 Eric D. Routh1‡
Eric D. Routh1‡ Amanda E. D. Van Swearingen1†‡
Amanda E. D. Van Swearingen1†‡ Maria J. Sambade1‡Steven Vensko1
Maria J. Sambade1‡Steven Vensko1 Marni B. McClure1,2†Mark G. Woodcock1,3Shengjie Chai1,4Luz A. Cuaboy1Amy Wheless1Amy Garrett1†Lisa A. Carey1,3
Marni B. McClure1,2†Mark G. Woodcock1,3Shengjie Chai1,4Luz A. Cuaboy1Amy Wheless1Amy Garrett1†Lisa A. Carey1,3 Alan P. Hoyle1Joel S. Parker1,5
Alan P. Hoyle1Joel S. Parker1,5 Benjamin G. Vincent1,3,4,6,7§
Benjamin G. Vincent1,3,4,6,7§ Carey K. Anders1,3*†§
Carey K. Anders1,3*†§- 1Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2National Cancer Center Research Institute, Tokyo, Japan
- 3Department of Medicine, Division of Medical Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 4Curriculum in Bioinformatics and Computational Biology, UNC School of Medicine, Chapel Hill, NC, United States
- 5Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 6Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 7Division of Hematology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Background: Triple negative breast cancer (TNBC) is an aggressive variant of breast cancer that lacks the expression of estrogen and progesterone receptors (ER and PR) and HER2. Nearly 50% of patients with advanced TNBC will develop brain metastases (BrM), commonly with progressive extracranial disease. Immunotherapy has shown promise in the treatment of advanced TNBC; however, the immune contexture of BrM remains largely unknown. We conducted a comprehensive analysis of TNBC BrM and matched primary tumors to characterize the genomic and immune landscape of TNBC BrM to inform the development of immunotherapy strategies in this aggressive disease.
Methods: Whole-exome sequencing (WES) and RNA sequencing were conducted on formalin-fixed, paraffin-embedded samples of BrM and primary tumors of patients with clinical TNBC (n = 25, n = 9 matched pairs) from the LCCC1419 biobank at UNC—Chapel Hill. Matched blood was analyzed by DNA sequencing as a comparison for tumor WES for the identification of somatic variants. A comprehensive genomics assessment, including mutational and copy number alteration analyses, neoantigen prediction, and transcriptomic analysis of the tumor immune microenvironment were performed.
Results: Primary and BrM tissues were confirmed as TNBC (23/25 primaries, 16/17 BrM) by immunohistochemistry and of the basal intrinsic subtype (13/15 primaries and 16/19 BrM) by PAM50. Compared to primary tumors, BrM demonstrated a higher tumor mutational burden. TP53 was the most frequently mutated gene and was altered in 50% of the samples. Neoantigen prediction showed elevated cancer testis antigen- and endogenous retrovirus-derived MHC class I-binding peptides in both primary tumors and BrM and predicted that single-nucleotide variant (SNV)-derived peptides were significantly higher in BrM. BrM demonstrated a reduced immune gene signature expression, although a signature associated with fibroblast-associated wound healing was elevated in BrM. Metrics of T and B cell receptor diversity were also reduced in BrM.
Conclusions: BrM harbored higher mutational burden and SNV-derived neoantigen expression along with reduced immune gene signature expression relative to primary TNBC. Immune signatures correlated with improved survival, including T cell signatures. Further research will expand these findings to other breast cancer subtypes in the same biobank. Exploration of immunomodulatory approaches including vaccine applications and immune checkpoint inhibition to enhance anti-tumor immunity in TNBC BrM is warranted.
Introduction
Triple-negative breast cancer (TNBC) lacks the expression of hormone receptors estrogen (ER) and progesterone (PR) as well as human epidermal growth factor receptor 2 (HER2). TNBC is also the most aggressive subtype of breast cancer, with a predilection for brain metastases; up to 50% of patients with metastatic TNBC will develop brain metastases (BrM) during their disease course (1). Patients with TNBC BrM face a poor prognosis with a median survival following diagnosis of less than 6 months (2). Despite progress in the treatment of ER+ and HER2+ breast cancer BrM with the advent of brain-penetrant targeted therapies, the prognosis for TNBC BrM remains largely unchanged over the past decade (3). Thus, studies to better understand the biology of TNBC BrM to identify new therapeutic targets are needed.
Previous studies have conducted sequencing of primary and metastatic tissues, including BrM, from melanoma (4), lung cancer (5), breast cancer (6–8), and multiple solid tumor types (9). A seminal work by Brastianos et al. demonstrated that some solid tumors (including BrM) undergo branched evolution during the metastatic process. These studies have led to a growing appreciation that BrM can be biologically different from not just their primary tumors but also extracranial metastases, including differential acquisition or loss of targetable alterations. Breast cancer brain metastasis (BCBrM) have demonstrated mutations and/or copy number alterations in clinically targetable genes and pathways such as HER2 (6, 7, 9), BRAF (8), PI3K/Akt (9), CDK (6), ATM (8), and CRYAB (10) not seen in primary tumors. BrM can also be metabolically different from primaries, with increased oxidative phosphorylation (4). Preclinical studies have demonstrated that these targets can alter the brain metastatic potential and/or growth of BrM in breast cancer models (4, 11–13). These findings have led to the first genomically guided clinical trial in BrM to match alterations present in BrM to an appropriate brain-penetrant inhibitor (NCT03994796).
While these and other prior studies have made significant progress in the genomic characterization of BrM in recent years, no studies have yet focused exclusively on TNBC BrM specifically, and few studies have looked at comprehensive RNA and DNA sequencing-derived features of the tumor immune microenvironment. Using the LCCC1419 Biobank of metastatic breast cancer samples, we have collected and analyzed BrM, matched primaries, and normal tissue from 25 patients with TNBC through both whole-exome sequencing (WES) and mRNA sequencing. We report the somatic mutational landscape of TNBC BrM compared to primary tumors and implement a comprehensive neoantigen prediction pipeline to elucidate potentially immunogenic peptides arising from traditional and alternative neoantigen sources. Utilizing mRNA gene expression, we evaluated the tumor immune microenvironment of TNBC BrM relative to primary tumors and correlate these features with overall survival. To our knowledge, this study represents the largest evaluation of TNBC BrM through WES and RNA-seq and is the first to analyze gene expression and immunogenomics in addition to the mutational landscape.
Materials and Methods
Patient Consent and Tissue Collection
Archival formalin-fixed, paraffin embedded (FFPE) tumor tissues were obtained from patients with clinically determined triple-negative breast cancer (TNBC) based on either a primary or metastatic site, with known metastasis to the brain. The patients consented to participation in either the UNC Health Registry (UNC IRB 09-0605), opened on 04/16/2009 and consented between 11/2014 and 06/2016, or to a clinically annotated biobank study at the University of North Carolina at Chapel Hill under an Institutional Review Board (IRB)-approved protocol (LCCC1419) which opened on 10/31/2014 and consented from 11/2014 to 11/2018. Brain metastases tissues were available from n = 19 patients (n = 19 with RNA, n = 17 with DNA), while primary tumor tissue was available from n = 16 patients (n = 15 with RNA, n = 13 with DNA). Matched whole blood samples were available for n = 22 patients (DNA). Of these cases, n = 9 included matched RNA primary TNBC and BrM tissue pairs from the same patient, and n = 6 had matched DNA triplet samples (primary, BrM, and blood), all n = 6 of which also had RNA for both primary and BrM samples.
DNA Whole-Exome Sequencing and Variant Calling
FFPE tumor/tissue blocks and normal fresh frozen blood samples were collected and inventoried through honest brokers in accordance with IRB standards through the UNC Health Registry. UNC patient samples were inventoried through UNC Surgical Pathology Core (SP), while patient samples from outside UNC were inventoried through UNC Tissue Procurement Facility. Tissue blocks were sectioned by UNC Translational Pathology Laboratory (TPL). Twenty-two sections were made at a time: two 5-μm sections at the beginning and end of sectioning for pathologist review and 20 10-μm sections for DNA/RNA isolation on glass slides. TPL pathologists reviewed the samples, circling areas with more than 50% tumor mass for DNA/RNA isolation. When required, the process was repeated to collect more DNA/RNA. UNC BioSpecimen Processing Facility performed all DNA/RNA isolations. DNA from tumor-enriched cores were extracted using the Maxwell 16 FFPE Tissue LEV DNA Purification Kit. DNA was exome-captured and amplified with Agilent SureSelect XT (G9611B) and capture library (5190-8881). WES was performed using the Illumina HiSeq 2500 or NextSeq 500 platform with multiple samples per lane using 2 × 100 paired-end chemistry. RNA was isolated from the same sections with QIAGEN AllPrep FFPE, and libraries were prepared with Illumina TruSeq Stranded with RiboZero Gold (RS-122-2301). mRNA-Seq libraries were run at 2 samples per lane on an Illumina HiSeq2500 sequencer in high-output mode using 2 × 50 paired-end chemistry.
WES was performed on FFPE tumor tissue, with peripheral blood mononuclear cells serving as a matched normal. Library preparation was performed with the SureSelect XT Human All Exon V6 + UTR kit (Agilent, Santa Clara, CA, USA), and pooled samples were sequenced on the HiSeq2500 platform (Illumina). The resulting somatic and germline WES sequencing files were aligned to Hg38 using bwa (v0.7.17), sorted, and indexed, and duplicates were marked using biobambam2 (v2.0.87). BAMs were re-aligned with Abra2 (v2.22), followed by somatic and germline variant detection with Strelka2 (v2.9.10), Cadabra (from Abra2 v2.22), and Mutect2 (GATK v4.1.4.0). The capture of exonic sequences was verified using the Picard (v2.21.1) CollectHsMetrics tool, and the quality of sequencing data was verified using FastQC (v0.11.8) and the Picard suite’s CollectAlignmentSummaryMetrics, CollectInsertSizeMetrics, QualityScoreDistribution, and MeanQualityByCycle tools. Variants with matched normals were filtered by the following criteria: protein-coding mutations only, Cadabra indel quality >10.5, Mutect2 indel quality >6.8, or (single-nucleotide variant) SNV quality >9.2, Strelka2 indel quality >15.2 or SNV quality >19.7. Additionally, Cadabra indels with quality <35 required a supporting high-quality call from either Strelka2 or Mutect2, and Strelka2 calls with SomaticEVS <20 similarly required a matching call from either Mutect2 or Cadabra. Variants for tumor-only samples were detected by Mutect2 and filtered to retain the protein-coding mutations. The remaining variants required at least 5 supporting reads and a minimum read depth of 40 or 10 supporting reads and a minimum read depth of 80 if MAF <5%. The variants with a MAF >5% in normal tissue were dropped, as were the variants appearing at rates above 1% in any subpopulation in either GnomAD or 1000 Genomes databases. To counter FFPE artifacts, C>T and G>A substitutions required a minimum MAF of 10%. Tumor mutational burden (TMB) was calculated from small indels and substitutions identified by WES and divided by the megabases adequately covered by sequencing reads. Whole-exome sequencing data for both tumor and germline was available for 28 samples (representing 22 unique patients) at baseline. Tumor-only whole-exome sequencing data—without matched normal—was available for a further 2 samples (representing 2 patients), for a total of 30 samples across 24 patients with WES data. Oncoplots were created in R using maftools v2.10.0, with variant genes limited to those implicated in breast cancer from the COSMIC Cancer Gene Census Tier 1 list (https://cancer.sanger.ac.uk/cosmic).
Copy Number Variation and Subclonal Heterogeneity Analysis
LCCC1419 patients with matched DNA normal and DNA tumor samples (n = 22) were processed through trim galore v. 0.6.2. The resulting trimmed FASTQs were aligned to the human reference genome FASTA file Homo_sapiens_assembly38.fasta (from the GATK hg38 file bundle) with the bwa mem command from BWA v. 0.7.17 with default parameters. The resulting SAM files were sorted, converted to BAM files, and indexed using SAMtools v. 1.9. The matched samples from each patient were then processed through the Sequenza v. 3.0.0 workflow (gc_wiggle with window size of 50 basepairs, bam2seqz, seqz_binning, sequenza.extract, sequenza.fit, and sequenza.results all with default parameters). The resulting patient-level segment files were then modified to conform to the format required by GISTIC. This conversion was performed with standard BASH scripting and included a log2(x) - 1 transformation of Sequenza’s raw depth ratio value for GISTIC. The resulting modified segment files were then concatenated across samples and ran through GISTIC v. 2.0.23 using the hg38.UCSC.add_miR.160920.refgene.mat file (from docker://shixiangwang/gistic:1.2) for the -refgene parameter. CNVKit was run using the “batch” mode for primary tumors and BrM separately (with their matched normal samples, respectively). All normal samples within each group were pooled together to generate a pan-sample normal control. Agilent’s SureSelect Human All Exon V6+UTRs probed bed file was provided for the –targets parameter, a standard hg38 refFlat.txt file was provided for annotations, and a k50 umap mappability BED file was provided for the –access parameter. Outputs from Sequenza were paired with each patient’s corresponding MuTect2 somatic variant calls to create PyClone-VI input files using Sequenza copy number information and MuTect2’s variant read support information. Files were created for each primary tumor and BrM sample separately, and paired input files were created for patients that had both primary tumor and BrM data available. The resulting files were run through PyClone-VI using default parameters.
Neoantigen Prediction Using Genomics Data
Tumor antigens were predicted from a comprehensive set of genomic sources (single-nucleotide variations, insertions/deletions, gene fusions, alternative splice variants, cancer testis antigens, overexpressed self-antigens, and viral and endogenous retroviral antigens) using methods developed by our group and implementations of methods developed by others (14–22). Briefly, whole-exome sequencing was used to identify tumor-specific genetic variants (single-nucleotide variations, insertions/deletions, gene fusions), and RNA sequencing was used to confirm the expression of these variants. RNA sequencing data alone were used to evaluate for expressed alternative splice variants, viral and endogenous retroviral antigens. Cancer testis antigens/overexpressed self-antigens were evaluated using RNAseq data, but WES data was used to incorporate germline variants. RNA sequencing data was also used to infer tumor MHC haplotypes via HLAProfiler, the most accurate tool for MHC haplotype inference (23). Peptide fragments generated in silico are evaluated for predicted binding affinity to tumor MHC alleles using NetMHCpan-4.1 (24). Peptides with predicted binding affinity <500 nM were considered positive binders (e.g., potential tumor antigens), while peptides with predicted binding affinity <50 nM considered strong binders and more likely to be tumor antigens (25).
RNA-Seq Data Processing
RNA-Seq Paired FASTQs were run through trim galore v. 0.6.2 using –paired parameter. STAR v. 2.7.0f was used to index the reference genome Homo_sapiens.GRCh38.dna_sm.primary assembly.fa from GATK and to map trimmed reads to reference (using parameters –quantMode TranscriptomeSAM – outSAMtype BAM SortedByCoordinate –sjdbGTFfile Homo_sapiens.GRCh38.100.gtf). Gffread v. 0.11.7 was used to create a transcriptome reference using the reference genome and the gtf file Homo_sapiens.GRCh38.100.gtf. The “toTranscriptome” alignments from STAR were used with Salmon v. 1.1.0 using “salmon quant -l a”. Sample quality was assessed using MultiQC v1.9 with Picard CollectRnaSeqMetrics, and samples with less than 20 M coding reads were excluded as this threshold has been found to approximate the minimal sequencing depth to achieve equivalent detection to microarrays (26). Counts were log2-transformed and the upper quartile normalized for further downstream analysis. Some patients’ tumors were sequenced multiple times (technical replicates), and in such cases, gene-level expression values were averaged across technical replicates.
Intrinsic Subtype Analysis
Intrinsic subtype analysis was performed according to the methods described in Picornell et al (27). The R package heatmaply (28) was used for heat map visualization with hierarchical clustering based on average linkage.
Immune Gene Signature Expression Analysis
Thirty-two immune gene signatures were chosen to reflect the diversity of tumor-infiltrating immune cell populations and to minimize redundancy (refer to associated.gmt file in the Supplementary Material). The binfotron R package (29) was used to compute the differential gene expression [along with the DESeq2 dependency (30)], produce a volcano plot, and calculate immune signature metagene scores (median log2 expression values) for downstream analysis. ssGSEA was performed using the R packages GSVA and GSEABase (31, 32). CIBERSORTx immune cell fraction imputation using the LM22 matrix was also performed (33).
TCR/BCR Repertoire Analysis
Immune chain inference was performed on RNA-Seq samples via MiXCR 2.1.2 for TCR chains (34) and V’DJer 0.12 (35) for BCR chains. The consensus BCR contigs from V’DJer were quantified using Salmon 0.13.11 (36). Repertoire diversity was calculated using a model-based approach, which improves estimations of diversity in part by minimizing known sources of estimate bias (37).
Survival Analyses
Multivariable Cox proportional hazards modeling was performed using the survival (38) R package. The model included cancer stage, age at primary tumor diagnosis, and race. Time from initial diagnosis, from diagnosis of any metastasis regardless of anatomical location, or from diagnosis of BrM to an event was interrogated. Hazard ratios and 95% confidence intervals were returned for gene signature covariates and visualized using the forestplot (39) R package.
Accession Numbers and Data Sharing
Sample information for RNA-seq and DNA-seq fastQ runs, including the clinical information, were uploaded to the NCBI’s dbGaP repository (accession no. phs002457.v1.p1) and SRA.
Results
Patient Cohort Characteristics
Tissues and blood from patients with clinical TNBC (n = 25) were included in this analysis, including BrM tissues (n = 19), primary breast tumors (n = 17), and whole blood samples (n = 22). The specimen numbers by tissue type and analysis (IHC, RNA, or DNA) are outlined in Supplementary Figure S1. Patient demographics are included in Table 1, with individual clinical–pathological characteristics and specimen availability presented in Supplementary Table S1. The majority of patients were Caucasian (n = 17, 68%), with African American, Asian, and other ethnicities represented [n = 6 (24%), n = 1 (4%), and n = 1 (4%), respectively]. One male was included in the cohort. Median age at breast cancer diagnosis was 46.7 years (range, 29–70.9), while median age at BrM diagnosis was 51.5 years (range, 31.7–72). The majority (n = 12, 48%) of patients were initially diagnosed with stage II disease prior to recurrence; a minority (n = 2, 8%) were diagnosed with de novo stage IV TNBC. In addition to BrM, other sites of disease included the liver (n = 8, 32%), bone (n = 15, 60%), lung (n = 16, 64%), and non-local lymph nodes (n = 18, 72%). Ten patients (40%) were initially diagnosed with a solitary BrM, while 7 patients were diagnosed with 5 or greater BrM (28%). BrM was supratentorial in n = 23 (92%) patients and infratentorial in n = 13 (52%) patients. The median progression-free survival (e.g., time from primary TNBC diagnosis to the diagnosis of any metastasis) was 1.8 years (range, 0–19.5). The median OS from primary TNBC diagnosis was 3.7 years (range, 0.9–19.8), while the median OS from BrM diagnosis was 1.2 years (range, 0 – 8.9).

Table 1 Relevant demographic, subtype, and clinical diagnostic information for the LCCC1419 TNBC cohort.
Intrinsic Subtype Classification of Primary TNBC and Brain Metastases
Intrinsic molecular subtype analysis using RNAseq data illustrated that the majority of clinically determined TNBC samples were of the basal subtype (Supplementary Figure S2). Of the BrM (n = 19), 16 were classified as basal (84%), with normal-like (n = 2) and HER2-enriched (n = 1) comprising a small fraction of the cohort. Similarly, primary tumors (n = 15) were predominantly classified as basal (n = 13, 87%), with the remaining tumors being normal-like (n = 2). Notably, the 4 samples that were called normal-like by PAM50 analysis had a basal subtype as the second highest identity probability. There were 2 cases with discordant receptor classification between primary tumor and BrM by immunohistochemistry (n = 1 ER+/PR+/HER2- Luminal A primary converted to a TNBC BrM, and n = 1 TNBC primary converted to an ER+/PR+/HER2- Luminal A BrM) (Table 1; Supplementary Figure S2). Despite the potential subtype switching between primary and BrM, these samples were included in the downstream analyses.
Mutational, Somatic Copy Number Alteration, and Subclonal Analyses of Primary TNBC Tumors and Brain Metastases
First, we analyzed the tumor mutational burden (TMB) of primary tumors (n = 13) relative to BrM (n = 17) using WES. On average, BrM harbored a greater mutational load than primary tumors (median 3.33 vs. 1.78 mutations/Mb, respectively, p < 0.05; Figure 1A). Upon analysis of matched primary-BrM WES pairs (n = 6), however, there was no significant difference between tumor location and TMB (median 2.80 vs. 1.88 mutations/Mb, respectively, p = 0.69; Figure 1B). An analysis of shared mutations within matched pairs revealed varying degrees of mutational conservation between anatomical locations (Figure 1C). The degree of variant sharing between matched pairs (Figure 1C) was generally greater than the degree of mutations shared between primaries and BrM globally (Supplementary Figure S3), highlighting interpatient tumor heterogeneity and mutational divergence. We assessed whether a survival benefit was conferred by increasing TMB, as TMB has been considered a proxy for neoantigen burden (40, 41). There was no significant association between TMB and survival (p = 0.07) from the time of primary TNBC diagnosis in the context of a model that included standard clinicopathological features (age at diagnosis of primary tumor, stage, and race) (Supplementary Figure S4). Next, we examined the mutational spectrum of genes with known associations to breast cancer development (42). We found that these genes were altered in 70% of combined primary and BrM samples, with TP53 being the most commonly mutated gene (mutated in 50% of samples, n = 15), in accordance with its known relevance to TNBC (43) (Figure 1D). The next most frequently altered genes were MAP3K13 and PIK3CA, which were mutated in 13% (n = 4) and 10% (n = 3) of samples, respectively; all other genes were mutated less frequently, occurring in 7% or less (n ≤ 2) of samples (Figure 1D).
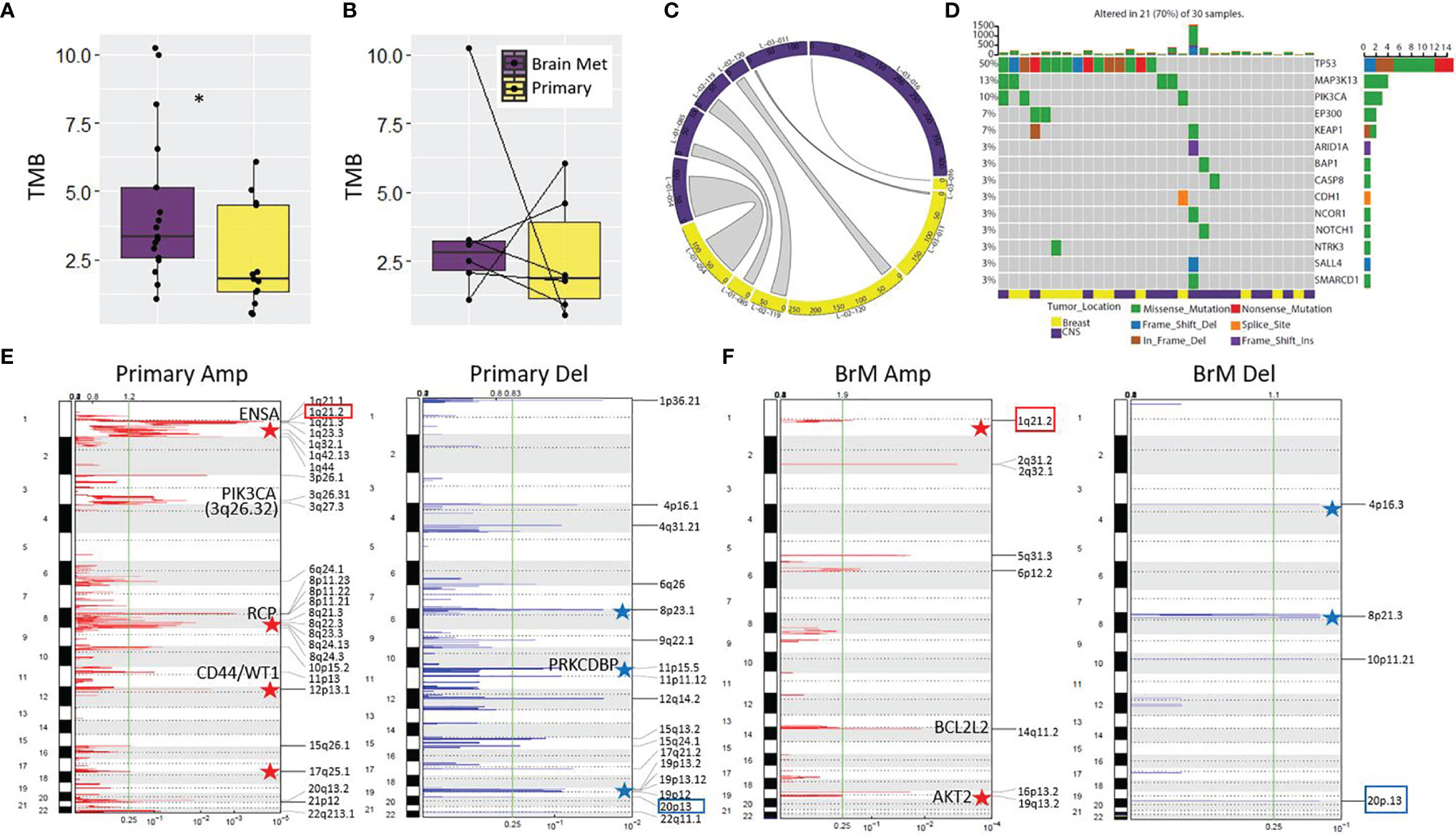
Figure 1 Mutational analysis of the LCCC1419 cohort. (A) Increased tumor mutational burden (TMB) was observed for BrM (n = 17) relative to primary (n = 13); *p <0.05, Wilcoxon rank-sum test. (B) No difference was observed between TMB for matched pairs (n = 6). (C) Circos plot showing the total number of variants and the proportion of variants shared between matched primary/BrM (n = 6). (D) Oncoprint displaying the mutational spectrum of cancer-associated genes with known etiology to breast cancer (COSMIC Tier 1, Sanger Institute). Representation of the somatic copy number alterations in (E) primary TNBC and (F) BrM as determined by CNVkit/GISTIC 2.0 analysis. Significant amplicons (Amp) or deleted (Del) regions are annotated (q <0.25, green line). The 1q21.2 amplification and the 20p13 deletion, which are shared genomic features of primary and BrM TNBC, are highlighted by colored boxes. Relevant oncogenes and tumor suppressors are annotated on the plots, and stars indicate genomic regions where copy number alterations are known to contribute to breast cancer/aggressive basal breast cancer.
We next analyzed the recurrent copy number alteration patterns in primary and BrM samples using Sequenza/GISTIC 2.0 (44, 45). The primary TNBC samples harbored 3 significant amplicons and 1 deleted region, whereas BrM was more profoundly altered with 15 significant amplicons and 12 regions of deletion (q < 0.25; Supplementary Figure S5). At this level of genomic resolution, only 2 regions were commonly altered between primary and BrM (11p13 amplicon and 13q11 deletion). In breast cancer, these two sites are previously known to be amplified or deleted, respectively (46, 47). Interestingly, a number of amplicons/deleted regions identified in this cohort are known to be associated with breast cancer/aggressive basal breast cancer, such as gains of 1q, 8p11-12, 8q, 12p13, 13q34, 17q, and 19q and deletions of 3p, 4p16.3, 8p, 11p15, 17p, and 19p13 (48–58). Documented breast cancer oncogenes (NOTCH2, ENSA, PIK3CA, CD44, WT1, BCL2L2, AKT2, and TFF3) and tumor suppressors (BRCA2 and PRKCDBP) reside or are in close proximity to some of these significantly amplified/deleted genomic regions, and these alterations potentially contribute to TNBC progression and BrM development.
Since somatic copy number alteration (SCNA) detection tools are prone to high false positive rates as well as issues with precision and accuracy (59, 60), we also performed SCNA calling using CNVkit (61), which combines all normal samples into a pooled reference to increase performance. In contrast to Sequenza, this method identified a greater number of SCNA in primary TNBC relative to BrM, showing that, as a collective group, primary TNBC samples harbored 27 significant amplicons and 17 deleted regions, whereas BrM had 8 significant amplicons and 4 regions of deletion (q < 0.25; Figures 1E, F). Despite notable differences between the two methods, there was a concordance in the results as well, with corroboration of 8p11.22 and 11p13 amplification in primary TNBC and validation of 14q11.2 and 19q13.2 amplification and 4p16.3 deletion in BrM (Figures 1E, F; Supplementary Figure S5). The CNVkit SCNA analysis also highlights the potential importance of oncogenes (e.g., RCP, CD44, WT1, BCL2L2, and AKT2) and tumor suppressors (e.g., PRKCDBP) to TNBC etiology and metastatic progression, as significant amplicons/deleted regions harbor these genes.
Finally, we examined the ploidy, tumor purity, and subclonal makeup of tumors in this cohort. No differences in cellular ploidy were noted between primary TNBC and BrM (median ploidy of 3.2 and 3.25, respectively; Supplementary Figure S6A). Similarly, no significant differences in tumor purity were observed between groups (median purity of 0.57 and 0.76, respectively; Supplementary Figure S6B). An analysis of subclonal tumoral architecture using Pyclone-VI (62) showed no difference between the number of clones per tumor in each location, where a median of 3.5 clones per tumor in primary TNBC (range, 1–5 clones/tumor) and a median of 4 clones per tumor in BrM (range, 3–6 clones/tumor) were observed (Supplementary Figure S6C). A subclonal assessment in patients with matched primary/BrM pairs showed that some pairs had similar clonal constituency between anatomical sites (e.g., patients L-01-054, L-01-085, L-02-119, and L-03-016), whereas other pairs showed signs of divergent clonal evolution (e.g., L-02-120 and L-03-011) (Supplementary Figure S6D). Interestingly, 5 of 6 matched pairs the dominant subclone harbored the highest mutational burden, which was reflected in the analysis of unmatched tumors as well (not shown), suggesting that increased mutational load may endow these subclones with a selective growth advantage.
Tumor Antigen Landscape
We next performed a comprehensive analysis of the neoantigen landscape in this cohort. We queried a range of neoantigen sources, including single-nucleotide variants (SNVs), insertion/deletion events (InDels), splice variants, structural fusion events, cancer testis antigens (CTAs)/self-antigens, endogenous retroviruses (ERVs), and viral sources excluding ERVs (63) (Figure 2). The predominant antigen sources in both primary TNBC and BrM were CTAs/self-antigens and ERVs (Figures 2A, B). Upon comparison of the number of predicted neoantigen-derived peptides, there were significantly more SNV-derived MHC class I-binding peptides in BrM as compared to primary TNBC (p = 0.005), with no differences seen between groups with respect to other neoantigen sources (Figure 2B). This analysis together shows that TNBC harbors a diverse set of potentially therapeutically actionable neoantigen-derived peptides.
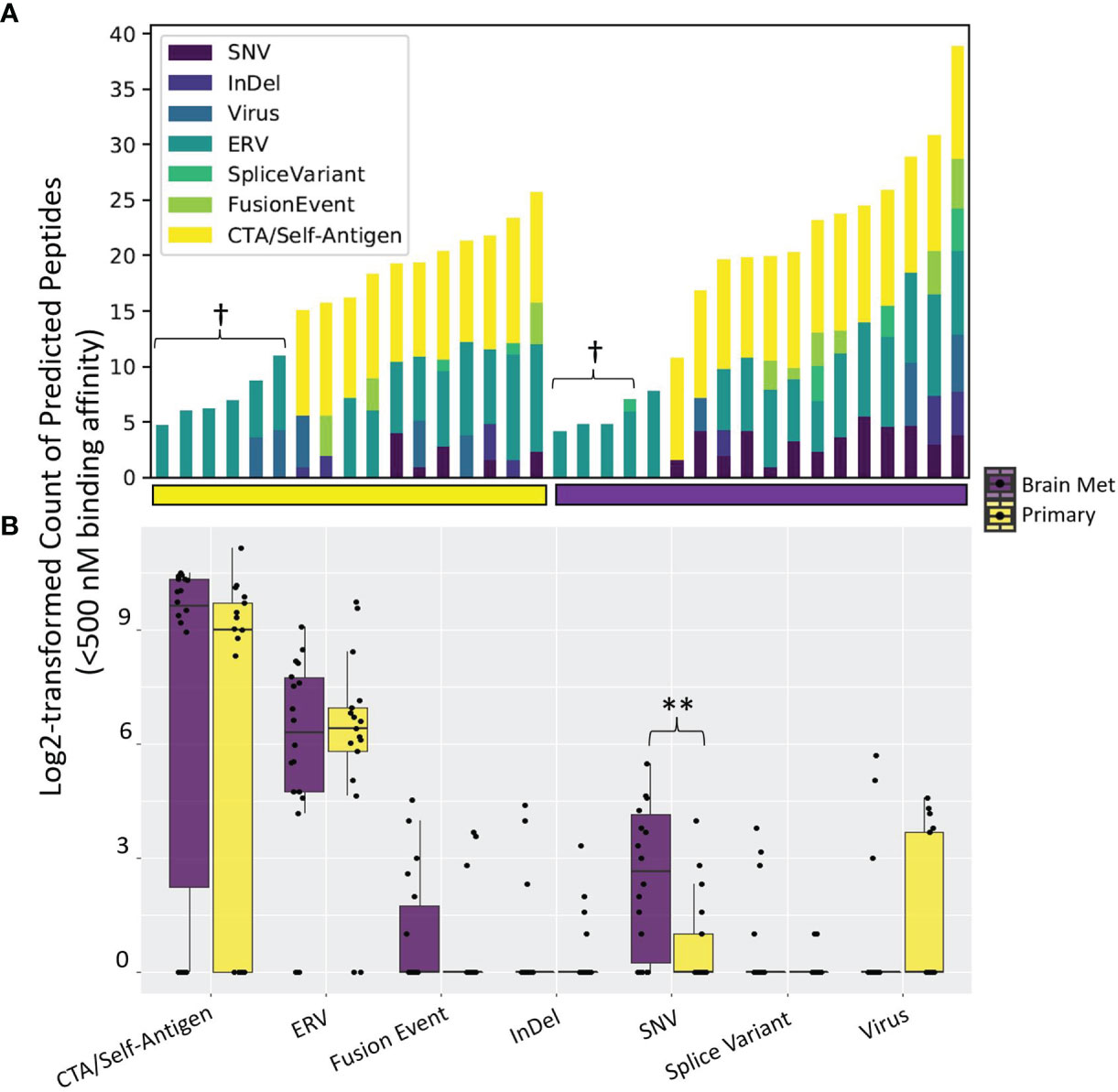
Figure 2 Tumor antigen sources among the LCCC1419 patients. A comprehensive bioinformatics prediction pipeline that exports (A) the number of neoantigen-derived MHC class I-binding peptides (Kd < 500 nM) broken down by antigen source was employed. Some patients’ tumors did not have associated tumor or normal whole-exome sequencing data, and as such, antigen sources that require DNA sequencing data (single-nucleotide variants, InDels, cancer testis antigens, or fusion events) are not able to be queried in these cases (denoted by †). (B) Distribution of the number of neoantigen-derived MHC class I-binding peptides (Kd < 500 nM) broken down by antigen source, corresponding to (A); **p <0.01 (Wilcoxon rank-sum test).
Comparison of Immune Gene Signatures Between Primary TNBC and BrM
Immune gene signatures (IGS) representing multiple components of the immune system, including B cells, T cells, natural killer cells, and innate immune cells along with immune cell phenotype frequencies, were evaluated between primary TNBC tumors (n = 15) and BrM (n = 19) using RNA-Seq (Figure 3; Supplementary Figure S7; Supplementary Data S1). The majority of IGS across each of these categories were lower in TNBC BrM compared to primary TNBC. A gene signature associated with fibroblast-associated wound healing [Chang_Serum_Response_Up (64)] was significantly higher in BrM relative to primary tumors (q < 0.05). RNAseq expression data from primary tumors and BrM were also assessed using CIBERSORTx (65) to determine relative frequencies of 22 immune cell subtypes (LM22) to tumor composition. In this analysis, naïve B cells and M1 macrophages were lower in BrM compared to primary tumors, while eosinophils and neutrophils were higher in BrM tissues (q < 0.05) (Supplementary Figure S8). The expression of the 20-gene immunologic constant of rejection signature [ICR (66)], which is representative of Th1-mediated immunity, cytotoxic function, and tissue-specific destruction (e.g., GVHD, autoimmunity, and allograft rejection), was also significantly reduced in BrM relative to primary TNBC (Supplementary Figure S9A). Additionally, the blood transcriptional modules reported by Rinchai et al. (67) were queried against our dataset and showed a significant reduction of B and T cell modules in BrM relative to primary TNBC (Supplementary Figure S9B), concordant with the abovementioned data. These results are together consistent with an overall immune-excluded brain tumor microenvironment (TME) in the context of TNBC BrM.
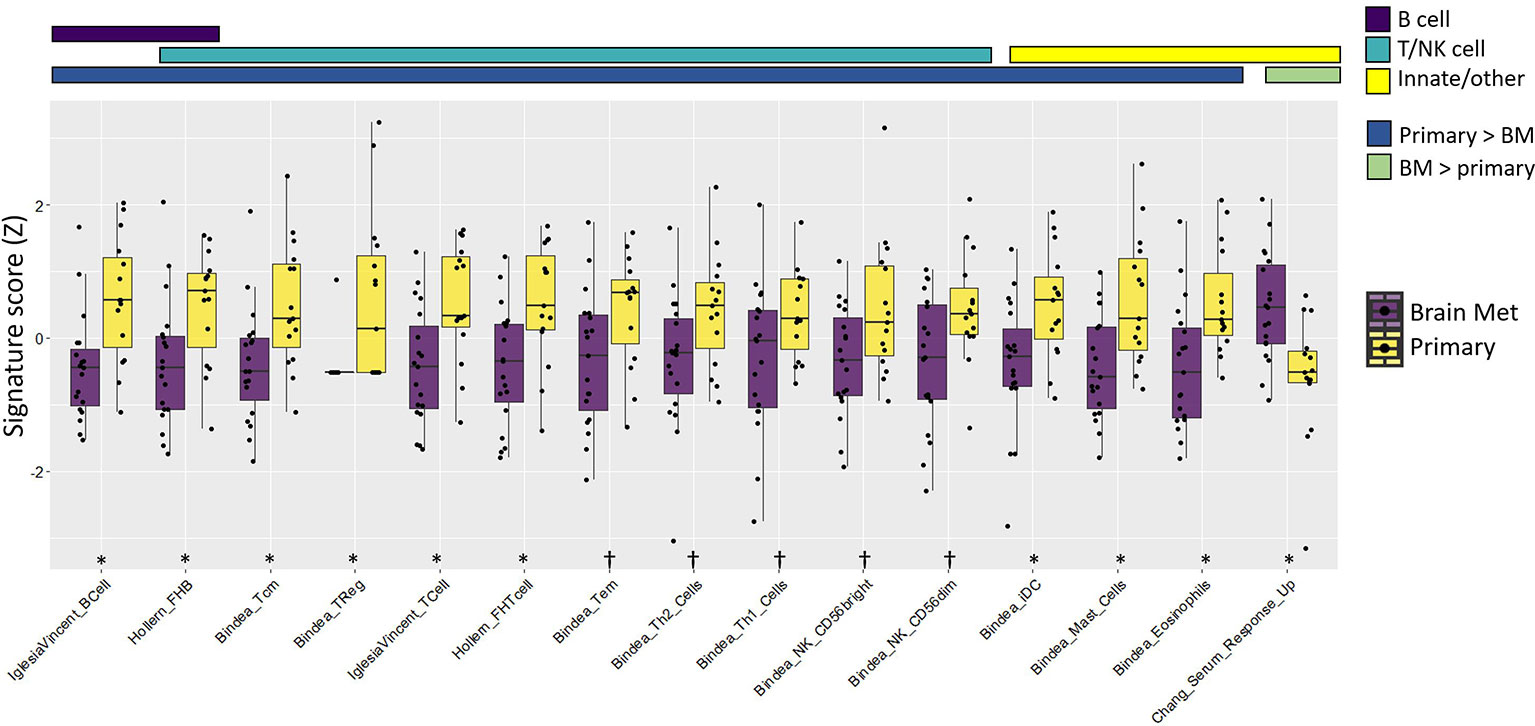
Figure 3 Immune gene signature metagene analysis showed an overall immune cell deficit in BrM relative to primary triple-negative breast cancer. The colored bars above the plot indicate both the immune cell category assigned to the respective signatures and whether the signatures were increased/decreased in the primary tumor relative to BrM. Wilcoxon rank-sum test was performed on Z-transformed signature scores to determine the statistical significance after false discovery rate correction. Significance codes: †q < 0.1, *q < 0.05.
T and B Cell Repertoire Analysis
We used RNA-Seq data from primary TNBC tumors (n = 15) and BrM (n = 19) to perform T cell and B cell repertoire (TCR/BCR) profiling. Relative to primary TNBC, TNBC BrM had lower read counts of T cell receptor alpha and beta (TRA, p < 0.001 and TRB, p < 0.01), with BCR heavy chain and light chain abundance showing trending but non-significant differences (Figure 4A). This result is in accordance with RNA-seq data that showed less T cell and B cell abundance in the primary samples relative to BrM (Figure 3). Repertoire diversity was indexed as modeled Shannon entropy (37), which is a diversity index that accounts for both the richness of the sample (e.g., the number of unique TCR/BCR sequences) and relative species abundance (evenness) (68, 69). Thus, a large Shannon entropy score reflects a more diverse distribution of TCR/BCR sequences. The modeled Shannon entropy (TCR/BCR diversity) was lower for BrM compared to primary tumors (TRA, p < 0.01 and TRB, p < 0.05) (Figure 4B). A comparison of matched BrM and primary TNBC pairs only, however, did not show a reduction of TCR/BCR read counts and modeled Shannon entropy (Figures 4C, D).
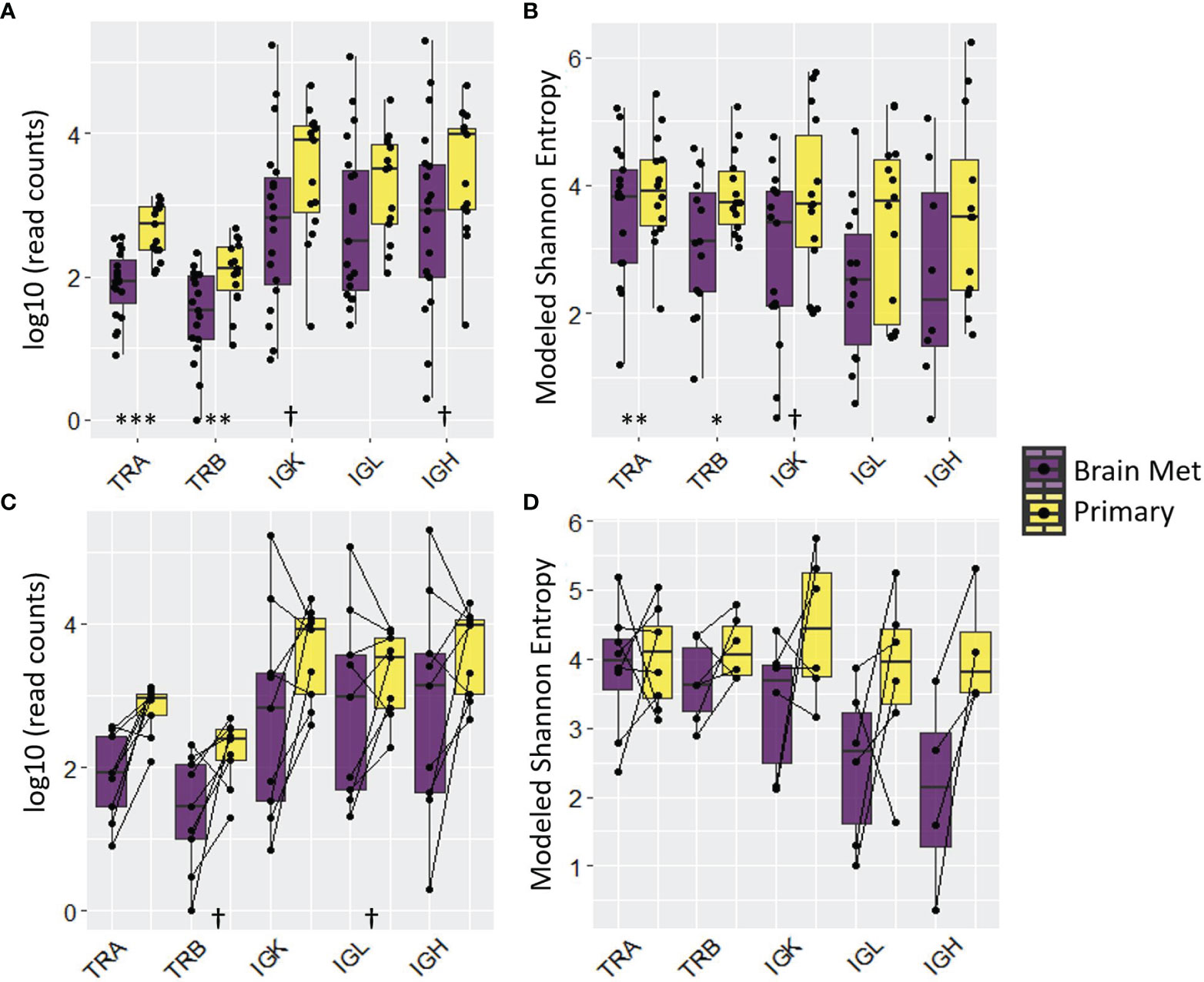
Figure 4 T cell and B cell repertoire analysis revealed adaptive immune cell deficit in BrM relative to primary triple-negative breast cancer. The distribution of read counts and modeled Shannon entropy for all samples is displayed in (A, B), respectively. The same information is displayed, respectively, in (C, D) for matched pairs (note that the number of matched pairs varies due to the presence/absence of relevant reads). Wilcoxon rank-sum test was performed on log10-transformed (read counts) or raw (modeled Shannon entropy) values to determine the statistical significance. Significance codes: †p < 0.1; *p < 0.05; **p < 0.01; ***p < 0.001.
Differential Gene Expression and Pathway Analysis Support an Immune Cell Deficit in TNBC BrM Relative to Primary TNBC Tumors
Gene expression was evaluated by utilizing RNA-Seq data between the primary tumor (n = 15) and BrM (n = 19) tissues. In total, there were 1,669 differentially expressed genes (DEGs) between these 2 groups, with 935 genes upregulated and 734 genes downregulated in the BrM tissues compared to primary tumors (q ≤ 0.1; Figure 5A; Supplementary Data 2). Gene ontology (GO) analysis of DEGs revealed a significant enrichment of immune-related terms in the primary TNBC tumors compared to the BrM (particularly terms reflecting adaptive immune system involvement), whereas GO terms associated with the nervous system were significantly higher in the BrM relative to the primary TNBC tumors (Figure 5B). Canonical pathway analysis (Ingenuity Pathway Analysis, IPA) of DEGs in primary tumors versus BrM illustrated a similar preponderance of immune signaling-related pathways as well as nervous system-related pathways associated with DEGs in BrM relative to primary tumors (Figure 5C). Upstream regulator analysis (IPA) further demonstrated an association of immune-related signaling activity with DEGs in primary tumors relative to BrM (e.g., IFNG, NFKB, CD3, CSF2, and IL-1β) and an association of potential oncogenic drivers (e.g., TCF7L2, mTOR, and SH3TC2) with regulation of BrM DEGs (Figure 5D; Supplementary Data S3).
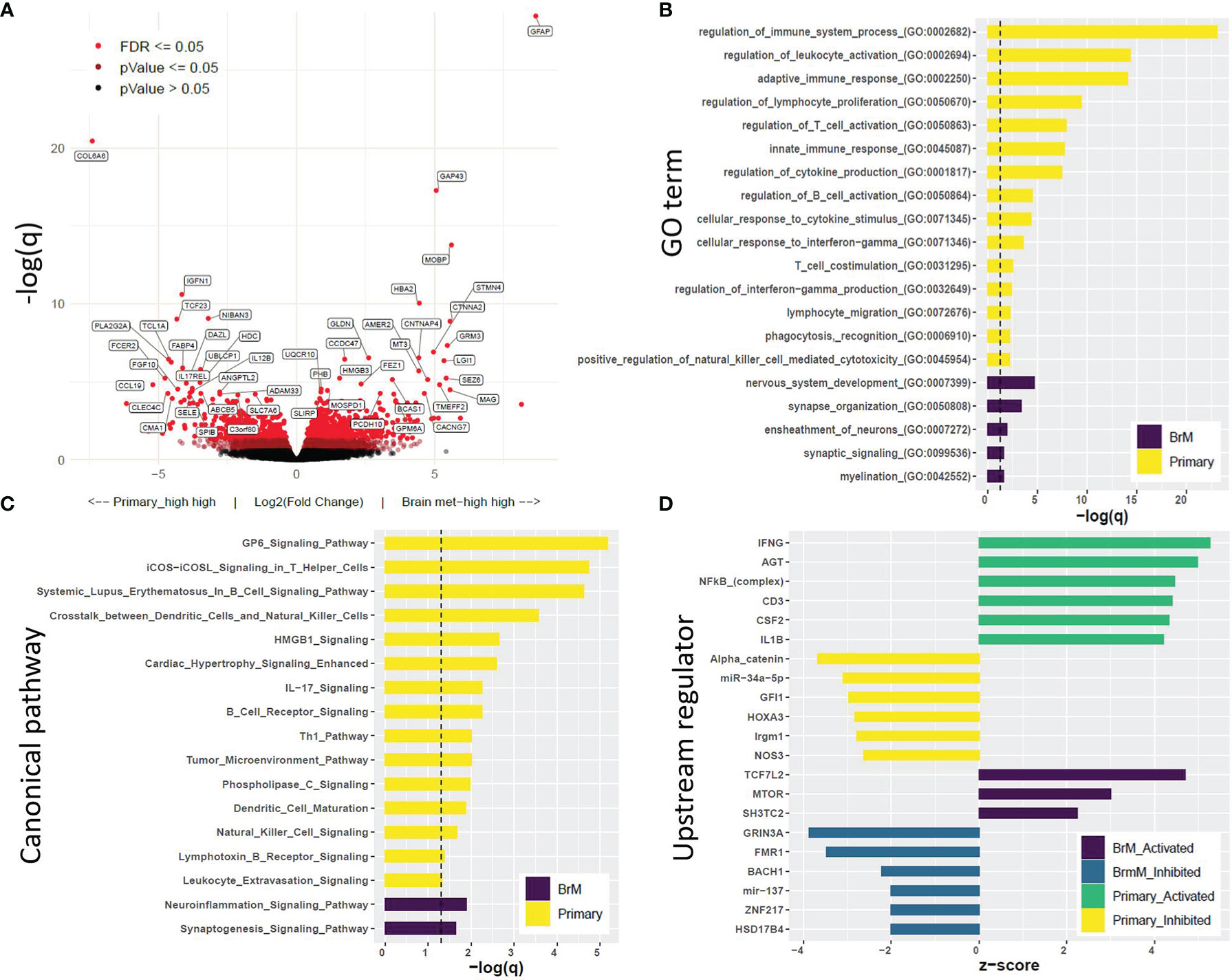
Figure 5 Differential gene expression analysis and Gene Set Enrichment Analysis further support the immune cell deficit in BrM relative to primary triple-negative breast cancer (TNBC). (A) Volcano plot displaying differentially expressed genes (DEGs) in primary TNBC relative to BrM, with the legend showing color-coded levels of significance. (B) Top Gene Ontology terms associated with DEGs. DEGs with LFC > |1| and q < 0.1 (equating to 468 genes for primary_vs_BrM and 463 genes for BrM_vs_primary) were subjected to PANTHER overrepresentation test (dotted line represents q = 0.1). (C) Canonical pathway analysis (Ingenuity Pathway Analysis, IPA) of DEGs. The pathways displayed were significant at q <0.05 (dotted line) and were associated with a significant z-score (z >|2|) which indicates associative activity. (D) Upstream regulator analysis (IPA) displaying top regulators (z >|2|, q < 0.1) identified to be associated with an active or inhibited state in primary versus BrM TNBC [see Supplementary Data S2, S3 for the full list of DEGs (q < 0.1) and upstream regulators (z >|2|, q <0.1).
Adaptive Immune Cell Signatures Are Associated With Improved Survival for Patients With TNBC BrM
We examined the survival association of standard clinicopathological variables (age at diagnosis of primary tumor, stage, and race) with different time metrics to event: (1) time from diagnosis of primary TNBC to death, (2) time from diagnosis of any metastatic disease to death, and (3) time from diagnosis of BrM to death. Of these variables, only older age was significantly associated with poor survival using each of these time metrics (Supplementary Figure S10), which was similar to other recent reports (70, 71). Next, survival associations relative to IGS expression were evaluated using multivariable CoxPH models in both primary TNBC and BrM. The IGS features in primary TNBC tumors which were associated with improved survival following metastatic diagnosis included T cell, B cell, and dendritic cell (DC) signatures (Supplementary Figure S11A). Interestingly, a fibroblast serum response/wound healing signature (64) (“Chang_Serum_Response_Up”) was associated with a significantly poorer survival (p = 0.025) in BrM (Supplementary Figure S11B) after a diagnosis of metastasis.
Discussion
In this study, we examined the genomic and transcriptomic landscape of TNBC BrM and primary tumors to further the understanding of TNBC BrM etiology and the tumor immune microenvironment. Despite recent progress in the treatment of ER+ and HER2+ BCBrM with newer brain-penetrant, targeted therapies, the treatment options for TNBC BrM remain largely restricted to chemotherapy and local therapy due to lack of known targets. A growing appreciation for the role of immunotherapy in the treatment of TNBC highlights the need to better understand the immune context of BrM as we consider incorporation of immunotherapy into the care of our patients (72).
Through whole-exome sequencing, we report that BrM, as a group, exhibited a greater TMB than primary tumors, though this observation was not recapitulated in matched tissue pairs. We suspect that this was due to underpowering of our study to assess matched pairs (n = 6) in the context of TNBC tumor biological heterogeneity (73). An analysis of shared variants showed that matched primary TNBC and BrM samples were more alike than inter-patient primaries and inter-patient BrM, showing that TNBC is a heterogeneous disease with potentially non-redundant mechanisms of tumorigenesis. A subclonal analysis of matched pairs also showed that some patients displayed patterns of divergent evolution between primary tumors and BrM. The mutational spectrum of genes with known causality to breast cancer tumorigenesis was also queried. TP53 was commonly mutated in this cohort (50%), supporting its causal role in the development of TNBC, while other genes such as MAP3K13 and PIK3CA were mutated at a lower frequency.
Copy number variation analysis revealed common and unique genomic alteration events between primary TNBC and BrM. 11p13 was commonly amplified in both primary tumors and BrM. This genomic location harbors the CD44 gene, which is used to discern breast CSCs, although it has been shown that it is not likely a driver of amplification of this region in basal breast cancer (74). WT1, which also resides at 11p13, has been shown to promote a mesenchymal phenotype in breast cancer cells as well as to elicit resistance to taxane therapy (47). Regions of 1q were also commonly amplified, which supports a known role for this genomic location in breast cancer development (48). The amplification and increased expression of ENSA (1q21.3) have recently been shown to drive TNBC progression via positive regulation of cholesterol biosynthesis (58). 13q11 was deleted in both primary and BrM TNBC, and this site is proximal to BRCA2 (located on 13q13.1). Whether or not the loss of 13q11 has any BRCA2-regulatory functionality is unknown, although deletions in 13q and 14q are common in BRCA2-mutated breast cancers (75). Common deletion of 20p13 was also observed. While this location is known to be deleted in colon cancer (76), its association with breast cancer has yet to be explored. Other common deletions identified included 1p36 and 11p15.5. In ductal breast carcinoma, the 1p36 deletion is associated with grade, ERBB2 loss, and loss of BCL2 expression (57) and is known to be a common feature underlying breast cancer development and the carcinogenesis of various cancer types (77). BrM-specific deletion at 11p15.5 (region harboring PRKCDBP) was also observed, and the chromosomal loss of this region is associated with BCBrM, with PRKCDBP identified as a putative tumor suppressor (53).
In primary TNBC, notable amplicons were associated with both arms of chromosomes 1 and 8. These locations are associated with breast cancer cytogenetics and pathology (48, 78, 79) and harbor genes (NOTCH2 and RCP, respectively) associated with breast cancer etiology (49, 80). There were also several notable alterations specific to BrM. Regions harboring the oncogenes BCL2L2 (14q11.2), AKT2 (19q13.2), and TFF3 (21q22.3) were amplified in BrM. BCL2L2 is an anti-apoptotic protein that has an oncogenic role in many solid tumor types, and it has been found to contribute to breast cancer progression through its upregulation via hypermethylation of the negative-regulatory miR-129-2 (81). While PIK3CA was only mutated in 10% of evaluated samples in this study, AKT2 upregulation via genomic amplification may have a significant impact on TNBC BrM progression. Dysregulation of the PI3K/AKT/mTOR axis is a common feature of TNBC (82), and this pathway represents a promising target in this disease context. TFF3 is also associated with breast cancer metastasis, where its expression predicts poor survival (83), and it is also associated with residual invasive disease following neoadjuvant chemotherapy in breast carcinoma (84). Interestingly, TFF3 was also found to be significantly upregulated in T cell-cold tumors of diverse tissue types, and it was in the top percentile of genes differentially expressed in T cell-cold versus T cell-hot breast cancers (85), which suggests its potential as an immunotherapy target. These mutational and copy number analyses together highlight potential causative genomic alterations contributing to TNBC progression and BrM.
A systematic evaluation of the neoantigen landscape in LCCC1419 was undertaken here. Using a suite of bioinformatics prediction software, we analyzed tumor-associated antigens (e.g., CTAs/self-antigens), traditional tumor-specific antigens (TSAs; e.g., SNVs), and alternative TSAs [e.g., derived from splice variants, chromosomal structural variants, InDels, ERVs, and other viral antigens (63)]. We found that both primary TNBC and BrM harbored substantial numbers of high-affinity MHC class I-binding peptides derived from CTAs and ERVs relative to other antigen sources. CTAs are known to be associated with aggressive hormone-negative breast cancers and poor survival; however, they have also been associated with robust immunogenicity in some contexts (86). ERVs, which are evolutionary remnants of viral insertional mutagenesis, are also potentially powerful immunogens (18). Although ERV transcriptional regulation is often epigenetically silenced in normal cells, tumor cell-specific derepression is known to occur and is associated with a response to immune checkpoint blockade (ICB) in multiple cancer types (18, 87, 88). As such, antigens derived from CTAs and ERVs may be invaluable immunotherapeutic targets for vaccine strategies targeting TNBC and BrM lesions. Relative to primary TNBC, we also found an elevated SNV mutational load associated with BrM. This augmented TSA burden in BrM also represents a potential vulnerability to be targeted by combination immunotherapeutic approaches, including neoantigen vaccine strategies.
A comprehensive analysis of transcriptomic data derived from this cohort was performed to further understand the difference between the tumor immune microenvironment of primary and BrM TNBC. We found that BrM lesions harbored significantly less immune infiltrate than primary tumors. This is not surprising, as the brain has historically been considered an immunologically protected organ (89). A recent study with RNA array data in BCBrM, agnostic to subtype of BC, has similarly reported reduced immune scores in BCBrM relative to primary tumors (90). The general dearth of immune involvement in the BrM spanned both adaptive (T and B cell) and innate (DC, eosinophils, and mast cells) immune populations, indicative of a broad immune deficit relative to primary tumors and again similar to recent reports (90). Interestingly, BrM displayed an elevated expression level of genes involved in a serum-induced fibroblast wound healing response (64). This finding may suggest that, relative to primary TNBC, BrM lesions are more reliant on aberrant wound healing properties, requiring increased levels of stromal involvement for growth and maintenance, as seminally put forth by Dvorak (91). We also observed that BrM had significantly decreased TCR (TRA/TRB) abundance and diversity as compared to primary tumors, and this association was verging on significance for certain immunoglobulin classes. These metrics are important, as increased TCR abundance and diversity have been associated with a response to ICB in multiple solid tumor types (92). DEGs between primary and BrM TNBC also reflected a BrM-specific immune deficit. Gene Ontology and canonical pathway analysis showed that genes that exhibited relatively lower expression levels in BrM versus primary tumors were enriched for terms related primarily to an adaptive immune response. An upstream regulator analysis further supported these findings, with IFNG being the putative regulator with the highest significance. In BrM, this upstream regulator analysis further demonstrated the importance of mTOR signaling but also showed that TCF7L2 and SH3TC2 may be important players in BrM development. TCF7L2 variants have been found to be associated with breast cancer incidence (93, 94). Additionally, this gene is a positive regulator of Wnt signaling, regulates the MYC oncogene, represses the cell cycle inhibitors CDKN2C/CDKN2D, and is a transcriptional driver of various oncogenes, contributing to the progression of colon cancer and other cancer types (95).
We performed survival analyses examining the prognostic potential of IGS in the context of standard clinicopathological features. Signatures representing levels of T cells, B cells, NK cells, and DC cells in the primary tumors were associated with improved survival. The collective association of these IGS with favorable survival is likely indicative of the high degree of expression correlation structure (and thus co-infiltration levels of the associated immune cell types) observed in this cohort (Supplementary Figure S5). Moreover, the favorable association between T cell, B cell, and DC IGS and survival may indicate that patients with higher anti-tumor immune infiltrate in their primary TNBC may have a higher propensity to develop long-lasting immunological memory that functions to stave off metastatic spread. Similarly, levels of signatures reflective of gamma-delta T cells and ICB responsiveness [e.g., Vincent_IPRES_Responder signature (96)] in BrM were associated with improved survival from the time of BrM diagnosis, indicating that elevated immune involvement in the brain TME may be beneficial to patient survival. Conversely, the aforementioned fibroblast wound healing signature [Chang_Serum_Response_Up (64)] was associated with poor survival in BrM, indicative of a deleterious quality of this signature and the underlying biology that it represents.
While this study represents the largest series focused on TNBC BrM to date, to our knowledge, it is mainly limited by low power, particularly regarding matched pairs (with only n = 6 matched WES and n = 9 matched RNA-seq pairs). An additional limitation is the inability to corroborate adaptive immune receptor repertoire inference with amplicon sequencing, which was precluded due to inadequate specimen nucleic acid abundance. Future work will expand these, and additional analyses to additional TNBC samples, as well as to other BC subtypes in the LCCC1419 biobank, including HER2+ and ER/PR+ BCBrM, to enable a comparison of BrM across the spectrum of BC. Utilization of in vivo murine models for testing the relevance of these findings, including the assessment of vaccine strategies and ICB as potential therapeutic approaches for TNBC BrM, is warranted.
In summary, we report the genomic characterization of BrM compared to primary tumors from TNBC patients, including some matched pairs, with a focus on the immune landscape. Utilizing both WES and RNA-seq analytical pipelines, we demonstrated that BrM exhibited increased TMB and SNV mutational load, reduced immune gene signature expression and TCR receptor abundance/diversity metrics, and increased expression of a wound healing signature. A prediction of elevated levels of CTA- and ERV-specific neoantigen peptides was confirmed in both anatomical locations, supporting the continued development of vaccine and immune checkpoint inhibition approaches in TNBC. IGS, including T cell-related immune signatures in primary and BrM TNBC, correlated with improved survival in this patient cohort. We expect that these results and the data reported herein will be valuable in understanding TNBC BrM biology going forward and provide further rationale for the application of immunotherapeutic approaches in this disease.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below:
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002457.v1.p1.
Ethics Statement
The studies involving human participants were reviewed and approved by the University of North Carolina at Chapel Hill Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conception: CA, BV, MM, and LCa. Design: ER, AVS, MS, MM, BV, SC, CA, and LiC. Acquisition/analysis: MS, LuC, LiC, AG, AW, ER, SV, SC, MW, JP, MM, AVS, BV, CA, and AH. Interpretation: ER, AVS, MS, SC, JP, MM, MW, CA, and BV. Manuscript preparation/editing: all authors. All authors contributed to the article and approved the submitted version.
Funding
This study received funding from Susan G. Komen Career Catalyst Award (BV), V Foundation for Cancer Research Translational Grant (BV and CA), AACR grant (CA), and Translating Duke Health (CA).
Conflict of Interest
JP is an inventor on patent applications for the Breast PAM50 assay. BV and JP report equity and consulting fees for GeneCentric Therapeutics. CA receives research funding from PUMA, Lilly, Merck, Seattle Genetics, Nektar, Tesaro, G1-Therapeutics, ZION, Novartis, and Pfizer; compensated consultant role: Genentech (1/2019-), Eisai (1/2019-), IPSEN (2/2019-), Seattle Genetics (11/15/2019-11/15/2020); Astra Zeneca (3/2020-6/2020), Novartis (5/2020-5/2022), Immunomedics (10/1/2020-9/22/2021), Elucida (9/2020); and royalties from UpToDate and Jones and Bartlett. LC has received institutional research funding from Syndax, Novartis, NanoString Technologies, AbbVie, Seattle Genetics, and Veracyte. An immediate family member has a royalty-sharing agreement and investorship interest in licensed IP to startup company Falcon Therapeutics that is designing neural stem-cell-based therapy for glioblastoma multiforme. She has uncompensated relationships with Sanofi, Novartis, G1 Therapeutics, Genentech/Roche, GlaxoSmithKline, AstraZeneca/Daiichi Sankyo, Aptitude Health, Exact Sciences and Eisai.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the following groups at UNC—Chapel Hill for their assistance with the creation, maintenance, processing, and analysis of this biobank and its samples: UNC Office of Clinical and Translational Research, UNC Health Registry team, UNC BioSpecimen Processing Facility, UNC High-Throughput Sequencing Facility, and UNC Bioinformatics Shared Resource. We also thank the patients in this study and their families, without whom this study would not have been possible. We thank the following sources for their funding support for this study: American Society of Clinical Oncology/Conquer Cancer Foundation, Advanced Clinical Research Award (10299 to CA).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.818693/full#supplementary-material
Supplemental Figure 1 | Samples included in immunohistochemical and sequencing (RNA and DNA) analyses. Primary breast tumor, BrM, and normal blood specimens from a total of 25 patients with BrM from TNBC were included in the analyses. Matched blood was analyzed by DNA sequencing as a comparison for tumor WES for identification of somatic variants. Final sample numbers by tissue and analysis type are provided.
Supplemental Table 1 | Clinicopathological characteristics and specimen availability by patient in the LCCC1419 study cohort. Grayed boxes indicate tumors that switched subtypes between the primary tumor and the BrM based on IHC (n=2 patients). “Y” indicates a specimen was analyzed by the indicated method.
Supplemental Figure 2 | PAM50 intrinsic molecular subtype analysis of RNA-Seq expression data. (A) Heatmap displaying that the majority of samples in 1419 were of the basal subtype. Subtype analysis was performed according to methods described in Picornell et al27. Red circles highlight samples from the two patients where IHC indicated subtype switching (see Table 1). The R package heatmaply97 was used for heatmap visualization with hierarchical clustering based on average linkage. (B) Stacked barplot displaying percentage of primary and BrM tumors belonging to respective molecular subtypes.
Supplemental Figure 3 | Intrasample variant sharing is minimal in primary and BrM TNBC. Circos plot showing total number of variants and proportion of variants shared between primary TNBC (yellow; n=13) and BrM (purple; n=17).
Supplemental Figure 4 | TMB is associated with survival in the LCCC1419 TNBC cohort. Multivariable survival analysis including age at diagnosis, race, stage, and TMB of primary tumor as covariates relative to time to event, which for this analysis was the time from diagnosis of the primary tumor to death. Patients with unknown race or stage were excluded from analysis (n=11 patients included). Additionally for this analysis stage I and II were binned, and stage III and IV were binned.
Supplemental Figure 5 | Somatic copy number alteration assessment. SCNA in (A) primary TNBC (n=12) and (B) BrM (n=16) as determined by Sequenza/GISTIC 2.0 analysis. Significant amplicons (Amp) or deleted (Del) regions are annotated (q<0.25, green line). 11p13 amplification and 13q11 deletion, which are shared genomic features of primary and BrM TNBC, are highlighted by colored boxes. Potential oncogenes and tumor suppressors are annotated on the plots, and stars indicate genomic regions where copy number alterations are known to contribute to breast cancer/aggressive basal breast cancer.
Supplemental Figure 6 | Ploidy, tumor purity, and subclonal heterogeneity assessment of LCCC1419. (A) Ploidy and (B) tumor purity were assessed using Sequenza, where no significant differences were observed between primary TNBC (n=12) and BrM (n=16); matched pairs shown on right of each panel (n=6). (C) Distribution of subclone number per tumor. (D) Cellular prevalence and mutational load of subclones in matched pairs (n=6).
Supplemental Figure 7 | Correlation matrix of expression of immune gene signatures (IGS) used in this study. (A) IGS (n=32; refer to Supplemental Data 1 for genes comprising respective signatures) were quantified from RNA-Seq expression data from primary TNBC (n=15) and BrM (n=19), and analyzed by Spearman correlation analysis. (B) IGS correlation matrix applied to the same dataset from (A) using a compendium of IGS (64 signatures; refer to Supplemental Data 1 for genes comprising respective signatures) curated by our group. Due to the high degree of correlation of some of the subsets of IGS, we culled this list to the 32 signatures shown in (A) so as to reduce signature redundancy while maintaining breadth of represented immune cell types/features. Color denotes Spearman rho, and X indicates a non-significant relationship (p>0.05). The R package Ggcorrplot98 was used to generate correlation plot.
Supplemental Figure 8 | Immune cell type deconvolution of RNA-Seq expression data. RNA-Seq expression data from primary TNBC (n=15) and BrM (n=19) was input into CIBERSORTx (33) (https://cibersortx.stanford.edu/) to determine relative contributions of 22 immune cell subtypes (LM22) to tumor composition. Wilcoxon rank-sum test was performed on Z-transformed cell fraction values to determine statistical significance. Significance codes: *q<0.05; **q<0.01.
Supplemental Figure 9 | Analysis of additional immune modules further demonstrate immune cell deficit in BrM relative to primary TNBC. (A) Expression of the 20-gene Immunologic Constant of Rejection (ICR) signature was significantly reduced in BrM (n=19) relative to primary TNBC (n=15). (B) Blood transcriptional module repertoires reported by Rinchai et al (DOI: 10.1093/bioinformatics/btab121) were applied to the LCCC1419 RNAseq dataset using the Bioconductor R package BloodGen3Module Groupcomparison function. Significant immune modules are annotated on the plot. This analysis corroborates data showing a reduction of B and T cells in the BrM relative to primary TNBC (q<0.1).
Supplemental Figure 10 | Multivariable survival analysis of clinicopathological variables. Clinicopathological variables (age at primary tumor diagnosis, race, and stage) were analyzed by multivariable CoxPH. Category labels indicate time metric to event that was applied. Patients with unknown race (including one Asian patient) or stage were excluded from analysis (n=21 patients included). Additionally for this analysis stage I and II were binned, and stage III and IV were binned. Significance codes: *q<0.05; **q<0.01; ns, not significant.
Supplemental Figure 11 | Multivariable CoxPH analysis of IGS in primary and BrM TNBC. Each of 32 IGS (refer to Supplemental Figure 7) was included as a covariate along with clinicopathological variables (age at diagnosis, race, stage), which yielded odds ratios (OR; hazard ratios) and p values. These p values were then FDR-adjusted. This analysis was performed using three different time metrics to death, as indicated in (A) primary TNBC and (B) BrM analyses. Only IGS that had unadjusted p values of < 0.1 are shown. Note that none of the IGS achieved significant association with survival after false discovery correction, which is likely due to low sample number.
References
1. Brosnan EM, Anders CK. Understanding Patterns of Brain Metastasis in Breast Cancer and Designing Rational Therapeutic Strategies. Ann Transl Med (2018) 6(9):163. doi: 10.21037/atm.2018.04.35
2. Niwinska A, Murawska M, Pogoda K. Breast Cancer Brain Metastases: Differences in Survival Depending on Biological Subtype, RPA RTOG Prognostic Class and Systemic Treatment After Whole-Brain Radiotherapy (WBRT). Ann Oncol (2010) 21(5):942–8. doi: 10.1093/annonc/mdp407
3. Diéras V, Weaver R, Tolaney SM, Bardia A, Punie K, Brufsky A, et al. Abstract PD13-07: Subgroup Analysis of Patients With Brain Metastases From the Phase 3 ASCENT Study of Sacituzumab Govitecan Versus Chemotherapy in Metastatic Triple-Negative Breast Cancer. Cancer Res (2021) 81(4 Supplement):PD13–07-PD13-07. doi: 10.1158/1538-7445.Sabcs20-pd13-07
4. Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov (2019) 9(5):628–45. doi: 10.1158/2159-8290.CD-18-1489
5. Shih DJH, Nayyar N, Bihun I, Dagogo-Jack I, Gill CM, Aquilanti E, et al. Genomic Characterization of Human Brain Metastases Identifies Drivers of Metastatic Lung Adenocarcinoma. Nat Genet (2020) 52(4):371–7. doi: 10.1038/s41588-020-0592-7
6. Salhia B, Kiefer J, Ross JT, Metapally R, Martinez RA, Johnson KN, et al. Integrated Genomic and Epigenomic Analysis of Breast Cancer Brain Metastasis. PloS One (2014) 9(1):e85448. doi: 10.1371/journal.pone.0085448
7. Priedigkeit N, Hartmaier RJ, Chen Y, Vareslija D, Basudan A, Watters RJ, et al. Intrinsic Subtype Switching and Acquired ERBB2/HER2 Amplifications and Mutations in Breast Cancer Brain Metastases. JAMA Oncol (2017) 3(5):666–71. doi: 10.1001/jamaoncol.2016.5630
8. Van Swearingen AED, Siegel MB, Deal AM, Sambade MJ, Hoyle A, Hayes DN, et al. LCCC 1025: A Phase II Study of Everolimus, Trastuzumab, and Vinorelbine to Treat Progressive HER2-Positive Breast Cancer Brain Metastases. Breast Cancer Res Treat (2018) 171(3):637–48. doi: 10.1007/s10549-018-4852-5
9. Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov (2015) 5(11):1164–77. doi: 10.1158/2159-8290.CD-15-0369
10. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map Format and SAMtools. Bioinformatics (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
11. Malin D, Strekalova E, Petrovic V, Deal AM, Al Ahmad A, Adamo B, et al. alphaB-Crystallin: A Novel Regulator of Breast Cancer Metastasis to the Brain. Clin Cancer Res (2014) 20(1):56–67. doi: 10.1158/1078-0432.CCR-13-1255
12. Ippen FM, Alvarez-Breckenridge CA, Kuter BM, Fink AL, Bihun IV, Lastrapes M, et al. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin Cancer Res (2019) 25(11):3374–83. doi: 10.1158/1078-0432.CCR-18-3049
13. Ippen FM, Grosch JK, Subramanian M, Kuter BM, Liederer BM, Plise EG, et al. Targeting the PI3K/Akt/mTOR Pathway With the Pan-Akt Inhibitor GDC-0068 in PIK3CA-Mutant Breast Cancer Brain Metastases. Neuro Oncol (2019) 21(11):1401–11. doi: 10.1093/neuonc/noz105
14. Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, et al. Claudin-Low Bladder Tumors Are Immune Infiltrated and Actively Immune Suppressed. JCI Insight (2016) 1(3):e85902. doi: 10.1172/jci.insight.85902
15. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-And-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol (2017) 18(8):1009–21. doi: 10.1016/S1470-2045(17)30516-8
16. Zhang J, Mardis ER, Maher CA. INTEGRATE-Neo: A Pipeline for Personalized Gene Fusion Neoantigen Discovery. Bioinformatics (2017) 33(4):555–7. doi: 10.1093/bioinformatics/btw674
17. Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep (2018) 23(1):270–281e273. doi: 10.1016/j.celrep.2018.03.052
18. Lansford JL, Dharmasiri U, Chai S, Hunsucker SA, Bortone DS, Keating JE, et al. Computational Modeling and Confirmation of Leukemia-Associated Minor Histocompatibility Antigens. Blood Adv (2018) 2(16):2052–62. doi: 10.1182/bloodadvances.2018022475
19. Saito R, Smith CC, Utsumi T, Bixby LM, Kardos J, Wobker SE, et al. Molecular Subtype-Specific Immunocompetent Models of High-Grade Urothelial Carcinoma Reveal Differential Neoantigen Expression and Response to Immunotherapy. Cancer Res (2018) 78(14):3954–68. doi: 10.1158/0008-5472.CAN-18-0173
20. Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous Retroviral Signatures Predict Immunotherapy Response in Clear Cell Renal Cell Carcinoma. J Clin Invest (2018) 128(11):4804–20. doi: 10.1172/jci121476
21. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity (2018) 48(4):812–830e814. doi: 10.1016/j.immuni.2018.03.023
22. Smith CC, Chai S, Washington AR, Lee SJ, Landoni E, Field K, et al. Machine-Learning Prediction of Tumor Antigen Immunogenicity in the Selection of Therapeutic Epitopes. Cancer Immunol Res (2019) 7(10):1591–604. doi: 10.1158/2326-6066.CIR-19-0155
23. Buchkovich ML, Brown CC, Robasky K, Chai S, Westfall S, Vincent BG, et al. HLAProfiler Utilizes K-Mer Profiles to Improve HLA Calling Accuracy for Rare and Common Alleles in RNA-Seq Data. Genome Med (2017) 9(1):86. doi: 10.1186/s13073-017-0473-6
24. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol (2017) 199(9):3360–8. doi: 10.4049/jimmunol.1700893
25. Hunsucker SA, McGary CS, Vincent BG, Enyenihi AA, Waugh JP, McKinnon KP, et al. Peptide/MHC Tetramer-Based Sorting of CD8(+) T Cells to a Leukemia Antigen Yields Clonotypes Drawn Nonspecifically From an Underlying Restricted Repertoire. Cancer Immunol Res (2015) 3(3):228–35. doi: 10.1158/2326-6066.CIR-14-0001
26. Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. Comparison of RNA-Seq by Poly (A) Capture, Ribosomal RNA Depletion, and DNA Microarray for Expression Profiling. BMC Genomics (2014) 15(1):419. doi: 10.1186/1471-2164-15-419
27. Picornell AC, Echavarria I, Alvarez E, Lopez-Tarruella S, Jerez Y, Hoadley K, et al. Breast Cancer PAM50 Signature: Correlation and Concordance Between RNA-Seq and Digital Multiplexed Gene Expression Technologies in a Triple Negative Breast Cancer Series. BMC Genomics (2019) 20(1):452. doi: 10.1186/s12864-019-5849-0
28. Galili T, O'Callaghan A, Sidi J, Sievert C. Heatmaply: An R Package for Creating Interactive Cluster Heatmaps for Online Publishing. Bioinformatics (2018) 34(9):1600–2. doi: 10.1093/bioinformatics/btx657
29. Bortone D. "Binfotron: Binfotron Bioinformatics Analysis Tools Suite" In (R package version 0.3-17 ed.). (2020).
30. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
31. Hänzelmann S, Castelo R, Guinney J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinf (2013) 14:7. doi: 10.1186/1471-2105-14-7
32. Morgan M, Falcon S, Gentleman R. "GSEABase: Gene Set Enrichmentdata Structures and Methods" In (R package version 1.48.0 ed.). (2019).
33. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust Enumeration of Cell Subsets From Tissue Expression Profiles. Nat Methods (2015) 12(5):453–7. doi: 10.1038/nmeth.3337
34. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat Methods (2015) 12(5):380–1. doi: 10.1038/nmeth.3364
35. Mose LE, Selitsky SR, Bixby LM, Marron DL, Iglesia MD, Serody JS, et al. Assembly-Based Inference of B-Cell Receptor Repertoires From Short Read RNA Sequencing Data With V'DJer. Bioinformatics (2016) 32(24):3729–34. doi: 10.1093/bioinformatics/btw526
36. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat Methods (2017) 14(4):417–9. doi: 10.1038/nmeth.4197
37. Bortone DS, Woodcock MG, Parker JS, Vincent BG. Improved T-Cell Receptor Diversity Estimates Associate With Survival and Response to Anti-PD-1 Therapy. Cancer Immunol Res (2021) 9(1):103–12. doi: 10.1158/2326-6066.CIR-20-0398
39. Max Gordon TL. "Forestplot: Advanced Forest Plot Using 'Grid' Graphics" In version 1.7.2 ed. (2017).
40. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic Identification of Personal Tumor-Specific Neoantigens in Chronic Lymphocytic Leukemia. Blood (2014) 124(3):453–62. doi: 10.1182/blood-2014-04-567933
41. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-Analysis of Tumor- and T Cell-Intrinsic Mechanisms of Sensitization to Checkpoint Inhibition. Cell (2021) 184(3):596–614e514. doi: 10.1016/j.cell.2021.01.002
42. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res (2019) 47(D1):D941–7. doi: 10.1093/nar/gky1015
43. Cancer Genome Atlas N. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
44. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol (2011) 12(4):R41. doi: 10.1186/gb-2011-12-4-r41
45. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: Allele-Specific Copy Number and Mutation Profiles From Tumor Sequencing Data. Ann Oncol (2015) 26(1):64–70. doi: 10.1093/annonc/mdu479
46. Klingbeil P, Natrajan R, Everitt G, Vatcheva R, Marchio C, Palacios J, et al. CD44 is Overexpressed in Basal-Like Breast Cancers But is Not a Driver of 11p13 Amplification. Breast Cancer Res Treat (2010) 120(1):95–109. doi: 10.1007/s10549-009-0380-7
47. Artibani M, Sims AH, Slight J, Aitken S, Thornburn A, Muir M, et al. WT1 Expression in Breast Cancer Disrupts the Epithelial/Mesenchymal Balance of Tumour Cells and Correlates With the Metabolic Response to Docetaxel. Sci Rep (2017) 7:45255. doi: 10.1038/srep45255
48. Rouault A, Banneau G, Macgrogan G, Jones N, Elarouci N, Barouk-Simonet E, et al. Deletion of Chromosomes 13q and 14q Is a Common Feature of Tumors With BRCA2 Mutations. PloS One (2012) 7(12):e52079. doi: 10.1371/journal.pone.0052079
49. Farabegoli F, Ceccarelli C, Santini D, Trere D, Baldini N, Taffurelli M, et al. Chromosome 1 Aneusomy With 1p36 Under-Representation Is Related to Histologic Grade, DNA Aneuploidy, High C-Erb B-2 and Loss of Bcl-2 Expression in Ductal Breast Carcinoma. Int J Cancer (1996) 69(5):381–5. doi: 10.1002/(SICI)1097-0215(19961021)69:5<381::AID-IJC5>3.0.CO;2-1
50. Shivapurkar N, Sood S, Wistuba II, Virmani AK, Maitra A, Milchgrub S, et al. Multiple Regions of Chromosome 4 Demonstrating Allelic Losses in Breast Carcinomas. Cancer Res (1999) 59(15):3576–80.
51. Martinez A, Walker RA, Shaw JA, Dearing SJ, Maher ER, Latif F. Chromosome 3p Allele Loss in Early Invasive Breast Cancer: Detailed Mapping and Association With Clinicopathological Features. Mol Pathol (2001) 54(5):300–6. doi: 10.1136/mp.54.5.300
52. Oesterreich S, Allredl DC, Mohsin SK, Zhang Q, Wong H, Lee AV, et al. High Rates of Loss of Heterozygosity on Chromosome 19p13 in Human Breast Cancer. Br J Cancer (2001) 84(4):493–8. doi: 10.1054/bjoc.2000.1606
53. Yang TL, Su YR, Huang CS, Yu JC, Lo YL, Wu PE, et al. High-Resolution 19p13.2-13.3 Allelotyping of Breast Carcinomas Demonstrates Frequent Loss of Heterozygosity. Genes Chromosomes Cancer (2004) 41(3):250–6. doi: 10.1002/gcc.20080
54. Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, et al. Frequency, Prognostic Impact, and Subtype Association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 Amplifications in Breast Cancers. BMC Cancer (2006) 6:245. doi: 10.1186/1471-2407-6-245
55. Melchor L, Saucedo-Cuevas LP, Munoz-Repeto I, Rodriguez-Pinilla SM, Honrado E, Campoverde A, et al. Comprehensive Characterization of the DNA Amplification at 13q34 in Human Breast Cancer Reveals TFDP1 and CUL4A as Likely Candidate Target Genes. Breast Cancer Res (2009) 11(6):R86. doi: 10.1186/bcr2456
56. Yu W, Kanaan Y, Bae YK, Gabrielson E. Chromosomal Changes in Aggressive Breast Cancers With Basal-Like Features. Cancer Genet Cytogenet (2009) 193(1):29–37. doi: 10.1016/j.cancergencyto.2009.03.017
57. Zhang J, Liu X, Datta A, Govindarajan K, Tam WL, Han J, et al. RCP is a Human Breast Cancer-Promoting Gene With Ras-Activating Function. J Clin Invest (2009) 119(8):2171–83. doi: 10.1172/JCI37622
58. Wikman H, Sielaff-Frimpong B, Kropidlowski J, Witzel I, Milde-Langosch K, Sauter G, et al. Clinical Relevance of Loss of 11p15 in Primary and Metastatic Breast Cancer: Association With Loss of PRKCDBP Expression in Brain Metastases. PloS One (2012) 7(10):e47537. doi: 10.1371/journal.pone.0047537
59. Chen YY, Ge JY, Zhu SY, Shao ZM, Yu KD. Copy Number Amplification of ENSA Promotes the Progression of Triple-Negative Breast Cancer via Cholesterol Biosynthesis. Nat Commun (2022) 13(1):791. doi: 10.1038/s41467-022-28452-z
60. Rieber N, Bohnert R, Ziehm U, Jansen G. Reliability of Algorithmic Somatic Copy Number Alteration Detection From Targeted Capture Data. Bioinformatics (2017) 33(18):2791–8. doi: 10.1093/bioinformatics/btx284
61. Tanner G, Westhead DR, Droop A, Stead LF. Benchmarking Pipelines for Subclonal Deconvolution of Bulk Tumour Sequencing Data. Nat Commun (2021) 12(1):6396. doi: 10.1038/s41467-021-26698-7
62. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization From Targeted DNA Sequencing. PloS Comput Biol (2016) 12(4):e1004873. doi: 10.1371/journal.pcbi.1004873
63. Gillis S, Roth A. PyClone-VI: Scalable Inference of Clonal Population Structures Using Whole Genome Data. BMC Bioinf (2020) 21(1):571. doi: 10.1186/s12859-020-03919-2
64. Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative Tumour-Specific Antigens. Nat Rev Cancer (2019) 19(8):465–78. doi: 10.1038/s41568-019-0162-4
65. Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene Expression Signature of Fibroblast Serum Response Predicts Human Cancer Progression: Similarities Between Tumors and Wounds. PloS Biol (2004) 2(2):E7. doi: 10.1371/journal.pbio.0020007
66. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining Cell Type Abundance and Expression From Bulk Tissues With Digital Cytometry. Nat Biotechnol (2019) 37(7):773–82. doi: 10.1038/s41587-019-0114-2
67. Bertucci F, Finetti P, Simeone I, Hendrickx W, Wang E, Marincola FM, et al. The Immunologic Constant of Rejection Classification Refines the Prognostic Value of Conventional Prognostic Signatures in Breast Cancer. Br J Cancer (2018) 119(11):1383–91. doi: 10.1038/s41416-018-0309-1
68. Rinchai D, Roelands J, Toufiq M, Hendrickx W, Altman MC, Bedognetti D, et al. BloodGen3Module: Blood Transcriptional Module Repertoire Analysis and Visualization Using R. Bioinformatics (2021). doi: 10.1093/bioinformatics/btab121
69. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive Assessment of T-Cell Receptor Beta-Chain Diversity in Alphabeta T Cells. Blood (2009) 114(19):4099–107. doi: 10.1182/blood-2009-04-217604
70. Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using Synthetic Templates to Design an Unbiased Multiplex PCR Assay. Nat Commun (2013) 4:2680. doi: 10.1038/ncomms3680
71. Gao YK, Kuksis M, Id Said B, Chehade R, Kiss A, Tran W, et al. Treatment Patterns and Outcomes of Women With Symptomatic and Asymptomatic Breast Cancer Brain Metastases: A Single-Center Retrospective Study. Oncologist (2021) 26(11):e1951-e1961. doi: 10.1002/onco.13965
72. Zimmerman BS, Seidman D, Cascetta KP, Ru M, Moshier E, Tiersten A. Prognostic Factors and Survival Outcomes Among Patients With Breast Cancer and Brain Metastases at Diagnosis: A National Cancer Database Analysis. Oncology (2021) 99(5):280–91. doi: 10.1159/000512212
73. Sammons S, Van Swearingen AED, Anders CK. The Promise of Immunotherapy for Breast Cancer Brain Metastases. Curr Breast Cancer Rep (2019). doi: 10.1007/s12609-019-00335-1
74. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J Clin Invest (2011) 121(7):2750–67. doi: 10.1172/JCI45014
75. Loizidou MA, Cariolou MA, Neuhausen SL, Newbold RF, Bashiardes E, Marcou Y, et al. Genetic Variation in Genes Interacting With BRCA1/2 and Risk of Breast Cancer in the Cypriot Population. Breast Cancer Res Treat (2010) 121(1):147–56. doi: 10.1007/s10549-009-0518-7
76. Giuli MV, Giuliani E, Screpanti I, Bellavia D, Checquolo S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J Oncol (2019) 2019:8707053. doi: 10.1155/2019/8707053
77. Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, et al. Association of Survival and Disease Progression With Chromosomal Instability: A Genomic Exploration of Colorectal Cancer. Proc Natl Acad Sci USA (2009) 106(17):7131–6. doi: 10.1073/pnas.0902232106
78. Titus AJ, Way GP, Johnson KC, Christensen BC. Deconvolution of DNA Methylation Identifies Differentially Methylated Gene Regions on 1p36 Across Breast Cancer Subtypes. Sci Rep (2017) 7(1):11594. doi: 10.1038/s41598-017-10199-z
79. Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, et al. Comprehensive Profiling of 8p11-12 Amplification in Breast Cancer. Mol Cancer Res (2005) 3(12):655–67. doi: 10.1158/1541-7786.MCR-05-0128
80. Mesquita B, Lopes P, Rodrigues A, Pereira D, Afonso M, Leal C, et al. Frequent Copy Number Gains at 1q21 and 1q32 are Associated With Overexpression of the ETS Transcription Factors ETV3 and ELF3 in Breast Cancer Irrespective of Molecular Subtypes. Breast Cancer Res Treat (2013) 138(1):37–45. doi: 10.1007/s10549-013-2408-2
81. Tang X, Tang J, Liu X, Zeng L, Cheng C, Luo Y, et al. Downregulation of miR-129-2 by Promoter Hypermethylation Regulates Breast Cancer Cell Proliferation and Apoptosis. Oncol Rep (2016) 35(5):2963–9. doi: 10.3892/or.2016.4647
82. Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front Pharmacol (2021) 12:662232. doi: 10.3389/fphar.2021.662232
83. Pandey V, Wu ZS, Zhang M, Li R, Zhang J, Zhu T, et al. Trefoil Factor 3 Promotes Metastatic Seeding and Predicts Poor Survival Outcome of Patients With Mammary Carcinoma. Breast Cancer Res (2014) 16(5):429. doi: 10.1186/s13058-014-0429-3
84. Al-Salam S, Sudhadevi M, Awwad A, Al Bashir M. Trefoil Factors Peptide-3 is Associated With Residual Invasive Breast Carcinoma Following Neoadjuvant Chemotherapy. BMC Cancer (2019) 19(1):135. doi: 10.1186/s12885-019-5316-y
85. Routh ED, Pullikuth AK, Jin G, Chifman J, Chou JW, D'Agostino RB Jr., et al. Transcriptomic Features of T Cell-Barren Tumors Are Conserved Across Diverse Tumor Types. Front Immunol (2020) 11:57. doi: 10.3389/fimmu.2020.00057
86. Mahmoud AM. Cancer Testis Antigens as Immunogenic and Oncogenic Targets in Breast Cancer. Immunotherapy (2018) 10(9):769–78. doi: 10.2217/imt-2017-0179
87. Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous Retrovirus Expression Is Associated With Response to Immune Checkpoint Blockade in Clear Cell Renal Cell Carcinoma. JCI Insight (2018) 3(16). doi: 10.1172/jci.insight.121522
88. Zhang SM, Cai WL, Liu X, Thakral D, Luo J, Chan LH, et al. KDM5B Promotes Immune Evasion by Recruiting SETDB1 to Silence Retroelements. Nature (2021) 598(7882):682–7. doi: 10.1038/s41586-021-03994-2
89. Louveau A, Harris TH, Kipnis J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol (2015) 36(10):569–77. doi: 10.1016/j.it.2015.08.006
90. Xiao L, Zhou J, Liu H, Zhou Y, Chen W, Cui W, et al. ). RNA Sequence Profiling Reveals Unique Immune and Metabolic Features of Breast Cancer Brain Metastases. Front Oncol (2021) 11:679262. doi: 10.3389/fonc.2021.679262
91. Dvorak HF. Tumors: Wounds That do Not Heal-Redux. Cancer Immunol Res (2015) 3(1):1–11. doi: 10.1158/2326-6066.CIR-14-0209
92. Kidman J, Principe N, Watson M, Lassmann T, Holt RA, Nowak AK, et al. Characteristics of TCR Repertoire Associated With Successful Immune Checkpoint Therapy Responses. Front Immunol (2020) 11:587014. doi: 10.3389/fimmu.2020.587014
93. Burwinkel B, Shanmugam KS, Hemminki K, Meindl A, Schmutzler RK, Sutter C, et al. Transcription Factor 7-Like 2 (TCF7L2) Variant Is Associated With Familial Breast Cancer Risk: A Case-Control Study. BMC Cancer (2006) 6:268. doi: 10.1186/1471-2407-6-268
94. Min W, Liu X, Lu Y, Gong Z, Wang M, Lin S, et al. Association of Transcription Factor 7-Like 2 Gene Polymorphisms With Breast Cancer Risk in Northwest Chinese Women. Oncotarget (2016) 7(47):77175–82. doi: 10.18632/oncotarget.12591
95. Wenzel J, Rose K, Haghighi EB, Lamprecht C, Rauen G, Freihen V, et al. Loss of the Nuclear Wnt Pathway Effector TCF7L2 Promotes Migration and Invasion of Human Colorectal Cancer Cells. Oncogene (2020) 39(19):3893–909. doi: 10.1038/s41388-020-1259-7
96. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell (2017) 168(3):542. doi: 10.1016/j.cell.2017.01.010
97. Galili T, O’Callaghan A, Sidi J, Sievert C. ). Heatmaply: An R Package for Creating Interactive Cluster Heatmaps for Online Publishing. Bioinformatics (2017) 34(9):1600–2. doi: 10.1093/bioinformatics/btx657
Keywords: triple-negative breast cancer, brain metastases, immunogenomics, whole-exome sequencing, mRNA sequencing, biobank
Citation: Routh ED, Van Swearingen AED, Sambade MJ, Vensko S, McClure MB, Woodcock MG, Chai S, Cuaboy LA, Wheless A, Garrett A, Carey LA, Hoyle AP, Parker JS, Vincent BG and Anders CK (2022) Comprehensive Analysis of the Immunogenomics of Triple-Negative Breast Cancer Brain Metastases From LCCC1419. Front. Oncol. 12:818693. doi: 10.3389/fonc.2022.818693
Received: 19 November 2021; Accepted: 30 May 2022;
Published: 27 July 2022.
Edited by:
Priscilla Brastianos, Harvard Medical School, United StatesReviewed by:
James M. Rae, University of Michigan, United StatesGanesh Rao, University of Texas MD Anderson Cancer Center, United States
Copyright © 2022 Routh, Van Swearingen, Sambade, Vensko, McClure, Woodcock, Chai, Cuaboy, Wheless, Garrett, Carey, Hoyle, Parker, Vincent and Anders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carey K. Anders, Y2FyZXkuYW5kZXJzQGR1a2UuZWR1
†Present Addresses: Amanda E. D. Van Swearingen, Duke Center for Brain and Spine Metastasis, Duke Cancer Institute, Duke University, Durham, NC, United States and Department of Medicine, Division of Medical Oncology, Duke University, Durham, NC, United States
Marni B. McClure, Department of Medicine, Johns Hopkins Hospital, Baltimore, MD, United States
Amy Garrett, ICON plc, Research Triangle Park, NC, United States
Carey K. Anders, Duke Center for Brain and Spine Metastasis, Duke Cancer Institute, Duke University, Durham, NC, United States and Department of Medicine, Division of Medical Oncology, Duke University, Durham, NC, United States
‡These authors have contributed equally to this work and share first authorship
§These authors have contributed equally to this work and share senior authorship