 Rui Guo1†
Rui Guo1† Tian Liu
Tian Liu Marzieh Dehghan Shasaltaneh
Marzieh Dehghan Shasaltaneh Saber Imani
Saber Imani QingLian Wen
QingLian Wen- 1Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 2Department of Biology, Faculty of Science, University of Zanjan, Zanjan, Iran
- 3China Regional Research Center, International Centre for Genetic Engineering and Biotechnology Taizhou, Jiangsu, China
The adenylate cyclase (ADCY) superfamily is a group of glycoproteins regulating intracellular signaling. ADCYs act as key regulators in the cyclic adenosine monophosphate (cAMP) signaling pathway and are related to cell sensitivity to chemotherapy and ionizing radiation. Many members of the superfamily are detectable in most chemoresistance cases despite the complexity and unknownness of the specific mechanism underlying the role of ADCYs in the proliferation and invasion of cancer cells. The overactivation of ADCY, as well as its upstream and downstream regulators, is implicated as a major potential target of novel anticancer therapies and markers of exceptional responders to chemotherapy. The present review focuses on the oncogenic functions of the ADCY family and emphasizes the possibility of the mediating roles of deleterious nonsynonymous single nucleotide polymorphisms (nsSNPs) in ADCY as a prognostic therapeutic target in modulating resistance to chemotherapy and immunotherapy. It assesses the mediating roles of ADCY and its counterparts as stress regulators in reprogramming cancer cell metabolism and the tumor microenvironment. Additionally, the well-evaluated inhibitors of ADCY-related signaling, which are under clinical investigation, are highlighted. A better understanding of ADCY-induced signaling and deleterious nsSNPs (p.E1003K and p.R1116C) in ADCY6 provides new opportunities for developing novel therapeutic strategies in personalized oncology and new approaches to enhance chemoimmunotherapy efficacy in treating various cancers.
1 Introduction
The latest global cancer burden data released by the International Agency for Research on Cancer (IARC) of the World Health Organization revealed 19.29 million new cancer cases and 9.96 million cancer deaths worldwide in 2020. The number of new cancer cases is predicted to increase to approximately 28.4 million worldwide in 2040, which is up by 47% compared to 2020. Following the development of targeted therapies and immunotherapy, cancer treatment modalities have long gone beyond surgery and radiotherapy. Outcomes for cancer patients are improved by applying tyrosine kinase, cell cycle-dependent kinase (CDK), cell cycle, integrin signaling, and immune checkpoint inhibitors, although their efficacy significantly varies due to a variety of limiting factors (1). The effectiveness of targeted therapy depends on the correct matching between drug and target. The targets are expressed to varying degrees on tumor cells and act as the oncogenic driver mutations that provide cancer cells with growth and survival advantages. Therefore, the specific mechanisms of tumorigenesis should be further explored to identify novel therapeutic targets meeting the challenge related to the precision of oncology.
Based on the results of many studies since 1971, adenylate cyclase (AC) activity and cAMP levels are associated with the normalization of tumor cell morphology and restoration of contact inhibition, as well as the reduced growth rate (2, 3). AC activation, which drives the production of cAMP from adenosine triphosphate (ATP), leading to elevated intracellular cAMP levels (4), is accompanied by cancer through protein kinase A (PKA)-dependent and independent pathways. In addition to being involved in cell cycle inhibition, apoptosis, and growth, cAMP regulates angiogenesis by inhibiting vascular endothelial growth factor (VEGF), TGF-β, and epidermal growth factor receptor (EGFR) pathways (5). Targeting the upstream and downstream of the cAMP signaling pathway exhibits a range of anticancer effects such as the induction of mesenchymal-to-epithelial transition (EMT), inhibition of cell growth and migration, and enhanced sensitivity of cancer cells to conventional anticancer drugs (6–8).
The adenylate cyclase (ADCY) gene family, which encodes AC proteins, plays important regulatory roles in a spectrum of biological processes like cell proliferation, apoptosis, migration, invasion, angiogenesis, abnormal metabolism, and immune escape (9–11). AC compounds are already in clinical use for certain diseases such as neuropathic pain, neurodegenerative diseases, congestive heart failure, asthma, and male contraception (12). However, the studies on the role of the various isoforms of the ADCY family in cancer have been mainly limited to the screening of biomarkers for different cancers and the discovery of interactions with other genes or small molecules (13). Certain AC isoforms are expressed more or less in specific cancers and are associated in tumor development as pro- or anticancer factors, although specific mechanisms are unclear. In general, the potential role of the family in cancer therapy warrants attention and can yield important new targets for the treatment. The present study reviewed the current understanding of the ADCY family and the mechanism of ADCY activation in response to various types of chemotherapy. It highlighted the role of the family in the different signaling pathways regulating resistance to chemotherapy in tumorigenesis.
2 ACs and cAMP Level Regulation
ACs are glycoproteins with important functions as intracellular signaling regulators. This large family contains 10 subtypes (AC I–IX and soluble AC (sAC)), each of which is encoded by an independent gene (ADCY1–ADCY10). ADCY1–ADCY18 are located on different chromosomes, while ADCY9 and ADCY7 are locateded on chromosome 16 (14) (Figure 1). Membrane-bound ACs can be divided into four types according to the various regulatory characteristics (Table 1), the first of which is composed of AC1, AC3, and AC8, which are activated by cationic calcium (Ca2+) and calmodulin (CAM). The second involves AC2, AC4, and AC7, which are activated by heterotrimeric GTP-binding protein (G-protein) βγ subunits, while they are insensitive to calcium. The next group, consisting of AC5 and AC6, is inhibited by Ca2+ and inhibitory G-protein (Gi), and AC9, as the only member of the fourth type, is the only isomer that is not activated by forskolin, while it responds to calcineurin (15). In addition, sAC is stimulated by bicarbonate, calcium, and ATP, while it is insensitive to forskolin and heterotrimeric G-protein (16). ACs serve as the effectors of the G-protein-coupled system. Furthermore, at least two heterotrimeric G-proteins are responsible for regulating the activity of transmembrane adenylate cyclase (tmAC), which include stimulatory G-protein (Gs) and Gi, which activate and inhibit AC, respectively (17). It is worth noting that the differential regulation and unique distribution of various AC isomers can create completely different cellular signals, making them key enzymes in the signaling pathway (18). ACs are widely distributed, the diverse subtypes of which perform various physiological functions, and each subtype has nonspecific cross-coordination or antagonism properties. The results of some studies demonstrated that the regulation of ADCY expression is closely related to animal health, while it lacks effective selectivity (19). Furthermore, the nonselective inhibition of ACs causes a series of potential adverse reactions like long-term memory (LTM) impairment (AC1 and AC8), motor dysfunction (AC3), nephrogenic diabetes insipidus (AC6), renal function decline (AC3, AC5, and AC6), and higher mortality rate during sympathetic nerve maintenance stimulation (other ACs) (19).
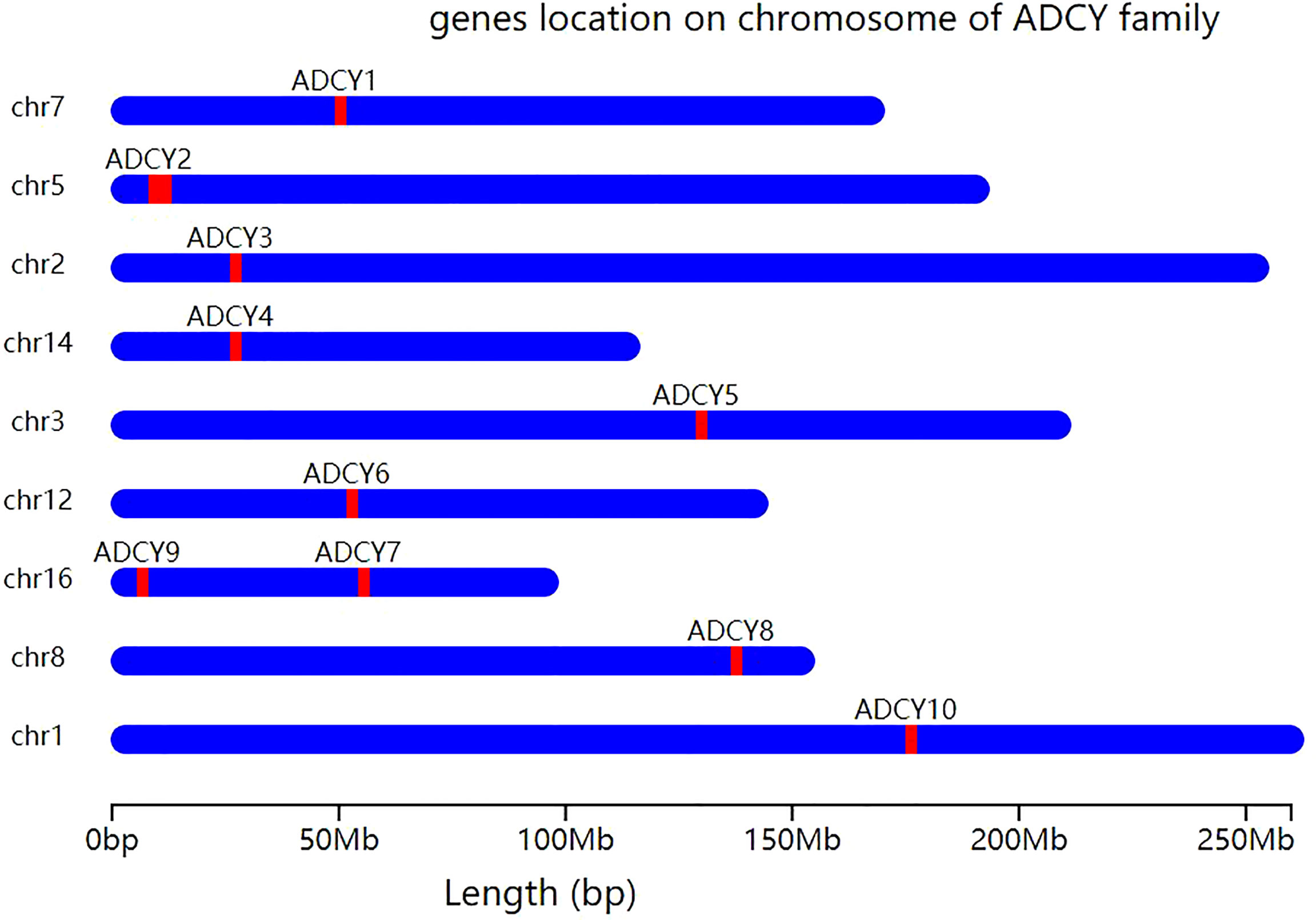
Figure 1 Chromosomal localization of ADCY gene isoforms. Except for ADCY7 and ADCY9, which are both located on chromosome 16, all other ADCY subtypes are located on different chromosomes. ADCY1 is located on chromosome 7, ADCY2 on chromosome 5, ADCY3 on chromosome 2, ADCY4 on chromosome 14, ADCY5 on chromosome 3, ADCY6 on chromosome 12, ADCY8 on chromosome 8, and ADCY10 on chromosome 1.
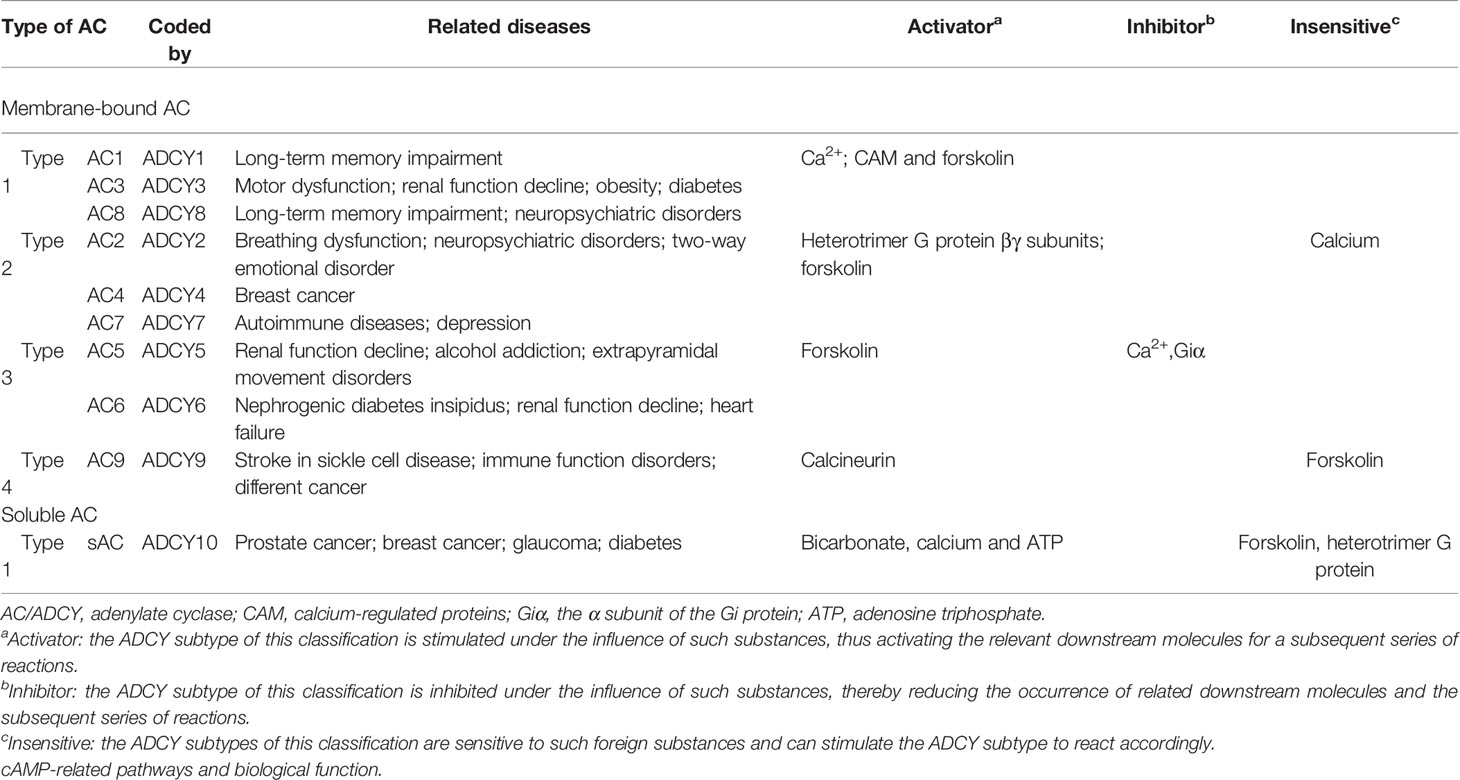
Table 1 Activators or inhibitors for AC subtypes and related diseases.
The cAMP directly regulates many cellular functions (20). Intracellular cAMP is produced from ATP during a reaction catalyzed by ACs, while cAMP degradation is mediated by cAMP phosphodiesterase (PDE), which hydrolyses cAMP to produce adenosine 5′-monophosphate (21). Thus, the dynamic balance of intracellular cAMP levels is maintained by a combination of AC stimulation and PDE-mediated degradation. Additionally, cAMP acts as an intracellular signal for many hormones, neurotransmitters, and other signaling molecules, the production and degradation of which are accompanied by its sensitivity to a wide range of extracellular and intracellular signals. Some heterologous G-protein-coupled receptors (GPCR) such as nicotinic acetylcholine, beta-adrenergic, or adenosine receptors activate AC, leading to a rise in cAMP production (22). Similarly, forskolin directly stimulates AC, and theophylline and caffeine promote intracellular cAMP levels by inhibiting PDE (16), and consequently affect a range of pathophysiological functions.
3 AC-Related Disorders
Today, AC subtypes are known to be associated with different diseases (Table 1), such as autoimmune ones and depression (23), as well as stroke in sickle cell disease, immune function disorders, and different cancers (AC9) (24). Furthermore, AC1 and AC4 are respectively accompanied by LTM impairment (25) and breast cancer (26), while AC3 is related to motor dysfunction, renal function decline, obesity, and diabetes (18). An association is found between AC8 with LTM impairment and neuropsychiatric disorders (27). Some researchers found a relationship between AC2 with breathing dysfunction and neuropsychiatric and bipolar disorders (28), as well as another between AC5 with renal function decline, alcohol addiction, and extrapyramidal movement disorders (29). Furthermore, AC6 is accompanied by nephrogenic diabetes insipidus, renal function decline, and heart failure (30). Finally, sAC is associated with prostate and breast cancer, glaucoma, and diabetes (31). Thus, AC subtypes are implied as important players in various diseases, suggesting that relevant diseases can be controlled by the specific targeting and regulation of AC isoforms.
4 cAMP-RELATED PATHWAYS AND BILOGICAL FUNCTION
The cAMP, as the first identified second messenger, regulates cellular transport by binding to PKAs, cAMP-activated guanine exchange factors (EPACs), and cyclic nucleotide-gated channels (CNGs) (32) (Figure 2). These cAMP sensors lead to the regulation of various physiological processes such as DNA degradation, cell proliferation, and apoptosis either alone or in cooperation with PKAs (33). PKAs and EPACs are the major targets of the cAMP signaling pathway (34). In addition, activated PKA-phosphatases regulate related metabolic and transcription factors like the activation of downstream cAMP response element-binding proteins (CREB) and consequently activate the WNT/β-catenin pathway, which is accompanied by cell cycle arrest, apoptosis, and survival (34). CREB regulates the inhibitory activity of apoptosis proteins (IAPs) to prevent apoptosis triggered by various stimuli and plays an important role in inhibiting cell death and regulating DNA damage in the cell cycle (35). Furthermore, the cAMP signaling pathway regulates sirtuin 6 expression through modulating the PKA-dependent Raf/mitogen-activated extracellular signal-regulated kinase/extracellular signal-regulated kinase (Raf-MEK-ERK) pathway as well as ubiquitin-protease-dependent degradation, causing apoptosis regulation (36). Similarly, the regulation of protein phosphatase 2A (PP2A) plays a crucial role in regulating apoptosis (37). PKA activation can protect cancer cells from apoptosis after exposure to radiation or chemotherapeutic agents by increasing parathyroid hormone-related protein expression (38). The higher PKA expression enhances the activity of the mitogen-activated protein kinase (MAPK) signaling pathway (39), which regulates cell proliferation. The results of a recent study demonstrated the important role of X-ray repair in complementing defective repair in repairing DNA in Chinese hamster cell 1 (XRCC1) and the ability of cAMP-mediated EPACs to repair DNA damage by regulating XRCC1 (40). Furthermore, EPACs regulate cell proliferation, differentiation, apoptosis, and inflammation as well as play a crucial role in endoskeletal remodeling, cell proliferation, adhesion, migration, and epithelial-mesenchymal transition by regulating downstream signaling molecules in the vascular cAMP signaling pathway (40). They can even act by preventing c-Jun N-terminal kinase (JNK) activation by inhibiting B-cell lymphoma-2 (Bcl-2) phosphorylation, downregulating cellular autophagy and phosphorylation, and subsequently leading to less ubiquitination and more histone deacetylase 8 (HDAC8) expression (41). Additionally, Bcl-2 is a well-known member of the antiapoptotic protein family, which serves by inhibiting the essential proapoptotic proteins that share multiple homology domains with Bcl-2 (42). It regulates mitochondrial outer membrane (MOM) permeability, which has a key role in apoptotic signaling and influences cell survival (43). Therefore, ACs regulate cAMP production, which results in modulating downstream signaling molecules to control various cellular physiological activities.
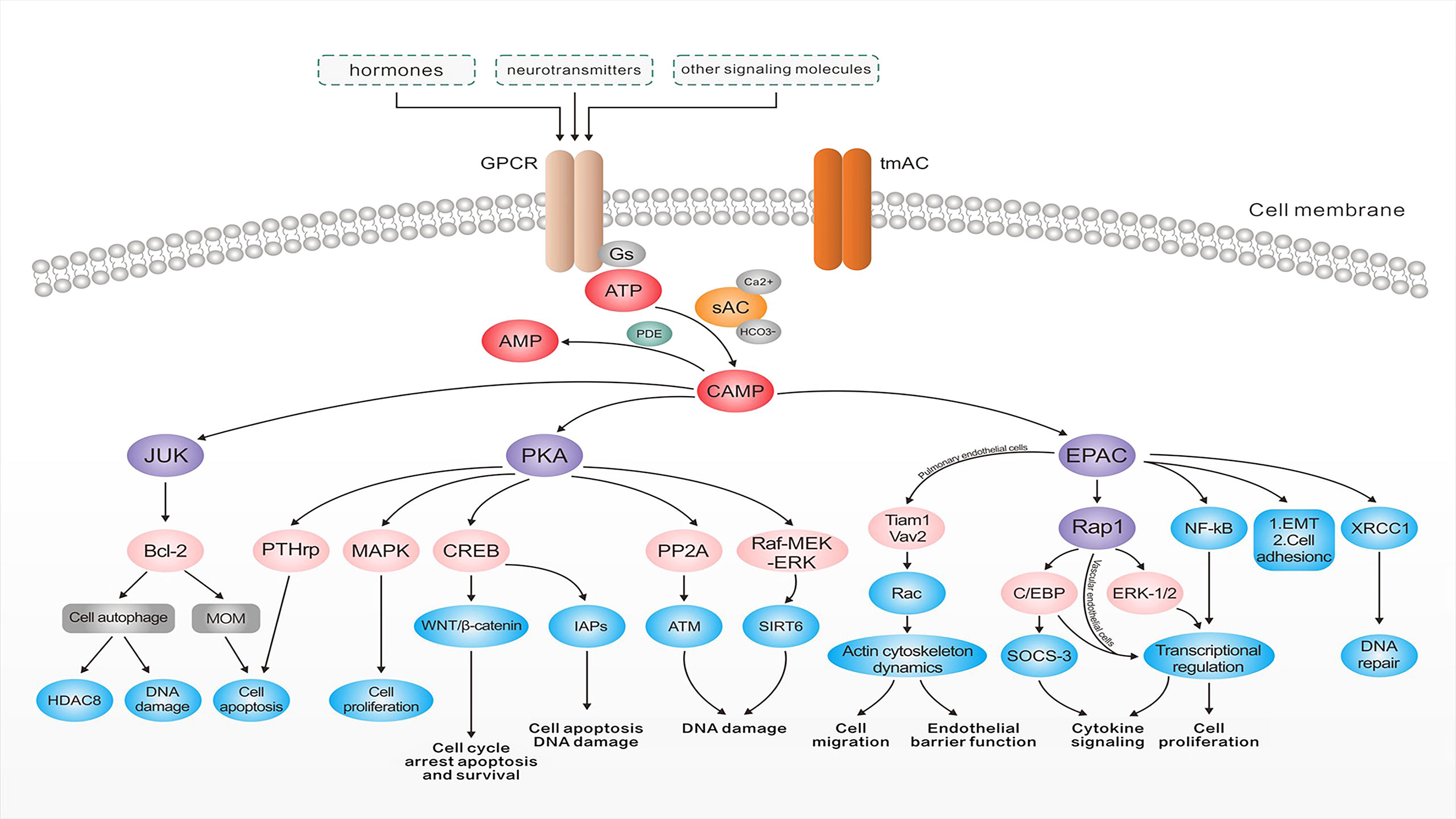
Figure 2 The roles of adenylate cyclases in tumorigenesis. After binding to G protein-coupled receptors, hormones, neurotransmitters, and a number of other signaling molecules, cyclic adenosine 3′,5′-monophosphate (cAMP) is generated from ATP by the action of adenylate cyclase and degraded to the 5′-cAMP by the action of cyclic nucleotide phosphodiesterase (PDE). cAMP can increase DNA damage via a range of pathways and plays an important role in endoskeletal remodeling, cell proliferation, adhesion, and EMT. It also regulates the permeability of the mitochondrial outer membrane, affecting cell survival, activates the WNT/β-catenin pathway, and regulates the processes of cell cycle arrest, apoptosis, survival, and DNA damage. cAMP signaling pathways can also protect cancer cells from apoptosis after exposure to radiation or chemotherapeutic agents through related pathways. Abbreviations: EGFR, epidermal growth factor receptor; tmAC, transmembrane adenylate cyclase; Gs, stimulatory G-protein; sAC, soluble adenylate cyclase; ATP, adenosine triphosphate; PDE, phosphodiesterase; cAMP, cyclic adenosine phosphate; PKA, protein kinase A; JNK, c-Jun N-terminal kinase; Bcl-2, B-cell lymphoma-2; MOM, mitochondrial outer membrane; HDAC8, histone deacetylase 8; MAPK, mitogen-activated protein kinase; CREB, cAMP response element-binding proteins; IAPs, inhibitor of apoptosis proteins; PP2A, protein phosphatase 2A; ATM, the ataxia–telangiectasia–mutated protein; Raf-MEK-ERK, Raf/mitogen-activated extracellular signal-regulated kinase/extracellular signal-regulated kinase; EPAC, cAMP-EGF-I/II; ERK, extracellular signal-regulated kinase; XRCC1, X-ray repair complementing defective repair in Chinese hamster cell 1; EMT, epithelial-mesenchymal transition; SOCS-3, suppressor of cytokine signaling 3; C/EBP, CCAAT/enhancer-binding protein.
5 Mutations in ADCY Family
5.1 Disease-Causing Effects of the Mutations in the ADCY Family
To date, at least 60 ADCY gene mutations have been reported in many genotype–phenotype databases, such as the Online Mendelian Inheritance in Man (OMIM) and Human Gene Mutation Database (HGMD), as well as the Database of Genotype and Phenotype (dbGAP). Mutations in ADCY1, ADCY2, ADCY3, ADCY4, ADCY6, and ADCY7 isoforms exhibit the most pathological effects and clinical significance. The disease-causing mutations are predominantly nonsense variants, partial and full deletions, and nonsense, insertion, and splice-site mutations. Nonsynonymous single nucleotide polymorphisms (nsSNPs, substitutions, and deletions) are considered the most common variants of ADCY1 and ADCY6. Furthermore, exons 22 (22,896 bp of variants/length) and 23 (148,977 bp of variants/length) are the most affected exons. Some ADCY mutations were initially identified incorrectly due to the similarity between their symptoms and the other cancer-driver mutations. Table 2 lists the most frequent disease-causing SNP mutation spectrums in ADCY1 and ADCY6. As shown, the nonsynonymous mutations of G>A are more widely observed and change highly conserved amino acids in ADCY1 and ADCY6. The deleterious and pathogenic aspects of mutations were predicted by using an in silico prediction pipeline consisting of polymorphism phenotyping v2 (PolyPhen-2), mutation taster, I-Mutant, sorting intolerant from tolerant (SIFT), and ExAC programs. Furthermore, several online programs were applied to predict the pathogenicity of the selected variants of ADCY1 and ADCY6. Based on the predictions, ADCY6 variants are disease-causing and damaging mutations, especially p.E1003K, p.R1116C, and p.Y992C, which were verified by more than two bioinformatics programs (Table 2). Three variants have a deleterious effect when using the SIFT and PolyPhen2 tools. Generally, the nsSNPs c. 3007G>A (p.E1003K), c. 3346C>T (p.R1116C), and c. 2975A>G (p.Y992C) are predicted to be pathogenic mutations with the potential as cancer-driver mutations.
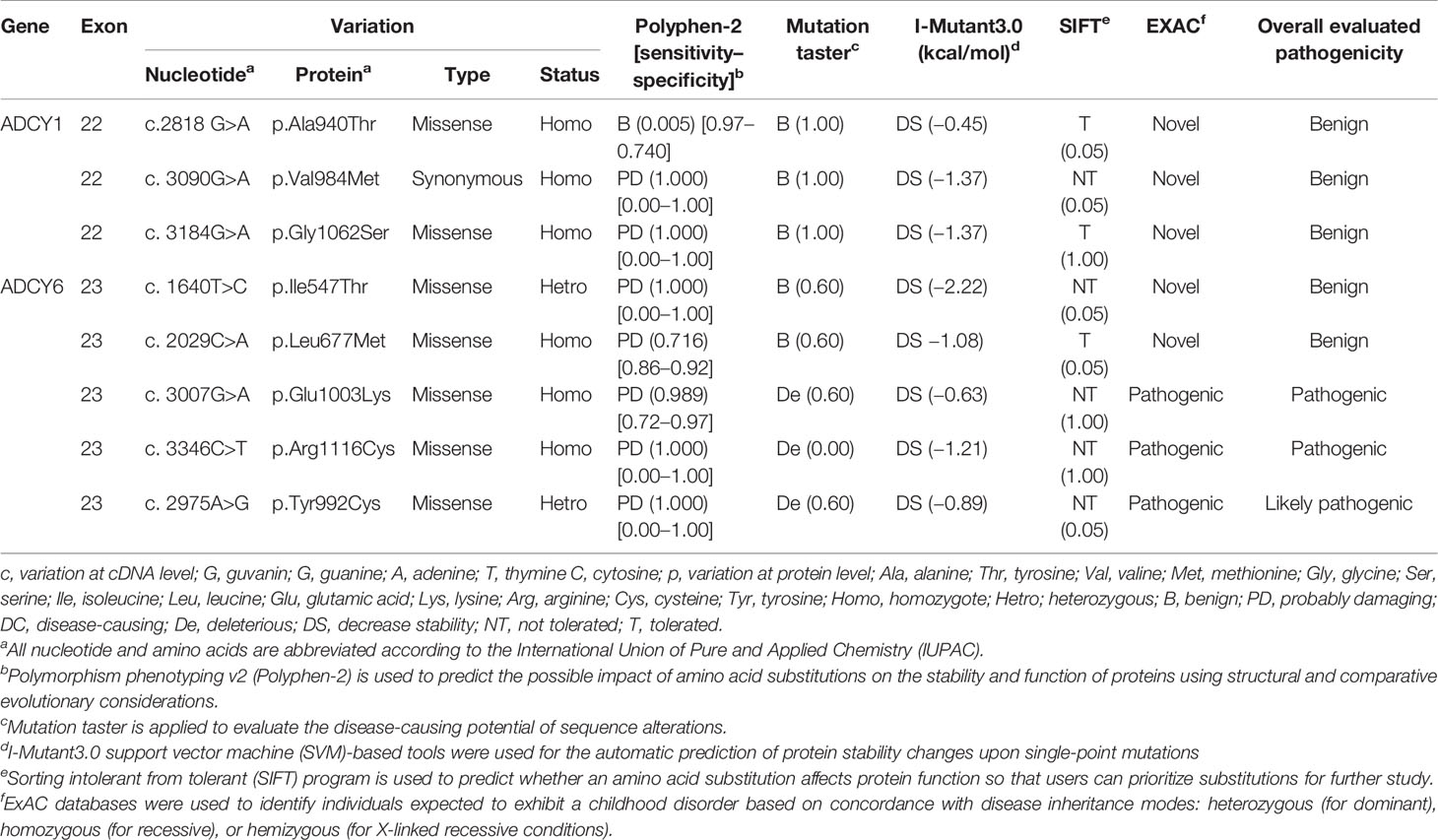
Table 2 Predicted protein structure and disease-causing effects of mutations in AC family.
5.2 Functional Effects of a Pathogenic Mutation in the ADCY Family
The analysis of the conserved domains of wild-type (WT) and mutant ADCY6 proteins is shown in Figure 3. In addition, the mutation sites were aligned to the ADCY6 protein sequence to explore the function-associated nsSNPs (Figure 3A). Most of the disease-driver mutations generate defective helicase proteins due to the changes in conserved residues (i.e., Y991, E1003, and R111), as well as the degradation of the improperly folded protein or truncated mRNA. Structural–functional analysis revealed that all three disease-driver nsSNPs (p.E1003K, p.R1116C, and p.Y992C) are located in the topological domain of the ADCY6 protein (914–1,168 amino acid residues in the cytoplasmic domain). Based on the sequence conservation alignment, the nsSNP Y991, E1003, and R1116 are highly conserved in all organisms (Figure 3A). As shown in Figure 3B, the predicted crystal structures of the WT (left panel), and E1003K, R1116C, and Y992C mutant (right panel) ADCY6 proteins exhibit the mutation of a glutamic acid residue to lysine, arginine to cysteine, and tyrosine to cysteine at conserved positions 1003, 1116, and 992, respectively. Structurally, the active site is situated at the interface of the two topological domains, locating the three residues in the ADCY6 enzyme active site. The comparison between the protein structure homology-modeling of the WT and ADCY6 mutant (p.E1003K) demonstrated a change from a smaller Glu to a larger Lys at position 1003, leading to charge variation (positive charge). However, the other mutations result in introducing cysteine residues, which form disulfide bonds in the protein structure. Furthermore, the WT and mutant proteins were significantly different in terms of hydrophobicity and hydrophilicity. Therefore, the mutation may cause the loss of hydrophobic interactions and hydrogen bonds with other molecules, which alters the enzyme activity. In general, ADCY6 catalyzes the formation of the signaling molecule cAMP downstream of G-protein-coupled receptors, which act in the signaling cascade downstream of beta-adrenergic receptors in the heart and vascular smooth muscle cells. Given the crystal structure, the p.E1003 and p.R1116C substitutions are suggested to trigger a change in substrate conformation slightly, with the potential to affect the enzyme active center. In the present review, it is hypothesized that the ADCY6 (p.E1003 and p.R1116C) mutant loses its activity and attenuates the hydroxylation of substrates such as those in the cAMP-PKA-PP2A (37) and WNT/β-catenin pathways.
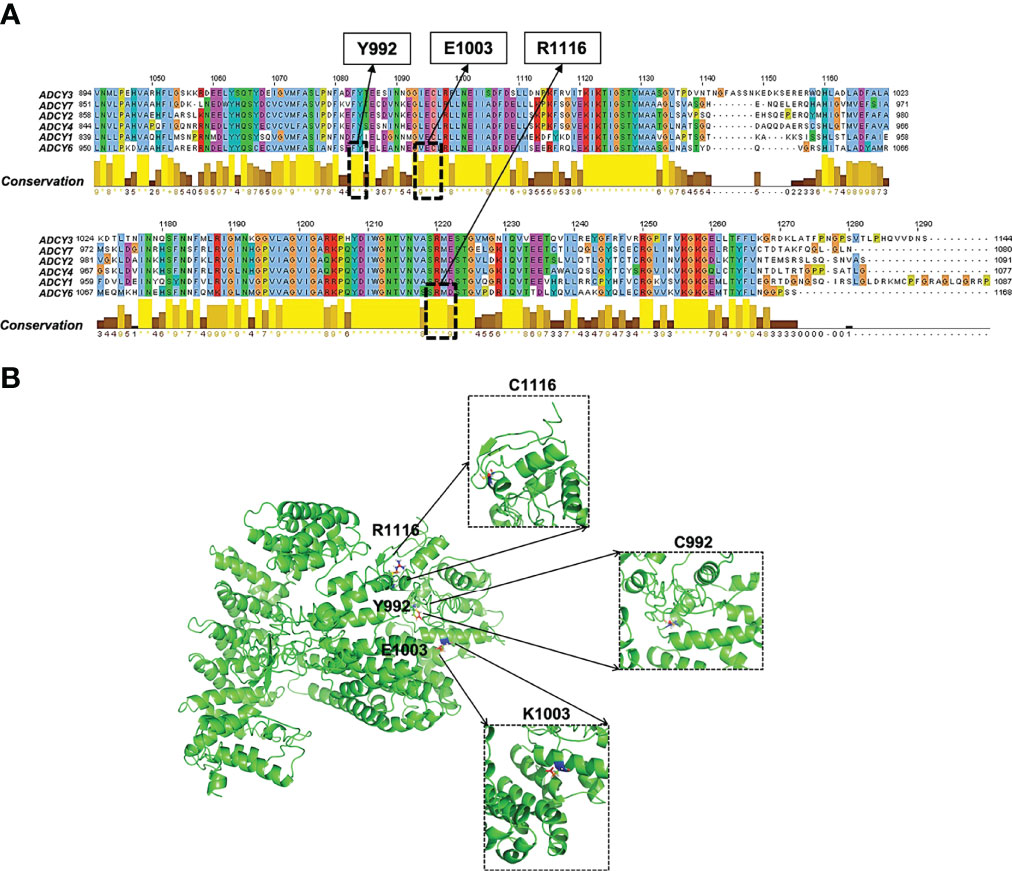
Figure 3 Functional effects of pathogenic ADCY6 SNPs. (A) Multiple sequence alignment of the ADCY protein family. (B) Surface and crystallographic imaging of the ADCY6 p.E1003K and p.R1116C SNPs located in the active site of the ADCY6 enzyme.
6 Crosstalk Between ADCYs and Cancer
The development, progression, metastasis, and drug resistance of most common cancers are mediated by the cAMP signal downstream of β-adrenergic receptors (β-ARs), which are coupled to stimulatory G-protein (Gs) (44). After hormones, neurotransmitters, and some other signaling molecules bind to G-protein-coupled receptors (Figure 4). Additionally, ATP is generated from cAMP through the action of AC and then involved in a range of pathophysiological activities. In contrast, 3′,5′-cAMP is degraded to 5′-cAMP by the action of PDE. The cAMP plays an important role in endoskeletal remodeling, cell proliferation, adhesion, and epithelial–mesenchymal transition (EMT) by preventing JNK activation, leading to the inhibition of Bcl-2 phosphorylation and, consequently, downregulation of cellular autophagy and phosphorylation. As a result, ubiquitination decreases, HDAC8 expression increases, and DNA damage increases (41). Furthermore, Bcl-2 regulates MOM permeability, which has a significant role in apoptotic signaling and affects cell survival (45). Activated PKA-phosphatases, which regulate related metabolic and transcription factors like CREB (34), are associated with cell cycle arrest, apoptosis, and survival through activating the WNT/β-catenin pathway. They control apoptosis and DNA damage by regulating the enhancer sequences of IAPs (35). Furthermore, the cAMP signaling pathway controls apoptosis and DNA damage by regulating the PKA-dependent Raf-MEK-ERK pathway as well as protecting cancer cells from apoptosis following exposure to radiation or chemotherapeutic agents by elevating the expression of parathyroid hormone-related proteins (38). A rise in PKA improves the activity of the MAPK signaling pathway, which regulates cell proliferation (39). In addition, EPACs regulate cell proliferation, migration, adhesion, EMT, and endoskeletal remodeling by regulating downstream signaling molecules. The cAMP-mediated EPACs can repair DNA damage by regulating XRCC1 (40).
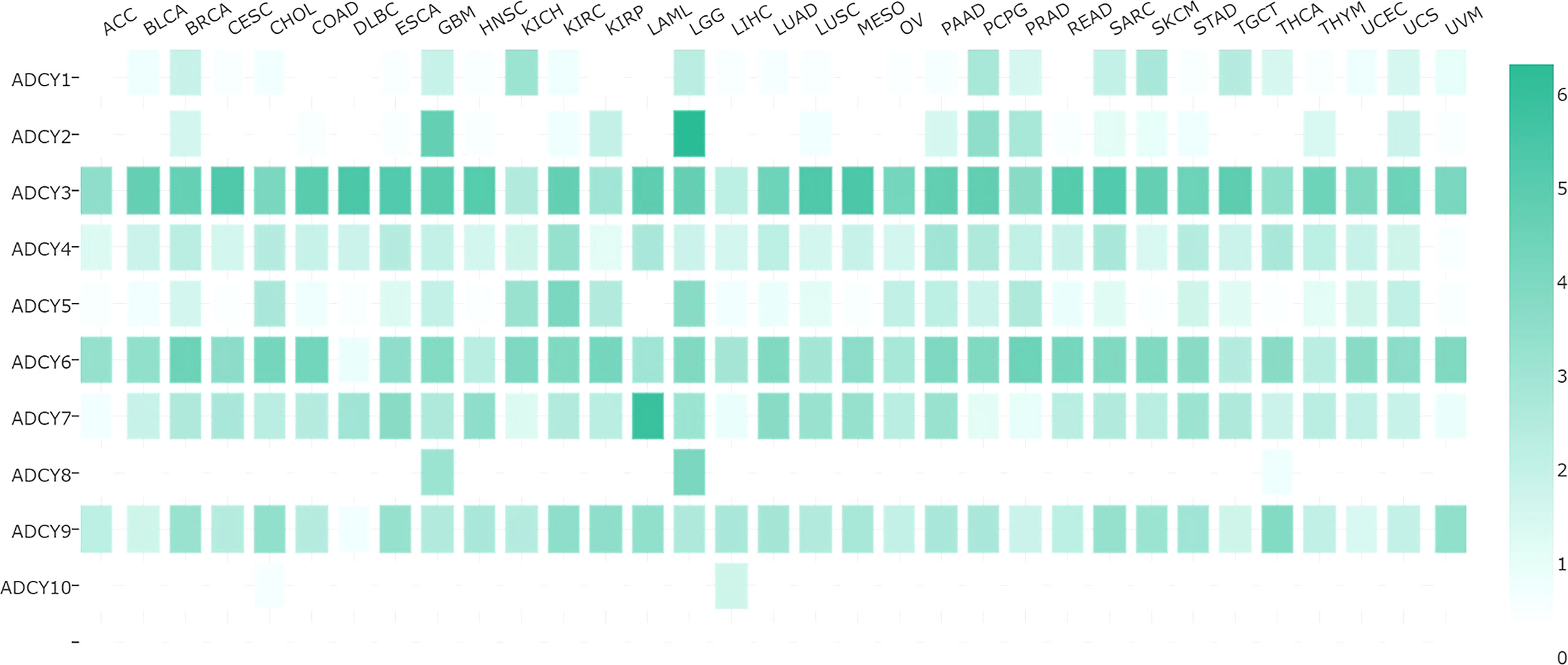
Figure 4 Comparison of the expression of various ACDY isoforms in different cancers. The adenylate cyclase gene is expressed differently in each cancer, with darker color indicating higher expression and vice versa. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower-grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
In this regard, AC activates the formation of intracellular cAMP-activated PKA and transcription factor CREB, which induces the overexpression of growth factors such as epidermal growth factor (EGF), VEGF, arachidonic acid (AA), and proinflammatory cytokines, and consequently stimulates the growth, development, metastasis, and drug resistance of many cancers (44). Furthermore, the Gi-coupled receptor can inhibit this signaling cascade by blocking the activation of the ACs catalyzing cAMP formation, and acting as a physiological inhibitor of the cascade (44). Thus, AC overexpression or silencing may be a key event in processes like tumorigenesis, cell proliferation, migration, and invasion.
6.1 ADCY Isoforms in Tumorigenesis
The cAMP-mediated activation of PKA in many cancer cells leads to cell cycle arrest and growth inhibition via the apoptotic pathway (46). The blocking of ERK (47), inhibition of antiapoptotic proteins Bcl2 and Bcl-xl (48), upregulation of tumor suppressor gene p53, and suppression of oncogenes c-myc and erb-2 (49) can be addressed as other ways in which cAMP is linked to growth inhibition. Furthermore, the regulation of the angiogenic pathway through preventing VEGF, transforming growth factor-β (TGF-β), and EGFR is associated with higher cAMP levels (50). However, a lower level of ADCY and cAMP is found in many cancers, which promotes cancer formation and cancer cell proliferation, decreases apoptosis, and even increases neovascularization, cancer cell migration, and invasion.
6.2 ADCY Family in Cancer Recurrence and Prognosis
As already mentioned, the ADCY family is closely related to cancer recurrence and prognosis (Figure 5). Cancer recurrence is a common phenomenon involving numerous complex mechanisms, in which the ADCY family plays a crucial role. The results of the previous studies indicated less AC7 expression among relapsed acute promyelocytic leukemia (APL) patients than the newly diagnosed APL ones. Additionally, miR-192, which directly targets AC7 expression, is relatively high in the relapsed APL individuals, which suggests the important role of miR-192-mediated AC7 in APL cell differentiation, as well as implicating AC7 and miR-192 as novel biomarkers and therapeutic targets for these patients (51). Individuals with primary or relapsed APL have a relatively lower level of AC9 expression compared to those with complete remission and nonleukemic patients (52). Furthermore, the expression is strongly accompanied by leukemogenesis in APL, proposing the potential of AC9 as a biomarker in clinical diagnosis and leukemia relapse treatment (52). The less expression of phosphorylated CREB protein produced through cAMP regulation is associated with the aggressive and metastatic recurrence of melanoma (53). Moreover, the ADCY family, as the key genes in important signaling pathways, influences patient prognosis through a variety of mechanisms. Based on the results of a cohort study, the level of ADCY9 is greater in colon cancer tissues than in the adjacent tissues (54). The low ADCY9 level in cancer tissues is attributed to longer disease-free survival (DFS), while high ADCY9 expression and distant metastasis indicate a poor prognosis after treatment (54).
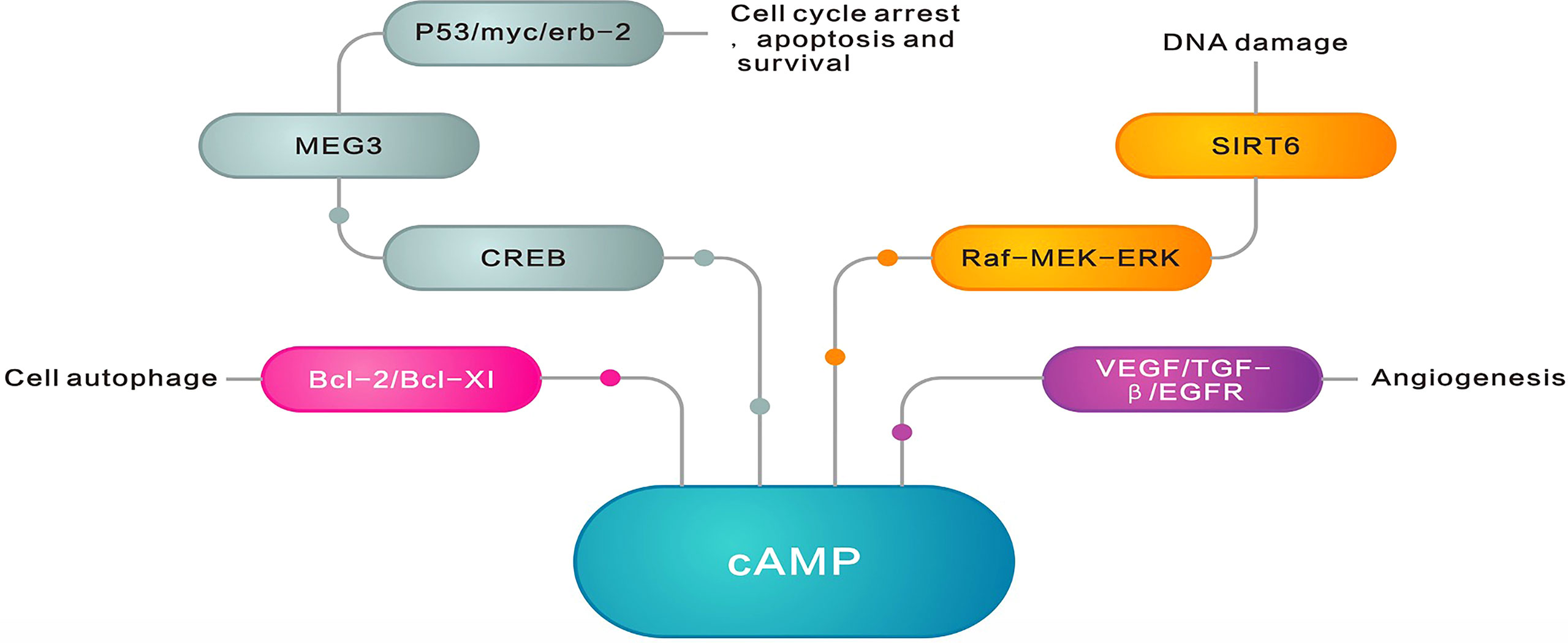
Figure 5 ADCY family roles in cancer recurrence and prognosis. cAMP inhibits cancer formation by blocking extracellular signal-regulated kinase (ERK), inhibiting the antiapoptotic proteins Bcl2 and Bcl-XI, upregulating the cancer suppressor gene p53, inhibiting the oncogenes c-myc and erbB-2, and inhibiting the regulation of the angiogenic pathway by VEGF, TGF-β, and EGFR. However, many cancers exhibit lower AC levels and lower cAMP levels, promoting cancer formation as well as cancer cell proliferation, reducing apoptosis, and even promoting neovascularization, cancer cell migration, and invasion.
6.3 ADCY Family in Chemo- and Radioresistance
Many treatment options are available for cancer, which include surgery, chemotherapy, radiotherapy, molecular targeted therapy, and immunotherapy. Specific cancer cell death induction and maximum remission are the ultimate goals of anticancer therapy. Currently, these goals still rely mainly on drug- or radiation-induced DNA-triggered death and antiproliferative signals. AC is activated by cAMP, which in turn activates PKA and CREB, which have roles in gene regulation, cell migration and proliferation, apoptosis, and mitochondrial homeostasis (55). In addition, forskolin, as an AC agonist, exerts several relevant anticancer effects such as inducing EMT, enhancing sensitivity to conventional antitumor drugs, and inhibiting the proliferation, motility, and migration of many types of cancer cells (50, 56).
In lung cancer, ADCY1 can modulate anticancer drugs and regulate apoptosis by regulating the expression of Bcl-2 family proteins, IAPs, and XRCC1 in human lung cancer cells (33). The expression of apoptosis inhibitors and inactivation of apoptosis promoters have been reported in human cancers. The results of the preclinical models suggested that functional defects in apoptosis signaling may translate into drug-resistant cells in cancer. The MOM permeability is known as a key step in apoptotic signaling, which is regulated by the Bcl-2 family of proteins, and targeting BCL in nonsmall cell lung cancer treatment leads to an initial improvement in chemoresistance (57). Further, ADCY1 catalyzes the elevation of cAMP and consequently inhibits the EPAC-dependent pathway degradation of XRCC1-induced DNA damage, DNA repair, and apoptosis in lung cancer cells (58). Ataxia-telangiectasia mutated (ATM) is a master regulator of the cellular response to the ionizing radiation-induced DNA damage, which can regulate multiple DNA damage responses like cell cycle, DNA repair, and apoptosis through activating downstream signaling pathways (59, 60). Gαs simulates ATM activation through the Gαs-cAMP-PKA-PP2A pathway, which results in declining the ATM protein-dependent nuclear factor kappa-B (NF-κB) activation, and increasing radiation-induced apoptosis in human and mouse lung cancer cells (37). Furthermore, the downregulation of cAMP-regulated Meg3 (an RNA gene) promotes cisplatin resistance in lung cancer cells by activating the WNT/β-catenin signaling pathway (61).
Breast cancer resistance to chemotherapeutic agent doxorubicin is generally accompanied by RAS/RAF/ERK activation (62). Additionally, AC activation can sensitize triple-negative breast cancer (TNBC) cells to doxorubicin via the cAMP/PKA-mediated mechanism of ERK inhibition, which greatly elevates doxorubicin-induced cell death (62). Forskolin can improve the sensitivity of MDA-MB-231 and MDA-MB-468 TNBC cells to 5-fluorouracil and taxol l (63).
Further, the cAMP signaling system inhibits DNA damage-induced apoptosis in leukemic cells by promoting p53 acetylation and turnover. The results of a study reflected an association between the upregulation of multidrug resistance gene (MDR1) expression through activating CREB by PKA with multidrug resistance in leukemia cells (64). EPAC1 and EPAC2 exhibit different expression patterns in mature and developing tissues. EPAC1 mRNA is widespread (65), while EPAC2 is mainly expressed in brain and endocrine tissues (66). Recently, some researchers have proposed the involvement of EPAC1 in regulating a variety of cancer cellular responses like cancer cell adhesion (67), proliferation (68), invasion (69), and migration (69, 70). Regarding ovarian cancer cells, roflumilast induces apoptosis and prevents tumor progression by activating the cAMP/PKA/CREB pathway and upregulating mitochondrial ferritin (FtMt) levels in the two types of cells (OVCAR3 and SKOV3) (55). Furthermore, ADCY4 is suggested to be accompanied by cetuximab insensitivity in colorectal cancer (71).
Table 3 presents an available spectrum of the multifunctional genes upregulated by ADCY in various radioresistant tumor cells. ADCY1 can modulate anticancer drugs and regulate cell apoptosis via the Bcl-2-MOM pathway in lung cancer (57). It can modulate cell apoptosis (35) and EPAC-XRCC1 through IAPs, both of which induce DNA damage (by increasing cell insensitivity to γ-ray), DNA repair, and apoptosis (40). In addition, it can enhance radiation-induced cell apoptosis via the cAMP-PKA-PP2A-ATM-NF-κB pathway (37) and cause more resistance to drugs such as cisplatin through the cAMP-Meg3-WNT/β-catenin pathway (61). All of the pathways in lung cancer involve changes in the sensitivity and effectiveness of chemotherapeutic drugs and radiotherapy. In breast cancer, it enhances the sensitivity of cancer cells to doxorubicin by improving the PKA-mediated activation of ERK to induce cell apoptosis and consequently promote the efficacy of the associated chemotherapy (63). Furthermore, it leads to the modulation of DNA damage-induced apoptosis via the cAMP-p53 pathway and cAMP/PKA/CREB1-MDR1 signaling axis in leukemia and is accompanied by multidrug resistance (64). In the case of ovarian cancer, it induces cell apoptosis and increases sensitivity to cisplatin, even reverses cisplatin resistance, through the cAMP/PKA/CREB-FtMt signaling axis (55). In general, the ADCY family results in improving the efficacy of cancer therapy because of modulating the downstream pathways which alter the sensitivity of cancer cells to chemotherapeutic agents and radiotherapy.
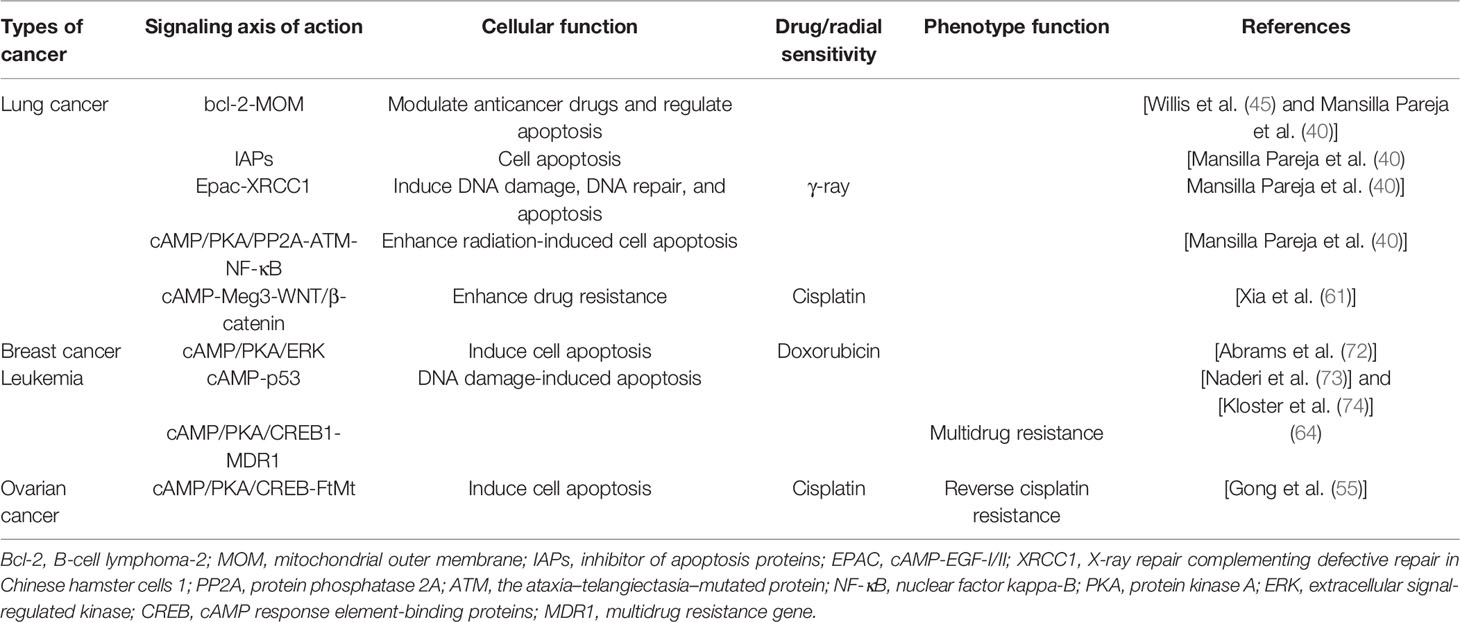
Table 3 Role of adenylate cyclase-related pathways in cancer drug or radiotherapy sensitivity.
6.4 Role of ADCY Isoforms in Various Cancers
The expression of ADCY subtypes varies in various cancers so that some are highly expressed in tumors and participate in tumor formation and development as oncogenes, while the others may have low, or even no expression. Analyses have revealed that this variation may lead to antitumor effects and may even be related to tumor prognosis. The expression of each subtype of the ADCY family in different cancers is demonstrated in Figure 4. As displayed, the most significant variation in expression is observed in ADCY4 and ADCY9. ADCY4 mRNA expression is significantly downregulated in breast cancer compared to the normal tissues, while the level more than triples in invasive breast cancer. Furthermore, ADCY9 expression diminishes in colorectal and lung cancers in comparison with the normal tissues (26). The epigenetic changes in the ADCY family, as a gene family regulating the extensive and global physiological activities of organisms, play a certain role in the biological behavior of tumors and other pathological processes. The results of recent gene sequencing-based studies have introduced a new role for ADCY1 mutations in influencing drug effectiveness in a variety of cancers like lung, esophageal, and colorectal ones (33). However, ADCY4 is significantly silenced and highly methylated in many cancers. ADCY5 has been less well studied, and the results of research knocking out the AC5 gene in mice indicated a significant reduction in angiogenesis and a rise in cancer cell apoptosis, both of which prevent cancer growth (75). ADCY6 expression is negatively correlated to the signaling pathways associated with immune processes, as well as the activation of immune checkpoint receptors and ligands (76). It is a potentially key gene in regulating immune cell infiltration in luminal-like carcinomas (76). Table 3 lists the cancers in which the AC subtype has been investigated so far.
6.4.1 Digest System Cancer
In gastric cancer cell lines and tissues, ADCY3 overexpression promotes tumorigenesis by upregulating the expression of matrix metalloproteinase-2 (MMP2) and metalloproteinase-9 (MMP9) via the cAMP/PKA/CREB pathway, which increases cell migration, invasion, proliferation, and colony formation (77, 78). ADCY3 serves as an oncogene in gastric cancer formation, as an oncogene, with high ADCY3 expression regulated by DNA methylation accompanied by low survival (78). Additionally, the ADCY3 gene is more expressed in primary pancreatic cancer and precancer tissues than the normal pancreatic tissues, while the expression of the ADCY2 gene is downregulated in primary tumors and adjacent nontumor tissues (79). Unlike gastric cancer, a greater cAMP level in pancreatic cancer inhibits cell migration and invasion (80, 81), although it does not affect cell proliferation (81). In fact, AC3 may enhance cAMP levels in response to forskolin stimulation, which causes PKA to phosphorylate CREB, a primary binding protein for the cAMP response, and consequently inhibits cell migration and invasion. Its inhibitory effect is accompanied by the rapid formation of AC3/adenylate cyclase-associated protein 1 the (CAP1)/G-actin complex, which prevents filopodia generation and cell motility (79).
Certain subtypes of the ADCY family (ADCY2, 3, 8, and 9) are mutated in hepatocellular carcinoma, which may be key events in the formation of the carcinoma (82). The results of the genomic analysis of a patient with lung metastasis from colorectal cancer suggested ADCY2 and ADCY9 as potential metastasis prognostic biomarkers (83).
6.4.2 Breast Cancer
Based on the results in Figure 4, ADCY4 expression significantly decreased in breast cancer. The results of the previous studies have shown that Giα is highly expressed in breast cancer cells, especially in metastatic ones. A potential relationship is found between ADCY4 and Giα (7, 84). Furthermore, the proximal targets of cAMP are proteins with diverse functions, which regulate the transcription rates of many genes, such as those playing a role in EMT and inhibiting cell growth and migration, as well as anticancer drug transcription factors with biological roles in anticancer drug sensitivity (7, 84). Unlike gastric cancer, ADCY4 may impose a tumor inhibitory effect in breast cancer, although this effect is terminated by the gene silencing caused by DNA methylation (26). Thus, ADCY4 is a potential biomarker and therapeutic target to predict the prognosis of human breast cancer.
Furthermore, ADCY6 may be implicated in breast cancer progression and affect breast cancer prognosis through calcium-regulated immune and molecular signaling pathways (85). An association is observed between downregulated ADCY6 gene expression and hypomethylation with a better prognosis in breast cancer patients (76).
Some researchers referred to the need for AC8 in breast cancer cell migration as well as the important role of the cAMP-PKA pathway in migrating MCF7 and MDA-MB-231 TNBC cells (86). In MDA-MB-231 cells, AC8 is required for focal adhesion kinase (FAK) phosphorylation, which may explain the role of AC8 in cell migration (87). AC8 and Orai1 interact to trigger the phosphorylation of Orai1 in MDA-MB-231 TNBCs, which results in inactivating Orai1 and stimulating breast cancer migration (86).
6.4.3 Leukemia
Previous studies have revealed the effectiveness of ADCY7 on intracellular cAMP levels and all-trans retinoic acid (ATRA)-induced cell differentiation in APL. The inhibition of ADCY7 elevates apoptosis, reduces cell growth, and declines c-myc expression (88). ADCY7 knockdown significantly inhibits inhibitor-mediated increases in CD11b expression and proliferation in NB4, an APL cell line (51). In addition, microRNA-192 (miRNA) directly targets AC7 expression, the knockdown of which promotes ATRA-induced APL cell differentiation by regulating AC7 expression (51).
Regarding acute myeloid leukemia (AML), CD300A (a type I transmembrane protein) is expressed in the myeloid cells (89). Furthermore, lymphoid lineages cause more proliferation and migration of U937 cells by improving the expression of platelet endothelial cell adhesion molecule (PECAM1) and ADCY7 and by activating the AKT/mTOR (autophagy classical pathway) signaling pathway (89).
6.4.4 Lung Cancer
ADCY1 is overexpressed in nonsmall cell lung cancer (90) and is only accompanied by patient prognosis. Furthermore, ADCY1-mediated cAMP can regulate multidrug resistance in lung cancer and other malignancies by regulating the specific long noncoding RNAs (lncRNAs) involved in different signaling pathways (33).
6.4.5 Melanoma
The low expression of miR-23a-3p (a miRNA) in a melanoma cell line is significantly related to poor prognosis, which significantly diminishes overall survival (OS) and DFS. MiR-23a-3p inhibits cAMP and MAPK signaling pathways because of its targeting of ADCY1 to prevent melanoma cell proliferation, migration, invasion, and tumorigenesis (9). Additionally, anal melanoma (MIS) is the most common malignant melanoma in the East Asian population. The sAC is mainly expressed in nuclei and nucleosomes, such as the deeply stained nuclei of melanoma cells, especially MIS, as well as the protruding nuclei in the vesicular nucleus (91). It may be associated with melanoma invasion, and cytological and nuclear changes in limbs end in melanoma progression (91). However, sAC can be implicated in melanocyte proliferation, apoptosis, and melanin synthesis through its catalytic product cAMP (91).
6.4.6 Laryngeal Cancer
In laryngeal cancer, ADCY6 is a biomarker with significantly different expressions in cancer and noncancer samples. Some researchers reported the involvement of ADCY6 in the cellular processes related to the cancer phenotype like cell cycle, apoptosis, DNA repair, protease inhibition, proteolysis, and transcriptional regulation (92).
6.4.7 Glioblastoma
ADCY8 polymorphisms are accompanied by the risk of glioma (brain tumor) in patients with neurofibromatosis type 1 (NF1), which is sex-specific (93). Based on the single nucleotide polymorphism (SNP) array analysis of cAMP pathway polymorphisms on DNA from NF1 patients with and without optic pathway glioma, polymorphisms in ADCY8 (AC8) lead to more glioma risk among female patients, although they exhibit a protective effect against glioma in male ones (94). Furthermore, ADCY activity and cAMP levels are higher in benign brain tumors, while the lower levels are associated with a greater degree of malignancy (95). Therefore, there is a good reason to believe that a rise in cAMP will be an important tool in brain tumor treatment.
6.4.8 Cervical Cancer
ADCY8 is the most widely reported subtype implicated in cervical intraepithelial neoplasia. Regarding cervical cancer, miR-181b (a miRNA) promotes the growth of cervical cancer cells and inhibits apoptosis by increasing mRNA degradation and decreasing intracellular cAMP levels, which may be caused by binding to the 30′UTR#2 of AC9 (96). MiR-181b has different roles in tumor cells (97, 98). AC9, which produces cAMP, has been identified as a potential target of miR-181b, which negatively regulates AC9 by reducing AC9 mRNA levels (96). Furthermore, the DNA methylation of ADCY8 in molecular Pap smears is considered a biomarker of high-grade cervical cytology (99).
6.4.9 Insulinoma
The expression of ADCY1 and calcium channel 2 (CACNA2) greatly enhances insulinoma with a YY1T372R mutation, while they are less expressed in normal β cells (100). The two gene products are involved in the key pathways regulating insulin secretion with the constitutive activation of cAMP and Ca2+ signaling pathways implied in insulin secretion (101). Accordingly, the higher expression of ADCY1 and CACNA2D2 may play a crucial role in the pathogenesis of insulinoma.
6.4.10 Prostate Cancer
Significant overexpression of sAC has been detected in prostate cancer. In addition, the inhibition of sAC activity prevents prostate cancer cell proliferation, leading to the release of lactate hydrogenase (LDH), as well as apoptosis. The regulation of sAC-dependent proliferation involves the EPAC/Rap1/B-RAF signaling pathway, and EPAC supports prostate cancer cell proliferation by promoting G2/M phase transition, while PKA has no role (99). The antiproliferative effect of sAC inhibition is seemingly ascribed to the inhibition of MEK1, a downstream target of Rap1/B-Raf signaling, which plays an important role in spindle organization and chromosome stabilization (102), and consequently, can impair spindle formation (103) and cause cell cycle arrest in the G2 phase (104).
7 Future Prospects
The ADCY family represents a wide range of functions and plays an important role in a variety of diseases such as neuropathic pain, neurodegenerative diseases, congestive heart failure, asthma, and male contraception. In addition, this family of proteins plays a crucial role in the development of different cancers (26). ADCY isoforms influence the biological behavior of cancer cells such as proliferation, differentiation, apoptosis, and invasive migration, and even have a critical role in cancer cell chemoresistance and sensitivity to radiation via cAMP-PKA and other pathways. In recent years, a relationship has been discovered between the family and cancer immunity. The high expression of some ADCY genes activates cancer-associated T-cell function and improves cancer cell immunity (105). This is another mechanism by which ADCYs exert their oncogenic effects, although the exact process is not well understood and should be clarified in continued research. However, better cancer immunity plays an important role in cancer therapy.
Furthermore, the family is expressed at varying levels in cancer cells, some of which are silenced as oncogenes due to DNA methylation. Therefore, a novel cancer treatment approach can involve the restoration of its expression and oncogenic effect by demethylating genes. Similarly, related protein products or specific protein activators can be developed as anticancer drugs to provide more treatment options for patients with various cancers. Currently, available AC agonists like forskolin exhibit some anticancer effects by elevating intracellular cAMP levels. Due to the extensive distribution of ADCYs and the wide range of involved biological functions, no targeted drugs are now available for ADCY isomers, and an urgent need is felt for the appropriate drugs for the various ADCY isomers that truly target cancer therapy.
The identification of a reagent that can be used to treat mutant ADCY indicates great scientific progress and provides important information for future targeted therapy. The present review introduced deleterious hotspot mutants p.E1003K and p.R1116C on ADCY6 as candidates for developing therapeutic strategies targeting cancer cells. The changes in amino acid positioning and H-bond strength can explain how the p.E1003K and p.R1116C mutations affect the complex interaction of ADCY6. Evidently, future studies on protein–protein docking, molecular dynamics, and conformational entropy, as well as principal component analysis, are required to reveal how the two hotspot mutants destroy the structure and function of the complex and increase the fluctuation range. This information presents an excellent basis for understanding the mechanism underlying an enhancement in p-Akt1 activity in carcinogenesis.
8 Conclusion
This review highlights the mechanistic functions of the AC family and cAMP in conferring chemoresistance, as well as cancer target therapy, which needs further comprehensive studies for full elucidation. The lower AC and cAMP levels have been reported in many cancers, which promote cancer formation and cancer cell proliferation, reduce apoptosis, and even increase neovascularization and cancer cell migration and invasion. Additionally, deleterious nsSNPs p.E1003K and p.R1116C on ADCY6 are candidates to develop potential therapeutic strategies targeting cancer cells. The possibility of targeting the AC family in oncology opens new opportunities for novel therapeutic strategy development and defines a new approach to improve chemoimmunotherapy efficacy in various cancers. The importance of immunotherapy and targeted therapies has become growing in cancer treatment, and the anticancer therapies targeting the ADCY family, certainly ADCY1 and ADCY6, are becoming increasingly possible. Furthermore studies are required to elucidate the paradoxical effects of the other members of the family pathway in response to cancer therapy. In particular, further investigations should be conducted to clarify the interplay between the overexpression of ADCY signaling and cAMP-dependent genes in various tumors. Therefore, targeting the different isoforms of the ADCY family may be a new strategy for treating various cancers.
Author Contributions
RG and TL checked the relevant literature and wrote the original draft of the manuscript. MD and XW performed the data interpretation and visualization in the form of diagrams and tables. SI and QW performed the project administration and funding acquisition. All authors have read and agreed to the published version of the manuscript and agreed to be accountable for all aspects of the work.
Funding
This research was funded by Southwest Medical University (SWMU), grant number HR21013.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully acknowledge all the co-workers who provided technical support for the images and tables in this paper. We sincerely apologize that we cannot cite all contributions due to space limitations.
References
1. Hamilton E, Infante JR. Targeting CDK4/6 in Patients With Cancer. Cancer Treat Rev (2016) 45:129–38. doi: 10.1016/j.ctrv.2016.03.002
2. Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, et al. Functional Expression of the Olfactory Signaling System in the Kidney. Proc Natl Acad Sci U.S.A. (2009) 106(6):2059–64. doi: 10.1073/pnas.0812859106
3. Aldehni F, Tang T, Madsen K, Plattner M, Schreiber A, Friis UG, et al. Stimulation of Renin Secretion by Catecholamines is Dependent on Adenylyl Cyclases 5 and 6. Hypertension (2011) 57(3):460–8. doi: 10.1161/HYPERTENSIONAHA.110.167130
4. Seamon KB, Padgett W, Daly JW. Forskolin: Unique Diterpene Activator of Adenylate Cyclase in Membranes and in Intact Cells. Proc Natl Acad Sci U.S.A. (1981) 78(6):3363–7. doi: 10.1073/pnas.78.6.3363
5. Tang G, Li S, Zhang C, Chen H, Wang N, Feng Y. Clinical Efficacies, Underlying Mechanisms and Molecular Targets of Chinese Medicines for Diabetic Nephropathy Treatment and Management. Acta Pharm Sin B (2021) 11(9):2749–67. doi: 10.1016/j.apsb.2020.12.020
6. Follin-Arbelet V, Torgersen ML, Naderi EH, Misund K, Sundan A, Blomhoff HK. Death of Multiple Myeloma Cells Induced by cAMP-Signaling Involves Downregulation of Mcl-1 via the JAK/STAT Pathway. Cancer Lett (2013) 335(2):323–31. doi: 10.1016/j.canlet.2013.02.042
7. Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C, et al. Activation of PKA Leads to Mesenchymal-to-Epithelial Transition and Loss of Tumor-Initiating Ability. Science (2016) 351(6277):aad3680–aad3680. doi: 10.1126/science.aad3680
8. Zhang H, Kong Q, Wang J, Jiang Y, Hua H. Complex Roles of cAMP-PKA-CREB Signaling in Cancer. Exp Hematol Oncol (2020) 9(1):32. doi: 10.1186/s40164-020-00191-1
9. Ma M, Dai J, Tang H, Xu T, Yu S, Si L, et al. MicroRNA-23a-3p Inhibits Mucosal Melanoma Growth and Progression Through Targeting Adenylate Cyclase 1 and Attenuating cAMP and MAPK Pathways. Theranostics (2019) 9(4):945–60. doi: 10.7150/thno.30516
10. Johann K, Bohn T, Shahneh F, Luther N, Birke A, Jaurich H, et al. Therapeutic Melanoma Inhibition by Local Micelle-Mediated Cyclic Nucleotide Repression. Nat Commun (2021) 12(1):5981. doi: 10.1038/s41467-021-26269-w
11. Sanchez-Collado J, Lopez JJ, Jardin I, Salido GM, Rosado JA. Cross-Talk Between the Adenylyl Cyclase/cAMP Pathway and Ca(2+) Homeostasis. Rev Physiol Biochem Pharmacol (2021) 179:73–116. doi: 10.1007/112_2020_55
12. Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol Rev (2017) 69(2):93–139. doi: 10.1124/pr.116.013078
13. Wen DY, Lin P, Liang HW, Yang X, Li HY, He Y, et al. Up-Regulation of CTD-2547G23.4 in Hepatocellular Carcinoma Tissues and its Prospective Molecular Regulatory Mechanism: A Novel qRT-PCR and Bioinformatics Analysis Study. Cancer Cell Int (2018) 18:74. doi: 10.1186/s12935-018-0566-3
14. Pierre S, Eschenhagen T, Geisslinger G, Scholich K. Capturing Adenylyl Cyclases as Potential Drug Targets. Nat Rev Drug Discovery (2009) 8(4):321–35. doi: 10.1038/nrd2827
15. Boran AD, Chen Y, Iyengar R. Identification of New Gβγ Interaction Sites in Adenylyl Cyclase 2. Cell Signal (2011) 23(9):1489–95. doi: 10.1016/j.cellsig.2011.05.002
16. Seamon KB, Daly JW, Metzger H, de Souza NJ, Reden J. Structure-Activity Relationships for Activation of Adenylate Cyclase by the Diterpene Forskolin and its Derivatives. J Med Chem (1983) 26(3):436–9. doi: 10.1021/jm00357a021
17. Sabbatini ME, Gorelick F, Glaser S. Adenylyl Cyclases in the Digestive System. Cell Signal (2014) 26(6):1173–81. doi: 10.1016/j.cellsig.2014.01.033
18. Bogard AS, Birg AV, Ostrom RS. Non-Raft Adenylyl Cyclase 2 Defines a cAMP Signaling Compartment That Selectively Regulates IL-6 Expression in Airway Smooth Muscle Cells: Differential Regulation of Gene Expression by AC Isoforms. Naunyn Schmiedebergs Arch Pharmacol (2014) 387(4):329–39. doi: 10.1007/s00210-013-0950-4
19. Tang T, Lai NC, Wright AT, Gao MH, Lee P, Guo T, et al. Adenylyl Cyclase 6 Deletion Increases Mortality During Sustained Beta-Adrenergic Receptor Stimulation. J Mol Cell Cardiol (2013) 60:60–7. doi: 10.1016/j.yjmcc.2013.04.005
20. Beavo JA, Brunton LL. Cyclic Nucleotide Research – Still Expanding After Half a Century. Nat Rev Mol Cell Biol (2002) 3(9):710–8. doi: 10.1038/nrm911
21. Omori K, Kotera J. Overview of PDEs and Their Regulation. Circ Res (2007) 100(3):309–27. doi: 10.1161/01.RES.0000256354.95791.f1
22. Dang N, Meng X, Song H. Nicotinic Acetylcholine Receptors and Cancer. BioMed Rep (2016) 4(5):515–8. doi: 10.3892/br.2016.625
23. Ho D, Yan L, Iwatsubo K, Vatner DE, Vatner SF. Modulation of Beta-Adrenergic Receptor Signaling in Heart Failure and Longevity: Targeting Adenylyl Cyclase Type 5. Heart Fail Rev (2010) 15(5):495–512. doi: 10.1007/s10741-010-9183-5
24. Lee YS, Marmorstein LY, Marmorstein AD. Soluble Adenylyl Cyclase in the Eye. Biochim Biophys Acta (2014) 1842(12 Pt B):2579–83. doi: 10.1016/j.bbadis.2014.07.032
25. Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, et al. Identification of an Adenylyl Cyclase Inhibitor for Treating Neuropathic and Inflammatory Pain. Sci Transl Med (2011) 3(65):65ra63. doi: 10.1126/scitranslmed.3001269
26. Fan Y, Mu J, Huang M, Imani S, Wang Y, Lin S, et al. Epigenetic Identification of ADCY4 as a Biomarker for Breast Cancer: An Integrated Analysis of Adenylate Cyclases. Epigenomics (2019) 11(14):1561–79. doi: 10.2217/epi-2019-0207
27. Zachariou V, Liu R, LaPlant Q, Xiao G, Renthal W, Chan GC, et al. Distinct Roles of Adenylyl Cyclases 1 and 8 in Opiate Dependence: Behavioral, Electrophysiological, and Molecular Studies. Biol Psychiatry (2008) 63(11):1013–21. doi: 10.1016/j.biopsych.2007.11.021
28. Li YR, Li J, Zhao SD, Bradfield JP, Mentch FD, Maggadottir SM, et al. Meta-Analysis of Shared Genetic Architecture Across Ten Pediatric Autoimmune Diseases. Nat Med (2015) 21(9):1018–27. doi: 10.1038/nm.3933
29. Niesor EJ, Benghozi R, Amouyel P, Ferdinand KC, Schwartz GG. Adenylyl Cyclase 9 Polymorphisms Reveal Potential Link to HDL Function and Cardiovascular Events in Multiple Pathologies: Potential Implications in Sickle Cell Disease. Cardiovasc Drugs Ther (2015) 29(6):563–72. doi: 10.1007/s10557-015-6626-1
30. Tang T, Lai NC, Wright AT, Gao MH, Lee P, Guo T, et al. Adenylyl Cyclase 6 Deletion Increases Mortality During Sustained β-Adrenergic Receptor Stimulation. J Mol Cell Cardiol (2013) 60:60–7. doi: 10.1016/j.yjmcc.2013.04.005
31. Holz GG, Leech CA, Chepurny OG. New Insights Concerning the Molecular Basis for Defective Glucoregulation in Soluble Adenylyl Cyclase Knockout Mice. Biochim Biophys Acta (2014) 1842(12 Pt B):2593–600. doi: 10.1016/j.bbadis.2014.06.023
32. Patel TB, Du Z, Pierre S, Cartin L, Scholich K. Molecular Biological Approaches to Unravel Adenylyl Cyclase Signaling and Function. Gene (2001) 269(1-2):13–25. doi: 10.1016/s0378-1119(01)00448-6
33. Zou T, Liu J, She L, Chen J, Zhu T, Yin J, et al. A Perspective Profile of ADCY1 in cAMP Signaling With Drug-Resistance in Lung Cancer. J Cancer (2019) 10(27):6848–57. doi: 10.7150/jca.36614
34. Bos JL. Epac: A New cAMP Target and New Avenues in cAMP Research. Nat Rev Mol Cell Biol (2003) 4(9):733–8. doi: 10.1038/nrm1197
35. Dong Z, Nishiyama J, Yi X, Venkatachalam MA, Denton M, Gu S, et al. Gene Promoter of Apoptosis Inhibitory Protein IAP2: Identification of Enhancer Elements and Activation by Severe Hypoxia. Biochem J (2002) 364(Pt 2):413–21. doi: 10.1042/bj20011431
36. Kim EJ, Juhnn YS. Cyclic AMP Signaling Reduces Sirtuin 6 Expression in non-Small Cell Lung Cancer Cells by Promoting Ubiquitin-Proteasomal Degradation via Inhibition of the Raf-MEK-ERK (Raf/mitogen-Activated Extracellular Signal-Regulated Kinase/Extracellular Signal-Regulated Kinase) Pathway. J Biol Chem (2015) 290(15):9604–13. doi: 10.1074/jbc.M114.633198
37. Cho EA, Kim EJ, Kwak SJ, Juhnn YS. cAMP Signaling Inhibits Radiation-Induced ATM Phosphorylation Leading to the Augmentation of Apoptosis in Human Lung Cancer Cells. Mol Cancer (2014) 13:36. doi: 10.1186/1476-4598-13-36
38. Luo K, Gu X, Liu J, Zeng G, Peng L, Huang H, et al. Inhibition of Disheveled-2 Resensitizes Cisplatin-Resistant Lung Cancer Cells Through Down-Regulating Wnt/beta-Catenin Signaling. Exp Cell Res (2016) 347(1):105–13. doi: 10.1016/j.yexcr.2016.07.014
39. Seino S, Shibasaki T. PKA-Dependent and PKA-Independent Pathways for cAMP-Regulated Exocytosis. Physiol Rev (2005) 85(4):1303–42. doi: 10.1152/physrev.00001.2005
40. Mansilla Pareja ME, Gaurón MC, Robledo E, Aguilera MO, Colombo MI. The cAMP Effectors, Rap2b and EPAC, are Involved in the Regulation of the Development of the Coxiella Burnetii Containing Vacuole by Altering the Fusogenic Capacity of the Vacuole. PloS One (2019) 14(2):e0212202. doi: 10.1371/journal.pone.0212202
41. Park JY, Juhnn YS. cAMP Signaling Increases Histone Deacetylase 8 Expression by Inhibiting JNK-Dependent Degradation via Autophagy and the Proteasome System in H1299 Lung Cancer Cells. Biochem Biophys Res Commun (2016) 470(2):336–42. doi: 10.1016/j.bbrc.2016.01.049
42. Khan N, Kahl B. Targeting BCL-2 in Hematologic Malignancies. Target Oncol (2018) 13(3):257–67. doi: 10.1007/s11523-018-0560-7
43. D'Orsi B, Mateyka J, Prehn JHM. Control of Mitochondrial Physiology and Cell Death by the Bcl-2 Family Proteins Bax and Bok. Neurochem Int (2017) 109:162–70. doi: 10.1016/j.neuint.2017.03.010
44. Schuller HM. Inhibitory Role of Gi-Coupled Receptors on cAMP-Driven Cancers With Focus on Opioid Receptors in Lung Adenocarcinoma and its Stem Cells. Vitam Horm (2019) 111:299–311. doi: 10.1016/bs.vh.2019.05.009
45. Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak. Science (2007) 315(5813):856–9. doi: 10.1126/science.1133289
46. Ferretti AC, Tonucci FM, Hidalgo F, Almada E, Larocca MC, Favre C. AMPK and PKA Interaction in the Regulation of Survival of Liver Cancer Cells Subjected to Glucose Starvation. Oncotarget (2016) 7(14):17815–28. doi: 10.18632/oncotarget.7404
47. Ostojić J, Yoon YS, Sonntag T, Nguyen B, Vaughan JM, Shokhirev M, et al. Transcriptional Co-Activator Regulates Melanocyte Differentiation and Oncogenesis by Integrating cAMP and MAPK/ERK Pathways. Cell Rep (2021) 35(7):109136. doi: 10.1016/j.celrep.2021.109136
48. Liu C, Du Q, Zhang X, Tang Z, Ji H, Li Y. Clematichinenoside Serves as a Neuroprotective Agent Against Ischemic Stroke: The Synergistic Action of ERK1/2 and cPKC Pathways. Front Cell Neurosci (2015) 9:517. doi: 10.3389/fncel.2015.00517
49. Sonabend AM, Musleh W, Lesniak MS. Oncogenesis and Mutagenesis of Pituitary Tumors. Expert Rev Anticancer Ther (2006) 6 Suppl 9:S3–14. doi: 10.1586/14737140.6.9s.S3
50. Sun HJ, Liu TY, Zhang F, Xiong XQ, Wang JJ, Chen Q, et al. Salusin-β Contributes to Vascular Remodeling Associated With Hypertension via Promoting Vascular Smooth Muscle Cell Proliferation and Vascular Fibrosis. Biochim Biophys Acta (2015) 1852(9):1709–18. doi: 10.1016/j.bbadis.2015.05.008
51. He B, Chang Y, Yang C, Zhang Z, Xu G, Feng X, et al. Adenylate Cyclase 7 Regulated by miR-192 Promotes ATRA-Induced Differentiation of Acute Promyelocytic Leukemia Cells. Biochem Biophys Res Commun (2018) 506(3):543–7. doi: 10.1016/j.bbrc.2018.10.125
52. Zhuang LK, Xu GP, Pan XR, Lou YJ, Zou QP, Xia D, et al. MicroRNA-181a-Mediated Downregulation of AC9 Protein Decreases Intracellular cAMP Level and Inhibits ATRA-Induced APL Cell Differentiation. Cell Death Dis (2014) 5:e1161. doi: 10.1038/cddis.2014.130
53. Rodríguez CI, Castro-Pérez E, Longley BJ, Setaluri V. Elevated Cyclic AMP Levels Promote BRAF(CA)/Pten(-/-) Mouse Melanoma Growth But pCREB is Negatively Correlated With Human Melanoma Progression. Cancer Lett (2018) 414:268–77. doi: 10.1016/j.canlet.2017.11.027
54. Li H, Liu Y, Liu J, Sun Y, Wu J, Xiong Z, et al. Assessment of ADCY9 Polymorphisms and Colorectal Cancer Risk in the Chinese Han Population. J Gene Med (2021) 23(2):e3298. doi: 10.1002/jgm.3298
55. Gong S, Chen Y, Meng F, Zhang Y, Li C, Zhang G, et al. Roflumilast Enhances Cisplatin-Sensitivity and Reverses Cisplatin-Resistance of Ovarian Cancer Cells via cAMP/PKA/CREB-FtMt Signalling Axis. Cell Prolif (2018) 51(5):e12474. doi: 10.1111/cpr.12474
56. Sapio L, Gallo M, Illiano M, Chiosi E, Naviglio D, Spina A, et al. The Natural cAMP Elevating Compound Forskolin in Cancer Therapy: Is It Time? J Cell Physiol (2017) 232(5):922–7. doi: 10.1002/jcp.25650
57. Wesarg E, Hoffarth S, Wiewrodt R, Kroll M, Biesterfeld S, Huber C, et al. Targeting BCL-2 Family Proteins to Overcome Drug Resistance in non-Small Cell Lung Cancer. Int J Cancer (2007) 121(11):2387–94. doi: 10.1002/ijc.22977
58. Ginsberg G, Angle K, Guyton K, Sonawane B. Polymorphism in the DNA Repair Enzyme XRCC1: Utility of Current Database and Implications for Human Health Risk Assessment. Mutat Res (2011) 727(1-2):1–15. doi: 10.1016/j.mrrev.2011.02.001
59. Lee Y, McKinnon PJ. ATM Dependent Apoptosis in the Nervous System. Apoptosis (2000) 5(6):523–9. doi: 10.1023/a:1009637512917
60. Derheimer FA, Kastan MB. Multiple Roles of ATM in Monitoring and Maintaining DNA Integrity. FEBS Lett (2010) 584(17):3675–81. doi: 10.1016/j.febslet.2010.05.031
61. Xia Y, He Z, Liu B, Wang P, Chen Y. Downregulation of Meg3 Enhances Cisplatin Resistance of Lung Cancer Cells Through Activation of the WNT/beta-Catenin Signaling Pathway. Mol Med Rep (2015) 12(3):4530–7. doi: 10.3892/mmr.2015.3897
62. Yang X, Shang P, Yu B, Jin Q, Liao J, Wang L, et al. Combination Therapy With Mir34a and Doxorubicin Synergistically Inhibits Dox-Resistant Breast Cancer Progression via Down-Regulation of Snail Through Suppressing Notch/NF-κb and RAS/RAF/MEK/ERK Signaling Pathway. Acta Pharm Sin B (2021) 11(9):2819–34. doi: 10.1016/j.apsb.2021.06.003
63. Illiano M, Sapio L, Salzillo A, Capasso L, Caiafa I, Chiosi E, et al. Forskolin Improves Sensitivity to Doxorubicin of Triple Negative Breast Cancer Cells via Protein Kinase A-Mediated ERK1/2 Inhibition. Biochem Pharmacol (2018) 152:104–13. doi: 10.1016/j.bcp.2018.03.023
64. Yamagishi N, Nakao R, Kondo R, Nishitsuji M, Saito Y, Kuga T, et al. Increased Expression of Sorcin is Associated With Multidrug Resistance in Leukemia Cells via Up-Regulation of MDR1 Expression Through cAMP Response Element-Binding Protein. Biochem Biophys Res Commun (2014) 448(4):430–6. doi: 10.1016/j.bbrc.2014.04.125
65. de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 Guanine-Nucleotide-Exchange Factor Directly Activated by Cyclic AMP. Nature (1998) 396(6710):474–7. doi: 10.1038/24884
66. Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A Family of cAMP-Binding Proteins That Directly Activate Rap1. Science (1998) 282(5397):2275–9. doi: 10.1126/science.282.5397.2275
67. Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, et al. Cyclic AMP Induces Integrin-Mediated Cell Adhesion Through Epac and Rap1 Upon Stimulation of the Beta 2-Adrenergic Receptor. J Cell Biol (2003) 160(4):487–93. doi: 10.1083/jcb.200209105
68. Misra UK, Pizzo SV. Epac1-Induced Cellular Proliferation in Prostate Cancer Cells is Mediated by B-Raf/ERK and mTOR Signaling Cascades. J Cell Biochem (2009) 108(4):998–1011. doi: 10.1002/jcb.22333
69. Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, et al. A Novel EPAC-Specific Inhibitor Suppresses Pancreatic Cancer Cell Migration and Invasion. Mol Pharmacol (2013) 83(1):122–8. doi: 10.1124/mol.112.080689
70. Baljinnyam E, Umemura M, De Lorenzo MS, Iwatsubo M, Chen S, Goydos JS, et al. Epac1 Promotes Melanoma Metastasis via Modification of Heparan Sulfate. Pigment Cell Melanoma Res (2011) 24(4):680–7. doi: 10.1111/j.1755-148X.2011.00863.x
71. Yu C, Hong H, Lu J, Zhao X, Hu W, Zhang S, et al. Prediction of Target Genes and Pathways Associated With Cetuximab Insensitivity in Colorectal Cancer. Technol Cancer Res Treat (2018) 17:1533033818806905. doi: 10.1177/1533033818806905
72. Abrams SL, Steelman LS, Shelton JG, Wong EW, Chappell WH, Basecke J, et al. The Raf/MEK/ERK Pathway Can Govern Drug Resistance, Apoptosis and Sensitivity to Targeted Therapy. Cell Cycle (2010) 9(9):1781–91. doi: 10.4161/cc.9.9.11483
73. Naderi EH, Jochemsen AG, Blomhoff HK, Naderi S. Activation of cAMP Signaling Interferes With Stress-Induced p53 Accumulation in ALL-Derived Cells by Promoting the Interaction Between p53 and HDM2. Neoplasia (2011) 13(7):653–63. doi: 10.1593/neo.11542
74. Kloster MM, Naderi EH, Haaland I, Gjertsen BT, Blomhoff HK, Naderi S. cAMP Signalling Inhibits p53 Acetylation and Apoptosis via HDAC and SIRT Deacetylases. Int J Oncol (2013) 42(5):1815–21. doi: 10.3892/ijo.2013.1853
75. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
76. Li W, Sang M, Hao X, Jia L, Wang Y, Shan B. Gene Expression and DNA Methylation Analyses Suggest That Immune Process-Related ADCY6 is a Prognostic Factor of Luminal-Like Breast Cancer. J Cell Biochem (2020) 121(7):3537–46. doi: 10.1002/jcb.29633
77. Hong SH, Goh SH, Lee SJ, Hwang JA, Lee J, Choi IJ, et al. Upregulation of Adenylate Cyclase 3 (ADCY3) Increases the Tumorigenic Potential of Cells by Activating the CREB Pathway. Oncotarget (2013) 4(10):1791–803. doi: 10.18632/oncotarget.1324
78. Zou J, Wu K, Lin C, Jie ZG. LINC00319 Acts as a microRNA-335-5p Sponge to Accelerate Tumor Growth and Metastasis in Gastric Cancer by Upregulating ADCY3. Am J Physiol Gastrointest Liver Physiol (2020) 318(1):G10–g22. doi: 10.1152/ajpgi.00405.2018
79. Quinn SN, Graves SH, Dains-McGahee C, Friedman EM, Hassan H, Witkowski P, et al. Adenylyl Cyclase 3/Adenylyl Cyclase-Associated Protein 1 (CAP1) Complex Mediates the Anti-Migratory Effect of Forskolin in Pancreatic Cancer Cells. Mol Carcinog (2017) 56(4):1344–60. doi: 10.1002/mc.22598
80. Burdyga A, Conant A, Haynes L, Zhang J, Jalink K, Sutton R, et al. cAMP Inhibits Migration, Ruffling and Paxillin Accumulation in Focal Adhesions of Pancreatic Ductal Adenocarcinoma Cells: Effects of PKA and EPAC. Biochim Biophys Acta (2013) 1833(12):2664–72. doi: 10.1016/j.bbamcr.2013.06.011
81. Zimmerman NP, Roy I, Hauser AD, Wilson JM, Williams CL, Dwinell MB. Cyclic AMP Regulates the Migration and Invasion Potential of Human Pancreatic Cancer Cells. Mol Carcinogenesis (2015) 54(3):203–15. doi: 10.1002/mc.22091
82. Zhang Y, Qiu Z, Wei L, Tang R, Lian B, Zhao Y, et al. Integrated Analysis of Mutation Data From Various Sources Identifies Key Genes and Signaling Pathways in Hepatocellular Carcinoma. PloS One (2014) 9(7):e100854. doi: 10.1371/journal.pone.0100854
83. Fang LT, Lee S, Choi H, Kim HK, Jew G, Kang HC, et al. Comprehensive Genomic Analyses of a Metastatic Colon Cancer to the Lung by Whole Exome Sequencing and Gene Expression Analysis. Int J Oncol (2014) 44(1):211–21. doi: 10.3892/ijo.2013.2150
84. Dong H, Claffey KP, Brocke S, Epstein PM. Inhibition of Breast Cancer Cell Migration by Activation of cAMP Signaling. Breast Cancer Res Treat (2015) 152(1):17–28. doi: 10.1007/s10549-015-3445-9
85. Liu S, Liu X, Wu J, Zhou W, Ni M, Meng Z, et al. Identification of Candidate Biomarkers Correlated With the Pathogenesis and Prognosis of Breast Cancer via Integrated Bioinformatics Analysis. Med (Baltimore) (2020) 99(49):e23153. doi: 10.1097/MD.0000000000023153
86. Sanchez-Collado J, Lopez JJ, Jardin I, Camello PJ, Falcon D, Regodon S, et al. Adenylyl Cyclase Type 8 Overexpression Impairs Phosphorylation-Dependent Orai1 Inactivation and Promotes Migration in MDA-MB-231 Breast Cancer Cells. Cancers (Basel) (2019) 11(11):1624–48. doi: 10.3390/cancers11111624
87. Abrams SL, Steelman LS, Shelton JG, Wong EW, Chappell WH, Bäsecke J, et al. The Raf/MEK/ERK Pathway can Govern Drug Resistance, Apoptosis and Sensitivity to Targeted Therapy. Cell Cycle (2010) 9(9):1781–91. doi: 10.4161/cc.9.9.11483
88. Sun X, Huang S, Wang X, Zhang X, Wang X. CD300A Promotes Tumor Progression by PECAM1, ADCY7 and AKT Pathway in Acute Myeloid Leukemia. Oncotarget (2018) 9(44):27574–84. doi: 10.18632/oncotarget.24164
89. Li C, Xie J, Lu Z, Chen C, Yin Y, Zhan R, et al. ADCY7 Supports Development of Acute Myeloid Leukemia. Biochem Biophys Res Commun (2015) 465(1):47–52. doi: 10.1016/j.bbrc.2015.07.123
90. He RQ, Li XJ, Liang L, Xie Y, Luo DZ, Ma J, et al. The Suppressive Role of miR-542-5p in NSCLC: The Evidence From Clinical Data and In Vivo Validation Using a Chick Chorioallantoic Membrane Model. BMC Cancer (2017) 17(1):655. doi: 10.1186/s12885-017-3646-1
91. Li H, Kim SM, Savkovic V, Jin SA, Choi YD, Yun SJ. Expression of Soluble Adenylyl Cyclase in Acral Melanomas. Clin Exp Dermatol (2016) 41(4):425–9. doi: 10.1111/ced.12730
92. Warrington NM, Sun T, Rubin JB. Targeting Brain Tumor cAMP: The Case for Sex-Specific Therapeutics. Front Pharmacol (2015) 6:153–60. doi: 10.3892/or_00000268
93. Warrington NM, Sun T, Rubin JB. Targeting Brain Tumor cAMP: The Case for Sex-Specific Therapeutics. Front Pharmacol (2015) 6:153. doi: 10.3389/fphar.2015.00153
94. Warrington NM, Sun T, Luo J, McKinstry RC, Parkin PC, Ganzhorn S, et al. The Cyclic AMP Pathway is a Sex-Specific Modifier of Glioma Risk in Type I Neurofibromatosis Patients. Cancer Res (2015) 75(1):16–21. doi: 10.1158/0008-5472.Can-14-1891
95. Held-Feindt J, Krisch B, Forstreuter F, Mentlein R. Somatostatin Receptors in Gliomas. J Physiol Paris (2000) 94(3-4):251–8. doi: 10.1016/s0928-4257(00)00213-8
96. Yang L, Wang YL, Liu S, Zhang PP, Chen Z, Liu M, et al. miR-181b Promotes Cell Proliferation and Reduces Apoptosis by Repressing the Expression of Adenylyl Cyclase 9 (AC9) in Cervical Cancer Cells. FEBS Lett (2014) 588(1):124–30. doi: 10.1016/j.febslet.2013.11.019
97. Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, et al. Hsa-Mir-181a and Hsa-Mir-181b Function as Tumor Suppressors in Human Glioma Cells. Brain Res (2008) 1236:185–93. doi: 10.1016/j.brainres.2008.07.085
98. Zhu W, Shan X, Wang T, Shu Y, Liu P. miR-181b Modulates Multidrug Resistance by Targeting BCL2 in Human Cancer Cell Lines. Int J Cancer (2010) 127(11):2520–9. doi: 10.1002/ijc.25260
99. Flacke JP, Flacke H, Appukuttan A, Palisaar RJ, Noldus J, Robinson BD, et al. Type 10 Soluble Adenylyl Cyclase is Overexpressed in Prostate Carcinoma and Controls Proliferation of Prostate Cancer Cells. J Biol Chem (2013) 288(5):3126–35. doi: 10.1074/jbc.M112.403279
100. Thorvaldsen JL, Weaver JR, Bartolomei MS. A YY1 Bridge for X Inactivation. Cell (2011) 146(1):11–3. doi: 10.1016/j.cell.2011.06.029
101. Cromer MK, Choi M, Nelson-Williams C, Fonseca AL, Kunstman JW, Korah RM, et al. Neomorphic Effects of Recurrent Somatic Mutations in Yin Yang 1 in Insulin-Producing Adenomas. Proc Natl Acad Sci U.S.A. (2015) 112(13):4062–7. doi: 10.1073/pnas.1503696112
102. Cao JN, Shafee N, Vickery L, Kaluz S, Ru N, Stanbridge EJ. Mitogen-Activated Protein/Extracellular Signal-Regulated Kinase Kinase 1act/Tubulin Interaction is an Important Determinant of Mitotic Stability in Cultured HT1080 Human Fibrosarcoma Cells. Cancer Res (2010) 70(14):6004–14. doi: 10.1158/0008-5472.Can-09-4490
103. Feinstein TN, Linstedt AD. Mitogen-Activated Protein Kinase Kinase 1-Dependent Golgi Unlinking Occurs in G2 Phase and Promotes the G2/M Cell Cycle Transition. Mol Biol Cell (2007) 18(2):594–604. doi: 10.1091/mbc.e06-06-0530
104. Hayne C, Tzivion G, Luo Z. Raf-1/MEK/MAPK Pathway is Necessary for the G2/M Transition Induced by Nocodazole. J Biol Chem (2000) 275(41):31876–82. doi: 10.1074/jbc.M002766200
Keywords: adenylate cyclase, cAMP signaling, molecular targeted therapy, chemoresistance, signaling pathway 4
Citation: Guo R, Liu T, Shasaltaneh MD, Wang X, Imani S and Wen Q (2022) Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front. Oncol. 12:829212. doi: 10.3389/fonc.2022.829212
Received: 05 December 2021; Accepted: 26 May 2022;
Published: 27 June 2022.
Edited by:
Yan-Yan Yan, Shanxi Datong University, ChinaReviewed by:
Silvio Naviglio, University of Campania Luigi Vanvitelli, ItalyShuanshuan Xie, Tongji University, China
Copyright © 2022 Guo, Liu, Shasaltaneh, Wang, Imani and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saber Imani, c2FiZXJpbWFuaUBzd211LmVkdS5jbg==; QingLian Wen, d3FsNzMxMTVAaG90bWFpbC5jb20=
†These authors have contributed equally to this work