 Anna Mária Tőkés
Anna Mária Tőkés Stefan Vári-Kakas2
Stefan Vári-Kakas2 Janina Kulka
Janina Kulka- 12nd Department of Pathology, Semmelweis University Budapest, Budapest, Hungary
- 2Department of Computers and Information Technology, Faculty of Electrical Engineering and Information Technology, University of Oradea, Oradea, Romania
- 3Department of Biochemistry, Semmelweis University Budapest, Budapest, Hungary
Breast cancer is characterized by considerable metabolic diversity. A relatively high percentage of patients diagnosed with breast carcinoma do not respond to standard-of-care treatment, and alteration in metabolic pathways nowadays is considered one of the major mechanisms responsible for therapeutic resistance. Consequently, there is an emerging need to understand how metabolism shapes therapy response, therapy resistance and not ultimately to analyze the metabolic changes occurring after different treatment regimens. The most commonly applied neoadjuvant chemotherapy regimens in breast cancer contain an anthracycline (doxorubicin or epirubicin) in combination or sequentially administered with taxanes (paclitaxel or docetaxel). Despite several efforts, drug resistance is still frequent in many types of breast cancer, decreasing patients’ survival. Understanding how tumor cells rapidly rewire their signaling pathways to persist after neoadjuvant cancer treatment have to be analyzed in detail and in a more complex system to enable scientists to design novel treatment strategies that target different aspects of tumor cells and tumor resistance. Tumor heterogeneity, the rapidly changing environmental context, differences in nutrient use among different cell types, the cooperative or competitive relationships between cells pose additional challenges in profound analyzes of metabolic changes in different breast carcinoma subtypes and treatment protocols. Delineating the contribution of metabolic pathways to tumor differentiation, progression, and resistance to different drugs is also the focus of research. The present review discusses the changes in glucose and fatty acid pathways associated with the most frequently applied chemotherapeutic drugs in breast cancer, as well the underlying molecular mechanisms and corresponding novel therapeutic strategies.
Introduction
Neoadjuvant systemic therapy (NeST) is routinely used to treat breast cancer, with significant geographical variation in its use across different countries (1, 2). When introduced, NeST was used mostly for the treatment of patients with locally advanced or inoperable breast cancer aiming to reduce the tumor size, enabling post-therapy breast-conservation surgery. Currently, the role of NeST has expanded and includes different patient groups with early-stage, operable breast cancer with the following benefits: checking the sensitivity of a tumor to therapy in vivo, reduction of postoperative complications such as lymphoedema, prolonged disease-free survival, and improved cosmetic outcomes (1, 2).
Since the first clinical trial (The National Surgical Adjuvant Breast and Bowel Project B-18 trial) from 1997 that compared the prognostic value of neoadjuvant and adjuvant use of the same chemotherapy regimen, results of several other clinical trials have been published reporting no significant differences in the long-term outcomes for early-stage and locally advanced breast cancer with either approach (1, 3, 4). Meanwhile, several studies confirmed and validated pathological complete response (pCR) to NeST as a strong predictive factor of favourable long-term outcomes. In contrast, patients with a significant residual cancer burden have a higher risk of distant metastases (5, 6). In unselected breast cancer patient groups treated with NeST approximately 20-26% of the cases can achieve pCR but this rate is significantly higher in HER2-positive and triple-negative breast cancer (TNBC) subtypes achieving pCR rates of up to 50-60% (7–9). Based on these data it is clear that a fraction of TNBC and HER2-positive breast carcinomas does not achieve pCR and some hormone receptor (HR)-positive/HER2-negative patients respond to NeST. It is also clear that new factors like tumor heterogeneity, tumor metabolic diversity have to be considered for future oncological strategies in breast cancer treatment (10).
Personalized therapeutical approaches in both the neoadjuvant and adjuvant setting play more and more important role in breast cancer management. Adding immunotherapies [anti-PD-L1 (durvalumab) or anti-PD-1 (pembrolizumab)] or PARP-inhibitor (talazoparib) to standard chemotherapy may improve the rate of pCR in TNBC and women with germline BRCA mutations (11, 12).
The classification of most frequently used chemotherapeutic drugs in neoadjuvant and adjuvant settings - mostly DNA alkylating, antimicrotubule, immunologic and hormonal agents, and antimetabolites - are presented in Table 1. The applied neoadjuvant chemotherapy regimens in breast carcinoma cases may differ across countries. Most commonly, anthracycline (AC) (doxorubicin or epirubicin) is administrated in combination or sequentially with taxanes (paclitaxel or docetaxel). To enhance cytotoxicity AC is frequently used in combination with cyclophosphamide with or without fluoropyrimidines (13).
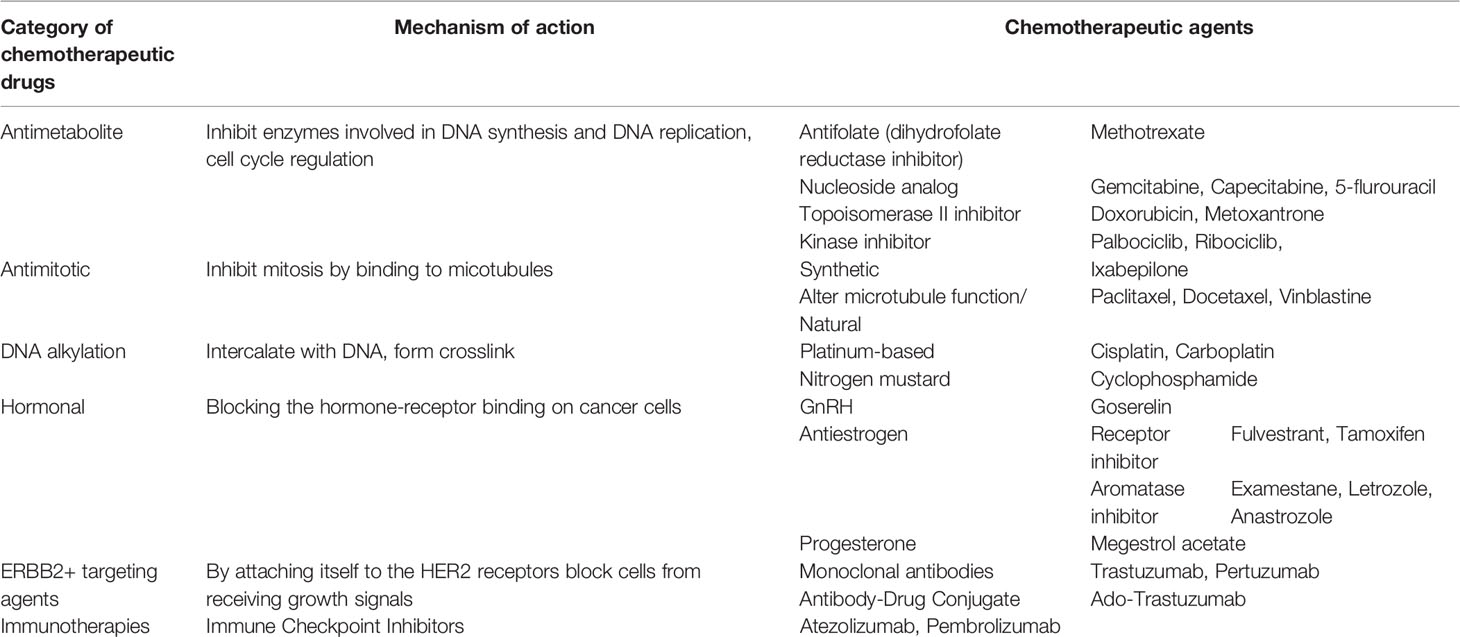
Table 1 Classification of most frequently used drugs in patients diagnosed with breast carcinomas based on the mode of action.
Considering the breast carcinoma subtypes in the UK the most commonly prescribed neoadjuvant chemotherapy regimes in breast cancers are: AC-containing combinations of fluorouracil, epirubicin and cyclophosphamide (FEC) with docetaxel/trastuzumab/pertuzumab for HER2-positive disease and FEC-docetaxel for HER2-negative disease (2).
Anthracylines are broadly used as anticancer agents. The first AC described was daunorubicin from which several derivatives were developed for clinical use, including doxorubicin (DOX) and epirubicin (Figure 1) (14).
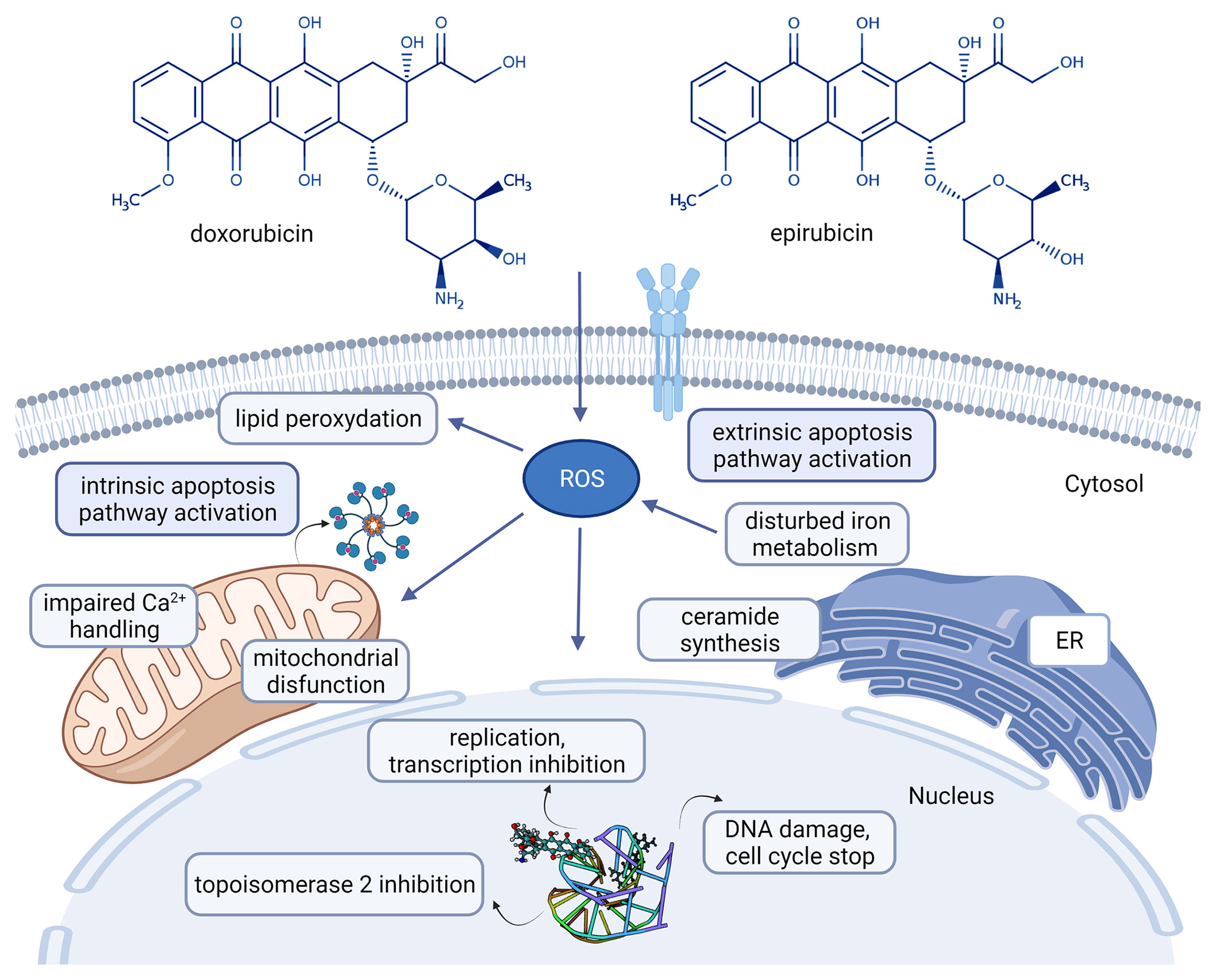
Figure 1 Chemical structures of the clinically used anthracyclines in breast cancer treatment and their mechanism of action [doxorubicin: CHEBI:28748, epirubicin CHEBI:47898 (14), adriamycin-DNA interaction: PDB ID 6KN4 (15)]. Created with BioRender.com. Agreement number: UB23MRCQE9.
Despite their efficacy treatment resistance (as a multifactorial clinical issue) and toxicity (especially cardiotoxicity) have to be considered in each patient before prescription (16, 17).
Several trials were and are designed to compare the effectiveness between ACs and/or taxane as neoadjuvant chemotherapy (1, 18) and repeatedly concluded that pCR is a prognostic marker of significantly higher disease-free survival and overall survival (4, 18). The rate of pCR highly depends on patients and drug selection but generally is lowest for hormone receptor (HR)-positive, HER2-negative tumors and increases approximately additively for HER2-positive/HR-negative tumors with considerable differences between different studies.
The Collaborative Trials in Neoadjuvant Breast Cancer group analyzing the pCR association with long-term outcome have found that the highest pCR was achieved in HER2-positive/HR-negative breast cancer (50.3%) after administration of trastuzumab followed by TNBC (33.6%) and by grade 3 HR-positive/HER2-negative breast cancer with a pCR rate of (16.2%) (1, 19).
To understand how the metabolic signature of a tumor is associated with drug resistance, first we have to understand the mode of action of that drug. ACs intercalate with DNA bases and stop the activity of polymerases blocking in this way the DNA and RNA synthesis (20). Recent studies demonstrated that after diffusion of AC through the plasma membrane, cytosolic AC forms a complex with proteasome that is later transferred into the nucleus, a process requiring ATP (21). ACs were also found to induce cytotoxicity through topoisomerase II (TOPOII) inhibition (block the catalytic activity of TOPOII, contributing to inhibition of DNA replication) (20). Free radicals damaging cell membranes as well as DNA and proteins are described under DOX treatment. The potential involvement of free radical generation in the cytotoxicity of AC is complex and not completely understood (16). ACs can also induce cell death mediated by ceramide. In 2012 Denard B et al. described that treatment of cancer cells with DOX-induced ceramide synthesis (a bioactive sphingolipid, N-acylsphingosine resulting from four consecutive reactions from the precursors palmitoyl-CoA and serine) (22). The schematic mechanism of actions of ACs is presented in Figure 1.
As detailed above ACs act on different pathways to induce tumor cell death. Accordingly, tumor resistance to ACs is also a multifactorial problem that involves several and diverse mechanisms. The main resistance mechanisms are (16): 1. PGp-dependent and PGp-independent multidrug resistance, often associated with upregulation or amplification of the MDR1 gene that encodes PGp. 2. DNA repair mechanisms. Spencer DM et al. demonstrated that nucleotide excision repair and homologous recombination are the most important mechanisms involved in DNA repair after ACs treatments (23). 3. Altered TOPOII activity. Mutation, abnormal expression of TOPOIIα subunit, suppression of TOPOII-mediated apoptotic signaling are associated with clinical resistance to ACs (24). 4. Cancer stemness. Several studies described a direct link between resistance to ACs and cancer stemness (25). 5. Metabolic changes associated with resistance.
Taxanes as microtubule-stabilizing cancer drugs were introduced in the treatment of breast carcinomas in the 1990s. Taxanes (paclitaxel (PTX) and docetaxel (DTX)) suppress microtubules involved in a variety of cellular processes like cell division, signaling, migration by binding to the β-subunit of the tubulin heterodimer (26). PTX was the first discovered member of the taxane family. DTX differs structurally (see Figure 2) and functionally from PTX (14, 27).
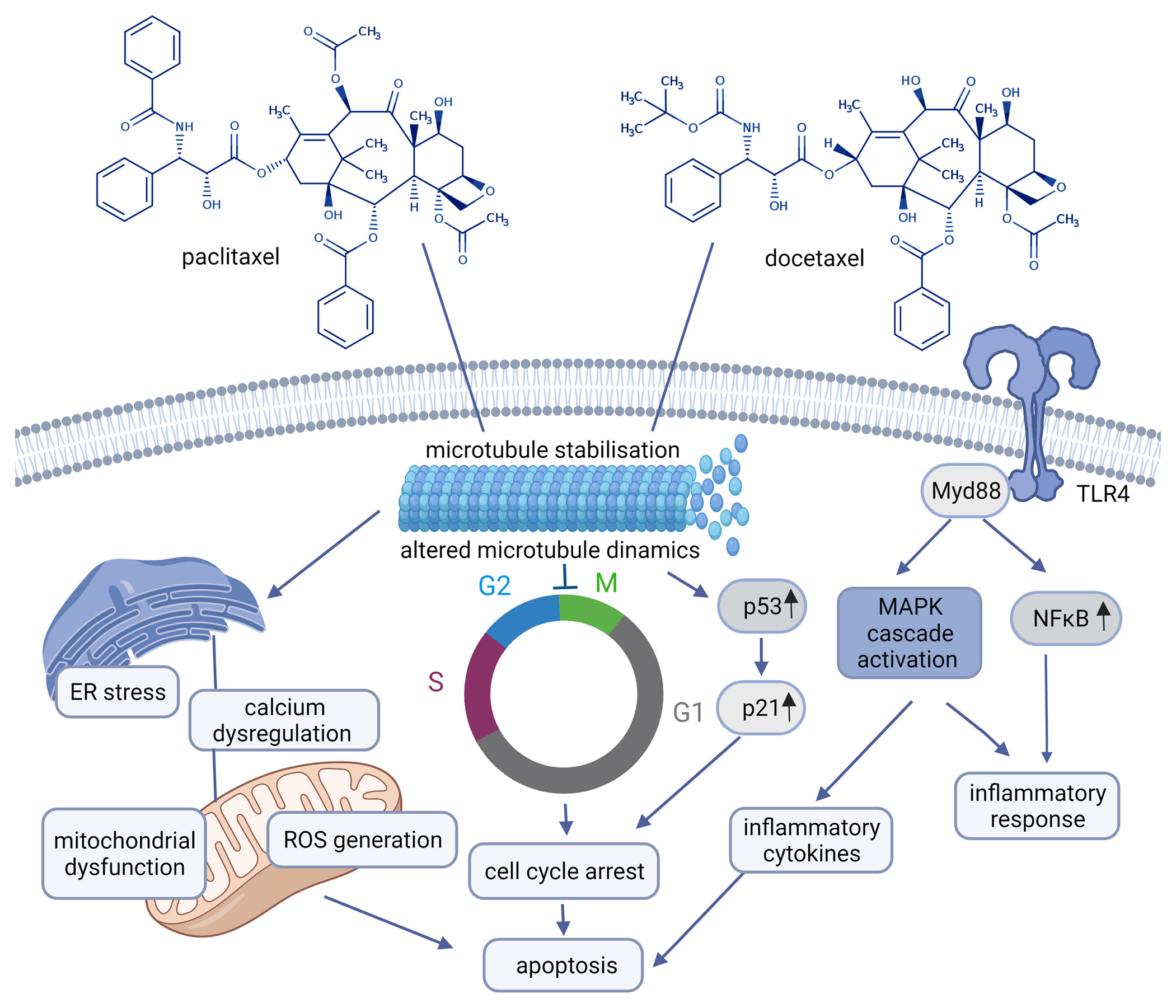
Figure 2 Chemical structures of paclitaxel, docetaxel and their mechanism of action (paclitaxel: CHEBI:45863, docetaxel: CHEBI:4672) Created with BioRender.com. Agreement number: UY23MRDFCE.
Due to drug resistance observed after PTX and DTX treatment, cabazitaxel (CBZ) as a novel taxane drug
was developed and approved by the FDA in 2010 and used as a second-line treatment for metastatic castration-resistant prostate cancer patients (28).
Though the initial response to taxanes is impressive, the resistance to these drugs is frequently observed and is supposed to be coordinated by: 1. alterations in tubulin, 2. proteins that interact with microtubules, 3. efflux pumps, 4. apoptotic proteins, 5. signal transduction pathways, 6. tumor metabolism. Accordingly, strategies to overcome resistance to PTX and DTX include inhibition of the efflux pumps, the use of novel taxane not interacting with drug efflux pumps, regulation of apoptotic and signal transduction pathways, and inhibition of metabolic pathways involved in drug resistance (28, 29). Tubulin is associated with taxane resistance in different ways: 1. mutation in tubulin (high number of point mutations in tubulin have been found in different cell lines selected for paclitaxel resistance analyzes), 2. changes in the expression of tubulin isomers, 3. several proteins that bind to tubulin like: microtubule associated protein 4. posttranslational modifications of tubulin like phosphorylation, acetylation, tyrosination etc. (30–32).
How the sequence order of ACs and taxanes used in neoadjuvant settings influence the pCR rate is under considerable debate. A recent study showed that sequence order did not influence the primary endpoint of pCR rate (19% for AC-Taxane vs. 21% for Taxane-AC) and pCR rate was higher in patients with TNBC (32% vs. 13% in hormone-positive cancers) (33).
Our knowledge about the impact of different AC-derived therapeutic drugs on the metabolic pathways in different cancers is mostly unknown. Achkar IW et al. in a very impressive study have analyzed the metabolic pathways affected by DOX treatment in chicken embryo tumor models (in ovo). By using two mass-spectrometry based platforms (broad metabolic profiling HD4 platform, and the Lipdyzer complex lipid platform) they finally have found that after DOX treatment statistically significant alteration in 127 metabolites (affecting the metabolism of amino acids, carbohydrates, cofactors and vitamins as well as nucleotides and lipids) can be documented on HD4 platform. Whereas on lipid platform they observed statistically significant decrease in 96 lipid molecules. Their results clearly indicate that DOX treatment generates metabolic rewiring (34).
Related to paclitaxel a study performed on human breast carcinoma cell lines demonstrated significant differences in 31 metabolites such as fructose-6-phosphate, citric acid, glycerophosphoinositol and glycerol 3-phosphate when compared metabolic alterations in paclitaxel treated MCF-7 cell lines to untreated ones (35).
Dysregulated cancer metabolism has been increasingly associated with acquired resistance to chemotherapeutic treatment, but no clear data are presented about how metabolic pathways are activated following oncological treatment. In addition to glycolysis that remains favored in resistant cancers undergoing AC treatment, experimental evidences also involve pentose phosphate pathway, fatty acid metabolism, nucleotide synthesis etc. However, there is an urgent need to analyze the complex system of tumor metabolism as the most pieces of evidence are often based on a single pathway without alternative explanation (34, 36).
Beside dysregulated metabolism observed in several cancer types, metabolic reprogramming and metabolic heterogeneity play critical role in carcinogenesis and in acquired resistance to different chemotherapeutic agents. As cancer cells with different metabolic profiles may not respond similarly to anticancer treatment current studies suggest that metabolic heterogeneity needs to be integrated in routine practice to precisely predict breast cancer response to different treatments (37). It is a further challenge in cancer therapy to consider the complex system of inter- and intra-tumour metabolic heterogeneity. Considering just the increased glycolysis as a hallmark of dysregulated metabolism it is well documented that differences in glucose utilization are not only seen between cancer cells, but also within other cells comprising the tumors (38).
Of the relatively few studies demonstrating that chemotherapy agents used in the current treatment regimens in breast carcinomas cause metabolic reprogramming in cancer cells Desbats MA et al. presented that ACs induce increased glycolysis, GLUT1 and glutaminase level whereas taxanes induce elevation in glycolysis, in lactate dehydrogenase A (LDHA), pyruvate kinase M (PKM2), pyruvate dehydrogenase kinase 2 (PDK2) (inhibitor of pyruvate dehydrogenase complex), glutamine uptake, fatty acid synthase (FASN), mitophagy, mitochondrial mass and oxidative phosphorylation (OXPHOS) (39).
Glucose Metabolism
Glycolysis is the major pathway of glucose metabolism occurring in the cytosol of the cells by which glucose is metabolized to pyruvate. Considering the aerobic and anaerobic conditions occurring in tumors and different organisms pyruvate transportation may follow two different pathways (36, 40).
Under aerobic conditions, pyruvate is transported into mitochondria, where it is converted to acetyl-CoA. Acetyl-CoA reacts with oxaloacetate to form citrate that later enters into the tricarboxylic acid cycle (TCA) and generates NADH and FADH2 (reducing equivalents) (41). These reducing equivalents later enter the electron transport chain leading to the production of 36-38 ATP per molecule of glucose.
Under anaerobic conditions, pyruvate instead of entering mitochondria follows a different pathway. In cytosol, the cytosolic enzyme lactate dehydrogenase (LDH) converts pyruvate to lactate. This reaction also allows for the regeneration of NAD+ (the primary oxidizing agent of glycolysis) from NADH (42).
Of the other pathways of carbohydrate metabolism (gluconeogenesis, glyoxylate cycle - typical for plants -, biosynthesis of oligosaccharides and glycoproteins, pentose phosphate pathway-PPP) the PPP pathway was described as highly affected in different tumor types (43).
Glycolysis is controlled by the following ten enzymes and enzymatic reactions (44) (Figure 3): 1. Hexokinase-HK (transfers a phosphoryl group from ATP to glucose to form glucose-6-phosphate-G6P), 2. phosphoglucose isomerase-PGI (converts G6P to fructose-6-phosphate-F6P), 3. phosphofructokinase-1-PFK-1 (phosphorylates F6P to fructose-1,6-bisphosphate-FBP), 4. aldolase (cleaves FBP to form the two trioses, glyceraldehyde-3-phosphate-GAP and dihydroxyacetone phosphate-DHAP), 5. triose phosphate isomerase-TPI (converts DHAP to GAP), 6. glyceraldehyde-3-phosphate dehydrogenase-GAPDH (catalyzes the reversible oxidative phosphorylation of GAP, and forms the first high-energy intermediate, 1,3 bisphosphoglycerate -1,3 BPG) 7. phosphoglycerate kinase-PGK (generates 3-phosphoglycerate-3PG and the first ATP) 8. phosphoglycerate mutase-PGM (converts 3PG to 2-phosphoglycerate -2PG), 9. enolase (dehydrates 2PG to phosphoenolpyruvate-PEP, 10. pyruvate kinase-PK (couple the free energy of PEP hydrolysis to the synthesis of ATP to form pyruvate).
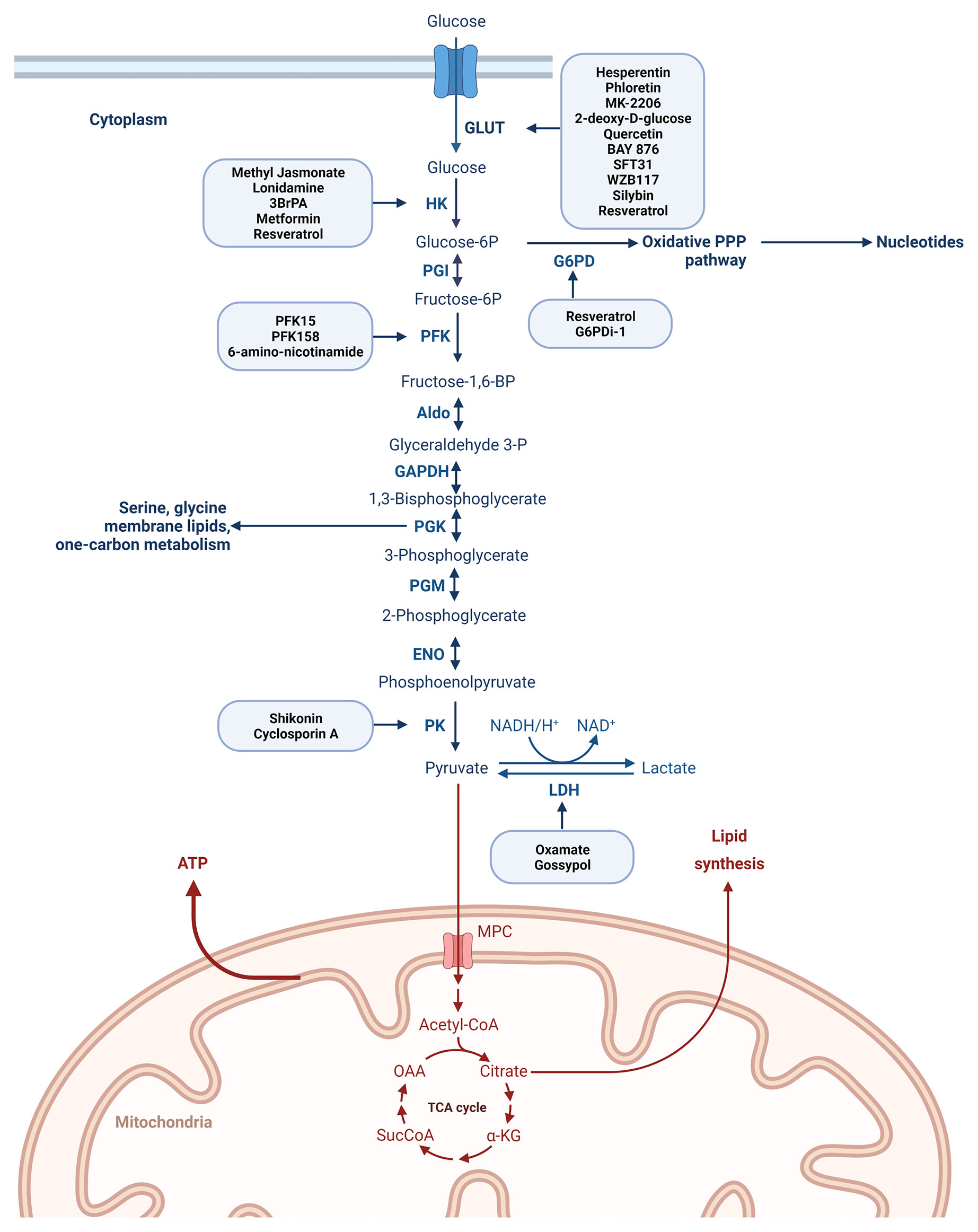
Figure 3 Degradation of glucose via the glycolytic pathway with the most representative enzymes playing a role in the process of glycolysis and with the main drugs described to inhibit different steps of the glucose metabolism. Created with BioRender.com. Agreement number: QP23MRDLKQ.
Glycolysis depends on a constant supply of glucose that enters by two different types of membrane- associated carrier proteins. Na+-glucose cotransporters (SGLTs) are present in the small intestine and the kidney, while passive transport occurs via ubiquitous glucose transporters, the GLUT family (45, 46).
The GLUT family consisting of 14 members facilitates the uptake of glucose through the cell membrane and plays crucial role in glycolysis (36, 46, 47). An increase in GLUT1–6 and 12 was reported in breast cancer (46, 48) and glucose uptake by GLUT1 is considered an important mechanism in breast carcinogenesis and plays an important role in the early phase of breast cancer development (49). Upregulation of GLUT1, 3, and 4 is associated with cancer resistance and inhibition of GLUT may sensitize the anticancer effect of chemotherapeutic drugs (50, 51). Several inhibitors of GLUT1 are reported in the literature and a non-exhaustive list is presented in Figure 3. Phloretin, a GLUT1 inhibitor re-sensitized colon and breast cancer cells to daunorubicin’s anticancer activity and apoptosis-inducing effects (51). GLUT1 inhibitors -WZB117 and SFT-31- inhibit cell proliferation and promote apoptosis in breast cancer cell lines and moreover it was shown that WZB117 increases the effectiveness of radiation (52). Another study presented that the combination of MK-2206 and WZB117 exerts a synergistic cytotoxic effect against breast cancer cells (53). BAY-876, a selective GLUT1 inhibitor analyzed on TNBC cell lines decreased glucose uptake (54). 2-deoxy-D-glucose (2-DG) (phosphorylated by hexokinase but not metabolized further) competes with glucose for binding GLUT reducing in this way glucose uptake in the MDA-MB-231 TNBC cell line (55).
As it is presented in Figure 3 each reaction in glycolysis is catalyzed by its own enzyme. Many of these key glycolytic enzymes are highly expressed in different cancers and correlated with chemoresistance.
A non-exhaustive list of drugs targeting glucose metabolism are presented in Table 2.
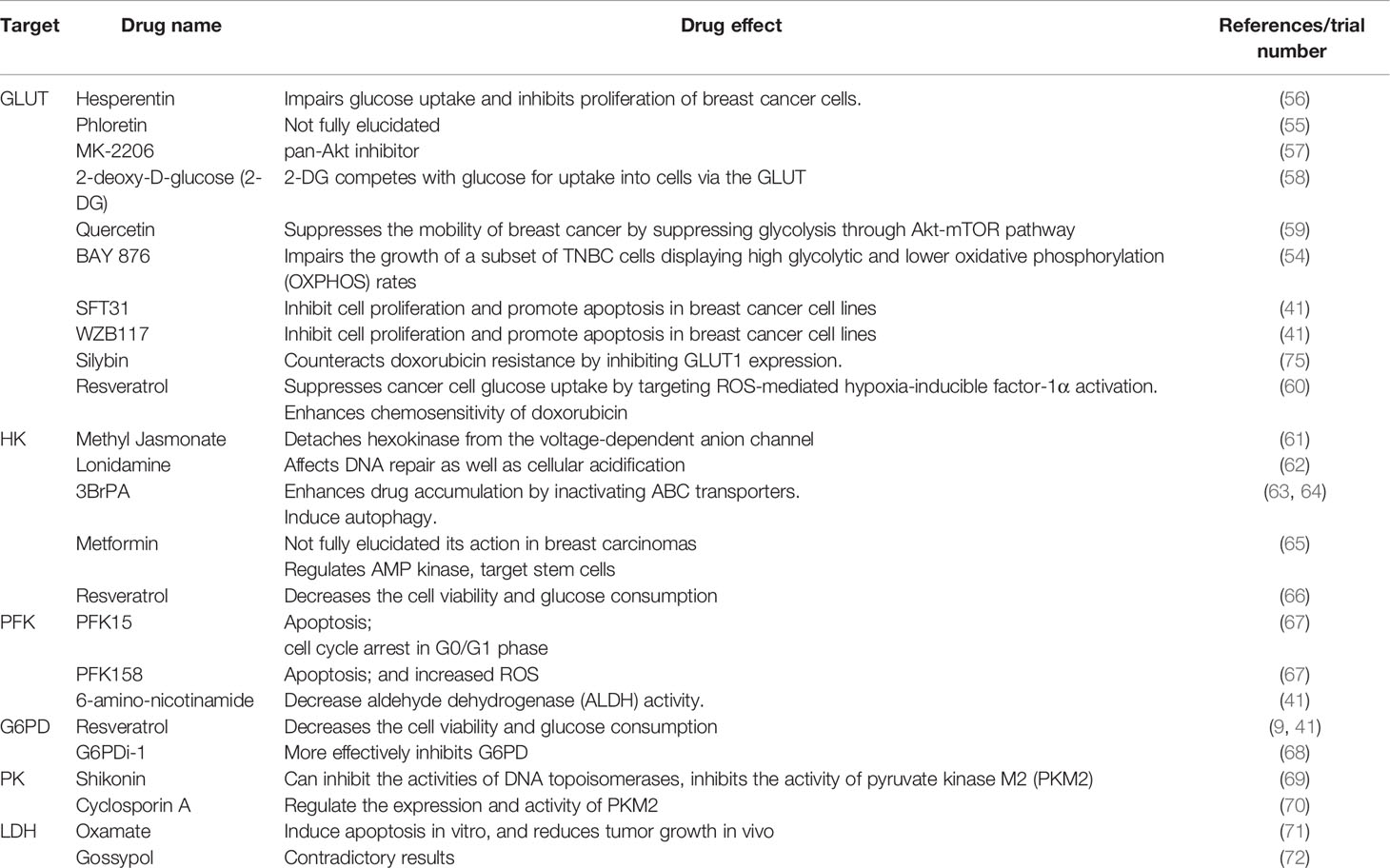
Table 2 Non-exhaustive list of drugs targeting glucose metabolism.
Hexokinase II (HKII)
HKII is considered as the first rate-limiting enzyme in the glycolytic pathway (73). HKII was shown to be upregulated in breast cancer tissues and found to contribute to PTX resistance (74, 75). Even if the major role of GLUT-1 and HKII in cellular metabolism is highly documented their expression after DOX or taxane treatment has not been systematically evaluated. Based on the studies of Zhou R et al. several chemotherapeutic agents may have a significant and direct effect on GLUT-1 and HKII expression (76). Of the HKII inhibitors 3-bromopyruvate (3-BrPA), and lonidamine (LND) have been used in preclinical experiments (36). 3-BrPA enhances drug accumulation by inactivating ABC transporters restoring the cytotoxic effects of DOX. It was shown that 3-BrPA used in combination with DOX significantly inhibited the growth of subcutaneous tumors in multiple myeloma mice (36, 63). The anticancer effect of 3-BrPA was also demonstrated on hepatocellular carcinoma both in in vitro and in in vivo studies and consequently, this drug has been approved by the FDA (63). Inhibition of HKII by LND enhanced the effects of DOX in rituximab-resistant lymphoma cell lines (RRCL) and used in combination with platinum and paclitaxel presented a good tolerability (77, 78).
However metformin, one of the most frequently used drugs in the treatment of type II diabetes mellitus demonstrated antitumor activity combined with or following other therapeutic agents in vitro and in vivo. The detailed mechanism of action of metformin in tumors is not fully elucidated but based on recent studies is mediated through regulation of AMP kinase (AMPK)/mammalian target of rapamycin (mTOR) and insulin/IGF-1 signaling pathways (65). Studies present several aspects of metformin activity in breast carcinomas like metformin downregulation of protein and lipid synthesis, modulation of mitochondrial respiration, induction of stem cell death etc. have been presented and discussed. The authors also outline that many of these anti-cancer effects are molecular subtype-specific being most potent in triple-negative breast carcinomas (79, 80). Nonetheless a study performed on 320,000 persons diagnosed with incident diabetes mellitus has not found any association between metformin treatment and the incidence of major cancers (81).
Glucose-6-Phosphate Dehydrogenase (G6PD)
The first step of the pentose phosphate pathway (PPP) is catalyzed by the G6PD enzyme. A relatively high number of studies have interrogated the role of G6PD in different cancer types but how the overexpression of G6PD contributes to the development of different tumors is largely unknown (82). Yang HC et al. in a very recent study have demonstrated that deregulated G6PD status and oxidative stress are highly related and are involved in cancer progression (83). Another study analyzing MDA-MB-231 cells implanted as orthotopic xenografts has found that the loss of G6PD modestly decreased primary site growth and the ability of breast cancer cells to colonize the lung (84). A relatively high number of G6PD inhibitors were documented in the last years but sometimes with contradictory results. 6-aminonicotinamide, was found to decrease mammosphere formation and aldehyde dehydrogenase activity (85, 86). Ghergurovich JM et al. presented that the most known G6PD antagonist, dehydroepiandrosterone, does not inhibit robustly G6PD in cells and as a consequence they have identified a small molecule (G6PDi-1) that more effectively inhibits G6PD and later the PPP pathway (68).
Phosphofructokinase (PFK)
Our understanding of PFK enzymes and their roles in cancer has developed significantly in the last years. The formation of fructose-1,6-bisphosphate from fructose-6-phosphate is catalyzed by PFK-1 in an irreversible step and therefore PKF-1 is considered as one of the most important rate-limiting enzyme in the process of glycolysis and is regulated by several effectors like fructose 2,6-bisphosphate (F2,6-BP), AMP and ATP. Moreover F2,6-BP levels are strongly associated with the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2, PFKFB) which display tissue-specific pattern. Four known isozymes of PFKFB are described (PFKFB1, PFKFB2, PFKFB3 and PFKFB4) with different kinase-to-phosphatase activities (87).
Of the PFKFB family members, PFKFB3 and PFKFB4 in particular, are overexpressed in breast cancers and in numerous other malignancies (87). PFKFB3 presents the highest kinase activity among the four isoforms and its inhibition resulted in the suppression of the growth of tumor cells by downregulating the glycolytic flux (74, 88).
Considering that standard chemotherapy inevitably is associated with the development of chemoresistance, the observation made by Kotowski K. et al. that PFKFB3 inhibition therapy concurs with carboplatin and paclitaxel in therapy-resistant cell lines of gynecological cancers to reduce tumor weight presents an important step in further therapeutic approach (87). Resveratrol (a natural product found in various plants) nowadays is also considered as a potential anti-tumoral drug that reduces glucose metabolism and viability in cancer cells. Gomez LS et al. have demonstrated that resveratrol decreases cell viability, glucose consumption and ATP content in the human breast cancer cell line MCF-7 (66).
Phosphoglycerate-Kinase (PGK)
Phosphoglycerate-kinase (PGK) catalyzes the first substrate-level phosphorylation in glycolysis while producing 3-PG. PGK1 activity is highly responsible for maintaining energy homeostasis and serine biosynthesis. Clinically, PGK1 was overexpressed in many types of tumors. The exact molecular mechanisms for PGK1-involved drug resistance are not fully clarified but were found to be upregulated in radioresistant astrocytomas and cisplatin-resistant ovarian cancers. Moreover, inhibition of PGK1 increased the sensitivity of gastric adenocarcinoma cells to chemotherapeutic drugs 5-fluorouracil and mitomycin-C (89–91). Sun S et al. by analyzing PGK1 at protein and mRNA level in breast carcinoma cases have found that high PGK1 expression is associated with worse overall survival and concluded that PGK1 may be considered as a predictive biomarker of chemoresistance to paclitaxel treatment in breast cancer (92).
Enolase (ENO)
Proteomics profiling revealed high enolase-1 (ENO-1) expression in ER+ breast carcinomas. The high ENO-1 status was correlated with poor prognosis and with positive nodal status (93). Qian X et al. by analyzing gastric cancers have found that elevated levels of ENO-1 proteins (or downregulation of ENO-1 targeting miR-22) were associated with shorter overall survival and ENO-1 is a novel biomarker to predict drug resistance and overall prognosis in gastric cancer. Based on their data targeting ENO-1 by chemical inhibitors or upregulating miR-22 could be valuable to overcome drug resistance (94). The ENO functions in different cancers and as a potential cancer biomarker are summarized in a very recent study by Almaguel FA et al. (95).
Pyruvate-Kinase (PK)
The conversion of phosphoenolpyruvate to pyruvate with generation of ATP is catalyzed by PK. Till now four PK isoforms are described: PKM1, PKM2, PKR, and PKL (96). Apart from regulating glucose metabolism, PKs are involved in the modulation of gene expression, cell cycle regulation and cell–cell communication (97). Both PKM1 and PKM2 protein expression showed high levels in TNBC and targeting PK inhibited the proliferation of TNBC MDA-MB-231 and MDA-MB-436 cells by involving the NFκB signaling pathway (98). In a study involving 296 invasive breast carcinoma samples a correlation between PKM2 expression and the prediction of chemosensitivity to epirubicin and 5-fluorouracil was demonstrated (99). Moreover, in ER+ breast carcinoma models using MCF-7 and T47D cells PKM2 enhanced chemotherapy resistance by promoting aerobic glycolysis (100). It was also shown that PKM2 expression correlated mostly with cisplatin resistance in breast carcinomas (74).
Lactate Dehydrogenase (LDH) and Drug Resistance
A large amount of literature has been published regarding LDH expression in different cancers, its effect on cancer development and progression, and its diagnostic and prognostic significance. Lactate production is an important phenomenon in the cancer microenvironment and is used as a mechanism of tumor escape from the immune response.
LDH is a tetrameric enzyme composed of two different subunits LDHA (M) and LDHB (H) (encoded in humans by LDHA and LDHB genes, which can assemble into five different isoenzymes as LDH1 or LDHB (H4), LDH2 (M1H3), LDH3 (M2H2), LDH4 (M3H1), and LDH5 or LDHA (M4) and is involved in the conversion of pyruvate to lactate. Deregulated levels of LDHs have been reported in multiple tumors (101, 102). In cancer cells, LDHA plays an important role in rapid conversion of pyruvate to lactate, minimizing in this way the pyruvate entry into TCA cycle in the mitochondria (103). LDHA is also involved in tumor cells proliferation, angiogenesis, tumor cells invasion and migration (103–105).
Based on the review by Mishra D et al. inhibition of LDHA and LDHB is of great interest and is unlikely to cause any possible side effect (103).
Data presenting the role of LDHA in drug resistance described that in chondrosarcoma inhibiting LDHA increased cancer cell sensitivity to DOX (106), in breast cancer cells, led to re-sensitization to paclitaxel (107). A link between LDHA and paclitaxel resistance was described by Varghese E et al. (74). Oxamate, an inhibitor of LDHA, combined with paclitaxel-induced apoptosis in paclitaxel-resistant breast carcinoma (MDA-MB-435 and MDA-MB-231) cells by inhibiting cellular glycolysis (107).
Inhibition of LDHA by small molecule inhibitors or its knockdown by siRNAs or shRNAs decreases tumorigenicity (107). LDHA seems to be a safe therapeutic target, as patients with hereditary LDHA gene deficiency only show symptoms (exertional myoglobinuria) after strenuous exercise but not under ordinary circumstances (108). In accordance with gene deficiency, LDHA inhibition was not proven to be harmful to normal cells (109). Studies showed that LDHB is up-regulated in triple-negative breast cancer (110). Recently, a highly selective LDHB inhibitor was identified (111).
PPP Pathway
In this pathway NADPH is generated by the oxidation of G6P via an alternative pathway to glycolysis. In PPP ribose-5-phosphate-R5P is synthesized as an essential precursor in nucleotide biosynthesis. PPP is made up of two branches: the oxidative and the non-oxidative. In the PPP oxidative phase, G6P is converted into ribulose 5-phosphate and CO2, leading to the synthesis of NADPH.The non-oxidative arm of the PPP composed of a series of reversible reactions generates pentose phosphates for ribonucleotide synthesis (112).
PPP activation has been described in different types of cancer and was associated with metastasis, angiogenesis, and response to chemotherapy and radiotherapy (113). Giacomini I et al. have found that in particular, the oxidative branch of PPP seems to be involved in cisplatin resistance (114). They also suggest that the possibility to selectively deliver into the same cancer cell an anticancer drug and a PPP inhibitor or drug affecting the glucose seems a strategy to overcome the problem of drug resistance (114). Goldman A et al. demonstrated that after taxane treatment metabolic reprogramming of breast cancer cells was observed and characterized by increased glycolytic and oxidative respiration and glucose flux through the PPP pathway (115). The association of PPP with cancer resistance to ACs has been described in several studies (16, 116). Polimeni M et al. (116) by comparing a DOX-resistant human colon cancer cell line (HT29-DX) with a DOX-sensitive (HT29 cells) have found increased PPP and G6PD activity in DOX-resistant cell lines (116).
Citric Acid Cycle
Alternatively known as the tricarboxylic acid cycle (TCA) or Krebs cycle is a hub of the metabolic system. Acetyl-CoA, a common product of carbohydrate, fatty acid and amino acid metabolism enters TCA cycle that oxidizes the acetyl group of acetyl-CoA to two molecules of CO2 with the generation of three NADH, one FADH2 and one ATP equivalent GTP.
The main enzymes involved in TCA cycle are: 1. citrate synthase (catalyzes the condensation of acetyl-CoA and oxaloacetate to yield citrate), 2. aconitase (isomerizes citrate), 3. isocitrate dehydrogenase (catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate with the coupled reduction of NAD+ to NADH and CO2 release), 4. α-ketoglutarate dehydrogenase (decarboxylates α-ketoglutarate to succinyl-CoA, releasing CO2 and NADH, 5. succinyl-CoA synthetase (converts succinyl-CoA to succinate with the coupled synthesis of a GTP), 6. succinate dehydrogenase (catalyzes the dehydrogenation of succinate yielding fumarate and FADH2, 7. fumarase (catalyzes the hydration of fumarates to yield malate) and 8. malate dehydrogenase (reforms oxaloacetate) (117) (Figure 4).
NADH and FADH2 are important products of citric acid cycle. Their reoxidation by O2 through the mediation of the electron-transport chain during OXPHOS completes the metabolic breakdown (117).
Several cancer types are characterized by drastic changes in TCA cycle enzymes compared to normal tissues. Accordingly components of the TCA cycle may be exploited therapeutically for the treatment of disease. Due to the importance of the TCA cycle in normal cell development, high toxicity of this approach have to be considered. By performing metabolomics profiling Ning Shen et al. have found significant changes in the TCA cycle and reactive oxygen species (ROS) related pathways in sensitive TNBC cells compared to resistant TNBC cells (118). Applying small molecule inhibitors to disturb the enhanced TCA for cancer treatment start to evolve CPI-613 (inhibiting both prolyl hydroxylases and α-ketoglutarate dehydrogenase complex- as a key regulator enzyme of TCA) is being tested in phase I and II clinical trials, as a single agent or in combination with standard chemotherapy to treat cancers (119). As many tumors utilize glutamine as a source for TCA cycle, suppression of glutaminolysis by small molecule inhibitors seems also an attractive approach to target tumors. CB-839 disrupts the conversion of glutamine to glutamate and alters TCA cycle, glutathione production, and amino acid synthesis (119).
OXPHOS
Oxidative phosphorylation is an active metabolic pathway in many cancers and there is great interest in targeting mitochondrial OXPHOS in cancer. Evans KW et al. performed RNA sequencing from pre-treatment biopsies of TNBC patients who received neoadjuvant chemotherapy. They demonstrated that the top canonical pathway associated with worse outcome showed higher expression of OXPHOS signature. They also found that inhibiting OXPHOS may be a novel approach to enhance efficacy of several targeted therapies in TNBCs and have validated the antitumor efficacy of the combination of palbociclib, a CDK4/6 inhibitor, and IACS-10759 (a selective OXPHOS inhibitor that blocks complex I) in vitro and in vivo (120). Another study has established that TNBC cell lines (MDA-MB-468 and MDA-MB-231) were highly dependent on OXPHOS when compared to HR+ cell line MCF-7 (121). Considering the ubiquitous necessity of OXPHOS in healthy cellular metabolism, a major barrier to mitochondrial-targeted drugs is the need for cancer-cell selectivity.
Future Challenges in Glucose Metabolism and Breast Cancer Treatment
The increased uptake of glucose, hyperactivated glycolysis and the accumulation of lactate are the main alterations in glucose metabolism associated with different tumor types. As future challenges the following questions must be addressed and discussed in more detail:
1. How the two frequently used ACs, DOX and epirubicin, generate distinct metabolic vulnerabilities in human breast cancer cells? This question is discussed in one recent study by McGuirk S et al. and they have found that in contrast to DOX-resistant cells, epirubicin-resistant cells present a drastic increase in OXPHOS and were more sensitive than DOX-resistant cells to treatment with phenformin. They have noticed that resistance to DOX was mostly associated with glutathione metabolism, whereas resistance to epirubicin to increased mitochondrial bioenergetic capacity (122). No similar data are available about the two frequently used taxanes (PTX and DTX) whether they elicit distinct primary metabolic vulnerabilities in human breast cancer cells.
2. Drug resistance is induced directly by one of the elements involved in the pathway (transporters, enzymes) or indirectly, through an overall increase of the glycolytic metabolism or both situations can occur? Marcucci F et al. in a recent review show that both situations can be present, and the upregulation of an individual enzyme is associated with enhanced overall elevation of the glycolytic metabolism (40).
3. What could be the importance of the fact that particular isoforms (ex. HK2, PFKFB isoform 3, PGK isoform 1 etc.) of the glycolytic enzymes expressed in tumor cells are involved in the induction of drug resistance and of the fact that some of these isoforms are associated with normal cells during embryonic development? (40).
4. How glucose-related metabolic changes are restored months or years after neoadjuvant therapy? Few studies address this question. According to a very recent study neoadjuvant chemotherapy worsens metabolic profile parameters (body mass index, total cholesterol, and fasting glucose) which are then recovered over 3 years. However, in the patients treated with neoadjuvant endocrine therapy there were no significant changes in fasting glucose and total cholesterol (123).
5. A hot topic in the oncological treatment is how selected metabolic inhibitors work alone or how they can be used in combination as potential treatments in breast cancer cases? How metabolic inhibitors used in combination with currently used drugs result in dose reduction of the chemotherapeutic agents known to be highly toxic? In a very recent study Draguet A et al. have evaluated the effect of selected metabolic inhibitors alone and in combination in two (MDA-MB-231 and MCF-7) breast cancer cell lines. Special attention was given to the analysis of metabolic inhibitors in combination with DOX. Their results are encouraging as the combination of CB-839 (glutaminase inhibitor) and Oxamate (lactate dehydrogenase inhibitor) and the combination of CB-839/Oxamate/D609 (a phosphatidylcholine-specific phospholipase C inhibitor) resulted in remarkable cell mortality and all the inhibitors improved the efficacy of DOX in cell lines. In addition, the same inhibitors improved the efficacy of DOX. Some of the metabolic inhibitors presented in their study like AZD3965 (a selective monocarboxylate transporter 1 inhibitor) are being evaluated in preclinical trials or in clinical trials (CB-839 - a glutaminase inhibitor) (124).
Lipid Metabolism
Obesity is considered as a risk factor of breast cancer especially in post-menopausal women correlating with a diminished therapeutic response and with worse disease outcome (125). By analyzing obese vs. lean mice it was shown that decreased efficacy of DOX corresponds with alterations of lipid metabolism markers (126, 127).
Elevated and dysregulated lipid metabolism is a common hallmark of cells surviving neoadjuvant therapy in breast cancer patients (128). Studies also suggested that aberrant lipid metabolism plays an important role in cancer cells’ adaptation to treatment-induced cellular stress (129).
With the advent of new and more effective tools to study lipids is more and more recognized that lipids are central players in cancer biology (are essential builders for membranes, serve as fuel to the highly energetic process of uncontrolled tumor cell division and regulate numerous cellular functions) (130).
It is intriguing to analyse whether the breast cancer subtype defined by the transcriptome is reflected in the lipidome of breast cancer cells or whether is there any relationship between lipidomic profile and response to different therapies. Eiriksson FF et al. by performing liquid chromatography mass spectrometry (LC-MS) to analyze the lipidome of six breast cancer cell lines of different subtypes have found differences in lipidomes within the previously defined subtypes and concluded that subtypes defined by the transcriptome are also reflected in differences in the lipidome (131).
As it is presented in some of the recently published papers it would be also important to discuss in further reviews the utility of circulating lipid metabolites [to enhance the accuracy of known tumor markers or to distinguish tumors with early-stage vs. benign tumors (132)], the importance of lysophosphatidic acids (LPAs) as their role in different cancers is of great interest from therapeutic view and the role of the polyunsaturated fatty acids (PUFA). Recent studies analysing the role of the arachidonic acid (AA), a representative PUFA in different tumors have found a link between AA and macrophage function in ovarian cancer and have demonstrated that high level of AA is associated with poor clinical outcome in ovarian cancers (133). Another recent study has presented that in chemoresistant malignant pleural mesothelioma AA is a mediator of the adaptive response to pemetrexed (134)
Based on preclinical models lipid metabolism inhibition reversed the resistance of cancer cells to anticancer drugs suggesting that lipid metabolism play important role in drug resistance (129). The question is how, and at which step lipid metabolic changes contribute to drug resistance. Germain N et al. have suggested that at least the following changes in lipid metabolism may contribute to anticancer drug resistance: 1. lipid metabolism counteracts oxidative and ER stress-induced by anticancer drugs, 2. reduces metabolic stress and genotoxicity induced by anticancer drugs and contributes to the maintenance of drug-resistant cancer stem cells (129).
Pharmacological inhibitors have been developed for some of the enzymes of lipid metabolism and some of the compounds are used in combination with conventional therapies (129).
The dysregulation of lipid metabolism may occur at different steps of the metabolic process. The most frequent aberrations associated with drug resistance are overactivation of fatty acid oxidation, elevated fatty acid biosynthesis, aberrant accumulation of lipid droplets (LD) and changes in lipid composition of cell membranes (135).
Changes in lipid metabolism of resistant cells are considered treatment-specific and may include changes both in de novo lipogenic synthesis and/or lipolytic pathway (129). Table 3 lists the most frequently used drugs in breast cancer treatment and the possible lipid metabolism pathway associated with resistance to different drugs.
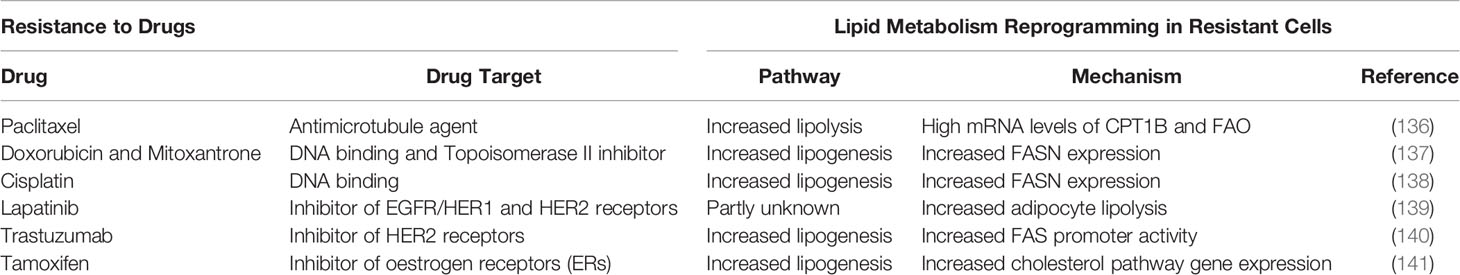
Table 3 The most frequently used drugs in breast cancer treatment and the possible lipid metabolism pathway associated with resistance to different drugs.
Fatty Acid Oxidation
Fatty acid β-oxidation (FAO) is a primary bioenergetic source by which fatty acids are broken down in a multistep process. Fatty acids enter cells through fatty acid transport proteins like: fatty acid translocase (FAT/CD36), tissue-specific fatty acid transport proteins (FATP), and plasma membrane-bound fatty acid-binding protein (FABPpm) (142, 143). In breast cancer loss-of-function studies have demonstrated that CD36 is critical in fatty acid uptake (144). Compared to CD36, FATPs and FABPpm have received far less attention (145).
Fatty acid transportation across the mitochondrial membrane is controlled by several enzymes. Once inside the cell, a CoA group is added to the fatty acid by acyl-CoA synthetase, forming long-chain acyl-CoA (146). The conversion of the long-chain acyl-CoA to long-chain acylcarnitine by carnitine palmitoyltransferase 1 (CPT1) is the step by which fatty acid is transported across the inner mitochondrial membrane, the process controlled by carnitine translocase (CAT). CPT2 enzyme converts back the long-chain acylcarnitine to long-chain acyl-CoA that enters the fatty acid β-oxidation pathway (146) (Figure 4).
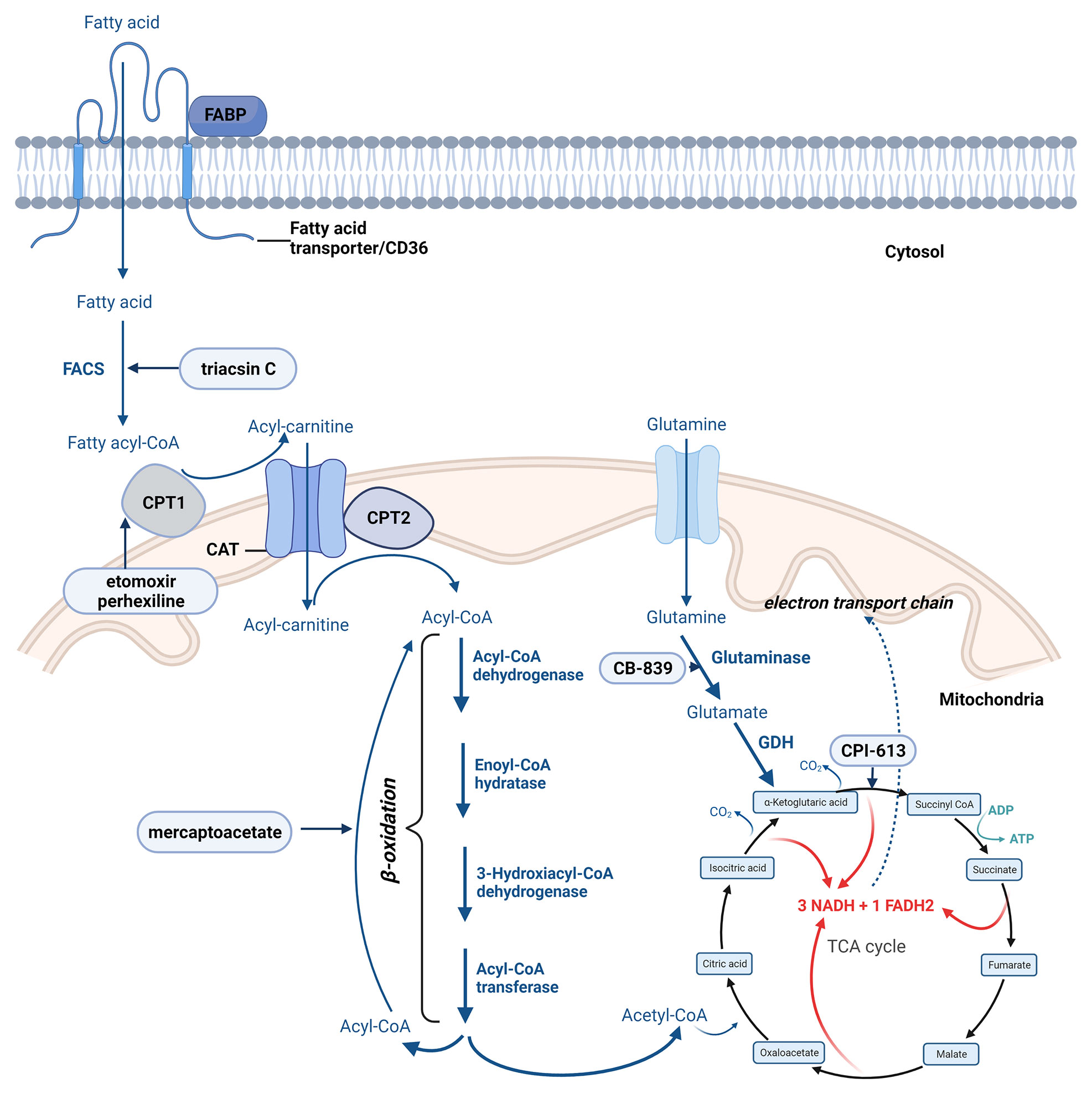
Figure 4 The most important steps of fatty acid oxidation. Created with BioRender.com. Agreement number: ZA23MRDTIU.
The transport of fatty acyl-CoAs into the mitochondria by CPT1 proteins is considered a rate-limiting step in the mitochondrial fatty acid β-oxidation pathway is (147). All three known isoforms of CPT1 enzyme (CPT1A, CPT1B, and CPT1C) have been recognized as important players in drug resistance. Using an integrated genomic strategy, Gatza ML et al. identified CPT1A as an important player also in cell proliferation especifically in hormone receptor-positive breast carcinomas (148).
The β-oxidation is a cyclic process responsible for the mitochondrial disintegration of long-chain acyl-CoA to acetyl-CoA. This process is controlled by following four main enzymes: acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and ketoacyl-CoA transferase (thiolase) (149). In each cycle, FAD-dependent dehydrogenation and NAD-dependent oxidation leads to the formation of FADH2 and NADH.
Complete oxidation of the produced acetyl-CoA, NADH, and FADH2 is accomplished by the TCA and OXPHOS (Figure 4) (146, 150).
Many types of cancer exhibit a high activity of FAO such as TNBC, KRAS mutant lung cancer, hepatitis B-induced hepatocellular carcinoma etc. (136, 151, 152). The mechanisms of FAO activation in different tumor cells are under serious debate and different mechanisms have been proposed to explain drug-induced FAO activation and the role played by FAO in drug resistance (153).
Studies have reported that prominent oncoproteins play important role in the activation of several FAO enzymes. Overexpression of c-Myc in TNBC resulted in FAO enzymes and metabolic intermediates upregulation whereas inhibition of FAO blocked Myc-driven tumorigenesis (154). Another study identified CPT1B as a downstream target of the JAK/STAT3 pathway in breast cancer (135, 136).
Hoy AJ et al. discuss several aspects of the association of tumor fatty acid metabolism and therapy resistance. In recurrent breast carcinomas they described enhanced CPT1B mRNA expression compared to tumours that did not recur (135). In patients with pancreatic and gastric cancers higher CPT1A expression was associated with chemoresistance and with shorter overall survival (135, 155, 156).
Studies suggest that pathways like PI3K/AKT/mTOR, JAK/STAT3 play a pertinent role in lipid metabolism regulation (157–159). Wang T et al. have found that leptins upregulate STAT3 and FAO activity and this metabolic switch promotes cancer stemness and chemoresistance. In in vivo conditions blocking FAO re-sensitized resistant cells to chemotherapy (136, 159). Another study described that inhibition of FAO by mercaptoacetate and etomoxir sensitizes paclitaxel-resistant lung adenocarcinoma cells (160).
Targeting FAO came into the focus of researches as a chemosensitization strategy given its key role in promoting tumor cell survival via energy generation. More and more studies suggest that addition of FAO inhibitors completely or partially inhibited drug resistance of cancer cells (160–162).
Fatty Acid Biosynthesis
Substantial efforts have been documented to develop strategies to target fatty acid biosynthesis since new and new studies have demonstrated that activation of de novo fatty acid synthesis is specific to some cancerous tissues (163).
In a standard way fatty acids are synthesized through the fatty acid synthesis cycle (Figure 5). The substrate for FA synthesis is cytoplasmic acetyl-CoA, which is obtained through different mechanisms (163). The synthesis of fatty acids from acetyl-CoA and malonyl-CoA involves two main steps: 1. carboxylation of acetyl-CoA by acetyl-CoA carboxylase (ACC) to form malonyl-CoA (ATP dependent) and 2. decarboxylation of the malonyl group in the condensation reaction catalyzed by the multifunctional FASN containing substrate shuttling domain ACP and six catalytic domains: malonyl/acetyl-CoA-ACP transacylase, β-ketoacyl-ACP synthase, β-ketoacyl-ACP reductase, β-hydroxyacyl-ACP dehydrase, enoyl-ACP reductase and thioesterase (163, 164).
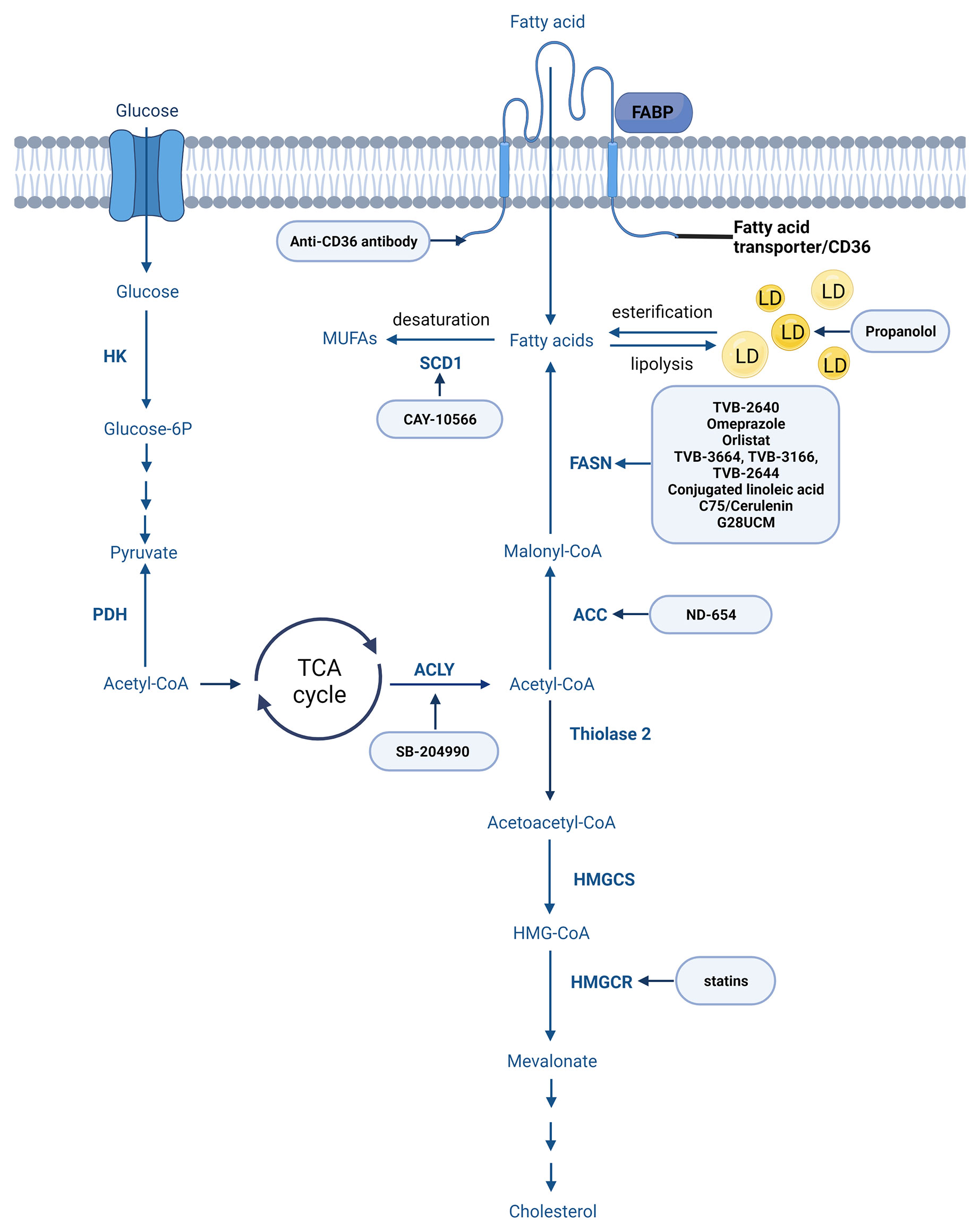
Figure 5 Main steps of fatty acid and cholesterol synthesis. Created with BioRender.com. Agreement number: YB23MRDXCR.
The enzymes playing role in FA biosynthesis are under the control of sterol regulatory element-binding proteins (SREBPs) (165). The three isoforms of SREBPs (SREBP1a, -1c, and -2) are encoded by two different genes (166). The importance of SREBP-1 in lipid metabolism and tumor prognosis is discussed in several papers (167, 168). In vitro studies performed on MDA-MB-231 and MCF7 breast cell lines revealed that the suppression of SREBP-1 significantly inhibited cell migration and invasion (168). Targeting SREBP could therefore be an efficient strategy to halt tumor growth (163). Fatostatin, a nonsteroidal diarylthiazole derivative inhibits the activation of SREBP1 and additionally inhibits cell proliferation (169).
FASN
Overexpression of FASN is considered as one of the major changes in lipid metabolism associated with drug resistance. FASN is overexpressed in breast carcinomas with over 70% of primary TNBC and in several other epithelial malignancies as demonstrated with immunohistochemistry. Breast cancers overexpressing FASN are more likely to recur and metastasize and present significantly shorter disease-free and overall survival (170, 171). FASN induces resistance to multiple DNA-damaging agents including DOX and cisplatin (170). The impact on sensitivity to microtubule agents such as taxane is contradictory. Sardesai SD et al. have found that FASN induces resistance to multiple DNA-damaging agents including DOX and cisplatin without impacting sensitivity to microtubule agents or antimetabolites whereas Menendez JA et al. have found that the inhibition of FASN activity strengthens the cytotoxicity of docetaxel in the HER2-overexpressing breast cancer cell lines (170, 171). FASN mediates drug resistance via palmitate production involving different pathways. Palmitate limits AC transmembrane uptake by cancer cells by modulating the transmembrane mobility of the drugs, a major way for AC entry inside the cells (16). FASN overexpression is also associated with the suppression of drug (DOX)-induced ceramide production by inhibiting the activity of sphingomyelinase (172).
Moreover FASN is an established therapeutic target. Wang W et al. in a recent study detailed the new therapeutic perspectives in cancers based on the lipid metabolic pathway (173).
Summarized data of the therapeutic challenges in lipid metabolism are presented in Table 4.

Table 4 Non-exhaustive list of drugs targeting lipid metabolism used in association with standard treatments in resistant cancer.
A real challenge in targeting lipid metabolism is the lack of selectivity. For example the first generation of FASN inhibitors such as Orlistat (presenting with lack of selectivity, poor metabolic stability) displayed considerable side effects (e.g. anorexia). The next-generation FASN inhibitors present limited systemic toxicity in a preclinical study, higher anti-tumor potential and higher specificity for FASN (173, 189). TVB-2640, a selective FASN inhibitor showed good tolerability and efficacy when combined with taxol in previously treated patients with advanced metastatic breast cancer. By inhibiting FASN a partial regression in ~20% of patients and stable disease in the remainder of patients was documented (189). Based on the very recent study of Sardesai SD et al. Omeprazole can be safely administered in doses that inhibit FASN. Further validation is needed but a promising pCR rate was diagnosed after the addition of Omeprazole to neoadjuvant AC-T (170).
Other enzymes involved in fatty acid synthesis might also be therapeutic targets in cancer. Inhibition of key enzymes for FAs synthesis, such as FASN (presented above), ATP-citrate lyase (ACLY) and ACC can decrease the proliferation and growth of different cancer cells. In hepatocellular carcinoma, ND-654, an ACC inhibitor, has been shown to suppress carcinogenesis (190). In human glioblastoma cells reduced proliferation and de novo lipogenesis was observed after the inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) (190, 191). However, Jeon SM et al. targeting ACCs by short interfering RNAs (siRNAs) have found that only ACC2 knockdown inhibited markedly cell death and H2O2 accumulation without decreasing O2 (192).
ACLY
ACLY, a cytoplasmic enzyme catalyzing citric acid breakdown to acetyl-CoA, was found to be overexpressed in several cancers like breast, colorectal, non-small cell lung cancers etc. (193). The ACLY inhibitor SB-204990 has been set out to inhibit the proliferation of lung adenocarcinoma cells in vivo and in vitro. Other ACLY inhibitors like difluorocitric acid and hydroxycitrate have been demonstrated to block the synthesis of FAs. Although some ACLY inhibitors performed well and have been validated, most of the studies to date are still in the preclinical phase (193–195).
Mitochondrial proteomics reveal acetyl-CoA acetyltransferase, hydroxacyl-CoA dehydrogenase and short chain enoyl-CoA hydratase overexpression in DOX-resistant compared to DOX-sensitive uterine sarcoma cells, providing potential diagnostic markers and therapeutic candidates (16).
SCD1
It is worth mentioning the stearoyl-CoA desaturase 1 (SCD1) enzyme as another rate-limiting enzyme in fatty acid synthesis that converts saturated acids to monounsaturated fatty acids (MUFAs) involved in many biological processes (are major constituent of biological structures such as membranes and can also function as or modify signaling molecules). Accordingly more and more studies highlight different roles of SCD1 like modulation of autophagy (196) others discussing the involvements of SCD1 in regulation of cancer stem cells (197) and in the promotion of cancer cell proliferation, migration and metastasis (198). Cancer cells presenting with high degree of membrane saturation are known to be less sensitive to oxidative stress induced by agents like ACs. It is intriguing and important to find combination of therapies that acts synergistically. Many inhibitors of SCD1 like CAY10566, MF-438 and CVT-11127 have been presented but only a few have progressed to clinical trials due to their adverse effects (198).
The Role of Membrane Lipid Composition in Chemotherapy Resistance
Reduced fluidity of lipid bilayers in the membranes are also characteristics of chemoresistant cancer cell lines. Membrane lipid composition has gained high importance in cancer research. The reduced membrane fluidity influence or disrupt drug uptake via passive diffusion or endocytosis (135). Plasma membrane cholesterol was reported to be elevated in AC-resistant MCF-7 breast cancer cells (199). The increased expression of sphingolipid metabolizing enzymes, specifically ceramide transport protein, sphingosine kinase 1 and 2 have been correlated with resistance to DOX and paclitaxel (39).
Studying the cholesterol metabolic reprogramming in cancer came in the focus of researches in the last years based in part on its important role in maintaining cellular homeostasis (providing essential hormones) as well as considering its role in forming lipid rafts, an indispensable cell membrane structure in cancer cells. As many tumor-related proteins are located in lipid rafts this cell component is an important platform for oncogenic signaling pathways. Considering other aspects like the metabolites obtained by de novo modification of cholesterol (mevalonic acid, farnesyl pyrophosphate and geranylgeranyl pyrophosphate) also playing an important role in cancer growth and/or oncogenic signaling pathways it is reasonable to consider the significant role played by cholesterol in tumor metabolism. Cholesterol deficiency in cell membranes has been shown to inhibit cancer progression (157) and cholesterol metabolic reprogramming in cancer cells is strongly linked to several aspects of drug resistance (157, 200).
Cholesterol is mostly synthetized through the mevalonate pathway (Figure 5) or acquired from the circulation via LDL receptor (LDLR)-mediated endocytosis. In the mevalonate pathway 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) is reduced to mevalonate by HMG-CoA-reductase enzyme considered as the primary control site for cholesterol biosynthesis (201). The enzyme activity and protein level is controlled by multiple regulatory mechanisms, its competitive inhibitors are among the most widely prescribed medications, collectively known as statins. Further a series of enzymatic reactions convert mevalonate to cholesterol (199, 202, 203).
The role of cholesterol in drug resistance has been demonstrated in several cancer types including breast cancer (199) and is mostly related to AC resistance given the role of the cell membrane permeability in AC drugs. It was shown that decreased cholesterol levels resulted in the increased uptake of DOX (204).
Some mechanisms through which cholesterol regulates drug resistance are lipid rafts, ABC transporters, drug uptake. These mechanisms are highly discussed in a recently published review of Yan A et al. (199).
The “membrane-lipid therapy” based on the modulation of membrane lipid composition is considered as an effective therapeutic strategy in several diseases. Preta G. mentions that instead of modifying membrane cholesterol/sphingolipids content new schemes like modulation of membrane bilayer properties (fluidity and elasticity) by inducing changes in the organization of lipid rafts are preferred (205).
The accumulation of lipid droplets is a less well-studied aspect of chemoresistant cancer cell lines (135). Lipid droplets are considered to directly contribute to chemoresistance by providing an extra source of lipids for FAO when nutrient stress occurs, or may play a role in hydrophobic drug sequestration (206).
Sirois I et al. in a very impressive study by analyzing a novel MDA-MB-436 cell-based model of chemoresistance characterized by a unique and complex morphologic phenotype with numerous lipid droplets and a set of primary chemoresistant TNBCs have identified several metabolic vulnerabilities. These include a dependence on perilipin family member perilipin 4 (PLIN4), a protein coating of the observed lipid droplets that play important role in fat mobilisation, expressed both in experimental conditions (in the chemoresistant TNBC cells) and in chemoresistant tumors treated with neoadjuvant DOX-based chemotherapy. Their results call attention to a novel mechanism of chemotherapy resistance that may have therapeutic consequences in the treatment of drug-resistant cancer (207).
Future Challenges in Lipid Metabolism and Breast Cancer Treatment
Based on the above-mentioned data related to lipid metabolism in breast carcinomas there are some intriguing questions to be analyzed in more detail:
1. How breast carcinoma treatment is going to be tailored based on the metabolic phenotypes/subtypes? In a review by Marie E. Monaco analyzing fatty acid metabolism based on mRNA expression data in breast cancer subtypes suggested that significant differences are observed. While the less aggressive, HR+ (luminal) subtypes depend on a balance between de novo fatty acid synthesis and oxidation as sources for energy requirements the TNBCs are characterized by overexpression of the genes involved in the utilization of exogenous fatty acids. They suggest that treatments have to be tailored to individual subtypes (147).
2. Lipid metabolism has been difficult to analyze partly due to technical problems. Considering the challenges (tumor heterogeneity, metabolic diversity, the rapidly changing environmental context, differences in nutrients use among different cell types, the cooperative or competitive relationships between cells, etc.) it is questionable how the new technologies could help in delineating the contribution of lipid metabolism to tumor differentiation, progression and resistance to different drugs. Matsushita Y et al. summarize the new technology currently used in lipidomics and confirm that recent innovations especially in mass spectrometry- and chromatography-based lipidomics technologies have improved our understanding of the role of lipids in cancer (208).
3. Cancer-associated adipocytes are poorly investigated cells in breast tumor microenvironment. What should be the role of the surrounding and breast cancer-associated adipocytes? A few studies suggest that cancer-associated adipocytes can also cause resistance to radiotherapy and to chemotherapeutic drugs (209, 210). Another study described that interaction between cancer-associated adipocytes and cancer cells seems to be more pronounced in obese patients (211).
4. How is lipid profile of patients restored after (neo)adjuvant chemotherapy? Tian W et al. suggested that different age groups showed different changes in lipid levels and in general the lipid profiles were restored to baseline levels approximately 6 months after chemotherapy completion (212).
Conclusions
Main metabolic alterations in breast cancers include the preference of the glycolytic pathway (the increased uptake of glucose, hyperactivated glycolysis), the enhanced oxidative phosphorylation pathway and dysregulation of fatty acid metabolism.
Current studies present the important aspect of tumor metabolism associated with anticancer drug resistance and the association of the partly unknown aspect of the metabolic plasticity in chemotherapy resistance. Efforts are made to combine chemotherapies with drugs targeting different steps of metabolic pathways. Compared to a huge amount of results related to the above-mentioned questions only a few studies present data related to changes in metabolic profile induced by currently applied chemotherapeutic agents used in neoadjuvant or adjuvant settings. There are also gaps in analysing the association of the metabolic changes induced by chemotherapies with different response rates to the applied treatment regimen or with tumor progression.
Considering just some of the points like: the regulation of lipid and glucose metabolism is also important for normal cells, the intra- and intertumoral metabolic heterogeneity in breast cancers, the different metabolic signatures observed in different breast carcinoma subtypes, the lack or imperfect technologies to study components of metabolic pathways, it is still a huge challenge to find substances that target different steps of glucose and lipid metabolism in the very heterogeneous group of breast tumors without affecting normal cells. Additionally, current metabolomic and especially lipidomic approaches have identified several partly unknown components of metabolic pathways paving the way to a better understanding of cancer biology. As exemplified in our and in recently published reviews there are relatively broad opportunities in targeting metabolism in cancers but it is a key question whether we have considered nearly all metabolic hubs as targets and whether we can rewire our mind to optimise the therapies based on the several new data.
Author Contributions
AMT: conceptualization, interpretation of data, writing original draft, review. JK: review, interpretation of data. SV-K: editing, preparing figures. BT: review, editing, preparing figures and supervision. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the CELSA 19/037 grant." FAT-MetBC: Facing Increased Adiposity in Treating Metastatic Breast Cancer.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All figures were created with BioRender.com.
Glossary

References
1. Asaoka M, Gandhi S, Ishikawa T, Takabe K. Neoadjuvant Chemotherapy for Breast Cancer: Past, Present, and Future. Breast Cancer (Auckl) (2020) 14:1178223420980377. doi: 10.1177/1178223420980377
2. Whitehead I, Irwin GW, Bannon F, Coles CE, Copson E, Cutress RI, et al. The Nest (Neoadjuvant Systemic Therapy in Breast Cancer) Study: National Practice Questionnaire of United Kingdom Multi-Disciplinary Decision Making. BMC Cancer (2021) 21(1):90. doi: 10.1186/s12885-020-07757-6
3. McGale P, Dodwell D, Gray R, Mannu G, Peto R, Taylor C, et al Long-Term Outcomes for Neoadjuvant Versus Adjuvant Chemotherapy in Early Breast Cancer: Meta-Analysis of Individual Patient Data From Ten Randomised Trials. Lancet Oncol (2018) 19(1):27–39. doi: 10.1016/S1470-2045(17)30777-5
4. Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, et al. Preoperative Chemotherapy: Updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J Clin Oncol (2008) 26(5):778–85. doi: 10.1200/JCO.2007.15.0235
5. Symmans WF, Wei C, Gould R, Yu X, Zhang Y, Liu M, et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J Clin Oncol (2017) 35(10):1049–60. doi: 10.1200/JCO.2015.63.1010
6. Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological Complete Response and Long-Term Clinical Benefit in Breast Cancer: The Ctneobc Pooled Analysis. Lancet (2014) 384(9938):164–72. doi: 10.1016/S0140-6736(13)62422-8
7. Spring LM, Fell G, Arfe A, Sharma C, Greenup R, Reynolds KL, et al. Pathologic Complete Response After Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive Meta-Analysis. Clin Cancer Res (2020) 26(12):2838–48. doi: 10.1158/1078-0432.CCR-19-3492
8. Asaoka M, Narui K, Suganuma N, Chishima T, Yamada A, Sugae S, et al. Clinical and Pathological Predictors of Recurrence in Breast Cancer Patients Achieving Pathological Complete Response to Neoadjuvant Chemotherapy. Eur J Surg Oncol (2019) 45(12):2289–94. doi: 10.1016/j.ejso.2019.08.001
9. Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol (2008) 26(8):1275–81. doi: 10.1200/JCO.2007.14.4147
10. Petovari G, Danko T, Tokes AM, Vetlenyi E, Krencz I, Raffay R, et al. In Situ Metabolic Characterisation of Breast Cancer and its Potential Impact on Therapy. Cancers (Basel) (2020) 12(9). doi: 10.3390/cancers12092492
11. Litton JK, Scoggins M, Ramirez DL, Murthy RK, Whitman GJ, Hess KR, et al. A Feasibility Study of Neoadjuvant Talazoparib for Operable Breast Cancer Patients With a Germline BRCA Mutation Demonstrates Marked Activity. NPJ Breast Cancer (2017) 3:49. doi: 10.1038/s41523-017-0052-4
12. Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, et al. Addition of the PARP Inhibitor Veliparib Plus Carboplatin or Carboplatin Alone to Standard Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (Brightness): A Randomised, Phase 3 Trial. Lancet Oncol (2018) 19(4):497–509. doi: 10.1016/S1470-2045(18)30111-6
13. Thompson AM, Moulder-Thompson SL. Neoadjuvant Treatment of Breast Cancer. Ann Oncol (2012) 23(Supplement 10):x231–6. doi: 10.1093/annonc/mds324
14. Hastings J, Owen G, Dekker A, Ennis M, Kale N, Muthukrishnan V, et al. Chebi in 2016: Improved Services and an Expanding Collection of Metabolites. Nucleic Acids Res (2016) 44(D1):D1214–9. doi: 10.1093/nar/gkv1031
15. Barthwal R, Raje S, Pandav K. Structural Basis for Stabilization of Human Telomeric G-Quadruplex [D-(TTAGGGT)]4 by Anticancer Drug Adriamycin. J Biomol Struct Dyn (2021) 39(3):795–815. doi: 10.1080/07391102.2020.1730969
16. Capeloa T, Benyahia Z, Zampieri LX, Blackman M, Sonveaux P. Metabolic and non-Metabolic Pathways That Control Cancer Resistance to Anthracyclines. Semin Cell Dev Biol (2020) 98:181–91. doi: 10.1016/j.semcdb.2019.05.006
17. Wallace KB, Sardao VA, Oliveira PJ. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res (2020) 126(7):926–41. doi: 10.1161/CIRCRESAHA.119.314681
18. von Minckwitz G, Raab G, Caputo A, Schutte M, Hilfrich J, Blohmer JU, et al. Doxorubicin With Cyclophosphamide Followed by Docetaxel Every 21 Days Compared With Doxorubicin and Docetaxel Every 14 Days as Preoperative Treatment in Operable Breast Cancer: The GEPARDUO Study of the German Breast Group. J Clin Oncol (2005) 23(12):2676–85. doi: 10.1200/JCO.2005.05.078
19. Nekljudova V, Loibl S, von Minckwitz G, Schneeweiss A, Gluck S, Crane R, et al. Trial-Level Prediction of Long-Term Outcome Based on Pathologic Complete Response (Pcr) After Neoadjuvant Chemotherapy for Early-Stage Breast Cancer (EBC). Contemp Clin Trials (2018) 71:194–8. doi: 10.1016/j.cct.2018.06.016
20. Meredith AM, Dass CR. Increasing Role of the Cancer Chemotherapeutic Doxorubicin in Cellular Metabolism. J Pharm Pharmacol (2016) 68(6):729–41. doi: 10.1111/jphp.12539
21. Kiyomiya K, Matsuo S, Kurebe M. Mechanism of Specific Nuclear Transport of Adriamycin: The Mode of Nuclear Translocation of Adriamycin-Proteasome Complex. Cancer Res (2001) 61(6):2467–71.
22. Denard B, Lee C, Ye J. Doxorubicin Blocks Proliferation of Cancer Cells Through Proteolytic Activation of CREB3L1. Elife (2012) 1:e00090. doi: 10.7554/eLife.00090
23. Spencer DM, Bilardi RA, Koch TH, Post GC, Nafie JW, Kimura K, et al. DNA Repair in Response to Anthracycline-DNA Adducts: A Role for Both Homologous Recombination and Nucleotide Excision Repair. Mutat Res (2008) 638(1-2):110–21. doi: 10.1016/j.mrfmmm.2007.09.005
24. Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, et al. Topoisomerase Levels Determine Chemotherapy Response in Vitro and in Vivo. Proc Natl Acad Sci U.S.A. (2008) 105(26):9053–8. doi: 10.1073/pnas.0803513105
25. Zheng Q, Zhang M, Zhou F, Zhang L, Meng X. The Breast Cancer Stem Cells Traits and Drug Resistance. Front Pharmacol (2020) 11:599965. doi: 10.3389/fphar.2020.599965
26. Martin M. Docetaxel, Doxorubicin and Cyclophosphamide (the TAC Regimen): An Effective Adjuvant Treatment for Operable Breast Cancer. Womens Health (Lond) (2006) 2(4):527–37. doi: 10.2217/17455057.2.4.527
27. Field JJ, Kanakkanthara A, Miller JH. Microtubule-Targeting Agents are Clinically Successful Due to Both Mitotic and Interphase Impairment of Microtubule Function. Bioorg Med Chem (2014) 22(18):5050–9. doi: 10.1016/j.bmc.2014.02.035
28. Maloney SM, Hoover CA, Morejon-Lasso LV, Prosperi JR. Mechanisms of Taxane Resistance. Cancers (Basel). (2020) 12(11). doi: 10.3390/cancers12113323
29. Fromes Y, Gounon P, Veitia R, Bissery MC, Fellous A. Influence of Microtubule-Associated Proteins on the Differential Effects of Paclitaxel and Docetaxel. J Protein Chem (1996) 15(4):377–88. doi: 10.1007/BF01886864
30. Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane Resistance in Breast Cancer: Mechanisms, Predictive Biomarkers and Circumvention Strategies. Cancer Treat Rev (2012) 38(7):890–903. doi: 10.1016/j.ctrv.2012.02.011
31. Nami B, Wang Z. Genetics and Expression Profile of the Tubulin Gene Superfamily in Breast Cancer Subtypes and its Relation to Taxane Resistance. Cancers (Basel). (2018) 10(8). doi: 10.3390/cancers10080274
32. Galletti E, Magnani M, Renzulli ML, Botta M. Paclitaxel and Docetaxel Resistance: Molecular Mechanisms and Development of New Generation Taxanes. ChemMedChem (2007) 2(7):920–42. doi: 10.1002/cmdc.200600308
33. Tesch ME, Chia SK, Simmons CE, LeVasseur N. Impact of Sequence Order of Anthracyclines and Taxanes in Neoadjuvant Chemotherapy on Pathologic Complete Response Rate in HER2-Negative Breast Cancer Patients. Breast Cancer Res Treat (2021) 187(1):167–76. doi: 10.1007/s10549-021-06110-0
34. Achkar IW, Kader S, Dib SS, Junejo K, Al-Bader SB, Hayat S, et al. Metabolic Signatures of Tumor Responses to Doxorubicin Elucidated by Metabolic Profiling in Ovo. Metabolites (2020) 10(7). doi: 10.3390/metabo10070268
35. Semreen MH, Alniss H, Cacciatore S, El-Awady R, Mousa M, Almehdi AM, et al. GC-MS Based Comparative Metabolomic Analysis of MCF-7 and MDA-MB-231 Cancer Cells Treated With Tamoxifen and/or Paclitaxel. J Proteomics (2020) 225:103875. doi: 10.1016/j.jprot.2020.103875
36. Ma L, Zong X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front Oncol (2020) 10:5. doi: 10.3389/fonc.2020.00005
37. Liu Y, Zhou Q, Song S, Tang S. Integrating Metabolic Reprogramming and Metabolic Imaging to Predict Breast Cancer Therapeutic Responses. Trends Endocrinol Metab (2021) 32(10):762–75. doi: 10.1016/j.tem.2021.07.001
38. Punekar S, Cho DC. Novel Therapeutics Affecting Metabolic Pathways. Am Soc Clin Oncol Educ Book (2019) 39:e79–87. doi: 10.1200/EDBK_238499
39. Desbats MA, Giacomini I, Prayer-Galetti T, Montopoli M. Metabolic Plasticity in Chemotherapy Resistance. Front Oncol (2020) 10:281. doi: 10.3389/fonc.2020.00281
40. Marcucci F, Rumio C. Glycolysis-Induced Drug Resistance in Tumors-a Response to Danger Signals? Neoplasia (2021) 23(2):234–45. doi: 10.1016/j.neo.2020.12.009
41. Shin E, Koo JS. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front Cell Dev Biol (2021) 9:728759. doi: 10.3389/fcell.2021.728759
42. Melkonian EA, Schury MP. Biochemistry, Anaerobic Glycolysis. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2021).
43. Cossu V, Bonanomi M, Bauckneht M, Ravera S, Righi N, Miceli A, et al. Two High-Rate Pentose-Phosphate Pathways in Cancer Cells. Sci Rep (2020) 10(1):22111. doi: 10.1038/s41598-020-79185-2
44. Zala D, Schlattner U, Desvignes T, Bobe J, Roux A, Chavrier P, et al. The Advantage of Channeling Nucleotides for Very Processive Functions. F1000Res (2017) 6:724. doi: 10.12688/f1000research.11561.2
45. Sano R, Shinozaki Y, Ohta T. Sodium-Glucose Cotransporters: Functional Properties and Pharmaceutical Potential. J Diabetes Investig (2020) 11(4):770–82. doi: 10.1111/jdi.13255
46. Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative Glucose Transporters: Implications for Cancer Detection, Prognosis and Treatment. Metabolism (2016) 65(2):124–39. doi: 10.1016/j.metabol.2015.10.007
47. Jiang T, Zhou ML, Fan J. Inhibition of GLUT-1 Expression and the PI3K/Akt Pathway to Enhance the Chemosensitivity of Laryngeal Carcinoma Cells in Vitro. Onco Targets Ther (2018) 11:7865–72. doi: 10.2147/OTT.S176818
48. Wuest M, Hamann I, Bouvet V, Glubrecht D, Marshall A, Trayner B, et al. Molecular Imaging of GLUT1 and GLUT5 in Breast Cancer: A Multitracer Positron Emission Tomography Imaging Study in Mice. Mol Pharmacol (2018) 93(2):79–89. doi: 10.1124/mol.117.110007
49. Wellberg EA, Johnson S, Finlay-Schultz J, Lewis AS, Terrell KL, Sartorius CA, et al. The Glucose Transporter GLUT1 is Required for Erbb2-Induced Mammary Tumorigenesis. Breast Cancer Res (2016) 18(1):131. doi: 10.1186/s13058-016-0795-0
50. Pliszka M, Szablewski L. Glucose Transporters as a Target for Anticancer Therapy. Cancers (Basel) (2021) 13(16). doi: 10.3390/cancers13164184
51. Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P, et al. Glucose Uptake Inhibitor Sensitizes Cancer Cells to Daunorubicin and Overcomes Drug Resistance in Hypoxia. Cancer Chemother Pharmacol (2007) 59(4):495–505. doi: 10.1007/s00280-006-0291-9
52. Xintaropoulou C, Ward C, Wise A, Marston H, Turnbull A, Langdon SP. A Comparative Analysis of Inhibitors of the Glycolysis Pathway in Breast and Ovarian Cancer Cell Line Models. Oncotarget (2015) 6(28):25677–95. doi: 10.18632/oncotarget.4499
53. Li YL, Weng HC, Hsu JL, Lin SW, Guh JH, Hsu LC. The Combination of MK-2206 and WZB117 Exerts a Synergistic Cytotoxic Effect Against Breast Cancer Cells. Front Pharmacol (2019) 10:1311. doi: 10.3389/fphar.2019.01311
54. Wu Q, Ba-Alawi W, Deblois G, Cruickshank J, Duan S, Lima-Fernandes E, et al. GLUT1 Inhibition Blocks Growth of RB1-Positive Triple Negative Breast Cancer. Nat Commun (2020) 11(1):4205. doi: 10.1038/s41467-020-18020-8
55. Barbosa AM, Martel F. Targeting Glucose Transporters for Breast Cancer Therapy: The Effect of Natural and Synthetic Compounds. Cancers (Basel) (2020) 12(1). doi: 10.3390/cancers12010154
56. Yang Y, Wolfram J, Boom K, Fang X, Shen H, Ferrari M. Hesperetin Impairs Glucose Uptake and Inhibits Proliferation of Breast Cancer Cells. Cell Biochem Funct (2013) 31(5):374–9. doi: 10.1002/cbf.2905
57. Chien AJ, Tripathy D, Albain KS, Symmans WF, Rugo HS, Melisko ME, et al. MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J Clin Oncol (2020) 38(10):1059–69. doi: 10.1200/JCO.19.01027
58. O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose Inhibits Aggressive Triple-Negative Breast Cancer Cells by Targeting Glycolysis and the Cancer Stem Cell Phenotype. Sci Rep (2019) 9(1):3788. doi: 10.1038/s41598-019-39789-9
59. Jia L, Huang S, Yin X, Zan Y, Guo Y, Han L. Quercetin Suppresses the Mobility of Breast Cancer by Suppressing Glycolysis Through Akt-Mtor Pathway Mediated Autophagy Induction. Life Sci (2018) :208:123–30. doi: 10.1016/j.lfs.2018.07.027
60. Jung KH, Lee JH, Thien Quach CH, Paik JY, Oh H, Park JW, et al. Resveratrol Suppresses Cancer Cell Glucose Uptake by Targeting Reactive Oxygen Species-Mediated Hypoxia-Inducible Factor-1alpha Activation. J Nucl Med (2013) 54(12):2161–7. doi: 10.2967/jnumed.112.115436
61. Cesari IM, Carvalho E, Figueiredo Rodrigues M, Mendonca Bdos S, Amoedo ND, Rumjanek FD. Methyl Jasmonate: Putative Mechanisms of Action on Cancer Cells Cycle, Metabolism, and Apoptosis. Int J Cell Biol (2014) 2014:572097. doi: 10.1155/2014/572097
62. Ozkan E, Bakar-Ates F. Potentiation of the Effect of Lonidamine by Quercetin in MCF-7 Human Breast Cancer Cells Through Downregulation of MMP-2/9 Mrna Expression. Acad Bras Cienc (2020) 92(4):e20200548. doi: 10.1590/0001-3765202020200548
63. Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, et al. Glycolysis Inhibition Inactivates ABC Transporters to Restore Drug Sensitivity in Malignant Cells. PloS One (2011) 6(11):e27222. doi: 10.1371/journal.pone.0027222
64. Zhang Q, Zhang Y, Zhang P, Chao Z, Xia F, Jiang C, et al. Hexokinase II Inhibitor, 3-Brpa Induced Autophagy by Stimulating ROS Formation in Human Breast Cancer Cells. Genes Cancer (2014) 5(3-4):100–12. doi: 10.18632/genesandcancer.9
65. Bao B, Azmi AS, Ali S, Zaiem F, Sarkar FH. Metformin may Function as Anti-Cancer Agent via Targeting Cancer Stem Cells: The Potential Biological Significance of Tumor-Associated Mirnas in Breast and Pancreatic Cancers. Ann Transl Med (2014) 2(6):59. doi: 10.3978/j.issn.2305-5839.2014.06.05
66. Gomez LS, Zancan P, Marcondes MC, Ramos-Santos L, Meyer-Fernandes JR, Sola-Penna M, et al. Resveratrol Decreases Breast Cancer Cell Viability and Glucose Metabolism by Inhibiting 6-Phosphofructo-1-Kinase. Biochimie (2013) 95(6):1336–43. doi: 10.1016/j.biochi.2013.02.013
67. Galindo CM, Oliveira Ganzella FA, Klassen G, Souza Ramos EA, Acco A. Nuances of PFKFB3 Signaling in Breast Cancer. Clin Breast Cancer (2022). doi: 10.1016/j.clbc.2022.01.002
68. Ghergurovich JM, Garcia-Canaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T, et al. A Small Molecule G6PD Inhibitor Reveals Immune Dependence on Pentose Phosphate Pathway. Nat Chem Biol (2020) 16(7):731–9. doi: 10.1038/s41589-020-0533-x
69. Li W, Liu J, Jackson K, Shi R, Zhao Y. Sensitizing the Therapeutic Efficacy of Taxol With Shikonin in Human Breast Cancer Cells. PloS One (2014) 9(4):e94079. doi: 10.1371/journal.pone.0094079
70. Jiang K, He B, Lai L, Chen Q, Liu Y, Guo Q, et al. Cyclosporine a Inhibits Breast Cancer Cell Growth by Downregulating the Expression of Pyruvate Kinase Subtype M2. Int J Mol Med (2012) 30(2):302–8. doi: 10.3892/ijmm.2012.989
71. Garcia-Castillo V, Lopez-Urrutia E, Villanueva-Sanchez O, Avila-Rodriguez MA, Zentella-Dehesa A, Cortes-Gonzalez C, et al. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J Cancer (2017) 8(2):178–89. doi: 10.7150/jca.16387
72. Mazzio E, Mack N, Badisa RB, Soliman KFA. Triple Isozyme Lactic Acid Dehydrogenase Inhibition in Fully Viable MDA-MB-231 Cells Induces Cytostatic Effects That are Not Reversed by Exogenous Lactic Acid. Biomolecules (2021) 11(12). doi: 10.3390/biom11121751
73. Liu C, Wang X, Zhang Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol Cancer Res Treat (2019) 18:1533033819871306. doi: 10.1177/1533033819871306
74. Varghese E, Samuel SM, Liskova A, Samec M, Kubatka P, Busselberg D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers (Basel). (2020) 12(8). doi: 10.3390/cancers12082252
75. Catanzaro D, Gabbia D, Cocetta V, Biagi M, Ragazzi E, Montopoli M, et al. Silybin Counteracts Doxorubicin Resistance by Inhibiting GLUT1 Expression. Fitoterapia (2018) 124:42–8. doi: 10.1016/j.fitote.2017.10.007
76. Zhou R, Vander Heiden MG, Rudin CM. Genotoxic Exposure is Associated With Alterations in Glucose Uptake and Metabolism. Cancer Res (2002) 62(12):3515–20.
77. De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, et al. Paclitaxel, Cisplatin and Lonidamine in Advanced Ovarian Cancer. A Phase II Study. Eur J Cancer (2001) 37(3):364–8. doi: 10.1016/S0959-8049(00)00400-7
78. Gu JJ, Singh A, Xue K, Mavis C, Barth M, Yanamadala V, et al. Up-Regulation of Hexokinase II Contributes to Rituximab-Chemotherapy Resistance and is a Clinically Relevant Target for Therapeutic Development. Oncotarget (2018) 9(3):4020–33. doi: 10.18632/oncotarget.23425
79. Pateliya B, Burade V, Goswami S. Combining Naringenin and Metformin With Doxorubicin Enhances Anticancer Activity Against Triple-Negative Breast Cancer in Vitro and in Vivo. Eur J Pharmacol (2021) 891:173725. doi: 10.1016/j.ejphar.2020.173725
80. Wahdan-Alaswad RS, Cochrane DR, Spoelstra NS, Howe EN, Edgerton SM, Anderson SM, et al. Metformin-Induced Killing of Triple-Negative Breast Cancer Cells is Mediated by Reduction in Fatty Acid Synthase via Mirna-193b. Horm Cancer (2014) 5(6):374–89. doi: 10.1007/s12672-014-0188-8
81. Dankner R, Agay N, Olmer L, Murad H, Keinan Boker L, Balicer RD, et al. Metformin Treatment and Cancer Risk: Cox Regression Analysis, With Time-Dependent Covariates, of 320,000 Persons With Incident Diabetes Mellitus. Am J Epidemiol (2019) 188(10):1794–800. doi: 10.1093/aje/kwz157
82. Nakamura M, Nagase K, Yoshimitsu M, Magara T, Nojiri Y, Kato H, et al. Glucose-6-Phosphate Dehydrogenase Correlates With Tumor Immune Activity and Programmed Death Ligand-1 Expression in Merkel Cell Carcinoma. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-001679
83. Yang HC, Stern A, Chiu DT. G6PD: A Hub for Metabolic Reprogramming and Redox Signaling in Cancer. BioMed J (2021) 44(3):285–92. doi: 10.1016/j.bj.2020.08.001
84. Ghergurovich JM, Esposito M, Chen Z, Wang JZ, Bhatt V, Lan T, et al. Glucose-6-Phosphate Dehydrogenase is Not Essential for K-Ras-Driven Tumor Growth or Metastasis. Cancer Res (2020) 80(18):3820–9. doi: 10.1158/0008-5472.CAN-19-2486
85. Debeb BG, Lacerda L, Larson R, Wolfe AR, Krishnamurthy S, Reuben JM, et al. Histone Deacetylase Inhibitor-Induced Cancer Stem Cells Exhibit High Pentose Phosphate Pathway Metabolism. Oncotarget (2016) 7(19):28329–39. doi: 10.18632/oncotarget.8631
86. Preuss J, Richardson AD, Pinkerton A, Hedrick M, Sergienko E, Rahlfs S, et al. Identification and Characterization of Novel Human Glucose-6-Phosphate Dehydrogenase Inhibitors. J Biomol Screen (2013) 18(3):286–97. doi: 10.1177/1087057112462131
87. Kotowski K, Rosik J, Machaj F, Supplitt S, Wiczew D, Jablonska K, et al. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers (Basel). (2021) 13(4). doi: 10.3390/cancers13040909
88. Lu L, Chen Y, Zhu Y. The Molecular Basis of Targeting PFKFB3 as a Therapeutic Strategy Against Cancer. Oncotarget (2017) 8(37):62793–802. doi: 10.18632/oncotarget.19513
89. Yan H, Yang K, Xiao H, Zou YJ, Zhang WB, Liu HY. Over-Expression of Cofilin-1 and Phosphoglycerate Kinase 1 in Astrocytomas Involved in Pathogenesis of Radioresistance. CNS Neurosci Ther (2012) 18(9):729–36. doi: 10.1111/j.1755-5949.2012.00353.x
90. Lincet H, Guevel B, Pineau C, Allouche S, Lemoisson E, Poulain L, et al. Comparative 2D-DIGE Proteomic Analysis of Ovarian Carcinoma Cells: Toward a Reorientation of Biosynthesis Pathways Associated With Acquired Platinum Resistance. J Proteomics (2012) 75(4):1157–69. doi: 10.1016/j.jprot.2011.10.030
91. Schneider CC, Archid R, Fischer N, Buhler S, Venturelli S, Berger A, et al. Metabolic Alteration–Overcoming Therapy Resistance in Gastric Cancer via PGK-1 Inhibition in a Combined Therapy With Standard Chemotherapeutics. Int J Surg (2015) 22:92–8. doi: 10.1016/j.ijsu.2015.08.020
92. Sun S, Liang X, Zhang X, Liu T, Shi Q, Song Y, et al. Phosphoglycerate Kinase-1 is a Predictor of Poor Survival and a Novel Prognostic Biomarker of Chemoresistance to Paclitaxel Treatment in Breast Cancer. Br J Cancer (2015) 112(8):1332–9. doi: 10.1038/bjc.2015.114
93. Tu SH, Chang CC, Chen CS, Tam KW, Wang YJ, Lee CH, et al. Increased Expression of Enolase Alpha in Human Breast Cancer Confers Tamoxifen Resistance in Human Breast Cancer Cells. Breast Cancer Res Treat (2010) 121(3):539–53. doi: 10.1007/s10549-009-0492-0
94. Qian X, Xu W, Xu J, Shi Q, Li J, Weng Y, et al. Enolase 1 Stimulates Glycolysis to Promote Chemoresistance in Gastric Cancer. Oncotarget (2017) 8(29):47691–708. doi: 10.18632/oncotarget.17868
95. Almaguel FA, Sanchez TW, Ortiz-Hernandez GL, Casiano CA. Alpha-Enolase: Emerging Tumor-Associated Antigen, Cancer Biomarker, and Oncotherapeutic Target. Front Genet (2020) 11:614726. doi: 10.3389/fgene.2020.614726
96. Chen X, Chen S, Yu D. Protein Kinase Function of Pyruvate Kinase M2 and Cancer. Cancer Cell Int (2020) 20(1):523. doi: 10.1186/s12935-020-01612-1
97. Hsu MC, Hung WC. Pyruvate Kinase M2 Fuels Multiple Aspects of Cancer Cells: From Cellular Metabolism, Transcriptional Regulation to Extracellular Signaling. Mol Cancer (2018) 17(1):35. doi: 10.1186/s12943-018-0791-3
98. Castagnoli L, Iorio E, Dugo M, Koschorke A, Faraci S, Canese R, et al. Intratumor Lactate Levels Reflect HER2 Addiction Status in HER2-Positive Breast Cancer. J Cell Physiol (2019) 234(2):1768–79. doi: 10.1002/jcp.27049
99. Lin Y, Lv F, Liu F, Guo X, Fan Y, Gu F, et al. High Expression of Pyruvate Kinase M2 is Associated With Chemosensitivity to Epirubicin and 5-Fluorouracil in Breast Cancer. J Cancer (2015) 6(11):1130–9. doi: 10.7150/jca.12719
100. Qian Y, Bi L, Yang Y, Wang D. Effect of Pyruvate Kinase M2-Regulating Aerobic Glycolysis on Chemotherapy Resistance of Estrogen Receptor-Positive Breast Cancer. Anticancer Drugs (2018) 29(7):616–27. doi: 10.1097/CAD.0000000000000624
101. Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, et al. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clin Cancer Res (2014) 20(10):2595–606. doi: 10.1158/1078-0432.CCR-13-2407
102. Kong L, Du W, Cui Z, Wang L, Yang Z, Zhang H, et al. Expression of Lactate Dehydrogenase C in MDAMB231 Cells and its Role in Tumor Invasion and Migration. Mol Med Rep (2016) 13(4):3533–8. doi: 10.3892/mmr.2016.4963
103. Mishra D, Banerjee D. Correction: Mishra, Et al.; Lactate Dehydrogenases as Metabolic Links Between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. Cancers (Basel) (2020) 12(4):932. doi: 10.3390/cancers12040932
104. Jiang F, Ma S, Xue Y, Hou J, Zhang Y. LDH-a Promotes Malignant Progression via Activation of Epithelial-to-Mesenchymal Transition and Conferring Stemness in Muscle-Invasive Bladder Cancer. Biochem Biophys Res Commun (2016) 469(4):985–92. doi: 10.1016/j.bbrc.2015.12.078
105. Arseneault R, Chien A, Newington JT, Rappon T, Harris R, Cumming RC. Attenuation of LDHA Expression in Cancer Cells Leads to Redox-Dependent Alterations in Cytoskeletal Structure and Cell Migration. Cancer Lett (2013) 338(2):255–66. doi: 10.1016/j.canlet.2013.03.034
106. Hua G, Liu Y, Li X, Xu P, Luo Y. Targeting Glucose Metabolism in Chondrosarcoma Cells Enhances the Sensitivity to Doxorubicin Through the Inhibition of Lactate Dehydrogenase-a. Oncol Rep (2014) 31(6):2727–34. doi: 10.3892/or.2014.3156
107. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg Effect in Chemosensitivity: Targeting Lactate Dehydrogenase-a Re-Sensitizes Taxol-Resistant Cancer Cells to Taxol. Mol Cancer (2010) 9:33. doi: 10.1186/1476-4598-9-33
108. Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K. Lactate Dehydrogenase M-Subunit Deficiency: A New Type of Hereditary Exertional Myopathy. Clin Chim Acta (1988) 173(1):89–98. doi: 10.1016/0009-8981(88)90359-2
109. Jeong DW, Cho IT, Kim TS, Bae GW, Kim IH, Kim IY. Effects of Lactate Dehydrogenase Suppression and Glycerol-3-Phosphate Dehydrogenase Overexpression on Cellular Metabolism. Mol Cell Biochem (2006) 284(1-2):1–8. doi: 10.1007/s11010-005-9004-7
110. McCleland ML, Adler AS, Shang Y, Hunsaker T, Truong T, Peterson D, et al. An Integrated Genomic Screen Identifies LDHB as an Essential Gene for Triple-Negative Breast Cancer. Cancer Res (2012) 72(22):5812–23. doi: 10.1158/0008-5472.CAN-12-1098
111. Shibata S, Sogabe S, Miwa M, Fujimoto T, Takakura N, Naotsuka A, et al. Identification of the First Highly Selective Inhibitor of Human Lactate Dehydrogenase B. Sci Rep (2021) 11(1):21353. doi: 10.1038/s41598-021-00820-7
112. Boros LG, Lee PW, Brandes JL, Cascante M, Muscarella P, Schirmer WJ, et al. Nonoxidative Pentose Phosphate Pathways and Their Direct Role in Ribose Synthesis in Tumors: Is Cancer a Disease of Cellular Glucose Metabolism? Med Hypotheses (1998) 50(1):55–9. doi: 10.1016/S0306-9877(98)90178-5
113. Alfarouk KO, Ahmed SBM, Elliott RL, Benoit A, Alqahtani SS, Ibrahim ME, et al. The Pentose Phosphate Pathway Dynamics in Cancer and its Dependency on Intracellular Ph. Metabolites (2020) 10(7). doi: 10.3390/metabo10070285
114. Giacomini I, Ragazzi E, Pasut G, Montopoli M. The Pentose Phosphate Pathway and its Involvement in Cisplatin Resistance. Int J Mol Sci (2020) 21(3). doi: 10.3390/ijms21030937
115. Goldman A, Khiste S, Freinkman E, Dhawan A, Majumder B, Mondal J, et al. Targeting Tumor Phenotypic Plasticity and Metabolic Remodeling in Adaptive Cross-Drug Tolerance. Sci Signal (2019) 12(595). doi: 10.1126/scisignal.aas8779
116. Polimeni M, Voena C, Kopecka J, Riganti C, Pescarmona G, Bosia A, et al. Modulation of Doxorubicin Resistance by the Glucose-6-Phosphate Dehydrogenase Activity. Biochem J (2011) 439(1):141–9. doi: 10.1042/BJ20102016
117. Haddad A, Mohiuddin SS. Biochemistry, Citric Acid Cycle. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2021).
118. Shen N, Korm S, Karantanos T, Li D, Zhang X, Ritou E, et al. DLST-Dependence Dictates Metabolic Heterogeneity in TCA-Cycle Usage Among Triple-Negative Breast Cancer. Commun Biol (2021) 4(1):1289. doi: 10.1038/s42003-021-02805-8
119. Anderson NM, Mucka P, Kern JG, Feng H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell (2018) 9(2):216–37. doi: 10.1007/s13238-017-0451-1
120. Evans KW, Yuca E, Scott SS, Zhao M, Paez Arango N, Cruz Pico CX, et al. Oxidative Phosphorylation is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res (2021) 81(21):5572–81. doi: 10.1158/0008-5472.CAN-20-3242
121. Pacheco-Velazquez SC, Robledo-Cadena DX, Hernandez-Resendiz I, Gallardo-Perez JC, Moreno-Sanchez R, Rodriguez-Enriquez S. Energy Metabolism Drugs Block Triple Negative Breast Metastatic Cancer Cell Phenotype. Mol Pharm (2018) 15(6):2151–64. doi: 10.1021/acs.molpharmaceut.8b00015
122. McGuirk S, Audet-Delage Y, Annis MG, Xue Y, Vernier M, Zhao K, et al. Resistance to Different Anthracycline Chemotherapeutics Elicits Distinct and Actionable Primary Metabolic Dependencies in Breast Cancer. Elife (2021) 10. doi: 10.7554/eLife.65150.sa2
123. Ryu HH, Ahn SH, Kim SO, Kim JE, Kim JS, Ahn JH, et al. Comparison of Metabolic Changes After Neoadjuvant Endocrine and Chemotherapy in ER-Positive, HER2-Negative Breast Cancer. Sci Rep (2021) 11(1):10510. doi: 10.1038/s41598-021-89651-0
124. Draguet A, Tagliatti V, Colet JM. Targeting Metabolic Reprogramming to Improve Breast Cancer Treatment: An in Vitro Evaluation of Selected Metabolic Inhibitors Using a Metabolomic Approach. Metabolites (2021) 11(8). doi: 10.3390/metabo11080556
125. Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA Cancer J Clin (2017) 67(5):378–97. doi: 10.3322/caac.21405
126. Bougaret L, Delort L, Billard H, Le Huede C, Boby C, de la Foye A, et al. Adipocyte/Breast Cancer Cell Crosstalk in Obesity Interferes With the Anti-Proliferative Efficacy of Tamoxifen. PloS One (2018) 13(2):e0191571. doi: 10.1371/journal.pone.0191571
127. Mentoor I, Nell T, Emjedi Z, van Jaarsveld PJ, de Jager L, Engelbrecht AM. Decreased Efficacy of Doxorubicin Corresponds With Modifications in Lipid Metabolism Markers and Fatty Acid Profiles in Breast Tumors From Obese vs. Lean Mice Front Oncol (2020) 10:306. doi: 10.3389/fonc.2020.00306
128. Havas KM, Milchevskaya V, Radic K, Alladin A, Kafkia E, Garcia M, et al. Metabolic Shifts in Residual Breast Cancer Drive Tumor Recurrence. J Clin Invest (2017) 127(6):2091–105. doi: 10.1172/JCI89914
129. Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P. Lipid Metabolism and Resistance to Anticancer Treatment. Biol (Basel). (2020) 9(12). doi: 10.3390/biology9120474
130. Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, et al. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv Drug Delivery Rev (2020) 159:245–93. doi: 10.1016/j.addr.2020.07.013
131. Eiriksson FF, Nohr MK, Costa M, Bodvarsdottir SK, Ogmundsdottir HM, Thorsteinsdottir M. Lipidomic Study of Cell Lines Reveals Differences Between Breast Cancer Subtypes. PloS One (2020) 15(4):e0231289. doi: 10.1371/journal.pone.0231289
132. Buas MF, Drescher CW, Urban N, Li CI, Bettcher L, Hait NC, et al. Quantitative Global Lipidomics Analysis of Patients With Ovarian Cancer Versus Benign Adnexal Mass. Sci Rep (2021) 11(1):18156. doi: 10.1038/s41598-021-97433-x
133. Dietze R, Hammoud MK, Gomez-Serrano M, Unger A, Bieringer T, Finkernagel F, et al. Phosphoproteomics Identify Arachidonic-Acid-Regulated Signal Transduction Pathways Modulating Macrophage Functions With Implications for Ovarian Cancer. Theranostics (2021) 11(3):1377–95. doi: 10.7150/thno.52442
134. Cioce M, Canino C, Pass H, Blandino G, Strano S, Fazio VM. Arachidonic Acid Drives Adaptive Responses to Chemotherapy-Induced Stress in Malignant Mesothelioma. J Exp Clin Cancer Res (2021) 40(1):344. doi: 10.1186/s13046-021-02118-y
135. Hoy AJ, Nagarajan SR, Butler LM. Tumour Fatty Acid Metabolism in the Context of Therapy Resistance and Obesity. Nat Rev Cancer (2021) 21(12):753–66. doi: 10.1038/s41568-021-00388-4
136. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid Beta-Oxidation is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27(1):136–50 e5. doi: 10.1016/j.cmet.2017.11.001
137. Liu H, Liu Y, Zhang JT. A New Mechanism of Drug Resistance in Breast Cancer Cells: Fatty Acid Synthase Overexpression-Mediated Palmitate Overproduction. Mol Cancer Ther (2008) 7(2):263–70. doi: 10.1158/1535-7163.MCT-07-0445
138. Al-Bahlani S, Al-Lawati H, Al-Adawi M, Al-Abri N, Al-Dhahli B, Al-Adawi K. Fatty Acid Synthase Regulates the Chemosensitivity of Breast Cancer Cells to Cisplatin-Induced Apoptosis. Apoptosis (2017) 22(6):865–76. doi: 10.1007/s10495-017-1366-2
139. Geneste A, Duong MN, Molina L, Conilh L, Beaumel S, Cleret A, et al. Adipocyte-Conditioned Medium Induces Resistance of Breast Cancer Cells to Lapatinib. BMC Pharmacol Toxicol (2020) 21(1):61. doi: 10.1186/s40360-020-00436-z
140. Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, et al. Inhibition of Fatty Acid Synthase (FAS) Suppresses HER2/Neu (Erbb-2) Oncogene Overexpression in Cancer Cells. Proc Natl Acad Sci U S A (2004) 101(29):10715–20. doi: 10.1073/pnas.0403390101
141. Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, Kangaspeska S, et al. Association of Tamoxifen Resistance and Lipid Reprogramming in Breast Cancer. BMC Cancer (2018) 18(1):850. doi: 10.1186/s12885-018-4757-z
142. Glatz JF, Luiken JJ, Bonen A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol Rev (2010) 90(1):367–417. doi: 10.1152/physrev.00003.2009
143. Schwenk RW, Holloway GP, Luiken JJ, Bonen A, Glatz JF. Fatty Acid Transport Across the Cell Membrane: Regulation by Fatty Acid Transporters. Prostaglandins Leukot Essent Fatty Acids (2010) 82(4-6):149–54. doi: 10.1016/j.plefa.2010.02.029
144. Feng WW, Wilkins O, Bang S, Ung M, Li J, An J, et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep (2019) 29(11):3405–20 e5. doi: 10.1016/j.celrep.2019.11.008
145. Nagarajan SR, Butler LM, Hoy AJ. The Diversity and Breadth of Cancer Cell Fatty Acid Metabolism. Cancer Metab (2021) 9(1):2. doi: 10.1186/s40170-020-00237-2
146. Talley JT, Mohiuddin SS. Biochemistry, Fatty Acid Oxidation. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2021).
147. Monaco ME. Fatty Acid Metabolism in Breast Cancer Subtypes. Oncotarget (2017) 8(17):29487–500. doi: 10.18632/oncotarget.15494
148. Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An Integrated Genomics Approach Identifies Drivers of Proliferation in Luminal-Subtype Human Breast Cancer. Nat Genet (2014) 46(10):1051–9. doi: 10.1038/ng.3073
149. Wanders RJ, Ruiter JP, L IJ, Waterham HR, Houten SM. The Enzymology of Mitochondrial Fatty Acid Beta-Oxidation and its Application to Follow-Up Analysis of Positive Neonatal Screening Results. J Inherit Metab Dis (2010) 33(5):479–94. doi: 10.1007/s10545-010-9104-8
150. Martinez-Reyes I, Chandel NS. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
151. Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, et al. Fatty Acid Oxidation Mediated by Acyl-Coa Synthetase Long Chain 3 is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep (2016) 16(6):1614–28. doi: 10.1016/j.celrep.2016.07.009
152. Wang MD, Wu H, Huang S, Zhang HL, Qin CJ, Zhao LH, et al. Hbx Regulates Fatty Acid Oxidation to Promote Hepatocellular Carcinoma Survival During Metabolic Stress. Oncotarget (2016) 7(6):6711–26. doi: 10.18632/oncotarget.6817
153. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
154. Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of Fatty Acid Oxidation as a Therapy for MYC-Overexpressing Triple-Negative Breast Cancer. Nat Med (2016) 22(4):427–32. doi: 10.1038/nm.4055
155. Luo J, Hong Y, Tao X, Wei X, Zhang L, Li Q. An Indispensable Role of CPT-1a to Survive Cancer Cells During Energy Stress Through Rewiring Cancer Metabolism. Tumour Biol (2016) 37:15795–804. doi: 10.1007/s13277-016-5382-6
156. He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, et al. MSC-Regulated Lncrna MACC1-AS1 Promotes Stemness and Chemoresistance Through Fatty Acid Oxidation in Gastric Cancer. Oncogene (2019) 38(23):4637–54. doi: 10.1038/s41388-019-0747-0
157. Fu Y, Zou T, Shen X, Nelson PJ, Li J, Wu C, et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm (2020) (2021) 2(1):27–59. doi: 10.1002/mco2.27
158. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP Activity is Regulated by Mtorc1 and Contributes to Akt-Dependent Cell Growth. Cell Metab (2008) 8(3):224–36. doi: 10.1016/j.cmet.2008.07.007
159. Feng WW, Kurokawa M. Lipid Metabolic Reprogramming as an Emerging Mechanism of Resistance to Kinase Inhibitors in Breast Cancer. Cancer Drug Resist (2020) 3(1). doi: 10.20517/cdr.2019.100
160. Li J, Zhao S, Zhou X, Zhang T, Zhao L, Miao P, et al. Inhibition of Lipolysis by Mercaptoacetate and Etomoxir Specifically Sensitize Drug-Resistant Lung Adenocarcinoma Cell to Paclitaxel. PloS One (2013) 8(9):e74623. doi: 10.1371/journal.pone.0074623
161. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells are Not Enriched for Leukemic Stem Cells But Require Oxidative Metabolism. Cancer Discovery (2017) 7(7):716–35. doi: 10.1158/2159-8290.CD-16-0441
162. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-Like Cells Through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab (2016) 23(1):206–19. doi: 10.1016/j.cmet.2015.12.004
163. Rohrig F, Schulze A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat Rev Cancer (2016) 16(11):732–49. doi: 10.1038/nrc.2016.89
164. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular Fatty Acid Metabolism and Cancer. Cell Metab (2013) 18(2):153–61. doi: 10.1016/j.cmet.2013.05.017
165. Bertolio R, Napoletano F, Mano M, Maurer-Stroh S, Fantuz M, Zannini A, et al. Sterol Regulatory Element Binding Protein 1 Couples Mechanical Cues and Lipid Metabolism. Nat Commun (2019) 10(1):1326. doi: 10.1038/s41467-019-09152-7
166. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
167. Dorotea D, Koya D, Ha H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front Pharmacol (2020) 11:265. doi: 10.3389/fphar.2020.00265
168. Bao J, Zhu L, Zhu Q, Su J, Liu M, Huang W. SREBP-1 is an Independent Prognostic Marker and Promotes Invasion and Migration in Breast Cancer. Oncol Lett (2016) 12(4):2409–16. doi: 10.3892/ol.2016.4988
169. Shao W, Machamer CE, Espenshade PJ. Fatostatin Blocks ER Exit of SCAP But Inhibits Cell Growth in a SCAP-Independent Manner. J Lipid Res (2016) 57(8):1564–73. doi: 10.1194/jlr.M069583
170. Sardesai SD, Thomas A, Gallagher C, Lynce F, Ottaviano YL, Ballinger TJ, et al. Inhibiting Fatty Acid Synthase With Omeprazole to Improve Efficacy of Neoadjuvant Chemotherapy in Patients With Operable TNBC. Clin Cancer Res (2021) 27(21):5810–7. doi: 10.1158/1078-0432.CCR-21-0493
171. Menendez JA, Lupu R, Colomer R. Inhibition of Tumor-Associated Fatty Acid Synthase Hyperactivity Induces Synergistic Chemosensitization of HER -2/Neu -Overexpressing Human Breast Cancer Cells to Docetaxel (Taxotere). Breast Cancer Res Treat (2004) 84(2):183–95. doi: 10.1023/B:BREA.0000018409.59448.60
172. Liu H, Wu X, Dong Z, Luo Z, Zhao Z, Xu Y, et al. Fatty Acid Synthase Causes Drug Resistance by Inhibiting TNF-Alpha and Ceramide Production. J Lipid Res (2013) 54(3):776–85. doi: 10.1194/jlr.M033811
173. Wang W, Bai L, Li W, Cui J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front Oncol (2020) 10:605154. doi: 10.3389/fonc.2020.605154
174. Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, et al. Orlistat as a FASN Inhibitor and Multitargeted Agent for Cancer Therapy. Expert Opin Investig Drugs (2018) 27(5):475–89. doi: 10.1080/13543784.2018.1471132
175. Aquino IG, Bastos DC, Cuadra-Zelaya FJM, Teixeira IF, Salo T, Coletta RD, et al. Anticancer Properties of the Fatty Acid Synthase Inhibitor TVB-3166 on Oral Squamous Cell Carcinoma Cell Lines. Arch Oral Biol (2020) 113:104707. doi: 10.1016/j.archoralbio.2020.104707
176. Zaytseva YY, Rychahou PG, Le AT, Scott TL, Flight RM, Kim JT, et al. Preclinical Evaluation of Novel Fatty Acid Synthase Inhibitors in Primary Colorectal Cancer Cells and a Patient-Derived Xenograft Model of Colorectal Cancer. Oncotarget (2018) 9(37):24787–800. doi: 10.18632/oncotarget.25361
177. Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, et al. Malonyl-Coenzyme-a is a Potential Mediator of Cytotoxicity Induced by Fatty-Acid Synthase Inhibition in Human Breast Cancer Cells and Xenografts. Cancer Res (2000) 60(2):213–8.
178. Puig T, Aguilar H, Cufi S, Oliveras G, Turrado C, Ortega-Gutierrez S, et al. A Novel Inhibitor of Fatty Acid Synthase Shows Activity Against HER2+ Breast Cancer Xenografts and is Active in Anti-HER2 Drug-Resistant Cell Lines. Breast Cancer Res (2011) 13(6):R131. doi: 10.1186/bcr3077
179. Krol SK, Kielbus M, Rivero-Muller A, Stepulak A. Comprehensive Review on Betulin as a Potent Anticancer Agent. BioMed Res Int (2015) 2015:584189. doi: 10.1155/2015/584189
180. Roelands J, Garand M, Hinchcliff E, Ma Y, Shah P, Toufiq M, et al. Long-Chain Acyl-Coa Synthetase 1 Role in Sepsis and Immunity: Perspectives From a Parallel Review of Public Transcriptome Datasets and of the Literature. Front Immunol (2019) 10:2410. doi: 10.3389/fimmu.2019.02410
181. Ding Y, Fan J, Fan Z, Zhang K. Gamma-Tocotrienol Reverses Multidrug Resistance of Breast Cancer Cells Through the Regulation of the Gamma-Tocotrienol-NF-Kappab-P-Gp Axis. J Steroid Biochem Mol Biol (2021) 209:105835. doi: 10.1016/j.jsbmb.2021.105835
182. Ding Y, Peng Y, Deng L, Fan J, Huang B. Gamma-Tocotrienol Reverses Multidrug Resistance of Breast Cancer Cells With a Mechanism Distinct From That of Atorvastatin. J Steroid Biochem Mol Biol (2017) 167:67–77. doi: 10.1016/j.jsbmb.2016.11.009
183. Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, et al. Inhibition of Farnesyltransferase Induces Regression of Mammary and Salivary Carcinomas in Ras Transgenic Mice. Nat Med (1995) 1(8):792–7. doi: 10.1038/nm0895-792
184. Brohee L, Demine S, Willems J, Arnould T, Colige AC, Deroanne CF. Lipin-1 Regulates Cancer Cell Phenotype and is a Potential Target to Potentiate Rapamycin Treatment. Oncotarget (2015) 6(13):11264–80. doi: 10.18632/oncotarget.3595
185. Liang Y, Han H, Liu L, Duan Y, Yang X, Ma C, et al. CD36 Plays a Critical Role in Proliferation, Migration and Tamoxifen-Inhibited Growth of ER-Positive Breast Cancer Cells. Oncogenesis (2018) 7(12):98. doi: 10.1038/s41389-018-0107-x
186. Ren XR, Wang J, Osada T, Mook RA Jr., Morse MA, Barak LS, et al. Perhexiline Promotes HER3 Ablation Through Receptor Internalization and Inhibits Tumor Growth. Breast Cancer Res (2015) 17:20. doi: 10.1186/s13058-015-0528-9
187. Mohammadzadeh F, Mosayebi G, Montazeri V, Darabi M, Fayezi S, Shaaker M, et al. Fatty Acid Composition of Tissue Cultured Breast Carcinoma and the Effect of Stearoyl-Coa Desaturase 1 Inhibition. J Breast Cancer (2014) 17(2):136–42. doi: 10.4048/jbc.2014.17.2.136
188. Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor Resistance to Ferroptosis Driven by Stearoyl-Coa Desaturase-1 (SCD1) in Cancer Cells and Fatty Acid Binding Protein-4 (FABP4) in Tumor Microenvironment Promote Tumor Recurrence. Redox Biol (2021) 43:102006. doi: 10.1016/j.redox.2021.102006
189. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a Novel Inhibitor of Fatty Acid Synthase With Antitumor Activity. Cancer Res (2004) 64(6):2070–5. doi: 10.1158/0008-5472.CAN-03-3645
190. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of Acetyl-Coa Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab (2019) 29(1):174–82 e5. doi: 10.1016/j.cmet.2018.08.020
191. Jones JE, Esler WP, Patel R, Lanba A, Vera NB, Pfefferkorn JA, et al. Inhibition of Acetyl-Coa Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of Egfrviii Human Glioblastoma Cells. PloS One (2017) 12(1):e0169566. doi: 10.1371/journal.pone.0169566
192. Jeon SM, Chandel NS, Hay N. AMPK Regulates NADPH Homeostasis to Promote Tumour Cell Survival During Energy Stress. Nature (2012) 485(7400):661–5. doi: 10.1038/nature11066
193. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. ATP Citrate Lyase: A Central Metabolic Enzyme in Cancer. Cancer Lett (2020) 471:125–34. doi: 10.1016/j.canlet.2019.12.010
194. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP Citrate Lyase Inhibition can Suppress Tumor Cell Growth. Cancer Cell (2005) 8(4):311–21. doi: 10.1016/j.ccr.2005.09.008
195. Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, et al. ATP Citrate Lyase (ACLY): A Promising Target for Cancer Prevention and Treatment. Curr Drug Targets (2015) 16(2):156–63. doi: 10.2174/1389450115666141224125117
196. Ascenzi F, De Vitis C, Maugeri-Sacca M, Napoli C, Ciliberto G, Mancini R. SCD1, Autophagy and Cancer: Implications for Therapy. J Exp Clin Cancer Res (2021) 40(1):265. doi: 10.1186/s13046-021-02067-6
197. Li H, Feng Z, He ML. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics (2020) 10(16):7053–69. doi: 10.7150/thno.41388
198. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-Coa Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers (Basel) (2019) 11(7). doi: 10.3390/cancers11070948
199. Yan A, Jia Z, Qiao C, Wang M, Ding X. Cholesterol Metabolism in Drugresistant Cancer (Review). Int J Oncol (2020) 57(5):1103–15. doi: 10.3892/ijo.2020.5124
200. Kopecka J, Trouillas P, Gasparovic AC, Gazzano E, Assaraf YG, Riganti C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist Updat (2020) 49:100670. doi: 10.1016/j.drup.2019.100670
201. Vogeli B, Engilberge S, Girard E, Riobe F, Maury O, Erb TJ, et al. Archaeal Acetoacetyl-Coa Thiolase/HMG-Coa Synthase Complex Channels the Intermediate via a Fused Coa-Binding Site. Proc Natl Acad Sci USA (2018) 115(13):3380–5. doi: 10.1073/pnas.1718649115
202. Berndt N, Hamilton AD, Sebti SM. Targeting Protein Prenylation for Cancer Therapy. Nat Rev Cancer (2011) 11(11):775–91. doi: 10.1038/nrc3151
203. Huang B, Song BL, Xu C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat Metab (2020) 2(2):132–41. doi: 10.1038/s42255-020-0174-0
204. Weber P, Wagner M, Schneckenburger H. Cholesterol Dependent Uptake and Interaction of Doxorubicin in Mcf-7 Breast Cancer Cells. Int J Mol Sci (2013) 14(4):8358–66. doi: 10.3390/ijms14048358
205. Preta G. New Insights Into Targeting Membrane Lipids for Cancer Therapy. Front Cell Dev Biol (2020) 8:571237. doi: 10.3389/fcell.2020.571237
206. Dubey R, Stivala CE, Nguyen HQ, Goo YH, Paul A, Carette JE, et al. Lipid Droplets can Promote Drug Accumulation and Activation. Nat Chem Biol (2020) 16(2):206–13. doi: 10.1038/s41589-019-0447-7
207. Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol Cancer Res (2019) 17(12):2492–507. doi: 10.1158/1541-7786.MCR-19-0264
208. Matsushita Y, Nakagawa H, Koike K. Lipid Metabolism in Oncology: Why it Matters, How to Research, and How to Treat. Cancers (Basel). (2021) 13(3). doi: 10.3390/cancers13030474
209. Isnaldi E, Richard F, De Schepper M, Vincent D, Leduc S, Maetens M, et al. Digital Analysis of Distant and Cancer-Associated Mammary Adipocytes. Breast (2020) 54:179–86. doi: 10.1016/j.breast.2020.10.004
210. Cao Y. Adipocyte and Lipid Metabolism in Cancer Drug Resistance. J Clin Invest (2019) 129(8):3006–17. doi: 10.1172/JCI127201
211. Lehuede C, Li X, Dauvillier S, Vaysse C, Franchet C, Clement E, et al. Adipocytes Promote Breast Cancer Resistance to Chemotherapy, a Process Amplified by Obesity: Role of the Major Vault Protein (MVP). Breast Cancer Res (2019) 21(1):7. doi: 10.1186/s13058-018-1088-6
Keywords: breast carcinoma, neoadjuvant and adjuvant chemotherapy, anthracycline, taxane, glucose and lipid metabolism
Citation: Tőkés AM, Vári-Kakas S, Kulka J and Törőcsik B (2022) Tumor Glucose and Fatty Acid Metabolism in the Context of Anthracycline and Taxane-Based (Neo)Adjuvant Chemotherapy in Breast Carcinomas. Front. Oncol. 12:850401. doi: 10.3389/fonc.2022.850401
Received: 07 January 2022; Accepted: 08 March 2022;
Published: 31 March 2022.
Edited by:
Domenica Scumaci, Magna Græcia University of Catanzaro, ItalyReviewed by:
Mario Cioce, Campus Bio-Medico University, ItalyLinchong Sun, South China University of Technology, China
Copyright © 2022 Tőkés, Vári-Kakas, Kulka and Törőcsik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Mária Tőkés, dG9rZXMuYW5uYV9tYXJpYUBtZWQuc2VtbWVsd2Vpcy11bml2Lmh1