
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 23 June 2022
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.858480
This article is part of the Research TopicFerroptosis in Malignant Brain TumorsView all 18 articles
 Luciano Ferrada1*María José Barahona1,2
Luciano Ferrada1*María José Barahona1,2 Katterine Salazar1,2
Katterine Salazar1,2 Alejandro S. Godoy3Matias Vera1
Alejandro S. Godoy3Matias Vera1 Francisco Nualart1,2*
Francisco Nualart1,2*Neuroblastomas are the main extracranial tumors that affect children, while glioblastomas are the most lethal brain tumors, with a median survival time of less than 12 months, and the prognosis of these tumors is poor due to multidrug resistance. Thus, the development of new therapies for the treatment of these types of tumors is urgently needed. In this context, a new type of cell death with strong antitumor potential, called ferroptosis, has recently been described. Ferroptosis is molecularly, morphologically and biochemically different from the other types of cell death described to date because it continues in the absence of classical effectors of apoptosis and does not require the necroptotic machinery. In contrast, ferroptosis has been defined as an iron-dependent form of cell death that is inhibited by glutathione peroxidase 4 (GPX4) activity. Interestingly, ferroptosis can be induced pharmacologically, with potential antitumor activity in vivo and eventual application prospects in translational medicine. Here, we summarize the main pathways of pharmacological ferroptosis induction in tumor cells known to date, along with the limitations of, perspectives on and possible applications of this in the treatment of these tumors.
Cancer is one of the most frequent pathologies worldwide; according to the World Health Organization (WHO) statistics, there were 18.1 million new cases and 9.6 million deaths related to this disease in 2018 (https://www.who.int/news-room/fact-sheets/detail/cancer). Cancers are difficult to treat because they employ multiple molecular mechanisms to evade different types of cell death, such as apoptosis, due to their overexpression of antiapoptotic proteins such as Bcl-2 and Bcl-xL and low expression of proapoptotic factors such as Bax, Bim and Puma (Figures 1A, B) (1). At the same time, it is known that the low efficacy of apoptosis induction with conventional therapies is due to the robust antioxidative defenses of tumor cells (2). Among the main antioxidants that confer apoptosis resistance on tumor cells is glutathione (GSH) (3, 4).

Figure 1 Survival programs in normal and tumor cells. (A) Under physiological conditions, normal cells maintain stable levels of death-executing proteins while maintaining a constant balance of nutrients and trace elements, promoting cell survival. (B) To avoid death, tumor cells activate various mechanisms, such as decreasing the expression of proapoptotic and necroptotic genes while increasing antioxidant defense by increasing GSH synthesis and GPX4 levels. In this way, ROS are efficiently eliminated, avoiding the damage produced by the accumulation of iron due to low FPN levels. This death evasion program makes many types of cancer highly difficult to treat, as classical apoptosis induction therapies fail because the machinery for the execution of this pathway is not available. FPN, Ferroportin; TfR1, Transferrin Receptor 1; DMT1, Divalent Metal Transporter 1; Cys2, Cystine; Cys, Cysteine; LIP, Labile Iron Pool.
Due to the high resistance of tumors to apoptosis, the induction of necroptosis was postulated to be a potential therapeutic approach (5, 6). In contrast to apoptosis, which does not generate an inflammatory response, necroptosis induces death by cellular explosion, which generates a microenvironment of proinflammatory signals that could favor tumor death (5, 6). Thus, necroptosis, a form of regulated necrosis dependent on RIPK1, RIPK3 and MLKL, was postulated as a potential therapy for cancer (Figures 1A, B) (7–9). Unfortunately, several tumor cells evade necroptosis efficiently by inhibiting the expression of RIPK3 via epigenetic control mechanisms (10–12).
In line with this idea, new and emerging forms of regulated cell death with characteristics of necrotic disintegration have been described and postulated as treatments for cancer; among these, ferroptosis is highlighted (13–19). Here, we describe the main pharmacological targets for the induction of ferroptosis with emphasis on the treatment of brain tumors.
System is an antiporter that imports cystine and exports glutamate from the cell in a 1:1 ratio (Figures 1A, B). System is composed of 2 subunits: the SLC7A11 subunit (also called xCT), with a transport function and solute carrier family 3 member 2 (SLC3A2; also called CD98hc or 4F2hc), a chaperone with a plasma membrane anchoring function (20–23). For the purposes of this review, we refer only to the SLC7A11 subunit, given the importance of cystine transport to the cell (Figures 1A, B). In this context, the uptake of cystine into the cell is essential to maintain the redox state, since the reduced form of this amino acid (nonessential) is necessary for the biosynthesis of the main intracellular antioxidant, glutathione (GSH) (Figure 1A). Interestingly, most cancer cells overexpress SLC7A11 (22), suggesting a strong dependence on GSH to maintain the levels of controlled reactive oxygen species (ROS) (Figure 1B); thus, SLC7A11 is an potential therapeutic target. Interestingly, in 2012, it was determined that the small molecule erastin (13, 24) targeted SLC7A11 for inhibition, which led to depletion of GSH, inducing a type of death dependent on iron and lipid ROS, called ferroptosis (14). This type of cell death was inhibited by radical trapping antioxidants (RTAs) such as Ferrostatin-1 (Fer-1), lipophilic antioxidants such as vitamin E or iron chelators such as Desferoxamine (DFO) (18, 25), with potential application in the treatment of cancer and other pathologies (Figure 2B) (26).
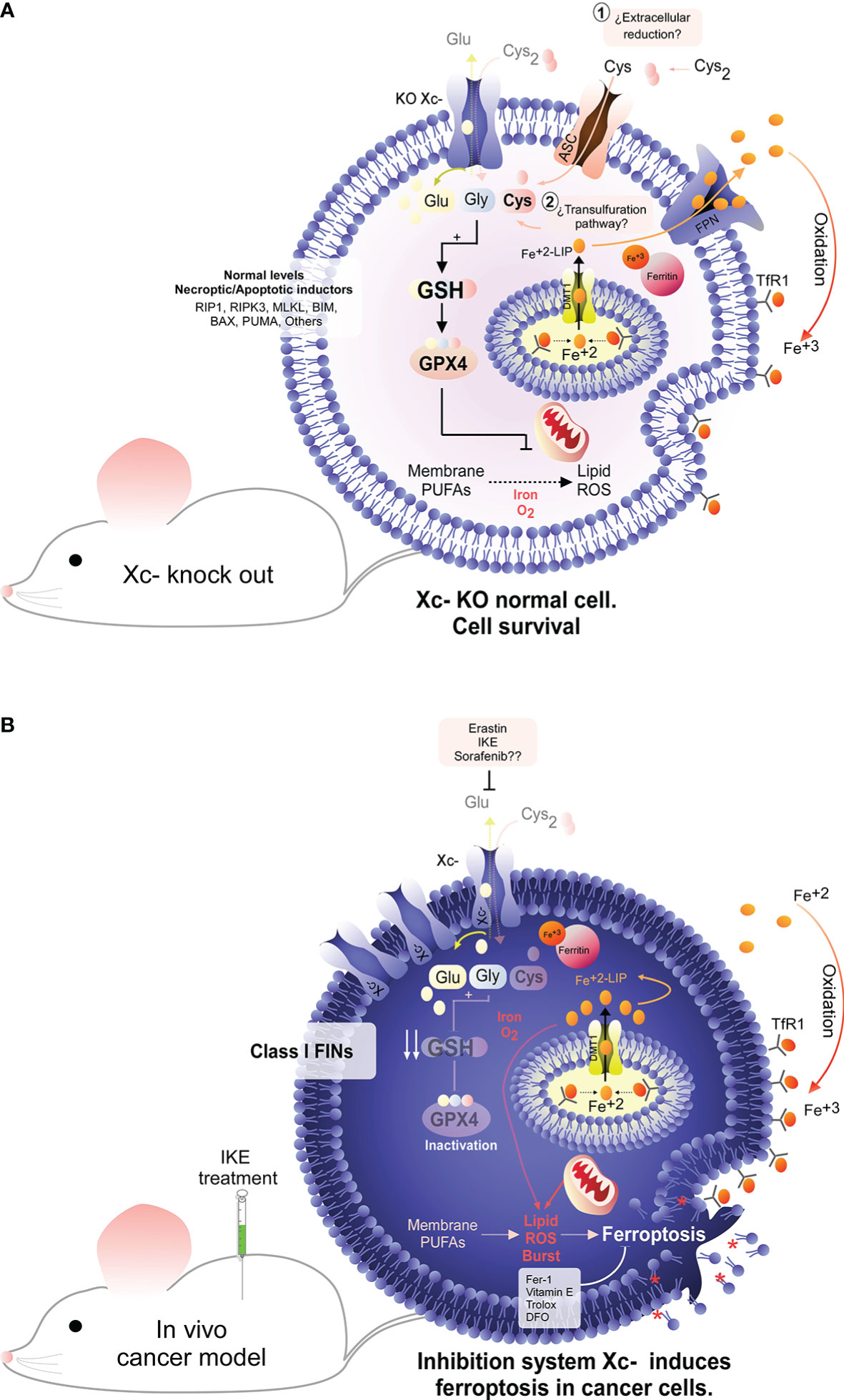
Figure 2 System dependence in cancer cells. Under physiological conditions, the nonessential amino acid cysteine is present as cystine due to the extracellular oxidative environment. To maintain a stable intracellular cysteine level, the presence of the cystine/glutamate antiporter (system ) is necessary. Interestingly, genetic deletion of system does not produce any damage in animals, suggesting that normal cells do not depend on this antiporter to maintain the intracellular cysteine level. In line with this idea, compensatory mechanisms, such as the transsulfuration pathway, may exist for the recovery of the intracellular cysteine level (A). Conversely, it has been widely described that tumor cells have a high dependence on system for the cellular uptake of cysteine (B). Pharmacological inhibition of this antiporter results in the depletion of intracellular cysteine, inducing an abrupt decrease in the GSH level, which ultimately triggers inactivation of GPX4, the main hydroperoxidase in the cell. Inactivation of GPX4 due to inhibition of system results in an overwhelming overload of lipid ROS that ultimately induces tumor death by ferroptosis (B). Interestingly, it has been determined that inhibition of system can induce tumor death both in vitro and in vivo, identifying this antiporter as a potential therapeutic target for cancer.
Thus, when tumor cells are incubated with erastin, cell death is induced independent of caspases (13) or mitochondrial oxidative stress but in a manner dependent on iron, ROS and lipid ROS (14). Even though there is evidence that mitochondria could be involved, regulating the “avidity” for ferroptosis induction (27–29), they are not necessary for activation of this pathway (30). Inhibition of system results in intracellular depletion of cysteine because extracellular cystine (Cys2) is imported through SLC7A11 and reduced intracellularly to cysteine (Figure 2B) (16, 31). Intracellular cysteine is necessary for the biosynthesis of GSH (16, 32). In turn, GSH is a cofactor for the selenoprotein GPX4, a hydroperoxidase responsible for detoxifying toxic hydroperoxides to alcohols (15). Therefore, erastin triggers indirect inhibition of GPX4 activity mediated by GSH depletion (Figure 2B).
Despite this apparent dependence of cells on system , animals with knockout of the slc7a11 gene are fertile and develop completely normally (33), which prompted the consideration of SLC7A11 inhibition as an eventual cancer therapy with few adverse effects (Figure 2A).
Thus, although many tumor cells can evade apoptosis and necroptosis due to their low expression of key genes for the activation of these pathways (Figure 1B) (1, 10, 11), RNA-seq data show that most cancer cells have high expression levels of SLC7A11 and GPX4 (https://portals.broadinstitute.org/ccle). Similarly, tumor cells are “addicted” to iron because they have decreased expression of ferroportin (FPN), the iron efflux pump, and overexpress the transferrin receptor (TfR1), the iron importer (Figure 1B) (34–37). Indeed, excess iron contributes to both tumor initiation and tumor growth (34). These observations indicate that SLC7A11, GPX4, iron and ferroptosis are potential therapeutic targets for cancer (Figures 2B, 3). However, there are cancer cells that do not express FPN (MCF-7 cells, among others) and therefore accumulate excess intracellular iron but are still resistant to ferroptosis (38, 39). An explanation for this phenomenon is the recent finding that in addition to GPX4 and iron, acyl-CoA synthetase long-chain family member 4 (ACSL4) is another component that dictates sensitivity to ferroptosis (39). Reinforcing this concept, ACSL4 is a key protein because it incorporates long polyunsaturated fatty acids (PUFAs) into membranes, which allows lipid peroxidation to proceed and ferroptosis to be carried out (39–41). In another context, the erastin analog imidazole ketone erastin (IKE) has been shown to be metabolically stable and a potent inducer of ferroptosis in tumor cells in vivo (19, 42). Thus, induction of ferroptosis in tumor cells through inhibition of SLC7A11 may be a promising treatment for use in patients.
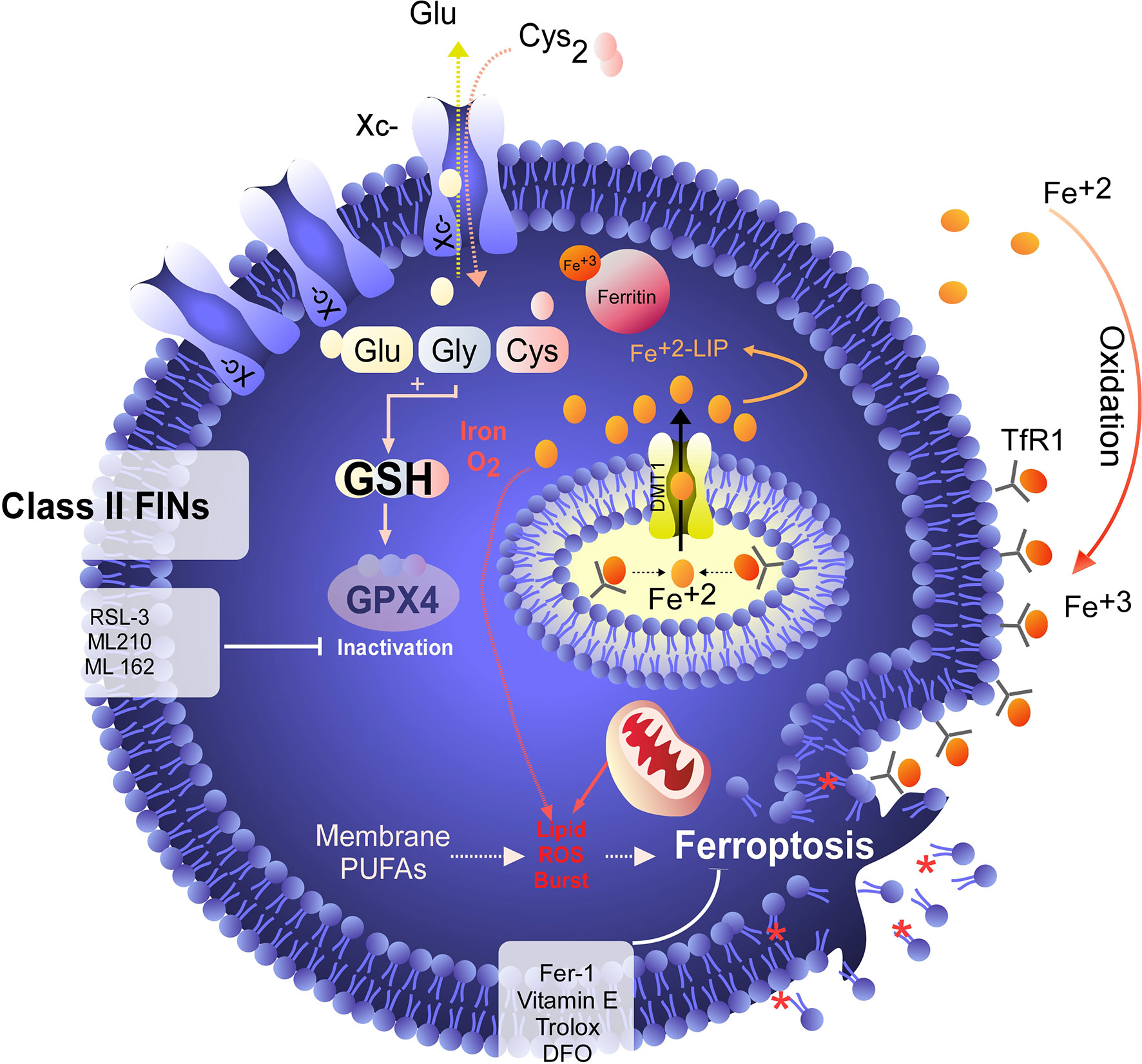
Figure 3 GPX4 as a target for ferroptosis induction. Unlike class I FINs, which indirectly inactivate GPX4, class II FINs such as RSL-3 directly inhibit GPX4, triggering ferroptosis independent of the GSH level. Direct inhibition of GPX4 results in rapid induction of ferroptosis, which can be inhibited by RTA or iron chelators. However, cell death is not inhibited by the recovery of cysteine uptake.
Interestingly, high doses of glutamate can inhibit system , emulating the effects induced by erastin (14, 43, 44). However, it is known that the responses to glutamate treatment are diverse and can induce cell death by apoptosis or necroptosis (45, 46) and eventually by other pathways of regulated necrosis. Thus, although high doses of glutamate can inhibit system , they are not necessarily a specific inducer of ferroptosis in tumor cells but could induce ferroptosis in normal tissues under pathophysiological conditions (44, 47–49).
Although the concept of ferroptosis was initially described in response to treatment with erastin, various ferroptosis inducers (FINs) have been developed to act independently of cystine uptake and GSH levels. FINs are currently classified into four classes (I-IV) (40, 50): class I FINs induce GSH depletion (Figure 2B), class II FINs inhibit GPX4 (Figure 3), class III FINs deplete GPX4 (Figure 4), and class IV FINs act through iron oxidation/iron overload (Figure 5) (summarized in Table 1). Interestingly, two research groups recently described a new player in the regulation of ferroptosis in parallel: ferroptosis suppressor protein 1 (FSP1) (Figure 4) (54, 55). Previously called apoptosis-inducing mitochondria-associated factor 2 (AIFM2), FSP1 is a flavoprotein with extramitochondrial oxidoreductase activity that can be recruited into the plasma membrane due to myristoylation. FSP1 in the plasma membrane catalyzes the conversion of ubiquinone (coenzyme Q10, CoQ10) to ubiquinol at the expense of NADPH (56).
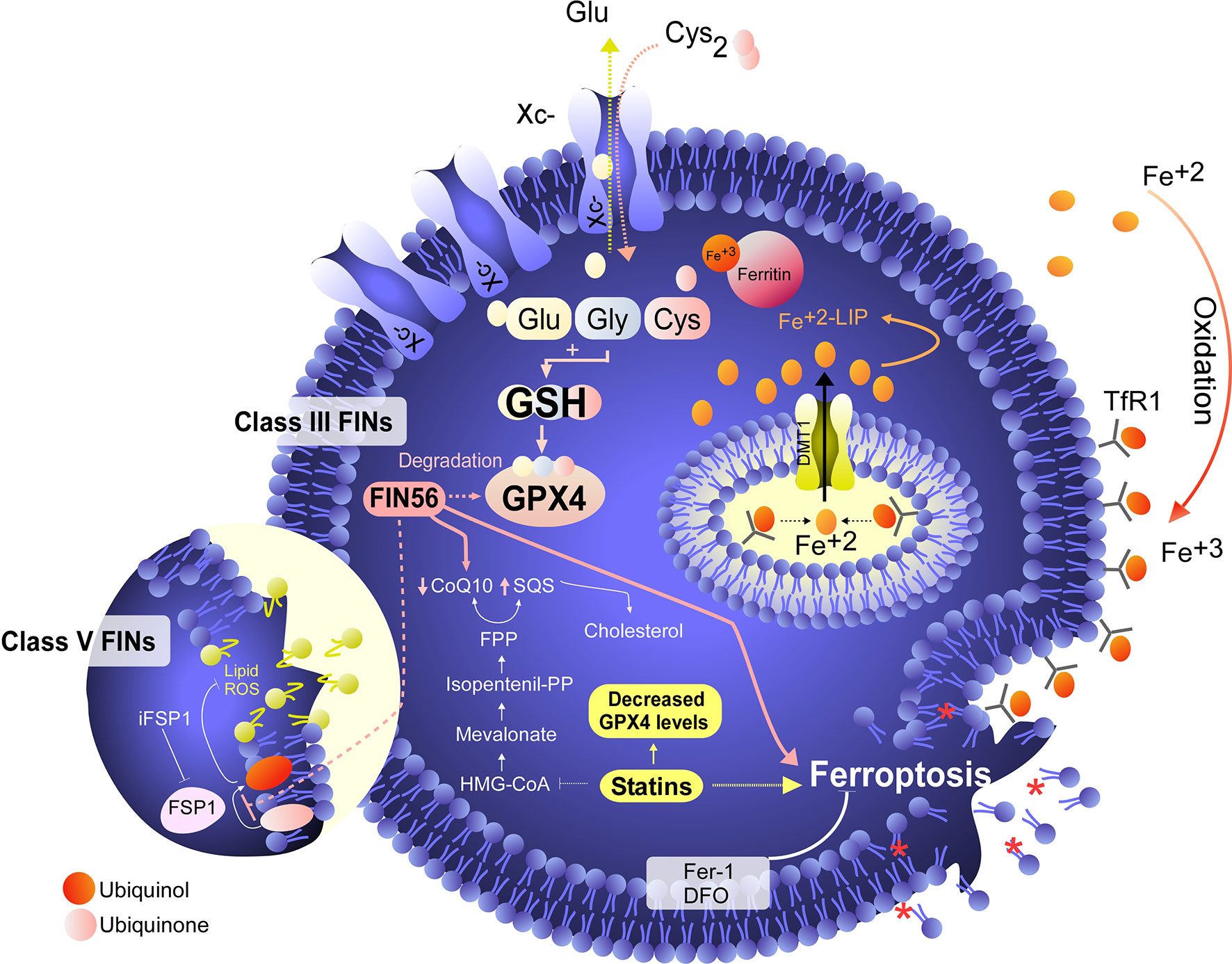
Figure 4 Degradation of GPX4/CoQ10 or inhibition of FSP1 induces ferroptosis in cancer cells. Class III FINs are molecules that act independently of system activity, the GSH level and direct inhibition of GPX4. These molecules, including FIN56, induce degradation of GPX4, which leads to ferroptosis induction. In addition to degrading GPX4, FIN56 also induces degradation of coenzyme Q10 (ubiquinone) by altering the mevalonate pathway. The importance of coenzyme Q10 degradation in the execution of ferroptosis is assumed because the function of a protein called FSP1 (a class V FIN) was recently described (33, 34). In this scenario, FSP1 converts extramitochondrial ubiquinone (the oxidized form of coenzyme Q10) to extramitochondrial ubiquinol (the reduced form of coenzyme Q10), and ubiquinol acts as an endogenous RTA that inhibits ferroptosis independent of the presence of GPX4. In this context, by inducing coenzyme Q10 degradation, FIN56 can inhibit the effects of FSP1 to confer resistance to ferroptosis. On the other hand, the inhibitor of FSP1 (iFSP1) controls ferroptosis without degrading CoQ10.
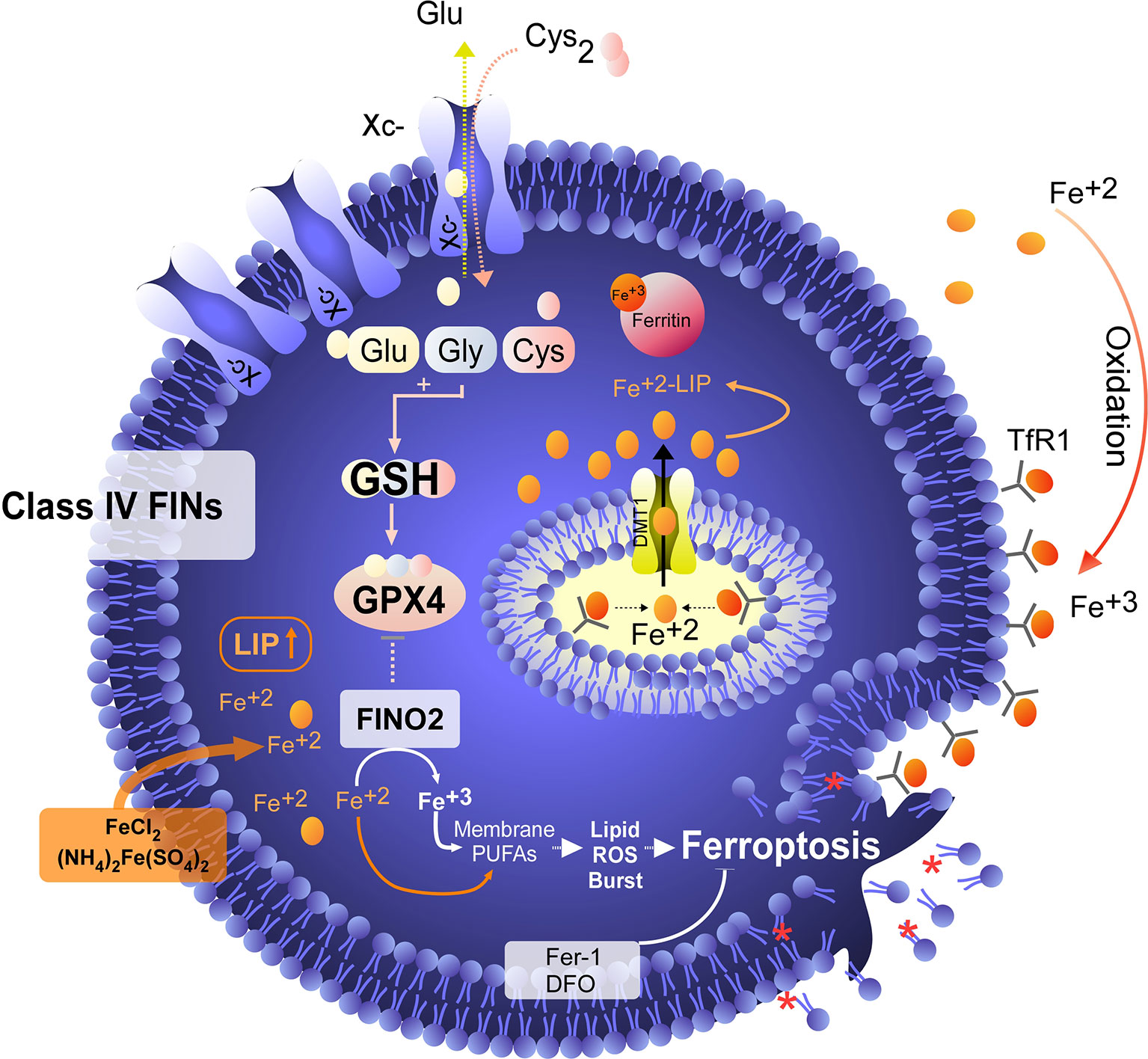
Figure 5 Iron overload or peroxidation induces ferroptosis in tumor cells. Class IV FINs are ferroptosis inducers that directly involve metabolism and iron levels in the cell. On the one hand, we have found synthetic molecules, such as FINO2, that alter the metabolism of iron, favoring its intracellular oxidation. In addition to promoting the oxidation of iron, FINO2 indirectly inhibits the activity of GPX4. On the other hand, when the labile iron pool (LIP) is increased by exogenous treatment with iron or iron nanoparticles, an overload of this metal is generated, which induces lipid peroxidation without the need for GPX4 inhibition. Class IV FINs are fairly attractive agents for the induction of ferroptosis because tumor cells are addicted to iron due to their low ferroportin (FPN) expression and high levels of transferrin receptor (TfR) expression, which favors an increase in the LIP. In this context, treatment with exogenous iron (e.g., FeCl2) in combination with FINO2 would eventually be a potent inducer of ferroptosis in tumor cells. Unfortunately, the development of FINO2 analogs for in vivo use is necessary to test whether the increases LIP and iron peroxidation are synergistic to specifically kill tumor cells.

Table 1 Principal Ferroptosis Inducers.
Thus, the FSP1-ubiquinone-ubiquinol axis inhibits lipid peroxidation and ferroptosis in parallel to GPX4 and independent of GSH levels (54, 55). An inhibitor of FSP1 (iFSP1) (54) that may stimulate the induction of ferroptosis was identified by drug screening. In this context, the iFSP does not fit within any class of FINs (I-IV) because it does not target GPX4 or iron metabolism. Thus, we suggest that FINs that do not target GPX4 or iron metabolism but, as their mechanism involves CoQ10, can be classified into class V (Figure 4). Because FIN56 depletes GPX4 and CoQ10 (52), this compound has a dual classification and should also be reclassified into class V. Despite the existence of various ferroptosis inducers, not all of them have therapeutic potential in vivo (40). However, it has been shown that the use of class IV inducers could have potential therapeutic effects in vivo to treat high-risk neuroblastomas (17).
Normal adult neurons are equipped to survive because they express low levels of proapoptotic proteins and high levels of antiapoptotic proteins (57, 58). Furthermore, it has been shown that as neurons mature, they lose chemosensitivity to staurosporine and doxorubicin (58). This evidence suggests that brain tumors would be highly resistant to conventional antineoplastic agents, given the preconditioning of this type of cell to efficiently evade apoptosis. At the same time, it has been shown that tumor cells of astroglial origin (T98G, U251 and A172) efficiently evade necroptosis induced by chemotherapeutic agents because they do not express RIPK3 due to epigenetic modifications (10, 11).
Thus, the development of new therapies for the treatment of brain tumors that do not involve the induction of apoptosis or necroptosis as the main strategy is urgently needed. In this sense, in recent years, the induction of ferroptosis has gained great relevance as a possible therapeutic approach to induce cell death in brain tumors (17, 50, 59, 60). Considering this concept in the following sections, we focus on the induction of ferroptosis in neuroblastoma (NB) and glioblastoma multiforme (GBM).
NB is the most common pediatric extracranial tumor, accounting for more than 15% of all cancer deaths in children (61). NB is classified as low-, intermediate- and high-risk (62). While low-risk and intermediate-risk NBs generally have a good prognosis given that they develop into benign ganglioneuromas or enter remission due to surgical or pharmacological treatment, high-risk NBs have few treatment options (17, 62, 63). The main diagnostic characteristics of high-risk NB are that it appears after 18 months of age, has MYCN amplification, or exhibits activation of telomere maintenance mechanisms (62, 63). In line with this observation, current therapies against the NB include treatment with cycles of cisplatin, etoposide, vincristine, doxorubicin, and cyclophosphamide (64), which are preferential inducers of apoptosis. However, this type of pharmacological treatment generates multidrug-resistant clones, which greatly hinders the eradication of this type of tumor and favors its relapse (64).
Considering that classical NB eradication therapies generally fail, it has been proposed that the induction of ferroptosis could be a feasible therapeutic approach. In this context, when the sensitivity of NB cell lines to classic ferroptosis inducers such as erastin or RSL-3 was studied (Figures 2B, 3), it was determined that most of the models (SHSY-5Y, SK-N-SH, NB69, SK-N -DZ, NLF, and CHP-134 cells, among others) are highly insensitive to SLC7A11 or GPX4 inhibition (17, 65, 66). At the same time, there is very little information on the potential use of iFSP1 as a possible strategy against NB, since this compound has only been tested in the IMR-5/75 cell line without major effects on viability (54). Based on this background, the scientific community has focused on the search for new strategies for ferroptosis induction in NB, not through the classical targets but instead through the use of combined therapies or noncanonical inducers of ferroptosis, as potential treatments for high-risk NB (17, 67).
Because NB generally presents resistance to Erastin and RSL-3, it is necessary to search for new ferroptosis inducers. To this end, it was recently determined that treatment with the natural compound withaferin A (WA) can eradicate high-risk NB (17) by inducing ferroptosis through the canonical pathway, this means with GPX4 as a direct target. On the other hand, via a noncanonical pathway, where keap1 is the target, thus favoring an increase in labile iron pool (LIP) (17). This dual behavior of WA, similar to that of a mixture of FIN56 and FINO2 (Figures 4, 5) (52, 53) (compounds that have not been tested in NB models), seems to render it a promising drug therapy for NB, since to date, it is unknown whether in vivo application of FINO2 is possible (40). Fortunately, WA has been shown to be effective in promoting the eradication of NB in vivo (17, 50). Interestingly, even though WA has been shown to induce iron-dependent lipid peroxidation and GPX4 depletion, Fer-1 treatment does not completely rescue NB cells from cell death (17). This suggests two alternatives; the first is that WA induces other types of ferroptosis-independent death in NB. However, WA induces lipid peroxidation, which is completely inhibited by treatment with DFO and partially inhibited by Fer-1, suggesting a strong iron dependence (17).
In this context, as a second alternative, the authors suggest that WA could eventually favor an overload of lipid ROS of various origins that may not necessarily be inhibited by Fer-1, such as lipid ROS generated by H2O2 (17). It is important to note that Fer-1 does not inhibit death induced by H2O2 treatment (14) or by extracellular H2O2 production mediated by pharmacological doses of ascorbic acid (68), because these treatments preferentially induce conventional necrosis. However, it has recently been determined that NADPH-cytochrome P450 reductase (POR) favors the induction of ferroptosis due to the cytoplasmic production of H2O2 (69), which is inhibited by Fer-1 or by the intracellular expression of catalase (69) but thus far, this finding is limited to cervical cancer cells (HeLa).
In this context, and considering the particularities of NB cells, it is likely that it is possible to classify the cell death induced in this type of tumor (or others) as ferroptosis, even when it is not inhibited by Fer-1, if it has other hallmarks, such as lipid peroxidation and iron dependence. Indeed, it has recently been determined that inhibition of lipid peroxidation mediated by liproxstatin-1 treatment is not sufficient to rescue SLC7A11 KO melanoma cells from death (70). Furthermore, it has also been shown that there is strong induction of lipid peroxidation during the activation of noncanonical pyroptosis that is not necessarily related to the direct execution of this death pathway (71). This evidence could limit lipid peroxidation as an exclusive hallmark of ferroptosis, driving the definition of ferroptosis toward a type of death dependent on lipid peroxidation (72).
In another context and emphasizing that MYCN is a protein overexpressed in NB, recent advances have been achieved to determine that MYCN favors an increase in intracellular iron per se, which could favor the pharmacological sensitization of NB to ferroptosis induction (66, 67). Thus, the authors determined that inhibition of SLC7A11 with sulfasalazine (14) or inhibition of GSH synthesis with Buthionine sulfoximine (BSO) favors the induction of ferroptosis in models of NB with MYCN amplification (67). This in vitro evidence from patient samples is closely related to an eventual clinical application, since the toxicity of BSO has been evaluated in a phase I clinical trial, as a possible treatment for NB in conjunction with other drugs (73). Despite being relatively well tolerated, the treated patients presented vomiting/nausea as adverse effects (73). However, there is also evidence indicating that the administration of BSO can trigger kidney failure in animal models (74) and patients (75). Thus, special precautions must be taken when trying to directly extrapolate in vitro findings to in vivo models or patients.
Curiously, some of the NB cell lines used in this study show partial resistance to death induced by the SLC7A11 inhibitor and GSH depletor erastin (17), which leads to an intriguing question: why are some NB cell lines resistant to erastin but sensitive to inhibition of GSH synthesis or inhibition of SLC7A11 mediated by sulfasalazine? In this scenario, it is important to highlight that in lung adenocarcinoma cells, it was recently determined that the SLC7A11 inhibitor HG106 preferentially induces GSH depletion and cell death by apoptosis, which is inhibited by the recovery of cysteine uptake, but without eventual induction of ferroptosis, since DFO treatment does not prevent cell death (76). This evidence suggests that although HG106 has the same target as erastin (SLC7A11), there are other off-targets that favor the induction of one type of death over another (apoptosis or ferroptosis) or the production of particular ROS that trigger differential cellular responses (22). Despite these pharmacological dichotomies, which induce different types of death even when the target is the same, or which have differential action mechanisms in response to treatment with SAS, erastin (IKE) or HG106, the message that remains the same: SLC7A11 is a potent therapeutic target for cancer (Figure 2B).
It was shown that NB cells with MYCN amplification are particularly sensitive to the induction of death mediated by treatment with auranofin (a rheumatoid arthritis drug) (67). Although the authors attribute the effect of auranofin to the induction of ferroptosis, treatment with Fer-1 only partially rescues cells from cell death, even when there is an increase in lipid peroxidation, and treatment with DFO effectively prevents cell death and ROS production (67). Again, this finding leads us to conclude that apparently in NBs, the lipid ROS generated are specific to this tumor type or there are parallel mechanisms of cell death, since Fer-1 is not capable of completely inhibiting cell death, even when the evidence points to iron and lipid ROS dependency. Accumulating evidence, the literature indicates that iron accumulation and increased LIP are strong candidates for exploiting the pharmacological sensitivity of high-risk NB to ferroptosis induction (Figure 5) (17, 50, 66, 67, 77). Thus, the use of compounds that promote the mobilization or uptake of iron in this type of tumor, in combination with ferroptosis inducers, could exploit the vulnerabilities of this tumor to favor its eradication. However, further studies are still needed to determine the potential lethal effects on nervous tissue and to assess whether these types of therapeutic agents can penetrate the blood–brain barrier.
Malignant gliomas are one of the most devastating and frequently diagnosed brain tumors in adults and are associated with a short life expectancy of only 12 to 15 months (78). The WHO classifies this type of tumor as grade I to IV, the latter being called glioblastoma multiforme (GBM), which corresponds to the most advanced stage and has a shorter life expectancy (78–80). The current incidence of GBM in the USA is approximately 7 per 100,000 inhabitants (79). Currently, therapy for GBM is based on surgery accompanied by radiation therapy and chemotherapy, since GBM cannot be completely removed surgically due to its infiltrative nature (78). Although radiotherapy increases the life expectancy of patients, 90% of GBMs exhibit recurrence at the original tumor site after therapy (81). Thus, all hopes for the treatment of this type of tumor are placed on the development of new agents or pharmacological strategies for successful chemotherapy. To date, the main pharmacological approaches for the treatment of GBM include the use of antiangiogenic therapies (bevacizumab, sunitinib, vandetanib), immunotherapy (anti-PD-1/PD-L1 antibodies) and various other molecular approaches, such as inhibitors of mTOR, EGFR, HSP90, and PI3K (78, 82). Unfortunately, GBMs acquire resistance to these types of treatment (78, 82). In this scenario, as a therapeutic strategy, one of the most commonly used compounds is temozolomide (TMZ), an oral alkylating agent (80, 83, 84) that targets the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT), which has been shown to prolong the life expectancy of patients when used in conjunction with radiotherapy (84–86). Unfortunately, most GBMs recur after 2 years with cell populations resistant to this type of therapy due to stem cell properties (87–89). Based on accumulating evidence and the strong resistance of GBM to multiple therapies, the development of new drugs for the treatment of these devastating tumors is urgently needed.
Based on the above premise, pharmacological induction of ferroptosis could exploit the vulnerabilities of GBM cells and sensitize them to death when used in combination with other antineoplastic compounds. In line with this idea, the evidence suggests that combined treatment with ferroptosis inducers plus other antineoplastic therapies (e.g., TMZ or radiation) could lead to sensitization to this type of death in GBM cells (60, 90). This is because most GBM cells are resistant to either SLC7A11 inhibition (erastin treatment) or GPX4 inhibition (RSL-3 treatment) (54, 91, 92), although they express ACSL4 (93).
In line with this observation, high levels of SLC7A11 expression are considered to predict poor survival in patients with malignant glioma (94). At the same time, high expression of SLC7A11 is associated with epileptic seizures, stem cell properties, increased migration and invasion, neurosphere formation and increased expression of Nanog, Sox-2 and Nestin, among other proteins (95, 96). Thus, the expression of this transporter is considered a possible biomarker for the diagnosis of GBM. In this scenario, it is tempting to speculate that SLC7A11 blockade could be an excellent therapy for GBM, since its high expression level indicates a strong dependence on its function.
However, current evidence has shown that GBM cells, such as U87, U251, and U373 cells, are highly insensitive to treatment with SAS or erastin (72, 97), a phenotype that could be related to resistance mechanisms mediated by ATF4 and Nrf-2 that favor overexpression of SLC7A11 (21, 97, 98). Furthermore, the use of SAS in a clinical trial against glioma did not show a response, and various adverse effects were observed (99), which greatly complicates its future use as a ferroptosis-inducing drug in patients. It is important to note that various studies have suggested that GBM cells (and cells of other lineages) have unique sensitivity to death (theoretically ferroptotic) mediated by sorafenib treatment (97, 100). However, recently, it was shown that sorafenib failed to induce ferroptosis in a wide panel of tumor cell lines (including GBM cell lines) (72), which leads us to take special care in the interpretation and specificity of sorafenib in triggering ferroptosis.
In this scenario, where GBM cells show great resistance to inhibition of system , it is possible to speculate that they obtain cysteine intracellularly from another source that implies mechanisms independent of the function of SLC7A11, which would explain the resistance to treatment with erastin or SAS. The main metabolic pathway that supplies cysteine intracellularly in tumor cells independent of the transport activity of SLC7A11 is the transsulfuration pathway (101). The transsulfuration pathway allows methionine to be used as a substrate for cysteine biosynthesis through various enzymatic reactions (101). At the same time, it has been shown that inhibition of this pathway in tumor cells makes it possible to recover sensitivity to erastin in certain cell lines other than GBM cell lines (102). Unfortunately, inhibition of the expression of cystathionine β-synthase (CBS), a key protein in the transsulfuration pathway, has been shown to promote GBM progression (103), while in other tumor models, CBS inhibition effectively causes cell death (104), which suggests that GBM cells could be resistant to ferroptosis induction, including that mediated through inhibition of system and the transsulfuration pathway.
On the other hand, one possible explanation for the strong resistance of GBM cells to the induction of ferroptosis is the protective effect exhibited by FSP1 in this type of tumor (Figure 4), since most GBM cells express high levels of this protein, and cotreatment with iFSP1 and RSL-3 strongly sensitizes them to ferroptosis (54, 72). However, cotreatment with erastin or SAS + iFSP1 fails to induce death in GBM cells (72). This evidence corroborates the findings that FSP1 acts independently of the GSH level (54, 55) and that it apparently can only have synergistic effects with direct GPX4 inhibitors such as RSL-3 or ML162.
Interestingly, the GPX4 depletor FIN56 (Figure 4) was recently shown to induce ferroptosis in in vitro and in vivo GBM models (105); this was the first study to use this compound in vivo. However, the trial was not carried out with tumors in nervous tissue but rather in nude mice with subcutaneous tumors, which makes it difficult to extrapolate the possible eventual effects of FIN56 on the brain, and it is not known whether this compound can cross the blood–brain barrier to be considered a potential therapy in the future.
Based on accumulating evidence and given the limitations of the use of direct GPX4 inhibitors for the treatment of tumors in vivo, the best therapeutic approach seems to be inhibition of SLC7A11. Along these lines, it has been demonstrated that cotreatment with IKE and radiation favors ROS production and induces cell death in GBM models (60). Concurrently, cotreatment with erastin and TMZ has been found to sensitize GBM cells to death (90). This eventual therapeutic strategy offered by treatment with SLC7A11 inhibitors should be exploited in the future in the search for compounds with synergistic activity that exploit the vulnerabilities of GBM cells.
Although there are several inducers of ferroptosis, the potential use of these drugs as cancer treatments is limited because they have little bioavailability for action in vivo. However, with the development of IKE, an avenue was opened for ferroptosis induction as an in vivo treatment by targeting system (19). To date, evidence suggests that inhibition of system could be a safe therapeutic approach as a tumor suppressor. Unfortunately, several types of tumors, including NB and GBM, are resistant to system inhibition for ferroptosis induction (13, 17, 92). Thus, combination therapies of other antineoplastic drugs with IKE may represent an option for the treatment of cancers highly resistant to cell death. However, ferroptosis dogma dictates that GPX4 is the key protein (15, 106); thus, all efforts have been focused on the development of new drugs for its inhibition. Although there are direct GPX4 inhibitors, such as RSL3 (15), they have little application in vivo (40), and GPX4 deletion in some types of cancer is not lethal (51), suggesting that there may be other mechanisms in addition to GPX4 inhibition to suppress lethal lipid peroxidation. FSP1, GCH1 and BH4/BH2 are proteins with the ability to inhibit ferroptosis independently of GPX4 and GSH levels (54, 55, 65), and FSP1 is a druggable protein (54). In line with this idea, a new avenue has been opened for the development of drugs that include SLC7A11, GPX4 and FSP1 inhibitors with potential in vivo application as a cancer treatment.
LF, MB, KS, AG and FN conceived the ideas and concepts. LF wrote the article. AG, KS, MV and FN critically revised the manuscript. MB generated the scientific illustrations. All authors approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a Fondecyt regular grants 1181243 and 1221147, ANID PIA ECM-12 grant (to FN), Fondecyt iniciacion 11200335 (LF), Fondecyt postdoctorado 3210076 (MB) and U.S. Department of Defense W81XWH-12-1-0341 (AG).
The authors declare that the research was conducted in the absence of any commercial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, et al. Cancer Cells Co-Opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell (2018) 33(6):985–1003.e7. doi: 10.1016/j.ccell.2018.05.001
3. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell (2015) 27(2):211–22. doi: 10.1016/j.ccell.2014.11.019
4. Lim JKM, Delaidelli A, Minaker SW, Zhang HF, Colovic M, Yang H, et al. Cystine/glutamate Antiporter xCT (SLC7A11) Facilitates Oncogenic RAS Transformation by Preserving Intracellular Redox Balance. Proc Natl Acad Sci Unite States Americ (2019) 116(19):9433–42. doi: 10.1073/pnas.1821323116
5. Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis Microenvironment Directs Lineage Commitment in Liver Cancer. Nature (2018) 562(7725):69–75. doi: 10.1038/s41586-018-0519-y
6. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination With Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep (2016) 15(2):274–87. doi: 10.1016/j.celrep.2016.03.037
7. Jiang YG, Peng Y, Koussougbo KS. Necroptosis: A Novel Therapeutic Target for Glioblastoma. Med Hypoth (2011) 76(3):350–2. doi: 10.1016/j.mehy.2010.10.037
8. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: Mechanisms and Relevance to Disease. Annu Rev Pathol (2017) 12:103–30. doi: 10.1146/annurev-pathol-052016-100247
9. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer Therapy in the Necroptosis Era. Cell Death Diff (2016) 23(5):748–56. doi: 10.1038/cdd.2016.8
10. Najafov A, Zervantonakis IK, Mookhtiar AK, Greninger P, March RJ, Egan RK, et al. BRAF and AXL Oncogenes Drive RIPK3 Expression Loss in Cancer. PLoS Biol (2018) 16(8):e2005756. doi: 10.1371/journal.pbio.2005756
11. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-Dependent Loss of RIP3 Expression in Cancer Represses Programmed Necrosis in Response to Chemotherapeutics. Cell Res (2015) 25(6):707–25. doi: 10.1038/cr.2015.56
12. Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, et al. Absence of RIPK3 Predicts Necroptosis Resistance in Malignant Melanoma. Cell Death Diseas (2015) 6:e1884. doi: 10.1038/cddis.2015.240
13. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature (2007) 447(7146):864–8. doi: 10.1038/nature05859
14. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
15. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
16. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat Rev Canc (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
17. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J Clin Invest (2018) 128(8):3341–55. doi: 10.1172/JCI99032
18. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
19. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol (2019) 26(5):623–33.e9. doi: 10.1016/j.chembiol.2019.01.008
20. Sato H, Tamba M, Ishii T, Bannai S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J Biol Chem (1999) 274(17):11455–8. doi: 10.1074/jbc.274.17.11455
21. Koppula P, Zhang Y, Shi J, Li W, Gan B. The Glutamate/Cystine Antiporter SLC7A11/xCT Enhances Cancer Cell Dependency on Glucose by Exporting Glutamate. J Biol Chem (2017) 292(34):14240–9. doi: 10.1074/jbc.M117.798405
22. Koppula P, Zhuang L, Gan B. Cystine Transporter SLC7A11/xCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
23. Koppula P, Zhang Y, Zhuang L, Gan B. Amino Acid Transporter SLC7A11/xCT at the Crossroads of Regulating Redox Homeostasis and Nutrient Dependency of Cancer. Cancer Commun (Lond) (2018) 38(1):12. doi: 10.1186/s40880-018-0288-x
24. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell (2003) 3(3):285–96. doi: 10.1016/S1535-6108(03)00050-3
25. Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A, et al. Insight Into the Mechanism of Ferroptosis Inhibition by Ferrostatin-1. Redox Biol (2019) 28:101328. doi: 10.1016/j.redox.2019.101328
26. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
27. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. Mol Cell (2019) 73(2):354–63.e3. doi: 10.1016/j.molcel.2018.10.042
28. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial Rescue Prevents Glutathione Peroxidase-Dependent Ferroptosis. Free Radical Biol Med (2018) 117:45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
29. Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID Links Ferroptosis to Mitochondrial Cell Death Pathways. Redox Biol (2017) 12:558–70. doi: 10.1016/j.redox.2017.03.007
30. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol (2018) 13(4):1013–20. doi: 10.1021/acschembio.8b00199
31. Ishii T, Bannai S, Sugita Y. Mechanism of Growth Stimulation of L1210 Cells by 2-Mercaptoethanol In Vitro. Role of the Mixed Disulfide of 2-Mercaptoethanol and Cysteine. J Biol Chem (1981) 256(23):12387–92. doi: 10.1016/S0021-9258(18)43284-X
32. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci (2017) 38(5):489–98. doi: 10.1016/j.tips.2017.02.005
33. Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox Imbalance in Cystine/Glutamate Transporter-Deficient Mice. J Biol Chem (2005) 280(45):37423–9. doi: 10.1074/jbc.M506439200
34. Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and Cancer. Annu Rev Nutr (2018) 38:97–125. doi: 10.1146/annurev-nutr-082117-051732
35. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron Addiction: A Novel Therapeutic Target in Ovarian Cancer. Oncogene (2017) 36(29):4089–99. doi: 10.1038/onc.2017.11
36. Rodriguez R, Schreiber SL, Conrad M. Persister Cancer Cells: Iron Addiction and Vulnerability to Ferroptosis. Mol Cell (2022) 82(4):728–40. doi: 10.1016/j.molcel.2021.12.001
37. Toyokuni S. Iron Addiction With Ferroptosis-Resistance in Asbestos-Induced Mesothelial Carcinogenesis: Toward the Era of Mesothelioma Prevention. Free Radical Biol Med (2019) 133:206–15. doi: 10.1016/j.freeradbiomed.2018.10.401
38. Pinnix ZK, Miller LD, Wang W, D'Agostino R Jr, Kute T, Willingham MC, et al. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci Transl Med (2010) 2(43):43ra56. doi: 10.1126/scitranslmed.3001127
39. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
40. Feng H, Stockwell BR. Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis? PLoS Biol (2018) 16(5):e2006203. doi: 10.1371/journal.pbio.2006203
41. Tang Y, Zhou J, Hooi SC, Jiang YM, Lu GD. Fatty Acid Activation in Carcinogenesis and Cancer Development: Essential Roles of Long-Chain Acyl-CoA Synthetases. Oncol Lett (2018) 16(2):1390–6. doi: 10.3892/ol.2018.8843
42. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular Interaction Dictates Cancer Cell Ferroptosis via NF2-YAP Signalling. Nature (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
43. Tang HM, Tang HL. Cell Recovery by Reversal of Ferroptosis. Biol Open (2019) 8(6):bio043182. doi: 10.1242/bio.043182
44. Huang L, McClatchy DB, Maher P, Liang Z, Diedrich JK, Soriano-Castell D, et al. Intracellular Amyloid Toxicity Induces Oxytosis/Ferroptosis Regulated Cell Death. Cell Death Diseas (2020) 11(10):828. doi: 10.1038/s41419-020-03020-9
45. Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, et al. Necrostatin-1 Protects Against Glutamate-Induced Glutathione Depletion and Caspase-Independent Cell Death in HT-22 Cells. J Neurochem (2007) 103(5):2004–14. doi: 10.1111/j.1471-4159.2007.04884.x
46. Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-Apoptotic Cell Death in Diseases of the Central Nervous System. Front Neurosci (2018) 12:214. doi: 10.3389/fnins.2018.00214
47. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
48. Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of Neuronal Ferroptosis Protects Hemorrhagic Brain. JCI Insight (2017) 2(7):e90777. doi: 10.1172/jci.insight.90777
49. Maher P, Currais A, Schubert D. Using the Oxytosis/Ferroptosis Pathway to Understand and Treat Age-Associated Neurodegenerative Diseases. Cell Chem Biol (2020) 27(12):1456–71. doi: 10.1016/j.chembiol.2020.10.010
50. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
51. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature (2017) 547(7664):453–7. doi: 10.1038/nature23007
52. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
53. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 Initiates Ferroptosis Through GPX4 Inactivation and Iron Oxidation. Nat Chem Biol (2018) 14(5):507–15. doi: 10.1038/s41589-018-0031-6
54. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a Glutathione-Independent Ferroptosis Suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
55. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
56. Stockwell BR. A Powerful Cell-Protection System Prevents Cell Death by Ferroptosis. Nature (2019) 575(7784):597–8. doi: 10.1038/d41586-019-03145-8
57. Kole AJ, Annis RP, Deshmukh M. Mature Neurons: Equipped for Survival. Cell Death Diseas (2013) 4:A. doi: 10.1038/cddis.2013.220
58. Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental Regulation of Mitochondrial Apoptosis by C-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell (2017) 31(1):142–56. doi: 10.1016/j.ccell.2016.11.011
59. Ye LF, Reznik E, Korn JM, Lin F, Yang G, Malesky K, et al. Patient-Derived Glioblastoma Cultures as a Tool for Small-Molecule Drug Discovery. Oncotarget (2020) 11(4):443–51. doi: 10.18632/oncotarget.27457
60. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes With Ferroptosis Inducers. ACS Chem Biol (2020) 15(2):469–84. doi: 10.1021/acschembio.9b00939
61. Joshi S. Targeting the Tumor Microenvironment in Neuroblastoma: Recent Advances and Future Directions. Cancers (2020) 12(8):2057. doi: 10.3390/cancers12082057
62. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J Clin Oncol (2009) 27(2):289–97. doi: 10.1200/JCO.2008.16.6785
63. Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A Mechanistic Classification of Clinical Phenotypes in Neuroblastoma. Science (2018) 362(6419):1165–70. doi: 10.1126/science.aat6768
64. Maris JM. Recent Advances in Neuroblastoma. N Engl J Med (2010) 362(23):2202–11. doi: 10.1056/NEJMra0804577
65. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis Through Lipid Remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
66. Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, et al. MYCN Mediates TFRC-Dependent Ferroptosis and Reveals Vulnerabilities in Neuroblastoma. Cell Death Diseas (2021) 12(6):511. doi: 10.1038/s41419-021-03790-w
67. Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res (2021) 81(7):1896–908. doi: 10.1158/0008-5472.CAN-20-1641
68. Lorincz T, Holczer M, Kapuy O, Szarka A. The Interrelationship of Pharmacologic Ascorbate Induced Cell Death and Ferroptosis. Pathol Oncol Res (2019) 25(2):669–79. doi: 10.1007/s12253-018-0539-9
69. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane Damage During Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024
70. Sato M, Onuma K, Domon M, Hasegawa S, Suzuki A, Kusumi R, et al. Loss of the Cystine/Glutamate Antiporter in Melanoma Abrogates Tumor Metastasis and Markedly Increases Survival Rates of Mice. Int J Canc (2020) 147(11):3224–35. doi: 10.1002/ijc.33262
71. Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive Phospholipid Peroxidation Distinguishes Ferroptosis From Other Cell Death Modes Including Pyroptosis. Cell Death Diseas (2020) 11(10):922. doi: 10.1038/s41419-020-03118-0
72. Zheng J, Sato M, Mishima E, Sato H, Proneth B, Conrad M. Sorafenib Fails to Trigger Ferroptosis Across a Wide Range of Cancer Cell Lines. Cell Death Diseas (2021) 12(7):698. doi: 10.1038/s41419-021-03998-w
73. Villablanca JG, Volchenboum SL, Cho H, Kang MH, Cohn SL, Anderson CP, et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatr Blood Canc (2016) 63(8):1349–56. doi: 10.1002/pbc.25994
74. Skapek SX, VanDellen AF, McMahon DP, Postels DG, Griffith OW, Bigner DD, et al. Melphalan-Induced Toxicity in Nude Mice Following Pretreatment With Buthionine Sulfoximine. Cancer Chemother Pharmacol (1991) 28(1):15–21. doi: 10.1007/BF00684950
75. Anderson CP, Matthay KK, Perentesis JP, Neglia JP, Bailey HH, Villablanca JG, et al. Pilot Study of Intravenous Melphalan Combined With Continuous Infusion L-S,R-Buthionine Sulfoximine for Children With Recurrent Neuroblastoma. Pediatr Blood Canc (2015) 62(10):1739–46. doi: 10.1002/pbc.25594
76. Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the SLC7A11/glutathione Axis Causes Synthetic Lethality in KRAS-Mutant Lung Adenocarcinoma. J Clin Invest (2020) 130(4):1752–66. doi: 10.1172/JCI124049
77. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of Ferroportin Accelerates Erastin-Induced Ferroptosis in Neuroblastoma Cells. Eur Rev Med Pharmacol Sci (2018) 22(12):3826–36. doi: 10.26355/eurrev_201806_15267
78. Wen PY, Kesari S. Malignant Gliomas in Adults. N Engl J Med (2008) 359(5):492–507. doi: 10.1056/NEJMra0708126
79. Francis SS, Ostrom QT, Cote DJ, Smith TR, Claus E, Barnholtz-Sloan JS. The Epidemiology of Central Nervous System Tumors. Hematol Oncol Clin North Am (2022) 36(1):23–42. doi: 10.1016/j.hoc.2021.08.012
80. Poon MTC, Bruce M, Simpson JE, Hannan CJ, Brennan PM. Temozolomide Sensitivity of Malignant Glioma Cell Lines - A Systematic Review Assessing Consistencies Between In Vitro Studies. BMC Canc (2021) 21(1):1240. doi: 10.1186/s12885-021-08972-5
81. Hochberg FH, Pruitt A. Assumptions in the Radiotherapy of Glioblastoma. Neurology (1980) 30(9):907–11. doi: 10.1212/WNL.30.9.907
82. Wang X, Lu J, Guo G, Yu J. Immunotherapy for Recurrent Glioblastoma: Practical Insights and Challenging Prospects. Cell Death Diseas (2021) 12(4):299. doi: 10.1038/s41419-021-03568-0
83. Allen BG, Bodeker KL, Smith MC, Monga V, Sandhu S, Hohl R, et al. First-In-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined With Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin Cancer Res (2019) 25(22):6590–7. doi: 10.1158/1078-0432.CCR-19-0594
84. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of Radiotherapy With Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
85. Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, et al. MGMT Promoter Methylation in Malignant Gliomas: Ready for Personalized Medicine? Nat Rev Neurol (2010) 6(1):39–51. doi: 10.1038/nrneurol.2009.197
86. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
87. deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, et al. Discordant Inheritance of Chromosomal and Extrachromosomal DNA Elements Contributes to Dynamic Disease Evolution in Glioblastoma. Nat Genet (2018) 50(5):708–17. doi: 10.1038/s41588-018-0105-0
88. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
89. Hu Z, Mi Y, Qian H, Guo N, Yan A, Zhang Y, et al. A Potential Mechanism of Temozolomide Resistance in Glioma-Ferroptosis. Front Oncol (2020) 10:897. doi: 10.3389/fonc.2020.00897
90. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin Sensitizes Glioblastoma Cells to Temozolomide by Restraining xCT and Cystathionine-Gamma-Lyase Function. Oncol Rep (2015) 33(3):1465–74. doi: 10.3892/or.2015.3712
91. Jara N, Ramirez E, Ferrada L, Salazar K, Espinoza F, Gonzalez-Chavarria I, et al. Vitamin C Deficient Reduces Proliferation in a Human Periventricular Tumor Stem Cell-Derived Glioblastoma Model. J Cell Physiol (2021) 236(8):5801–17. doi: 10.1002/jcp.30264
92. Quartararo CE, Reznik E, deCarvalho AC, Mikkelsen T, Stockwell BR. High-Throughput Screening of Patient-Derived Cultures Reveals Potential for Precision Medicine in Glioblastoma. ACS Med Chem Lett (2015) 6(8):948–52. doi: 10.1021/acsmedchemlett.5b00128
93. Tan S, Hou X, Mei L. Dihydrotanshinone I Inhibits Human Glioma Cell Proliferation via the Activation of Ferroptosis. Oncol Lett (2020) 20(4):122. doi: 10.3892/ol.2020.11980
94. Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, et al. SLC7A11 Expression is Associated With Seizures and Predicts Poor Survival in Patients With Malignant Glioma. Sci Transl Med (2015) 7(289):289ra86. doi: 10.1126/scitranslmed.aaa8103
95. Sorensen MF, Heimisdottir SB, Sorensen MD, Mellegaard CS, Wohlleben H, Kristensen BW, et al. High Expression of Cystine-Glutamate Antiporter xCT (SLC7A11) Is an Independent Biomarker for Epileptic Seizures at Diagnosis in Glioma. J Neurooncol (2018) 138(1):49–53. doi: 10.1007/s11060-018-2785-9
96. Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Aboody KS. SLC7A11 Overexpression in Glioblastoma Is Associated With Increased Cancer Stem Cell-Like Properties. Stem Cells Dev (2017) 26(17):1236–46. doi: 10.1089/scd.2017.0123
97. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 Promotes Angiogenesis and Neuronal Cell Death and Confers Ferroptosis in a xCT-Dependent Manner. Oncogene (2017) 36(40):5593–608. doi: 10.1038/onc.2017.146
98. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
99. Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, et al. Early Termination of ISRCTN45828668, A Phase 1/2 Prospective, Randomized Study of Sulfasalazine for the Treatment of Progressing Malignant Gliomas in Adults. BMC Canc (2009) 9:372. doi: 10.1186/1471-2407-9-372
100. Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 Drive Resistance to Sorafenib in Hepatocellular Carcinoma by Preventing Ferroptosis. EMBO Mol Med (2021) 13(12):e14351. doi: 10.15252/emmm.202114351
101. Zhu J, Berisa M, Schworer S, Qin W, Cross JR, Thompson CB. Transsulfuration Activity Can Support Cell Growth Upon Extracellular Cysteine Limitation. Cell Metab (2019) 30(5):865–76.e5. doi: 10.1016/j.cmet.2019.09.009
102. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. Cell Death Diff (2016) 23(2):270–8. doi: 10.1038/cdd.2015.93
103. Takano N, Sarfraz Y, Gilkes DM, Chaturvedi P, Xiang L, Suematsu M, et al. Decreased Expression of Cystathionine Beta-Synthase Promotes Glioma Tumorigenesis. Mol Cancer Res (2014) 12(10):1398–406. doi: 10.1158/1541-7786.MCR-14-0184
104. Wang L, Cai H, Hu Y, Liu F, Huang S, Zhou Y, et al. A Pharmacological Probe Identifies Cystathionine Beta-Synthase as a New Negative Regulator for Ferroptosis. Cell Death Diseas (2018) 9(10):1005. doi: 10.1038/s41419-018-1063-2
105. Zhang X, Guo Y, Li H, Han L. FIN56, A Novel Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization in a TFEB-Dependent Manner in Glioblastoma. J Canc (2021) 12(22):6610–9. doi: 10.7150/jca.58500
Keywords: ferroptosis, cancer cell, brain tumors, GPX4, system , lipid ROS, iron
Citation: Ferrada L, Barahona MJ, Salazar K, Godoy AS, Vera M and Nualart F (2022) Pharmacological targets for the induction of ferroptosis: Focus on Neuroblastoma and Glioblastoma. Front. Oncol. 12:858480. doi: 10.3389/fonc.2022.858480
Received: 20 January 2022; Accepted: 19 May 2022;
Published: 23 June 2022.
Edited by:
Nicolai Savaskan, Independent researcher, Berlin, GermanyReviewed by:
Jiri Neuzil, Griffith University, AustraliaCopyright © 2022 Ferrada, Barahona, Salazar, Godoy, Vera and Nualart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Nualart, ZnJudWFsYXJ0QHVkZWMuY2w=; Luciano Ferrada, THVmZXJyYWRhQHVkZWMuY2w=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.