 Jeremy M. Kelm1†
Jeremy M. Kelm1† Amirreza Samarbakhsh1†
Amirreza Samarbakhsh1† Athira Pillai1Pamela S. VanderVere-Carozza2
Athira Pillai1Pamela S. VanderVere-Carozza2 Hariprasad Aruri1
Hariprasad Aruri1 Deepti S. Pandey1
Deepti S. Pandey1 Katherine S. Pawelczak3
Katherine S. Pawelczak3 John J. Turchi2,3,4
John J. Turchi2,3,4 Navnath S. Gavande1,5*
Navnath S. Gavande1,5*- 1Department of Pharmaceutical Sciences, Eugene Applebaum College of Pharmacy and Health Sciences, Wayne State University, Detroit, MI, United States
- 2Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, United States
- 3NERx Biosciences, Indianapolis, IN, United States
- 4Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, United States
- 5Molecular Therapeutics Program, Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, MI, United States
The vast majority of cancer patients receive DNA-damaging drugs or ionizing radiation (IR) during their course of treatment, yet the efficacy of these therapies is tempered by DNA repair and DNA damage response (DDR) pathways. Aberrations in DNA repair and the DDR are observed in many cancer subtypes and can promote de novo carcinogenesis, genomic instability, and ensuing resistance to current cancer therapy. Additionally, stalled or collapsed DNA replication forks present a unique challenge to the double-strand DNA break (DSB) repair system. Of the various inducible DNA lesions, DSBs are the most lethal and thus desirable in the setting of cancer treatment. In mammalian cells, DSBs are typically repaired by the error prone non-homologous end joining pathway (NHEJ) or the high-fidelity homology directed repair (HDR) pathway. Targeting DSB repair pathways using small molecular inhibitors offers a promising mechanism to synergize DNA-damaging drugs and IR while selective inhibition of the NHEJ pathway can induce synthetic lethality in HDR-deficient cancer subtypes. Selective inhibitors of the NHEJ pathway and alternative DSB-repair pathways may also see future use in precision genome editing to direct repair of resulting DSBs created by the HDR pathway. In this review, we highlight the recent advances in the development of inhibitors of the non-phosphatidylinositol 3-kinase-related kinases (non-PIKKs) members of the NHEJ, HDR and minor backup SSA and alt-NHEJ DSB-repair pathways. The inhibitors described within this review target the non-PIKKs mediators of DSB repair including Ku70/80, Artemis, DNA Ligase IV, XRCC4, MRN complex, RPA, RAD51, RAD52, ERCC1-XPF, helicases, and DNA polymerase θ. While the DDR PIKKs remain intensely pursued as therapeutic targets, small molecule inhibition of non-PIKKs represents an emerging opportunity in drug discovery that offers considerable potential to impact cancer treatment.
Introduction
DSB Repair Pathways
DNA double-strand breaks (DSB)s are considered the most lethal of all DNA lesions. DSBs may be induced by various exogenous and endogenous factors, such as ionizing or ultraviolet radiation, genotoxic chemicals/chemotherapeutic agents, replication errors or collapsed replication forks, reactive oxygen species (ROS), free radicals, V(D)J recombination and abortive enzymatic activity (1–4). Unrepaired DSBs can lead to cell death, as persistent DSBs can trigger apoptosis (5–7). Moreover, misrepair or inaccurate repair of DSBs can lead to pathological genomic alterations resulting in senescence, loss of heterozygosity or chromosomal translocations which can ultimately result in oncogenesis (8). Interestingly, DSBs are routinely generated in the process of V(D)J recombination in naïve B- and T-lymphocytes to generate a diverse array of immunoglobulins and T-cell receptors, and the role of DSB repair in these processes has recently been reviewed (9). Aside from posing risk for cancer, DSBs are implicated in premature aging, and DSB repair capacity generally declines with age (10). In mammalian cells, the majority of DSBs are repaired via non-homologous end joining (NHEJ) or the homology directed repair (HDR)/homologous recombination (HR) pathways (Figure 1). In addition to NHEJ and HDR, less frequently involved or backup pathways including single-strand annealing (SSA) and alternative NHEJ (alt-NHEJ) also contribute to DSB repair (11–13). Alt-NHEJ is also called microhomology-mediated end joining (MMEJ) and more recently referred to as polymerase theta-mediated end joining (TMEJ) as recently reviewed by Ramsden et al. (14). Canonical or classical NHEJ is the predominant pathway in human cells and is active throughout the cell cycle, rapidly repairing up to ∼80% of all DSBs (15, 16). HDR is a much slower DSB repair process and is restricted exclusively to S and G2 phases of the cell cycle due to the requirement for a homologous DNA sequence or availability of sister chromatid as a template for the repair process (3).
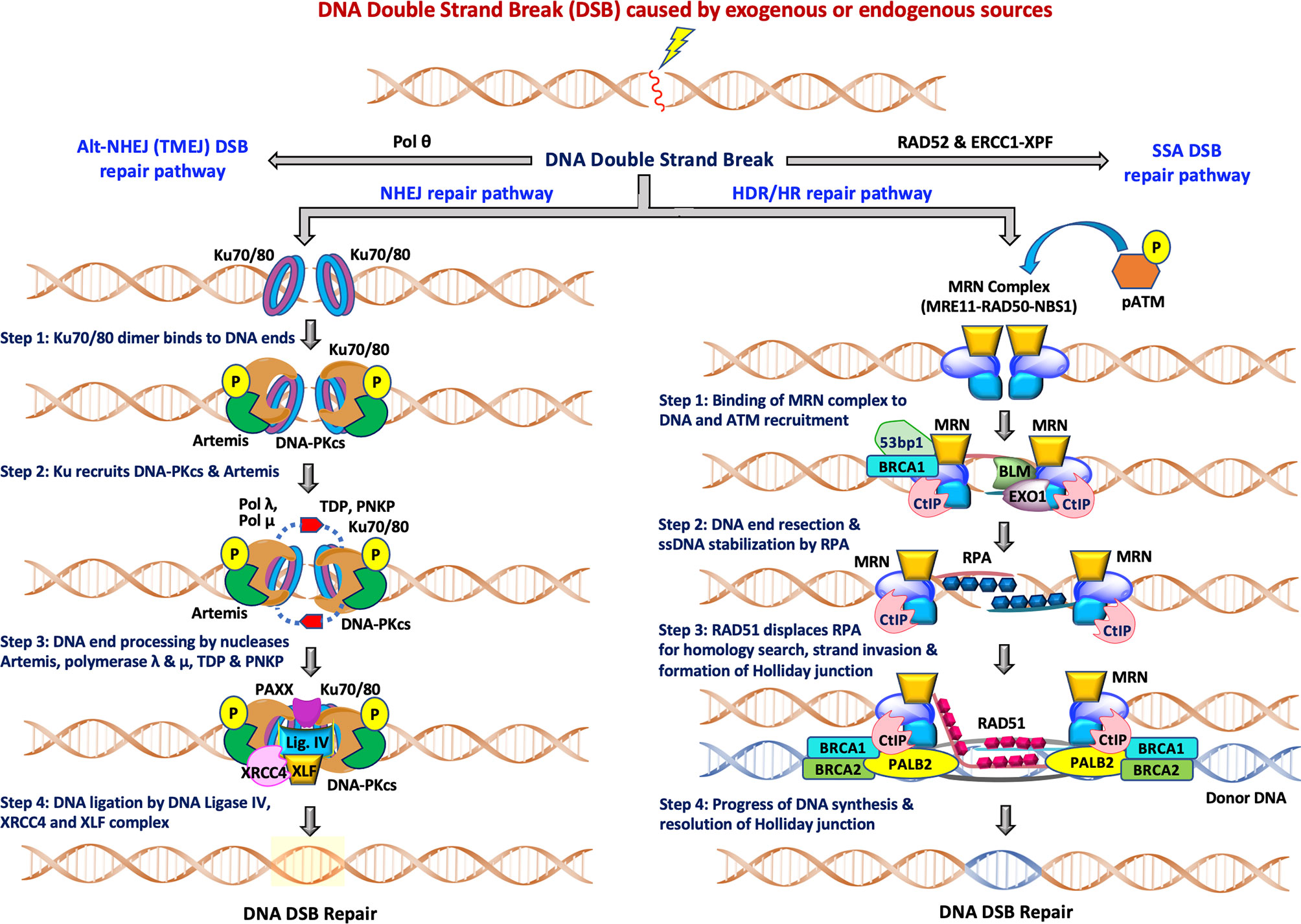
Figure 1 The two major pathways of DNA double-strand break repair: During NHEJ, the DNA double strand sites are initially recognized by heterodimeric Ku70/80. This is followed by recruitment of DNA-PKcs and Artemis, DNA end processing by Artemis, polymerase λ and μ, TDP and PNKP and finally ligation of DSB breaks by Ligase IV/XRCC4/XLF complex for completion of the repair pathway. The other accessory proteins like APLF, PAXX and XLF also participate in the repair functions. During HDR/HR, DSBs are recognized and resected by the MRN complex to generate a 3’ overhang. BRCA2/RAD51, along with other RAD51 paralogs, binds to the RPA coated ssDNA tails after which RAD51 replaces RPA in a BRCA1-and BRCA2-dependent process, forming a presynaptic filament. Upon strand invasion, D-loop formation and DNA repair synthesis can be resolved through Holliday junction, after which distinct independent pathways can operate to complete the HDR repair pathway. NHEJ is available throughout interphase while HDR is restricted to S/G2 phases of the cell cycle.
Figure 1 (left) depicts the various steps in the NHEJ repair pathway, and these can be summarized into four specific steps which include: (i) DNA termini recognition by Ku70/80; (ii) bridging of the two DNA ends also known as formation of the synaptic complex; (iii) DNA end processing, and finally (iv) DNA ligation (16–20). Following the induction of DSB by exogenous sources like ionizing radiation (IR) or chemotherapeutics, the NHEJ pathway is initiated by the binding of the heterodimeric Ku70/80 to the end of DNA break which recruits DNA-dependent protein kinase catalytic subunits (DNA-PKcs) to form the DNA-PK holoenzyme (DNA-PK). The DNA-PKcs serine/threonine protein kinase activity is activated once bound to a DNA terminus in the presence of Ku70/80. The formation of the DNA-PK complex stabilizes the two DNA ends at the site of the break by forming a synaptic complex that holds the two DNA termini together (21, 22). DNA-PK catalyzes both autophosphorylation and phosphorylation of other target proteins including Ku70/80, Artemis, polynucleotide kinase 3′-phosphatase (PNKP) and XRCC4. When required, DNA end processing relies on the kinase activity of DNA-PKcs, endonuclease cleavage activity of Artemis, nucleotide addition and modification by DNA polymerases (Pol X family polymerases such as pol λ and pol μ), tyrosyl-DNA phosphodiesterase (TDP), and PNKP. Finally, the DNA Ligase IV/XRCC4/XLF complex is recruited to DNA termini and catalyzes ligation of the DNA DSB.
The HDR pathway is depicted in stepwise fashion in Figure 1 (right) and can be summarized as: (i) binding of the MRN complex to each of the damaged dsDNA ends; (ii) end resection by the MRN complex, CtIP, EXO1, BLM and stabilization of the ssDNA overhangs by RPA binding; (iii) RAD51 displacement of RPA and formation of the Holliday junction with a homologous sequence; and (iv) resolution of the Holliday junction. The MRN complex (MRE11-RAD50-NBS1) is crucial for recognition of homologous sequences, performing end resections to generate ssDNA tails and nucleofilament formation by Replication Protein A (RPA) which is eventually replaced by RAD51. BRCA1 and BRCA2 also facilitate RAD51 filament nucleation. After recruitment of RAD51, a homology search can now be performed that when successful allows invasion of the non-resected strand into the homologous template and resulting D-loop formation of the displaced template strand. Capture of the D-loop by the broken dsDNA produces a Holliday junction that is later resolved by endonuclease activity, completing HDR. The distinct independent pathways that can operate to complete the HDR repair pathway are reviewed elsewhere (23, 24). Decades of investigation have established the importance of NHEJ, HDR, TMEJ and SSA pathways, the roles of the various factors/proteins involved in these pathways and how these factors coordinate and regulate distinct steps of these pathways at the molecular level. More detailed descriptions of these pathways can be found in recent reviews (11, 14, 16, 25, 26).
It is worth mentioning that a prerequisite to repair of DSBs is that the lesion is accessible which typically requires histone modifications and reorganization of chromatin (27). Acetylation of histones promotes DNA unraveling by electronegative repulsion which enables the DNA repair machinery access to the DSB. Targeting histone deacetylases (HDACs) with small molecule drugs or through promoting their degradation by inhibition of the deubiquitinase conferring HDAC stability are other strategies to enhance radiosensitivity (28, 29). To complicate matters further, the chromatin state must be returned to the pre-existing state after DSB repair.
Telomeres are repetitive DNA elements that protect chromosomal termini and prevent their false recognition as DSBs which could activate a deleterious DDR such as NHEJ-mediated chromosomal fusion or cyclization (30, 31). DDR activation and maintenance at telomeres depends on the biogenesis and functions of the site-specific small non-coding RNAs, also known as DNA damage response RNAs (DDRNAs) (32). Telomeres shorten with cell division during replicative senescence (due to the end-replication problem). Excessive telomeric erosion has been shown to contribute to a persistent DDR and have been implicated in the ageing process and disease development alongside with a host of lifestyle factors, stresses, and environmental exposures (33). The maintenance of telomere homeostasis is critical for chromosome stability in proliferating cancer cells which usually have higher telomerase activity compared to normal cells (34). In general, cancer cells maintain telomeres at shorter lengths compared to normal cells. Besides preventing chromosome shortening, telomerase also intervenes to thwart the DSB response through protein-protein interactions with specialized telomere-binding proteins. There are several proteins involved in the DSB response which are also localized to telomeres and participate in telomere homeostasis (34). For example, the Ku protein has been demonstrated to be localized to telomeres and serves to protect the telomere against fusions. Particularly, depletion of the Ku heterodimer leads to severe telomere erosion and loss of cell viability (35, 36). Overall, telomerase has been an attractive target for the development of effective cancer therapeutics as it has shown overexpression in the majority of human cancers. The anti-telomerase therapeutics can provide selective destruction of cancer cells while noncancerous cells are predominantly spared owing to telomerase silencing in most normal somatic cells (37).
RecQ and MCM (Minichromosome Maintenance) helicases, a family of DNA unwinding enzymes, play important roles in genomic stability through diverse roles in DNA recombination, replication and repair (38, 39). RecQ proteins can function both at early and late stages during repair of DSB. In addition, RecQ helicase proteins BLM (Bloom syndrome) and WRN (Werner syndrome) are also involved in telomere homeostasis as well as the processing and re-initiation of stalled replication forks (40, 41). The CMG helicase complex composed of three replication factors (Cdc45/Mcm2-7/GINS) is required to unwind dsDNA to generate the ssDNA template during DNA replication (42). A stalled replication forks by MCM helicases can lead to a DSB as well as chromosomal rearrangements, which can eventually recruit RecQ proteins for repair due to their functional connections (39, 43). Targeting these helicases has immense importance in developing new therapeutics against various cancers.
The recent advances in the field of the mitochondrial DNA damage response (mtDDR) warrant consideration of the nonnuclear genome in the development of inhibitors of non-PIKKs in DSB repair, as several non-PIKKs are now implicated in the mtDDR (44–46). In humans, mitochondria contain a polyploid genome comprised of a heterogenous mixture of ~16.5 kbp circular DNA chromosomes. Consistent with endosymbiotic ancestry from proteobacteria, the mitochondrial DNA (mtDNA) replication, transcription, and DDR machinery includes gene products evolutionary derived from eukarya, bacteria, and T7-like bacteriophages (47). mtDNA replication, transcription, and damage repair occur independently of their nuclear counterparts. The maternally inherited mitochondrial genome includes 37 genes encoding all required mitochondrial tRNAs and rRNAs and 13 core proteins of complexes I, III, IV, and V of the electron transport chain (ETC).
mtDNA is subjected to damage by the same sources as nuclear DNA, although the proximity of mtDNA to the ETC complexes heightens the risk for ROS-induced DNA damage. Aberrations of mtDNA including mutations or deletions are associated with the development of diseases including Kearns-Sayre syndrome, Pearson syndrome, cancer, aging, Alzheimer’s disease, and diabetes among others (44–46). Unlike the nucleus, there appears to be no role for classical NHEJ in the mitochondria where DSBs are predominantly repaired by the alt-NHEJ pathway. Mitochondrial alt-NHEJ proceeds independent of Ku70/80 and is dependent on Ligase III and MRE11 among others (48). DNA polymerase θ also appears to play a role in mitochondrial alt-NHEJ but in an error-prone manner unlike in the nucleus where fidelity is high (49, 50). Besides alt-NHEJ there is now mounting evidence to suggest that HDR may also repair DSBs in mtDNA, although possibly with nuances and a requirement for additional proteins. Four-way junctions and HDR mediators RAD51, RAD51C, XRCC3, and MRE11 have been detected in the mitochondria, and functional assays have demonstrated DSB repair in mitochondria consistent with HDR (51–54). Unrepaired mtDNA may be compensated for by undamaged mitochondrial chromosomes or trigger mitochondrial translesion synthesis, fusion, fission, or mitophagy to manage or purge the damaged mtDNA (44). There is evidence to suggest that mtDNA damage is sufficient to induce apoptosis or enhanced immunogenicity independent of nuclear DNA damage (55–58). Given the roles of non-PIKKs in the mtDDR, inhibitors should be assessed for their effects on mitochondrial HDR and alt-NHEJ. In similar fashion, the proapoptotic and immunogenic effects seen with targeted mtDNA damage highlights a potential for mitochondrial-selective anticancer drugs.
The innate immune response to cancer is favored by heightened DNA damage such as by unrepaired DSBs, although the adaptive immune response effectors B cells and T cells require intact DSB repair to repair the DSBs they routinely generate in V(D)J recombination. Accordingly, the strategy for targeting DSB repair in cancer will need to balance these opposing effects on the immune system. A precondition to repairing DSBs is access to the lesion by the DDR machinery through chromatin modification, and inhibition of HDACs may provide a way to block DSB repair upstream of DDR effector scaffolding at the damaged site.
An emerging area of research within the field of DNA damage and repair is the role of non-coding RNAs (ncRNAs) in the DDR to DSBs which has recently been reviewed (59–61). ncRNAs are classified as being either long ncRNAs (lncRNAs) or short ncRNAs (sncRNAs) depending on whether length exceeds 200 nucleotides. A subclass of lncRNAs is damage-induced lncRNAs (dilncRNAs), which are transcribed bidirectionally from DSBs after the arrival of the MRN complex and promote HDR by localizing RAD51, BRCA1, and BRCA2 to the lesion (59, 60). Micro RNAs (miRNAs), a subclass of sncRNAs, are typically derived from RNases such as DICER or Drosha and in the DDR regulate gene expression post-transcriptionally to select the DSB repair pathway employed, induce cell cycle arrest, and promote apoptosis where indicated (61). More broadly, ncRNAs are thought to serve as an alert to the presence of DSBs, to recruit DDR effectors to the lesion, and to temporarily bridge the broken ends in proximity, among other functions (59, 60). However, an improved understanding of ncRNAs in DSB repairs may produce additional opportunities for RNA-targeted therapeutic intervention. Overall, the multifunctional role of DNA repair and DDR pathways increases the complexity and difficulty of targeting DNA repair pathways for a positive clinical outcome.
Biological Impacts of DSB Repair in Cancer
DNA repair pathways play a central role in protecting cells against genomic instability and mutations. Moreover, DNA repair pathways play a multifaceted role in cancer onset, progression, metastasis, and ultimately on clinical outcome of cancer therapeutic strategies. Aberrations of DNA repair proteins or genes can predispose the cells to carcinogenesis and this vulnerability can be therapeutically exploited to preferentially kill tumor cells. The relative functionalities of the DNA repair and DNA damage response (DDR) pathways, whether defective, deficient, or hyperactive, as well as the ability of cancer therapeutics to inhibit or activate DNA repair, all can influence a patient’s response to therapy (5, 62, 63). The upregulated DNA repair and DDR activity can promote disease progression and make cancer cells resistant to the treatment or cause post-treatment relapse.
Many well-known anticancer chemotherapeutics and IR impart their clinical efficacy by inducing DNA damage. DNA damaging agents such as etoposide, bleomycin, doxorubicin and IR (radiotherapy) exert their therapeutic efficacy by inducing DNA DSBs. Approximately 50% of all cancer patients worldwide with common epithelial malignancies (including lung, prostate, breast, colon, head and neck, and esophageal cancers) are subjected to radiation therapy as a component of their treatment regimen. Radiotherapy is very cost effective and in combination with other medical treatments has contributed to improved long-term survival in subsets of cancer patients. Despite advanced technical improvements and the fact that radiotherapy is one of the most effective forms of cancer treatment, many patients still suffer from detrimental locally recurrent disease or long-term chronic side effects after radiotherapy due to being treated with higher doses of radiation (64–66). Most importantly, radiotherapy and DNA damaging chemotherapeutics often lead to poor clinical response due to the development of intrinsic or extrinsic resistance. There are multiple factors involved in IR and drug resistance, among them increased capacity of DNA DSB repair is one of the major primary concerns, and in many cases, resistance to therapy is an adaptive response linked to hyperactive DSB repair mechanisms (64, 67). The overexpression or loss of function due to polymorphisms, mutations of core and processing NHEJ proteins such as Ku70/80, DNA-PKcs, Ligase IV/XRCC4, and HDR proteins such as MRN, BRCA1/2 and RAD51 have been implicated in reduced therapeutic efficacy of IR and DNA damaging chemotherapeutics. Research within cancer genomics, proteomics, and metabolomics has led to a deeper understanding of the molecular mechanisms driving the development of resistance. In response to DNA damage, the affected cells recruit functional proteins to initiate the DSB repair pathway that enhances the DNA lesion repair which ultimately leads to drug resistance.
The targeted inhibition of repair pathways is a novel and effective strategy to induce persistent DSBs and increase apoptosis of cancer cells. This strategy is particularly promising in the setting of combination therapy with DSB-inducing treatments such as radiotherapy or radiomimetic drugs or in combination with other DNA damaging drugs. However, where unrepaired DSBs fail to directly induce cell death, induction of the innate immune response may ensue. The interplay of the DDR and the innate immune response has recently been reviewed (9). Cytosolic DNA arising from damaged nuclear or mitochondrial DNA is recognized as a pathogen- or damage-associated molecular pattern (PAMP/DAMP) and ultimately induces stimulator of interferon genes (STING)-dependent signaling. The resulting production of interferons enhances the cellular antitumor response by the immune system. Intriguingly, several of the non-PIKKs targeted by ligands reviewed here such as MRN complex, DNA ligase IV, and XRCC4 appear to have dual roles in stimulating the innate immune response aside from their classical roles in DSB repair.
As cancer cells frequently harbor defect in genes of a DNA repair pathway, they may be increasingly reliant on the remaining available pathways to repair DNA damage occurring endogenously or in response to treatment. Defects in DNA repair in cancer cells thus presents a vulnerability to exploit synthetical lethal interactions where noncancerous cells would remain resilient. In cancer treatment, this has been typified using PARP inhibitors in cancers that are HDR-deficient. The synthetic lethality approaches have provided novel mechanisms to specifically target cancer cells while noncancerous cells can tolerate or repair the damage which is anticipated to reduce toxicity associated with treatment. The availability of DNA repair inhibitors targeting a variety of DSB repair and DDR mediators will allow the strategy of synthetic lethal interactions to be more broadly applied clinically and with greater efficacy.
In this review, we focus specifically on recent advances in the development of non-PIKKs (PI3 kinase-like kinases) DSB repair targeted inhibitors that can be exploited for effective chemo- or radio-sensitization and to enhance the efficiency of precise genome editing as well. PIKKs such as ATM, ATR, and DNA-PK which are involved in DNA repair and DDR, have received considerable attention recently as pharmacological targets, and several inhibitors have risen to clinical trials. The progress pertaining to the development of these inhibitors is reviewed elsewhere (62, 68–70).
Recent Advances in the Development of Non-Pikks DSB Repair Inhibitors
The rare mutations and altered expression levels of key NHEJ and HDR proteins, mainly Ku70/80, DNA-PK, Artemis, Ligase IV, XRCC4, XLF, MRE11, RAD51, RPA and RAD52 can lead to predisposition to cancer, whereas increased capacity of DNA repair and DDR can be clinically exploited by targeting repair pathways to overcome resistance and enhance chemo- or radiosensitivity in cancer patients (5, 71–75). DNA repair inhibitors can be used to specifically target proteins involved in key steps of NHEJ, HDR, MMEJ and SSA as well as core or processing proteins involved in DDR signaling pathways. Developing drugs aimed at modulating DNA DSB repair activity are most likely to have a profound impact on the efficacy of radio- and chemotherapy. Therefore, targeting these key proteins in the DNA DSB repair pathways (Figure 2) has recently become a popular approach for potential cancer treatments.
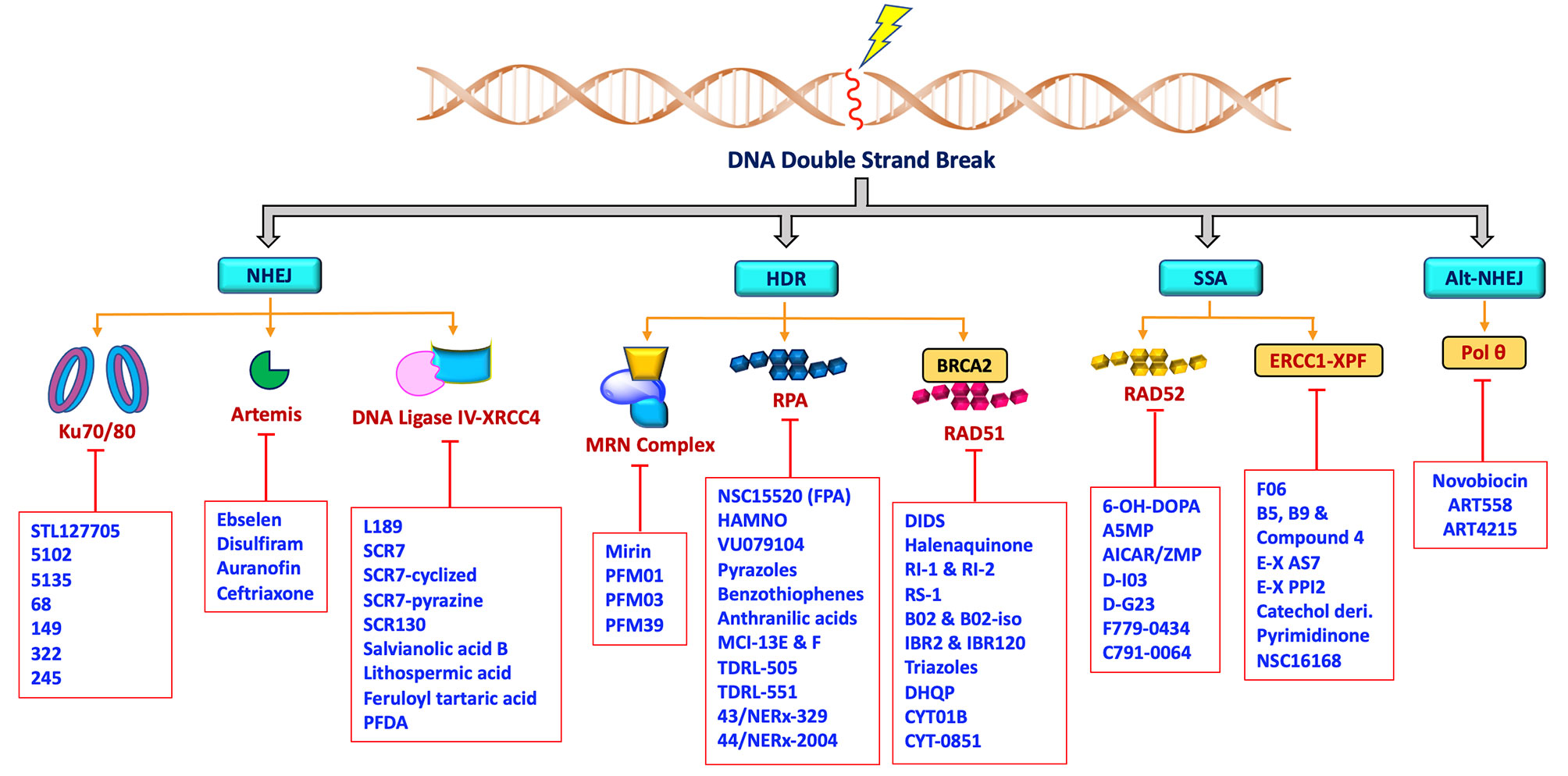
Figure 2 Schematic representation showing non-PIKKs DSB repair inhibitors that target key/core and accessory proteins involved in DSB repair pathways.
Inhibitors Targeting NHEJ Pathway
Ku 70/80 Inhibitors
There has been considerable progress made in the development of DNA-PK inhibitors and several of them are in various stages of clinical trials (NCT02644278, NCT04172532, NCT03907969), but less attention has been placed on the upstream and most essential Ku70/80 heterodimer that recruits DNA-PKcs (68, 69). In the absence of heterodimeric Ku subunits, DNA-PKcs binding affinity to DNA DSB is significantly weak, resulting in halting of the repair process (76). DNA-PK has a unique mechanism of activation that requires binding to DNA termini, and this strong binding interaction is solely dependent on a protein-protein interaction with the Ku70/80 heterodimeric complex for the subsequent NHEJ activation (77, 78). Being the primary sensor and core regulator of this pathway, Ku is absolutely required for DNA DSBs repair by NHEJ (79–81). Inhibition of Ku subunits could therefore produce reduced DNA-PK and NHEJ activity. Therefore, Ku has a high potential for therapeutic outcomes in oncology.
Recent studies have demonstrated a significant increase in expression levels of Ku70 and Ku80 after chemo- and radiotherapy which correlates with poor prognosis in patients with rectal and cervical cancers (82–84). Further studies have also demonstrated that overexpression of Ku70/Ku80 is directly correlated with chemotherapy and radiotherapy resistance in various cancers (82). Previously, shRNA depletion of Ku70 or Ku80 produced cytotoxicity and radiosensitization in pancreatic cancer cells (85). In addition, Ku70 or Ku80 null cells exhibited enhanced chemo sensitization to DNA damaging agents including bleomycin, doxorubicin, and etoposide (86). Ku is also involved in several other DNA metabolism processes and in telomere maintenance (35, 36, 87). Despite the crucial role of Ku subunits early in the NHEJ pathway, there are currently a limited number of Ku70/80 inhibitors developed so far. In 2016, Weterings et al. identified STL127705 (compound L) (Figure 3) by computational screening of a commercial library that disrupts Ku-DNA binding activity in micro-molar range and has potential to sensitize cancer cells to IR (88). However, the ability of STL127705 to block NHEJ catalyzed DNA DSB repair is not documented to date.
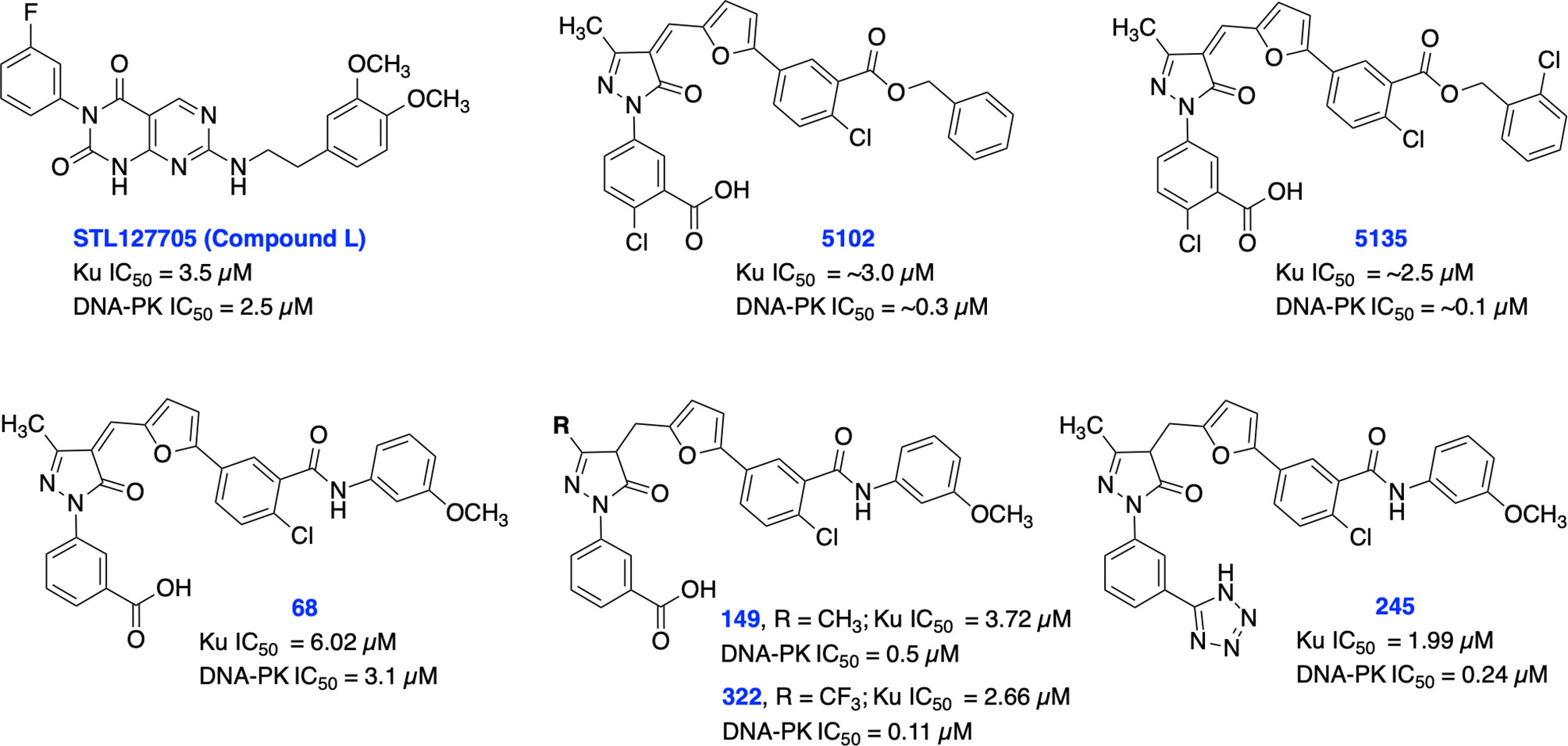
Figure 3 Small molecule inhibitors of Ku70/80 and their respective IC50 values for disruption of DNA-binding by Ku70/80 and DNA-PK activity.
Initially, our group identified arylalkyl esters of arylpyrazolone carboxylic acid derivatives, 5102 and 5135, through screening of a commercial library and both inhibitors displayed high potency in both Ku-DNA EMSA and DNA-PK kinase assays (Figure 3) (89). Retaining the core scaffold employed in 5102 and 5135, we recently further expanded our structure-guided synthetic chemistry efforts with the aim of improving Ku inhibitory potency, selectivity, and cellular activity while simultaneously improving solubility among other physicochemical properties (90). The structure activity relationship (SAR) from this study showed that an amide moiety increased both the solubility and the inhibition of Ku-DNA interaction by 4-fold over the ester group. Compounds 68, 149, 322 and 245 exhibited a high potency and specificity towards Ku and DNA-PK. Moreover, these compounds also showed improved chemical properties including solubility and stability. These Ku-DNA binding inhibitors (Ku-DBi’s) directly interact with Ku and inhibit in vitro NHEJ, cellular NHEJ, and potentiate the cellular activity of radiomimetic agents and IR. Further analysis demonstrated that Ku-null cells are insensitive to Ku-DBi’s however, Ku-DBi’s potentiate cellular sensitivity to DSB-inducing agents in cancer cells. Molecular docking studies indicated that compounds 149 and 245 possess high affinity towards the Ku binding site (Figure 4). Inhibiting Ku interactions with DNA ends can efficiently block NHEJ catalyzed repair which is anticipated to increase efficiency of HDR-mediated recombination events. Therefore, we performed CRISPR-Cas9-mediated genome editing in the presence of Ku-DBi 245 where we observed a 6-fold increase in HDR mediated insertion at a DSB at the target site compared to the controls (90). These data suggests that Ku-DBi’s could be effective to reduce off-target, potentially mutagenic events that have hampered CRISPR mediated therapeutic applications.
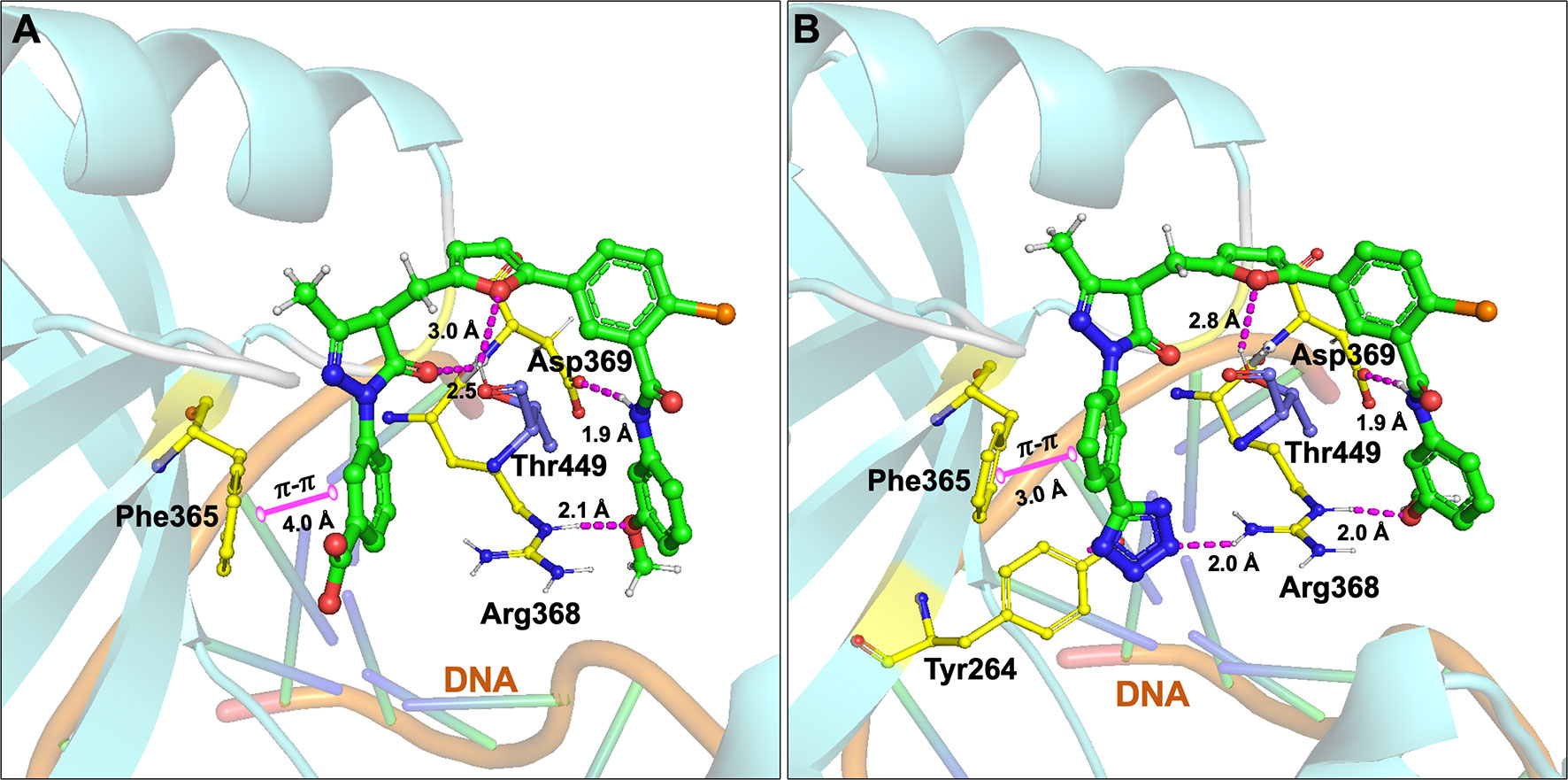
Figure 4 Molecular interactions of (A) compound 149 and (B) 245 (all in green carbon) with Ku70/80 heterodimer (key amino acids are shown in yellow carbon (Ku70), blue carbon (Ku80) and cartoon is shown in cyan color). Interaction with amino acid side chains is indicated with the dashed magenta lines and π – π stacking interactions are shown in solid magenta dumbbell. The DNA helical structure is depicted in greenish blue sticks and light orange cartoon. Interaction distances indicated in Å.
Further development of Ku70/80 inhibitors has a considerable potential to impact cancer therapy as well as precise genome editing.
Artemis Inhibitors
Artemis is a structure specific endonuclease with critical roles in DSB repair by NHEJ, in the development of B- and T- lymphocytes via cleaving a hairpin intermediate during V(D)J recombination and has also been implicated to play a role in the maintenance of genomic stability (91–94). Artemis was first reported after investigators implicated its deficiency in severe combined immunodeficiency (SCID) as causative for observed phenotypes in this disorder including impaired V(D)J recombination and enhanced IR sensitivity, supporting the mechanistic role of Artemis in these pathways. Mouse embryonic fibroblasts (MEFs) derived from Artemis defective mice have increased chromosomal abnormalities, suggesting a role for Artemis in genome stability maintenance (95). In NHEJ, DNA-PKcs undergo autophosphorylation and activate the endonuclease activity of Artemis at DNA ends (93). Artemis’ C-terminal region influences V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs, suggesting that the Artemis-binding site on Ligase IV also has physiological relevance to potentially disrupting NHEJ complex formation (96, 97). Artemis is the main nuclease known to remove DNA single-strand overhangs and 3′-phosphoglycolate groups from DNA termini generated by IR with its endonuclease activity (98). It is well documented that IR-induced DSBs require Artemis for repair (94, 95, 99, 100).
Recently, Yosaatmadja et al. generated a model for Artemis DNA binding based on their zinc bound Artemis crystal structure and another recently reported structure of the Artemis catalytic domain (101, 102). This unique zinc-finger-like motif has not been reported in other metallo-β-lactamase (MBL) enzymes (the super family to which Artemis belongs) and presents a possible novel targeting location. Further, they have screened thiol reactive compounds using this unique zinc-finger like motif of Artemis and identified that ebselen and disulfiram are able to inhibit Artemis endonuclease activity in the low micro-molar range (IC50s = 8.5 uM and 10.8 uM, respectively), while auranofin and ceftriaxone are less potent (IC50s = 46 uM and 65 uM, respectively) (Figure 5).
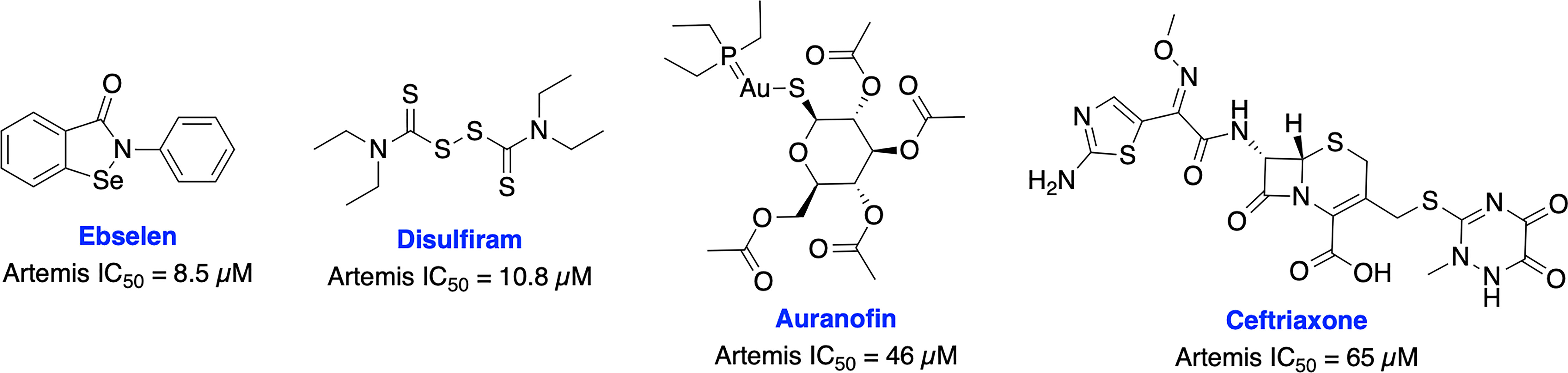
Figure 5 Small molecule inhibitors targeting Artemis and their respective IC50 values for disruption of endonuclease activity.
The recent crystal structures of Artemis and these inhibitors provide useful information for structure-based design of inhibitors to generate more selective and potent Artemis inhibitors, either binding at the active site or the unique zinc finger motif of Artemis. The key roles of Artemis within DNA repair make it an attractive target for a variety of therapeutic avenues. Artemis inhibitors have the potential to be used as radiosensitizers in various tumor types, demonstrated biologically by the sensitivity to IR seen in SCID patients. Because of its clear role in DNA repair and genome stability, Artemis targeted inhibitors also have the potential to synergize well with other DDR targeted inhibitors. There is the potential for impacts on immune cell maturation with long term clinical Artemis inhibition which could result in compromised immune function, thus monitoring immune system function will be critical as Artemis targeted agents progress to the clinic.
DNA Ligase IV Inhibitors
After DNA end processing, the final step in NHEJ pathway is ligation which is a crucial step in the repair of DNA DSBs and is an attractive target for inhibition of the DSB repair pathway. This is demonstrated by various Ligase IV deficient mutants and knockout studies, that have been shown to have significantly reduced NHEJ activity (103–105). Upon activation of kinase activity by DNA-PKcs, Ku heterodimer translocates internally to make DSB ends accessible to a specific ligation complex, which is composed of DNA ligase IV and its partnering proteins, XRCC4 and XLF (106).
In 2008, Chen et al. identified a competitive and non-specific ligase inhibitor, L189 through a computational drug design strategy which showed equipotent inhibitory activity against Ligase I, III, and IV (Figure 6) (107). Raghavan and co-workers in 2012 developed SCR7, a derivative of L189, which was initially suggested to be more selective for Ligase IV (108). Further extensive structural analysis by Greco et al. revealed that parental SCR7 is nonspecific, only exists in the more stable cyclized SCR7 pyrazine form and failed to inhibit DNA ligase IV-dependent V(D)J recombination in a cell-based ligation assay (109). On the contrary, Raghavan and co-workers in 2018 showed both intramolecular cyclized SCR7 (SCR7-cyclized) and further oxidized product (SCR7-pyrazine) could inhibit Ligase IV-mediated end joining and V(D)J recombination (110). Further studies showed that the SCR7-cyclized is Ligase IV specific and SCR7-pyrazine induced nonspecific cytotoxicity at higher concentrations in Ligase IV-null cells. Recently, Raghavan and co-workers developed a new ligase IV-specific inhibitor, SCR130, which exhibited 20-fold improved cytotoxicity compared to SCR7 and potentiated radiosensitivity in cancer cells (111). Furthermore, SCR7 produced enhanced HDR-mediated repair for CRISPR mediated genome editing by inhibiting NHEJ at lower concentrations (1 μM) (103), but cellular toxicity was observed with concentrations above 1 μM (104), suggesting that cell-dependent toxicity or off-target effects associated with the inhibitor (112). The higher IC50s and inconsistent results could be explained by instability of parental SCR7 and its analogs. Given the conflicting results and unclear therapeutic and toxicological mechanisms of action, more research is required on this area. There is also a great need within medicinal chemistry to identify novel scaffolds apart from SCR7 to target Ligase IV as this will broaden the chemical space available to develop Ligase IV inhibitors.
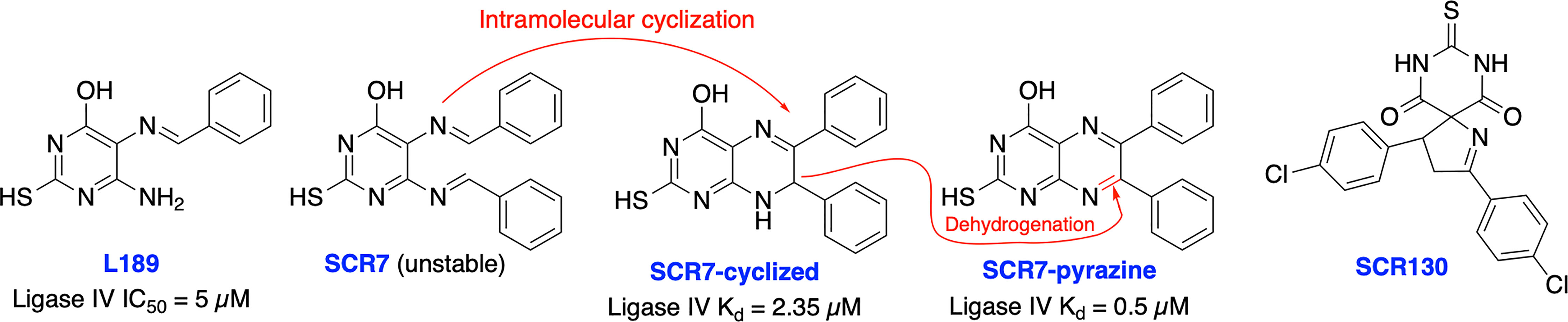
Figure 6 Small molecule inhibitors targeting DNA Ligase IV and their IC50 values for either inhibition of Ligase IV adenylation or Ligase IV end-joining.
XRCC4 Inhibitors
XRCC4 and its paralog, PAXX are responsible for the recruitment of other NHEJ factors to the damage site and XRCC4 is also a key regulator of DNA ligase IV activity in the NHEJ ligation step (17, 113–115). XRCC4 holds a potential to enhance chemo- and radiosensitivity of current therapeutics. Early attempts to inhibit XRCC4 resulted in the development of compounds salvianolic acid B, lithospermic acid, and 2-O-feruloyl tartaric acid (Figure 7); however, potential in vitro and in vivo effects of these agents is not documented to date (116). Recently, Liu et al. identified perfluorodecanoic acid (PFDA), a common persistent environmental pollutant, as a XRCC4 inhibitor which was able to sensitize gastric cancer cells to chemotherapy; however, mechanism of action, target engagement with XRCC4 and the toxicity profile of the inhibitor needed to be explored in more details (117).
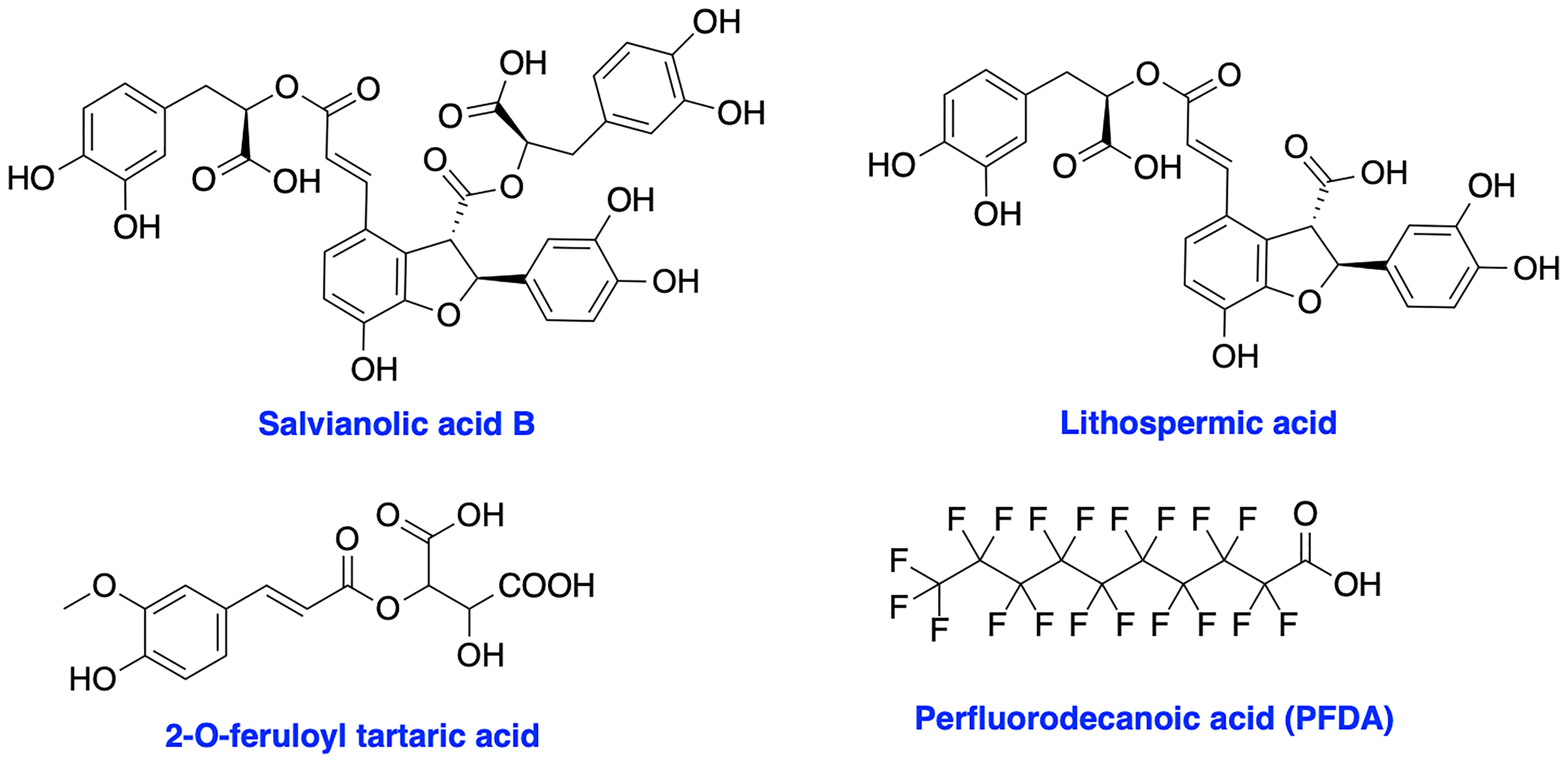
Figure 7 Structures of XRCC4 inhibitors.
Inhibitors Targeting HDR Pathway
MRN Complex (MRE11-RAD50-NBS1) Inhibitors
In case canonical NHEJ pathway fails to enact timely DNA repair, DSBs are subjected to end resection leading to the generation of 3′ ss-DNA that interfere with Ku recruitment and promote high-fidelity repair process by HDR (11, 118). Homologous recombination occurs between homologous DNA sequences through the MRN-RPA-RAD51 axis which facilitates repair of the damaged sequence without loss of genetic information. The DSB recognition and DNA end resection are mediated by MRE11-RAD50-NBS1 (MRN) complex which further recruits and activates ATM kinase immediately after detection of DSB. Simultaneously, RPA mediates the recruitment of ATR/ATRIP (11, 119–121). An additional oncogenic role of the MRN complex involves promoting telomere lengthening via alternative telomere lengthening (ALT) by homologous recombination (122, 123). In this case, the chromosomal ends are first resected 5’→3’ and then treated as broken ends for HDR. Additional mechanistic insight into HDR in response to telomeric DSBs and telomere lengthening has recently been reported (124).
The crucial role of the MRN complex in DSB repair and its potential as a target for cancer therapy has been widely explored in various types of cancers. The high-level expression of MRN complex is associated with chemo- and radio-resistance in breast cancer, glioblastoma and NSCLC as well as correlated with worse disease-free (DFS) and poor overall survival (OS) in rectal, prostate, gastric and NSCLC patients. However, the consequences of defects and/or altered expression level of MRN complex are still controversial with respect to its dual roles in tumorigenesis and prognosis (125–128).
Initial attempts to inhibit MRE11 resulted in Mirin as the first MRE11 inhibitor from high-throughput screening (HTS). Mirin blocks Mre11 exonuclease activity, prevents MRN-dependent ATM activation without affecting its kinase activity and abolishes the G2/M checkpoint and homology-dependent repair in mammalian cells (129). Mirin displayed inhibition of androgen-dependent transcription and growth of prostate cancer cells, MYCN-amplified neuroblastoma cells and enhanced chemosensitivity to DNA damaging agents in glioblastoma cells (130–132). Further structural modification of Mirin resulted in PFM01 and PFM03 as selective endonuclease inhibitors and PFM39 which selectively block the exonuclease activity of MRE11 (Figure 8), while their potential function in cancer therapy remains poorly explored (133). Further mechanistic studies revealed MRE11 exo- or endonuclease inhibitors confer distinct DSB repair mechanisms. Inhibition of endonuclease activity of MRE11 drives the cell to NHEJ repair pathway over HDR, while blocking the exonuclease activity of MRE11 results in a repair defect. These studies demonstrate the potential impact of targeting MRN complex for cancer therapy; however, the lack of HDR specificity and the broad spectrum of activity restricted further development of these inhibitors.
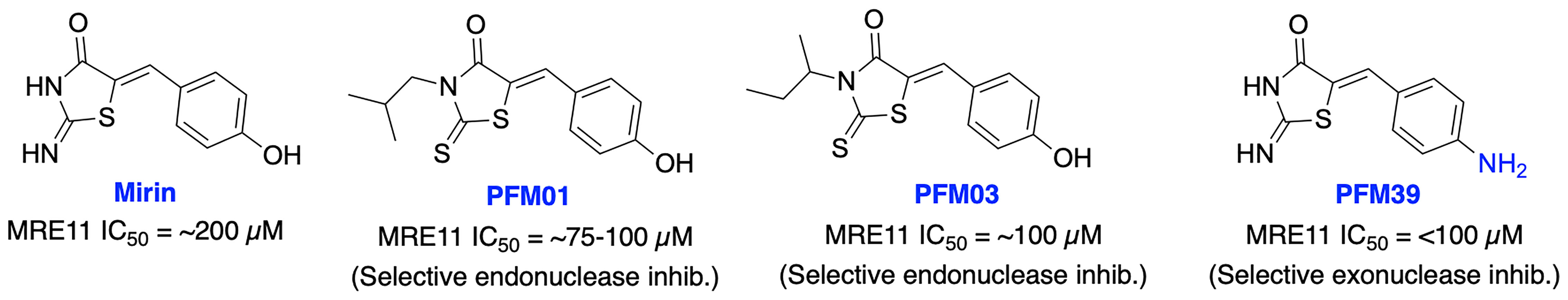
Figure 8 Small molecule inhibitors targeting MRE11 with their respective IC50 values for inhibition of nuclease activity.
To date, there is no inhibitor developed targeting RAD50 and NBS1 despite their crucial role in MRN complex mediated repair pathway and targeting protein-protein interactions in the MRN complex could also provide a potential chemotherapeutic strategy.
RPA Inhibitors
Replication protein A (RPA) is the major human single stranded DNA (ssDNA)-binding protein and plays critical roles in a variety of DNA metabolic pathways including DNA replication, repair, recombination, checkpoint activation and DDR. RPA interacts with several functional proteins to regulate DNA metabolism for the maintenance of genomic stability. RPA’s integral and non-redundant roles in both nucleotide excision repair (NER) and homology directed repair (HDR) DNA repair pathways have been well studied. Beyond NER and HDR, RPA is involved in the process of replication fork reversal and other DNA maintenance pathways such as DNA mismatch repair (MMR) and base excision repair (BER) (134–137). In NER, the recognition and verification of bulky adduct DNA damage requires RPA in conjunction with XPA while in HDR, RPA ssDNA-binding activity is required to promote RAD51 filament formation in preparation for strand invasion. RPA binding drives a chain of cooperative events that results in the recruitment of HDR repair proteins (including BRCA1 and BRCA2) at the site of DSB DNA damage. RPA acts as a key sensor to elicit cell cycle arrests at checkpoints and potentiate the activation of the ATR kinase mediated DNA damage signaling/DDR by following cellular exposure to genotoxic stresses (135, 138). Each of these roles requires binding of RPA to ssDNA, making the RPA-ssDNA interaction a promising target for cancer therapy. RPA has been shown to be over-expressed in several cancers including lung, ovarian, breast, colon, bladder, gastric, hepatic, and esophageal and these solid tumors may rely on RPA to mitigate the replication stress associated with these cancers (135, 139, 140).
RPA is a heterotrimeric complex consisting of 70 kDa (RPA70), 32 kDa (RPA32), and 14 kDa (RPA14) subunits (141). The 70 kDa subunit contains the two major high affinity ssDNA binding domains A and B, in addition to domains C and F. The F-domain located on the N-terminal of the 70 kDa subunit (RPA70N) of RPA does not bind ssDNA with high affinity; however, it is involved in a series of protein–protein interactions. The development of small molecule inhibitors of RPA has been pursued by either targeting the (i) N-terminal region of the 70 kDa subunit (RPA70N) to disrupt its interactions with key DDR proteins or (ii) the DNA-binding A and B domains of RPA to prevent binding of ssDNA. Early attempts to develop N-terminal RPA70N targeted inhibitors resulted in NSC15520 (Fumaropimaric acid, FPA) and HAMNO (Figure 9); however, their further progress is restricted due to limited cellular uptake, specificity, or metabolic instability (142–144). Fesik and co-workers exploited fragment-based NMR, HTS screening approaches, and further structure-based optimization efforts which led to the discovery of nanomolar or sub micromolar stapled helix peptides, thiazolothienopyrimidinone- (VU079104), anthranilic acid-, chlorobenzothiophene-, pyrazole-based inhibitors targeting RPA70-N-terminal domain (62, 137, 145). High binding affinity, good in vitro potency and cellular uptake observed with some of these inhibitors suggest potential for further development, albeit neither cellular activity nor specificity is documented to date.
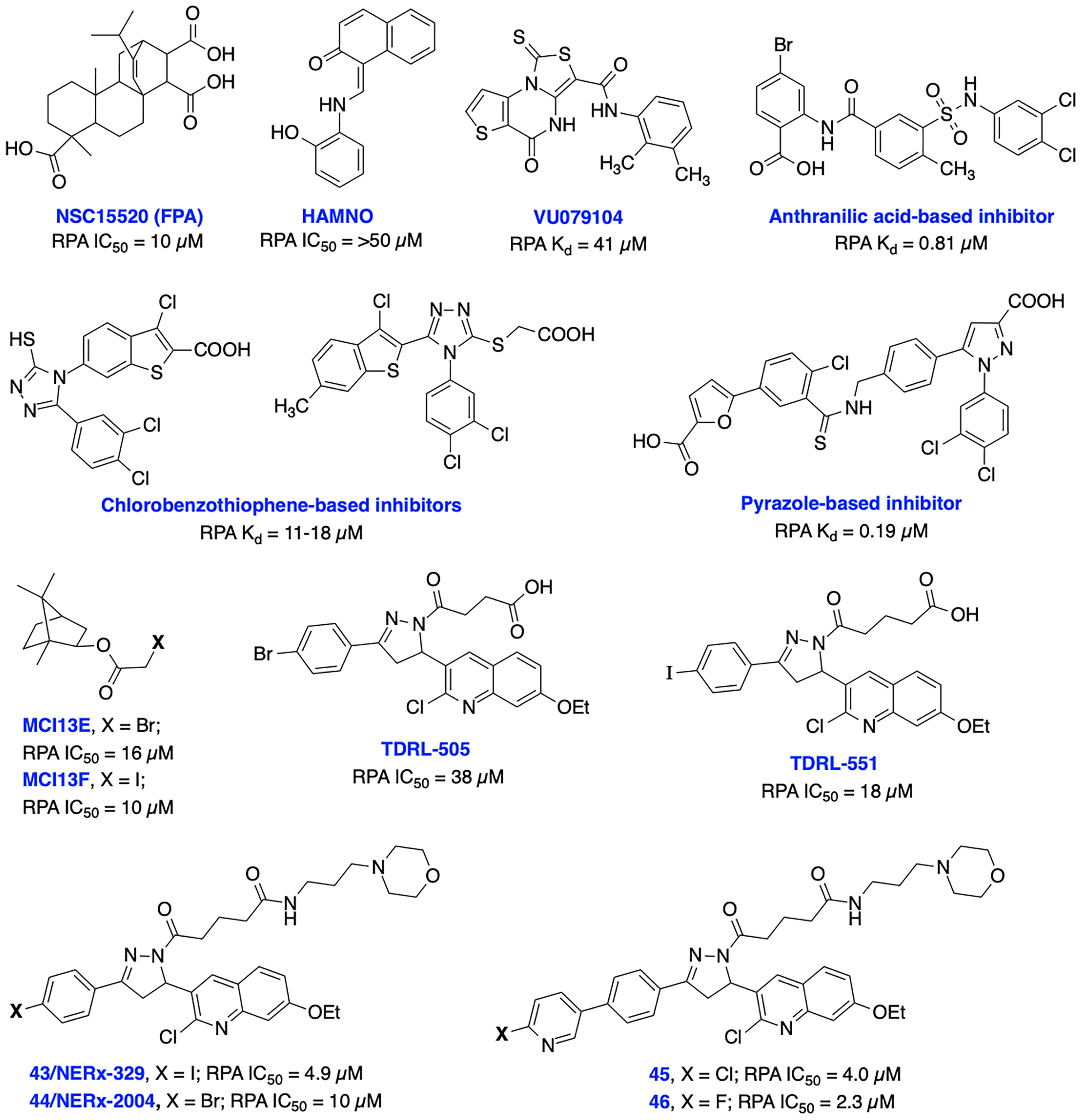
Figure 9 Small molecule inhibitors targeting RPA N-terminal protein-protein interactions and RPA-DNA interactions with their respective Kd/IC50 values.
Recent advances in the development of inhibitors targeting protein-DNA interactions hold considerable promise and opened an entirely new class of ‘druggable’ targets for therapeutic intervention. Earlier, we identified isoborneol haloacetate MCI13E and MCI13F as potent RPA inhibitors and biochemical analysis revealed an irreversible mechanism of inhibition involving covalent modification of RPA with these inhibitors. MCI13E showed cytotoxicity, induced apoptosis and demonstrated synergy with cisplatin in lung cancer cell line models (62, 146). Toward identifying reversible inhibitors of the RPA-DNA interactions, we identified TDRL-505 through HTS screening using a fluorescence polarization-based assay (147, 148). Further SAR studies with TDRL-505 scaffold generated several analogs and among them TDRL-551 was identified as the most potent compound (149). This proof-of-concept study identified that both inhibitors were capable of blocking the RPA-DNA interaction, resulting in cell cycle arrest, cytotoxicity, and increased the efficacy of the chemotherapeutic drugs cisplatin and etoposide in vitro. Moreover, TDRL-551 displays modest single agent activity in lung and ovarian cancer cell lines and synergy in combination with cisplatin and etoposide. Recently, we performed systematic structural modification of TDRL-551 in our laboratory by utilizing a structure-based drug design strategy and identified a series of novel chemical inhibitors (43/NERx-329, 44/NERx-2004 and 45-46) with improved RPA inhibitory potency, solubility, and cellular uptake for preclinical settings (150). Moreover, NERx-329 exhibited single agent activity in a broad spectrum of cancer cells, synergism with DNA damaging agents (cisplatin, etoposide and bleomycin) and DDR inhibitors (BMN673, NU7441 and VE821) in lung cancer cells and single agent anticancer activity in lung cancer xenograft models. DNA fiber analysis showed degradation of replication forks upon stalling and RPA exhaustion by NERx-329 and other known DDR inhibitors (151). Overall, a multifaceted role of RPA mediated DNA damage repair through NER, DSB repair through HDR, DNA damage signaling/DDR, replication fork dynamics and its interaction with other proteins holds the potential to fine tune the pathway and it’s response to chemotherapy or radiotherapy induced DNA damage toward maximizing efficacy, overcoming resistance, and reducing the toxicities associated with existing cancer therapeutics.
RAD51 Inhibitors
RAD51 is essential for promoting the HDR pathway as RAD51 binds to ssDNA by displacing RPA with the help of BRCA2 and other accessory factors to allow homology search and strand invasion.
Both RAD51 and RPA also are essential regulators of replication forks stability including in regulating fork restart and reversal through management of ssDNA. RAD51 and RPA function early in the processing of stalled forks, before the formation of a DSB, to facilitate fork reversal and protection that help maintain genome stability during DNA replication (152, 153, 136). RAD51 overexpression is observed in several cancers, including pancreatic, soft tissue sarcoma, breast, NSCLC, prostate cancer, glioblastoma and leukemia (152). Overexpression of RAD51 enhanced DNA repair HDR activity and helps cancer cells to survive and develop resistance to DNA damaging agents (154, 155). Depletion of RAD51 expression or inhibition heightened sensitivity to DSB inducing agents including IR in various cancer cells. Therefore, developing RAD51 inhibitors could lead to persistent DNA damage, G2/M arrest, apoptosis in the cancer cells and overcome resistance associated with current DSB inducing agents. Additionally, making HDR-proficient tumor cells HR-deficient by inhibiting RAD51 could prove useful in restoring synthetic lethality in tumors that have developed resistance with PARP inhibitors (PARPi).
Currently, several RAD51 inhibitors have been developed to further exploit the HDR pathway as a therapeutic target for cancer therapy. RAD51 has been explored as a pharmacological target in two different ways, first, in cancers known to overexpress RAD51, compounds with single-agent activity have been described that exploit overexpression by inducing formation of toxic RAD51 complexes on undamaged DNA. The second of which is as a component of combination therapy where disruption of RAD51’s ssDNA binding activity synergizes DNA damaging therapies. Ishida et al. identified DIDS as a competitive RAD51 inhibitor that prevents RAD51-ssDNA and RAD51-dsDNA binding, RAD51-mediated strand exchange and homologous pairing (Figure 10). However, the elevated human cell toxicity of DIDS has restricted its further development (156). A natural compound, halenaquinone was identified through an extensive screen of marine sponge extracts which directly inhibit RAD51-dsDNA binding, but it does not alter RAD51 affinity for ssDNA (157). Furthermore, halenaquinone-treated cells showed a reduction of IR induced RAD51 foci formation at DSB sites probably by preventing the DNA homologous pairing step of the HDR pathway. Chloromaleimide derivative RI-1 was identified as a potent RAD51 inhibitor and its biochemical analysis revealed an irreversible mechanism of inhibition involving covalent modification of the thiol group on the C319 residue of human RAD51 (158). In order to avoid off-target effects associated with covalent inhibitors and improve metabolic stability of the compound in biological systems, the reversible RAD51 inhibitor RI-2 was developed by introducing an aromatic ring at maleimide ring. RI-2 displayed a 6-fold decrease in potency compared to RI-1 and specifically inhibited HDR efficiency and sensitize human cancer cells to mitomycin C (MMC)-induced synthetic lethality (159).
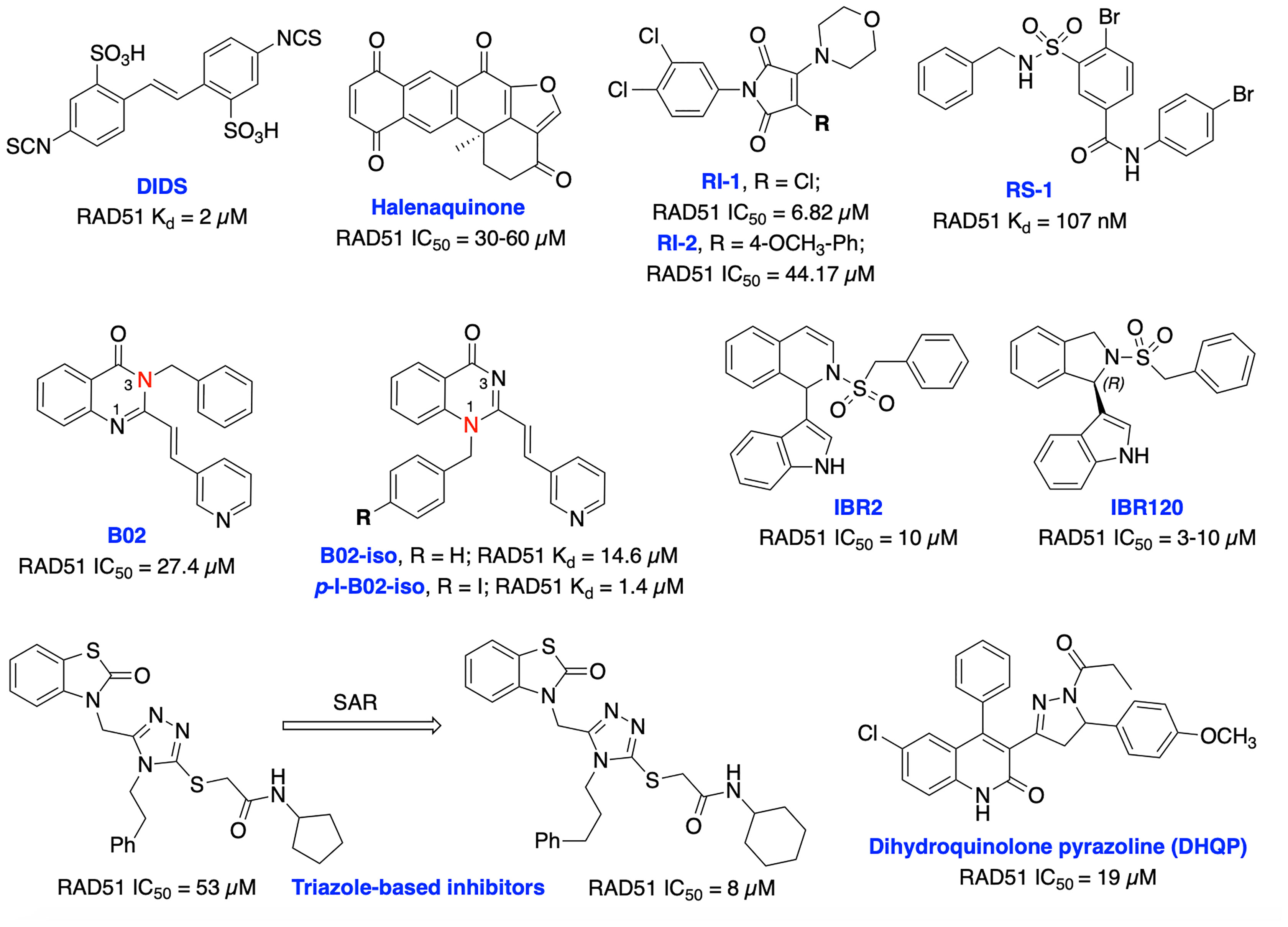
Figure 10 Small molecule inhibitors targeting RAD51 with their respective Kd/IC50 values for either disruption of RAD51 binding or RAD51 mediated D-loop formation.
The small molecule RS-1 was developed as an allosteric effector to exploit overexpression of RAD51 activity by further stimulating the formation of toxic RAD51 complexes on undamaged chromatin as a potential cancer therapy (160, 161). RS-1 was able to stimulate binding of RAD51 to ssDNA and dsDNA and enhanced recombination activities of RAD51 by locking its active conformation, without affecting ATP hydrolysis. RS-1 demonstrated a single agent activity in tumor cell lines which have more ssDNA due to increased replication, that leads to cytotoxicity while sparing normal cells (162). RS-1 also showed significant anticancer activity in a prostate cancer xenograft animal model (161). RS-1 exhibited inconsistent HDR efficiency in CRISPR/Cas9 precision genome editing in various other organisms and cell types (163, 164), suggesting that either different species may respond differently, or RAD51 may not be the most reliable target for improving precision genome engineering applications. Mazin and co-workers identified B02 as a highly specific RAD51 inhibitor that directly binds to RAD51, increases sensitivity to IR and several DNA damaging agents including etoposide and doxorubicin by inducing DSBs and subsequent blocking of HDR repair (154, 165, 166). Recently, they have carried out further structural analysis of B02 and identified B02-iso and p-I-B02-iso as substantially stronger inhibitors of RAD51 and HDR than the parent compound. B02-iso significantly increased the sensitivity of BRCA-proficient triple-negative breast cancer (TNBC) MDA-MB-231 cells to the PARPi, olaparib through synthetic lethality (167).
Zhu et al. targeted protein–protein interaction sites of RAD51 by developing IBR2 which disrupts the RAD51-BRCA interaction and RAD51 multimerization and enhances proteasomal degradation of RAD51 (168). Further structural optimization of IBR2 generated the stereo selective inhibitor IBR20 which also disrupts RAD51 multimerization, impairs HDR activity and increases cytotoxic activity in a variety of cancer cell lines (169). Utilizing high throughput docking and further SAR optimization, Cavalli and co-workers identified a series of triazoles that mimic BRCA2 mutations by disrupting the RAD51-BRCA2 interaction. Further, these compounds inhibited DSB repair and exhibited synergy with olaparib in pancreatic cancer cells with functional BRCA2 (170, 171). Recently, the same research group identified a dihydroquinolone pyrazoline (DHQP)-based inhibitor which also disrupted the RAD51-BRCA2 interaction, inhibited HDR activity and showed synergy with olaparib in pancreatic cancer to trigger synthetic lethality (172). However, further structural optimization is needed to improve potency, solubility, cytotoxicity and true synthetic lethality outcome of both triazole- and DHQP-based inhibitors. The fatty acid nitroalkene 10-nitro-octadec-9-enoic acid (OA-NO2) inhibited RAD51-ABL1 complex formation by alkylating RAD51 Cys-319 residue and decreased HDR activity. It also increased the sensitivity of doxorubicin, olaparib, IR and cisplatin in TNBC cells (68, 173). CYT01B and CYT-0851 (structures are not disclosed), are orally bioavailable small molecule RAD51 inhibitors, being developed by Cyteir Therapeutics. Both inhibitors blocked HDR activity and have demonstrated anticancer activity in cells expressing activation-induced cytidine deaminase (AID), a protein that promotes formation of DSBs. Preclinical data showed synergy with PARP and ATR inhibitors in various models, suggesting these inhibitors have the ability to overcome resistance of PARPi (174, 175). CYT-0851 is currently in Phase 1/2 clinical trials demonstrated promising antitumor activity in vitro and in vivo models across different tumor types including both hematologic malignancies and solid tumors (NCT03997968, https://clinicaltrials.gov/ct2/show/NCT03997968).
The development of either RAD51 inhibitors or modulators can be safe and effective for clinical use and it is an exciting approach for cancer therapy.
Inhibitors Targeting SSA and Alt-NHEJ (TMEJ) Pathways
SSA is a RAD51-independent DSB repair pathway which joins two homologous repetitive sequences oriented in the same direction through annealing. SSA shares DNA end resection and RPA displacement steps with HDR to reveal complementary homologous sequences. RAD52 is the central protein for SSA which is recruited to anneal each ssDNA with two repetitive sequences. After the annealing step, the sequences between the homologous repeats are flanked out on either side. These flanked ends are then cleaved off by nucleases, preferentially by ERCC1/XPF endonuclease and finally the ssDNA gap is closed by ligation (11, 176).
Alt-NHEJ (MMEJ/TMEJ) utilizes short microhomologies to join the two DNA strands. PARP1 is involved in promoting DNA end synapsis and recruiting the DNA polymerase θ (Pol θ) to DSB ends. Pol θ eventually stabilizes microhomology-mediated joints between the two DNA ends and flaps extending from these joints are cleaved off by either ERCC1-XPF or Flap endonuclease 1 (FEN1), followed by a ligation step (5). However, both SSA and alt-NHEJ DSB repair pathways serve primarily as backup pathways in mammalian cells which are deficient of either NHEJ or HDR pathways.
RAD52 Inhibitors
RAD52 plays essential roles in homology dependent DSB repair. RAD52 binds to ssDNA, promotes DNA annealing in the SSA pathway while it interacts with RAD51 to modulate its DNA strand-exchange activity in the HDR pathway. In addition, RAD52 protects stalled replication forks from degradation (177–180). RAD52-mediated annealing of large regions of a homologous sequence, independent of RAD51-mediated strand invasion is key for the SSA (181). The N-terminal region of RAD52 is involved in the oligomeric ring formation leading to RAD52-ssDNA binding (182). The ring structure is crucial during different repair pathways by promoting annealing of complementary DNA strands. RAD52 also has a second DNA binding site that binds to dsDNA (183). Several studies demonstrated that unlike normal cells, RAD52 is required for the survival of cancer cells with loss-of-function mutation in genes such as BRCA1, BRCA2, PALB2, and RAD51 paralogs (184–186). Therefore, this differential effect facilitates RAD52 as a promising target to trigger synthetic lethality in BRCA-deficient tumor cells without affecting normal cells.
To date, there have been several RAD52 inhibitors identified by various research groups (Figure 11) (179). Chandramouly et al. identified 6-OH-DOPA as a specific inhibitor to RAD52 ring structure formation through HTS. Notably, 6-OH-DOPA disrupts the heptamer and undecamer ring of truncated RAD52 (residues 1-209) into dimers (187), leading to abolished recruitment of RAD52 to ssDNA damage sites. 6-OH DOPA disrupted the association of ssDNA with RAD52 and consistently inhibited SSA in cells while having a minimal effect on HR and NHEJ in BRCA-proficient cells while increased level of apoptosis and DNA damage observed in BRCA1/2-deficient cells. In addition, 6-OH DOPA selectively halted proliferation of BRCA1/2 deficient TNBC cells, pancreatic cancer cells and patient-derived AML and CML cells. Another study reported Adenosine 5’-monophosphate (A5MP), its mimics 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and 5’ phosphate (ZMP) as RAD52 inhibitors through virtual computer screening of FDA and NCI drug libraries (188). All three inhibitors inhibited RAD52-ssDNA binding, while cell permeable AICAR disrupted SSA repair and reduced cisplatin-induced RAD52-ssDNA foci formation in BRCA1-deficient leukemic cells. Both A5MP and AICAR exerted anti-tumor activity against BRCA-deficient cancer cells by triggering synthetic lethality. Huang et al. identified 17 putative inhibitors of RAD52 through HTS. Among these, D-G09 and D-I03 showed exquisite selectivity against RAD51 and anticancer activity in BRCA1/2 deficient pancreas, ovarian, and TNBC cells with no effect in BRCA1/2 proficient cells (189). Further biochemical studies confirmed that both inhibitors bind directly to RAD52, impairs its ssDNA-annealing activity and DNA pairing activity of RAD52 (D-loop formation) in the sub-micromolar range. D-I03 showed no significant effect on cisplatin-induced RAD51 foci formation although this compound significantly reduced level of SSA repair without influencing HDR indicating specific targeting of RAD52. In addition, structurally distinct compounds, D-G23, D-I05 and D-K17 also inhibited RAD52 ssDNA annealing, DNA pairing activities of RAD52 and preferentially inhibited at least two BRCA1/2-defficient cell lines.
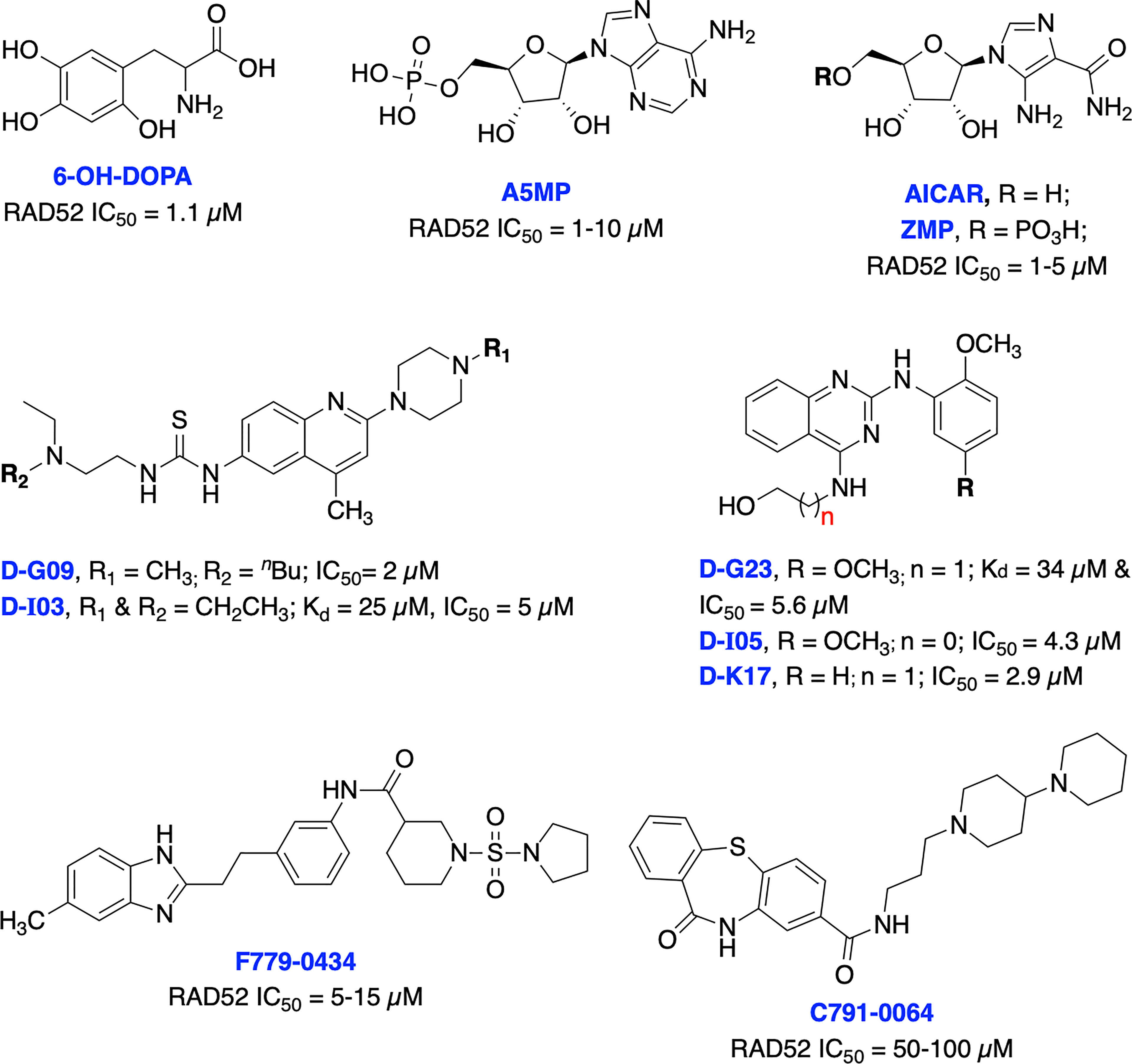
Figure 11 Small molecule inhibitors targeting RAD52 with their respective Kd/IC50 values for either RAD52 binding or ssDNA annealing activity.
Li et al. identified several RAD52 inhibitors through virtual HTS and docking studies with top compounds F779-0434 and C791-0064 inhibiting RAD52-ssDNA association and disrupting single strand annealing activity of RAD52, respectively and inducing synthetic lethality by suppressing the proliferation of BRCA2-deficient cancer cells at high concentrations (190, 191). Hengel et al. identified natural products (−)-epigallocatechin, epigallocatechin-3-monogallate and NP-004255 (RAD52 IC50s = 1.8, 0.277 and 1.5 μM, respectively) as potent inhibitors of RAD52 by utilizing HTS and FRET-based assays. Both (−)-epigallocatechin and epigallocatechin-3-monogallate inhibited DSB repair and significantly reduced proliferation of BRCA2 and MUS81 deficient cells under conditions of replication stress (192).
While clearly in the developmental stages, each of the RAD52 inhibitors could offer potential for further development of effective treatment to improve therapeutic outcome of BRCA deficient malignancies in combination with PARPi.
ERCC1-XPF Inhibitors
The structure-specific heterodimeric endonuclease ERCC1-XPF complex is primarily involved in NER but has roles in SSA and alt-NHEJ mediated DSB repair as well as interstrand cross-link (ICL) repair pathways due to its unique catalytic incision properties (193, 194). ERCC1 regulates DNA-protein and protein-protein interactions and is catalytically inactive while XPF which contains an inactive helicase-like motif, is involved in protein-protein interactions and DNA binding, and provides the endonuclease activity. The overexpression of ERCC1-XPF has been linked with poor responses to chemotherapy in various cancers including NSCLC, squamous cell carcinoma, ovarian cancer and melanoma while low ERCC1-XPF expression observed in testicular cancer has extended overall survival of cancer patients (195, 196). Further, ERCC1 deficient melanoma cells exhibited around 10-fold more sensitivity to cisplatin than ERCC1-proficient cells and in a xenograft mouse model as well (197). ERCC1-XPF became an interesting target to investigate in order to overcome resistance to chemotherapeutic agents due to its involvement in multiple key repair pathways.
The heterodimerization and localization of ERCC1 and XPF is required to constitute a functional and stable complex and essential for endonuclease activity. ERCC1-XPF interaction through their double helix–hairpin–helix (HhH2) domains is an essential requirement to stabilize ERCC1-XPF complex to promote catalytic activity (198, 199). Therefore, several research groups are targeting ERCC1-XPF HhH2 domain protein-protein interaction to develop novel inhibitors to increase sensitivity of existing therapies whose DNA-damaging effects are primarily repaired by ERCC1-XPF-dependent pathways.
Jordheim et al. identified F06/NERI02 (NSC130813) through in silico screening, as a small molecule inhibitor targeting ERCC1-XPF heterodimerization and demonstrated modest affinity for XPF and sensitized cancer cells to MMC and cisplatin (Figure 12) (200). In addition, F06 exhibited a synergy with PARPi olaparib in BRCA1-deficient breast cancer cells. However, suboptimal potency, toxicity and off-target effects of F06 restricted further biochemical and cellular studies (62). Recently, West and co-workers rationally modified the structure of F06 by utilizing computer-aided drug design (CADD) to identify potential binding interactions and further SAR studies to improve inhibition of ERCC1-XPF endonuclease activity. The lead compounds B5/B9 and compound 4 showed 3-fold improvement in inhibition activity compared to F06. The sensitivity to UV radiation and cyclophosphamide also increased significantly in reducing proliferation of metastatic colorectal cancer (201–203). Moreover, compound 4 showed lower lipophilicity and greater metabolic stability which makes this compound an interesting candidate for further advancement. McNeil et al. targeted three sites of the ERCC1-XPF HhH2 domain to identify possible inhibitors for the heterodimer by utilizing an in silico screening approach (204). They identified E–X AS7 which binds to ERCC1-XPF through a metal-based interaction, inhibits NER in low micromolar concentrations and specifically increases the cisplatin sensitivity of NER-proficient human and mouse cells. E-X PPI2 inhibited the NER activity in melanoma cells, showed marginal sensitivity to cisplatin treatment but caused significant reduction in the level of ERCC1-XPF heterodimer levels in ovarian cancer cells. However, the medium-high micromolar range binding affinity (Kd) and inhibitory potency (IC50) makes these compounds unsuitable for further studies. A series of highly potent and selective catechols, hydroxylimides/hydroxy pyrimidinones have been identified as ERCC1-XPF inhibitors through in silico HTS and SAR approach (205, 206). Most of the compounds from these series showed good selectivity for ERCC1-XPF against FEN-1 and DNase I; however, potential in vitro and in vivo effects of these compounds are not documented yet. Patrick and co-workers targeted the active site on the XPF nuclease domain and identified NSC16168 as a potent ERCC1-XPF inhibitor by performing a HTS using the NCI-DTP (National Cancer Institute Developmental Therapeutics Program) diversity database. NSC16168 significantly enhanced cisplatin antitumor activity in a lung cancer xenograft model (207).
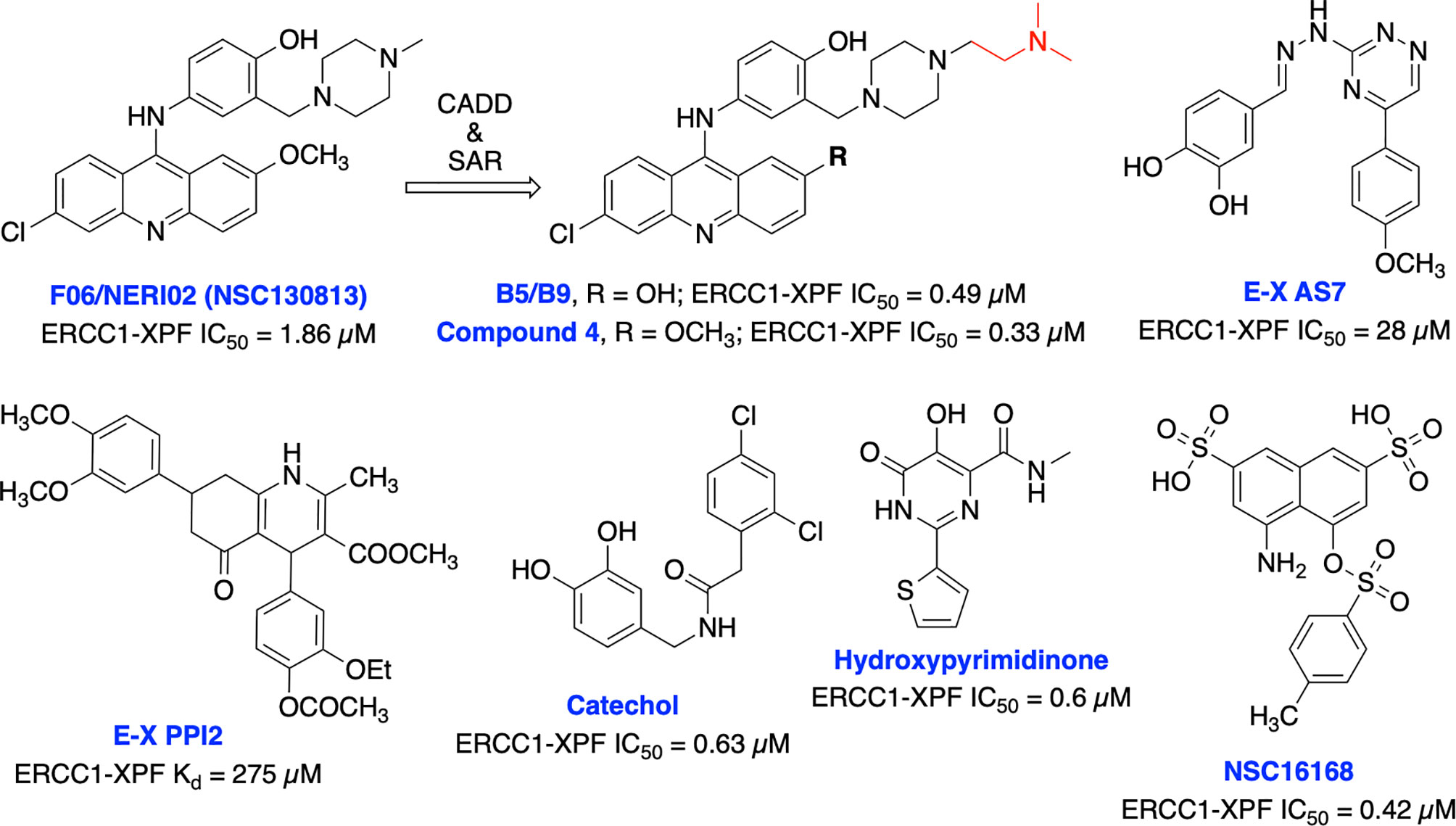
Figure 12 Small molecule inhibitors targeting ERCC1-XPF with their respective Kd/IC50 values for inhibition of ERCC1-XPF endonuclease activity.
Overall efforts resulted in several potent ERCC1-XPF endonuclease inhibitors which are capable to diminish NER activity and enhance the cytotoxicity of platinum-based chemotherapeutics although these inhibitors are not explored in targeting DSB repair and its defects for cancer therapy. Moreover, the lack of structural insights, selectivity against other endonucleases and most importantly limited utilization of these inhibitors in targeting DSB repair restricts their further advancement into the clinic.
DNA Polymerase Theta (Pol θ) Inhibitors
Pol θ (gene name, PolQ) belongs to the error-prone A family of DNA polymerases and is a critical component of the alt-NHEJ (MMEJ or TMEJ) repair pathway of resected DSBs. Biochemical and mechanistic studies have shown that the helicase domain of Pol θ displaces RPA bound to the ssDNA overhang and facilitates joining of short microhomologies to the two DNA strands that flank a DSB. The polymerase domain of Pol θ initiates DNA synthesis to fill in the DNA gaps, prior to the ligation step employed by DNA Ligase I or III. In addition, Pol θ also plays an important role in joining unprotected telomeres in alt-NHEJ pathway (14, 208–211). Alt-NHEJ serves as an essential backup pathway to repair DSBs when HDR and NHEJ pathways are compromised in cancer cells such as germline BRCA-gene deficient cancer cells. Recently, Pol θ emerged as a new promising drug target to trigger the synthetic lethality between loss of the PolQ gene and deficiencies in DSB DNA repair-related tumor suppressor genes including BRCA1/2, ATM and FANCD2 for the treatment of HDR-deficient tumors (212–214). The expression of Pol θ is particularly high in subtypes of breast and ovarian cancers featuring loss of HDR activity and Pol θ-depletion reduced the survival of HR-deficient cancer cells in the presence of PARPi, cisplatin, or MMC (214). Pol θ overexpression also found in other cancers, including stomach, lung and colon (215). In addition, the higher expression of Pol θ is correlated with shorter relapse-free survival compared to patients with relatively lower expression of Pol θ. Feng et al. employed CRISPR-based genetic screening and identified 140 genes that are synthetically lethal with Pol θ, highlighting the impact of Pol θ inhibitor for cancer therapy (216).
Recently, Zhou et al. identified antibiotic novobiocin (NVB) as a specific potent inhibitor of human Pol θ (Figure 13) which inhibited alt-NHEJ repair and selectively killed HDR-deficient (both BRCA1- and BRCA2-deficient) cells over wild-type cells and significantly enhanced the cytotoxic effect of PARPi in HDR-deficient tumor cells in cellular as well as in xenograft and PDX mouse models (217). Most importantly NVB also killed HDR-deficient, PARPi-resistant tumor cells. Artios Pharma in collaboration with the Institute of Cancer Research (UK) identified ART558 as a highly potent and specific small molecule Pol θ inhibitor (213). ART558 exhibited not only BRCA-gene synthetic lethality, but also targets cells with PARPi resistance caused by defects in 53BP1/Shieldin DNA repair complex. There is a possibility that Pol θ inhibitors might be a more suitable treatment option than PARPi for combination with existing DNA-damaging chemotherapies. Several biopharmaceutical companies are currently pursuing Pol θ as a therapeutic target and the first orally bioavailable Pol θ inhibitor ART4215 (structure is not disclosed) is currently in Phase 1/2 clinical trials where it is being investigated as a monotherapy and in combination with PARPi talazoparib in patients with advanced or metastatic solid tumors (NCT04991480, https://clinicaltrials.gov/ct2/show/NCT04991480).
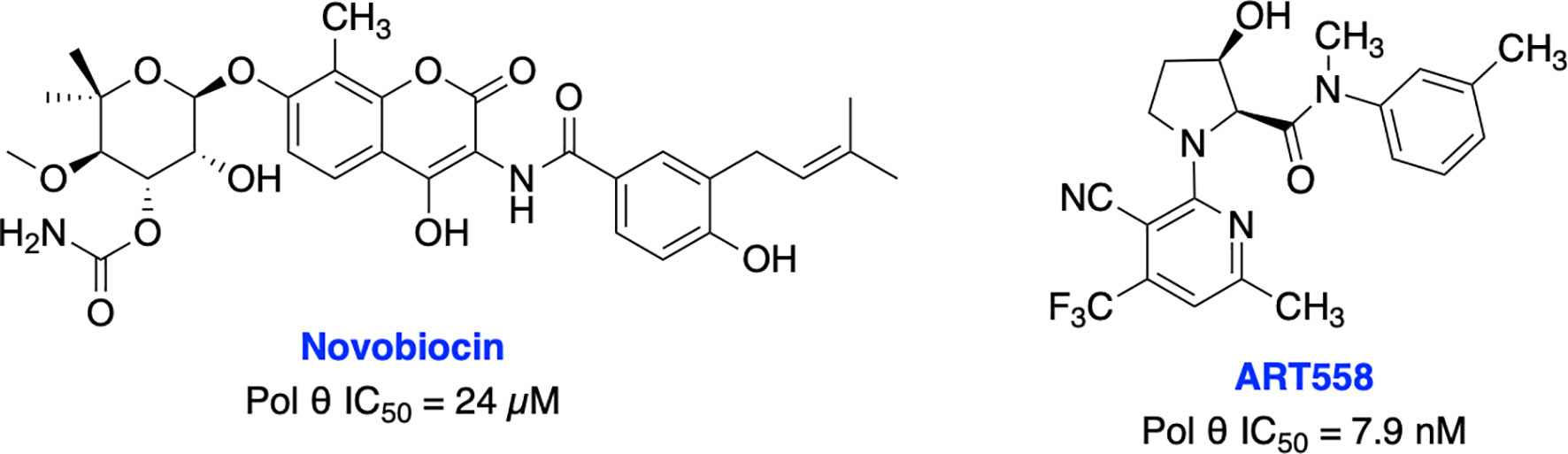
Figure 13 Small molecule inhibitors targeting Pol θ with their respective IC50 values for inhibition of polymerase activity.
RecQ and MCM Helicases Inhibitors
It is well established that RecQ helicases play an important role in DSB repair and the maintenance of genome stability. However, a direct or passive role of each RecQ helicase’s enzymatic activity in NHEJ, HDR, TMEJ and SSA mediated DSB repair pathway is yet to be elucidated (43). RecQ proteins are highly conserved from bacteria to humans, and the reduced RecQ helicases activity is associated with cancer predisposition, metastasis and premature aging. In contrast, overexpression of RecQ helicases may promote carcinogenesis and RecQ helicases are highly upregulated in various cancers (218, 219). Aggarwal et al. identified NSC 19630 and NSC 617145 (Figure 14) as WRN inhibitors through HTS of the National Cancer Institute (NCI) diversity set of compounds. Both NSC compounds dramatically impaired growth and proliferation, induced apoptosis in a WRN-dependent manner, and DSBs and chromosomal abnormalities in cellular models (220, 221). However, the presence of the maleimide group in both compounds may restrict their further development due to its propensity for non-specific covalent interactions. The same group recently identified several non-specific reversible and irreversible helicase inhibitors through HTS using a larger library of approximately 350,000 small molecules (222). Several studies identified WRN synthetic lethal vulnerability in cancers with microsatellite instability (218, 223, 224), suggesting specific WRN inhibitors hold great potential to target microsatellite instability tumors to enable a clear stratification path in the clinic.
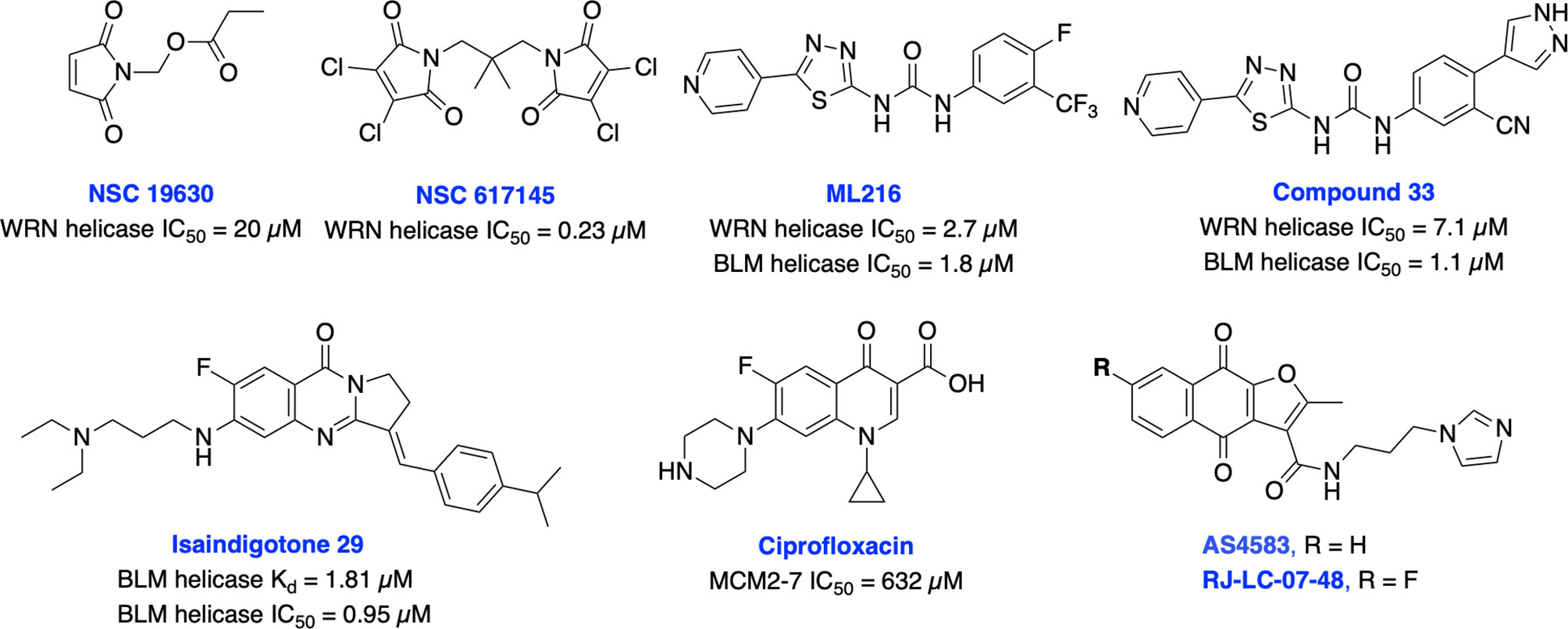
Figure 14 Small molecule inhibitors targeting WRN, BLM and MCM helicases with their respective IC50 values.
BLM helicase plays a multifaceted role in HDR pathway as it is required for the early phase of the pathway to stimulate resection of DSB ends or displacement of the invading strand of DNA displacement loops as well as at the terminal steps in dissolution of double Holliday junctions (43). Nguyen et al. identified the first BLM inhibitor by utilizing HTS and further structural optimization efforts yielding ML216 and compound 33 as potent inhibitors of the DNA unwinding activity of both BLM and WRN (225, 226). ML216 exhibited cellular induction of sister chromatid exchanges and demonstrated selective antiproliferative activity in BLM-positive cells but not those lacking BLM. However, further preclinical studies may be restricted due to poor selectivity, solubility, and cell permeability of these inhibitors. Recently, Yin et al. identified isaindigotone derivatives as novel BLM helicase inhibitors that disrupted the recruitment of BLM at DNA DSB sites. BLM inhibition by their lead compound promoted accumulation of RAD51, regulated HDR repair, and synergized cytotoxicity of cisplatin and the RAD51 inhibitor, RI-1 (227). BLM and other helicases are attractive targets for the development of cancer therapeutics which rely on synthetic lethality effects for targeting tumors with preexisting DNA repair deficiencies. Minichromosome maintenance (MCM) complex is a family of six proteins 2-7 (MCM2-7) that are activated by forming a holo-helicase CMG complex with Cdc45 and the hetero-tetrameric GINS complex (Cdc45-MCM2-7-GINS). CMG complex is cell-cycle regulated and responsible for unwinding DNA forks during DNA replication. MCM2-7 proteins have essential roles in DNA replication particularly under replicative stress where they activate dormant replication origins which allows for continued genome replication in spite of replication stress. Increased levels of MCM2-7 protein expression have been observed in a variety of cancers (39, 42). Initially, Simon et al. identified ciprofloxacin which preferentially inhibits MCM2-7 at higher concentrations than its normal therapeutic range (228). However, most recently inhibition of MCM2-7 activity by ciprofloxacin significantly delayed neuroendocrine prostate cancer (NEPC) cell growth and migration in vitro, exhibited potent anti-tumor effects in an NEPC xenograft model, and partially reversed neuroendocrine features (229). Alshahrani et al. identified UEFS99, UEFS137 and UEFS428 as MCM7 inhibitors from the natural compounds databases using in silico computational screening, however further in vitro and in vivo studies are needed to validate target engagement (230). A furanonaphthoquinone-based small molecule, AS4583 was identified as an MCM2 inhibitor through phenotypic screening and target deconvolution (231). Further mechanistic studies revealed that AS4583 inhibited cell-cycle progression and reduced DNA replication by inducing proteasomal degradation of MCM complex which ultimately contributed to the death of NSCLC cells. Subsequently, structural optimization of AS4583 led to compound RJ-LC-07-48 which showed greater potency in drug-resistant NSCLC cells and in mice bearing H1975 tumor xenografts. Overall, MCM complex can serve as a potential target for cancer therapy. Further exploration of design, screening and medicinal chemistry efforts are needed to develop MCM2-7 complex-specific inhibitors for better clinical outcomes.
DSB Repair Inhibitors for Combination Therapy, Induction of Synthetic Lethality and Precision Genome Editing
While there remain no FDA approved inhibitors of non-PIKKs within the DSB repair pathways, the future applications of such compounds may include use within combination chemotherapy regimens, as chemo- or radiosensitizers, induction of synthetic lethality in HDR-deficient cancer subtypes, and as an adjuvant therapy in precision genome editing. As described in the above corresponding sections, each of the non-PIKK pharmacological targets, whether involve directly or indirectly mediate repair of DSBs induced by either DNA damaging agents or IR, and combination therapy with either DNA damaging agents or IR has been the natural step towards maximizing synergistic efficacy, overcoming resistance, and reducing the toxicities associated with existing chemo- and radiotherapy. Provided that the preponderance of cancer patients receives DNA-damaging drugs or IR and later experience disease progression, the therapeutic potential for agents that augment the response to such therapies is large.
Synthetic lethality refers to any scenario whereby loss of two gene products produces cellular death, but loss of either individually is non-lethal. In the setting of cancer treatment, synthetic lethality is a term usually used in reference to disruption of the repair of DNA nicks in HDR-deficient cancers which yields DSBs that are repaired by error-prone pathways resulting in cell death or senescence (232). The clinically available PARP inhibitors (PARPis) olaparib, rucaparib, talazoparib, and niraparib operate by this mechanism and are frequently employed in cancers where HDR-deficiency is conferred by BRCA mutations. PARPis remain the sole class of approved anticancer drugs capable of exploiting this unique vulnerability. However, more than 40% patients with BRCA mutations fail to respond to PARPis and resistance mechanisms have been described indicating new classes of medications capable of inducing synthetic lethality are needed (233). Two particularly noteworthy non-PIKKs targets within the DSB repair pathways whose inhibition have been probed for synthetic lethality in the setting of PARPi resistance include DNA polymerase θ and RAD52. The activity of DNA polymerase θ offers an escape pathway beyond NHEJ via alt-NHEJ in the setting of BRCA mutations (70, 208). An siRNA knockdown of DNA polymerase θ produces synthetic lethality in BRCA2 mutation variants (214) and the aforementioned DNA polymerase θ inhibitor ART558 retains preclinical efficacy even in the presence of 53bp1 mutations which are known to confer PARPi resistance (213). The RAD52 deficiency leads to loss of a compensatory DNA repair pathway resulting in genomic instability and persistent cell death in BRCA1/2-deficient cells. Intriguingly, RAD52 is thought to be capable of orchestrating HDR even in BRCA1/2 mutants and thus may also play a role in PARPi resistance. Targeting RAD52 for the induction of synthetic lethality could potentially improve the therapeutic outcome of BRCA-deficient malignancies treated with PARPi and restrict the emergence of drug-induced toxicity to normal tissues (179, 208).
CRISPR-Cas9 genome editing offers the possibility to prevent, treat, or even cure human diseases that are initiated by or maintained by genetic aberrations (234). Notwithstanding other barriers to the clinical application of CRISPR-Cas9 for genome editing such as selective delivery and the requirement for protospacer adjacent motifs (PAM) at the targeted region of a locus, a major challenge this platform faces is management of the DSBs created both on-target and off-target (235, 236). Provided that HDR is only available in the G2/S phases of the cell cycle because of the requirement for a sister chromatid, the predominant NHEJ pathway must be regulated in precision genome editing to preempt chromosomal rearrangements and large indels. DNA DSB repair inhibitors could help in enhancing precision genome editing as well as improving the safety of gene targeting. NHEJ inhibitors and HDR modulators can be exploited to increase the current efficiency of nuclease-based HDR mediated gene editing alongside CRISPR towards the more precise HDR mediated repair while decreasing inaccurate integration events. However, specificity, efficacy and toxicity associated with DSB repair inhibitors targeting NHEJ pathway restricted utilization of these inhibitors in CRISPR-Cas9 genome editing (89, 103, 104). Future availability of an arsenal of DSB-repair inhibitors capable of directing DSB repair by HDR will foster the arrival of precision genome editing within clinical practice.
We have summarized a list of targeted proteins and their respective inhibitors, mechanism of their action, binding affinity or in vitro potency, indication along with cellular activity and their phase of development in Table 1.
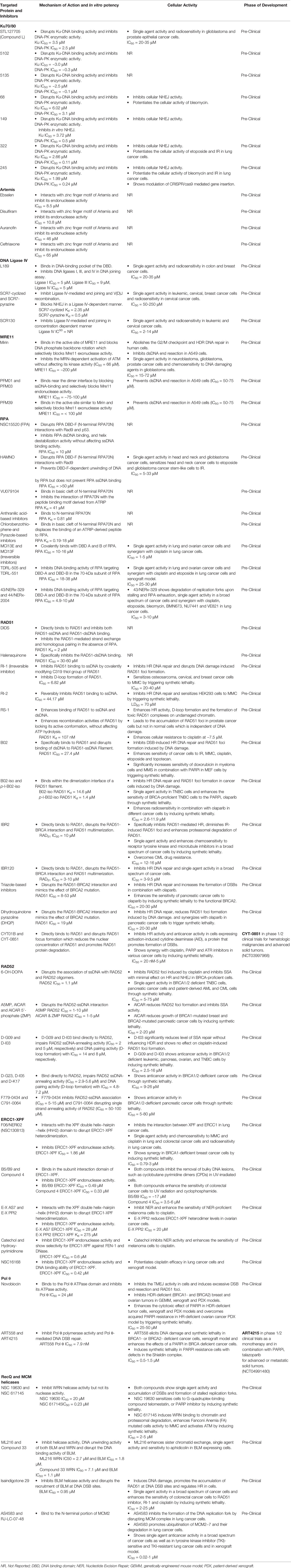
Table 1 A summary of non-PIKKs DSB Repair inhibitors.
Conclusions
DSBs are the most lethal of all DNA lesions and the cadre of proteins that respond to repair of DSBs represent a diverse array of proteins and enzymes of which a small portion are in fact kinases. Combinational therapy of DSB repair inhibitors with existing DSB inducing agents has been the most effective strategy. Careful consideration of the sequence of combination drug administration and optimizing drug scheduling will likely be needed to optimize synergistic effects of combination therapy while sparing normal cells. DSB repair deficiency and mutation can increase the immunogenicity of cancers and combination of selective DSB repair inhibitors with immunotherapy could be a useful strategy in treating subsets of cancer patients. The identification of useful synthetic lethal interactions to enhance the sensitivity to widely prescribed chemotherapeutics is expected to allow more selective and efficient tumor killing with reduced toxicity. However, stratification of clinically relevant biomarkers along with extensive medicinal chemistry efforts are needed to develop novel compounds that can be exploited to discover synthetic lethal interactions with other DNA repair and DDR genes.
In the last two decades, our understanding of DSB repair pathways has improved dramatically, however, development of small molecule inhibitors targeting these repair pathways are only now being pursued in earnest and recent high-resolution protein structures of many of these putative targets can enhance these efforts. Even though, there is still an urgent need for rapid expansion of DNA repair targeted agents to move from the lab to the clinic through drug discovery and development efforts. The interdependencies between DNA repair pathways can lead to potential druggable vulnerabilities but may increase the mutagenic lesions in surviving cells and drug resistance to DSB inhibitors so a cautious approach is warranted. Thus, development of potent and selective inhibitors for each of the DSB repair proteins accompanied by robust clinical trials will have new treatment modalities for a wide range of tumors and ultimately confer benefit to human health.
Author Contributions
All authors listed have made a substantial and direct contribution to the work. All authors have given approval to the final version of the manuscript.
Funding
This work is supported by NIH grant R01 CA247370 (JT and NG), Wayne State University Start-up funds (NG) and the Tom and Julie Wood Family Foundation (JT).
Conflict of Interest
KSP is a Vice-President of Research and JJT is a cofounder and CSO of NERx Biosciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Diana Ainembabazi and Olusayo Ogunyemi for critical reading and comments on the manuscript. We apologize to authors whose work could not be cited due to space limitations.
References
1. Ray U, Raghavan SC. Inhibitors of DNA Double-Strand Break Repair at the Crossroads of Cancer Therapy and Genome Editing. Biochem Pharmacol (2020) 182:114195.
2. Mehta A, Haber JE. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb Perspect Biol (2014) 6:1–17.
3. Chapman JR, Taylor MR, Boulton SJ. Playing the End Game: DNA Double Strand Break Repair Pathway Choice. Mol Cell (2012) 47:497–510.
4. O’Driscoll M, Jeggo PA. The Role of Double-Strand Break Repair-Insights From Human Genetics. Nat Rev Genet (2006) 7:45–54.
5. Trenner A, Sartori AA. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front Oncol (2019) 9:1388. doi: 10.3389/FONC.2019.01388
6. Jeggo PA, Löbrich M. DNA Double-Strand Breaks: Their Cellular and Clinical Impact? Oncogene (2007) 26:7717–9. doi: 10.1038/sj.onc.1210868
7. Helleday T, Lo J, van Gent DC, Engelward BP. DNA Double-Strand Break Repair: From Mechanistic Understanding to Cancer Treatment. DNA Repair (Amst) (2007) 6:923–35. doi: 10.1016/j.dnarep.2007.02.006
8. Bunting SF, Nussenzweig A. End Joining, Translocations and Cancer. Nat Rev Cancer (2013) 13:443–54.
9. Ye Z, Shi Y, Lees-Miller SP, Tainer JA. Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front Immunol (2021) 12:797880–0.
10. Gorbunova V, Seluanov A. DNA Double Strand Break Repair, Aging and the Chromatin Connection. Mutat Res - Fundam Mol Mech Mutagenesis (2016) 788:2–6.
11. Scully R, Panday A, Elango R, Willis NA. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat Rev Mol Cell Biol (2019) 20:698–714.
12. Sfeir A, Symington LS. Microhomology-Mediated End Joining: A Back-Up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci (2015) 40:701–14.
13. Bhargava R, Onyango DO, Stark JM. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet (2016) 32:566–75.
14. Ramsden DA, Carvajal-Garcia J, Gupta GP. Mechanism, Cellular Functions and Cancer Roles of Polymerase-Theta-Mediated DNA End Joining. Nat Rev Mol Cell Biol (2021) 23:125–40. doi: 10.1038/s41580-021-00405-2. ahead of print.
15. Karanam K, Kafri R, Loewer A, Lahav G. Quantitative Live Cell Imaging Reveals a Gradual Shift DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol Cell (2012) 47:320–29.
16. Stinson BM, Loparo JJ. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu Rev Biochem (2021) 90:137–64.
17. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat Rev Mol Cell Biol (2017) 18:495–506.
18. Pawelczak KS, Bennett SM, Turchi JJ. Coordination of DNA-PK Activation and Nuclease Processing of DNA Termini in NHEJ. Antioxid Redox Signal (2011) 14:2531–43.
19. Pawelczak KS, Turchi JJ. A Mechanism for DNA-PK Activation Requiring Unique Contributions From Each Strand of a DNA Terminus and Implications for Microhomology-Mediated Nonhomologous DNA End Joining. Nucleic Acids Res (2008) 36:4022–31.
20. Pawelczak KS, Andrews BJ, Turchi JJ. Differential Activation of DNA-PK Based on DNA Strand Orientation and Sequence Bias. Nucleic Acids Res (2005) 33:152–61.
21. Hammel M, Tainer JA. X-Ray Scattering Reveals Disordered Linkers and Dynamic Interfaces in Complexes and Mechanisms for DNA Double-Strand Break Repair Impacting Cell and Cancer Biology. Protein Sci (2021) 30:1735–56. doi: 10.1002/pro.4133
22. Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR. The Essential Elements for the Noncovalent Association of Two DNA Ends During NHEJ Synapsis. Nat Commun (2019) 10:3588. doi: 10.1038/s41467-019-11507-z
23. San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem (2008) 77:229–57.
24. Li X, Heyer W-D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res (2008) 18:99–113.
25. Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The Molecular Basis and Disease Relevance of non-Homologous DNA End Joining. Nat Rev Mol Cell Biol (2020) 12:765–81. doi: 10.1038/s41580-020-00297-8
26. Huang R, Zhou PK. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct Target Ther (2021) 6:254. doi: 10.1038/s41392-021-00648-7
27. Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K. Chromatin Modifications and the DNA Damage Response to Ionizing Radiation. Front Oncol (2013) 2012:2:214–214.
28. Camphausen K, Tofilon PJ. Inhibition of Histone Deacetylation: A Strategy for Tumor Radiosensitization. J Clin Oncol (2007) 25:4051–6.
29. Hu C, Zhang M, Moses N, Hu C, Polin L, Chen W. The USP10-HDAC6 Axis Confers Cisplatin Resistance in non-Small Cell Lung Cancer Lacking Wild-Type P53. Cell Death Dis (2020) 11:328. doi: 10.1038/s41419-020-2519-8
30. Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-Targeted Therapies. Genome Med (2016) 8:69. doi: 10.1186/s13073-016-0324-x
31. Marcand S. How do Telomeres and NHEJ Coexist? Mol Cell Oncol (2014) 1(3):1–6. doi: 10.4161/23723548.2014.963438
32. Rossiello F, Aguado J, Sepe S, Iannelli F, Nguyen Q, Pitchiaya S, et al. DNA Damage Response Inhibition at Dysfunctional Telomeres by Modulation of Telomeric DNA Damage Response RNAs. Nat Commun (2017) 8:13980. doi: 10.1038/ncomms13980
33. Nelson CB, Alturki TM, Luxton JJ, Taylor LE, Maranon DG, Muraki K, et al. Telomeric Double Strand Breaks in G1 Human Cells Facilitate Formation of 5’ C-Rich Overhangs and Recruitment of TERRA. Front Genet (2021) 12:644803:644803. doi: 10.3389/fgene.2021.644803
34. Galli M, Frigerio C, Longhese MP, Clerici M. The Regulation of the DNA Damage Response at Telomeres: Focus on Kinases. Biochem Soc Trans (2021) 49(2):933–43. doi: 10.1042/BST20200856
35. Sui J, Zhang S, Chen BPC. DNA-Dependent Protein Kinase in Telomere Maintenance and Protection. Cell Mol Biol Lett (2020) 25:2. doi: 10.1186/s11658-020-0199-0
36. Wang Y, Ghosh G, Hendrickson EA. Ku86 Represses Lethal Telomere Deletion Events in Human Somatic Cells. Proc Natl Acad Sci USA (2009) 106:12430–5. doi: 10.1073/pnas.0903362106
37. Guterres AN, Villanueva J. Targeting Telomerase for Cancer Therapy. Oncogene (2020) 39:5811–24. doi: 10.1038/s41388-020-01405-w
38. Bochman ML. Roles of DNA Helicases in the Maintenance of Genome Integrity. Mol Cell Oncol (2014) 1(3):1–9. doi: 10.4161/23723548.2014.963429
39. Daniel DC, Dagdanova AV, Johnson EM. The MCM and RecQ Helicase Families: Ancient Roles in DNA Replication and Genomic Stability Lead to Distinct Roles in Human Disease. In: The Mechanisms of DNA Replication. London, United Kingdom:IntechOpen (2013). doi: 10.5772/52961
40. Suhasini AN, Brosh RM Jr. Fanconi Anemia and Bloom’s Syndrome Crosstalk Through FANCJ-BLM Helicase Interaction. Trends Genet (2012) 28:7–13. doi: 10.1016/j.tig.2011.09.003
41. Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA Helicases in DNA Repair. Annu Rev Genet (2010) 44:393–417. doi: 10.1146/annurev-genet-102209-163602
42. Seo YS, Kang YH. The Human Replicative Helicase, the CMG Complex, as a Target for Anti-Cancer Therapy. Front Mol Biosci (2018) 5:26. doi: 10.3389/fmolb.2018.00026
43. Lu H, Davis AJ. Human RecQ Helicases in DNA Double-Strand Break Repair. Front Cell Dev Biol (2021) 9:640755:640755. doi: 10.3389/fcell.2021.640755
44. Rong Z, Tu P, Xu P, Sun Y, Yu F, Tu N, et al. The Mitochondrial Response to DNA Damage. Front Cell Dev Biol (2021) 9:669379–9.
45. Dahal S, Raghavan SC. Mitochondrial Genome Stability in Human: Understanding the Role of DNA Repair Pathways. Biochem J (2021) 478:1179–97.
46. Yasukawa T, Kang D. An Overview of Mammalian Mitochondrial DNA Replication Mechanisms. J Biochem (Tokyo) (2018) 164:183–93.
47. Shutt TE, Gray MW. Bacteriophage Origins of Mitochondrial Replication and Transcription Proteins. Trends Genet (2006) 22:90–5.
48. Tadi SK, Sebastian R, Dahal S, Babu RK, Choudhary B, Raghavan SC. Microhomology-Mediated End Joining is the Principal Mediator of Double-Strand Break Repair During Mitochondrial DNA Lesions. Mol Biol Cell (2016) 27:223–35.
49. Wisnovsky S, Sack T, Pagliarini DJ, Laposa RR, Kelley SO. DNA Polymerase θ Increases Mutational Rates in Mitochondrial DNA. ACS Chem Biol (2018) 13:900–8.
50. Krasich R, Copeland WC. DNA Polymerases in the Mitochondria: A Critical Review of the Evidence. Front Biosci - Landmark (2017) 22:692–709.
51. Dahal S, Dubey S, Raghavan SC. Homologous Recombination-Mediated Repair of DNA Double-Strand Breaks Operates in Mammalian Mitochondria. Cell Mol Life Sci: CMLS (2017) 2018:75:1641–1655.
52. Mishra A, Saxena S, Kaushal A, Nagaraju G. RAD51C/XRCC3 Facilitates Mitochondrial DNA Replication and Maintains Integrity of the Mitochondrial Genome. Mol Cell Biol (2018) 2017:38.
53. Sage JM, Gildemeister OS, Knight KL. Discovery of a Novel Function for Human Rad51: Maintenance of the Mitochondrial Genome. J Biol Chem (2010) 285:18984–90.
54. Pohjoismäki JLO, Goffart S, Tyynismaa H, Wilcox S, Ide T, Kang D. Human Heart Mitochondrial DNA Is Organized in Complex Catenated Networks Containing Abundant Four-Way Junctions and Replication Forks. J Biol Chem (2009) 284:21446–57.
55. Tann AW, Boldogh I, Meiss G, Qian W, Van Houten B, Mitra S. Apoptosis Induced by Persistent Single-Strand Breaks in Mitochondrial Genome: Critical Role of EXOG (5’-EXO/endonuclease) in Their Repair. J Biol Chem (2011) 286(37):31975–83. doi: 10.1074/jbc.M110.215715
56. Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G. Mitochondrial DNA Drives Abscopal Responses to Radiation That are Inhibited by Autophagy. Nat Immunol (2020) 21:1160–71.
57. Buondonno I, Gazzano E, Jean SR, Audrito V, Kopecka J, Fenelli M. Mitochondria-Targeted Doxorubicin: A New Therapeutic Strategy Against Doxorubicin-Resistant Osteosarcoma. Mol Cancer Ther (2016) 15:2640–52.
58. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nat (London) (2015) 520:553–7.
59. Durut N, Mittelsten Scheid O. The Role of Noncoding RNAs in Double-Strand Break Repair. Front Plant Sci (2019) 10:1155–5.
60. Statello L, Guo C, Chen L, Huarte M. Gene Regulation by Long non-Coding RNAs and its Biological Functions. Nat Rev Mol Cell Biol (2021) 2020:22:96–118.
61. Chowdhury D, Choi YE, Brault ME. DNA DAMAGE - OPINION Charity Begins at Home: non-Coding RNA Functions in DNA Repair. Nat Rev Mol Cell Biol (2013) 14:181–9.
62. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA Repair Targeted Therapy: The Past or Future of Cancer Treatment? Pharmacol Ther (2016) 160:65–83.
63. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA Repair Pathways as Targets for Cancer Therapy. Nat Rev Cancer (2008) 8:193–204.
64. Begg AC, Stewart FA, Vens C. Strategies to Improve Radiotherapy With Targeted Drugs. Nat Rev Cancer (2011) 11:239–53.
66. Barnett GC, West CML, Dunning AM, Elliott RM, Coles CE, Pharoah PDP, et al. Normal Tissue Reactions to Radiotherapy: Towards Tailoring Treatment Dose by Genotype. Nat Rev Cancer (2009) 9:134–42.
67. Jeggo P, Lavin MF. Cellular Radiosensitivity: How Much Better do We Understand it? Int J Radiat Biol (2009) 85:1061–81.
68. Myers SH, Ortega JA, Cavalli A. Synthetic Lethality Through the Lens of Medicinal Chemistry. J Med Chem (2020) 63:14151–83. doi: 10.1021/acs.jmedchem.0c00766
69. Suwen H, Zi H, Frédéric L, Carmen G, Xiang-Yang Y, Tian X. Small Molecule DNA-PK Inhibitors as Potential Cancer Therapy: A Patent Review (2010–Present). Expert Opin Ther Pat (2021) 31:435–52. doi: 10.1080/13543776.2021.1866540
70. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discovery (2017) 7:20–37. doi: 10.1158/2159-8290.CD-16-0860
71. Woodbine L, Gennery AR, Jeggo PA. The Clinical Impact of Deficiency in DNA non-Homologous End-Joining. DNA Repair (2014) 16:84–96. doi: 10.1016/j.dnarep.2014.02.011
72. Van der Burg M, IJspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs Mutation in a Radiosensitive T-B- SCID Patient Inhibits Artemis Activation and Nonhomologous End-Joining. J Clin Invest (2009) 119:91–8. doi: 10.1172/JCI37141
73. Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche MC, Sanal O, et al. Cernunnos, a Novel Nonhomologous End-Joining Factor, is Mutated in Human Immunodeficiency With Microcephaly. Cell (2006) 124:287–99. doi: 10.1016/j.cell.2005.12.030
74. Marangoni E, Foray N, O’Driscoll M, Douc-Rasy S, Bernier J, Bourhis J, et al. A Ku80 Fragment With Dominant Negative Activity Imparts a Radiosensitive Phenotype to CHO-K1 Cells. Nucleic Acids Res (2000) 28:4778–82. doi: 10.1093/nar/28.23.4778
75. Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a Defect in DNA Ligase IV in a Radiosensitive Leukaemia Patient. Curr Biol (1999) 9:699–702. doi: 10.1016/s0960-9822(99)80311-x
76. Downs JA, Jackson SP. A Means to a DNA End: The Many Roles of Ku. Nat Rev Mol Cell Biol (2004) 5:367–78.
77. Walker JR, Corpina RA, Goldberg J. Structure of the Ku Heterodimer Bound to DNA and its Implications for Double-Strand Break Repair. Nature (2001) 412:607–14.
78. Bennett SM, Woods DS, Pawelczak KS, Turchi JJ. Multiple Protein-Protein Interactions Within the DNA-PK Complex are Mediated by the C-Terminus of Ku 80. Int J Biochem Mol Biol (2012) 3:36–45.
79. Fell VL, Schild-Poulter C. The Ku Heterodimer: Function in DNA Repair and Beyond. Mutat Res (2015) 763:15–29.
80. Kragelund BB, Weterings E, Hartmann-Petersen R, Keijzers G. The Ku70/80 Ring in Non Homologous End-Joining: Easy to Slip on Hard to Remove. Front Biosci (Landmark Ed) (2016) 21:514–27.
81. Osipovich O, Duhe RJ, Hasty P, Durum SK, Muegge K. Defining Functional Domains of Ku80: DNA End Binding and Survival After Radiation. Biochem Biophys Res Commun (1999) 261:802–7.
82. Baptistella AR, Landemberger MC, Dias MVS, Giudice FS, Rodrigues BR, Da Silva PPCE. Rab5C Enhances Resistance to Ionizing Radiation in Rectal Cancer. J Mol Med (Berl) (2019) 97:855–69.
83. Pucci S, Polidoro C, Joubert A, Mastrangeli F, Tolu B, Benassi M. Ku70, Ku80, and Sclusterin: A Cluster of Predicting Factors for Response to Neoadjuvant Chemoradiation Therapy in Patients With Locally Advanced Rectal Cancer. Int J Radiat Oncol Biol Phys (2017) 97:381–8.
84. Beskow C, Skikuniene J, Holgersson A, Nilsson B, Lewensohn R, Kanter L, et al. Radioresistant Cervical Cancer Shows Upregulation of the NHEJ Proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer (2009) 101:816–221.
85. Li YH, Wang X, Pan Y, Lee DH, Chowdhury D, Kimmelman AC. Inhibition of non-Homologous End Joining Repair Impairs Pancreatic Cancer Growth and Enhances Radiation Response. PloS One (2012) 7:e39588.
86. Kim SH, Kim D, Han JS, Jeong CS, Chung BS, Kang CD. Ku Autoantigen Affects the Susceptibility to Anticancer Drugs. Cancer Res (1999) 59:4012–7.
87. Zahid S, Seif El Dahan M, Iehl F, Fernandez-Varela P, Le Du MH, Ropars V, et al. The Multifaceted Roles of Ku70/80. Int J Mol Sci (2021) 22:1–23. doi: 10.3390/ijms22084134
88. Weterings E, Gallegos AC, Dominick LN, Cooke LS, Bartels TN, Vagner J, et al. A Novel Small Molecule Inhibitor of the DNA Repair Protein Ku70/80. DNA Repair (Amst) (2016) 43:98–106. doi: 10.1016/j.dnarep.2016.03.014
89. Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ. Modulating DNA Repair Pathways to Improve Precision Genome Engineering. ACS Chem Biol (2018) 13:389–96. doi: 10.1021/acschembio.7b00777
90. Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Mendoza-Munoz P, Vernon TL, Hanakahi LA, et al. Discovery and Development of Novel DNA-PK Inhibitors by Targeting the Unique Ku-DNA Interaction. Nucleic Acids Res (2020) 48:11536–50. doi: 10.1093/nar/gkaa934
91. Pannunzio NR, Watanabe G, Lieber MR. Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J Bio Chem (2018) 293:10512–23.
92. Löbrich M, Jeggo P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem Sci (2017) 9:690–701. doi: 10.1016/j.tibs.2017.06.011
93. Gu J, Li S, Zhang X, Wang LC, Niewolik D, Schwarz K, et al. DNA-PKcs Regulates a Single-Stranded DNA Endonuclease Activity of Artemis. DNA Repair (Amst) (2010) 9:429–37.
94. Moshous D, Callebaut I, De Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a Novel DNA Double-Strand Break Repair/V(D)J Recombination Protein, is Mutated in Human Severe Combined Immune Deficiency. Cell (2001) 105:177–86.
95. Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, et al. Defective DNA Repair and Increased Genomic Instability in Artemis-Deficient Murine Cells. J Exp Med (2003) 197:553–65. doi: 10.1084/jem.20021891
96. Ochi T, Gu X, Blundell TL. Structure of the Catalytic Region of DNA Ligase IV in Complex With an Artemis Fragment Sheds Light on Double-Strand Break Repair. Structure (2013) 21:672–9. doi: 10.1016/j.str.2013.02.014
97. Malu S, De Ioannes P, Kozlov M, Greene M, Francis D, Hanna M, et al. Artemis C-Terminal Region Facilitates V(D)J Recombination Through its Interactions With DNA Ligase IV and DNA-PKcs. J Exp Med (2012) 209:955–63. doi: 10.1084/jem.20111437
98. Povirk LF, Zhou T, Zhou R, Cowan MJ, Yannone SM. Processing of 3’-Phosphoglycolate-Terminated DNA Double Strand Breaks by Artemis Nuclease. J Biol Chem (2007) 282:3547–58.
99. Riballo E, Kühne M, Rief M, Doherty A, Smith GCM, Recio MJ. A Pathway of Double-Strand Break Rejoining Dependent Upon ATM, Artemis, and Proteins Locating to γ-H2AX Foci. Mol Cell (2004) 16:715–24.
100. Kurosawa A, Koyama H, Takayama S, Miki K, Ayusawa D, Fujii M. The Requirement of Artemis in Double-Strand Break Repair Depends on the Type of DNA Damage. DNA Cell Biol (2008) 27:55–61.
101. Yosaatmadja Y, Baddock HT, Newman JA, Bielinski M, Gavard AE, Mukhopadhyay SMM, et al. Structural and Mechanistic Insights Into the Artemis Endonuclease and Strategies for its Inhibition. Nucleic Acids Res (2021) 49:9310–26. doi: 10.1093/nar/gkab693
102. Karim MF, Liu S, Laciak AR, Volk L, Koszelak-Rosenblum M, Lieber MR, et al. Structural Analysis of the Catalytic Domain of Artemis Endonuclease/SNM1C Reveals Distinct Structural Features. J Biol Chem (2020) 295:12368–77. doi: 10.1074/jbc.RA120.014136
103. Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, et al. Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-Induced Precise Gene Editing in Mammalian Cells. Nat Biotechnol (2015) 32:543–8. doi: 10.1038/nbt.3198
104. Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the Efficiency of Precise Genome Editing With CRISPR-Cas9 by Inhibition of Nonhomologous End Joining. Nat Biotechnol (2015) 33:538–42. doi: 10.1038/nbt.3190
105. Adachi N, Ishino T, Ishii Y, Takeda S, Koyama H. DNA Ligase IV-Deficient Cells are More Resistant to Ionizing Radiation in the Absence of Ku70: Implications for DNA Double-Strand Break Repair. Proc Natl Acad Sci USA (2001) 98:12109–13. doi: 10.1073/pnas.201271098
106. Davis AJ, Chen DJ. DNA Double Strand Break Repair via non-Homologous End-Joining. Transl Cancer Res (2013) 2:130–43.
107. Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, et al. Rational Design of Human DNA Ligase Inhibitors That Target Cellular DNA Replication and Repair. Cancer Res (2008) 68:3169–77. doi: 10.1158/0008-5472.CAN-07-6636
108. Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, et al. An Inhibitor of Nonhomologous End-Joining Abrogates Double-Strand Break Repair and Impedes Cancer Progression. Cell (2012) 151:1474–87.
109. Greco GE, Matsumoto Y, Brooks RC, Lu Z, Lieber MR, Tomkinson AE. SCR7 is Neither a Selective Nor a Potent Inhibitor of Human DNA Ligase Iv. DNA Repair (2016) 43:18–23.
110. Vartak SV, Swarup HA, Gopalakrishnan V, Gopinatha VK, Ropars V, Nambiar M, et al. Autocyclized and Oxidized Forms of SCR7 Induce Cancer Cell Death by Inhibiting Nonhomologous DNA End Joining in a Ligase IV Dependent Manner. FEBS J (2018) 285:3959–76.
111. Ray U, Raul SK, Gopinatha VK, Ghosh D, Rangappa KS, Mantelingu K, et al. Identification and Characterization of Novel SCR7-Based Small-Molecule Inhibitor of DNA End-Joining, SCR130 and its Relevance in Cancer Therapeutics. Mol Carcinog (2020) 59:618–28.
112. Ray U, Raghavan SC. Modulation of DNA Double-Strand Break Repair as a Strategy to Improve Precise Genome Editing. Oncogene (2020) 39:6393–405. doi: 10.1038/s41388-020-01445-2
113. Koike M, Yutoku Y, Koike A. Establishment of Hamster Cell Lines With EGFP-Tagged Human XRCC4 and Protection From Low-Dose X-Ray Radiation. J Vet Med Sci (2012) 74:1269–75. doi: 10.1292/jvms.12-0112
114. Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, et al. DNA Repair. PAXX, a Paralog of XRCC4 and XLF, Interacts With Ku to Promote DNA Double-Strand Break Repair. Science (2015) 347:185–8.
115. Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku Heterodimer With the DNA Ligase IV/Xrcc4 Complex and its Regulation by DNA-Pk. DNA Repair (2007) 6:712–22.
116. Sun MF, Chen HY, Tsai FJ, Lui SH, Chen CY, Chen CY. Search for Novel Remedies to Augment Radiation Resistance of Inhabitants of Fukushima and Chernobyl Disasters: Identifying DNA Repair Protein XRCC4 Inhibitors. J Biomol Struct Dyn (2011) 29:325–37. doi: 10.1080/07391102.2011.10507388
117. Liu F, Fan Z, Song N, Han M, Yan M, Guo LH, et al. XRCC4, Which is Inhibited by PFDA, Regulates DNA Damage Repair and Cell Chemosensitivity. J Cell Biochem (2019) 120:12665–76. doi: 10.1002/jcb.28534
118. Jeggo PA, Löbrich M. How Cancer Cells Hijack DNA Double-Strand Break Repair Pathways to Gain Genomic Instability. Biochem J (2015) 471:1–11. doi: 10.1042/BJ20150582
119. Zhao F, Kim W, Kloeber JA, Lou Z. DNA End Resection and its Role in DNA Replication and DSB Repair Choice in Mammalian Cells. Exp Mol Med (2020) 52:1705–14. doi: 10.1038/s12276-020-00519-1
120. Kopa P, Macieja A, Galita G, Witczak ZJ, Poplawski T. DNA Double Strand Breaks Repair Inhibitors: Relevance as Potential New Anticancer Therapeutics. Curr Med Chem (2019) 26:1483–93. doi: 10.2174/0929867325666180214113154
121. Lee JH, Paull TT. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science (2005) 308:551–4. doi: 10.1126/science.1108297
122. Zhang J, Zou L. Alternative Lengthening of Telomeres: From Molecular Mechanisms to Therapeutic Outlooks. Cell Biosci (2020) 10:30–0.
123. Reddel RR, Cesare AJ. Alternative Lengthening of Telomeres: Models, Mechanisms and Implications. Nat Rev Genet (2010) 11:319–30.
124. Nelson CB, Alturki TM, Luxton JJ, Taylor LE, Maranon DG, Muraki K. Telomeric Double Strand Breaks in G1 Human Cells Facilitate Formation of 5′ C-Rich Overhangs and Recruitment of TERRA. Front Genet (2021) 12:644803–3.
125. Wang YY, Hung AC, Lo S, Hsieh YC, Yuan SF. MRE11 as a Molecular Signature and Therapeutic Target for Cancer Treatment With Radiotherapy. Cancer Lett (2021) 28:514:1–11. doi: 10.1016/j.canlet.2021.05.013
126. Bian L, Meng Y, Zhang M, Li D. MRE11-RAD50-NBS1 Complex Alterations and DNA Damage Response: Implications for Cancer Treatment. Mol Cancer (2019) 18:169. doi: 10.1186/s12943-019-1100-5
127. Wang J, Xu WH, Wei Y, Zhu Y, Qin XJ, Zhang HL. Elevated MRE11 Expression Associated With Progression and Poor Outcome in Prostate Cancer. J Cancer (2019) 10:4333–40. doi: 10.7150/jca.31454
128. Cheng L, Wu Q, Huang Z, Guryanova OA, Huang Q, Shou W, et al. L1CAM Regulates DNA Damage Checkpoint Response of Glioblastoma Stem Cells Through Nbs1. EMBO J (2011) 30:800–13.
129. Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11-Rad50-Nbs1 Complex. Nat Chem Biol (2008) 4:119–25. doi: 10.1038/nchembio.63
130. Jividen K, Kedzierska KZ, Yang CS, Szlachta K, Ratan A, Paschal BM. Genomic Analysis of DNA Repair Genes and Androgen Signaling in Prostate Cancer. BMC Cancer (2018) 18:960. doi: 10.1186/s12885-018-4848-x
131. Petroni M, Sardina F, Infante P, Bartolazzi A, Locatelli E, Fabretti F, et al. MRE11 Inhibition Highlights a Replication Stress-Dependent Vulnerability of MYCN-Driven Tumors. Cell Death Dis (2018) 9:895. doi: 10.1038/s41419-018-0924-z
132. Berte N, Piée-Staffa A, Piecha N, Wang M, Borgmann K, Kaina B, et al. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated With Alkylating Drugs. Mol Cancer Ther (2016) 15:2665–78. doi: 10.1158/1535-7163.MCT-16-0176
133. Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, et al. DNA Double-Strand Break Repair Pathway Choice is Directed by Distinct MRE11 Nuclease Activities. Mol Cell (2014) 53:7–18. doi: 10.1016/j.molcel.2013.11.003
134. Par S, Vaides S, VanderVere-Carozza PS, Pawelczak KS, Stewart J, Turchi JJ. OB-Folds and Genome Maintenance: Targeting Protein-DNA Interactions for Cancer Therapy. Cancers (Basel) (2021) 13:3346. doi: 10.3390/cancers13133346
135. Dueva R, Iliakis G. Replication Protein A: A Multifunctional Protein With Roles in DNA Replication, Repair and Beyond. NAR Cancer (2020) 2:1–24. doi: 10.1093/narcan/zcaa022
136. Bhat KP, Cortez D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat Struct Mol Biol (2018) 25:446–53. doi: 10.1038/s41594-018-0075-z
137. Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Turchi JJ. Targeting the Nucleotide Excision Repair Pathway for Therapeutic Applications. In: Kelley M, editor. DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications. Amsterdam, The Netherlands: Elsevier Academic Press (2016). p. 135–50.
138. Maréchal A, Zou L. RPA-Coated Single-Stranded DNA as a Platform for Post-Translational Modifications in the DNA Damage Response. Cell Res (2015) 25:9–23. doi: 10.1038/cr.2014.147
139. Jekimovs C, Bolderson E, Suraweera A, Adams M, O’Byrne KJ, Richard DJ. Chemotherapeutic Compounds Targeting the DNA Double-Strand Break Repair Pathways: The Good, the Bad, and the Promising. Front Oncol (2014) 4:86. doi: 10.3389/fonc.2014.00086
140. Planchard D, Domont J, Taranchon E, Monnet I, Tredaniel J, Caliandro R, et al. The NER Proteins are Differentially Expressed in Ever Smokers and in Never Smokers With Lung Adenocarcinoma. Ann Oncol (2009) 20:1257–63. doi: 10.1093/annonc/mdn785
141. Fan J, Pavletich NP. Structure and Conformational Change of a Replication Protein A Heterotrimer Bound to ssDNA. Genes Dev (2012) 26:2337–47. doi: 10.1101/gad.194787.112
142. Glanzer JG, Liu S, Oakley GG. Small Molecule Inhibitor of the RPA70 N-Terminal Protein Interaction Domain Discovered Using in Silico and In Vitro Methods. Bioorg Med Chem (2011) 19:2589–95. doi: 10.1016/j.bmc.2011.03.012
143. Glanzer JG, Carnes KA, Soto P, Liu S, Parkhurst LJ, Oakley GG. A Small Molecule Directly Inhibits the P53 Transactivation Domain From Binding to Replication Protein a. Nucleic Acids Res (2013) 41:2047–59. doi: 10.1093/nar/gks1291
144. Glanzer JG, Liu S, Wang L, Mosel A, Peng A, Oakley GG. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res (2014) 74:5165–72. doi: 10.1158/0008-5472.CAN-14-0306
145. Patrone JD, Waterson AG, Fesik SW. Recent Advancements in the Discovery of Protein-Protein Interaction Inhibitors of Replication Protein a. Medchemcomm (2016) 8:259–67. doi: 10.1039/c6md00460a
146. Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel Irreversible Small Molecule Inhibitors of Replication Protein A Display Single-Agent Activity and Synergize With Cisplatin. Mol Cancer Ther (2011) 10:1796–806. doi: 10.1158/1535-7163.MCT-11-0303
147. Shuck SC, Turchi JJ. Targeted Inhibition of Replication Protein A Reveals Cytotoxic Activity, Synergy With Chemotherapeutic DNA-Damaging Agents, and Insight Into Cellular Function. Cancer Res (2010) 70:3189–98. doi: 10.1158/0008-5472.CAN-09-3422
148. Andrews BJ, Turchi JJ. Development of a High-Throughput Screen for Inhibitors of Replication Protein A and its Role in Nucleotide Excision Repair. Mol Cancer Ther (2004) 3:385–91.
149. Mishra AK, Dormi SS, Turchi AM, Woods DS, Turchi JJ. Chemical Inhibitor Targeting the Replication Protein A-DNA Interaction Increases the Efficacy of Pt-Based Chemotherapy in Lung and Ovarian Cancer. Biochem Pharmacol (2015) 93:25–33. doi: 10.1016/j.bcp.2014.10.013
150. Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Vernon TL, Jordan MR, Turchi JJ. Structure-Guided Optimization of Replication Protein A (RPA)-DNA Interaction Inhibitors. ACS Med Chem Lett (2020) 11:1118–24. doi: 10.1021/acsmedchemlett.9b00440
151. VanderVere-Carozza PS, Gavande NS, Jalal SI, Pollok KE, Ekinci KE, Heyza J, et al. In Vivo Targeting Replication Protein A for Cancer Therapy. Front Oncol (2022) 12:1–13. doi: 10.3389/fonc.2022.826655. Article in Press.
152. Laurini E, Marson D, Fermeglia A, Aulic S, Fermeglia M, Pricl S. Role of Rad51 and DNA Repair in Cancer: A Molecular Perspective. Pharmacol Ther (2020) 208:107492. doi: 10.1016/j.pharmthera.2020.107492
153. Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, et al. RAD51 Interconnects Between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res (2017) 45:4590–605. doi: 10.1093/nar/gkx126
154. Grundy MK, Buckanovich RJ, Bernstein KA. Regulation and Pharmacological Targeting of RAD51 in Cancer. NAR Cancer (2020) 2:1–14. doi: 10.1093/narcan/zcaa024
155. Makino E, Fröhlich LM, Sinnberg T, Kosnopfel C, Sauer B, Garbe C, et al. Targeting Rad51 as a Strategy for the Treatment of Melanoma Cells Resistant to MAPK Pathway Inhibition. Cell Death Dis (2020) 11:581. doi: 10.1038/s41419-020-2702-y
156. Ishida T, Takizawa Y, Kainuma T, Inoue J, Mikawa T, Shibata T, et al. DIDS, a Chemical Compound That Inhibits RAD51-Mediated Homologous Pairing and Strand Exchange. Nucleic Acids Res (2009) 37:3367–76. doi: 10.1093/nar/gkp200
157. Takaku M, Kainuma T, Ishida-Takaku T, Ishigami S, Suzuki H, Tashiro S, et al. Halenaquinone, a Chemical Compound That Specifically Inhibits the Secondary DNA Binding of RAD51. Genes Cells (2011) 16:427–36. doi: 10.1111/j.1365-2443.2011.01494.x
158. Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, et al. RI-1: A Chemical Inhibitor of RAD51 That Disrupts Homologous Recombination in Human Cells. Nucleic Acids Res (2012) 40:7347–57. doi: 10.1093/nar/gks353
159. Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, et al. An Optimized RAD51 Inhibitor That Disrupts Homologous Recombination Without Requiring Michael Acceptor Reactivity. J Med Chem (2013) 56(1):254–63. doi: 10.1021/jm301565b
160. Jayathilaka K, Sheridan SD, Bold TD, Bochenska K, Logan HL, Weichselbaum RR, et al. A Chemical Compound That Stimulates the Human Homologous Recombination Protein Rad51. Proc Natl Acad Sci USA (2008) 105:15848–53. doi: 10.1073/pnas.0808046105
161. Mason JM, Logan HL, Budke B, Wu M, Pawlowski M, Weichselbaum RR, et al. The RAD51-Stimulatory Compound RS-1 can Exploit the RAD51 Overexpression That Exists in Cancer Cells and Tumors. Cancer Res (2014) 74:3546–55. doi: 10.1158/0008-5472.CAN-13-3220
162. Mason JM, Dusad K, Wright WD, Grubb J, Budke B, Heyer WD, et al. RAD54 Family Translocases Counter Genotoxic Effects of RAD51 in Human Tumor Cells. Nucleic Acids Res (2015) 43:3180–96. doi: 10.1093/nar/gkv175
163. Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 Enhances CRISPR/Cas9- and TALEN-Mediated Knock-In Efficiency. Nat Commun (2016) 7:10548. doi: 10.1038/ncomms10548
164. Zhang JP, Li XL, Li GH, Chen W, Arakaki C, Botimer GD, et al. Efficient Precise Knocking With a Double Cut HDR Donor After CRISPR/Cas9-Mediated Double-Stranded DNA Cleavage. Genome Biol (2017) 18:35. doi: 10.1186/s13059-017-1164-8
165. Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of Specific Inhibitors of Human RAD51 Recombinase Using High-Throughput Screening. ACS Chem Biol (2011) 6:628–35. doi: 10.1021/cb100428c
166. Alagpulinsa DA, Ayyadevara S, Shmookler Reis RJ. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front Oncol (2014) 4:289. doi: 10.3389/fonc.2014.00289
167. Shkundina IS, Gall AA, Dick A, Cocklin S, Mazin AV. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes (Basel) (2021) 12:920. doi: 10.3390/genes12060920
168. Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, et al. A Novel Small Molecule RAD51 Inactivator Overcomes Imatinib-Resistance in Chronic Myeloid Leukaemia. EMBO Mol Med (2013) 5:353–65. doi: 10.1002/emmm.201201760
169. Zhu J, Chen H, Guo XE, Qiu XL, Hu CM, Chamberlin AR, et al. Synthesis, Molecular Modeling, and Biological Evaluation of Novel RAD51 Inhibitors. Eur J Med Chem (2015) 96:196–208. doi: 10.1016/j.ejmech.2015.04.021
170. Roberti M, Schipani F, Bagnolini G, Milano D, Giacomini E, Falchi F, et al. Rad51/BRCA2 Disruptors Inhibit Homologous Recombination and Synergize With Olaparib in Pancreatic Cancer Cells. Eur J Med Chem (2019) 165:80–92. doi: 10.1016/j.ejmech.2019.01.008
171. Falchi F, Giacomini E, Masini T, Boutard N, Di Ianni L, Manerba M, et al. Synthetic Lethality Triggered by Combining Olaparib With BRCA2-Rad51 Disruptors. ACS Chem Biol (2017) 12:2491–7. doi: 10.1021/acschembio.7b00707
172. Bagnolini G, Milano D, Manerba M, Schipani F, Ortega JA, Gioia D, et al. Synthetic Lethality in Pancreatic Cancer: Discovery of a New RAD51-BRCA2 Small Molecule Disruptor That Inhibits Homologous Recombination and Synergizes With Olaparib. J Med Chem (2020) 63:2588–619. doi: 10.1021/acs.jmedchem.9b01526
173. Asan A, Skoko JJ, Woodcock CC, Wingert BM, Woodcock SR, Normolle D, et al. Electrophilic Fatty Acids Impair RAD51 Function and Potentiate the Effects of DNA-Damaging Agents on Growth of Triple-Negative Breast Cells. J Biol Chem (2019) 294:397–404. doi: 10.1074/jbc.AC118.005899
174. Maclay T, Day M, Mills K. Abstract 363: CYT01B, a Novel RAD51 Inhibitor, Acts Synergistically With PARP Inhibitors. Cancer Res (2019) 79:363.
175. Lynch RC, Bendell JC, Advani RJ, Falchook GS, Munster PN, Patel MR. First-In-Human Phase I/II Study of CYT-0851, a First-In-Class Inhibitor of RAD51-Mediated Homologous Recombination in Patients With Advanced Solid and Hematologic Cancers. J Clin Oncol (2021) 39:3006–6.
176. Shinohara A, Shinohara M, Ohta T, Matsuda S, Ogawa T. Rad52 Forms Ring Structures and Co-Operates With RPA in Single-Strand DNA Annealing. Genes Cells (1998) 3:145–56. doi: 10.1046/j.1365-2443.1998.00176.x
177. Rossi MJ, DiDomenico SF, Patel M, Mazin AV. RAD52: Paradigm of Synthetic Lethality and New Developments. Front Genet (2021) 12:780293. doi: 10.3389/fgene.2021.780293
178. Nogueira A, Fernandes M, Catarino R, Medeiros R. RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy. Cancers (Basel) (2019) 11:1622. doi: 10.3390/cancers11111622
179. Toma M, Sullivan-Reed K, Śliwiński T, Skorski T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers (Basel) (2019) 11:1561. doi: 10.3390/cancers11101561
180. Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, et al. Rad52 Prevents Excessive Replication Fork Reversal and Protects From Nascent Strand Degradation. Nat Commun (2019) 10:1412. doi: 10.1038/s41467-019-09196-9
181. Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC. Rad51 Protein Controls Rad52-Mediated DNA Annealing. J Biol Chem (2008) 283:14883–92. doi: 10.1074/jbc.M801097200
182. Velic D, Couturier AM, Ferreira MT, Rodrigue A, Poirier GG, Fleury F, et al. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules (2015) 5:3204–59. doi: 10.3390/biom5043204
183. Kagawa W, Kagawa A, Saito K, Ikawa S, Shibata T, Kurumizaka H, et al. Identification of a Second DNA Binding Site in the Human Rad52 Protein. J Biol Chem (2008) 283:24264–73. doi: 10.1074/jbc.M802204200
184. Lok BH, Carley AC, Tchang B, Powell SN. RAD52 Inactivation is Synthetically Lethal With Deficiencies in BRCA1 and PALB2 in Addition to BRCA2 Through RAD51-Mediated Homologous Recombination. Oncogene (2013) 32:3552–8. doi: 10.1038/onc.2012.391
185. Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, et al. Rad52 Inactivation is Synthetically Lethal With BRCA2 Deficiency. Proc Natl Acad Sci USA (2011) 108:686–91. doi: 10.1073/pnas.1010959107
186. Chun J, Buechelmaier ES, Powell SN. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol Cell Biol (2013) 33:387–95. doi: 10.1128/MCB.00465-12
187. Chandramouly G, McDevitt S, Sullivan K, Kent T, Luz A, Glickman JF, et al. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem Biol (2015) 22:1491–504. doi: 10.1016/j.chembiol.2015.10.003
188. Sullivan K, Cramer-Morales K, McElroy DL, Ostrov DA, Haas K, Childers W, et al. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PloS One (2016) 11:e0147230. doi: 10.1371/journal.pone.0147230
189. Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, et al. Targeting BRCA1- and BRCA2-Deficient Cells With RAD52 Small Molecule Inhibitors. Nucleic Acids Res (2016) 44:4189–99. doi: 10.1093/nar/gkw087
190. Li J, Yang Q, Zhang Y, Huang K, Sun R, Zhao Q. Compound F779-0434 Causes Synthetic Lethality in BRCA2-Deficient Cancer Cells by Disrupting RAD52–SsDNA Association. RSC Adv (2018) 8:18859–69. doi: 10.1039/C8RA01919C
191. Yang Q, Li Y, Sun R, Li J. Identification of a RAD52 Inhibitor Inducing Synthetic Lethality in BRCA2-Deficient Cancer Cells. Front Pharmacol (2021) 12:637825. doi: 10.3389/fphar.2021.637825
192. Hengel SR, Malacaria E, Folly da Silva Constantino L, Bain FE, Diaz A, Koch BG, et al. Small-Molecule Inhibitors Identify the RAD52-ssDNA Interaction as Critical for Recovery From Replication Stress and for Survival of BRCA2 Deficient Cells. Elife (2016) 5:e14740. doi: 10.7554/eLife.14740
193. Faridounnia M, Folkers GE, Boelens R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules (2018) 23:3205. doi: 10.3390/molecules23123205
194. Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, et al. ERCC1-XPF Endonuclease Facilitates DNA Double-Strand Break Repair. Mol Cell Biol (2008) 28:5082–92. doi: 10.1128/MCB.00293-08
195. McNeil EM, Melton DW. DNA Repair Endonuclease ERCC1-XPF as a Novel Therapeutic Target to Overcome Chemoresistance in Cancer Therapy. Nucleic Acids Res (2012) 40:9990–10004. doi: 10.1093/nar/gks818
196. Bowden NA. Nucleotide Excision Repair: Why is it Not Used to Predict Response to Platinum-Based Chemotherapy? Cancer Lett (2014) 346:163–71. doi: 10.1016/j.canlet.2014.01.005
197. Song L, Ritchie AM, McNeil EM, Li W, Melton DW. Identification of DNA Repair Gene Ercc1 as a Novel Target in Melanoma. Pigment Cell Melanoma Res (2011) 24:966–71. doi: 10.1111/j.1755-148X.2011.00882.x
198. Tsodikov OV, Enzlin JH, Schärer OD, Ellenberger T. Crystal Structure and DNA Binding Functions of ERCC1, a Subunit of the DNA Structure-Specific Endonuclease XPF-Ercc1. Proc Natl Acad Sci USA (2005) 102:11236–41. doi: 10.1073/pnas.0504341102
199. Tripsianes K, Folkers G, Ab E, Das D, Odijk H, Jaspers NG, et al. The Structure of the Human ERCC1/XPF Interaction Domains Reveals a Complementary Role for the Two Proteins in Nucleotide Excision Repair. Structure (2005) 13:1849–58. doi: 10.1016/j.str.2005.08.014
200. Jordheim LP, Barakat KH, Heinrich-Balard L, Matera EL, Cros-Perrial E, Bouledrak K, et al. Small Molecule Inhibitors of ERCC1-XPF Protein-Protein Interaction Synergize Alkylating Agents in Cancer Cells. Mol Pharmacol (2013) 84:12–24. doi: 10.1124/mol.112.082347
201. Elmenoufy AH, Gentile F, Jay D, Karimi-Busheri F, Yang X, Soueidan OM, et al. Targeting DNA Repair in Tumor Cells via Inhibition of ERCC1-XPF. J Med Chem (2019) 62:7684–96. doi: 10.1021/acs.jmedchem.9b00326
202. Gentile F, Elmenoufy AH, Ciniero G, Jay D, Karimi-Busheri F, Barakat KH, et al. Computer-Aided Drug Design of Small Molecule Inhibitors of the ERCC1-XPF Protein-Protein Interaction. Chem Biol Drug Des (2020) 95:460–71. doi: 10.1111/cbdd.13660
203. Elmenoufy AH, Gentile F, Jay D, Karimi-Busheri F, Yang X, Soueidan OM, et al. Design, Synthesis and In Vitro Cell-Free/Cell-Based Biological Evaluations of Novel ERCC1-XPF Inhibitors Targeting DNA Repair Pathway. Eur J Med Chem (2020) 204:112658. doi: 10.1016/j.ejmech.2020.112658
204. McNeil EM, Astell KR, Ritchie AM, Shave S, Houston DR, Bakrania P, et al. Inhibition of the ERCC1-XPF Structure-Specific Endonuclease to Overcome Cancer Chemoresistance. DNA Repair (Amst) (2015) 31:19–28. doi: 10.1016/j.dnarep.2015.04.002
205. Chapman TM, Gillen KJ, Wallace C, Lee MT, Bakrania P, Khurana P, et al. Catechols and 3-Hydroxypyridones as Inhibitors of the DNA Repair Complex ERCC1-XPF. Bioorg Med Chem Lett (2015) 19:4097–103. doi: 10.1016/j.bmcl.2015.08.031
206. Chapman TM, Wallace C, Gillen KJ, Bakrania P, Khurana P, Coombs PJ, et al. N-Hydroxyimides and Hydroxypyrimidinones as Inhibitors of the DNA Repair Complex ERCC1-XPF. Bioorg Med Chem Lett (2015) 25:4104–8. doi: 10.1016/j.bmcl.2015.08.024
207. Arora S, Heyza J, Zhang H, Kalman-Maltese V, Tillison K, Floyd AM, et al. Identification of Small Molecule Inhibitors of ERCC1-XPF That Inhibit DNA Repair and Potentiate Cisplatin Efficacy in Cancer Cells. Oncotarget (2016) 7:75104–17. doi: 10.18632/oncotarget.12072
208. Patel PS, Algouneh A, Hakem R. Exploiting Synthetic Lethality to Target BRCA1/2-Deficient Tumors: Where We Stand. Oncogene (2021) 40:3001–14. doi: 10.1038/s41388-021-01744-2
209. Carvajal-Garcia J, Cho JE, Carvajal-Garcia P, Feng W, Wood RD, Sekelsky J, et al. Mechanistic Basis for Microhomology Identification and Genome Scarring by Polymerase Theta. Proc Natl Acad Sci USA (2020) 117(15):8476–85. doi: 10.1073/pnas.1921791117
210. Seol JH, Shim EY, Lee SE. Microhomology-Mediated End Joining: Good, Bad and Ugly. Mutat Res (2018) 809:81–7. doi: 10.1016/j.mrfmmm.2017.07.002
211. Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of Microhomology-Mediated End-Joining Promoted by Human DNA Polymerase θ. Nat Struct Mol Biol (2015) 22:230–7. doi: 10.1038/nsmb.2961
212. Higgins GS, Boulton SJ. Beyond PARP-Polθ as an Anticancer Target. Science (2018) 359:1217–8. doi: 10.1126/science.aar5149
213. Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat Commun (2021) 12:3636. doi: 10.1038/s41467-021-23463-8
214. Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-Recombination-Deficient Tumours are Dependent on Polθ-Mediated Repair. Nature (2015) 518:258–62. doi: 10.1038/nature14184
215. Wood RD, Doublié S. DNA Polymerase θ (POLQ), Double-Strand Break Repair, and Cancer. DNA Repair (Amst) (2016) 44:22–32. doi: 10.1016/j.dnarep.2016.05.003
216. Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE, et al. Genetic Determinants of Cellular Addiction to DNA Polymerase Theta. Nat Commun (2019) 10:4286. doi: 10.1038/s41467-019-12234-1
217. Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A First-In-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat Cancer (2021) 2:598–610. doi: 10.1038/s43018-021-00203-x
218. Newman JA, Gileadi O. RecQ Helicases in DNA Repair and Cancer Targets. Essays Biochem (2020) 64:819–30. doi: 10.1042/EBC20200012
219. Kaiser S, Sauer F, Kisker C. The Structural and Functional Characterization of Human RecQ4 Reveals Insights Into its Helicase Mechanism. Nat Commun (2017) 8:15907. doi: 10.1038/ncomms15907
220. Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM Jr.. Inhibition of Helicase Activity by a Small Molecule Impairs Werner Syndrome Helicase (WRN) Function in the Cellular Response to DNA Damage or Replication Stress. Proc Natl Acad Sci U.S.A. (2011) 108:1525–30. doi: 10.1073/pnas.1006423108
221. Aggarwal M, Banerjee T, Sommers JA, Iannascoli C, Pichierri P, Shoemaker RH, et al. Werner Syndrome Helicase has a Critical Role in DNA Damage Responses in the Absence of a Functional Fanconi Anemia Pathway. Cancer Res (2013) 73(17):5497–507. doi: 10.1158/0008-5472.CAN-12-2975
222. Sommers JA, Kulikowicz T, Croteau DL, Dexheimer T, Dorjsuren D, Jadhav A, et al. A High-Throughput Screen to Identify Novel Small Molecule Inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PloS One (2019) 14(1):1–23. doi: 10.1371/journal.pone.0210525
223. Picco G, Cattaneo CM, van Vliet EJ, Crisafulli G, Rospo G, Consonni S. Werner Helicase Is a Synthetic-Lethal Vulnerability in Mismatch Repair-Deficient Colorectal Cancer Refractory to Targeted Therapies, Chemotherapy, and Immunotherapy. Cancer Discovery (2021) 11:1923–37. doi: 10.1158/2159-8290.CD-20-1508
224. Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N. WRN Helicase is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature (2019) 568:551–6. doi: 10.1038/s41586-019-1102-x
225. Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, et al. A Small Molecule Inhibitor of the BLM Helicase Modulates Chromosome Stability in Human Cells. Chem Biol (2013) 20:55–62. doi: 10.1016/j.chembiol.2012.10.016
226. Rosenthal AS, Dexheimer TS, Gileadi O, Nguyen GH, Chu WK, Hickson ID, et al. Synthesis and SAR Studies of 5-(Pyridin-4-Yl)-1,3,4-Thiadiazol-2-Amine Derivatives as Potent Inhibitors of Bloom Helicase. Bioorg Med Chem Lett (2013) 23:5660–6. doi: 10.1016/j.bmcl.2013.08.025
227. Yin QK, Wang CX, Wang YQ, Guo QL, Zhang ZL, Ou TM, et al. Discovery of Isaindigotone Derivatives as Novel Bloom’s Syndrome Protein (BLM) Helicase Inhibitors That Disrupt the BLM/DNA Interactions and Regulate the Homologous Recombination Repair. J Med Chem (2019) 62:3147–62. doi: 10.1021/acs.jmedchem.9b00083
228. Simon N, Bochman ML, Seguin S, Brodsky JL, Seibel WL, Schwacha A. Ciprofloxacin is an Inhibitor of the Mcm2-7 Replicative Helicase. Biosci Rep (2013) 33:783–95. doi: 10.1042/BSR20130083
229. Hsu EC, Shen M, Aslan M, Liu S, Kumar M, Garcia-Marques F, et al. MCM2-7 Complex is a Novel Druggable Target for Neuroendocrine Prostate Cancer. Sci Rep (2021) 11:13305. doi: 10.1038/s41598-021-92552-x
230. Alshahrani MY, Alshahrani KM, Tasleem M, Akeel A, Almeleebia TM, Ahmad I, et al. Computational Screening of Natural Compounds for Identification of Potential Anti-Cancer Agents Targeting MCM7 Protein. Molecules (2021) 26(19):5878. doi: 10.3390/molecules26195878
231. Lin CY, Wu HY, Hsu YL, Cheng TR, Liu JH, Huang RJ, et al. Suppression of Drug-Resistant Non-Small-Cell Lung Cancer With Inhibitors Targeting Minichromosomal Maintenance Protein. J Med Chem (2020) 63:3172–87. doi: 10.1021/acs.jmedchem.9b01783
232. O’Neil NJ, Bailey ML, Hieter P. Synthetic Lethality and Cancer. Nat Rev Genet (2017) 18:613–23. doi: 10.1038/nrg.2017.47
233. Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP Inhibitor Resistance: The Underlying Mechanisms and Clinical Implications. Mol Cancer (2020) 19:107. doi: 10.1186/s12943-020-01227-0
234. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct Target Ther (2020) 5:1. doi: 10.1038/s41392-019-0089-y
235. Liu M, Zhang W, Xin C, Yin J, Shang Y, Ai C, et al. Global Detection of DNA Repair Outcomes Induced by CRISPR-Cas9. Nucleic Acids Res (2021) 49(15):8732–42. doi: 10.1093/nar/gkab686
Keywords: DNA repair and DNA damage response (DDR), DNA double-strand break (DSB) repair, non-PIKKs inhibitors, non-homologous end joining (NHEJ), homology directed repair (HDR), single-strand annealing (SSA), polymerase theta-mediated end joining (TMEJ), synthetic lethality
Citation: Kelm JM, Samarbakhsh A, Pillai A, VanderVere-Carozza PS, Aruri H, Pandey DS, Pawelczak KS, Turchi JJ and Gavande NS (2022) Recent Advances in the Development of Non-PIKKs Targeting Small Molecule Inhibitors of DNA Double-Strand Break Repair. Front. Oncol. 12:850883. doi: 10.3389/fonc.2022.850883
Received: 08 January 2022; Accepted: 22 February 2022;
Published: 06 April 2022.
Edited by:
Maria Felice Brizzi, University of Turin, ItalyReviewed by:
Roopa Thapar, Rice University, United StatesNoritaka Adachi, Yokohama City University, Japan
Copyright © 2022 Kelm, Samarbakhsh, Pillai, VanderVere-Carozza, Aruri, Pandey, Pawelczak, Turchi and Gavande. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Navnath S. Gavande, bmdhdmFuZGVAd2F5bmUuZWR1; orcid.org/0000-0002-2413-0235
†These authors have contributed equally to this work