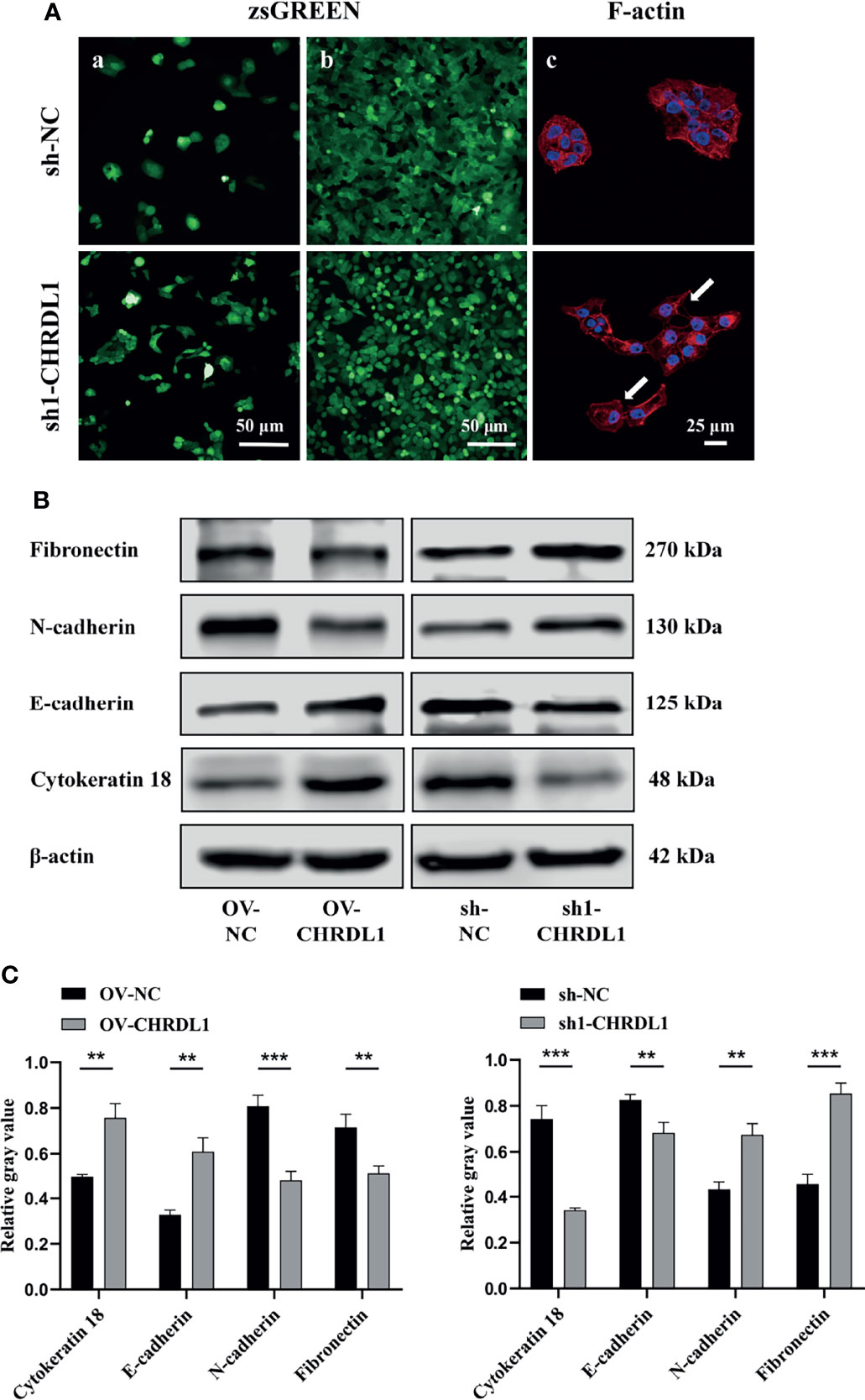
Figure 4 Depletion of CHRDL1 induces the EMT phenotype in CAL27 cells. (A) Representative fluorescence images of sh-NC/sh1-CHRDL1 CAL27 cells under low (A) and high (b) cell density conditions. (c) Rhodamine-conjugated phalloidin staining of F-actin in sh-NC/sh1-CHRDL1 CAL27 cells, examined using confocal microscopy. The arrows indicate filopodia. (B, C) The expression of EMT markers (Fibronectin, N-cadherin, E-cadherin, and Cytokeratin 18) in CAL27 cells after lentivirus transfection was detected by Western blot and quantitatively analyzed. All experiments were performed three times, and one representative experiment is shown. Error bars represent the standard deviation. **p < 0.01, ***p < 0.001.
CHRDL1 Regulates the MAPK Signaling Pathway in CAL27 Cells
To identify genes differentially expressed specifically in the context of high versus low CHRDL1 levels, RNA sequencing was performed in OV-NC/OV-CHRDL1 and sh-NC/sh1-CHRDL1 CAL27 cells. A fold-change >2.0 or <−2.0 and padj < 0.05 were chosen as cutoff criteria and visualized using the heatmap. A total of 163 upregulated and 179 downregulated genes in OV-CHRDL1 CAL27 cells compared to OV-NC cells were identified by gene expression analysis (Figure 5A, left). In addition, 1067 genes were upregulated and 952 genes were downregulated in sh1-CHRDL1 CAL27 cells compared to sh-NC cells (Figure 5A, right). Then, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed on the differentially expressed genes (DEGs) identified in OV-CHRDL1 CAL27 cells and sh1-CHRDL1 CAL27 cells. The top 10 most significant GO biological processes (BP) are shown in Figure 5B. Cell morphogenesis and regulation of cell adhesion were identified in sh1-CHRDL1 CAL27 cells, consistent with the in vitro results. The top 10 enriched KEGG pathways are shown in Figure 5C. Interestingly, MAPK signaling was enriched in both OV-CHRDL1 CAL27 cells and sh1-CHRDL1 CAL27 cells. Thus, MAPK signaling pathway components were further probed by WB (Figures 5D, E). The protein expression of phosphorylated JNK (p-JNK) was upregulated, and that of phosphorylated ERK (p-ERK1/2) and p38 (p-p38) was downregulated, in OV-CHRDL1 CAL27 cells compared to OV-NC CAL27 cells. In contrast, the protein expression of p-JNK was downregulated and that of p-ERK and p-p38 was upregulated in sh1-CHRDL1 CAL27 cells compared to sh-NC CAL27 cells. These results indicated that CHRDL1 may influence the MAPK pathway in CAL27 cells to regulate cell metastasis.
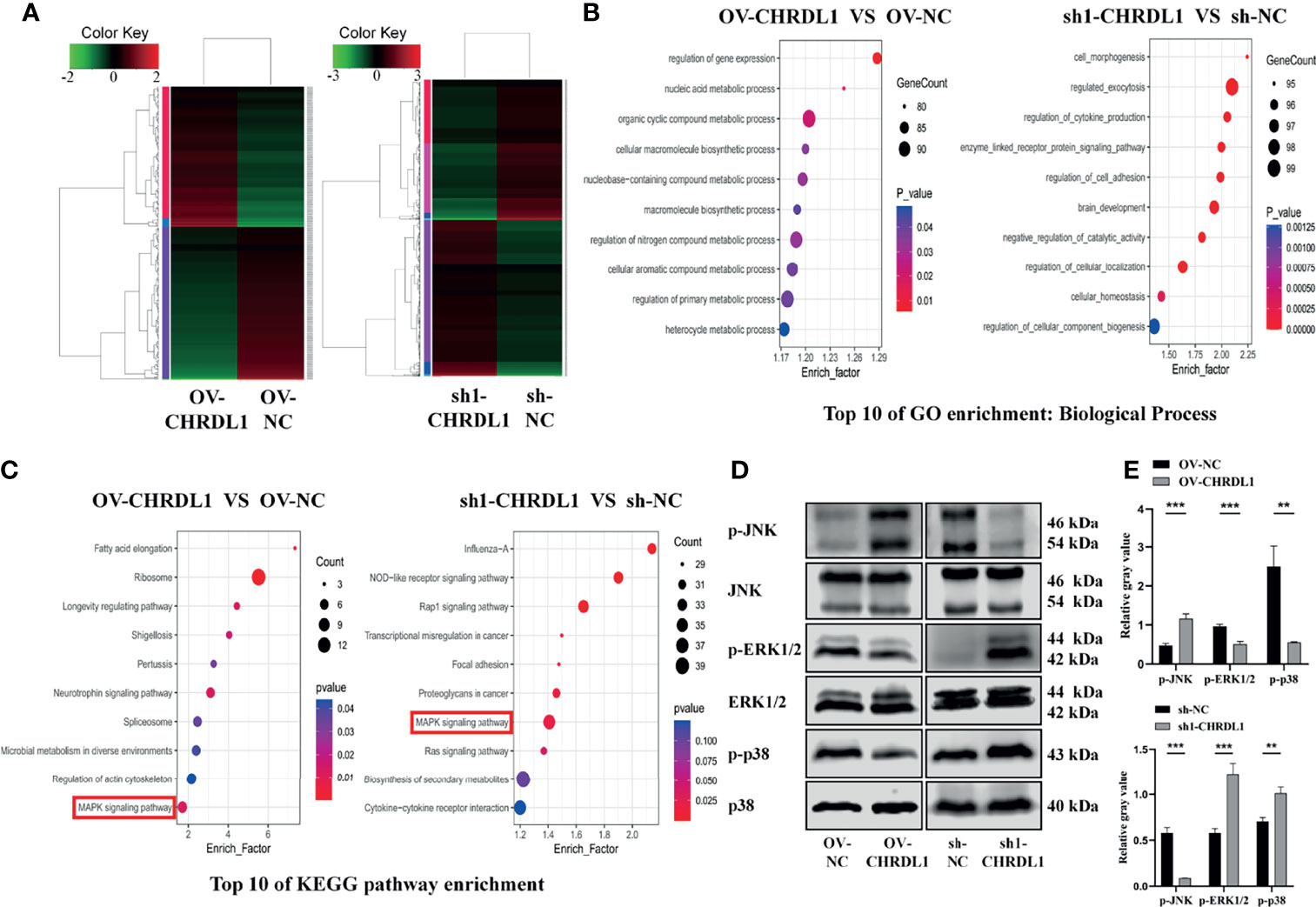
Figure 5 CHRDL1 regulates the MAPK signaling pathway in CAL27 cells. (A) Heatmap of differentially expressed genes between OV-CHRDL1 and OV-NC (left) and between sh1-CHRDL1 and sh-NC (right). Green indicates downregulated genes, and red indicates upregulated genes. The thresholds for up- and downregulation were a +2.0-fold and -2.0-fold change, respectively, and a padj < 0.05. (B) Top 10 GO-BP terms enriched among DEGs from OV-CHRDL1 CAL27 cells and sh1-CHRDL1 CAL27 cells. (C) Top 10 KEGG pathways enriched among DEGs from OV-CHRDL1 CAL27 cells and sh1-CHRDL1 CAL27 cells. (D, E) Protein expression of MAPK signaling pathway-related markers in CAL27 cells after lentivirus transfection. Error bars represent the standard deviation. **p < 0.01, ***p < 0.001.
CHRDL1 Regulates Tumor Metastasis In Vivo
To determine the metastatic effect of CHRDL1 in vivo, OV-NC/OV-CHRDL1 and sh-NC/sh1-CHRDL1 CAL27 cells were injected into 4-week-old male BALB/c nude mice via the tail vein, and their lung colonization ability was analyzed. Samples were collected after 9 weeks to determine the number of pulmonary tumor colonies and weight changes. As shown in Figure 6A, the number of lung tumors (red arrow) derived from OV-CHRDL1 CAL27 cells was significantly decreased compared to that derived from OV-NC CAL27 cells. The number of lung tumors (red arrow) derived from sh1-CHRDL1 CAL27 cells was significantly increased compared to that derived from sh-NC CAL27 cells. The lung weights of the mice injected with OV-CHRDL1 CAL27 cells were lower than those of the mice injected with OV-NC CAL27 cells. In contrast, the lungs from the mice injected with sh1-CHRDL1 CAL27 cells weighed more than those of the mice injected with sh-NC CAL27 cells (Figure 6B). Furthermore, hematoxylin-eosin staining of lung tissues showed that mice injected with OV-CHRDL1 CAL27 cells had fewer lung colonies, while mice injected with sh1-CHRDL1 CAL27 cells showed more lung colonies, compared to the control group (Figure 6C). These results demonstrated that CHRDL1 silencing could promote tumor metastasis and enhance lung colonization in vivo, while CHRDL1 overexpression could restrain tumor metastasis and enhance lung colonization in vivo.
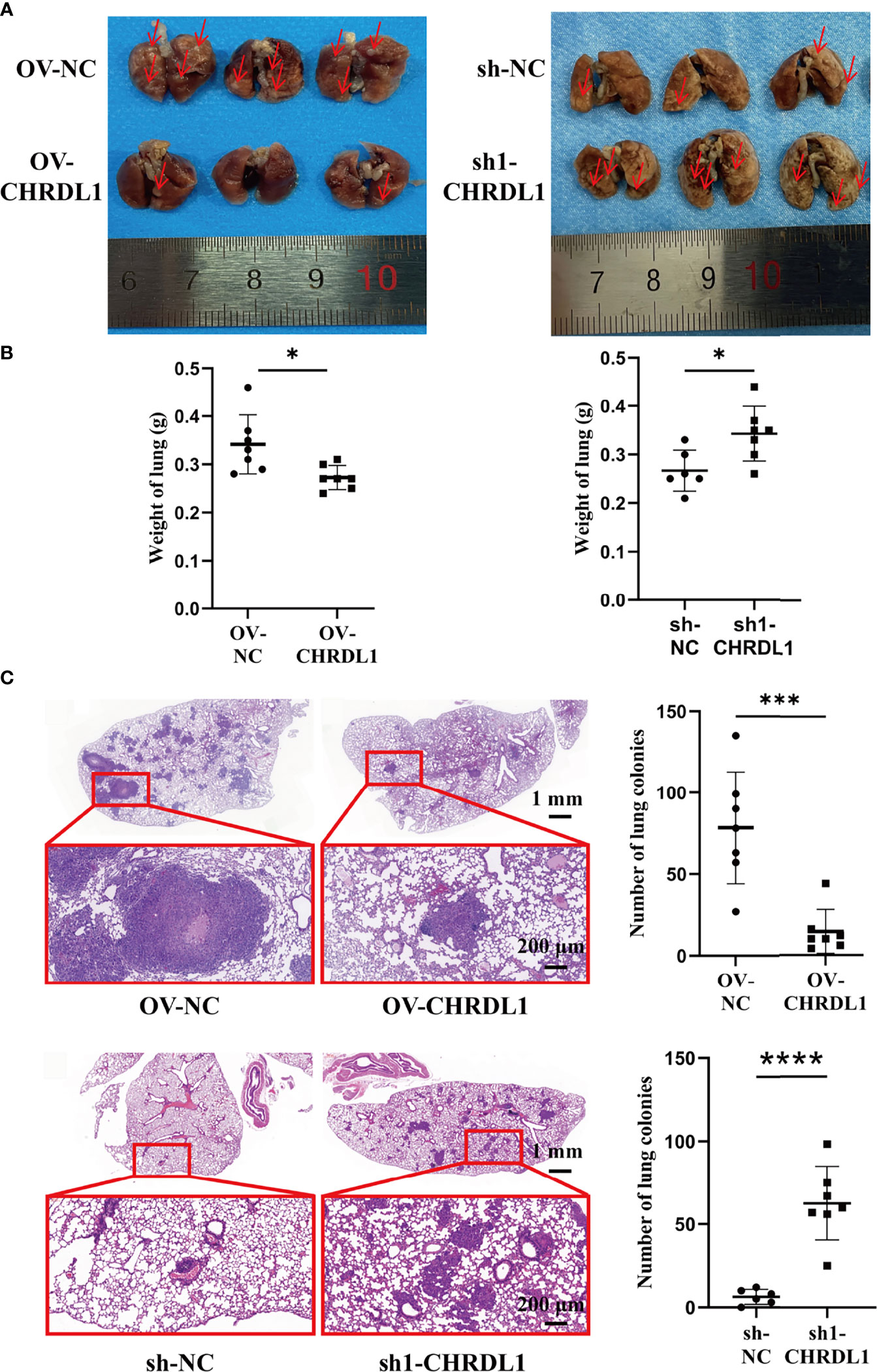
Figure 6 Effects of CHRDL1 on CAL27 cells in vivo. (A) Representative images of the lungs of BALB/c nude mice at 9 weeks after inoculation with OV-NC/OV-CHRDL1 (left) or sh-NC/sh1-CHRDL1 (right) CAL27 cells by tail vein injection. The red arrows show nodules on the lung surfaces. (B) Weights of the lungs of BALB/c-nude mice inoculated with OV-NC/OV-CHRDL1 (left) or sh-NC/sh1-CHRDL1 (right) CAL27 cells by tail vein injection. (C) Photomicrographs of hematoxylin-and-eosin-stained lungs of mice injected with OV-NC/OV-CHRDL1 (up) or sh-NC/sh1-CHRDL1 (down) CAL27 cells. Error bars represent the standard deviation. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Discussion
The roles of CHRDL1, a BMPS antagonist associated with various cancers, should be more deeply explored. In the present study, we focused on the effect of CHRDL1 on OSCC in vitro and in vivo and further revealed its function and potential mechanisms.
The expression of CHRDL1 is dysregulated in different types of tumors. CHRDL1 was reported to be downregulated in T-ALL (35), gastric cancer (29), thyroid cancer (31–34), lung cancer (25–28), malignant melanoma (36), and breast cancer (22–24). However, Mock etal. (42) reported an upregulation of CHRDL1, which is activated by the transcription factor ZEB1 and correlated with bone metastasis rather than brain or lung metastasis, in breast cancer. Cha etal. (37) used array-based comparative genomic hybridization (aCGH) and multiplex ligation-dependent probe amplification (MLPA) to screen human genome-wide alterations in seven OSCC tissues and their resection margins. They found that CHRDL1 was one of the eleven genes with the highest amplification frequencies. No additional expression analysis was performed. However, we found lower CHRDL1 expression in OSCC tissues than in adjacent tissues by bioinformatic analysis, RT-qPCR and WB confirmation. The inconsistent findings regarding the expression of CHRDL1 in OSCC might be attributable to various factors, including differences in controls and populations, small sample sizes, and the heterogeneity of OSCC. The GEPIA database (43) was searched to analyze CHRDL1 expression between the tumor and normal groups (Supplementary Figure 2). CHRDL1 was downregulated in head and neck squamous cell carcinoma (HNSC), which was consistent with our results. Furthermore, CHRDL1 was also downregulated in esophageal carcinoma (ESCA). It is possible that HNSCC (including OSCC) and ESCA share a common histological origin (i.e., stratified squamous epithelium) (44) and a similar genomic characterization (45–47). CHRDL1 is downregulated in most malignant tumors (except for thymoma), despite their differing histologic origins. We also found that CHRDL1 expression was decreased in OSCC. These findings suggest that CHRDL1 downregulation may regulate the initiation of malignant tumors across different tissues.
CHRDL1 knockdown significantly facilitated migration, invasion, and adhesion, but not proliferation, in OSCC cells. CHRDL1 overexpression reversed these cellular phenotypes. It has been demonstrated that both endogenous CHRDL1 and recombinant CHRDL1 suppress the migration and invasion induced by BMP4 signaling in different breast cancer cell lines (21). CHRDL1 knockdown promotes the proliferation and migration of gastric cancer cells through BMPR II by activating Akt, Erk, and β-catenin (29). Mithani etal. (36) demonstrated growth-suppressive properties with CHRDL1 transfection into melanoma-derived cell lines. Tumor metastasis in vivo can be simplified into an ordered process consisting of local invasion, intravasation, survival in the circulation, extravasation, and colonization (48). In an in vivo nude mouse model, CHRDL1 knockdown in CAL-27 cells significantly increased the weight of the lungs and the metastatic nodules of the lungs. Overexpression of CHRDL1 reversed this effect.
The initiation of cancer cell metastasis is highly associated with the aberrant activation of the EMT process, which allows cancer cells to disseminate from the primary tumor to the surrounding tissues (49). EMT involves the decreased expression of epithelial markers such as E-cadherin and cytokeratin 18 and the increased expression of mesenchymal markers such as fibronectin and N-cadherin (50). Activation of EMT was also found in gastric cancer and breast cancer cells after CHRDL1 silencing (29, 42). It is worth noting that we observed an EMT-like phenotype in CHRDL1-depleted CAL27 cells.
The dysregulated signaling pathway after CHRDL1 dysregulation was further explored in CAL-27 cells. Mitogen-activated protein kinase (MAPK) signaling pathway members, including p38, ERK1/2, and JNKs, regulate all critical phases of cell growth, including proliferation, differentiation, and apoptosis. The deregulation of the MAPK pathway has been reported in several types of tumors, including human epithelial carcinogenesis, prostate cancers, renal cell carcinoma, hepatocellular carcinoma, glial neoplasms, and breast cancer (51).
Various lines of evidence in OSCC have reported that the phosphorylated activation of p38 promotes cell proliferation, migration, and invasion (52–55). In addition, inhibition of p38 signaling reduces proliferation, angiogenesis, lymphangiogenesis and tumor-induced inflammation (52, 56) and increases apoptosis and autophagy in OSCC tumor cells (57). ERK1/2 phosphorylation appears to promote OSCC cell progression, migration, proliferation, and metastasis (58, 59). Notably, the role of JNK signaling in OSCC is complex and remains controversial. Some authors have argued that downregulation of JNK1/2 pathways inhibits OSCC cell metastasis (60–63) and increases apoptosis and autophagy (57). In contrast, some reports have concluded that reducing the phosphorylation of JNK promotes OSCC cell progression and migration (64–66), which is consistent with our results.
The effects of CHRDL1 on the MAPK signaling pathway have rarely been reported. We found that silencing CHRDL1 in an OSCC cell line could activate the MAPK signaling pathway, leading to the promotion of malignant behaviors in OSCC. In contrast, overexpression of CHRDL1 in CAL27 cells inhibited the expression of phosphorylated ERK1/2 and p38 and increased the level of phosphorylated JNK, leading to the malignant characteristics of OSCC. Several studies of OSCC have indicated that activation of the MAPK signaling pathway induces EMT to increase cellular malignancy (67–70). In addition, MAPK inhibitors can restore the original morphology of epithelial cells or block the expression of EMT-related factors (71, 72). These findings supported the hypothesis that CHRDL1 is associated with EMT and aggressive behaviors through the MAPK pathway in OSCC. MAPK inhibitors should be used to confirm the involvement of ERK1/2, p38, and JNK in CHRDL1-related phenotypes in future studies.
Furthermore, noninvasive detection of salivary, serum, and plasma factors is an important method for OSCC prediction (73–76). Our study is limited by the fact that CHRDL1 levels were not measured in the saliva and serum of OSCC patients due to the lack of such samples.
Conclusion
In conclusion, our study revealed the modulatory role of CHRDL1 in the malignant characteristics of OSCC as well as the molecular mechanism. The downregulation of CHRDL1 in OSCC may induce EMT and promote metastasis via the MAPK signaling pathway, while upregulation of CHRDL1 could reverse this effect. Our study provides the new perspective that CHRDL1 has possible potential as a molecular target in OSCC therapy.
Data Availability Statement
The data presented in the study are deposited in the NCBI SRA database, accession number: PRJNA801461.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Stomatology Hospital of Guangzhou Medical University (Approval No. KY2019026). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Guangzhou Medical University of China (Approval No. 2017-086).
Author Contributions
QW, ZZ, and LW contributed to the conception and design of the study. QW, ZZ, MX, XX, AW, TZ, SH, and SX performed the experiments. QW, JWZ, ZP, YQ, JYZ, and HC performed the statistical analysis. QW wrote the first draft of the manuscript. QW, LW, ZZ, and KO critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 31801152) and the University Student Laboratory Open Project of Guangzhou Medical University (Grant No. 2020-27).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.862751/full#supplementary-material
References
1. Tang J, Fang X, Chen J, Zhang H, Tang Z. Long Non-Coding RNA (lncRNA) in Oral Squamous Cell Carcinoma: Biological Function and Clinical Application. Cancers (Basel) (2021) 13:5944. doi: 10.3390/cancers13235944
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer Statistics 2008. CA Cancer J Clin (2008) 58:71–96. doi: 10.3322/CA.2007.0010
4. Todd R, Donoff RB, Wong DT. The Molecular Biology of Oral Carcinogenesis: Toward a Tumor Progression Model. J Oral Maxillofac Surg (1997) 55:613–623; discussion 623-615. doi: 10.1016/s0278-2391(97)90495-x
5. Patel V, Leethanakul C, Gutkind JS. New Approaches to the Understanding of the Molecular Basis of Oral Cancer. Crit Rev Oral Biol Med (2001) 12:55–63. doi: 10.1177/10454411010120010401
6. Nana AW, Yang PM, Lin HY. Overview of Transforming Growth Factor Beta Superfamily Involvement in Glioblastoma Initiation and Progression. Asian Pac J Cancer Prev (2015) 16:6813–23. doi: 10.7314/apjcp.2015.16.16.6813
7. Kobayashi T, Lyons KM, Mcmahon AP, Kronenberg HM. BMP Signaling Stimulates Cellular Differentiation at Multiple Steps During Cartilage Development. Proc Natl Acad Sci USA (2005) 102:18023–7. doi: 10.1073/pnas.0503617102
8. Secondini C, Wetterwald A, Schwaninger R, Thalmann GN, Cecchini MG. The Role of the BMP Signaling Antagonist Noggin in the Development of Prostate Cancer Osteolytic Bone Metastasis. PloS One (2011) 6:e16078. doi: 10.1371/journal.pone.0016078
9. Sanders AJ, Ye L, Li J, Mason MD, Jiang WG. Tumour Angiogenesis and Repulsive Guidance Molecule B: A Role in HGF- and BMP-7-Mediated Angiogenesis. Int J Oncol (2014) 45:1304–12. doi: 10.3892/ijo.2014.2508
10. Clement JH, Raida M, Sanger J, Bicknell R, Liu J, Naumann A, et al. Bone Morphogenetic Protein 2 (BMP-2) Induces In Vitro Invasion and In Vivo Hormone Independent Growth of Breast Carcinoma Cells. Int J Oncol (2005) 27:401–7. doi: 10.3892/ijo.27.2.401
11. Kim MJ, Kim KM, Kim J, Kim KN. BMP-2 Promotes Oral Squamous Carcinoma Cell Invasion by Inducing CCL5 Release. PloS One (2014) 9:e108170. doi: 10.1371/journal.pone.0108170
12. Kim BR, Oh SC, Lee DH, Kim JL, Lee SY, Kang MH, et al. BMP-2 Induces Motility and Invasiveness by Promoting Colon Cancer Stemness Through STAT3 Activation. Tumour Biol (2015) 36:9475–86. doi: 10.1007/s13277-015-3681-y
13. Herrera B, Van Dinther M, Ten Dijke P, Inman GJ. Autocrine Bone Morphogenetic Protein-9 Signals Through Activin Receptor-Like Kinase-2/Smad1/Smad4 to Promote Ovarian Cancer Cell Proliferation. Cancer Res (2009) 69:9254–62. doi: 10.1158/0008-5472.CAN-09-2912
14. Rothhammer T, Poser I, Soncin F, Bataille F, Moser M, Bosserhoff AK. Bone Morphogenic Proteins are Overexpressed in Malignant Melanoma and Promote Cell Invasion and Migration. Cancer Res (2005) 65:448–56.
15. Yin J, Jung JE, Choi SI, Kim SS, Oh YT, Kim TH, et al. Inhibition of BMP Signaling Overcomes Acquired Resistance to Cetuximab in Oral Squamous Cell Carcinomas. Cancer Lett (2018) 414:181–9. doi: 10.1016/j.canlet.2017.11.013
16. Sakuta H, Suzuki R, Takahashi H, Kato A, Shintani T, Iemura S, et al. Ventroptin: A BMP-4 Antagonist Expressed in a Double-Gradient Pattern in the Retina. Science (2001) 293:111–5. doi: 10.1126/science.1058379
17. Webb TR, Matarin M, Gardner JC, Kelberman D, Hassan H, Ang W, et al. X-Linked Megalocornea Caused by Mutations in CHRDL1 Identifies an Essential Role for Ventroptin in Anterior Segment Development. Am J Hum Genet (2012) 90:247–59. doi: 10.1016/j.ajhg.2011.12.019
18. Davidson AE, Cheong SS, Hysi PG, Venturini C, Plagnol V, Ruddle JB, et al. Association of CHRDL1 Mutations and Variants With X-Linked Megalocornea, Neuhauser Syndrome and Central Corneal Thickness. PloS One (2014) 9:e104163. doi: 10.1371/journal.pone.0104163
19. Mangialavori D, Colao E, Carnevali A, Bruzzichessi D, Grillone T, Perrotti N, et al. Novel Mutation in the CHRDL1 Gene Detected in Patients With Megalocornea. Cornea (2015) 34:976–9. doi: 10.1097/ICO.0000000000000472
20. Pfirrmann T, Emmerich D, Ruokonen P, Quandt D, Buchen R, Fischer-Zirnsak B, et al. Molecular Mechanism of CHRDL1-Mediated X-Linked Megalocornea in Humans and in Xenopus Model. Hum Mol Genet (2015) 24:3119–32. doi: 10.1093/hmg/ddv063
21. Cyr-Depauw C, Northey JJ, Tabaries S, Annis MG, Dong Z, Cory S, et al. Chordin-Like 1 Suppresses Bone Morphogenetic Protein 4-Induced Breast Cancer Cell Migration and Invasion. Mol Cell Biol (2016) 36:1509–25. doi: 10.1128/MCB.00600-15
22. Jiang YZ, Liu YR, Xu XE, Jin X, Hu X, Yu KD, et al. Transcriptome Analysis of Triple-Negative Breast Cancer Reveals an Integrated mRNA-lncRNA Signature With Predictive and Prognostic Value. Cancer Res (2016) 76:2105–14. doi: 10.1158/0008-5472.CAN-15-3284
23. Li JW, Zhou J, Shi ZT, Li N, Zhou SC, Chang C. Sonographic Features of Triple-Negative Breast Carcinomas Are Correlated With mRNA-lncRNA Signatures and Risk of Tumor Recurrence. Front Oncol (2020) 10:587422:587422. doi: 10.3389/fonc.2020.587422
24. Wang X, Gao C, Feng F, Zhuang J, Liu L, Li H, et al. Construction and Analysis of Competing Endogenous RNA Networks for Breast Cancer Based on TCGA Dataset. BioMed Res Int (2020) 2020:4078596. doi: 10.1155/2020/4078596
25. Chen L, Zhuo D, Chen J, Yuan H. Screening Feature Genes of Lung Carcinoma With DNA Microarray Analysis. Int J Clin Exp Med (2015) 8:12161–71.
26. Deng H, Hang Q, Shen D, Zhang Y, Chen M. Low Expression of CHRDL1 and SPARCL1 Predicts Poor Prognosis of Lung Adenocarcinoma Based on Comprehensive Analysis and Immunohistochemical Validation. Cancer Cell Int (2021) 21:259. doi: 10.1186/s12935-021-01933-9
27. Deng B, Chen X, Xu L, Zheng L, Zhu X, Shi J, et al. Chordin-Like 1 is a Novel Prognostic Biomarker and Correlative With Immune Cell Infiltration in Lung Adenocarcinoma. Aging (Albany NY) (2022) 14:389–409. doi: 10.18632/aging.203814
28. Wu Y, Yang L, Zhang L, Zheng X, Xu H, Wang K, et al. Identification of a Four-Gene Signature Associated With the Prognosis Prediction of Lung Adenocarcinoma Based on Integrated Bioinformatics Analysis. Genes (Basel) (2022) 13:238. doi: 10.3390/genes13020238
29. Pei YF, Zhang YJ, Lei Y, Wu WD, Ma TH, Liu XQ. Hypermethylation of the CHRDL1 Promoter Induces Proliferation and Metastasis by Activating Akt and Erk in Gastric Cancer. Oncotarget (2017) 8:23155–66. doi: 10.18632/oncotarget.15513
30. Hu C, Zhou Y, Liu C, Kang Y. A Novel Scoring System for Gastric Cancer Risk Assessment Based on the Expression of Three CLIP4 DNA Methylation-Associated Genes. Int J Oncol (2018) 53:633–43. doi: 10.3892/ijo.2018.4433
31. Shen Y, Dong S, Liu J, Zhang L, Zhang J, Zhou H, et al. Identification of Potential Biomarkers for Thyroid Cancer Using Bioinformatics Strategy: A Study Based on GEO Datasets. BioMed Res Int (2020) 2020:9710421. doi: 10.1155/2020/9710421
32. Pan Y, Wu L, He S, Wu J, Wang T, Zang H. Identification of Hub Genes in Thyroid Carcinoma to Predict Prognosis by Integrated Bioinformatics Analysis. Bioengineered (2021) 12:2928–40. doi: 10.1080/21655979.2021.1940615
33. Ren H, Liu X, Li F, He X, Zhao N. Identification of a Six Gene Prognosis Signature for Papillary Thyroid Cancer Using Multi-Omics Methods and Bioinformatics Analysis. Front Oncol (2021) 11:624421:624421. doi: 10.3389/fonc.2021.624421
34. Sun T, Guan Q, Wang Y, Qian K, Sun W, Ji Q, et al. Identification of Differentially Expressed Genes and Signaling Pathways in Papillary Thyroid Cancer: A Study Based on Integrated Microarray and Bioinformatics Analysis. Gland Surg (2021) 10:629–44. doi: 10.21037/gs-20-673
35. Nagel S, Ehrentraut S, Meyer C, Kaufmann M, Drexler HG, Macleod RA. Repressed BMP Signaling Reactivates NKL Homeobox Gene MSX1 in a T-ALL Subset. Leuk Lymphoma (2015) 56:480–91. doi: 10.3109/10428194.2014.924119
36. Mithani SK, Smith IM, Califano JA. Use of Integrative Epigenetic and Cytogenetic Analyses to Identify Novel Tumor-Suppressor Genes in Malignant Melanoma. Melanoma Res (2011) 21:298–307. doi: 10.1097/CMR.0b013e328344a003
37. Cha JD, Kim HJ, Cha IH. Genetic Alterations in Oral Squamous Cell Carcinoma Progression Detected by Combining Array-Based Comparative Genomic Hybridization and Multiplex Ligation-Dependent Probe Amplification. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2011) 111:594–607. doi: 10.1016/j.tripleo.2010.11.020
38. Guo R, Berry LD, Aisner DL, Sheren J, Boyle T, Bunn PA Jr., et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data From a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J Thorac Oncol (2019) 14:1666–71. doi: 10.1016/j.jtho.2019.06.009
39. Paschalis A, Sheehan B, Riisnaes R, Rodrigues DN, Gurel B, Bertan C, et al. Prostate-Specific Membrane Antigen Heterogeneity and DNA Repair Defects in Prostate Cancer. Eur Urol (2019) 76:469–78. doi: 10.1016/j.eururo.2019.06.030
40. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia Induced EMT: A Review on the Mechanism of Tumor Progression and Metastasis in OSCC. Oral Oncol (2018) 80:23–32. doi: 10.1016/j.oraloncology.2018.03.004
41. Vallina C, Lopez-Pintor RM, Gonzalez-Serrano J, De Vicente JC, Hernandez G, Lorz C. Genes Involved in the Epithelial-Mesenchymal Transition in Oral Cancer: A Systematic Review. Oral Oncol (2021) 117:105310. doi: 10.1016/j.oraloncology.2021.105310
42. Mock K, Preca BT, Brummer T, Brabletz S, Stemmler MP, Brabletz T. The EMT-Activator ZEB1 Induces Bone Metastasis Associated Genes Including BMP-Inhibitors. Oncotarget (2015) 6:14399–412. doi: 10.18632/oncotarget.3882
43. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45:W98–W102. doi: 10.1093/nar/gkx247
44. Xu TT, Zeng XW, Wang XH, Yang LX, Luo G, Yu T. Cystatin-B Negatively Regulates the Malignant Characteristics of Oral Squamous Cell Carcinoma Possibly Via the Epithelium Proliferation/Differentiation Program. Front Oncol (2021) 11:707066:707066. doi: 10.3389/fonc.2021.707066
45. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science (2011) 333:1157–60. doi: 10.1126/science.1208130
46. Cancer Genome Atlas, N. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
47. Dotto GP, Rustgi AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell (2016) 29:622–37. doi: 10.1016/j.ccell.2016.04.004
48. Tsuji T, Ibaragi S, Hu GF. Epithelial-Mesenchymal Transition and Cell Cooperativity in Metastasis. Cancer Res (2009) 69:7135–9. doi: 10.1158/0008-5472.CAN-09-1618
49. De Craene B, Berx G. Regulatory Networks Defining EMT During Cancer Initiation and Progression. Nat Rev Cancer (2013) 13:97–110. doi: 10.1038/nrc3447
50. Wang J, Jiang C, Li N, Wang F, Xu Y, Shen Z, et al. The Circepsti1/Mir-942-5p/LTBP2 Axis Regulates the Progression of OSCC in the Background of OSF via EMT and the PI3K/Akt/mTOR Pathway. Cell Death Dis (2020) 11:682. doi: 10.1038/s41419-020-02851-w
51. Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, et al. Activated Extracellular Signal-Regulated Kinases: Association With Epidermal Growth Factor Receptor/Transforming Growth Factor Alpha Expression in Head and Neck Squamous Carcinoma and Inhibition by Anti-Epidermal Growth Factor Receptor Treatments. Cancer Res (2001) 61:6500–10.
52. Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS. A Role for P38 MAPK in Head and Neck Cancer Cell Growth and Tumor-Induced Angiogenesis and Lymphangiogenesis. Mol Oncol (2014) 8:105–18. doi: 10.1016/j.molonc.2013.10.003
53. Lin Y, Mallen-St Clair J, Wang G, Luo J, Palma-Diaz F, Lai C, et al. P38 MAPK Mediates Epithelial-Mesenchymal Transition by Regulating P38ip and Snail in Head and Neck Squamous Cell Carcinoma. Oral Oncol (2016) 60:81–9. doi: 10.1016/j.oraloncology.2016.06.010
54. Ma C, Wang J, Fan L, Guo Y. Inhibition of CD147 Expression Promotes Chemosensitivity in HNSCC Cells by Deactivating MAPK/ERK Signaling Pathway. Exp Mol Pathol (2017) 102:59–64. doi: 10.1016/j.yexmp.2017.01.002
55. Katase N, Nishimatsu SI, Yamauchi A, Yamamura M, Fujita S. DKK3 Knockdown Confers Negative Effects on the Malignant Potency of Head and Neck Squamous Cell Carcinoma Cells via the PI3K/Akt and MAPK Signaling Pathways. Int J Oncol (2019) 54:1021–32. doi: 10.3892/ijo.2018.4667
56. Simon C, Goepfert H, Boyd D. Inhibition of the P38 Mitogen-Activated Protein Kinase by SB 203580 Blocks PMA-Induced Mr 92,000 Type IV Collagenase Secretion and In Vitro Invasion. Cancer Res (1998) 58:1135–9.
57. Hsieh MJ, Lin CW, Chen MK, Chien SY, Lo YS, Chuang YC, et al. Inhibition of Cathepsin S Confers Sensitivity to Methyl Protodioscin in Oral Cancer Cells via Activation of P38 MAPK/JNK Signaling Pathways. Sci Rep (2017) 7:45039. doi: 10.1038/srep45039
58. Wang L, Liu T, Nishioka M, Aguirre RL, Win SS, Okada N. Activation of ERK1/2 and Cyclin D1 Expression in Oral Tongue Squamous Cell Carcinomas: Relationship Between Clinicopathological Appearances and Cell Proliferation. Oral Oncol (2006) 42:625–31. doi: 10.1016/j.oraloncology.2005.11.002
59. Lakshmanachetty S, Balaiya V, High WA, Koster MI. Loss of TP63 Promotes the Metastasis of Head and Neck Squamous Cell Carcinoma by Activating MAPK and STAT3 Signaling. Mol Cancer Res (2019) 17:1279–93. doi: 10.1158/1541-7786.MCR-18-1355
60. O-charoenrat P, Wongkajornsilp A, Rhys-Evans PH, Eccles SA. Signaling Pathways Required for Matrix Metalloproteinase-9 Induction by Betacellulin in Head-and-Neck Squamous Carcinoma Cells. Int J Cancer (2004) 111:174–83. doi: 10.1002/ijc.20228
61. Chang CM, Chang PY, Tu MG, Lu CC, Kuo SC, Amagaya S, et al. Epigallocatechin Gallate Sensitizes CAL-27 Human Oral Squamous Cell Carcinoma Cells to the Anti-Metastatic Effects of Gefitinib (Iressa) via Synergistic Suppression of Epidermal Growth Factor Receptor and Matrix Metalloproteinase-2. Oncol Rep (2012) 28:1799–807. doi: 10.3892/or.2012.1991
62. Chuang JY, Huang YL, Yen WL, Chiang IP, Tsai MH, Tang CH. Syk/JNK/AP-1 Signaling Pathway Mediates Interleukin-6-Promoted Cell Migration in Oral Squamous Cell Carcinoma. Int J Mol Sci (2014) 15:545–59. doi: 10.3390/ijms15010545
63. Lin FY, Hsieh YH, Yang SF, Chen CT, Tang CH, Chou MY, et al. Resveratrol Suppresses TPA-Induced Matrix Metalloproteinase-9 Expression Through the Inhibition of MAPK Pathways in Oral Cancer Cells. J Oral Pathol Med (2015) 44:699–706. doi: 10.1111/jop.12288
64. Gorogh T, Berwig J, Scola N, Lippert BM. Differential Regulation of MAPK (JNK 3) Gene Expression in Human Head and Neck Squamous Cell Carcinomas. Onkologie (2004) 27:353–7. doi: 10.1159/000079087
65. Liu FY, Safdar J, Li ZN, Fang QG, Zhang X, Xu ZF, et al. CCR7 Regulates Cell Migration and Invasion Through MAPKs in Metastatic Squamous Cell Carcinoma of Head and Neck. Int J Oncol (2014) 45:2502–10. doi: 10.3892/ijo.2014.2674
66. Chan LP, Liu C, Chiang FY, Wang LF, Lee KW, Chen WT, et al. IL-8 Promotes Inflammatory Mediators and Stimulates Activation of P38 MAPK/ERK-NF-kappaB Pathway and Reduction of JNK in HNSCC. Oncotarget (2017) 8:56375–88. doi: 10.18632/oncotarget.16914
67. Chen SF, Nieh S, Jao SW, Liu CL, Wu CH, Chang YC, et al. Quercetin Suppresses Drug-Resistant Spheres via the P38 MAPK-Hsp27 Apoptotic Pathway in Oral Cancer Cells. PloS One (2012) 7:e49275. doi: 10.1371/journal.pone.0049275
68. Nguyen PT, Tsunematsu T, Yanagisawa S, Kudo Y, Miyauchi M, Kamata N, et al. The FGFR1 Inhibitor PD173074 Induces Mesenchymal-Epithelial Transition Through the Transcription Factor AP-1. Br J Cancer (2013) 109:2248–58. doi: 10.1038/bjc.2013.550
69. Cheng Z, Xing D. Ginsenoside Rg3 Inhibits Growth and Epithelial-Mesenchymal Transition of Human Oral Squamous Carcinoma Cells by Down-Regulating miR-221. Eur J Pharmacol (2019) 853:353–63. doi: 10.1016/j.ejphar.2019.03.040
70. Kim SA, Lee KH, Lee DH, Lee JK, Lim SC, Joo YE, et al. Receptor Tyrosine Kinase, RON, Promotes Tumor Progression by Regulating EMT and the MAPK Signaling Pathway in Human Oral Squamous Cell Carcinoma. Int J Oncol (2019) 55:513–26. doi: 10.3892/ijo.2019.4836
71. Wang D, Shen Q, Chen YQ, Wang MH. Collaborative Activities of Macrophage-Stimulating Protein and Transforming Growth Factor-Beta1 in Induction of Epithelial to Mesenchymal Transition: Roles of the RON Receptor Tyrosine Kinase. Oncogene (2004) 23:1668–80. doi: 10.1038/sj.onc.1207282
72. Wang D, Shen Q, Xu XM, Chen YQ, Wang MH. Activation of the RON Receptor Tyrosine Kinase Attenuates Transforming Growth Factor-Beta1-Induced Apoptotic Death and Promotes Phenotypic Changes in Mouse Intestinal Epithelial Cells. Carcinogenesis (2005) 26:27–36. doi: 10.1093/carcin/bgh284
73. Chakraborty D, Natarajan C, Mukherjee A. Advances in Oral Cancer Detection. Adv Clin Chem (2019) 91:181–200. doi: 10.1016/bs.acc.2019.03.006
74. Lu Z, Liang J, He Q, Wan Q, Hou J, Lian K, et al. The Serum Biomarker Chemerin Promotes Tumorigenesis and Metastasis in Oral Squamous Cell Carcinoma. Clin Sci (Lond) (2019) 133:681–95. doi: 10.1042/CS20181023
75. Khan T, Relitti N, Brindisi M, Magnano S, Zisterer D, Gemma S, et al. Autophagy Modulators for the Treatment of Oral and Esophageal Squamous Cell Carcinomas. Med Res Rev (2020) 40:1002–60. doi: 10.1002/med.21646
Keywords: chordin-like 1, oral squamous cell carcinoma (OSCC), epithelial-to-mesenchymal transition (EMT), MAPK, metastasis
Citation: Wu Q, Zheng Z, Zhang J, Piao Z, Xin M, Xiang X, Wu A, Zhao T, Huang S, Qiao Y, Zhou J, Xu S, Cheng H, Wu L and Ouyang K (2022) Chordin-Like 1 Regulates Epithelial-to-Mesenchymal Transition and Metastasis via the MAPK Signaling Pathway in Oral Squamous Cell Carcinoma. Front. Oncol. 12:862751. doi: 10.3389/fonc.2022.862751
Received: 26 January 2022; Accepted: 18 March 2022;
Published: 14 April 2022.
Edited by:
Ira Ida Skvortsova, Innsbruck Medical University, AustriaReviewed by:
Gaoxiang Ma, China Pharmaceutical University, ChinaQingqing Wu, Southern Medical University, China
Vimi Mutalik, University of Manitoba, Canada
Copyright © 2022 Wu, Zheng, Zhang, Piao, Xin, Xiang, Wu, Zhao, Huang, Qiao, Zhou, Xu, Cheng, Wu and Ouyang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kexiong Ouyang, RHIub3V5YW5na2V4aW9uZ0Bob3RtYWlsLmNvbQ==; Lihong Wu, d2NhbmhvbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship