 Nuofan Wang
Nuofan Wang Xinying Huo
Xinying Huo Baoguo Zhang
Baoguo Zhang Xiaoxiang Chen
Xiaoxiang Chen Shuli Zhao
Shuli Zhao Xuesong Shi
Xuesong Shi Hao Xu
Hao Xu Xiaowei Wei
Xiaowei Wei- 1Department of General Surgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 2Department of Oncology, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
- 3General Clinical Research Center, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
- 4Department of General Surgery, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
The role of methyltransferase-like 3 (METTL3), which participates in catalyzing N-methyladenosine (m6A) RNA modification, in gastric cancer (GC) is unclear. Here, we found that METTL3 was overexpressed in human GC. Functionally, we verified that METTL3 promoted tumor cell proliferation and angiogenesis through a series of phenotypic experiments. Subsequently, ADAMTS9 was identified as the downstream effector of METTL3 in GC, which could be degraded by the YTHDF2-dependent pathway. Finally, the data suggested that METTL3 might facilitate GC progression through the ADAMTS9-mediated PI3K/AKT pathway. Our study unveiled the fundamental mechanisms of METTL3 in GC progression. The clinical value of METTL3 in GC deserves further exploration.
Introduction
GC is the fifth most prevalent gastrointestinal malignancy globally (1). Although advancements in the treatment of GC have been achieved in recent years (2, 3), the prognosis of GC patients is still suboptimal. To date, the molecular pathogenesis of GC has not been well illustrated. It is essential to explore any critical cancer-related event that may contribute to the development of novel effective targeted therapies.
Increasing studies have shown that epigenetic modification strikingly contributes to GC progression, while major studies are focused on the transcriptional level (4–6). An increasing amount of evidence has demonstrated that m6A is the prevalent posttranscriptional modification in RNAs (7). Similar to other epigenetic processes, m6A modification is regulated by specific enzymes, including writers (methyltransferases), erasers (demethylases), and readers (m6A-binding proteins) (8). Aberrant expression of m6A regulators has been extensively identified in numerous diseases, including malignancies. For example, the core components of m6A writers, e.g., METTL14 and WTAP, can promote the progression of pancreatic and liver cancers (9–11). Erasers, including ALKBH5, have been reported to be tumor-suppressor genes (12, 13). Similarly, m6A reader proteins, such as YTHDF1/2/3, also participate in complex pathophysiological processes by identifying and binding to m6A motifs (14–20).
As a core component of the m6A writer, METTL3 is vital for m6A modification and has been identified as an oncogene in some hematological diseases and solid tumors (21–25). In myeloid leukemia, the inhibition of METTL3 has been proven to be a great potential medical strategy in the future. In the present study, we elucidated the promoting role of METTL3 in cell proliferation and angiogenesis, which are considered important in GC.
Methods and Materials
Clinical Samples and Public Data Collection
A total of 20 specimens, including tumor and adjacent normal tissues (ANTs), were obtained from GC patients receiving gastrectomy at Nanjing First Hospital from July 2019 to July 2020. No patient received antitumor treatment before surgery. The project was approved by the ethics committee of Nanjing First Hospital.
The public transcriptome data of METTL3 in GC were downloaded from The Cancer Genome Atlas (TCGA) database. The prognostic value of METTL3 in GC was evaluated in the Kaplan–Meier plotter database (http://kmplot.com/analysis/).
Cell Transfection
Specific small hairpin RNAs (shRNAs) targeting METTL3 (shMETTL3#1, shMETTL3#2) or YTHDF2 (shYTHDF2) and the control group (shNC) were synthesized (GenePharma, Shanghai, China). pcDNA3.1 (Invitrogen, Carlsbad, CA, USA) was used to construct pcDNA-METTL3 and pcDNA-ADAMTS9. Lipofectamine 2000 (Invitrogen, USA) was employed to transfect cells following the manufacturer’s instructions. The sequence of shRNAs is listed in Supplementary File 1.
Construction of Stable Knockdown and Overexpression Cells
Stable METTL3 knockdown and overexpression models were established by using a lentivirus vector system. Briefly, plasmid-cloned RelB cDNA and siRNA for knocking down METTL3 were transfected into AGC and HCG-27 cell lines according to the manufacturer’s instructions.
Animal Experiments
For the formation of xenograft tumors, 5 × 106 AGS cells mixed in Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) were subcutaneously injected into BALB/c nude mice (5-week-old male). Tumor volumes were routinely measured every 5 days according to the following equation: V = 0.5 × (length × width2). One month later, the xenografts were surgically dissected after sacrificing the mice.
Chick Chorioallantoic Membrane Assay
The chorioallantoic membrane (CAM) assay was employed to estimate the potential role of METTL3 in angiogenesis. The eggs were incubated in a humidified atmosphere of 80% at 38°C for 1 week. On embryonic day 8, 100 µl of a suspension of 1 × 106 AGS cells in culture medium mixed with Matrigel was deposited on the surface of the CAM. The images were then taken, and the vessel areas were calculated.
RNA-seq and meRIP-seq
For RNA-seq, RNA-seq of stable METTL3 knockdown AGS cells and their controls (NC) were performed with HiSeq-2500. The PolyA+ messenger RNA (mRNA) library was estimated with RNA-seq (H/M/R) Library Prep Kit. KEGG analysis was detected with DAVID.
For meRIP-seq, RNA fragmentation was performed by using NEBNext Magnesium RNA Fragmentation Module (NEB, Cambridge, UK). About 1/10 of the fragmented RNA was used as input control for RNA-seq by Genesky (Shanghai, China). The others were incubated with m6A antibody, and the enrichment of m6A containing mRNA was analyzed using high-throughput sequencing by Genesky (Shanghai, China).
Statistical Analysis
All the data analyses were performed using SPSS 22.0 (IBM, Armonk, NY, USA). Our data are described as the mean ± S.D. Continuous variables with a normal distribution were analyzed using Student’s t-test. For multiple comparisons, the significant difference test was applied following ANOVA. The correlations were analyzed using Pearson’s correlation coefficients. A P value < 0.05 indicated statistical significance.
Cell culture, qRT-PCR, cell proliferation assays, tube formation assays, RNA stability, Western blots, RNA immunoprecipitation (RIP), m6A quantification, RNA-seq, and MeRIP-Seq are listed in Supplementary File 1.
Results
METTL3 Is Overexpressed and Related to Poor Prognosis in GC
In both clinical and TCGA samples, METTL3 was observed to be highly expressed in tumors (Figures 1A–C). Similar results were also detected in different GC cell lines (Figure 1D). The bioinformatics analysis revealed that high METTL3 levels were markedly associated with poor PFS, OS, and post-progression survival (PPS) in GC patients (Figures 1E–G).
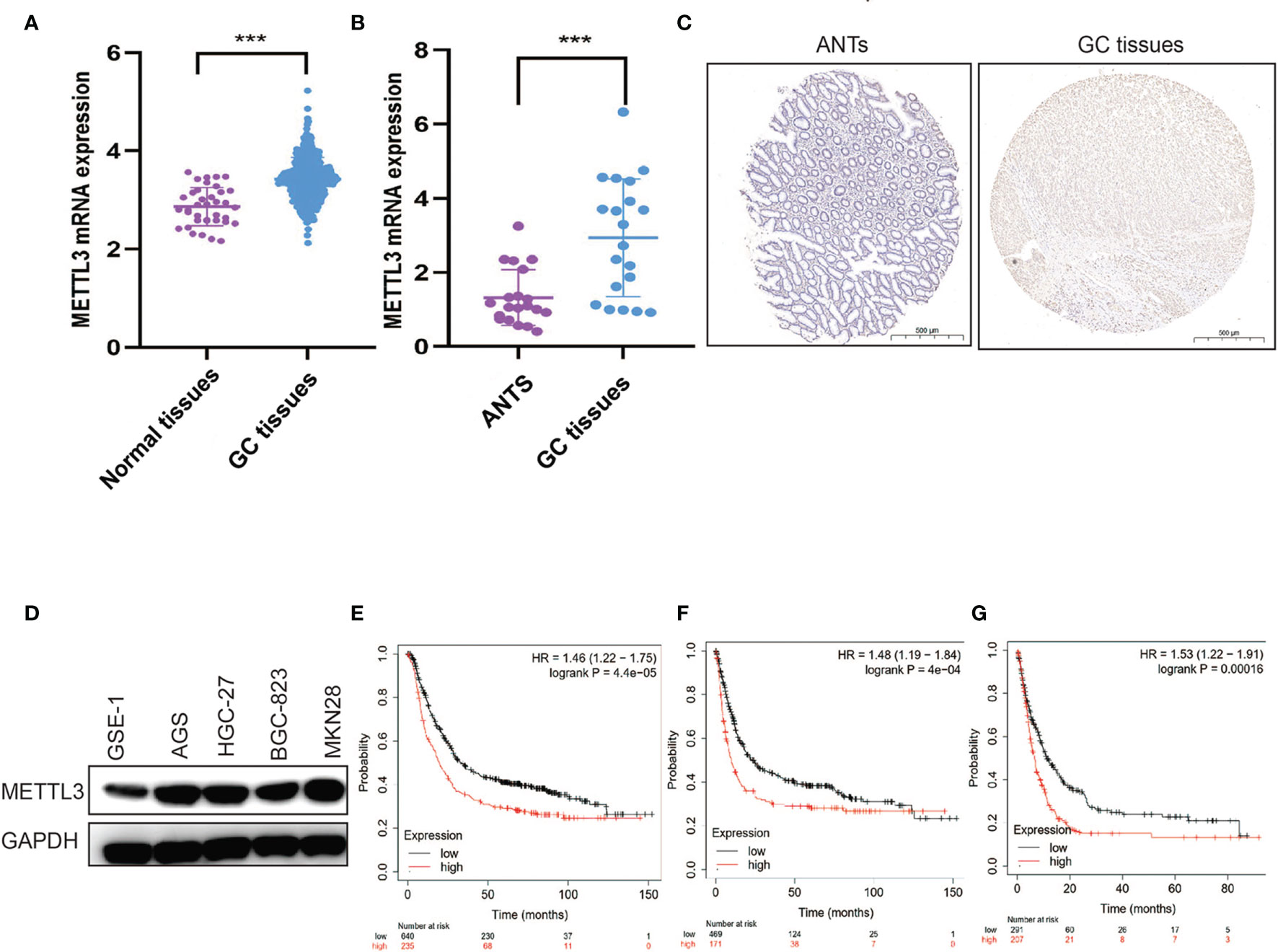
Figure 1 METTL3 is overexpressed and related to poor prognosis in GC. (A) METTL3 mRNA expression was evaluated in TCGA GC cohort. (B) METTL3 mRNA expression was evaluated in clinical GC samples. (C) Representative IHC stains of METTL3 in clinical GC samples. (D) METTL3 expression was measured in gastric epithelial cell and GC cell lines. (E–G) OS (E), PFS (F), and PPS (G) were analyzed in GC patients according to METTL3 expression. ***P < 0.001.
METTL3 Promotes GC Growth
To confirm the role of METTL3 in human GC, we established stable METTL3 knockdown and overexpression GC cell lines (Figure S1A). As expected, METTL3 depletion significantly reduced the m6A levels in GC cells (Figure S1B). The CCK-8 and colony formation assays showed that METTL3 overexpression significantly improved GC cell proliferative capacity, while METTL3 deficiency markedly inhibited GC cell survival (Figures 2A, B and Figures S1C, D). To further confirm our findings in vitro, a tumor-bearing mouse model was used to verify the effect of METTL3 on GC. As shown in Figures 2C, D, the tumor growth rate was positively correlated with METTL3 expression. Our data confirmed the function of METTL3 in facilitating GC tumorigenicity.

Figure 2 METTL3 promotes GC growth in vitro and in vivo. (A, B) The proliferative ability of GC cells was evaluated after METTL3 knockdown or overexpression by colony formation assay. (C, D) The tumor formation was verified after METTL3 knockdown or overexpression in a xenograft model. **P < 0.01, ***P < 0.001.
METTL3 Promotes GC Angiogenesis
The role of METTL3 in angiogenesis was investigated by using tube formation and CAM assays. As shown in Figures 3A, B, HUVEC tube formation was markedly impaired by conditioned medium (CM) from GC cells with METTL3 knockdown, while CM from METTL3-overexpressing GC cells promoted tube formation. In the CAM model, treatment with CM from METTL3 knockdown GC cells obviously decreased the vessel number in comparison with its control group (Figure 3C), whereas treatment with CM from METTL3 overexpression GC cells obviously elevated the vessel number in comparison with its control group (Figure 3D).
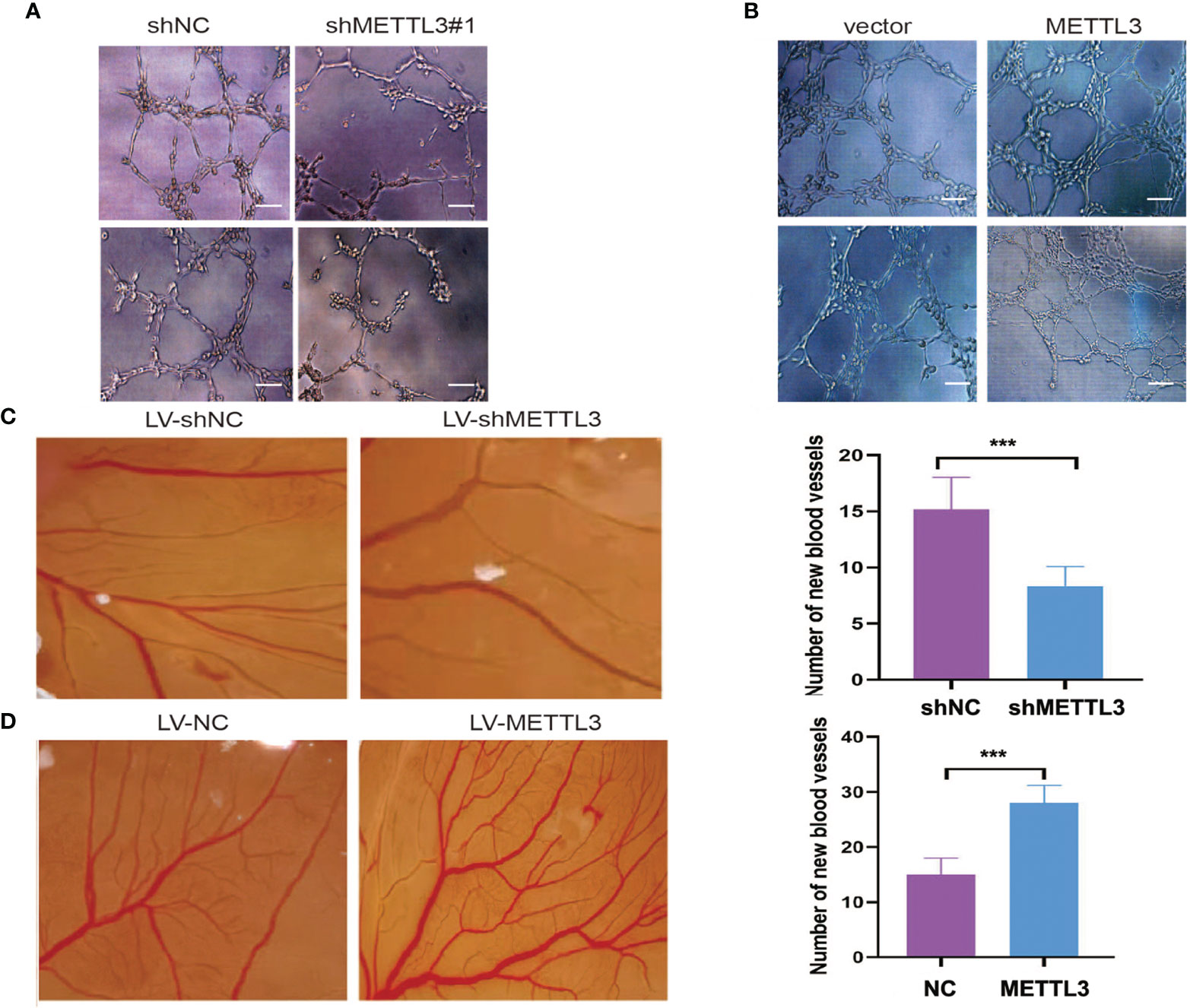
Figure 3 METTL3 promoted GC angiogenesis. (A, B) HUVECs cultured with medium obtained from GC cells with METTL3 deficiency or overexpression. (C) Representative images of the CAM experiment from LV-shMETTL3 and LV-shNC group. (D) Representative images of the CAM experiment from LV-METTL3 and LV-NC group. The average number of new vessels was quantified. Scar bar = 50 μm, ***P < 0.001.
ADAMTS9 Is Identified as a Downstream Target of METTL3
In this part, we identified 174 genes that were significantly upregulated in METTL3-deficient GC cells by using RNA-seq (Figure 4A). To investigate whether these differentially expressed genes were ascribed to METTL3-mediated m6A modification, MeRIP-seq was employed to map the m6A modification in stable METTL3-knockdown and control GC cells, and 397 peaks were found to be decreased in AGS cells with stable METTL3 knockdown (Figure 4A and Supplementary File 1). After filtering the upregulated genes and reduced m6A peaks, two genes (ADAMTS9 and POSTN) overlapped between the MeRIP-seq and RNA-seq data (Figure 4A). Of the two potential genes, ADAMTS9, which acts as a potential tumor-suppressor gene, was selected for further research (26). Subsequently, an m6A peak was found in ADAMTS9 mRNA in control AGS cells and was decreased upon METTL3 knockdown (Figure 4B). Furthermore, we verified that ADAMTS9 was downregulated by METTL3 in GC cells (Figure 4C).
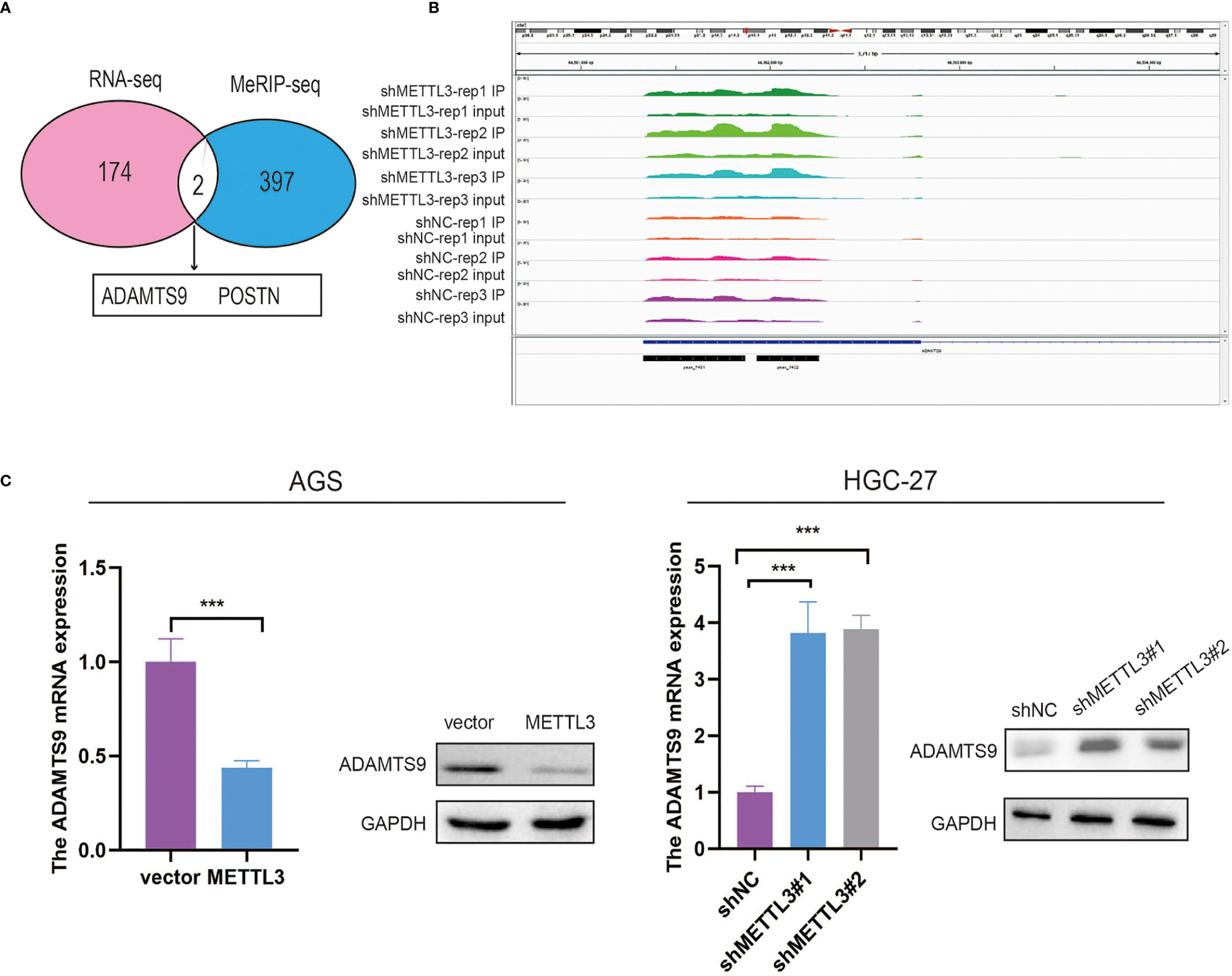
Figure 4 MeRIP-seq and RNA-seq identified ADAMTS9 as a downstream target of METTL3. (A) ADAMTS9 and POSTN were selected as candidate genes after filtering the upregulated genes and reduced m6A peaks. (B) The m6A abundances on ADAMTS9 mRNA in AGS cells with METTL3 knockdown or not as examined by MeRIP-seq were plotted. (C) The expression level of ADAMTS9 in METTL3-decificent and -overexpression GC cells. ***P < 0.001.
METTL3 Decreased ADAMTS9 mRNA Stability Through an m6A-YTHDF2-Dependent Pathway
MeRIP-seq data were validated to demonstrate the m6A modification of ADAMTS9 mRNA by using MeRIP-qPCR. Our results demonstrated that the m6A-specific antibody markedly enriched ADAMTS9 mRNA in comparison with the IgG control, METTL3 knockdown markedly decreased the m6A levels of ADAMTS9 mRNA in AGS cells, and METTL3 overexpression obviously elevated the m6A levels of ADAMTS9 mRNA in HGC-27 cells (Figure 5A). Next, we established luciferase reporters, including the wild-type (WT) and mutant (Mut) reporters. The adenosine (A) bases in the m6A consensus sequence (RRACH) were replaced by cytosine (C) in the mutant form of ADAMTS9. As shown in Figure 5B, the luciferase activity of the WT reporter was obviously increased upon METTL3 knockdown, while the Mut reporter exhibited no response to METTL3 knockdown, indicating that the modulation of ADAMTS9 mRNA expression was regulated by METTL3-mediated m6A modification. Previous studies have shown that the m6A-binding proteins YTHDF1/2/3 can target many mRNA transcripts by recognizing m6A motifs (27). In this study, our findings revealed that only knockdown of YTHDF2 could augment ADAMTS9 mRNA expression levels, while YTHDF1/3 had no remarkable effect (Figure 5C and Figure S1E).
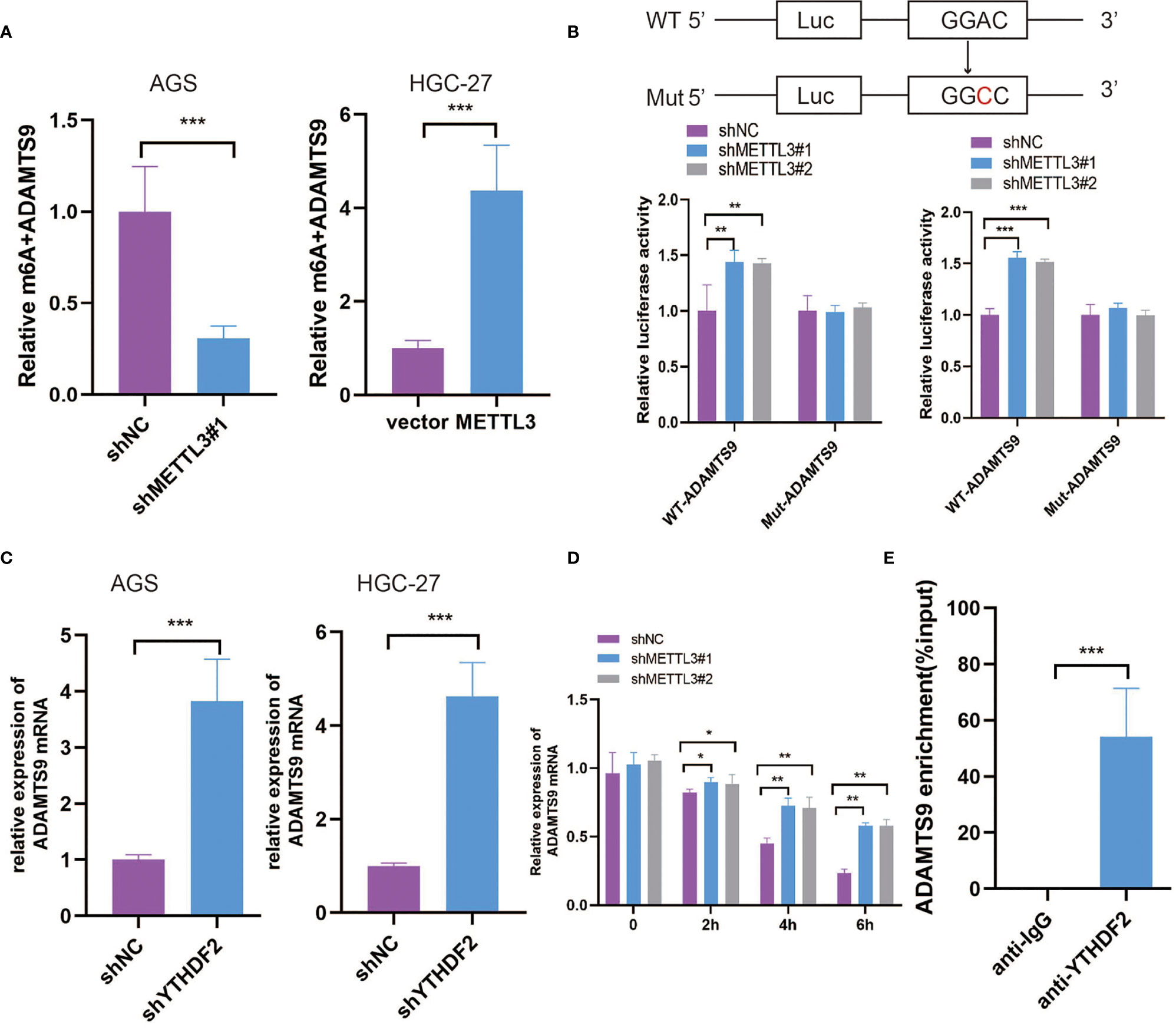
Figure 5 METTL3 decreased ADAMTS9 mRNA stability through m6A-YTHDF2 dependent pathway. (A) The regulation of METTL3 on ADAMTS9 m6A modification was evaluated in different GC cells. (B) The post-transcription level of ADAMTS9 was detected in METTL3-knockdown GC cells by using firefly luciferase reporter assay. (C) The level of ADAMTS9 mRNA was detected in YTHDF2 knockdown GC cells by qRT-PCR. (D) ADAMTS9 mRNA was analyzed at indicated times in METTL3 knockdown and control AGS cells with Act-D treatment. (E) RIP-qPCR assay was used to detect the enrichment of YTHDF2 binding to ADAMTS9 m6A modification sites. *P < 0.05, **P < 0.01, ***P < 0.001.
Subsequently, METTL3-knockdown AGS cells were treated with actinomycin D (Act-D) to block RNA transcription. As shown in Figure 5D, METTL3 knockdown significantly prolonged the half-life of ADAMTS9 mRNA, which indicated the promoting effect of METTL3 on ADAMTS9 mRNA degradation. YTHDF2-RIP was performed to prove that ADAMTS9 was a target of YTHDF2 (Figure 5E), which was reported to target and facilitate the degradation of mRNAs (28). Our data indicated that METTL3 may inhibit ADAMTS9 expression via YTHDF2-dependent mRNA decay.
ADAMTS9 Reversed the Effects of METTL3 in GC
To further confirm the importance of ADAMTS9 in our study, we suppressed ADAMTS9 expression in METTL3-knockdown GC cells (Figure 6A). The rescue experiments showed that ADAMTS9 inhibition enhanced the proliferative ability of METTL3-knockdown GC cells (Figures 6B, C). In addition, ADAMTS9 suppression also reversed the inhibition of HUVEC tube formation affected by METTL3 deficiency (Figure 6D). Therefore, our work demonstrated that METTL3 facilitates angiogenesis and carcinogenesis in GC by reducing the expression of ADAMTS9.
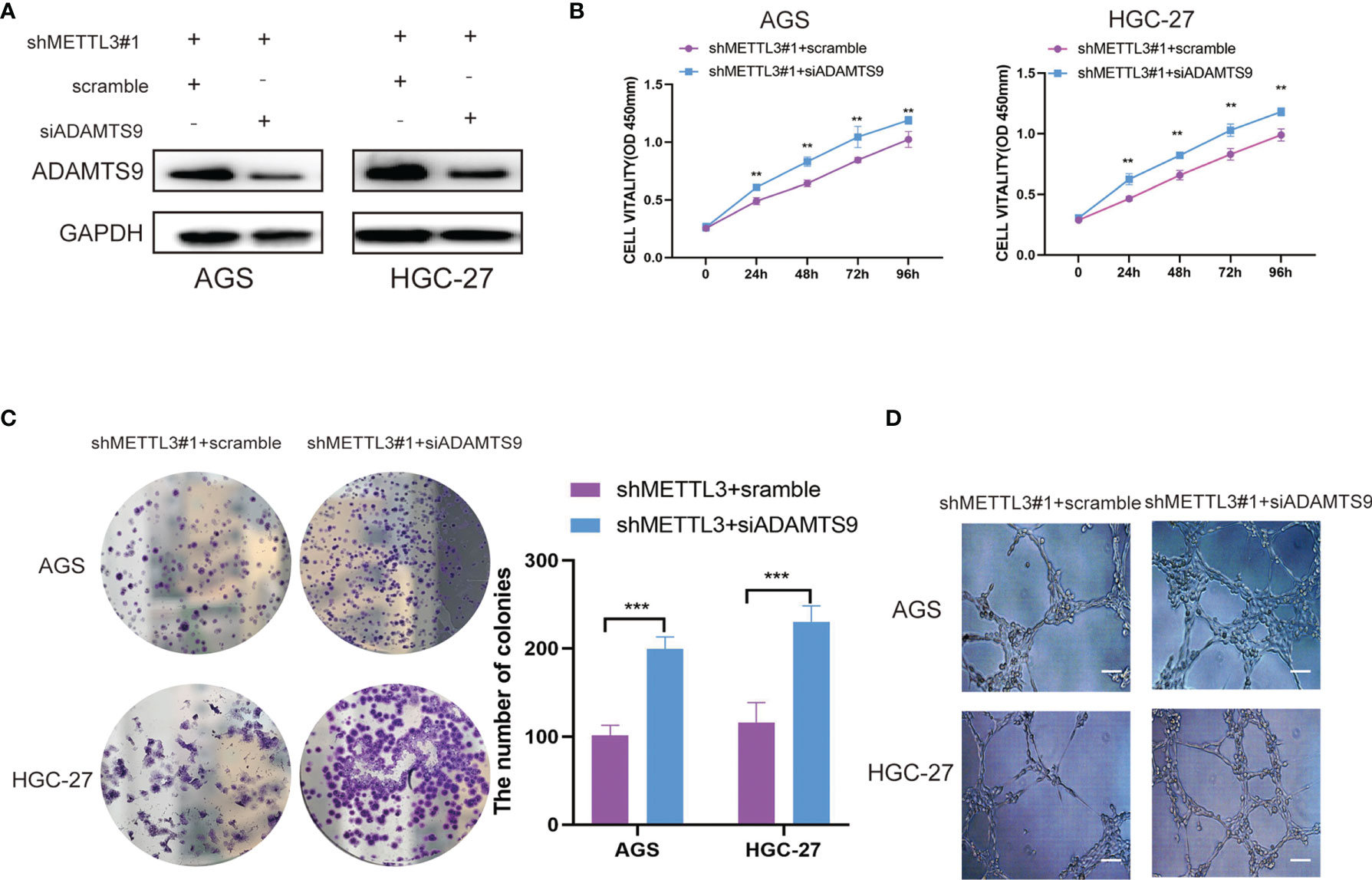
Figure 6 ADAMTS9 reversed the effects of METTL3 in GC. (A) The ADAMTS9 expressions were artificially reduced in METTL3 knockdown GC cells. (B) The cell viability of METTL3-knockdown GC cells was detected after ADAMTS9 suppression by CCK-8 assay. (C) The proliferation of METTL3-knockdown GC cells were detected after ADAMTS9 suppression by colony formation assay. (D) The angiogenesis ability of METTL3-knockdown GC cells were evaluated after ADAMTS9 suppression by tube formation assay. Scar bar = 50 μm, **P < 0.01, ***P < 0.001.
METTL3 Promotes GC Progression Through ADAMTS9-Mediated PI3K/AKT Signaling
Through RNA-seq analysis, we found that the downregulation of METTL3 could cause a large number of changes in gene expression levels, and the top 10 KEGG pathways of differentially expressed genes caused by METTL3 knockdown are shown in Figure 7A. Among them, the PI3K/AKT signaling pathway was included. A previous study showed that ADAMTS9 exerted antitumor effects by inhibiting the PI3K/AKT/mTOR pathway in GC (26). In our work, the PI3K/AKT signaling pathway was significantly inactivated due to the downregulation of PI3K and AKT phosphorylation in METTL3-knockdown GC cells. Moreover, ADAMTS9 inhibition reactivated the phosphorylation of PI3K and AKT (Figure 7B). These results indicated that METTL3 might facilitate GC progression through ADAMTS9-mediated PI3K/AKT signaling.
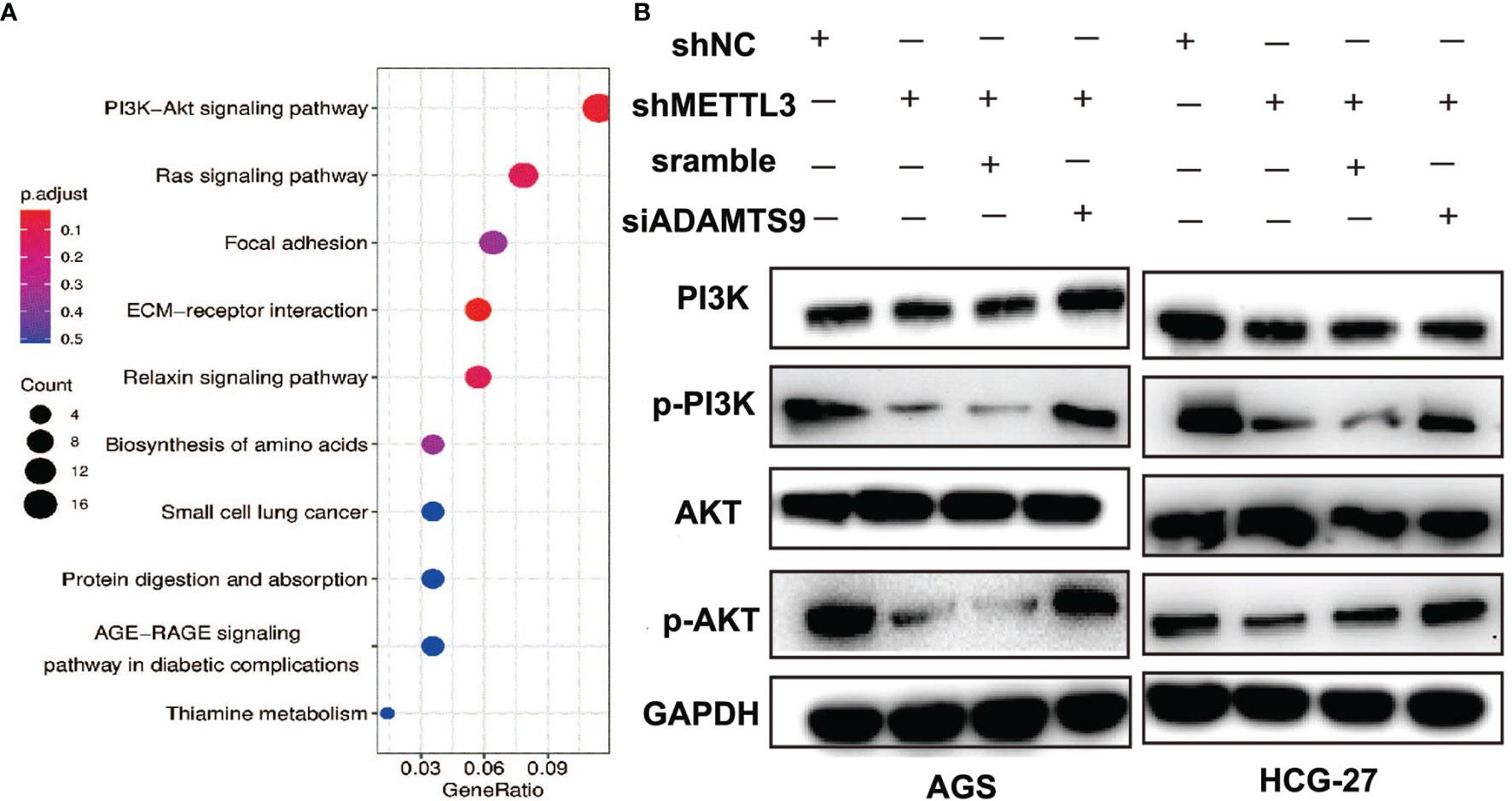
Figure 7 METTL3 promotes GC progression through ADAMTS9-mediated PI3K/AKT signaling. (A) Top 10 enrichment KEGG pathways of dysregulated genes in METTL3-knockdown GC cells. (B) The activation of PI3K/AKT signal pathway was evaluated using Western blot in GC cells with indicated gene knockdown. GAPDH was used as a loading control.
Discussion
The clinical relevance and functional role of METTL3 in malignancies are controversial. In most studies, METTL3 has been recognized as a potential oncogene in human cancers (29). However, few studies have also provided the converse findings regarding METTL3 expression and its tumor-suppressing role in endometrial and breast cancer (30, 31). A recent study indicated that METTL3 is overexpressed in GC patients who have a shortened survival time (32). Another study demonstrated that the METTL3/BATF2/p53/ERK axis suppresses GC development by modulating cell-cycle progression and extracellular matrix degradation (33). Consistently, our findings support METTL3 as a candidate oncogene that is markedly upregulated and dramatically associated with poor outcome in GC.
Previous studies have found a series of molecular targets of METTL3 involved in the carcinogenesis of GC, such as HDGF (34) and SEC62 (35). In a recent study, the METTL3/SPHK2/KLF2 axis was demonstrated to promote the proliferation and metastasis of GC cells (36). Another study revealed that THAP7-AS1, modified by METTL3-mediated m6A, plays an oncogenic role in GC cells by repressing the transcription of miR-320a and miR-22-3p (37). In the present study, our data suggested that ADAMTS9 could be a new potential downstream target of METTL3 involved in GC carcinogenesis and angiogenesis. ADAMTS9 belongs to the ADAMTS protein family and is frequently downregulated as a result of promoter hypermethylation in a variety of human cancers (38). In addition, ADAMTS9 is considered to be an independent prognostic factor of GC (26). Our findings might be conducive to gaining a better understanding of the mechanisms of METTL3 in GC pathogenesis.
YTHDF1/2/3 are the core reading proteins for m6A modification. The relationship between ADAMTS9 and YTHDF family proteins is unclear. In a recent study, ADAMTS9 was found to regulate mRNA decay via the m6A/mRNA pathway as a target of YTHDF2 in spermatogenesis (39). Here, we consistently confirmed the regulatory role of YTHDF2, but not YTHDF1/3, on ADAMTS9 in GC. Briefly, YTHDF2 modulated the ADAMTS9 mRNA half-life in a METTL3-dependent manner, which may interfere with the antitumor functions of ADAMTS9. Our work provides novel insights into METTL3-mediated GC growth and angiogenesis via the YTHDF2/ADAMTS9 axis.
In addition, ADAMTS9 has been identified to inhibit tumor progression by regulating the PI3K/AKT/mTOR pathway (40, 41). The latest studies have shown that PI3K/AKT/mTOR can also be activated by METTL3-mediated M6A modification in ovarian cancer (42) and retinoblastoma (43). Here, by analyzing differentially expressed genes caused by METTL3 knockdown, the PI3K/AKT signaling pathway was proven to be correlated with METTL3 expression in GC. Further experiments verified that p-PI3K and p-AKT were downregulated after METTL3 knockdown, while ADAMTS9 inhibition increased the expression of p-PI3K and p-AKT in GC cells. Our results indicated that METTL3 might facilitate GC progression through the ADAMTS9-mediated PI3K/AKT signaling pathway.
In conclusion, we demonstrated a new METTL3-related signaling pathway that facilitates angiogenesis and carcinogenesis in GC. In brief, METTL3 overexpression in GC promoted the m6A modification of ADAMTS9, a tumor-suppressor gene, and epigenetically prevented its transcription in a YTHDF2-dependent manner. This work sheds light on the pathological role and molecular mechanism of METTL3 and further supports that METTL3 could be a candidate prognostic biomarker and therapeutic target for GC.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Nanjing First Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the ethics committee of Nanjing First Hospital.
Author Contributions
XW, HX, and XS designed the experiments. NW, XH, and XC performed the experiments. NW, SZ, and XH analyzed and confirmed all data and wrote the manuscript. BZ assessed all content. All authors reviewed the manuscript. XW and HX made a final approval. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Grants from the National Natural Science Foundation of China (Grant No. 81773240), The Natural Science Foundation of Jiangsu Province (Grant No. BK20181118), Jiangsu Provincial Special Program of Medical Science (BE2019617), and Nanjing Medical Science and Technique Development Foundation (Grant No. QRX17062).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YY declared a shared parent affiliation with the authors to the handling editor at the time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.861807/full#supplementary-material
Supplementary Figure 1 | (A) Efficient knockdown and overexpression of METTL3 were established in GC cells. (B) The m6A content of total RNAs in METTL3 knockdown GC cells. (C) The proliferative ability of GC cells after METTL3 knockdown. (D) The proliferative ability of GC cells after METTL3 overexpression. (E) The ADAMTS9 mRNA expression levels were evaluated in GC cells after YTHDF1 or 3 knockdown.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
2. Zeng Y, Jin RU. Molecular Pathogenesis, Targeted Therapies, and Future Perspectives for Gastric Cancer. Semin Cancer Biol (2021). doi: 10.1016/j.semcancer.2021.12.004
3. Deng Y, Cai S, Shen J, Peng H. Tetraspanins: Novel Molecular Regulators of Gastric Cancer. Front Oncol (2021) 11:702510. doi: 10.3389/fonc.2021.702510
4. Moro H, Hattori N, Nakamura Y, Kimura K, Imai T, Maeda M, et al. Epigenetic Priming Sensitizes Gastric Cancer Cells to Irinotecan and Cisplatin by Restoring Multiple Pathways. Gastric Cancer (2020) 23:105–15. doi: 10.1007/s10120-019-01010-1
5. Tian Y, Ma R, Sun Y, Liu H, Zhang H, Sun Y, et al. SP1-Activated Long Noncoding RNA lncRNA GCMA Functions as a Competing Endogenous RNA to Promote Tumor Metastasis by Sponging miR-124 and miR-34a in Gastric Cancer. Oncogene (2020) 39:4854–68. doi: 10.1038/s41388-020-1330-4
6. Vedeld HM, Goel A, Lind GE. Epigenetic Biomarkers in Gastrointestinal Cancers: The Current State and Clinical Perspectives. Semin Cancer Biol (2018) 51:36–49. doi: 10.1016/j.semcancer.2017.12.004
7. Desrosiers R, Friderici K, Rottman F. Identification of Methylated Nucleosides in Messenger RNA From Novikoff Hepatoma Cells. Proc Natl Acad Sci USA (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
8. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The Role of M6a Modification in the Biological Functions and Diseases. Signal Transduct Target Ther (2021) 6:74. doi: 10.1038/s41392-020-00450-x
9. Wang M, Liu J, Zhao Y, He R, Xu X, Guo X, et al. Upregulation of METTL14 Mediates the Elevation of PERP mRNA N(6) Adenosine Methylation Promoting the Growth and Metastasis of Pancreatic Cancer. Mol Cancer (2020) 19:130. doi: 10.1186/s12943-020-01249-8
10. Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP Facilitates Progression of Hepatocellular Carcinoma via M6a-HUR-Dependent Epigenetic Silencing of ETS1. Mol Cancer (2019) 18:127. doi: 10.1186/s12943-019-1053-8
11. Lan T, Li H, Zhang D, Xu L, Liu H, Hao X, et al. KIAA1429 Contributes to Liver Cancer Progression Through N6-Methyladenosine-Dependent Post-Transcriptional Modification of GATA3. Mol Cancer (2019) 18:186. doi: 10.1186/s12943-019-1106-z
12. Cui YH, Yang S, Wei J, Shea CR, Zhong W, Wang F, et al. Autophagy of the M(6)A mRNA Demethylase FTO is Impaired by Low-Level Arsenic Exposure to Promote Tumorigenesis. Nat Commun (2021) 12:2183. doi: 10.1038/s41467-021-22469-6
13. Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 Suppresses Malignancy of Hepatocellular Carcinoma via M(6)A-Guided Epigenetic Inhibition of LYPD1. Mol Cancer (2020) 19:123. doi: 10.1186/s12943-020-01239-w
14. Wang H, Luo Q, Kang J, Wei Q, Yang Y, Yang D, et al. YTHDF1 Aggravates the Progression of Cervical Cancer Through M(6)A-Mediated Up-Regulation of RANBP2. Front Oncol (2021) 11:650383. doi: 10.3389/fonc.2021.650383
15. Shen J, Feng XP, Hu RB, Wang H, Wang YL, Qian JH, et al. N-Methyladenosine Reader YTHDF2-Mediated Long Noncoding RNA FENDRR Degradation Promotes Cell Proliferation in Endometrioid Endometrial Carcinoma. Lab Invest (2021) 101:775–84. doi: 10.1038/s41374-021-00543-3
16. Zhang Y, Wang X, Zhang X, Wang J, Ma Y, Zhang L, et al. RNA-Binding Protein YTHDF3 Suppresses Interferon-Dependent Antiviral Responses by Promoting FOXO3 Translation. Proc Natl Acad Sci USA (2019) 116:976–81. doi: 10.1073/pnas.1812536116
17. Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A Novel Hypoxic Long Noncoding RNA KB-1980E6.3 Maintains Breast Cancer Stem Cell Stemness via Interacting With IGF2BP1 to Facilitate C-Myc mRNA Stability. Oncogene (2021) 40:1609–27. doi: 10.1038/s41388-020-01638-9
18. Regue L, Zhao L, Ji F, Wang H, Avruch J, Dai N. RNA M6a Reader IMP2/IGF2BP2 Promotes Pancreatic Beta-Cell Proliferation and Insulin Secretion by Enhancing PDX1 Expression. Mol Metab (2021) 48:101209. doi: 10.1016/j.molmet.2021.101209
19. Yang Z, Wang T, Wu D, Min Z, Tan J, Yu B. RNA N6-Methyladenosine Reader IGF2GF3 Regulates Cell Cycle and Angiogenesis in Colon Cancer. J Exp Clin Cancer Res (2020) 39:203. doi: 10.1186/s13046-020-01714-8
20. Guo H, Wang B, Xu K, Nie L, Fu Y, Wang Z, et al. M(6)A Reader HNRNPA2B1 Promotes Esophageal Cancer Progression via Up-Regulation of ACLY and ACC1. Front Oncol (2020) 10:553045. doi: 10.3389/fonc.2020.553045
21. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-Molecule Inhibition of METTL3 as a Strategy Against Myeloid Leukaemia. Nature (2021) 593:597–601. doi: 10.1038/s41586-021-03536-w
22. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. MRNA Circularization by METTL3-EIF3H Enhances Translation and Promotes Oncogenesis. Nature (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
23. Zheng W, Dong X, Zhao Y, Wang S, Jiang H, Zhang M, et al. Multiple Functions and Mechanisms Underlying the Role of METTL3 in Human Cancers. Front Oncol (2019) 9:1403. doi: 10.3389/fonc.2019.01403
24. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression Through YTHDF2-Dependent Posttranscriptional Silencing of SOCS2. Hepatology (2018) 67:2254–70. doi: 10.1002/hep.29683
25. Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in Cancer: Mechanisms and Therapeutic Targeting. J Hematol Oncol (2020) 13:117. doi: 10.1186/s13045-020-00951-w
26. Du W, Wang S, Zhou Q, Li X, Chu J, Chang Z, et al. ADAMTS9 is a Functional Tumor Suppressor Through Inhibiting AKT/mTOR Pathway and Associated With Poor Survival in Gastric Cancer. Oncogene (2013) 32:3319–28. doi: 10.1038/onc.2012.359
27. Dai XY, Shi L, Li Z, Yang HY, Wei JF, Ding Q. Main N6-Methyladenosine Readers: YTH Family Proteins in Cancers. Front Oncol (2021) 11:635329. doi: 10.3389/fonc.2021.635329
28. Wang JY, Lu AQ. The Biological Function of M6a Reader YTHDF2 and its Role in Human Disease. Cancer Cell Int (2021) 21:109. doi: 10.1186/s12935-021-01807-0
29. Cai Y, Feng R, Lu T, Chen X, Zhou X, Wang X. Novel Insights Into the M(6)A-RNA Methyltransferase METTL3 in Cancer. biomark Res (2021) 9:27. doi: 10.1186/s40364-021-00278-9
30. Shi Y, Zheng C, Jin Y, Bao B, Wang D, Hou K, et al. Reduced Expression of METTL3 Promotes Metastasis of Triple-Negative Breast Cancer by M6a Methylation-Mediated COL3A1 Up-Regulation. Front Oncol (2020) 10:1126. doi: 10.3389/fonc.2020.01126
31. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. M(6)A mRNA Methylation Regulates AKT Activity to Promote the Proliferation and Tumorigenicity of Endometrial Cancer. Nat Cell Biol (2018) 20:1074–83. doi: 10.1038/s41556-018-0174-4
32. Yang DD, Chen ZH, Yu K, Lu JH, Wu QN, Wang Y, et al. METTL3 Promotes the Progression of Gastric Cancer via Targeting the Myc Pathway. Front Oncol (2020) 10:115. doi: 10.3389/fonc.2020.00115
33. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-Mediated N6-Methyladenosine Modification is Critical for Epithelial-Mesenchymal Transition and Metastasis of Gastric Cancer. Mol Cancer (2019) 18:142. doi: 10.1186/s12943-019-1065-4
34. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-Mediated M(6)A Modification of Hdgf Mrna Promotes Gastric Cancer Progression and has Prognostic Significance. Gut (2020) 69:1193–205. doi: 10.1136/gutjnl-2019-319639
35. He H, Wu W, Sun Z, Chai L. MIR-4429 Prevented Gastric Cancer Progression Through Targeting METTL3 to Inhibit M(6)A-Caused Stabilization of SEC62. Biochem Biophys Res Commun (2019) 517:581–7. doi: 10.1016/j.bbrc.2019.07.058
36. Huo FC, Zhu ZM, Zhu WT, Du QY, Liang J, Mou J. METTL3-Mediated M(6)a Methylation of SPHK2 Promotes Gastric Cancer Progression by Targeting KLF2. Oncogene (2021) 40:2968–81. doi: 10.1038/s41388-021-01753-1
37. Liu HT, Zou YX, Zhu WJ, Sen-Liu, Zhang GH, Ma RR, et al. LncRNA THAP7-AS1, Transcriptionally Activated by SP1 and Post-Transcriptionally Stabilized by METTL3-Mediated M6a Modification, Exerts Oncogenic Properties by Improving CUL4B Entry Into the Nucleus. Cell Death Differ (2021) 27:627-41. doi: 10.1038/s41418-021-00879-9
38. Chen J, Cheng L, Zou W, Wang R, Wang X, Chen Z. ADAMTS9-AS1 Constrains Breast Cancer Cell Invasion and Proliferation via Sequestering Mir-301b-3p. Front Cell Dev Biol (2021) 9:719993. doi: 10.3389/fcell.2021.719993
39. Huang T, Liu Z, Zheng Y, Feng T, Gao Q, Zeng W. YTHDF2 Promotes Spermagonial Adhesion Through Modulating MMPs Decay via M(6)a/Mrna Pathway. Cell Death Dis (2020) 11:37. doi: 10.1038/s41419-020-2235-4
40. Duan S, Huang W, Liu X, Liu X, Chen N, Xu Q, et al. IMPDH2 Promotes Colorectal Cancer Progression Through Activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 Signaling Pathways. J Exp Clin Cancer Res (2018) 37:304. doi: 10.1186/s13046-018-0980-3
41. Tan X, Zhang Z, Yao H, Shen L. TIM-4 Promotes the Growth of Colorectal Cancer by Activating Angiogenesis and Recruiting Tumor-Associated Macrophages via the PI3K/AKT/mTOR Signaling Pathway. Cancer Lett (2018) 436:119–28. doi: 10.1016/j.canlet.2018.08.012
42. Bi X, Lv X, Liu D, Guo H, Yao G, Wang L, et al. METTL3-Mediated Maturation of miR-126-5p Promotes Ovarian Cancer Progression via PTEN-Mediated PI3K/AKT/mTOR Pathway. Cancer Gene Ther (2021) 28:335–49. doi: 10.1038/s41417-020-00222-3
Keywords: gastric cancer, N6-methyladenosine, METTL3, YTHDF2, ADAMTS9
Citation: Wang N, Huo X, Zhang B, Chen X, Zhao S, Shi X, Xu H and Wei X (2022) METTL3-Mediated ADAMTS9 Suppression Facilitates Angiogenesis and Carcinogenesis in Gastric Cancer. Front. Oncol. 12:861807. doi: 10.3389/fonc.2022.861807
Received: 25 January 2022; Accepted: 22 March 2022;
Published: 28 April 2022.
Edited by:
Tian Li, Independent Researcher, Xi’an, ChinaReviewed by:
Tristan M. Carland, Q² Solutions, United StatesYang Yang, Nanjing Drum Tower Hospital, China
Copyright © 2022 Wang, Huo, Zhang, Chen, Zhao, Shi, Xu and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuesong Shi, c3hzNTE5M0BxcS5jb20=; Hao Xu, aHh1QG5qbXUuZWR1LmNu; Xiaowei Wei, Z3N3eHdAbmptdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship