Corrigendum: Murine models of acute myeloid leukemia
 Kristen J. Kurtz
Kristen J. Kurtz Shannon E. Conneely
Shannon E. Conneely Madeleine O’Keefe1
Madeleine O’Keefe1 Rachel E. Rau
Rachel E. Rau- 1Department of Pediatrics, Baylor College of Medicine, Texas Children’s Hospital, Houston, TX, United States
- 2Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States
Acute myeloid leukemia (AML) is a phenotypically and genetically heterogeneous hematologic malignancy. Extensive sequencing efforts have mapped the genomic landscape of adult and pediatric AML revealing a number of biologically and prognostically relevant driver lesions. Beyond identifying recurrent genetic aberrations, it is of critical importance to fully delineate the complex mechanisms by which they contribute to the initiation and evolution of disease to ultimately facilitate the development of targeted therapies. Towards these aims, murine models of AML are indispensable research tools. The rapid evolution of genetic engineering techniques over the past 20 years has greatly advanced the use of murine models to mirror specific genetic subtypes of human AML, define cell-intrinsic and extrinsic disease mechanisms, study the interaction between co-occurring genetic lesions, and test novel therapeutic approaches. This review summarizes the mouse model systems that have been developed to recapitulate the most common genomic subtypes of AML. We will discuss the strengths and weaknesses of varying modeling strategies, highlight major discoveries emanating from these model systems, and outline future opportunities to leverage emerging technologies for mechanistic and preclinical investigations.
1 Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy. The heterogeneity of AML has been understood for as long as the disease has been described and led to efforts to categorize the disease into similarly behaving subgroups (1). The earliest divisions were based on microscopic visualization. Perhaps the most well-known is the French-American-British classification system, first outlined in 1976. This system divided AML into eight subtypes based on morphology and cytochemical properties of leukemic blasts.
Advances in chromosome banding visualization techniques in the 1970s allowed for the first identification of genetic changes associated with AML. Several common, non-random cytogenetic abnormalities were found to correlate with clinical behavior, morphology, and patient outcomes with high predictability (2). These patterns included: favorable survival with inv (16) and t (8;21), increased early hemorrhage with t (15;17), and correlation of the poor-prognosis monosomy 5/7 or del 5q/7q with history of previous malignancy or carcinogenic exposures. Though not known at the time if these changes were drivers of or resulting from leukemia, these correlations provided some of the earliest evidence that the clinical heterogeneity of AML may be explained by underlying genetic heterogeneity.
It is now understood that approximately 55% of adult AML and 75% of pediatric AML is driven by a cytogenetic aberration (3–5). Presently, assessment of cytogenetic abnormalities is performed by karyotyping and fluorescent in situ hybridization for specific recurrent rearrangements of clinical significance (6). There are currently seven subclasses of AML that are defined by their translocations or inversions as defined by the World Health Organization (7). The most common are the same that were first identified in the 1970s. Now known as core binding factor (CBF) leukemias, those with t(8;21)(q22;q22) with resulting RUNX1-RUNX1T1 fusion (formerly AML1-ETO) and inv(16)(p13q22)/t(16;16)(p13;q22) with resulting CBFB-MYH11 fusion gene carry favorable prognoses. CBF AML is more prevalent in the pediatric population, estimated at 20-25% compared to 13% of AML in adults (4, 6). Acute promyelocytic leukemia (APL) defined by t (15;17) (q24;21) and resulting in a PML-RARA fusion comprises about 13% of adult and 10% of pediatric AML cases (4, 6). APL is associated with severe, sometimes life-threatening, coagulopathy at presentation. Prompt treatment with all-trans retinoic acid is critical to preventing early death during this high-risk time. With early recognition and treatment, survival is excellent for patients with APL, with long term remission rates as high as 85-90% (6). Monosomy 5/7 or del 5q/7q are rare and typically confer poor prognosis. Though estimated to represent only 4% of adult AML, KMT2A rearrangements (KMT2Ar) are found in 20% of pediatric AML and are especially common in the infant population (4). The prognosis of KMT2Ar AML depends on the fusion partner, over 100 of which have been identified, but is often poor (8).
In addition to cytogenetic abnormalities, extensive sequencing efforts have revealed driving gene mutations in nearly all cases of AML (6, 9–13). The mutational landscape of pediatric AML differs from that in adults, and though mutational burden increases with age overall AML has one of the lowest mutational rates amongst malignancies (13, 14). In adults the most commonly mutated genes include fms-like tyrosine kinase 3 (FLT3), nucleophosmin 1 (NPM1), and DNA methyltransferase 3A (DNMT3A), each occurring in approximately one third of patients (10, 12, 13, 15). While FLT3 mutations also occur in about 30% of pediatric AML cases, NPM1 is seen in only 10% of pediatric patients and DNMT3A mutations are almost never identified (13, 16–18). Along with FLT3, other signaling pathway-affecting mutations such as Ras pathway mutations (NRAS, KRAS, HRAS, NF1, CBL and PTPN11) and KIT are some of the most common in pediatric AML, and less commonly seen in adults (6, 10, 12, 13).
Many mutations have prognostic implications, and some may represent therapeutic targets. For example, FLT3 mutations most often confer poor prognosis, depending on the allelic fraction and co-occurring mutations. Importantly, FLT3 is a targetable tyrosine kinase receptor. The addition of FLT3-targeting tyrosine kinase inhibitors has significantly improved the outcome of these patients (3). DNMT3A, along with other methylation associated genes including TET2, IDH1, and IDH2, have been associated with poor prognosis in adults but are infrequently found in children (16). Each newly identified driver mutation presents the opportunity for targeted treatment with Food and Drug Administration approval of IDH inhibitors as a recent example.
Our understanding of the genomic landscape of AML continues to expand. Ongoing investigations are focused on co-occurring mutations and their effects on patient outcomes and responses to treatment. Much has been uncovered over the last several decades, and much remains to be discovered. To adequately address these remaining questions, in addition to rigorous clinical investigations of large, uniformly-treated patient populations, the availability of faithful model systems will be essential. While not all encompassing, here we will review several recent murine models selected for their relevance to human AML, with similar genetic lesions modeled, characteristic disease phenotypes, and that have advanced AML research over the last several decades. Additionally, we have included papers that highlight a spectrum of techniques used to generate murine model systems to allow for comparison of the advantages and disadvantages of each strategy that may aid in selecting the appropriate model system to address a specific research question.
2 History of Mouse Modeling Hematologic Malignancy
The goals of biomedical research of leukemia include predicting disease behavior to subclassify and risk stratify and developing or refining therapies to achieve the best efficacy. Animal models of human disease are vital to understanding disease pathogenesis and development of novel therapeutic strategies. The mouse (Mus musculus) is the most widely used animal model of human disease because of its genetic and physiologic similarity to humans. Humans and mice share approximately 80% of their genes with conservation of tissue-specific gene expression across species (19, 20). They also have remarkably similar organ systems. Other advantages to murine model systems include their small size, short lifespan, and rapid breeding, making them ideal models for scientific research. Murine hematopoiesis has been well-characterized over the years such that the similarities and differences to human hematopoiesis can be considered when creating and interpreting mouse models of leukemia.
When using murine models, it is crucial that each model be validated and evaluated for its similarities and potential differences to human disease. Some mouse strains harbor characteristic background lesions or are prone to diseases that are related to mouse biology rather than the model of human disease they are meant to represent. These details must be teased out so as not to attribute unrelated sequelae to the model of human disease. Differences in the human and murine genome as well as differences in hematopoiesis, aging and general development may impact the evolution and behavior of leukemia within the mouse model and must be considered. There are innate differences in hematopoiesis between humans and mice that may influence interpretation of mouse models. For example, mice exhibit lymphocyte predominance in the circulating leukocyte population while humans exhibit neutrophil predominance (21). There may also be subtle differences in the hematopoietic niche that affect leukemia development and progression that are unidentified and should be considered when interpreting results from mouse models.
The earliest mouse models of leukemia were created via exposure to external carcinogens. These included carcinogenic chemicals, irradiation, and viruses. The first transplantable leukemic mouse cell lines (L1210 and p388) were isolated from DBA/2 mice following exposure to 3-methylcholantrene (22, 23). These mouse models were highly valuable for exploring drug efficacy and developing strategies to overcome drug resistance (24). However, such chemically induced models can be inefficient and imprecise when used to recapitulate a specific malignant process and more often resulted in lymphoblastic rather than myeloid leukemia (25–27). Once the link between radiation and leukemia was established from observational studies involving individuals exposed to excessive radiation from nuclear attacks, the RF mouse model of myeloid leukemia was created via exposure to ionizing radiation (28). While this method effectively mirrored an actual environmental trigger and subsequent leukemic process to establish this causal relationship, there was a 6–8-month latency to leukemia onset and a low incidence of leukemia in the exposed mice, rendering it inefficient to create a robust cohort of leukemic mice for further study. Alternatively, viruses have been used to induce a more efficient animal model of leukemia. Murine leukemia viruses (MuLV) are retroviruses that have been used to induce myeloid leukemias in mice with a relatively short latency since the 1950s (29–31). Virally-induced mouse models of AML led to important discoveries of previously unknown oncogenes and the underlying pathogenesis of leukemia (32–34).
Gene editing has recently revolutionized the way in which mouse models of leukemia are generated. Advancements during the 1970-80s introduced techniques that allowed scientists to deliver engineered genetic material into the murine genome, thus creating transgenic mouse models. As advancements in high-throughput sequencing led to a wealth of new data on the genetic underpinnings of hematologic malignancy, gene editing paved the way for novel animal models of these newly defined genetic subtypes of disease. This led to more precise models that better mimicked the progression and behavior of subgroups of human leukemias. These techniques were first used to introduce proto-oncogenes under the control of a constitutively activated promoter to define their role in leukemogenesis in the 1980s (35, 36). Since then, the field of gene editing has advanced significantly to allow for a variety of constitutive and inducible models that can be integrated to more precisely recapitulate leukemogenesis.
3 Mouse Models of AML Fusions
Classic cytogenetic rearrangements found in AML are associated with the generation of fusion genes which demonstrate altered function compared to their wild-type components (Table 1). Here, we will review murine models used to study these classic fusion genes as well as newly described fusion genes not identified via standard cytogenetic testing.
TABLE 1
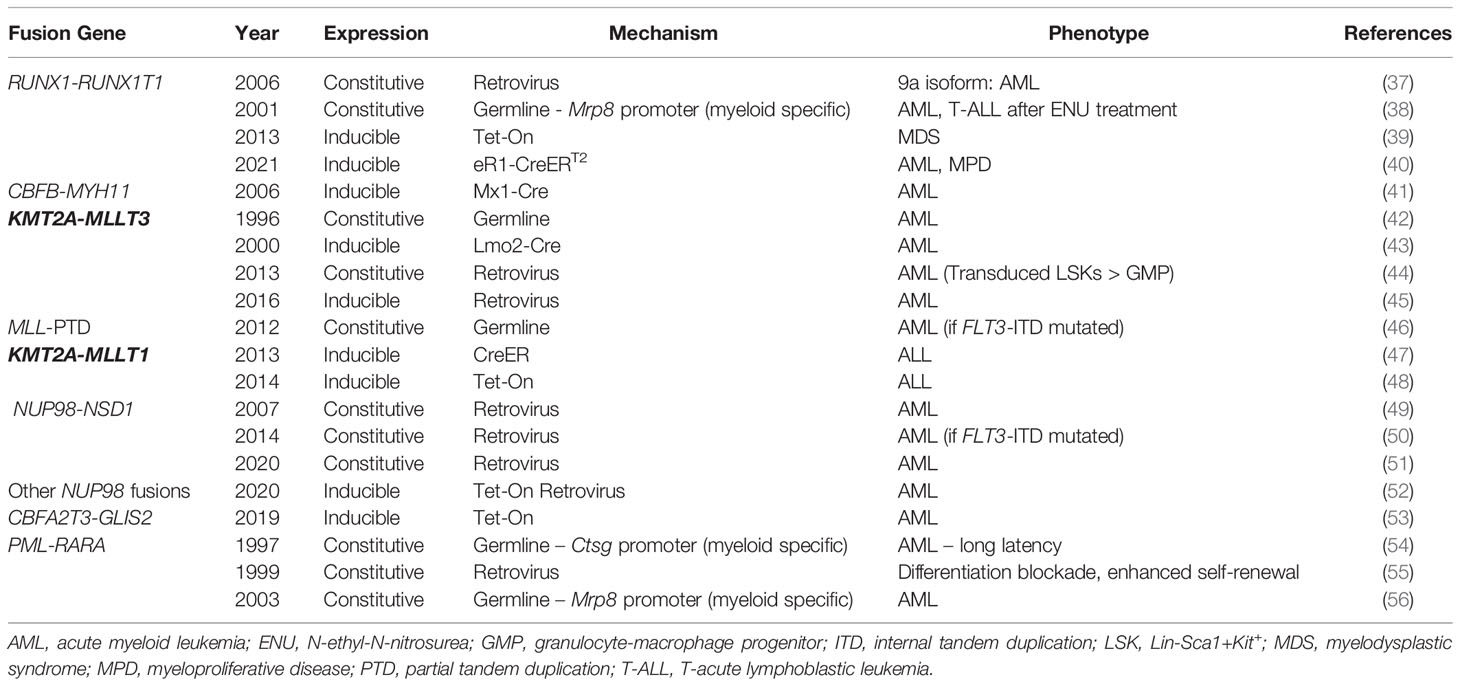
Table 1 Mouse models of fusion genes.
3.1 Core Binding Factor Leukemias
CBF AML encompasses patients with t(8;21) or inv(16) cytogenetic rearrangements or their associated fusion genes, RUNX1-RUNX1T1 and CBFB-MYH11, respectively (57). The chimeric proteins which result from CBF AML fusion genes function as dominant negative inhibitors of the CBF transcription factor, composed of RUNX1 and CBFβ, which are essential for normal myeloid cell development. The CBF AML fusion genes are associated with a favorable prognosis and share a common pathogenic mechanism, though RUNX1-RUNX1T1 and CBFB-MYH11 driven leukemias are distinct from one another.
Pioneering work from the labs of Drs. Dong Er Zhang, Nancy Speck and others have generated various murine model systems that have enhanced our understanding of this common AML fusion. Initial attempts to model t (8;21) AML utilized a germline knock-in of the RUNX1-RUNX1T1 fusion gene (58). However, embryonic expression of RUNX1-RUNX1T1proved to be embryonic lethal due to central nervous system hemorrhage and failed hematopoiesis. While this method failed to create a model of t (8;21) leukemia, these early studies helped to establish the role of RUNX1-RUNX1T1 as an inhibitor of normal RUNX1 function and highlighted the need for alternative murine models with delayed RUNX1-RUNX1T1 expression.
Current murine models of t (8;21) AML rely on delayed introduction or expression of the RUNX1-RUNX1T1 fusion through various means (Figure 1). One method utilizes retroviral transduction to introduce constitutive expression of RUNX1-RUNX1T1 from a retroviral vector incorporated into the DNA of a cell of interest. Retroviral expression of the full length 752 amino acid protein in murine hematopoietic stem cells (HSCs) failed to produce leukemia or signs of altered hematopoiesis when transplanted into lethally irradiated recipient mice, however, and led to the conclusion that RUNX1-RUNX1T1 alone is insufficient for leukemogenesis (59). Yan et al. subsequently identified a splice variant which generates a 575 amino acid protein, termed RUNX1-RUNX1T19a, that is variably expressed in human AML and generates leukemia when transduced into murine fetal liver cells (37). All mice transplanted with RUNX1-RUNX1T19a expressing cells developed leukemia within 16 weeks of transplant. As this is the only retroviral model to establish a t(8;21)-like AML without the addition of cooperating mutations, it is commonly used for in vivo modeling of RUNX1-RUNX1T1 driven disease, though the full-length construct remains the preferred model for in vitro experiments.
FIGURE 1
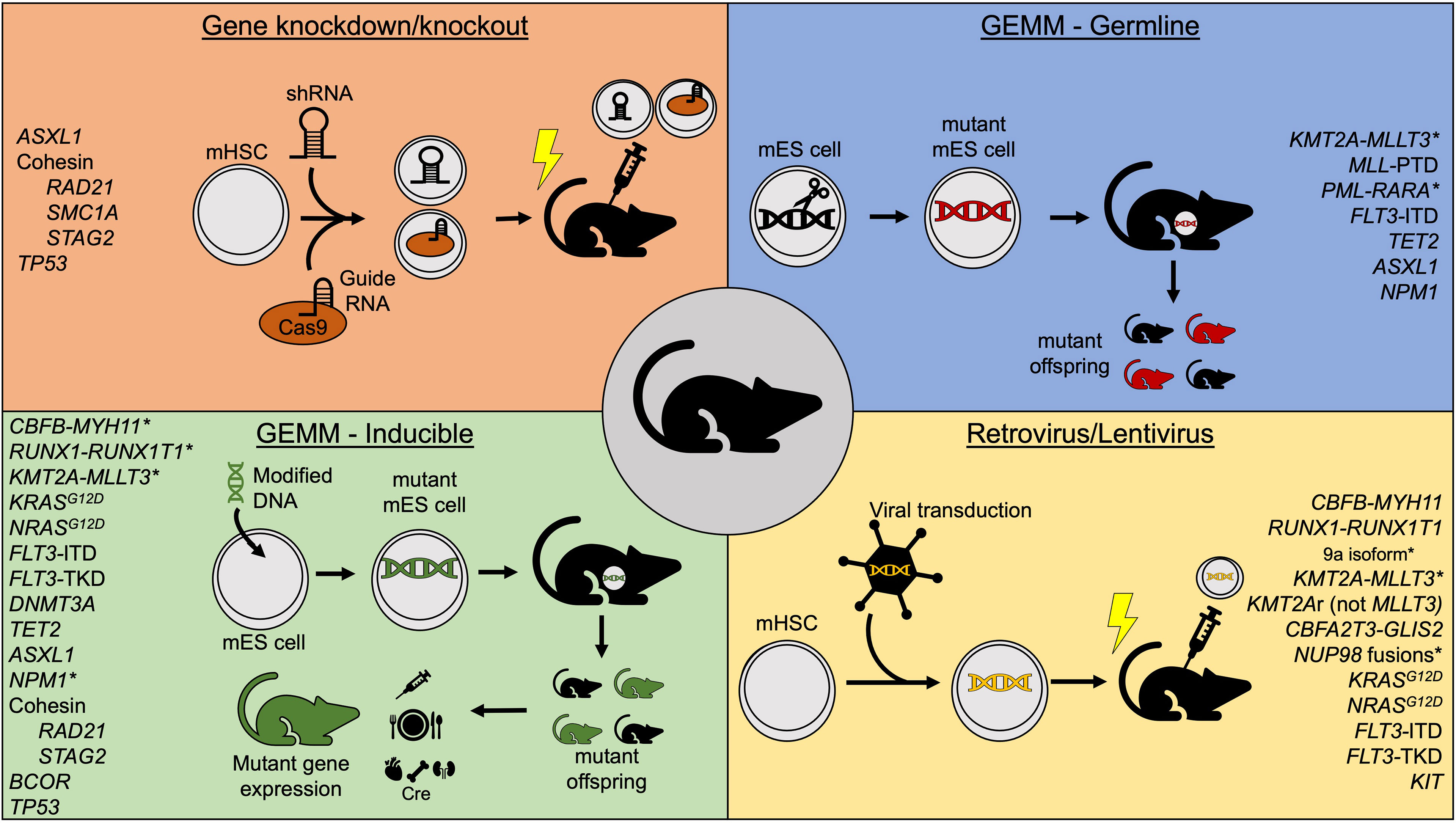
Figure 1 Summary of commonly used mouse model methods to study acute myeloid leukemia. *Indicates models that independently generate leukemia without cooperating mutations. GEMM, Genetically-engineered mouse model; mHSC, murine hematopoietic stem cell; mES, murine embryonic stem cell; shRNA, short hairpin RNA.
Transgenic models in which RUNX1-RUNX1T1 is transcribed in mice only in the presence of specific drivers have also been developed. One example is the MRP8-AE mouse in which RUNX1-RUNX1T1 is expressed in myeloid cells under control of the MRP8 promoter (38). Consistent with results from retroviral models, these mice remain healthy during their lifetime unless secondary mutations are induced with the alkylating agent N-ethyl-N-nitrosourea. However, only 55% of leukemia generated via this method was AML, while the remaining 45% was T-cell acute leukemia. A recent study published by Abdallah et al. used a model in which the RUNX1-RUNX1T1 downstream of a lox-stop-lox cassette was knocked-in to the ROSA26 locus such that Cre mediated recombination leads to excision of the stop codon and induced expression of the knocked-in fusion gene in HSCs at varying ages (40). They demonstrated that earlier expression of RUNX1-RUNX1T1, as early as postnatal day 3, resulted in a higher AML penetrance and lower incidence of non-AML disease compared to mice where RUNX1-RUNX1T1 expression was induced at 8-16 weeks of age. This is the only transgenic model and full-length isoform to consistently produce AML in the absence of other mutations. An alternative model uses a ‘tet-On’ method to express RUNX1-RUNX1T1 [ROSA26-iM2-tetrO-GFP/TgPtet-AML1-ETO (R26/AE)] (39). R26/AE mice that also carry a reverse tetracycline transactivator (rtTa) gene will express RUNX1-RUNX1T1 from a tetracycline-responsive promoter when exposed to doxycycline in their food or water. However, transplantation of bone marrow from R26/AE rtTa+/- mice into wild-type recipients is required to isolate expression to hematopoietic cells. Prolonged expression of RUNX1-RUNX1T1 in this system led to expansion of myeloid progenitor cells and produced myelodysplastic syndrome (MDS) with peripheral blasts without overt leukemia.
Similar to the effects of RUNX1-RUNX1T1 expression in embryos, germline knock-in of CBFB-MYH11 is embryonic lethal due to multiple fetal hemorrhages and failed hematopoiesis (60). However, early models showed that induced expression of CBFB-MYH11 in the hematopoietic system could independently induce leukemia in mice. This was robustly demonstrated by Kuo et al. who developed a conditional knock-in of the Cbfb-Myh11 gene (CBFB56M) (41). Following induction of Cre recombinase expression in Mx1-Cre+ CBFB56M mice with polyinosinic: polycytidilic acid (pIpC), the fusion gene is knocked in to the native Cbfb locus. Treated mice develop AML 11-21 weeks following the fusion gene restoration. This model has been used extensively to characterize inv (16) AML, cooperating mutations, and essential partners in leukemogenesis.
3.2 KMT2A Fusions
KMT2Ar AML poses a particular challenge for mouse modeling strategies, as over 100 different fusion genes are known to partner with KMT2A as a result of 11q23 rearrangements (61). Over 90% of KMT2Ar AML is caused by 9 specific fusions, and the frequency of these fusions varies with patient age. Thus, established murine models represent only the most common KMT2A fusions.
KMT2A-MLLT3 (formerly MLL-AF9) fusions are the most common KMT2A rearrangement in AML and account for 25-40% of KMT2Ar AML across all age groups (61). Murine models have all demonstrated that KMT2A-MLLT3 is a potent oncogene with the ability to induce leukemic transformation. Early studies produced genetically engineered germline Kmt2a-MLLT3 mutants in which the fusion is expressed in all cells throughout development (42). Although this model did lead to development of AML, or less commonly acute lymphoid leukemia, there are known effects of Kmt2a haploinsufficiency during murine development that cannot be controlled using this method, such as anemia, thrombocytopenia, and decreased B-cells (62). Thus, a Cre-loxP system was introduced such that intrachromosomal recombination of Kmt2a and Mllt3 could be induced via Cre-mediated recombination (43). Although effective in generating AML when Kmt2a-Mllt3 recombination was induced in primitive progenitor cells via progenitor-specific Cre, Lmo2-Cre, off-target expression of Cre recombinases poses concerns regarding specificity for cellular populations of interest (63). This is particularly true in KMT2Ar AML, where cell of origin can dictate the resultant type of leukemia and gene expression pattern (44).
To restrict expression of KMT2A-MLLT3 to specific stem and progenitor populations and study the differing effects on each, retroviral transduction of isolated bone marrow stem/progenitor cells with KMT2A-MLLT3 followed by transplantation has become a popular model. Lineage-Sca-1+Kit+ (LSK) cells expressing KMT2A-MLLT3 are more potent inducers of leukemic transformation than KMT2A-MLLT3 expressing granulocyte-macrophage progenitors (44). Interestingly, leukemia generated from transduced LSK cells is also more chemoresistant than that originating from granulocyte-macrophage progenitors, is more highly methylated, and carries a gene expression signature associated with poor prognosis in patients with AML (44). Selection of appropriate cell population for retroviral transduction and attendant gene expression is therefore integral to the interpretation of results from KMT2Ar models and relating them to human AML. One concern regarding retroviral transduction is the possibility of supraphysiologic expression of KMT2A-MLLT3 which may significantly impact study results. Recently, doxycycline-inducible genetic KMT2A-MLLT3 models have helped to address these concerns. These models demonstrate dose-dependent expression of KMT2A-MLLT3 resulting in 10-20-fold lower protein amounts compared to retroviral transduction models, allowing improved control over fusion gene expression (45). As with retroviral models, cells of interest can be isolated and transplanted into recipients, though expression of KMT2A-MLLT3 can be induced before or after transplantation. These studies have confirmed effects of KMT2A-MLLT3 expression on different stem and progenitor populations identified from studies using retroviral transduction and demonstrate their utility in future mouse modeling of KMT2A-MLLT3 AML.
As KMT2A-MLLT3 is the most common KMT2Ar fusion gene in AML, it is also the most studied. After KMT2A-MLLT3, the next most common fusion partners are MLLT10, ELL, AFDN, MLLT1, and SEPT6 (61). Murine models of these KMT2A fusions depend primarily on retroviral transduction of murine HSCs or human hematopoietic cells isolated from cord blood (64–66). As an alternative, immortalized murine cells lines expressing KMT2A-MLL10 and KMT2A-ELL have been created which engraft into syngeneic recipients and cause disease (67, 68). In addition to recurrent KMT2A fusions, partial tandem duplications (PTD) of KMT2A are also common in adult AML while nearly absent from infant and pediatric cases (61). A germline knock-in model of Kmt2a-PTD exists but requires cooperating mutations, such as a Flt3 internal tandem duplication (ITD), to produce disease (46). Transgenic mouse models of KMT2A-MLLT1 have also been created and characterized including one which utilizes Cre recombination to induce expression of KMT2A-MLLT1 and the other with doxycycline-inducible expression of KMT2A-MLLT1 (47, 69). However, KMT2A-MLLT1 fusions are much more common in ALL and occur rarely in AML, limiting the use of these models in AML research. Finally, CRISPR/Cas9-mediated genome editing has been used to create a model of Kmt2a-Mllt3 AML via dual single-guide RNAs simultaneous targeting the breakpoint cluster region of Kmt2a and Mllt3 (70). Unique to KMT2Ar AML, close attention must be paid to the model system and cell of origin in studying KMT2Ar disease, as similar KMT2A fusions can be found across hematologic malignancies, and selection of methods will have profound effects on the resulting disease.
The development of these varying murine models of KMT2Ar AML have helped identify potential novel therapeutic strategies for this poor prognosis subset of AML. These include Dr. Katherine Bernt and colleagues’ identification of the histone 3, lysine 79 methylase, DOT1L, as a critical dependency and therapeutic vulnerability in KMT2Ar AML (48). Other investigations have revealed preclinical efficacy of BET bromodomain inhibitors in KMT2Ar AML (71). Perhaps the most exciting, pioneering work by Dr. Jolanta Grembecka and colleagues has led to the development and validation of inhibitors of the MENIN-MLL interaction as highly promising agents for the treatment of KMT2Ar leukemias (72). Results from these seminal investigations have led to early phase clinical trials and ultimately could result in meaningful improvements in outcomes for patients with KMT2Ar AML.
3.3 NUP98 Fusions
While relatively uncommon in AML, NUP98 gene fusions carry a dismal prognosis even with stem cell transplant. Thus, establishment of mouse models is a high priority to identify novel treatments for this chemoresistant AML. Retroviral models of NUP98 fusions have served as the predominant mouse models to date. In 2007 Wang et al. (49) showed that retroviral transduction of NUP98-NSD1, the most common NUP98 fusion in pediatric AML, into murine hematopoietic progenitors followed by transplantation independently produced AML with an average survival of 126 days post-transplant (68). Mohanty et al. created a similar retroviral model of NUP98-NSD1 and demonstrated significant disease acceleration when NRASG12D is co-expressed. Furthermore, the authors found that upregulation of Hox genes, specifically Hoxa7, Hoxa9, and Hoxa10 was a major contributor to disease development, while other studies have demonstrated a dependence on interaction between NUP98-NSD1 and SMARCA5 for leukemic transformation (51, 73). A separate study introduced three different AML-specific NUP98 fusions (NUP98-NSD1, NUP98-KDM5A, and NUP98-DDX10) into murine fetal liver cells via retrovirus with doxycycline-inducible expression of the fusion gene tagged with GFP (52). Following transplantation into recipient mice and induction with doxycycline, all 3 tested NUP98 fusions rapidly produced leukemia. This also identified cyclin-dependent kinase 6 (CDK6) as an integral disease driver and that pharmacologic inhibition of CDK6 could prolong survival in these mice. Interestingly, CDK6 is also an important driver in KMT2Ar AML (74). Other murine-based models of NUP98 fusions suggest that NUP98 fusion proteins interact with MLL1, suggesting a common link between these two genetic AML subtypes (75). Ex vivo drug sensitivity assays have also been performed using retroviral transduction models, identifying BRD2/4 inhibitors, topoisomerase II inhibitors, and gemcitabine as effective in NUP98-KDM5A AML (76). Finally, Heikamp et al. recently used retroviral transduction of murine hematopoietic cells with NUP98-HOXA9 or NUP98-KDM5A followed by transplantation to generate pre-clinical models which they used to define MENIN-MLL1 inhibition as a potential new treatment strategy for this refractory disease (77). It is important to note that, like KMT2Ar AML, the cell of origin of gene expression significantly impacts disease phenotype. NUP98-KDM5A is often associated with acute erythroid or megakaryoblastic leukemia in patients, but when expressed in mouse HSPCs then transplanted into syngeneic recipients leads to an AML-like myeloid phenotype with blasts expressing the mature myleoid markers, Cd11b and Gr1in transplant models (52, 78).
3.4 CBFA2T3-GLIS2
Lopez et al. (53) developed a novel transgenic model using tetracycline-inducible expression of CBFA2T3-GLIS2 in which nearly all mice developed a lethal hematologic malignancy, 20% of which displayed megakaryoblastic markers with a disease latency of 164 days, whereas the remaining mice displayed heterogeneous non-megakaryoblastic immunophenotypes with a significantly longer disease latency (79). As this fusion is predominantly found in pediatric AML, the authors sought to investigate the role of developmental stage on leukemia onset. They found that transplantation of fetal liver cells from their inducible model led to a shorter disease latency with a predominant megakaryoblastic population compared to mice transplanted with bone marrow from adult mice, demonstrating that developmental stage significantly impacts disease phenotype. The Lopez et al. (53) transgenic model was a substantial technical advancement as prior to its description, murine models had been largely limited to patient-derived xenografts (PDXs) (80).
3.5 PML-RARA
APL, characterized by the PML-RARA gene fusion, is a unique disease entity that requires vastly different treatment than standard AML and has significantly superior outcomes. Murine models have been integral to understanding the pathophysiology of APL and critical tools for the development of current targeted treatment strategies. Retroviral transduction models used to express PML-RARA in hematopoietic cells demonstrate increased cellular self-renewal and differentiation blockade but fail to generate leukemia when transplanted into recipient mice (55). Transgenic models have therefore become preferred with multiple models available, each with slightly different features. The earliest transgenic models expressed PML-RARA under control of sequences that regulate the expression of the human cathepsin G gene in myeloid cells, though leukemia penetrance was low at 30% and disease latency was prolonged (54). This model was improved upon with PML-RARA under control of the murine cathepsin G (MRP8) promoter, yielding higher expression of the fusion product and 90% leukemia penetrance with continued prolonged disease latency (56). This MRP8-PML/RARA model is the predominant model currently used in APL research. However, while AML does occur, differentiation arrest characteristic of APL varies and is less pronounced than in human correlates. Prior efforts have attempted to improve on this model by selectively expressing the fusion in promyelocytes to mimic the suspected origin cell population in human APL but fail to induce leukemia or demonstrate enhanced self-renewal properties of PML-RARA-expressing cells despite a distinct gene expression signature (81).
4 Mutations That Activate Signal Transduction Pathways
Mutation profiling studies have subsequently identified activating mutations in genes involved in signal transduction pathways as commonly mutated in both adult and pediatric AML and key targetable lesions (Table 2). These mutations generally occur at hotspot locations within the gene and are thus ideal to recapitulate with murine models that mimic activation of these pathways. Here we will review the murine models currently used to study these activating mutations.
TABLE 2
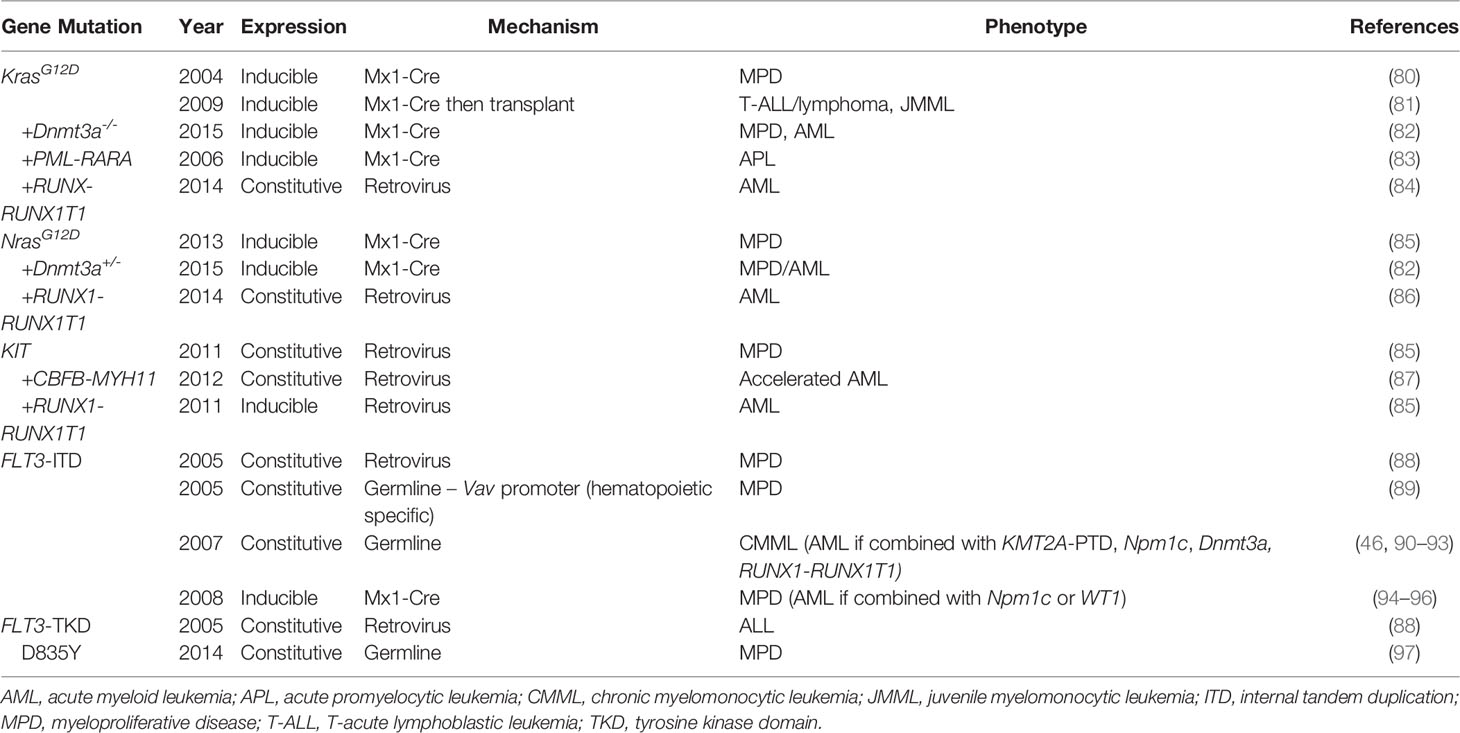
Table 2 Mouse models of signal transduction pathways.
4.1 Ras Pathway
The Rat Sarcoma virus (Ras) family of genes represents critical regulators of cell proliferation and differentiation and is mutated across a variety of cancers. Activating mutations in Ras family genes including NRAS, KRAS, NF1, PTPN11 and CBL occur in up to 50% of pediatric AML but are less common in adult AML. The predominant model used to study KRAS mutations is used in both hematologic and solid malignancies and was originally developed by Dr. Erica Jackson and colleagues as a model for lung cancer (100). This model has lox-stop-lox KrasG12D knocked into the native Kras locus leading to expression of mutant Kras following Cre recombination. When combined with tissue-specific Cre drivers, the mutation is expressed only in the tissue of interest. Induction of KrasG12D expression in the hematopoietic system via Mx1-Cre leads to a rapid and highly penetrant myeloproliferative disease (MPD) but not overt AML (82). Concurrent homozygous loss of Dnmt3a cooperates with KrasG12D to accelerate disease progression and leads to development of AML in approximately 30% of mice, though heterozygous loss of Dnmt3a had no effect (84). KrasG12D also cooperates with PML-RARA to produce malignancy consistent with APL with a short latency of only 37 days (85). Despite the ability to target specific cell populations with tissue-specific Cre drivers, this model has also been used in transplantation experiments where induced bone marrow from primary transgenic mice is transplanted into recipient mice along with supportive bone marrow (83). Interestingly, transplant recipients predominantly develop T-cell leukemia or lymphoma and occasionally juvenile myelomonocytic leukemia in contrast to the MPD described above, thus limiting the use of transplant models to study AML biology (83).
NRAS is another Ras pathway gene that is commonly mutated in AML. As with KRAS mutations, NRAS mutations are activating mutations that have been modeled using genetic mouse models. When expressed in the hematopoietic system, NrasG12D alone induces a MPD similar to KrasG12D but with significantly longer disease latency and lower penetrance (88). However, NrasG12D cooperates with heterozygous loss of Dnmt3a to promote AML development in one third of induced mice (84). As DNMT3A mutations are predominantly heterozygous in human disease, this may represent a more biologically relevant model of AML.
Retroviral transduction models of Ras mutants have also been employed to study cooperating mutations in AML. Zhao et al. used co-transduction of KRASG12D or NRASG12D and RUNX1-RUNX1T1 followed by transplantation to demonstrate cooperation between activating Ras mutations and RUNX1-RUNX1T1 in leukemogenesis (86). Retroviral expression of either KRASG12D or NRASG12D alone did not induce leukemia or an identified MPD, yet both cooperated with RUNX1-RUNX1T1 to accelerate development of AML with NRAS serving as a more potent inducer of disease in this context. Retroviral expression of NRASG12D has also been shown to cooperate with loss of Dnmt3a in mouse models to generate AML (101). Retroviral expression of Ras mutants may therefore serve as a reasonable method when transplantation models using AML-specific drivers are preferred.
4.2 KIT
Tyrosine protein kinase KIT, encoded by the gene KIT (formerly c-KIT), is a proto-oncogene which plays a critical role in signaling pathways that promote cellular proliferation, particularly within HSCs. Activating mutations in KIT have been found at several gene loci in patients with AML, and such mutations occur in nearly 40% of patients with CBF AML (102). These mutations are predominantly found in exons 8 and 17 with amino acids D816 and N822 serving as recurrent mutation hotspots. As with other tyrosine kinase activating mutations, studies have shown that activating mutations in KIT are not sufficient to cause leukemia but cooperate with other driver mutations to transform leukemic cells. Retroviral transduction of the two recurrent exon 17 KIT mutant genes into CBFB-MYH11-expressing bone marrow cells shortened survival of transplant recipients compared to those which expressed wild-type KIT (89). In a model of t (8;21) AML in which retrovirus expression KIT N822 and RUNX1-RUNX1T1 were co-transduced, expression of the KIT mutant alone led to MPD, but overt leukemia was observed when KITN822 was expressed in conjunction with RUNX1-RUNX1T1 (87).
4.3 FLT3
FLT3 is a commonly mutated gene in AML, leading this receptor tyrosine kinase to be constitutively active driving uninhibited cell growth. Internal tandem duplications (FLT3-ITD) of the juxtamembrane domain encompass a majority of FLT3 mutations, though activating mutations in the tyrosine kinase domain (FLT3-TKD) also occur (10, 13). Early retroviral transduction models of both FLT3-ITD and FLT3-TKD mutations revealed that expression of these activating mutations lead to distinctly different disease phenotypes (90). Transplantation of FLT3-ITD expressing bone marrow led to MPD, whereas transplantation of FLT3-TKD expressing cells led to a lymphoid disease with longer latency, consistent with the finding that TKD mutations are more common in acute lymphoblastic leukemia than AML (103). In addition, these studies revealed that FLT3-ITD mutations, and not FLT3-TKD, lead to activation of STAT5 signaling.
Several transgenic models of FLT3-ITD have been developed, each with slightly different advantages, though they predominantly produce MPD. First, a transgenic model expressing FLT3-ITD under the control of the hematopoietic-specific Vav promoter was created by the lab of Dr. D. Gary Gilliland which developed MPD at 6-12 months (91). Subsequently, the same group developed a model in which a humanized ITD is knocked into the native murine Flt3 gene (92). This model minimizes the effects of gene overexpression on disease phenotype inherent to retroviral transduction or use of heterologous promoters, as expression levels of FLT3 in both wild-type and mutant forms have a significant impact on disease. In the absence of cooperating mutations, this model leads to the development of chronic myelomonocytic leukemia (CMML) and not AML. However, when used in combination with other common AML lesions, such as KMT2A-PTD, Npm1c mutation, Dnmt3a deletion, and RUNX1-RUNX1T1, expression of Flt3-ITD is capable of producing AML, though with relatively long latency (46, 93–95). A similar model in which an 18-base pair ITD was knocked into the juxtamembrane domain of native Flt3 gene was also developed by Dr. Li in the lab of Dr. Donald Small. This model is characterized by the development of MPD with a median survival of 10 months (96), and cooperates with other common AML mutations, including NPM1c and mutant Wt1, to generate AML or accelerate the MPD disease process (97, 98).
FLT3-TKD mutations also occur in AML but are less common and not as strongly associated with prognosis compared to ITD mutations. Plasmids conferring the expression of various FLT3-TKD mutants have been developed but are predominantly used to transform cell lines, and murine models of FLT3-TKD mutants are lacking (99, 104, 105). Dr. Emily Bailey and colleagues previously generated a knock-in of the most common TKD mutant, D835Y, which developed predominantly MPD but with a longer latency than ITD models (106). Thus, new murine models which cover the spectrum of relevant FLT3 aberrations are needed to adequately study this common mutation in AML.
5 Mutations That Affect Transcription Factors or Epigenetic Modifiers
Genomic landscape studies have revealed critical epigenetic modifiers that are frequently mutated in myeloid malignancies (Table 3). Here we will review data from some of the best characterized murine models of these epigenetic modifier mutations.
TABLE 3
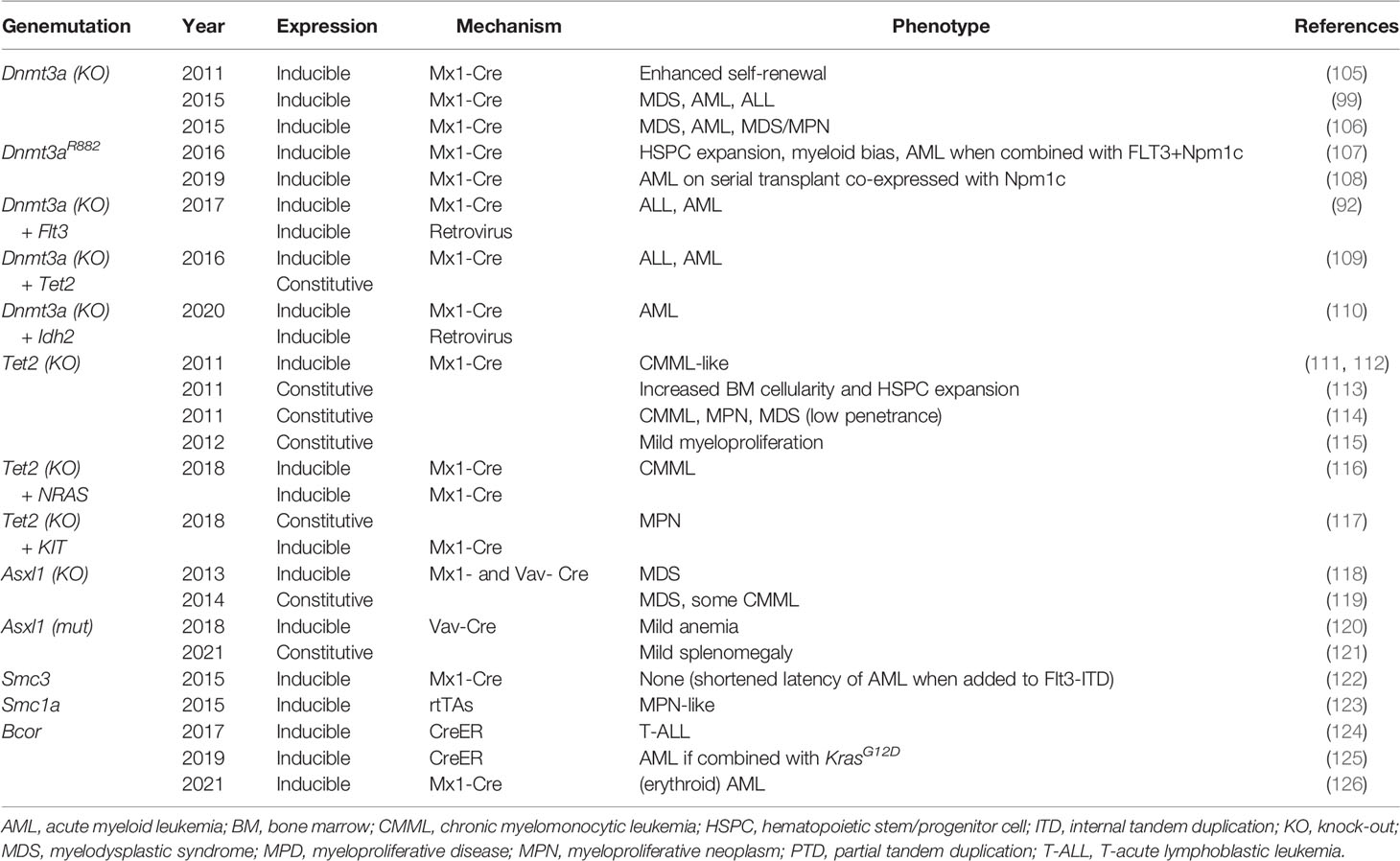
Table 3 Mouse models of epigenetic regulators.
5.1 DNMT3A
DNA methyltransferase 3A (DNMT3A) is a de novo DNA methyltransferase that methylates cytosine moieties of CpG dinucleotides (129). HSCs frequently acquire DNMT3A mutations which act as pre-leukemic lesions and in turn lead to clonal hematopoiesis which, in some cases, ultimately progresses to leukemia. Up to 22% of adult de novo AML cases (130, 131) and 10% of MDS (107, 132) harbor somatic mutations in DNMT3A, most occurring at arginine 882 (R882) in the DNMT3A methyltransferase domain. Since DNMT3A variants were first reported in AML in 2010, a variety of mouse modeling techniques have been used to clarify the role of DNMT3A in HSCs and the precise role that loss of function of DNMT3A plays in leukemogenesis.
In 2011, the lab of Dr. Margaret Goodell reported the effects of Dnmt3a deficiency in vivo using an inducible Dnmt3a knock-out (KO) murine model (133). Transgenic animals carrying hematopoietic tissue-specific Mx1-Cre were crossed with mice carrying loxP-flanked copies of Dnmt3a, and Dnmt3a loss was then induced via serial intraperitoneal injections of pIpC. In competitive transplant experiments, equal parts purified HSCs from non-induced Dnmt3afl/fl and wild-type mice were transplanted into primary recipients followed by induced deletion of Dnmt3a. They found no difference in the represented proportion of HSCs before or after Dnmt3a deletion was induced in primary recipients. However, in secondary competitive transplants, there was a significant increase in Dnmt3a-null HSCs compared to wild-type, demonstrating that loss of Dnmt3a led to enhanced stem cell self-renewal. No mice developed MPD or overt leukemia suggesting that additional cooperating mutations are necessary for leukemogenesis.
Similar inducible models have since been used in non-competitive transplants resulting in a variety of hematologic malignancies, including MDS, AML, primary myelofibrosis, and T- and B-cell acute lymphoblastic lymphoma. Dr. Allison Mayle and colleagues demonstrated that with non-competitive transplantation of Dnmt3a-null bone marrow, all transplanted mice died between 200- and 400-days post-transplant from a variety of hematologic malignancies (101). Challen et al. reported similar findings of bone marrow failure resulting in death with 100% penetrance following transplantation of Dnmt3a-null whole bone marrow into sublethally irradiated mice (134). The majority of moribund mice met diagnostic criteria for MDS with 2/15 mice developing frank AML and 4/25 developing an intermediate MDS/myeloproliferative neoplasm. This inducible Dnmt3a KO model has also been combined with a variety of murine models harboring genetic lesions of genes that are commonly co-mutated in human DNMT3A-mutant AML, including FLT3, TET2, IDH2, and KIT, identifying critical cooperative mechanisms that drive leukemogenesis (94, 108, 111, 112).
In AML, the most common DNMT3A mutation affects amino acid R882 (DNMT3AR882) in the methyltransferase domain. DNMT3AR882 encodes a mutant protein that is hypomorphic and exerts a dominant negative effect, interfering with wild-type DNMT3A function resulting in severely reduced cellular methyltransferase activity (135, 136). Several inducible models have been created to specifically replicate DNMT3AR882 and elucidate the precise role of this dominant negative acting mutant in leukemogenesis. Dr. Olga Guryanova and colleagues utilized a model in which mutant Dnmt3aR878H (the mouse homolog to DNMT3AR882H) is expressed from the endogenous Dnmt3a locus. To do so, they replaced endogenous Dnmt3a exon 23 with a Lox-Stop-Lox cassette followed by exon 23 and 24 with the point mutation affecting amino acid R878. In this model, prior to Cre recombination, the modified allele functions as a null allele, and after Cre recombination leads to expression of the mutant mRNA and protein. When Dnmt3aR878 is conditionally expressed in the hematopoietic system by induction of the hematopoietic-specific Mx-Cre by pIpC injections, hematopoietic stem/progenitor cell expansion and myeloid bias in differentiation were observed, but no overt leukemia (109). They were able to create a fully penetrant and robust model of AML via co-expression of Dnmt3aR878H, Flt3ITD and Npm1c. The lab of Dr. Jennifer Trowbridge recently created a similar conditional model of Dnmt3aR878H (110). However, an advantage of the Trowbridge model over that used by Guryanova is that prior to Cre-mediated recombination, wild-type Dnmt3a is expressed from the modified allele whereas the modified allele in the Guryanova model is null prior to recombination. This strategy eliminates the possible confounding impact of constitutive haploinsufficiency of Dnmt3a in the hematopoietic system prior to recombination.
Our group recently used CRISPR/Cas9-mediated gene editing of murine embryonic stem cells to create constitutive models germline Dnmt3a lesions that have been previously described in patients with the rare overgrowth, intellectual disability syndrome, Tatton-Brown-Rahman syndrome. We found that each model recapitulated the distinct growth, behavioral and hematologic phenotypes observed in their human counterparts including increased risk of hematologic malignancy (137). Dr. Amanda Smith in the lab of Dr. Timothy Ley similarly found that a germline Dnmt3aR878 model also recapitulated the features of TBRS including risk of hematologic malignancy development (138). These and additional models that recapitulate specific point mutations observed in human disease will help us establish the connection between alterations in DNMT3A function and pathogenesis.
Further development of mouse models that utilize inducible gene deletion strategies to incorporate mutations that are known to co-occur with DNMT3A in human disease will help to further define the role of DNMT3A in leukemic transformation and more closely mirror the natural disease process.
5.2 TET2
TET2 is a member of the TET family of proteins with dioxygenase enzymatic activity resulting in oxidation of the methyl group at the 5-position of cytosine. The precise effect of TET2’s action has not been proven, but it is hypothesized that this modified locus prevents DNMT1-mediated methylation during DNA replication, thus leading to passive loss of DNA methylation. Loss of function TET2 mutations are among the most common drivers of clonal hematopoiesis and have been detected in 10-20% of de novo AML (139) and up to 50% of cases of CMML (140). As one of the most prevalent mutations affecting hematopoiesis, the development of faithful murine models of TET2 loss has been pursued by several research groups.
Multiple Tet2 KO mouse models have been created, all demonstrating expansion of the HSC compartment following ablation of Tet2 expression secondary to increased self-renewal capacity (113–117). In 2011, Moral-Crusio et al. developed a novel conditional Tet2 KO murine model that resulted in MPD (113). This group utilized homologous recombination to introduce two loxP sites flanking exon 3 of Tet2 in embryonic stem cells that were then injected into blastocysts. The resulting mice (Tet2fl/fl) were then crossed with transgenic Vav-Cre mice, resulting in Tet2 deletion in the hematopoietic system in utero, leading to the development of a CMML-like disease by 20 weeks of life with monocyte–predominant leukocytosis and splenomegaly.
Another group developed a novel constitutive model via construction of a Tet2-targeting vector that disrupted the endogenous ATG start codon, leading to silencing of Tet2 gene expression (116). The resultant mice with germline deletion of Tet2 are viable and fertile but develop a CMML-like disease with leukocytosis, neutrophilia and monocytosis along with increased bone marrow cellularity, splenomegaly, and moderate liver enlargement. Approximately one-third of the Tet2-/- mice died within a year due to myeloid malignancy. These mice exhibited two distinct phenotypes: one with a population of erythroblasts infiltrating hematopoietic organs and the other with an aberrant population of mature myeloid cells including myeloblasts, monocytes/macrophages and neutrophils. Both malignant myeloid phenotypes caused massive hepatosplenomegaly and anemia.
These models of Tet2 loss have proven useful for studies of collaborative leukemogenesis. A number of labs have conducted important studies combining Tet2 deletion with other lesions that frequently co-occur in human AML, such as Dnmt3a as previously mentioned, NRAS and KIT (111, 118, 119). These models are crucial for clarifying the interplay between cooperative mutations providing models that closely mirror human disease.
5.3 ASXL1
Additional sex comb-like 1 (ASXL1) is a polycomb group protein that interacts with BAP1 and PRC2 to remodel chromatin thus regulate gene expression. ASXL1 mutations have been reported in 5-11% of de novo AML (141–143). These mutations occur with increased frequency among older patients and are frequently detected in clonal hematopoiesis (142).
Abdel-Wahab et al. developed a novel murine model of Asxl1 knockdown via retroviral transduction to introduce short hairpin RNA (shRNA) constructs into mouse bone marrow cells that were then transplanted into lethally irradiated recipient mice (144). They found that knockdown of Asxl1 in a mouse expressing the oncogene NrasG12D accelerated the expected MPD, resulting in more severe symptoms of anemia and organomegaly as well as decreased lifespan. The same group then created a conditional KO model via insertion of two loxP sites flanking exons 5-10 of Asxl1 (120). They crossed Asxl1fl/fl mice with transgenic mice harboring various tissue-specific Cre recombinase systems. Ella-Cre+ Asxlfl/fl mice had germline deletion of Asxl1 and resulted in 100% embryonic lethality. Heterozygous germline deletion of Asxl1 led to viable embryos at expected Mendelian ratios, though 35% of these mice exhibited craniofacial dysmorphism. Hematopoietic-specific deletion of Asxl1 using either Mx1-Cre or Vav-Cre resulted in MDS with progressive anemia and leukopenia compared with littermate controls. They observed morphological dysplasia of peripheral myeloid cells and erythroid precursors with bone marrow hypocellularity.
A constitutive Asxl1-null murine model was created by Wang et al. the following year (121). This group replaced part of Asxl1 exon 1 with nlacZ/nGFP reporter to disrupt the endogenous ATG and inhibit transcription of Asxl1. They used this technique to generate both Asxl1+/- and Asxl1-/- mice to compare the effects of haploinsufficiency versus complete loss of Asxl1. Complete loss of Asxl1 resulted in an estimated 80% embryonic lethality, similar to the high rate observed in the inducible germline model that was previously published. Of the Asxl1-null mice, 80% that survived to birth died within the first day of life, and the remainder survived for 18-42 days. Surviving mice exhibited cytopenias and myeloid dysplasia, consistent with MDS-like disease. Interestingly, heterozygous deletion of Asxl1 was also sufficient for development of a similar MDS-like disease. The disease phenotype became increasingly severe with age in Asxl+/- mice with worsening cytopenias and dysplasia, and progressed to a CMML-like disease in 22% (4 of 18 mice).
An alternative approach was taken to better mimic the truncating mutations that typically occur in human disease. A conditional knock-in mouse model was created by introducing a floxed mutant Asxl1 allele via homologous recombination to mimic p.E635RfsX15 that is well-described in human disease (122). These mice were crossed with transgenic Vav-Cre mice in which recombination occurs in the hematopoietic system. Modest anemia was observed along with skewing of hematopoietic bone marrow population toward megakaryocyte progenitors and away from erythrocyte progenitors, but no frank MDS or AML was observed. However, when the authors incorporated concurrent expression of a mutant form of RUNX1 into their mouse model, they found that co-expression of these two mutations led to development of frank MDS/AML with a short latency period following induced expression of the mutations (median survival of 160 days). They also performed retroviral insertional mutagenesis and found that all mice in this experimental cohort developed AML within a 1.5-year observation period.
Another knock-in model was recently created using CRISPR/Cas9 to introduce the most common MDS-associated mutation in humans, ASXLl1G643W, into murine embryonic stem cells resulting in expression of a truncated ASXL1 protein, mirroring that which occurs in humans (123, 145). This solo model again produced only mild impacts on hematopoiesis with skewing towards the myeloid lineage and mild splenomegaly. However, combination with an inducible model of Cepba haploinsufficiency drove the development of AML. Together these findings demonstrate the utility of a mouse model of Asxl1 that recapitulates the pre-leukemic effect of this lesion and can be used to study its interaction with cooperating mutations in the progression to leukemia.
5.4 Cohesin Complex
The cohesin complex is a ring-like structure made up of various protein components including SMC1, SMC3, RAD21, and STAG1/2. Cohesin mediates the approximation of DNA fragments and plays a crucial role in sister chromatid cohesion, homologous recombination, and DNA looping. In turn, cohesin regulates gene expression via the approximation of distal enhancers and promoters. Somatic cohesin mutations have been identified in 12% of myeloid malignancies and often co-occur with genetic lesions such as t (8;21) and mutations in genes such as TET2, NPM1, and ASXL1 (146, 147). Cohesin mutations are mutually exclusive, suggesting that reduction of one component leads to insufficiency of the entire complex. Therefore, the specific cohesin gene targeted in various murine models should not affect the disease. It is believed that cohesin’s vital role in sister chromatid cohesion makes complete cohesin loss incompatible with life. This hypothesis has been supported by the universally lethal effect of homozygous Rad21 deletion in mice (148).
Two groups published mouse models of cohesin loss in 2015 using different genetic engineering techniques. Viny et al. (124) created a conditional knock-out model in which exon 4 is flanked by loxP sites. When crossed with transgenic Mx1-Cre mice, they induced complete loss of SMC3 in hematopoietic tissues via pIpC injections. Biallelic loss of Smc3 resulted in 100% lethality within 11 days of Cre recombinase activation due to CNS hemorrhage and multiorgan failure. They then created a model of conditional Smc3 haploinsufficiency via Cre-mediated deletion of a single Smc3 allele in hematopoietic tissues and found a resultant increase in self-renewal capacity of HSCs. Alone, Smc3 haploinsufficiency did not lead to phenotypic changes suggestive of MDS or leukemia, but the addition of Flt3-ITD expression within the mouse model of cohesin insufficiency led to AML with shortened latency compared to Flt3-ITD alone.
Alternatively, Mullenders et al. introduced shRNA to silence expression of cohesin components in a mouse model (125). They introduced a GFP transgene that housed a single copy of their engineered shRNA downstream of a tetracycline (Tet)-responsive element and crossed them with rtTA-transgenic animals to create an inducible and reversible model of cohesin knockdown. shRNAs were engineered to knockdown three of the core cohesin proteins, Rad21, Smc1a and Stag2. Mice were exposed to doxycycline starting at 6 weeks of age to induce shRNA expression. Rad21 (shRNA/+), Smc1a (shRNA/+), Stag2 (shRNA/+) mice all demonstrated a skewing toward the myeloid lineages. Interestingly, Smc1a-targeting shRNA expression resulted in a significant decrease in all core cohesin proteins rather than just in SMC1A. Over time, a subset of these cohesin knockdown mice developed a myeloproliferative neoplasm-like phenotype characterized by blood and bone marrow myeloid hyperplasia, hypocellular bone marrow, and splenomegaly.
shRNA knockdown and Cre recombinase-mediated deletion remain popular methods of modeling cohesin loss, particularly in novel models that combine cohesin insufficiency with other gene mutations to mirror leukemogenesis in humans. Both strategies effectively reduce gene expression and can be induced at specific timepoints to elucidate the precise order of acquisition that is hypothesized to occur in subsets of leukemia.
5.5 BCOR
BCOR is a tumor suppressor gene that encodes the transcription repressor BCL6 corepressor (BCOR). Over the past decade mutations in BCOR have been identified in 3.4-5% of cytogenetically normal AML in adults and 1.2% of pediatric AML (149–152). In 2017, Tanaka et al. created a transgenic mouse model using tamoxifen-inducible Cre-ERT in the ROSA26 locus to delete exon 4 of Bcor resulting in a truncated protein that lacks the BCL6 binding site (126). HSCs harvested from this model exhibited impaired repopulating capabilities. Half of their cohort developed T lymphoblastic leukemia but they did not observe development of myeloid malignancy.
Dr. Kelly and colleagues similarly generated a model in which exons 9 and 10 of the murine Bcor gene is flanked by loxP sites. When crossed to mice with transgenic HSC-SCL-Cre-ERT recombinase, Cre activation by tamoxifen injections lead to deletion of exons 9 and 10 of Bcor (termed BcorΔE9-10) specifically in HSCs, resulting in low level expression of a truncated protein that lacks the C-terminal PCGF Ub-like fold discriminator domain (127). BcorΔE9-10 mice did not exhibit an abnormal hematopoietic phenotype but did demonstrate expansion of the myeloid progenitor compartment. The authors then found that combination of BcorΔE9-10 and KrasG12D results in a highly penetrant model of myeloid leukemia with significantly decreased survival compared to leukemia resulting from KrasG12D alone.
Findings from recent Bcor-mutated murine models of AML have established a strong role for Bcor in the development of acute erythroid leukemia (AEL). Drs. Brunangelo Falini, Margaret Goodell, and colleagues collaborated to generate a double Dnmt3a-/- and Bcor-/- mouse model (128). They first created conditional Bcor knockout in which exons 8-10 are flanked by loxP sites crossed with transgenic Mx1-Cre to induce Bcor knock out in the hematopoietic system. These mice were then mated with others carrying two floxed Dnmt3a alleles and induced full knockout of both genes, which resulted in a fully penetrant and lethal AEL. They then used this model to demonstrate the efficacy of the demethylating agent decitabine, which exhibited better control of tumor burden than cytarabine alone. Dr. Charles Mullighan and colleagues used multiplex genome editing in HSPCs that were transplanted into primary recipient mice and similarly found that co-occurrence of mutations in Bcor and Trp53 strongly promoted development of AEL (76).
These existing murine models recapitulate what is observed in humans and will help to better understand the precise role of BCOR loss as it cooperates with leukemogenic mutations in other cellular pathways as well as the role it plays in therapeutic response.
6 Other Recurrent AML Mutations
There are several recurrently mutated genes that do not fall into the specific categories discussed above which have distinct roles in leukemia pathophysiology. These mutations will be reviewed below.
6.1 NPM1
Mutations of the gene NPM1 are among the most common mutations in adult AML, found in up to 35% of patients (15). While less common in childhood AML (8-10%), like in adults, NPM1-mutant AML is generally associated with a favorable prognosis, co-occurrence with FLT3-ITD mutations, and HOX gene overexpression (18). NPM1 is a molecular chaperone with roles in centrosome duplication, ribosome biogenesis, and stress-induced regulation of P53 (153). In its wild-type state, NPM1 shuttles rapidly between the nucleus and cytoplasm, and predominantly localizes to the nucleoli. In AML, frameshift mutations of NPM1 lead to its aberrant cytoplasmic localization, thus the common designation, NPMc+ (15). Given the prevalence of NPM1 mutations in AML, there has been much interest in generating murine models with which to study the biology of NPM1-mutant AML.
In 2010, researchers from the lab of Dr. Pier Paolo Pandolfi published their work developing and characterizing a murine model in which mutant NPM1 was transgenically expressed under the control of the hMRP8 promoter, resulting in overexpression of mutant NPM1 in myeloid progenitors. These mice developed myeloproliferation in the bone marrow and spleens but did not develop overt AML (154). This model was later crossed to mice with a heterozygous germline Flt3-ITD mutation, demonstrating cooperativity between these commonly co-occurring lesions (98). In 2012, another group developed a model in which they knocked-in TCTG into murine Npm1 exon 11, similar to the most common frameshift mutation found in human AML. While the resultant mutant protein differed slightly from human NPMc+ protein, the group still found excess cytoplasmic expression. This strategy led to an incompletely penetrant MPD, but like the transgenic model from the Pandolfi group, did not develop AML (155). Around this same time, Vassiliou et al. developed the first conditional NPMc+ model, by knocking-in a ‘humanized’ exon 11 frameshift mutation (the most common mutation in AML) just downstream of murine exon 11. They also knocked-in loxP sites flanking murine exon 11, such that Cre-mediated recombination leads to excision of the native exon 11 and exclusive expression of humanized mutant Npm1c. As expected, when induced in the hematopoietic system by pIpC activation of Mx1-Cre, the mutant protein localized predominantly to the cytoplasm. Expression of mutant NPM1c led to Hox gene overexpression, enhanced stem cell self-renewal and myeloid skewing with ~1/3 of the mice developing a delayed onset AML (156). When crossed to mice constitutively expressing Flt3-ITD mutation, mice with induced Npm1c expression developed a rapid onset AML, confirming molecular synergy (157). This model has also proved useful for pre-clinical investigations including studies demonstrating the therapeutic potential of inhibitors of the MENIN-MLL interaction for the treatment of NPMc+ AML (158).
More recently, the lab of Dr. Jennifer Trowbridge designed a humanized, inducible Npm1c mutation similar to that developed by Vassiliou et al., but instead of a Cre-inducible system, utilized an Flp-recombinase inducible system. They crossed these mice with transgenic mice in which the tamoxifen-inducible FlpoER is knocked into the Gt (ROSA) 26Sor locus. This strategy allowed for the sequential induction of both a Cre-inducible Dnmt3aR878 mutation (see DNMT3A section above) and the FLP-inducible expression of Npm1c. A potential disadvantage of this strategy is that because FlpoER is knocked into the Rosa26 locus it is expressed ubiquitously; to isolate the hematologic effects of these combined mutations, hematopoietic transplantation was necessary. In isolation, induction of the Npm1c mutation in the hematopoietic system led to a low penetrance MPD but no overt AML. However, when induction of Npm1c was preceded by induction of the Dnmt3a mutation, the mice developed a highly penetrant MDS and/or MPD. Interestingly, the disease latency was inversely correlated with the length of time between induction of the Dnmt3a mutation and the Npm1c mutation (i.e., the longer the mouse had expression of mutant Dnmt3a prior to induction of Npm1c expression, the shorter the disease latency) (110).
6.2 TP53
Greater than 50% of human cancers carry mutations in the well-established tumor suppressor gene TP53 (159). Interestingly TP53 mutations are only detected in less than 10% of de novo AML but confer high risk disease with an extremely poor prognosis (10, 160). Creation of a mouse model of TP53-mutant AML is important for further therapeutic development in this traditionally hard to treat subset. Isolated mutations in Trp53 have proven insufficient to induce leukemogenesis in mouse models but can be combined with commonly co-occurring mutations to generate models of myeloid leukemia that can be used to define the role of cooperative mutations and provide an environment for the development of new therapeutic targets and strategies.
Several transgenic mouse models have shown that Trp53 cooperates with other genetic aberrations to hasten the development of AML. In the development of these AML models it was important to utilize non-germline methods to reduce p53 expression as Trp53 null mice are prone to developing T cell malignancies. Stoddart et al. created a novel model of therapy-related AML using a Trp53-targeting shRNA to knock down expression of p53 in bone marrow cells harvested from transgenic mice harboring heterozygous loss of Erg1 and Apc, two genes located on the long arm of chromosome 5, and thus lost in AML with 5q deletion (161). While they were successful in generating a novel model that may mirror the behavior of therapy-related AML, the low penetrance of disease (17%) likely indicates that additional genes lost with 5q deletion play an important role in the transformation to AML. This is supported by work done by Yang et al., who recently generated dual transgenic mice with Gilliland Flt3-ITD knock-in model and either heterozygous or homozygous Trp53 deletion (162). Heterozygous Trp53 knockout significantly increased the penetrance and lethality of myeloid leukemia compared to Flt3-ITD alone, and homozygous Trp53 knockout led to a further decrease in median survival.
Additional models have been created to recapitulate the common co-occurrence of mutations in TP53 and Ras family genes and determine their cooperative roles in leukemogenesis. Zhou et al. found that Trp53 depletion accelerated AML development in mice expressing KrasG12D (163). Members of Dr. Jing Zhang’s lab then created a conditional transgenic model of AML using Mx1-Cre to induce homozygous loss of Trp53 in the hematopoietic system and concurrently induce expression of the oncogenic NrasG12D (164). They used RNA sequencing to define the transcriptome of the leukemic cells and determined that Trp53 loss cooperated with mutant Nras within the megakaryocyte-erythroid progenitors to result in AML.
Together, these findings have shown the utility of generating mouse models of Trp53-mutant AML to advance our understanding of and develop new treatment strategies for this aggressive subcategory of AML.
7 Patient Derived Xenografts
The murine models of AML discussed above are essential tools to study leukemia development and unfold molecular mechanisms related to the disease. Despite sophisticated advancements in transgenic mouse modeling, there are differences in the murine and human leukemic phenotype that require humanized models to better understand nuances of human disease. Ex vivo models with human AML blasts have been developed for these purposes but are limited due to the missing interaction of the human AML blasts with the bone marrow microenvironment which plays an important role in leukemia development and therapeutic response. Here we review patient derived xenograft models that have been developed and improved over time to overcome these limitations.
The development of immunocompromised mouse models was essential for the study of patient-derived tumors and preclinical discovery of new compounds for cancer treatment (Table 4). The first immunodeficient mouse model was described in 1966 by Flanagan (165). These nude athymic (nu/nu) mice were T-cell-deficient due to a homozygous Foxn1 mutation and enabled the study of human cancer in mice (166). However, this model has its limitations for studying AML since the intact murine B- and natural killer (NK)-cell populations lead to poor human AML engraftment (167). The severe combined immune deficiency (SCID) phenotype was first described in C.B-17 mice in 1983, caused by a homozygous mutation in the Scid gene (scid/scid). The SCID immunodeficient mouse model proved superior to nude mice for in vivo studies of human cancers (168). The Scid mutation affects the VDJ recombinase system and results in a lack of mature T- and B-lymphocytes. In the early 1990s, multiple groups showed the engraftment of human myeloid leukemia cell lines and patient AML cells in SCID mice, indicating that this PDX model is a useful tool for studying AML. However, the SCID model still had technical limitations as some human AMLs failed to engraft without exogenous cytokine treatment (169–171). Reports by Carrol et al. and Riggs et al. suggested that this mouse model is “leaky” as mice showed the presence of mature lymphocytes, likely explaining the relative engraftment barrier (182, 183).
TABLE 4
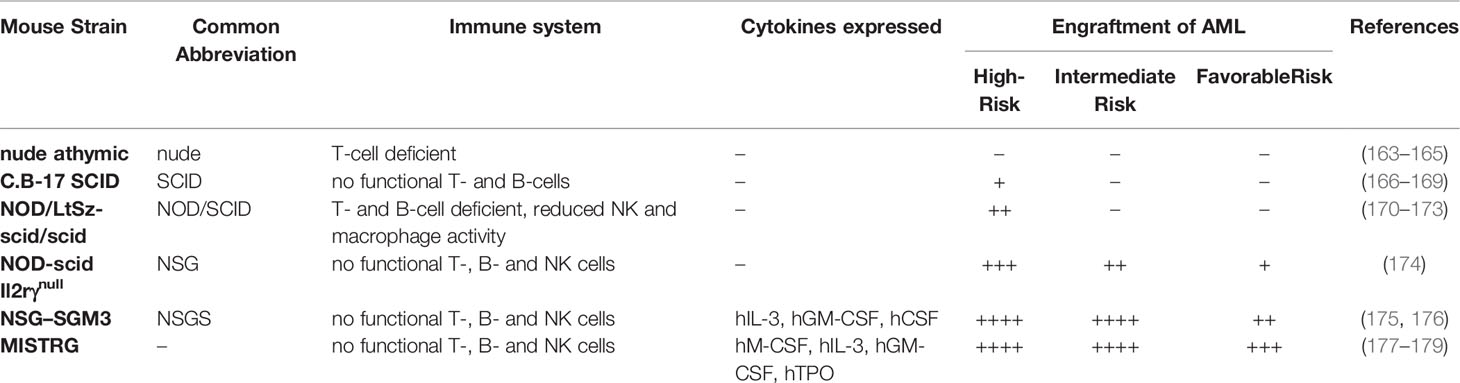
Table 4 Mouse models available for patient-derived xenografts of AML.
Throughout the years more severe immunodeficient mouse models have been developed to improve engraftment, including the non-obese diabetic NOD/SCID (NOD/LtSz-scid/scid) mouse. SCID mice were backcrossed onto the NOD/Lt strain background resulting in a strain that is deficient in B- and T-lymphocytes and shows reduced NK-cell and macrophage activity (172). In contrast to SCID mice, normal human bone marrow cells and patient-derived AML cells can be engrafted with much higher efficiency in NOD/SCID mice using fewer cells (173, 174, 184, 185). A robust engraftment of primitive CD34+CD38- leukemic progenitors, also known as NOD/SCID leukemia-initiating cells (NOD/SL-IC) or leukemic stem cells (LSC), was observed in NOD/SCID mice. Serial transplantation of LSCs and their potential to differentiate into leukemic blasts provides the opportunity to study those leukemia initiating clones more closely and identify potential drugs against them. However, while the NOD/SCID xenograft model has shown efficient engraftment for high-risk AML cases, it is still limited by poor engraftment for favorable and intermediate-risk AML (175).
The introduction of a deletion of the interleukin 2 (IL2) receptor gamma chain (Il2rγ) gene led to an even more immune compromised mouse model, the NOD-scid Il2rγnull (NSG) mouse, that improved engraftment of patient AML cells regardless of the French-American-British classification or cytogenetic features (176). Better engraftment and longer lifespan (>16 months) made NSG mice the preferred immunodeficient model for AML research over NOD/SCID mice. Studies in NSG mice showed that human LSCs can home to the bone marrow niche of the mouse resulting in quiescent LSC that are resistant to chemotherapy (186). Those features of the NSG PDX model that recapitulates human AML facilitate the study of chemotherapy resistance conferring mechanisms. The use of NSG PDX models has also led to a better understanding of the heterogeneity of leukemia-initiating cells (LIC) as reports identified the existence of not only CD34+CD38- LIC cells but also CD34+CD38+ and even CD34- populations capable of initiating AML (187, 188).
Over time more strains have been developed to overcome the limitations of the previous models. Wunderlich et al. generated a mouse strain that transgenically expresses the human cytokines stem cell factor (SCF), GM-CSF, and IL-3 (SGM3) in the NOD/SCID background (177). Usage of NOD/LtSz-scid IL2RG–SGM3 mice (NSGS) led to superior engraftment of AML cells compared to other strains (177, 178). Even AML samples from favorable risk groups like CBF AML, NPM1-mutated/FLT3 wild type, or CEBPA-mutated AML, that are difficult to engraft in previous immunocompromised strains, were capable of engrafting in NSGS mice (177, 178).
An even more advanced immunodeficient mouse strain was developed by Dr. Flavell and colleagues (179). MISTRG mice are Rag2- and IL2rγ-deficient, with genes for four human cytokines (hM-CSF, hIL-3, hGM-CSF, and hTPO) knocked-in to their respective mouse loci. Because they are under the control of their endogenous promoters, they are expressed at physiologic levels as opposed to the supraphysiologic levels in NSGS mice. The MISTRG mice also express human SIRPa which binds to human CD47 and results in inhibition of phagocytosis of human cells and hence supports better engraftment (180). Ellegast et al. reported successful engraftment of NPM1 mutated and inv (16) AML samples using this advanced mouse model (181). They also reported an important role for M-CSF expression in mice using MSTRG (knock-in for only hMCSF and hTPO) as an important factor for inv (16) AML engraftment. The successful engraftment of t (8;21) AML cells in MISTRG mice that leads to leukemia has not yet been shown.
The development of new immune compromised mouse models over the last few decades has led to increasingly efficient engraftment of human AML samples. This gave rise to new insights into AML hierarchy, genetic and functional characteristics of human AML, and the option for drug testing on human AML cells in an in vivo model. Yet, there are still challenges that need to be addressed in the future such as efficient engraftment of less aggressive AML subtypes. Recently, it has been shown by Reinisch et al. that establishing a human bone marrow niche in the mouse by transplanting human bone marrow-mesenchymal stem cell (MSC)-derived ossicles results in robust and superior engraftment of human AML samples (189). In another study, hMSC were seeded in a gelatin-based porous scaffold and cultured in vitro (190). Before implanting the scaffolds into NSG mice, patient-derived AML cells were pre-seeded into the scaffold. This method led to successful engraftment of AML samples that failed to engraft in NSG mice after intravenous injection of the leukemic cells. This approach has also been tested for other stromal cell types like endothelial cells and osteoblasts. Since there is crosstalk between leukemia cells and the bone marrow niche, these kinds of models help to study the leukemia bone marrow microenvironment and possible cell-extrinsic mechanisms that may contribute to chemotherapy resistance.
Another xenograft mouse model to study leukemia has been developed using isolated human CD34+ hematopoietic cells from healthy donors that are transduced with a viral vector expressing system or have been genetically engineered which are then transplanted into immunodeficient mice. Wei et al. used the retroviral transduction method to express the KMT2A-MLLT3 fusion protein in human CD34+ cord blood (huCB) cells resulting in indefinite proliferation of the cells in vivo and in vitro (191). Transplanting the KMT2A-MLLT3 expressing cells into different immunodeficient mouse models led to AML in NSGS mice and AML, B-ALL or mixed lineage leukemia in NSG and NSG beta 2 microglobulin (NS-B2M) mice showing that the immune microenvironment plays an important role in leukemia development. In this approach both copies of the wild-type KMT2A gene are present which is not found in KMT2Ar leukemias resulting in unknown regulatory effects by the wild-type protein. Dr. Corinna Buechele and colleagues used transcription activator-like effector nucleases (TALENs) gene editing tool to introduce DNA double strand breaks at known breakpoint cluster regions in the KMT2A gene and nucleofected with DNA templates with KMT2A homology flanking the sites targeted by TALENs, fusion partner (MLLT3 or MLLT1) cDNA, and a fluorescent tag. The strategy resulting in the expression of the designed KMT2A fusion proteins in huCB cells (192). The model was sufficient to initiate leukemia in NSG mice and recapitulated many clinical features of KMT2Ar leukemias. A model for AML with t(6;9)(p22;q34) was successfully established by transplanting human CD34+ cells transduced with the DEK-NUP214 fusion gene into NSGS mice resulting in AML development and showing phenotypic and genetic features of human t(6;9) AML (193). Genetic alteration of human CD34+ hematopoietic cells and transplantation into immunodeficient mice provides a humanized mouse model to study the leukemic potential of mutations and chromosomal aberrations found in AML without the potential confounding effects of the co-occurring mutations inherent to human AML cell lines and primary patient samples.
8 Conclusion
For AML, like many other human diseases, mouse models are indispensable research tools. Genetically engineered models of common AML driver lesions have provided invaluable insight into disease mechanisms, led to identification of therapeutic targets, and enhanced our understanding of the intricate interactions between collaborating genetic and epigenetic events. Engraftment of human AML cells into immunocompromised mice has expanded our understanding of tumor heterogeneity, shaped the definition of the leukemia initiating cell and allowed for critical pre-clinical investigations of promising novel therapeutics.
Over just the last few years, research employing murine models of AML has resulted in several highly significant advancements in the field. For example, we have learned that not just the combination of mutations matter in the genesis of AML, but the order of mutation acquisition, the age at which the mutations are acquired, and even the length of time between collaborating mutations critically impacts disease development (40, 118). Additionally, efforts to develop mouse models of rare genetic disease entities, such as NUP98-rearranged and BCOR mutant AML have shed new mechanistic light on previously poorly understood genetic drivers and revealed novel therapeutic vulnerabilities (51, 52, 75, 77, 128). Further, ongoing improvements in immunodeficient mouse strains coupled with efficient genome engineering tools have allowed for not only the engraftment of virtually all subsets of human AML cells into mice, but also for the transformation of normal human hematopoietic cells into leukemic cells capable of engraftment into immunocompromised mice (179, 189, 191–193).
While undeniably powerful, limitations of murine models still must be acknowledged including potential differences in murine and human biology, differences in the malignancies that develop in mice compared to humans, and remaining challenges in engraftment of certain human leukemias into immunodeficient murine recipients. However, with numerous labs around the world working to optimize existing and generate new and relevant murine models, the field will undoubtedly continue to advance, paving the way for ongoing improvements in the care of patients with AML.
Author Contributions
RR, SC, KK and MO’K participated in the conception and content planning for the review. KK and SC contributed equally to the composition of the manuscript. MO’K, KW and RR contributed substantially to the fabrication of the paper. RR reviewed and edited the manuscript as last author. All authors contributed to the article and approved the submitted version.
Funding
KK was supported by CPRIT RP210027 - Baylor College of Medicine Comprehensive Cancer Training Program. SC was supported by the St. Baldrick’s Foundation and Children’s Cancer Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sakurai M, Sandberg AA. Prognosis of Acute Myeloblastic Leukemia: Chromosomal Correlation. Blood (1973) 41(1):93–104. doi: 10.1182/blood.V41.1.93.93
2. Keating MJ, Cork A, Broach Y, Smith T, Walters RS, McCredie KB, et al. Toward a Clinically Relevant Cytogenetic Classification of Acute Myelogenous Leukemia. Leuk Res (1987) 11(2):119–33. doi: 10.1016/0145-2126(87)90017-8
3. Voso MT, De Bellis E, Ottone T. Diagnosis and Classification of AML: WHO 2016. In: Acute Myeloid Leukemia. Cham: Springer (2021). p. [23–54].
4. Quessada J, Cuccuini W, Saultier P, Loosveld M, Harrison CJ, Lafage-Pochitaloff M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes (Basel) (2021) 12(6):924. doi: 10.3390/genes12060924
5. Creutzig U, Zimmermann M, Reinhardt D, Rasche M, von Neuhoff C, Alpermann T, et al. Changes in Cytogenetics and Molecular Genetics in Acute Myeloid Leukemia From Childhood to Adult Age Groups. Cancer (2016) 122(24):3821–30. doi: 10.1002/cncr.30220
6. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
7. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
8. Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Novel Prognostic Subgroups in Childhood 11q23/MLL-Rearranged Acute Myeloid Leukemia: Results of an International Retrospective Study. Blood (2009) 114(12):2489–96. doi: 10.1182/blood-2009-04-215152
9. Moarii M, Papaemmanuil E. Classification and Risk Assessment in AML: Integrating Cytogenetics and Molecular Profiling. Hematol Am Soc Hematol Educ Program (2017) 2017(1):37–44. doi: 10.1182/asheducation-2017.1.37
10. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
11. Chen X, Zhu H, Qiao C, Zhao S, Liu L, Wang Y, et al. Next-Generation Sequencing Reveals Gene Mutations Landscape and Clonal Evolution in Patients With Acute Myeloid Leukemia. Hematology (2021) 26(1):111–22. doi: 10.1080/16078454.2020.1858610
12. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional Genomic Landscape of Acute Myeloid Leukaemia. Nature (2018) 562(7728):526–31. doi: 10.1038/s41586-018-0623-z
13. Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. Publisher Correction: The Molecular Landscape of Pediatric Acute Myeloid Leukemia Reveals Recurrent Structural Alterations and Age-Specific Mutational Interactions. Nat Med (2019) 25(3):530. doi: 10.1038/s41591-019-0369-7
14. Conneely SE, Rau RE. The Genomics of Acute Myeloid Leukemia in Children. Cancer Metastasis Rev (2020) 39(1):189–209. doi: 10.1007/s10555-020-09846-1
15. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia With a Normal Karyotype. N Engl J Med (2005) 352(3):254–66. doi: 10.1056/NEJMoa041974
16. Ho PA, Kutny MA, Alonzo TA, Gerbing RB, Joaquin J, Raimondi SC, et al. Leukemic Mutations in the Methylation-Associated Genes DNMT3A and IDH2 are Rare Events in Pediatric AML: A Report From the Children's Oncology Group. Pediatr Blood Cancer (2011) 57(2):204–9. doi: 10.1002/pbc.23179
17. Juhl-Christensen C, Ommen HB, Aggerholm A, Lausen B, Kjeldsen E, Hasle H, et al. Genetic and Epigenetic Similarities and Differences Between Childhood and Adult AML. Pediatr Blood Cancer (2012) 58(4):525–31. doi: 10.1002/pbc.23397
18. Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, et al. The Incidence and Clinical Significance of Nucleophosmin Mutations in Childhood AML. Blood (2007) 110(3):979–85. doi: 10.1182/blood-2007-02-076604
19. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial Sequencing and Comparative Analysis of the Mouse Genome. Nature (2002) 420(6915):520–62. doi: 10.1038/nature01262
20. Zheng-Bradley X, Rung J, Parkinson H, Brazma A. Large Scale Comparison of Global Gene Expression Patterns in Human and Mouse. Genome Biol (2010) 11(12):R124. doi: 10.1186/gb-2010-11-12-r124
21. O'Connell KE, Mikkola AM, Stepanek AM, Vernet A, Hall CD, Sun CC, et al. Practical Murine Hematopathology: A Comparative Review and Implications for Research. Comp Med (2015) 65(2):96–113.
22. LW L, Taormina V, Boyle PJ. Response of Acute Lymphocytic Leukemias to the Purine Antagonist 6-Mercaptopurine. Ann N Y Acad Sci (1954) 60(2):244–50. doi: 10.1111/j.1749-6632.1954.tb40015.x
23. Skipper HE, Perry S. Kinetics of Normal and Leukemic Leukocyte Populations and Relevance to Chemotherapy. Cancer Res (1970) 30(6):1883–97.
24. Skipper HE, Schabel FM, Wilcox WS. Experimental Evaluation of Potential Anticancer Agents. XXI. Scheduling of Arabinosylcytosine to Take Advantage of Its S-Phase Specificity Against Leukemia Cells. Cancer Chemother Rep (1967) 51(3):125–65.
25. Casazza AM, Pratesi G, Giuliani F, Di Marco A. Antileukemic Activity of 4-Demethoxydaunorubicin in Mice. Tumori (1980) 66(5):549–64. doi: 10.1177/030089168006600503
26. Law LW, Dunn TB. Observations on the Effect of a Folic-Acid Antagonist on Transplantable Lymphoid Leukemias in Mice. J Natl Cancer Inst (1949) 10(1):179–92.
27. Kawasaki Y, Hirabayashi Y, Kaneko T, Kanno J, Kodama Y, Matsushima Y, et al. Benzene-Induced Hematopoietic Neoplasms Including Myeloid Leukemia in Trp53-Deficient C57BL/6 and C3H/He Mice. Toxicol Sci (2009) 110(2):293–306. doi: 10.1093/toxsci/kfp107
28. Furth J, Seibold H, Rathbone R. Experimental Studies on Lymphomatosis of Mice. Am J Cancer (1933) 19(3):521–604.
29. Gross L. “Spontaneous” Leukemia Developing in C3H Mice Following Inoculation in Infancy, With AK-Leukemic Extracts, or AK-Embrvos. Proc Soc Exp Biol Med (1951) 76(1):27–32.
30. Siegler R, Rich MA. Pathogenesis of Virus-Induced Myeloid Leukemia in Mice. J Natl Cancer Inst (1967) 38(1):31–50.
31. McGarry MP, Steeves RA, Eckner RJ, Mirand EA, Trudel PJ. Isolation of a Myelogenous Leukemia-Inducing Virus From Mice Infected With the Friend Virus Complex. Int J Cancer (1974) 13(6):867–78. doi: 10.1002/ijc.2910130614
32. Bedigian HG, Johnson DA, Jenkins NA, Copeland NG, Evans R. Spontaneous and Induced Leukemias of Myeloid Origin in Recombinant Inbred BXH Mice. J Virol (1984) 51(3):586–94. doi: 10.1128/jvi.51.3.586-594.1984
33. Copeland NG, Buchberg AM, Gilbert DJ, Jenkins NA. Recombinant Inbred Mouse Strains: Models for Studying the Molecular Genetic Basis of Myeloid Tumorigenesis. Curr Top Microbiol Immunol (1989) 149:45–57. doi: 10.1007/978-3-642-74623-9_4
34. Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, Nakamura T, et al. Leukaemia Disease Genes: Large-Scale Cloning and Pathway Predictions. Nat Genet (1999) 23(3):348–53. doi: 10.1038/15531
35. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The C-Myc Oncogene Driven by Immunoglobulin Enhancers Induces Lymphoid Malignancy in Transgenic Mice. Nature (1985) 318(6046):533–8. doi: 10.1038/318533a0
36. Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic Mice Bearing the Human C-Myc Gene Activated by an Immunoglobulin Enhancer: A Pre-B-Cell Lymphoma Model. Proc Natl Acad Sci USA (1988) 85(16):6047–51. doi: 10.1073/pnas.85.16.6047
37. Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, et al. A Previously Unidentified Alternatively Spliced Isoform of T(8;21) Transcript Promotes Leukemogenesis. Nat Med (2006) 12(8):945–9. doi: 10.1038/nm1443
38. Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, et al. AML1-ETO Expression Is Directly Involved in the Development of Acute Myeloid Leukemia in the Presence of Additional Mutations. Proc Natl Acad Sci U S A (2001) 98(18):10398–403. doi: 10.1073/pnas.171321298
39. Cabezas-Wallscheid N, Eichwald V, de Graaf J, Löwer M, Lehr HA, Kreft A, et al. Instruction of Haematopoietic Lineage Choices, Evolution of Transcriptional Landscapes and Cancer Stem Cell Hierarchies Derived From an AML1-ETO Mouse Model. EMBO Mol Med (2013) 5(12):1804–20. doi: 10.1002/emmm.201302661
40. Abdallah MG, Niibori-Nambu A, Morii M, Yokomizo T, Ideue T, Kubota S, et al. RUNX1-ETO (RUNX1-RUNX1T1) Induces Myeloid Leukemia in Mice in an Age-Dependent Manner. Leukemia (2021) 35(10):2983–8. doi: 10.1038/s41375-021-01268-4
41. Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, et al. Cbf Beta-SMMHC Induces Distinct Abnormal Myeloid Progenitors Able to Develop Acute Myeloid Leukemia. Cancer Cell (2006) 9(1):57–68. doi: 10.1016/j.ccr.2005.12.014
42. Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, et al. An Mll-AF9 Fusion Gene Made by Homologous Recombination Causes Acute Leukemia in Chimeric Mice: A Method to Create Fusion Oncogenes. Cell (1996) 85(6):853–61. doi: 10.1016/S0092-8674(00)81269-6
43. Collins EC, Pannell R, Simpson EM, Forster A, Rabbitts TH. Inter-Chromosomal Recombination of Mll and Af9 Genes Mediated by cre-loxP in Mouse Development. EMBO Rep (2000) 1(2):127–32. doi: 10.1093/embo-reports/kvd021
44. Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of Origin Determines Clinically Relevant Subtypes of MLL-Rearranged AML. Leukemia (2013) 27(4):852–60. doi: 10.1038/leu.2012.363
45. Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell (2016) 30(1):43–58. doi: 10.1016/j.ccell.2016.05.011
46. Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, et al. Mll Partial Tandem Duplication and Flt3 Internal Tandem Duplication in a Double Knock-in Mouse Recapitulates Features of Counterpart Human Acute Myeloid Leukemias. Blood (2012) 120(5):1130–6. doi: 10.1182/blood-2012-03-415067
47. Ono R, Masuya M, Nakajima H, Enomoto Y, Miyata E, Nakamura A, et al. Plzf Drives MLL-Fusion-Mediated Leukemogenesis Specifically in Long-Term Hematopoietic Stem Cells. Blood (2013) 122(7):1271–83. doi: 10.1182/blood-2012-09-456665
48. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-Rearranged Leukemia is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell (2011) 20(1):66–78. doi: 10.1016/j.ccr.2011.06.010
49. Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol (2007) 9(7):804–12. doi: 10.1038/ncb1608
50. Thanasopoulou A, Tzankov A, Schwaller J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica (2014) 99(9):1465–71. doi: 10.3324/haematol.2013.100917
51. Mohanty S, Jyotsana N, Sharma A, Kloos A, Gabdoulline R, Othman B, et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers (Basel) (2020) 12(10):2766. doi: 10.3390/cancers12102766
52. Schmoellerl J, Barbosa IAM, Eder T, Brandstoetter T, Schmidt L, Maurer B, et al. CDK6 is an Essential Direct Target of NUP98 Fusion Proteins in Acute Myeloid Leukemia. Blood (2020) 136(4):387–400. doi: 10.1182/blood.2019003267
53. Lopez CK, Noguera E, Stavropoulou V, Robert E, Aid Z, Ballerini P, et al. Ontogenic Changes in Hematopoietic Hierarchy Determine Pediatric Specificity and Disease Phenotype in Fusion Oncogene-Driven Myeloid Leukemia. Cancer Discov (2019) 9(12):1736–53. doi: 10.1158/2159-8290.CD-18-1463
54. Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered Myeloid Development and Acute Leukemia in Transgenic Mice Expressing PML-RAR Alpha Under Control of Cathepsin G Regulatory Sequences. Blood (1997) 89(2):376–87. doi: 10.1182/blood.V89.2.376
55. Du C, Redner RL, Cooke MP, Lavau C. Overexpression of Wild-Type Retinoic Acid Receptor Alpha (RARalpha) Recapitulates Retinoic Acid-Sensitive Transformation of Primary Myeloid Progenitors by Acute Promyelocytic Leukemia RARalpha-Fusion Genes. Blood (1999) 94(2):793–802. doi: 10.1182/blood.V94.2.793
56. Westervelt P, Lane AA, Pollock JL, Oldfather K, Holt MS, Zimonjic DB, et al. High-Penetrance Mouse Model of Acute Promyelocytic Leukemia With Very Low Levels of PML-RARalpha Expression. Blood (2003) 102(5):1857–65. doi: 10.1182/blood-2002-12-3779
57. Speck NA, Gilliland DG. Core-Binding Factors in Haematopoiesis and Leukaemia. Nat Rev Cancer (2002) 2(7):502–13. doi: 10.1038/nrc840
58. Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M, et al. Embryonic Lethality and Impairment of Haematopoiesis in Mice Heterozygous for an AML1-ETO Fusion Gene. Nat Genet (1997) 15(3):303–6. doi: 10.1038/ng0397-303
59. Downing JR. The Core-Binding Factor Leukemias: Lessons Learned From Murine Models. Curr Opin Genet Dev (2003) 13(1):48–54. doi: 10.1016/S0959-437X(02)00018-7
60. Castilla LH, Wijmenga C, Wang Q, Stacy T, Speck NA, Eckhaus M, et al. Failure of Embryonic Hematopoiesis and Lethal Hemorrhages in Mouse Embryos Heterozygous for a Knocked-in Leukemia Gene CBFB-Myh11. Cell (1996) 87(4):687–96. doi: 10.1016/S0092-8674(00)81388-4
61. Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, et al. The MLL Recombinome of Acute Leukemias in 2017. Leukemia (2018) 32(2):273–84. doi: 10.1038/leu.2017.213
62. Hess JL, Yu BD, Li B, Hanson R, Korsmeyer SJ. Defects in Yolk Sac Hematopoiesis in Mll-Null Embryos. Blood (1997) 90(5):1799–806. doi: 10.1182/blood.V90.5.1799
63. Drynan LF, Pannell R, Forster A, Chan NM, Cano F, Daser A, et al. Mll Fusions Generated by Cre-loxP-Mediated De Novo Translocations can Induce Lineage Reassignment in Tumorigenesis. EMBO J (2005) 24(17):3136–46. doi: 10.1038/sj.emboj.7600760
64. Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, et al. Abrogation of MLL-AF10 and CALM-AF10-Mediated Transformation Through Genetic Inactivation or Pharmacological Inhibition of the H3K79 Methyltransferase Dot1l. Leukemia (2013) 27(4):813–22. doi: 10.1038/leu.2012.327
65. Moriya K, Suzuki M, Watanabe Y, Takahashi T, Aoki Y, Uchiyama T, et al. Development of a Multi-Step Leukemogenesis Model of MLL-Rearranged Leukemia Using Humanized Mice. PLoS One (2012) 7(6):e37892. doi: 10.1371/journal.pone.0037892
66. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic Transformation by the MLL-AF6 Fusion Oncogene Requires the H3K79 Methyltransferase Dot1l. Blood (2013) 121(13):2533–41. doi: 10.1182/blood-2012-11-465120
67. Fu JF, Hsu CL, Shih LY. MLL/AF10(OM-LZ)-Immortalized Cells Expressed Cytokines and Induced Host Cell Proliferation in a Mouse Bone Marrow Transplantation Model. Int J Cancer (2010) 126(7):1621–9. doi: 10.1002/ijc.24867
68. Li Z, Luo RT, Mi S, Sun M, Chen P, Bao J, et al. Consistent Deregulation of Gene Expression Between Human and Murine MLL Rearrangement Leukemias. Cancer Res (2009) 69(3):1109–16. doi: 10.1158/0008-5472.CAN-08-3381
69. Ugale A, Norddahl GL, Wahlestedt M, Säwén P, Jaako P, Pronk CJ, et al. Hematopoietic Stem Cells are Intrinsically Protected Against MLL-ENL-Mediated Transformation. Cell Rep (2014) 9(4):1246–55. doi: 10.1016/j.celrep.2014.10.036
70. Sarrou E, Richmond L, Carmody RJ, Gibson B, Keeshan K. CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis. Int J Mol Sci (2020) 21(12):4266. doi: 10.3390/ijms21124266
71. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature (2011) 478(7370):529–33. doi: 10.1038/nature10509
72. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL Inhibitors Reverse Oncogenic Activity of MLL Fusion Proteins in Leukemia. Nat Chem Biol (2012) 8(3):277–84. doi: 10.1038/nchembio.773
73. Jevtic Z, Matafora V, Casagrande F, Santoro F, Minucci S, Garre' M, et al. SMARCA5 Interacts With NUP98-NSD1 Oncofusion Protein and Sustains Hematopoietic Cells Transformation. J Exp Clin Cancer Res (2022) 41(1):34. doi: 10.1186/s13046-022-02248-x
74. Placke T, Faber K, Nonami A, Putwain SL, Salih HR, Heidel FH, et al. Requirement for CDK6 in MLL-Rearranged Acute Myeloid Leukemia. Blood (2014) 124(1):13–23. doi: 10.1182/blood-2014-02-558114
75. Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, et al. NUP98 Fusion Proteins Interact With the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer Cell (2016) 30(6):863–78. doi: 10.1016/j.ccell.2016.10.019
76. Iacobucci I, Qu C, Varotto E, Janke LJ, Yang X, Seth A, et al. Modeling and Targeting of Erythroleukemia by Hematopoietic Genome Editing. Blood (2021) 137(12):1628–40. doi: 10.1182/blood.2020009103
77. Heikamp EB, Henrich JA, Perner F, Wong EM, Hatton C, Wen Y, et al. The Menin-MLL1 Interaction is a Molecular Dependency in NUP98-Rearranged AML. Blood (2021) 139(6):894–906. doi: 10.1182/blood.2021012806
78. Iacobucci I, Wen J, Meggendorfer M, Choi JK, Shi L, Pounds SB, et al. Genomic Subtyping and Therapeutic Targeting of Acute Erythroleukemia. Nat Genet (2019) 51(4):694–704. doi: 10.1038/s41588-019-0375-1
79. Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-Encoded CBFA2T3-GLIS2 Fusion Protein Defines an Aggressive Subtype of Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell (2012) 22(5):683–97. doi: 10.1016/j.ccr.2012.10.007
80. Dang J, Nance S, Ma J, Cheng J, Walsh MP, Vogel P, et al. AMKL Chimeric Transcription Factors are Potent Inducers of Leukemia. Leukemia (2017) 31(10):2228–34. doi: 10.1038/leu.2017.51
81. Wartman LD, Welch JS, Uy GL, Klco JM, Lamprecht T, Varghese N, et al. Expression and Function of PML-RARA in the Hematopoietic Progenitor Cells of Ctsg-PML-RARA Mice. PLoS One (2012) 7(10):e46529. doi: 10.1371/journal.pone.0046529
82. Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional Expression of Oncogenic K-Ras From its Endogenous Promoter Induces a Myeloproliferative Disease. J Clin Invest (2004) 113(4):528–38. doi: 10.1172/JCI20476
83. Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, et al. Oncogenic Kras-Induced Leukemogeneis: Hematopoietic Stem Cells as the Initial Target and Lineage-Specific Progenitors as the Potential Targets for Final Leukemic Transformation. Blood (2009) 113(6):1304–14. doi: 10.1182/blood-2008-01-134262
84. Chang YI, You X, Kong G, Ranheim EA, Wang J, Du J, et al. Loss of Dnmt3a and Endogenous Kras(G12D/+) Cooperate to Regulate Hematopoietic Stem and Progenitor Cell Functions in Leukemogenesis. Leukemia (2015) 29(9):1847–56. doi: 10.1038/leu.2015.85
85. Chan IT, Kutok JL, Williams IR, Cohen S, Moore S, Shigematsu H, et al. Oncogenic K-Ras Cooperates With PML-RAR Alpha to Induce an Acute Promyelocytic Leukemia-Like Disease. Blood (2006) 108(5):1708–15. doi: 10.1182/blood-2006-04-015040
86. Zhao S, Zhang Y, Sha K, Tang Q, Yang X, Yu C, et al. KRAS (G12D) Cooperates With AML1/ETO to Initiate a Mouse Model Mimicking Human Acute Myeloid Leukemia. Cell Physiol Biochem (2014) 33(1):78–87. doi: 10.1159/000356651
87. Wang YY, Zhao LJ, Wu CF, Liu P, Shi L, Liang Y, et al. C-KIT Mutation Cooperates With Full-Length AML1-ETO to Induce Acute Myeloid Leukemia in Mice. Proc Natl Acad Sci U S A (2011) 108(6):2450–5. doi: 10.1073/pnas.1019625108
88. Wang J, Kong G, Liu Y, Du J, Chang YI, Tey SR, et al. Nras(G12D/+) Promotes Leukemogenesis by Aberrantly Regulating Hematopoietic Stem Cell Functions. Blood (2013) 121(26):5203–7. doi: 10.1182/blood-2012-12-475863
89. Zhao L, Melenhorst JJ, Alemu L, Kirby M, Anderson S, Kench M, et al. KIT With D816 Mutations Cooperates With CBFB-MYH11 for Leukemogenesis in Mice. Blood (2012) 119(6):1511–21. doi: 10.1182/blood-2011-02-338210
90. Grundler R, Miething C, Thiede C, Peschel C, Duyster J. FLT3-ITD and Tyrosine Kinase Domain Mutants Induce 2 Distinct Phenotypes in a Murine Bone Marrow Transplantation Model. Blood (2005) 105(12):4792–9. doi: 10.1182/blood-2004-11-4430
91. Lee BH, Williams IR, Anastasiadou E, Boulton CL, Joseph SW, Amaral SM, et al. FLT3 Internal Tandem Duplication Mutations Induce Myeloproliferative or Lymphoid Disease in a Transgenic Mouse Model. Oncogene (2005) 24(53):7882–92. doi: 10.1038/sj.onc.1208933
92. Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, et al. FLT3 Mutations Confer Enhanced Proliferation and Survival Properties to Multipotent Progenitors in a Murine Model of Chronic Myelomonocytic Leukemia. Cancer Cell (2007) 12(4):367–80. doi: 10.1016/j.ccr.2007.08.031
93. Yun H, Narayan N, Vohra S, Giotopoulos G, Mupo A, Madrigal P, et al. Mutational Synergy During Leukemia Induction Remodels Chromatin Accessibility, Histone Modifications and Three-Dimensional DNA Topology to Alter Gene Expression. Nat Genet (2021) 53(10):1443–55. doi: 10.1038/s41588-021-00925-9
94. Yang L, Rodriguez B, Mayle A, Park HJ, Lin X, Luo M, et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell (2016) 29(6):922–34. doi: 10.1016/j.ccell.2016.05.003
95. Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO Fusion Gene and the FLT3 Length Mutation Collaborate in Inducing Acute Leukemia in Mice. J Clin Invest (2005) 115(8):2159–68. doi: 10.1172/JCI24225
96. Li L, Piloto O, Nguyen HB, Greenberg K, Takamiya K, Racke F, et al. Knock-In of an Internal Tandem Duplication Mutation Into Murine FLT3 Confers Myeloproliferative Disease in a Mouse Model. Blood (2008) 111(7):3849–58. doi: 10.1182/blood-2007-08-109942
97. Annesley CE, Rabik C, Duffield AS, Rau RE, Magoon D, Li L, et al. Knock-In of the. Oncotarget (2018) 9(82):35313–26. doi: 10.18632/oncotarget.26238
98. Rau R, Magoon D, Greenblatt S, Li L, Annesley C, Duffield AS, et al. NPMc+ Cooperates With Flt3/ITD Mutations to Cause Acute Leukemia Recapitulating Human Disease. Exp Hematol (2014) 42(2):101–13.e5. doi: 10.1016/j.exphem.2013.10.005
99. Lee HK, Kim HW, Lee IY, Lee J, Jung DS, Lee SY, et al. G-749, a Novel FLT3 Kinase Inhibitor, can Overcome Drug Resistance for the Treatment of Acute Myeloid Leukemia. Blood (2014) 123(14):2209–19. doi: 10.1182/blood-2013-04-493916
100. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of Lung Tumor Initiation and Progression Using Conditional Expression of Oncogenic K-Ras. Genes Dev (2001) 15(24):3243–8. doi: 10.1101/gad.943001
101. Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Dnmt3a Loss Predisposes Murine Hematopoietic Stem Cells to Malignant Transformation. Blood (2015) 125(4):629–38. doi: 10.1182/blood-2014-08-594648
102. Duployez N, Marceau-Renaut A, Boissel N, Petit A, Bucci M, Geffroy S, et al. Comprehensive Mutational Profiling of Core Binding Factor Acute Myeloid Leukemia. Blood (2016) 127(20):2451–9. doi: 10.1182/blood-2015-12-688705
103. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 Mutations in Childhood Acute Lymphoblastic Leukemia. Blood (2004) 103(9):3544–6. doi: 10.1182/blood-2003-07-2441
104. Yu Z, Du J, Hui H, Kan S, Huo T, Zhao K, et al. LT-171-861, a Novel FLT3 Inhibitor, Shows Excellent Preclinical Efficacy for the Treatment of FLT3 Mutant Acute Myeloid Leukemia. Theranostics (2021) 11(1):93–106. doi: 10.7150/thno.46593
105. Janke H, Pastore F, Schumacher D, Herold T, Hopfner KP, Schneider S, et al. Activating FLT3 Mutants Show Distinct Gain-of-Function Phenotypes In Vitro and a Characteristic Signaling Pathway Profile Associated With Prognosis in Acute Myeloid Leukemia. PloS One (2014) 9(3):e89560. doi: 10.1371/journal.pone.0089560
106. Bailey E, Li L, Duffield AS, Ma HS, Huso DL, Small D. FLT3/D835Y Mutation Knock-in Mice Display Less Aggressive Disease Compared With FLT3/internal Tandem Duplication (ITD) Mice. Proc Natl Acad Sci U S A (2013) 110(52):21113–8. doi: 10.1073/pnas.1310559110
107. Xu F, Wu LY, He Q, Wu D, Zhang Z, Song LX, et al. Exploration of the Role of Gene Mutations in Myelodysplastic Syndromes Through a Sequencing Design Involving a Small Number of Target Genes. Sci Rep (2017) 7:43113. doi: 10.1038/srep43113
108. Celik H, Mallaney C, Kothari A, Ostrander EL, Eultgen E, Martens A, et al. Enforced Differentiation of Dnmt3a-Null Bone Marrow Leads to Failure With C-Kit Mutations Driving Leukemic Transformation. Blood (2015) 125(4):619–28. doi: 10.1182/blood-2014-08-594564
109. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A Mutations Promote Anthracycline Resistance in Acute Myeloid Leukemia via Impaired Nucleosome Remodeling. Nat Med (2016) 22(12):1488–95. doi: 10.1038/nm.4210
110. Loberg MA, Bell RK, Goodwin LO, Eudy E, Miles LA, SanMiguel JM, et al. Sequentially Inducible Mouse Models Reveal That Npm1 Mutation Causes Malignant Transformation of Dnmt3a-Mutant Clonal Hematopoiesis. Leukemia (2019) 33(7):1635–49. doi: 10.1038/s41375-018-0368-6
111. Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, et al. DNMT3A and TET2 Compete and Cooperate to Repress Lineage-Specific Transcription Factors in Hematopoietic Stem Cells. Nat Genet (2016) 48(9):1014–23. doi: 10.1038/ng.3610
112. Zhang X, Wang X, Wang XQD, Su J, Putluri N, Zhou T, et al. Dnmt3a Loss and Idh2 Neomorphic Mutations Mutually Potentiate Malignant Hematopoiesis. Blood (2020) 135(11):845–56. doi: 10.1182/blood.2019003330
113. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell (2011) 20(1):11–24. doi: 10.1016/j.ccr.2011.06.001
114. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and is a Recurrent Event During Human Lymphomagenesis. Cancer Cell (2011) 20(1):25–38. doi: 10.1016/j.ccr.2011.06.003
115. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. Proc Natl Acad Sci U S A (2011) 108(35):14566–71. doi: 10.1073/pnas.1112317108
116. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
117. Kunimoto H, Fukuchi Y, Sakurai M, Sadahira K, Ikeda Y, Okamoto S, et al. Tet2 Disruption Leads to Enhanced Self-Renewal and Altered Differentiation of Fetal Liver Hematopoietic Stem Cells. Sci Rep (2012) 2:273. doi: 10.1038/srep00273
118. Kunimoto H, Meydan C, Nazir A, Whitfield J, Shank K, Rapaport F, et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell (2018) 33(1):44–59.e8. doi: 10.1016/j.ccell.2017.11.012
119. Palam LR, Mali RS, Ramdas B, Srivatsan SN, Visconte V, Tiu RV, et al. Loss of Epigenetic Regulator TET2 and Oncogenic KIT Regulate Myeloid Cell Transformation via PI3K Pathway. JCI Insight (2018) 3(4):e94679. doi: 10.1172/jci.insight.94679
120. Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 Results in Myelodysplasia and Severe Developmental Defects In Vivo. J Exp Med (2013) 210(12):2641–59. doi: 10.1084/jem.20131141
121. Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome-Like Disease in Mice. Blood (2014) 123(4):541–53. doi: 10.1182/blood-2013-05-500272
122. Nagase R, Inoue D, Pastore A, Fujino T, Hou HA, Yamasaki N, et al. Expression of Mutant Asxl1 Perturbs Hematopoiesis and Promotes Susceptibility to Leukemic Transformation. J Exp Med (2018) 215(6):1729–47. doi: 10.1084/jem.20171151
123. D'Altri T, Wilhelmson AS, Schuster MB, Wenzel A, Kalvisa A, Pundhir S, et al. The ASXL1-G643W Variant Accelerates the Development of CEBPA Mutant Acute Myeloid Leukemia. Haematologica (2021) 106(4):1000–7. doi: 10.3324/haematol.2019.235150
124. Viny AD, Ott CJ, Spitzer B, Rivas M, Meydan C, Papalexi E, et al. Dose-Dependent Role of the Cohesin Complex in Normal and Malignant Hematopoiesis. J Exp Med (2015) 212(11):1819–32. doi: 10.1084/jem.20151317
125. Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, et al. Cohesin Loss Alters Adult Hematopoietic Stem Cell Homeostasis, Leading to Myeloproliferative Neoplasms. J Exp Med (2015) 212(11):1833–50. doi: 10.1084/jem.20151323
126. Tanaka T, Nakajima-Takagi Y, Aoyama K, Tara S, Oshima M, Saraya A, et al. Internal Deletion of BCOR Reveals a Tumor Suppressor Function for BCOR in T Lymphocyte Malignancies. J Exp Med (2017) 214(10):2901–13. doi: 10.1084/jem.20170167
127. Kelly MJ, So J, Rogers AJ, Gregory G, Li J, Zethoven M, et al. Bcor Loss Perturbs Myeloid Differentiation and Promotes Leukaemogenesis. Nat Commun (2019) 10(1):1347. doi: 10.1038/s41467-019-09250-6
128. Sportoletti P, Sorcini D, Guzman AG, Reyes JM, Stella A, Marra A, et al. Bcor Deficiency Perturbs Erythro-Megakaryopoiesis and Cooperates With Dnmt3a Loss in Acute Erythroid Leukemia Onset in Mice. Leukemia (2021) 35(7):1949–63. doi: 10.1038/s41375-020-01075-3
129. Butler JS, Dent SY. The Role of Chromatin Modifiers in Normal and Malignant Hematopoiesis. Blood (2013) 121(16):3076–84. doi: 10.1182/blood-2012-10-451237
130. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med (2010) 363(25):2424–33. doi: 10.1056/NEJMoa1005143
131. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome Sequencing Identifies Somatic Mutations of DNA Methyltransferase Gene DNMT3A in Acute Monocytic Leukemia. Nat Genet (2011) 43(4):309–15. doi: 10.1038/ng.788
132. Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A Mutations in Patients With Myelodysplastic Syndromes. Leukemia (2011) 25(7):1153–8. doi: 10.1038/leu.2011.44
133. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is Essential for Hematopoietic Stem Cell Differentiation. Nat Genet (2011) 44(1):23–31. doi: 10.1182/blood.V118.21.386.386
134. Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell (2014) 15(3):350–64. doi: 10.1016/j.stem.2014.06.018
135. Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A Mutation Common in AML Exhibits Dominant-Negative Effects in Murine ES Cells. Blood (2013) 122(25):4086–9. doi: 10.1182/blood-2013-02-483487
136. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, et al. The R882H DNMT3A Mutation Associated With AML Dominantly Inhibits Wild-Type DNMT3A by Blocking its Ability to Form Active Tetramers. Cancer Cell (2014) 25(4):442–54. doi: 10.1016/j.ccr.2014.02.010
137. Tovy A, Rosas C, Gaikwad AS, Medrano G, Zhang L, Reyes JM, et al. Perturbed Hematopoiesis in Individuals With Germline DNMT3A Overgrowth Tatton-Brown-Rahman Syndrome. Haematologica (2021) 107(4):887–98. doi: 10.3324/haematol.2021.278990
138. Smith AM, LaValle TA, Shinawi M, Ramakrishnan SM, Abel HJ, Hill CA, et al. Functional and Epigenetic Phenotypes of Humans and Mice With DNMT3A Overgrowth Syndrome. Nat Commun (2021) 12(1):4549. doi: 10.1038/s41467-021-24800-7
139. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in Myeloid Cancers. N Engl J Med (2009) 360(22):2289–301. doi: 10.1056/NEJMoa0810069
140. Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 Gene Mutation Is a Frequent and Adverse Event in Chronic Myelomonocytic Leukemia. Haematologica (2009) 94(12):1676–81. doi: 10.3324/haematol.2009.011205
141. Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CA, et al. Acquired Mutations in ASXL1 in Acute Myeloid Leukemia: Prevalence and Prognostic Value. Haematologica (2012) 97(3):388–92. doi: 10.3324/haematol.2011.051532
142. Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, et al. ASXL1 Mutations in Younger Adult Patients With Acute Myeloid Leukemia: A Study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica (2015) 100(3):324–30. doi: 10.3324/haematol.2014.114157
143. Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D, et al. Spectrum and Prognostic Relevance of Driver Gene Mutations in Acute Myeloid Leukemia. Blood (2016) 128(5):686–98. doi: 10.1182/blood-2016-01-693879
144. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 Mutations Promote Myeloid Transformation Through Loss of PRC2-Mediated Gene Repression. Cancer Cell (2012) 22(2):180–93. doi: 10.1016/j.ccr.2012.06.032
145. Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, et al. Distinct Clinical and Biological Features of De Novo Acute Myeloid Leukemia With Additional Sex Comb-Like 1 (ASXL1) Mutations. Blood (2010) 116(20):4086–94. doi: 10.1182/blood-2010-05-283291
146. Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, et al. Genetic Alterations of the Cohesin Complex Genes in Myeloid Malignancies. Blood (2014) 124(11):1790–8. doi: 10.1182/blood-2014-04-567057
147. Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, et al. Mutations in the Cohesin Complex in Acute Myeloid Leukemia: Clinical and Prognostic Implications. Blood (2014) 123(6):914–20. doi: 10.1182/blood-2013-07-518746
148. Xu H, Balakrishnan K, Malaterre J, Beasley M, Yan Y, Essers J, et al. Rad21-Cohesin Haploinsufficiency Impedes DNA Repair and Enhances Gastrointestinal Radiosensitivity in Mice. PLoS One (2010) 5(8):e12112. doi: 10.1371/journal.pone.0012112
149. de Rooij JD, van den Heuvel-Eibrink MM, Hermkens MC, Verboon LJ, Arentsen-Peters ST, Fornerod M, et al. BCOR and BCORL1 Mutations in Pediatric Acute Myeloid Leukemia. Haematologica (2015) 100(5):e194–5. doi: 10.3324/haematol.2014.117796
150. Terada K, Yamaguchi H, Ueki T, Usuki K, Kobayashi Y, Tajika K, et al. Usefulness of BCOR Gene Mutation as a Prognostic Factor in Acute Myeloid Leukemia With Intermediate Cytogenetic Prognosis. Genes Chromosomes Cancer (2018) 57(8):401–8. doi: 10.1002/gcc.22542
151. Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-Exome Sequencing Identifies Somatic Mutations of BCOR in Acute Myeloid Leukemia With Normal Karyotype. Blood (2011) 118(23):6153–63. doi: 10.1182/blood-2011-07-365320
152. Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, et al. Whole-Exome Sequencing Reveals the Spectrum of Gene Mutations and the Clonal Evolution Patterns in Paediatric Acute Myeloid Leukaemia. Br J Haematol (2016) 175(3):476–89. doi: 10.1111/bjh.14247
153. Rau R, Brown P. Nucleophosmin (NPM1) Mutations in Adult and Childhood Acute Myeloid Leukaemia: Towards Definition of a New Leukaemia Entity. Hematol Oncol (2009) 27(4):171–81. doi: 10.1002/hon.904
154. Cheng K, Sportoletti P, Ito K, Clohessy JG, Teruya-Feldstein J, Kutok JL, et al. The Cytoplasmic NPM Mutant Induces Myeloproliferation in a Transgenic Mouse Model. Blood (2010) 115(16):3341–5. doi: 10.1182/blood-2009-03-208587
155. Chou SH, Ko BS, Chiou JS, Hsu YC, Tsai MH, Chiu YC, et al. A Knock-in Npm1 Mutation in Mice Results in Myeloproliferation and Implies a Perturbation in Hematopoietic Microenvironment. PLoS One (2012) 7(11):e49769. doi: 10.1371/journal.pone.0049769
156. Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant Nucleophosmin and Cooperating Pathways Drive Leukemia Initiation and Progression in Mice. Nat Genet (2011) 43(5):470–5. doi: 10.1038/ng.796
157. Mupo A, Celani L, Dovey O, Cooper JL, Grove C, Rad R, et al. A Powerful Molecular Synergy Between Mutant Nucleophosmin and Flt3-ITD Drives Acute Myeloid Leukemia in Mice. Leukemia (2013) 27(9):1917–20. doi: 10.1038/leu.2013.77
158. Kühn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov (2016) 6(10):1166–81. doi: 10.1158/2159-8290.CD-16-0237
159. Kastenhuber ER, Lowe SW. Putting P53 in Context. Cell (2017) 170(6):1062–78. doi: 10.1016/j.cell.2017.08.028
160. Barbosa K, Li S, Adams PD, Deshpande AJ. The Role of TP53 in Acute Myeloid Leukemia: Challenges and Opportunities. Genes Chromosomes Cancer (2019) 58(12):875–88. doi: 10.1002/gcc.22796
161. Stoddart A, Fernald AA, Wang J, Davis EM, Karrison T, Anastasi J, et al. Haploinsufficiency of Del(5q) Genes, Egr1 and Apc, Cooperate With Tp53 Loss to Induce Acute Myeloid Leukemia in Mice. Blood (2014) 123(7):1069–78. doi: 10.1182/blood-2013-07-517953
162. Yang M, Pan Z, Huang K, Büsche G, Liu H, Göhring G, et al. A Unique Role of P53 Haploinsufficiency or Loss in the Development of Acute Myeloid Leukemia With FLT3-ITD Mutation. Leukemia (2022) 36(3):675–86. doi: 10.1038/s41375-021-01452-6
163. Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, et al. P53 Loss Promotes Acute Myeloid Leukemia by Enabling Aberrant Self-Renewal. Genes Dev (2010) 24(13):1389–402. doi: 10.1101/gad.1940710
164. Zhang J, Kong G, Rajagopalan A, Lu L, Song J, Hussaini M, et al. P53-/- Synergizes With Enhanced NrasG12D Signaling to Transform Megakaryocyte-Erythroid Progenitors in Acute Myeloid Leukemia. Blood (2017) 129(3):358–70. doi: 10.1182/blood-2016-06-719237
165. Flanagan SP. 'Nude', A New Hairless Gene With Pleiotropic Effects in the Mouse. Genet Res (1966) 8(3):295–309. doi: 10.1017/s0016672300010168
166. Pantelouris EM. Absence of Thymus in a Mouse Mutant. Nature (1968) 217(5126):370–1. doi: 10.1038/217370a0
167. Nara N, Miyamoto T. Direct and Serial Transplantation of Human Acute Myeloid Leukaemia Into Nude Mice. Br J Cancer (1982) 45(5):778–82. doi: 10.1038/bjc.1982.120
168. Bosma GC, Custer RP, Bosma MJ. A Severe Combined Immunodeficiency Mutation in the Mouse. Nature (1983) 301(5900):527–30. doi: 10.1038/301527a0
169. Cesano A, Hoxie JA, Lange B, Nowell PC, Bishop J, Santoli D. The Severe Combined Immunodeficient (SCID) Mouse as a Model for Human Myeloid Leukemias. Oncogene (1992) 7(5):827–36.
170. Sawyers CL, Gishizky ML, Quan S, Golde DW, Witte ON. Propagation of Human Blastic Myeloid Leukemias in the SCID Mouse. Blood (1992) 79(8):2089–98. doi: 10.1182/blood.V79.8.2089.2089
171. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
172. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple Defects in Innate and Adaptive Immunologic Function in NOD/LtSz-Scid Mice. J Immunol (1995) 154(1):180–91.
173. Bonnet D, Dick JE. Human Acute Myeloid Leukemia is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
174. Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth Characteristics of Acute Myelogenous Leukemia Progenitors That Initiate Malignant Hematopoiesis in Nonobese Diabetic/Severe Combined Immunodeficient Mice. Blood (1999) 94(5):1761–72. doi: 10.1182/blood.V94.5.1761.417k23_1761_1772
175. Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, et al. AML Engraftment in the NOD/SCID Assay Reflects the Outcome of AML: Implications for Our Understanding of the Heterogeneity of AML. Blood (2006) 107(3):1166–73. doi: 10.1182/blood-2005-06-2325
176. Sanchez PV, Perry RL, Sarry JE, Perl AE, Murphy K, Swider CR, et al. A Robust Xenotransplantation Model for Acute Myeloid Leukemia. Leukemia (2009) 23(11):2109–17. doi: 10.1038/leu.2009.143
177. Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML Xenograft Efficiency is Significantly Improved in NOD/SCID-IL2RG Mice Constitutively Expressing Human SCF, GM-CSF and IL-3. Leukemia (2010) 24(10):1785–8. doi: 10.1038/leu.2010.158
178. Krevvata M, Shan X, Zhou C, Dos Santos C, Habineza Ndikuyeze G, Secreto A, et al. Cytokines Increase Engraftment of Human Acute Myeloid Leukemia Cells in Immunocompromised Mice But Not Engraftment of Human Myelodysplastic Syndrome Cells. Haematologica (2018) 103(6):959–71. doi: 10.3324/haematol.2017.183202
179. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and Function of Human Innate Immune Cells in a Humanized Mouse Model. Nat Biotechnol (2014) 32(4):364–72. doi: 10.1038/nbt.2858
180. Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, Philbrick W, et al. Transgenic Expression of Human Signal Regulatory Protein Alpha in Rag2-/-Gamma(C)-/- Mice Improves Engraftment of Human Hematopoietic Cells in Humanized Mice. Proc Natl Acad Sci U S A (2011) 108(32):13218–23. doi: 10.1073/pnas.1109769108
181. Ellegast JM, Rauch PJ, Kovtonyuk LV, Müller R, Wagner U, Saito Y, et al. Inv(16) and NPM1mut AMLs Engraft Human Cytokine Knock-in Mice. Blood (2016) 128(17):2130–4. doi: 10.1182/blood-2015-12-689356
182. Carroll AM, Hardy RR, Bosma MJ. Occurrence of Mature B (IgM+, B220+) and T (CD3+) Lymphocytes in Scid Mice. J Immunol (1989) 143(4):1087–93.
183. Riggs JE, Stowers RS, Mosier DE. Adoptive Transfer of Neonatal T Lymphocytes Rescues Immunoglobulin Production in Mice With Severe Combined Immune Deficiency. J Exp Med (1991) 173(1):265–8. doi: 10.1084/jem.173.1.265
184. Pflumio F, Izac B, Katz A, Shultz LD, Vainchenker W, Coulombel L. Phenotype and Function of Human Hematopoietic Cells Engrafting Immune-Deficient CB17-Severe Combined Immunodeficiency Mice and Nonobese Diabetic-Severe Combined Immunodeficiency Mice After Transplantation of Human Cord Blood Mononuclear Cells. Blood (1996) 88(10):3731–40. doi: 10.1182/blood.V88.10.3731.bloodjournal88103731
185. Wang JC, Doedens M, Dick JE. Primitive Human Hematopoietic Cells are Enriched in Cord Blood Compared With Adult Bone Marrow or Mobilized Peripheral Blood as Measured by the Quantitative In Vivo SCID-Repopulating Cell Assay. Blood (1997) 89(11):3919–24. doi: 10.1182/blood.V89.11.3919
186. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-Resistant Human AML Stem Cells Home to and Engraft Within the Bone-Marrow Endosteal Region. Nat Biotechnol (2007) 25(11):1315–21. doi: 10.1038/nbt1350
187. Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 Antibody-Mediated Clearance of Human Repopulating Cells Masks the Heterogeneity of Leukemia-Initiating Cells. Blood (2008) 112(3):568–75. doi: 10.1182/blood-2007-10-118331
188. Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-Initiating Cells From Some Acute Myeloid Leukemia Patients With Mutated Nucleophosmin Reside in the CD34(-) Fraction. Blood (2010) 115(10):1976–84. doi: 10.1182/blood-2009-02-206565
189. Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A Humanized Bone Marrow Ossicle Xenotransplantation Model Enables Improved Engraftment of Healthy and Leukemic Human Hematopoietic Cells. Nat Med (2016) 22(7):812–21. doi: 10.1038/nm.4103
190. Abarrategi A, Foster K, Hamilton A, Mian SA, Passaro D, Gribben J, et al. Versatile Humanized Niche Model Enables Study of Normal and Malignant Human Hematopoiesis. J Clin Invest (2017) 127(2):543–8. doi: 10.1172/JCI89364
191. Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment Determines Lineage Fate in a Human Model of MLL-AF9 Leukemia. Cancer Cell (2008) 13(6):483–95. doi: 10.1016/j.ccr.2008.04.020
192. Buechele C, Breese EH, Schneidawind D, Lin CH, Jeong J, Duque-Afonso J, et al. MLL Leukemia Induction by Genome Editing of Human CD34+ Hematopoietic Cells. Blood (2015) 126(14):1683–94. doi: 10.1182/blood-2015-05-646398
Keywords: acute myeliod leukemia, AML, transgenic mouse, genetically engineered mice (GEM), core binding factor acute myeliod leukemia, KMT2a (MLL) rearrangements, NUP98 fusion, patient-derived xenograft (PDX)
Citation: Kurtz KJ, Conneely SE, O’Keefe M, Wohlan K and Rau RE (2022) Murine Models of Acute Myeloid Leukemia. Front. Oncol. 12:854973. doi: 10.3389/fonc.2022.854973
Received: 14 January 2022; Accepted: 16 May 2022;
Published: 08 June 2022.
Edited by:
Paraskevi Panagopoulou, Aristotle University of Thessaloniki, GreeceReviewed by:
Stefan Heinrichs, Essen University Hospital, GermanyIlaria Iacobucci, St. Jude Children’s Research Hospital, United States
Copyright © 2022 Kurtz, Conneely, O’Keefe, Wohlan and Rau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel E. Rau, Rachel.rau@bcm.edu
†These authors have contributed equally to this work and share first authorship