 Yuanming Jing1
Yuanming Jing1 Zongliang Huang
Zongliang Huang- 1Department of Gastrointestinal Surgery, Shaoxing People’s Hospital (Shaoxing Hospital, Zhejiang University School of Medicine), Shaoxing, China
- 2 Department of Orthopaedics, Zhoushan Hospital of Traditional Chinese Medicine Affiliated to Zhejiang Chinese Medical University, Zhoushan, China
- 3Department of Pharmacy, Shaoxing People’s Hospital, Shaoxing Hospital, Zhejiang University School of Medicine, Shaoxing, China
- 4Department of Radiology, Tongji Hospital, School of Medicine, Tongji University, Shanghai, China
Cancer stem cells (CSCs) modify and form their microenvironment by recruiting and activating specific cell types such as mesenchymal stem cells (MSCs). Tumor-infiltrating MSCs help to establish a suitable tumor microenvironment for the restoration of CSCs and tumor progression. In addition, crosstalk between cancer cells and MSCs in the microenvironment induces a CSC phenotype in cancer cells. Many mechanisms are involved in crosstalk between CSCs/cancer cells and MSCs including cell-cell interaction, secretion of exosomes, and paracrine secretion of several molecules including inflammatory mediators, cytokines, and growth factors. Since this crosstalk may contribute to drug resistance, metastasis, and tumor growth, it is suggested that blockade of the crosstalk between MSCs and CSCs/cancer cells can provide a new avenue to improving the cancer therapeutic tools. In this review, we will discuss the role of MSCs in the induction of cancer stem cell phenotype and the restoration of CSCs. We also discuss targeting the crosstalk between MSCs and CSCs/cancer cells as a therapeutic strategy.
1 Introduction
Cancer stem cells (CSCs), which have been shown to play a vital role in tumor origin, are considered to be responsible for tumor progression, drug resistance, and metastasis (1). CSCs can form their microenvironment by recruiting and activating specific cell types such as mesenchymal stem cells (MSCs). Then, MSCs can modify the stroma and establish a unique tissue microenvironment that is suitable for the restoration of CSCs and tumor progression (2). In addition, crosstalk between cancer cells and MSCs in the microenvironment can induce a CSC phenotype in cancer cells. Many mechanisms are involved in crosstalk between tumor cells and MSCs including cell-cell interaction, secretion of exosomes, and paracrine secretion of several molecules including inflammatory mediators, cytokines, and growth factors (3). Since crosstalk between tumor cells and MSCs may contribute to drug resistance, metastasis, and tumor growth, it is suggested that blockade of the crosstalk between MSCs and tumor cells can provide a new avenue to improving the cancer therapeutic tools.
Many studies show the crosstalk between tumor cells and MSCs. For instance, transforming growth factor (TGF)-β-stimulated MSCs can induce a metastatic phenotype by upregulating Jagged-1, a major ligand of Notch signaling, in tumor cells (4). Indeed, activation of the Notch signaling pathway induces epithelial-mesenchymal transition (EMT) and promotes a cancer stem cell phenotype. This phenomenon is supported by other studies that show the relationship between the EMT process and CSCs (5). In another study, in hepatocellular carcinoma, treating MSCs with tumor necrosis factor-α (TNF-α) and interferon γ (IFNγ) causes an increase in production of TGFβ by MSCs which in turn could promote tumor metastasis by inducing EMT in cancer cells (6). Luo, et al. have reported that the increased metastatic phenotype of prostate cancer (PCa) cells could be due to an increase in the PCa stem cell population. They showed that increase in the stem cell population is mediated by MSCs through alteration of the CCL5–AR signaling pathway (7). Indeed, the upregulation of CCL5 in bone marrow mesenchymal stem cells (BM-MSCs) and PCa cells, after MSCs infiltrated into PCa microenvironment, lead to downregulation of the androgen receptor (AR) signaling pathway (7). Increasing in the PCa stem cell populations led to the upregulation of CXCR4, ZEB-1, matrix metallopeptidase 9 (MMP-9), and CD133 that these molecules promote the metastatic phenotype of PCa cells (7). Recently, Hossain et al. reported that in glioblastomas, tumor-associated mesenchymal stromal cells promote the proliferative and tumorigenic phenotype of glioma cancer stem cells (gCSCs) through the IL-6/STAT3 signaling pathway (8). The aggressiveness of gCSCs is enhanced in co-culture with MSCs, and these observations were supported by reduced survival in orthotopic xenograft mouse models, increased cell counts in vitro, enhanced angiogenesis, and tumor size in vivo (9). Gene expression analysis of cancer-associated (CA)-MSCs revealed that they can alter synthesis levels of bone morphogenetic protein (BMP) signaling pathway proteins. BMP2 can significantly increase the number of CSCs in primary ovarian tumor cells and ovarian cancer cell lines (10). In vivo and in vitro suppression of the BMP signaling pathway with Noggin suppress the capability of CA-MSCs to support tumor growth and tumor stemness (10). Thus, MSCs can enhance tumorigenesis, at least in part, through the promotion of the BMP signaling pathway (10). On the other hand, Vulcano et al. reported that Wharton’s jelly of umbilical cord (WJMSC) exert, both in vivo and in vitro, conflicting impacts on lung cancer stem cells derived from various lung cancer subtypes (11).
In this review, we will discuss the role of MSCs in the induction of cancer stem cell phenotype and the restoration of CSCs. We also discuss targeting the crosstalk between MSCs and CSCs/cancer cells as a therapeutic strategy.
2 MSCs Mediated Mechanisms of Increasing CSC Population
Various mechanisms are involved in inducing the stem cell phenotype in tumor cells and restoring of CSCs including cell fusion, direct transformation of MSCs into CSCs, crosstalk of MSCs with CSCs/tumor cells mediated by secretory factors, exosomes, etc. We will go into the details of the mentioned mechanisms in the following.
2.1 MSCs Secreted Factors/Cancer Cell Contact and CSCs
Plenty of studies has been performed to indicate how cellular components of the cancer microenvironment participate in cancer development. CSCs by recruiting and activating specific cell types establish their microenvironment. MSCs are one of the main cellular components which release various cytokines that have both autocrine and paracrine functions in the cancer milieu (2).
2.1.1 MSCs Secreted Factors/Cancer Cell Contact and Induction of CSC Phenotype
MSCs promote EMT by the secretion of cytokines and growth factors such as TGFβ (Figure 1). These factors stimulate transcriptional regulators, such as Zeb1, Twist, Slug, Snail, and others which are related to EMT (12, 13). For instance, in hepatocellular carcinoma, treatment of MSCs with IFNγ and TNFα leads to a high expression level of TGFβ which in turn induces EMT-related properties in tumor cells (6). In another study, TGFβ-induced MSCs in pancreatic cancer increase the metastatic potential by upregulating Jagged-1, a major ligand of Notch signaling in cancer cells (4). In turn, stimulation of the Notch signaling pathway promotes EMT and induces a CSC phenotype. Some other studies supported the role of EMT in the induction of CSC phenotype (5). An alternative mechanism of MSC-induced CSC phenotype has been indicated in gastric cancer. MSCs are recruited by gastric mucosal cells infected with Helicobacter which then transform into gastric cells expressing epithelial biomarkers including TFF2 and KRT1-19. Therefore, chronic inflammation induces the CSC properties of gastric cancer by inducing the EMT and metastatic phenotype (14).
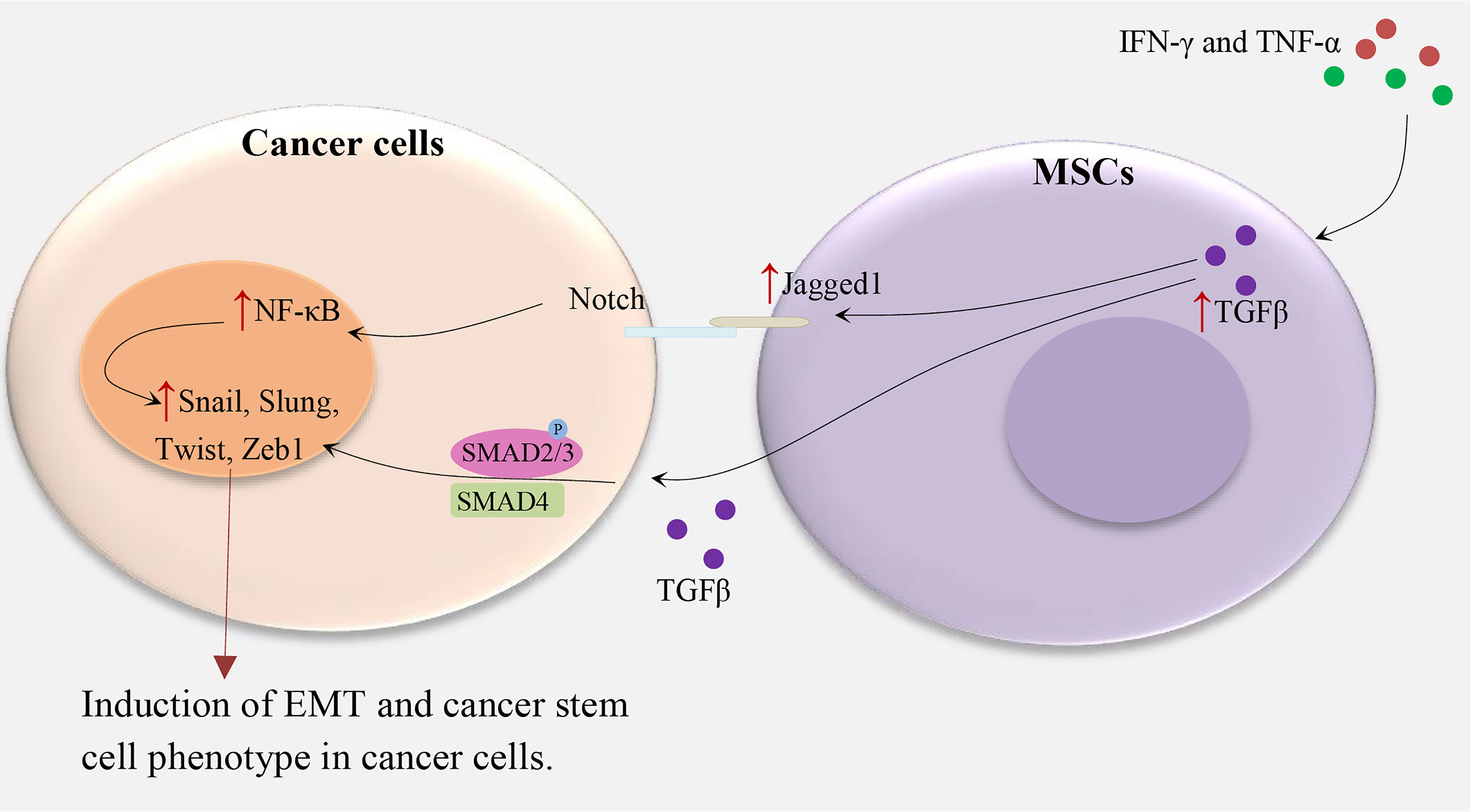
Figure 1 MSCs and induction of CSC phenotype in cancer cells. IFN-γ and TNF-α lead to TGFβ overexpression in MSCs, subsequently, TGFβ upregulates Notch signaling and TGF-β/Smad signaling pathways in cancer cells and induce the cancer stem cell phenotype by upregulating Zeb1, Twist, Slug, Snail, and others which are related to EMT (4, 6, 12, 13).
The MSC-induced paracrine effect of TGF-β1 and autocrine effects of WNT5A on the restoration of CD133+ CSC populations show the importance of the tumor microenvironment for the maintenance of CSCs (15), as illustrated in Figure 2. The autocrine effects of WNT5A in gastric carcinoma cells can contribute to the activation of the WNT-β-catenin signaling pathway (15). It has been shown that both TGF-β and WNT5A play a crucial role in the stimulation of EMT in tumor cells: WNT5A stable melanoma cells transfectants indicate a spindle shape accompanied by increased vimentin expression and decreased E-cadherin expression (16), and TGF-beta mediated EMT is regulated by the SNAIL1-SMAD3/4 transcriptional complex, which acts as a suppressor of E-cadherin expression (17). Indeed, WNT5A and TGF-β significantly enhance the expression of the Snail-family transcription factors, including Slug, Snail, Twist1, and Twist2 (15). These studies suggest a direct relationship between the EMT and the gain of CSC phenotype (18).
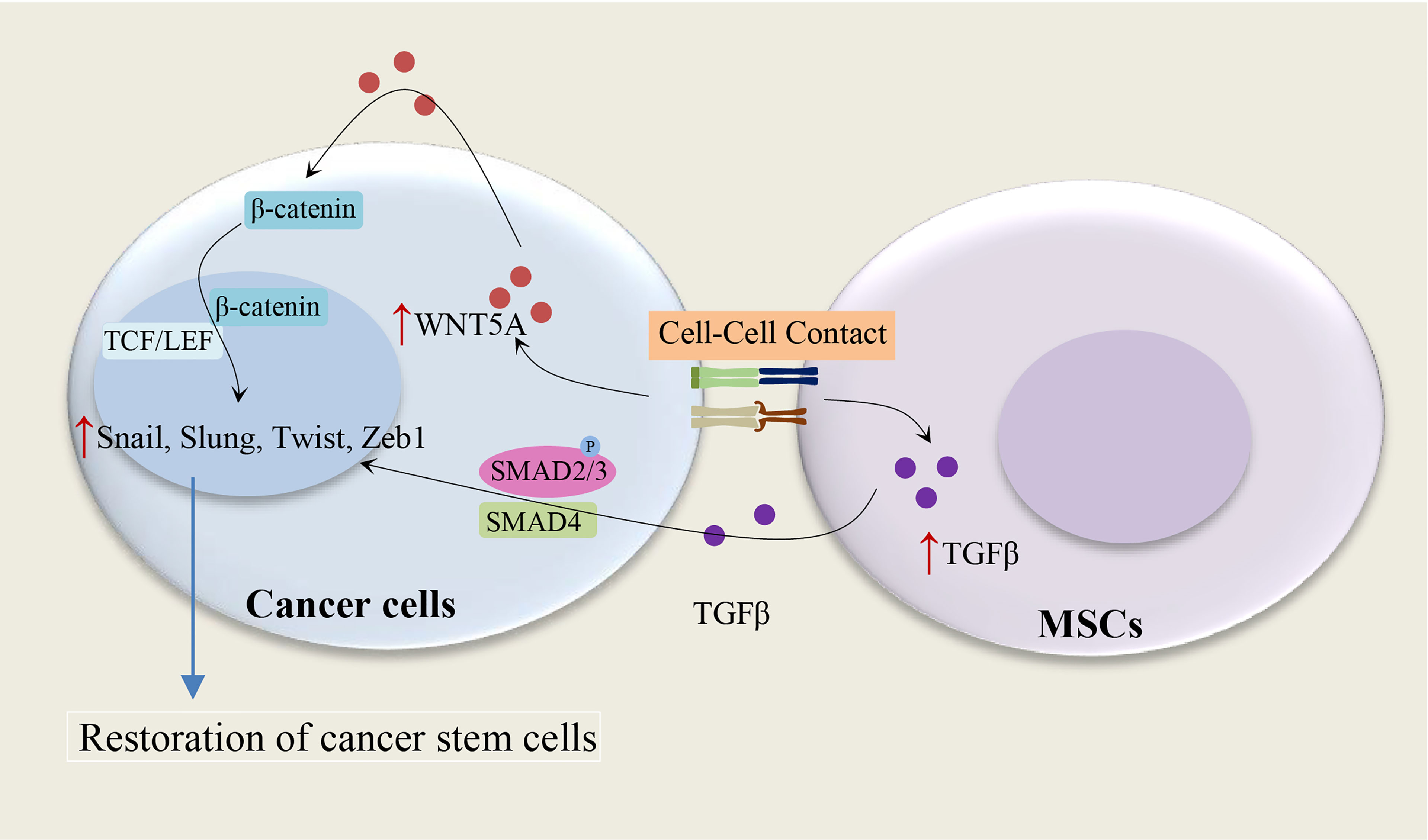
Figure 2 Cell-cell contact and restoration of CSC populations. Cell contact between MSCs and cancer cells leads to an increase of TGF-β1 in MSCs and WNT5A in cancer cells, subsequently, the paracrine effect of TGF-β1 and autocrine effects of WNT5A on cancer cells result in the restoration of CSC populations. WNT5A and TGF-β enhance the expression of the Snail-family transcription factors, including Slug, Snail, Twist1, and Twist2 (15–17).
2.1.2 MSCs Secreted IL-1, IL-6, PEG-2, and Induction of CSCs
It has been shown that IL-1 released by head and neck squamous cell carcinoma (HNSCC) cells stimulates prostaglandin-E2 (PGE-2) from fibroblasts (19), as illustrated in Figure 3. It has been also reported that cancer cells are able to provoke a strong stimulation of the cyclooxygenase-2 (COX-2)/microsomal Prostaglandin-E synthase-1 (mPGES-1)/PGE 2 axis in MSCs recruited to the cancer-associated stroma by releasing IL-1 (20). The tumor-promoting effects of COX-2 are mostly related to its role in inducing PGE-2, which has pleiotropic effects on invasiveness, angiogenesis, motility, survival, and cell proliferation (21). It is found that IL-1 plays a critical role in the cancer cell-induced COX-2/mPGES1/PGDH/PGE 2 response in MSCs that is necessary for tumor development (20). In colorectal cancer, MSCs release PGE-2 in response to IL-1 secreted by tumor cells, PGE-2 in an autocrine fashion promotes the expression of IL-8, IL-6, CXCL1, RANTES, and GRO-α, which together stimulate the formation of CSCs (20). PGE-2, which can trigger the EMT phenotype, promote both the frequency of cancer initiation and the number of CSCs (20). Li et al. (20) showed the partial EMT phenotype induced by PGE 2 suffices to increase CSCs by inducing a stem cell-like phenotype in cancer cells by suppressing cell-cell junctions without stimulating mesenchymal traits (20). Previous studies have shown the role of prostaglandin E2 in increasing the number of CD44+ cancer cells (22, 23). Observations show that other MSC-derived cytokines, compared to PEG-2, have marginal effects on the increasing tumor-initiating cell frequency (20). The PGE-2 and cytokines act in a paracrine fashion on the tumor cells to stimulate the β-catenin signaling pathway and formation of CSCs. Therefore, MSCs and derived cell types construct a CSC niche and promote cancer progression via the secretion of PGE-2 and other cytokines (20). IL-1 blocking therapies are used in the clinic to control inflammatory and infectious diseases and have a remarkable safety record (24). IL-1 blocking may provide a promising alternative to COX-2 inhibitors in cancer therapy (20).
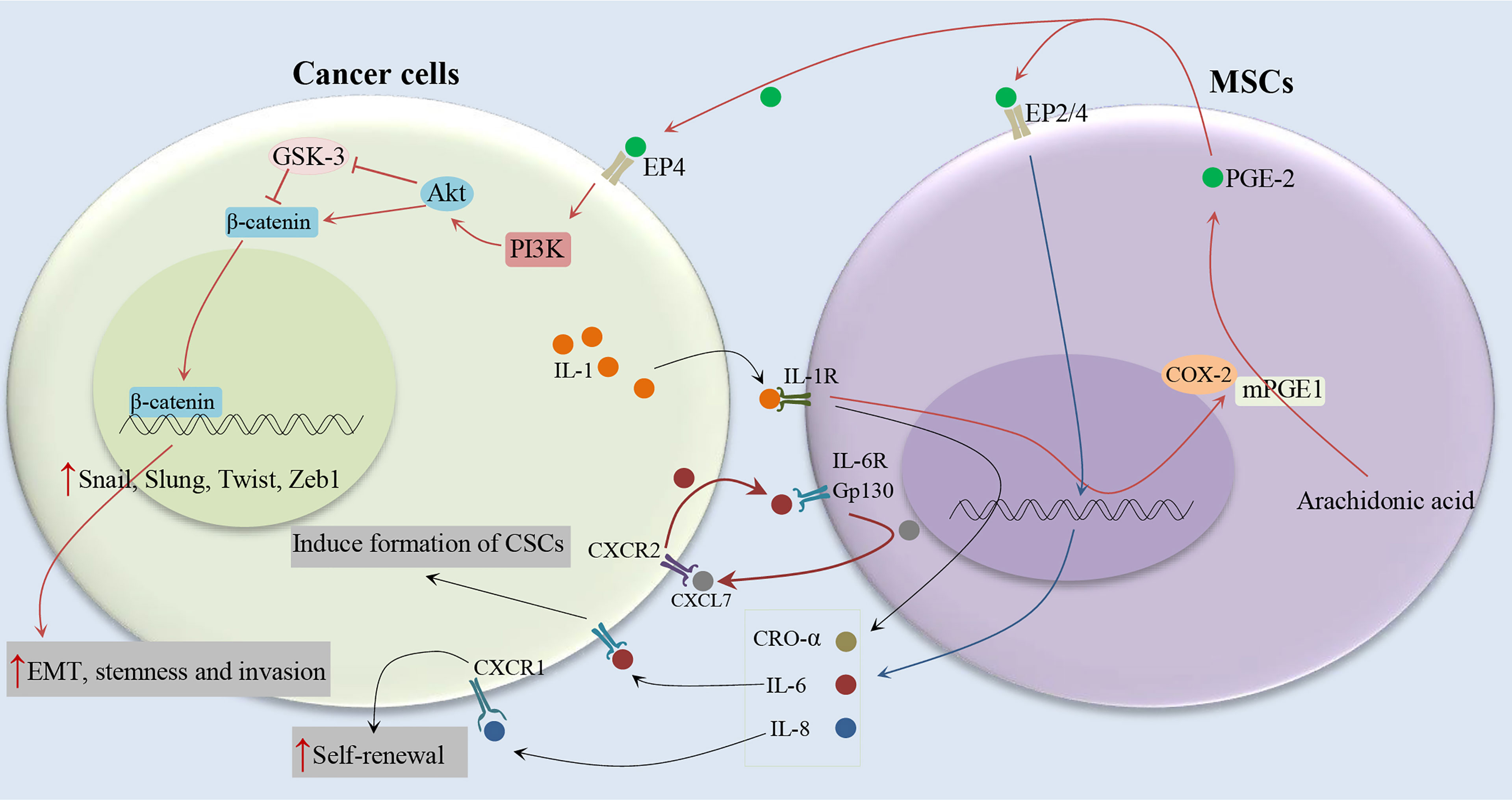
Figure 3 Cytokine networks between MSCs and cancer cells and induction of CSC phenotype. Cancer cells stimulate the COX-2/mPGES-1/PGE 2 axis in MSCs by releasing IL-1. MSCs release PGE-2 in response to IL-1, PGE-2 in an autocrine fashion promote the expression of IL-8, IL-6, CXCL1, RANTES, and GRO-α, which together stimulate the formation of CSCs (19–21).
It has been indicated the association between serum IL-6 levels and poor clinical outcomes of breast cancer patients (25, 26). IL-6 acts as a direct regulator of CSC self-renewal, a process triggered by the IL-6 receptor/GP130 complex through the activation of STAT3 (27). On the other hand, It has been reported that MSCs can secrete IL-6 (28, 29), and induce tumor growth through the paracrine function (30). Liu et al. (31) indicated that the communication between CSCs and MSCs is facilitated by a positive feedback cytokine loop in which CXCL7 and IL6 play essential roles (31), as illustrated in Figure 3. This loop needs the simultaneous presence of these cells but does not require direct interactions between cell surfaces as indicated by trans-well and conditioned medium experiments (31). IL6 secreted by tumor cells interacts with gp130 and interleukin 6 receptor (IL-6R) on ALDEFLUOR-positive mesenchymal cells (MCs) and can promote the homing of MSCs to the tumor sites, as well as stimulate CXCL7 expression by these cells. Then, MSC-derived CXCL7 interacts with tumor cells through the CXCR2 receptor (32), where it stimulates the production of some cytokines, such as IL-8 and IL-6 (32). IL-6 secreted by tumor cells interacts with MSCs and further enhances their CXCL7 expression, therefore generate a positive feedback loop. This cytokine loops between BCSCs and MSCs promote the self-renewal of BCSCs (31). Moreover, it has been shown that CXCL7 transfection increases the invasive ability of cancer cells (33), consistent with the previous findings that showed an increase in metastatic and invasive properties of CSCs (34). Furthermore, Sethi et al. indicated that IL-6–mediated Jagged1/Notch signaling induces breast cancer bone metastasis (35). These findings introduce IL-6 and its receptor as attractive therapeutic targets. It has been shown that CXCR1, a receptor for IL-8, and IL-8 can induce their self-renewal (36). In addition, it has been reported that the interaction of IL-8 with the CXCR1 (highly expressed on breast cancer stem cells), on CSCs increases their invasive and self-renewal properties (36, 37). Blocking the CXCR1 in mouse xenograft models significantly decreases the number of BCSCs, leading to reduced metastasis and tumorigenicity.
2.1.3 Crosstalk Between CSCs and MSCs Mediated by Exosomes
The crosstalk between stromal cells and CSCs are facilitated by cell-cell interaction and paracrine factors (2). In addition, cellular crosstalk has also been reported to be facilitated by the secretion of extracellular vesicles (EVs) that can transfer nucleic acids, lipids, and proteins and are able to induce epigenetic changes in target cells (38–40). This EV-mediated crosstalk is associated with chemoresistance, tumor development, and the capacity of evading from immune surveillance (41–44). Numerous studies have shown that cancer EVs are not limited only to the cancer microenvironment but there are also in body fluids such as blood circulation, emphasizing the idea that these vesicles can also influence the cells in other tissues (45–48). Peinado et al. indicated that exosomes secreted by melanoma can “educated” bone marrow progenitor cells to promote metastatic phenotype (49). Stem cells likely can alter the expression of genes in neighboring cells through exosomes containing microRNAs (miRNAs) (50).
It has been shown that stimulated MSCs with CSC-derived EVs considerably increase the migratory ability in response to cancer chemo-attractive stimuli. Indeed, CSC-EVs increase the expression of migration process-related genes. CXCR4, which is increased after CSC-EV stimulation, enhances the migratory capacity of MSCs toward the tumor site through an SDF-1 concentration gradient secreted by cancer cells (51–53). Stimulated MSCs also show an increased CXCR7 expression, an SDF-1 receptor associated with paracrine actions of MSCs (51, 54, 55). The ability of MSCs to modify extracellular matrix within the cancer microenvironment has been confirmed by increased expression of MMP1, 2, and 3 after stimulation with CSC-EVs. MMPs are proteolytic enzymes that are associated with metastasis processes, invasion, tumor growth, and angiogenesis (56). Tumor matrix remodeling activity of stimulated MSCs has also been confirmed by the increase of COL4A3 gene expression (51). This gene is involved in regulating cell adhesion, migration, and metastasis in various cancer types (57–59). EVs secreted from CSC rather than total cancer cell population show a central role in inducing pro-tumorigenic phenotype in MSCs (51).
Upon stimulation with CSC-EVs, the secretory profile of MSCs is changed and expression of IL-8, myeloperoxidase (MPO), and osteopontin (OPN) are increased (51). MPO has been shown to be involved in oxidative stress response and the anti-apoptotic process by converting nitric oxide (NO) into NO(+) that induces an S-nitrosylation of caspase-3, inhibiting its activity (60, 61). In clear cell renal cell carcinoma, OPN by stimulating of NF-κB and protecting cells from apoptosis induces tumor development (62). OPN also shows an autocrine function on MSCs by stimulating extracellular signal-regulated kinase (ERK) and focal adhesion kinase (FAK) signaling pathways via β1-integrin activation. This leads to the increase of MSCs motility and subsequently promotes the migration of MSC (63).
The anti-tumor activity of MSCs may depend on the type or even stage of cancer. Whereas naive MSCs may demonstrate an anti-cancer activity (64–66), indeed pre-conditioning of MSCs by cancer EVs may change their phenotype and function. Therefore, it is recommended that the secretion of cancer EVs be pharmacologically inhibited for preventing their unwanted effects before the administration of MSCs in cell-based cancer therapy approaches (67, 68). The phenotypic changes in MSCs mediated by CSC-derived EVs are maintained even after removal of stimulation, suggesting a persistent change in MSC phenotype (51).
2.1.4 CAFs Originated From MSCs and Cancer Stemness
Although the majority of tumor tissue cancer-associated fibroblasts (CAFs) may originate from resident stromal fibroblasts, but now many studies show that a significant percentage of CAFs in cancers are originated from BM-MSCs. For instance, MSCs in mouse models of gastric cancer, breast cancer, and PDAC, BM-MSCs are recruited to the cancer site where they differentiate into CAFs (69–71). It has been reported that in a gastric cancer model, about 20% of CAFs in tumor sites are derived from BM-MSCs which have been recruited into the tumor microenvironment in a TGF-β and CXCL-12 dependent manner (72).
The pro-stemness abilities of CAFs are one of the main mechanisms that increase oncogenesis. A specific subpopulation of CAFs has been identified that are proficient in secreting pro-stemness paracrine factors (73–79), thereby supporting the stemness properties and the self-renewal of tumor resident CSCs or increasing the transformation of tumor cells into CSCs. When exposed to cytotoxic stress such as chemotherapy, CAFs are further stimulated to produce pro-stemness cytokines or obtain a senescence-like secretory phenotype and secrete large amounts of pro-stemness chemokines that increase cancer stemness and aggressiveness following cancer treatment (80, 81). Therefore, blockade of the crosstalk of CSCs/cancer cells with pro-stemness CAFs and MSCs may introduce a new tool to improving the clinical outcome of solid tumors.
2.2 Cell Fusion and CSC-Like Phenotype
It has been shown that the biological phenomenon of cell fusion plays an important role in several pathological and physiological processes (82, 83). Cell fusion of stromal cells with tumor cells has been confirmed in human tumors and animal models (84–86). Studies have demonstrated that hybrid cells resulting from a spontaneous fusion between cancer cells and MSCs indicate metastatic, tumorigenic, and stem cell-like properties (87–89) and this phenomenon leads to nuclear reprogramming (90). In addition, the theory of cell fusion may clarify the origin of CSCs and the mechanism for cancer metastasis and carcinogenesis. He et al. (91) reported that cell fusion of gastric epithelial cells with MSCs promotes proliferation, migration, and invasion capabilities compared with the parental cells (91). The fusion of MSCs with HepG2 also promotes the malignant properties of in vivo metastasis models (92). In contrast, some findings demonstrate that the fusion of MSCs with esophageal carcinoma cells suppresses tumorigenicity (93). It has been also reported that the fusion of tumor cells with normal fibroblasts inhibits the tumorigenicity through cell cycle arrest effects (94). Similar results have been shown in cell fusion studies of stem cells and tumor cells (93, 95). The cell fusion hypothesis of the CSCs suggests that the fusion process may lead to the EMT of tumor cells and simultaneously promote the generation of CSCs (96). Zhang et al. demonstrated that fusion between MSCs and lung tumor cells may directly activate EMT of the hybrids, which promotes the invasion and migration properties (97). Meanwhile, the overexpression of stem cell surface markers (CD44 and CD133), and stem cell transcription factors (Kif4, Oct4, Sox2, Nanog, and Bmi1) in hybrids, show that the hybrids may obtain CSC properties after cell fusion (97). Although the hybrids show stem cell-like properties, further studies are required to determine whether the hybrids are the origin of CSCs.
2.3 Transformation of MSCs Into CSCs
The origin of cancer-initiating cells or CSCs has not been clearly determined. CSCs may be derived either from dedifferentiated mature cells or from transformed somatic stem cells (98–104). It has been shown that specific methylation of tumor suppressor genes, HIC1 and RasF1A, in MSCs can lead to the transformation of MSCs into CSCs. When MSCs transform into CSCs, they can increase drug resistance and allow tumor recurrence after treatment cessation (105). Indeed, abnormal DNA methylation of RassF1A and HIC1 is involved in the transformation of MSCs to cancer-like stem cells. Concurrent methylation of RassF1A and HIC1 has been reported in advanced ovarian cancer (106), and HIC1 demonstrates enhanced concordant hypermethylation with other genes in advanced myelodysplasia syndrome (107), suggesting that disruption of HIC1-associated networks may be critical for cancer initiation. Unlike RassF1A, which can be inactivated by either epigenetic or genetic mechanisms, suppression of the HIC1 gene is mainly caused by DNA methylation (108). Thus, specific methylation of HIC1 could predispose cells to tumor development. A subsequent epigenetic/genetic hit, such as RassF1A methylation, may then permit more efficient tumor progression. Concordant silencing of RassF1A and HIC1 may synergistically interrupt the p53 Pathway for apoptosis signaling and involve in the observed tumorigenic capability of MSCs. Teng et al. (105) showed that forced epigenetic silencing of RassF1A and HIC1 is adequate to induce malignant properties, including migration ability, enhanced colony formation, chemoresistance, loss of contact inhibition, and tumor formation in normal somatic stem cells (105). In addition, MSCs also reprogram toward CSCs, due to the aberrant changes of tumor microenvironments, which leads to the tumor development through the increased production of Oct4, Sox2, Nanog and the activation of Hedgehog, Wnt, Akt/mTOR, and NF-kB signaling pathways (31, 109–112). It has been reported that EWS-FLI-1 fusion protein modulates the expression of CSC signature proteins such as Oct4, Nanog, and Sox2 in MSCs that can reprogram these cells toward Ewing sarcoma CSCs (112). It has been also shown that MSCs cultured with cancer cell-soluble factors show a cancer stem cell-like state (113).
3 Cancer Treatment by Targeting CSCs, MSCs, and Their Crosstalk
MSCs help to establish a suitable tumor microenvironment for the restoration of CSCs and tumor progression, as well as crosstalk between cancer cells and MSCs in the microenvironment induces a CSC phenotype in cancer cells. Since this communication can contribute to drug resistance, metastasis, and tumor growth, it is suggested that blockade of the crosstalk between MSCs and CSCs/cancer cells can provide a new avenue to improving the cancer therapeutic tools. Here, we discussed various strategies in targeting the crosstalk between MSCs and CSCs/cancer cells (Tables 1, 2).

Table 1 Targeting approaches of crosstalk between MSCs and CSCs.
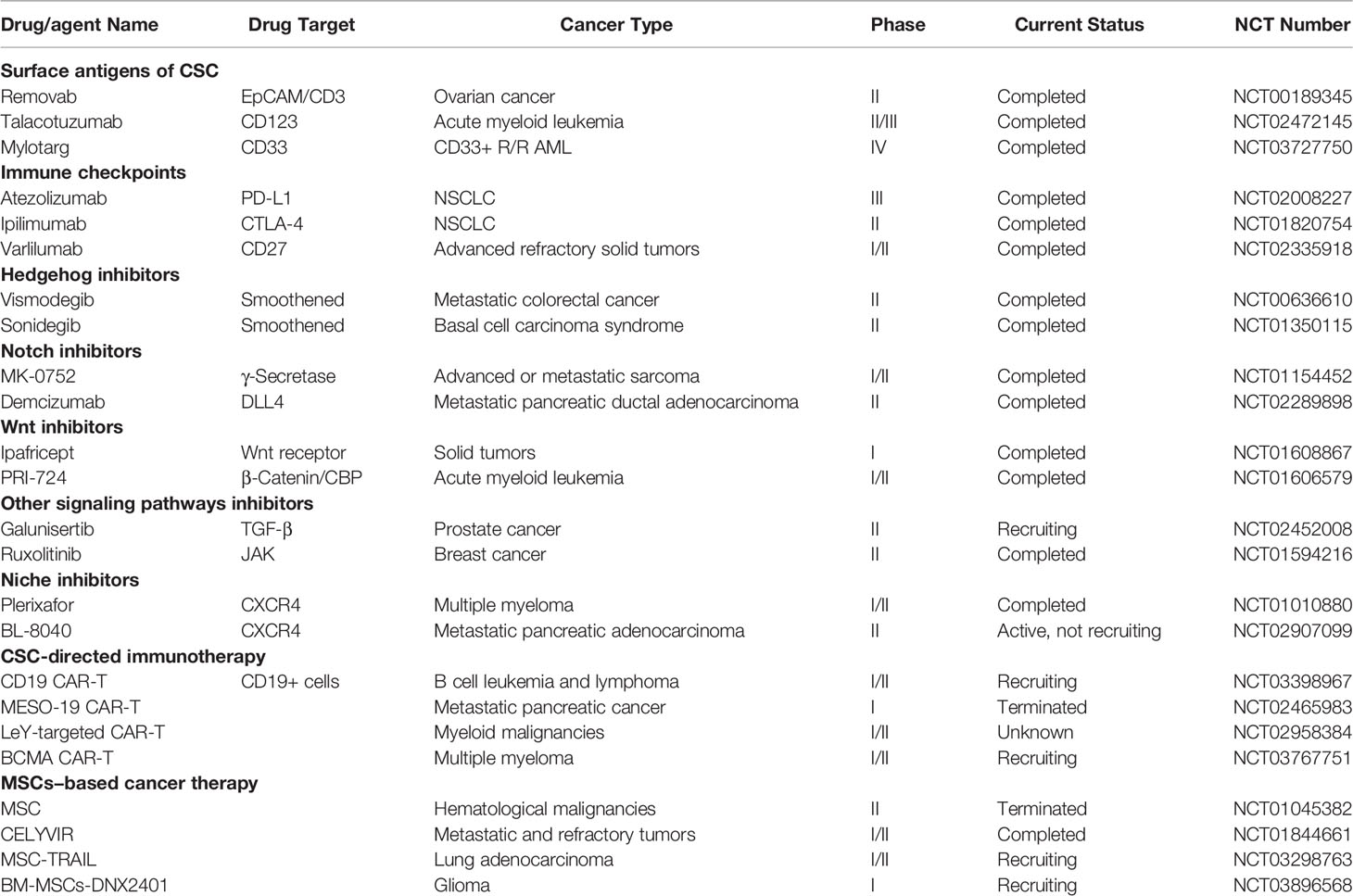
Table 2 Agents targeting CSC, MSC, and CSC-MSC crosstalk in clinical trials.
3.1 Chemotherapy-Educated MSCs and CSCs
Exposure of MSCs to cytotoxic agents resulted in a physiological response in these cells that eventually supports chemoresistance by enriching the CSC population. It has been reported that MSCs are recruited in large numbers to the tumor site in response to gemcitabine treatment. Gemcitabine-exposed MSCs, which are located near CSCs and support the CSC niche, significantly increase the secretion of CXCL10, which in turn induces CSCs proliferation as they overexpress CXCR3, the CXCL10 receptor. These events ultimately lead to increased tumor growth and drug resistance (114). It has been also shown that in a mouse xenograft model of PDAC, the number of BM-MSCs significantly increases following gemcitabine therapy in the tumor stroma (114). These gemcitabine-educated MSCs present a positive regulatory effect on cancer stem cells through the STAT-3: CXCL-10: CXCR-3 paracrine signaling axis. Similarly, MSCs secrete IL-8, IL-6, IL-7, IGF, and EGF, which promote chemoresistance following hyperthermia or paclitaxel therapy (127, 128). It has been also reported that cisplatin-exposed MSCs release specific polyunsaturated fatty acids which in turn increase the regrowth of cancers following treatment (129). In addition, exposure with cisplatin changes phosphorylation of several tyrosine kinases, including c-Jun, WNK-1, p53, and STAT3 in MSCs, and promotes MSC survival and secretion of IL-8 and IL-6. In turn, these events induce chemoresistance of cancer cells (130). However, the exact mechanisms by which MSCs promote chemoresistance have not been identified. Altogether, ample evidence emphasizes the central role of MSCs and CAFs in the expansion and maintenance of CSCs. Thus, targeting these cells may provide a new way to improve the clinical outcome of desmoplastic cancers.
3.2 Targeting the Crosstalk Between MSCs and CSCs
Due to the pro-tumorigenic activities of MSCs, a number of studies had been conducted to target MSCs as a therapeutic method in cancer (131). Because tumor-infiltrating MSCs can directly support cancer stem cells through several paracrine signaling pathways, including IL-7, IL-6, Jagged-1, PGE-2, CXCL-1, and CXCL-10 (4, 20, 114, 132), blockade of this paracrine communication between CSCs and MSCs may be potentially valuable in inhibiting tumor stemness in solid tumors. Indeed, a recent study has demonstrated the potential application of this approach (114). In a mouse model of PDAC, MSCs are located near CSCs, the CSC niche, following gemcitabine treatment. Gemcitabine-exposed MSCs release high levels of CXCL-10 that stimulate its receptor CXCR-3 on CSCs, triggering the STAT-3 signaling pathway and supporting the survival of CSCs (114). It has been reported that systemic injection of the CXCR3 antagonist (AMG487) with “nano-ghost (NG)”, MSC-derived membrane-based nanoparticles, leads to its accumulation in the CSC niche, thus decreasing the number of CSCs and enhancing the therapeutic efficacy of gemcitabine (114). The direct depletion of MSCs may be an alternative approach to preventing their communication with CSCs. However, the negative effects of the removal of MSCs on a person’s health remain an open question. Alternatively, it has been shown that low-dose metronomic (LDM) gemcitabine therapy regimen can decrease therapy-induced secretion of pro-stemness factors from CAFs in PDAC (81). Therefore, it is likely that LDM chemotherapy may also at least reduce chemotherapy-induced stimulation of MSCs and their production of pro-stemness chemokines such as CXCL-10. This possibility needs further investigation. On the other hand, since MSCs release the pro-stemness cytokine IL-6 (20), the various STAT-3 inhibitors and/or anti-IL-6 antibodies are exploited to blockade the CAF/MSC–CSC crosstalk (74, 115, 116, 133). It is anticipated that dual targeting of CAFs and MSCs may have a synergistic effect and maximize the anticancer efficacy in the treatment of desmoplastic cancers (121). Targeting approaches of crosstalk between MSCs and CSCs are summarized in Tables 1, 2.
3.3 Targeting Pro-Stemness CAFs and MSCs
Unlike direct targeting of CSCs, which poses substantial challenges such as dynamic and plastic properties of CSCs, targeting the MSCs or CAFs along with the pro-stemness niches they create can have several advantages in cancer therapy. First and foremost, there is ample evidence to show that CSCs are very plastic and heterogeneous and the transformation between different CSC populations plays a key role in cancer development and treatment response (122). For example, breast cancer CSCs can transition between epithelial-like states and mesenchymal-like (134–136). CSCs can also be originated from differentiated tumor cells through transdifferentiation or cellular reprogramming (73), which can be facilitated by cytotoxic agents such as ionizing radiation and chemotherapy (81, 137). It has been shown that eradicating LGR-5+ CSCs suppresses tumor growth, whereas the regrowth of cancer occurs following the removal of the cell death inducers due to the regeneration of CSCs from differentiated cancer cells (137, 138). Unlike CSCs, CAFs are both phenotypically and genetically stable; thus, CAF-targeted treatments may result in a more stable anti-CSC effect compared with direct targeting of CSCs. Second, identification of specific subpopulations of pro-stemness CAFs will facilitate CAF-targeted therapy, and they not only render new therapeutic targets, such as GPR-77 (123) but also provide more specific and safer therapies than the non-specific targeting of CAFs (139). In desmoplastic cancers such as pancreatic cancer, CAFs are present in large numbers in the stroma, which can account for more than 90% of the total cancer size (118, 119). Accordingly, MSC- or CAF-targeted therapies may be more effective than CSC-targeted therapy in desmoplastic cancers. Furthermore, CAFs are often localized to the margin of the glands or the cancer cell nests and close to blood vessels, therefore, drug delivery to these cells is easy (120). By contrast, CSCs are located farther away from blood vessels in desmoplastic cancers. In fact, CAFs per se is a major barrier for the delivery of nanoparticles and drugs to tumor cells (140, 141). Indeed, studies have been shown the importance of the spatial distribution of cells in the treatment of desmoplastic cancers (142). Collectively, in the treatment of desmoplastic cancers, targeting the communication between MSCs or CAFs with CSCs is more reasonable, possible, and clinically promising than the direct targeting of CSCs (121).
3.4 Specifically Targeting of CSCs by TRAIL-Expressing MSCs
By genetic engineering of MSCs, specific cancer cells can be targeted. For instance, it has been shown that TNF-related apoptosis-inducing ligand (TRAIL)-expressing MSCs specifically target tumor cells in lung carcinoma, therefore reducing chemoresistance, cancer aggressiveness, and relapse (124). TRAIL is a member of the TNF ligand family that can cause apoptosis through the interaction of its death receptors. TRAIL selectively initiates apoptosis of a variety of cancer cells and transformed cells, but not most normal cells, and therefore it has attracted great interest as a promising factor in cancer therapy (143). Several studies have shown the ability of TRAIL-expressing MSCs homing to the tumor site (144–146). TRAIL is a protein that causes apoptosis of tumor cells, without injuring the normal cells, by binding to specific TRAIL receptors and stimulation of the extrinsic apoptosis pathway (147). The activation of the NF-κB signaling pathway (148) and the overexpression of the TRAIL decoy receptors can contribute toward TRAIL resistance in normal cells (149). TRAIL-induced apoptosis has been shown in CD133-positive glioma cells (125). However, Capper et al. showed that CD133-positive neuro-sphere-forming glioma cells were completely resistant to TRAIL (150). It has been reported that TRAIL-expressing MSCs can target both stem-like, side population (SP) cells, and non-SP cells, and in combination with traditional chemotherapies show a synergistic effect in apoptosis induction (151). It has been also shown that physiological levels of TRAIL in MSC-EV was not effective in inducing apoptosis in NSCLC cells (152). High expression of death receptor 4 (DR4) and DR5 were observed in liver and lung cancer-derived CSCs, representing their contribution to CSCs TRAIL sensitivity (153, 154). Activation of both intrinsic and extrinsic apoptosis pathways through extracellular stimulation by TRAIL may induce further effects, especially for chemo-resistant CSCs that show resistance to intrinsic apoptosis pathways (155).
3.5 Exosome-Based Cancer Therapy
Numerous miRNAs are differentially expressed in CSCs, which can be used as potential targets in the treatment of cancer (156). It has been recently shown that upregulation of miR-150 and miR-142-3p in BCSCs compared to non-tumorigenic tumor cells can be related to clonogenicity and tumorigenicity of BCSCs (157). Thus, using complementary miR-142 inhibitors in BCSCs could reduce tumor growth. Exosomes derived from MSCs can act as an extracellular messenger to introduce exogenous LNA-antimiR-142-3p to breast cancer stem-like cells to inhibit miR-142-3p and decrease the tumorigenicity, proliferation, and colony formation ability of the cancer stem-like cells (126). MSCs are one of the main sources of exosomes that are especially considered in clinical applications. Indeed, the biological activity of MSCs-derived exosomes is likely akin to the effects mediated by MSCs themselves. Thus, unlike MSCs, the exosomes can be exploited as cell-free carriers, which do not have a risk of tumorigenesis (158). Targeting the CSCs by using the exosomes can introduce a novel tool for destroying CSCs in anti-cancer therapies (126).
3.6 CSCs Targeting by Immunotherapy
Recently, immunotherapy has gained great attention in cancer treatment (159). Many studies have used immunotherapeutic approaches to target cancer stem cells. Immune checkpoint inhibitors, antibody-based and adoptive cell therapy approaches are used for CSCs targeting (Table 2). CAR-T cell therapy, as an adoptive cell therapy method, is used for CSC-directed immunotherapy by targeting CD20 (NCT03398967), CD123 (NCT02937103), CD19 (NCT03398967) positive cells. Various immune checkpoint blocking agents, such as CTLA-4 inhibitors (Ipilimumab that is approved by the FDA) (1) and PD1/PD-L1 inhibitors: atezolizumab (2), avelumab (3), cemiplimab (4), and nivolumab (5) are undergoing clinical trials. Targeting surface antigens of CSC, such as EpCAM/CD3 (NCT00189345), CD123 (NCT02472145), CD33 (NCT03727750) are other strategies in CSC-directed immunotherapy.
3.7 The Clinical Challenges in MSC-Based Therapies
Various factors affect the clinical outcome of MSCs-based therapies. One of the influencing factors is variables related to the preparation of the MSC product. Donor variations such as genetics, age, health status, gender can affect the potency of MSCs (160). In addition, MSCs tissue of origin (161), isolation methods (162), the culture conditions (163), cryopreservation, and thaw/culture rescue protocols (164, 165) causing additional variations in potency of MSCs. The administration of MSCs is another variable that can affect the residence time, viability, and homing of MSCs. This variable includes the following: the inoculation site (dense/non-dense tissue), administration route (local/systemic), injection/infusion buffer, injection device features (needle size/geometry), and cell carrier materials (166, 167). MSC recipients are the third important variable that can affect the therapeutic outcome. Which can refer to the following: host cytotoxic responses against MSCs (168), and the host disease/severity which can lead to highly variable microenvironmental factors such as hypoxia, inflammation status, and ECM that influence the function of MSCs (169).
4 Conclusions and Perspective
MSCs can modify the stroma, and helping to establish a tissue microenvironment that favors the restoration of CSCs and tumor progression, as well as crosstalk between cancer cells and MSCs in the microenvironment, promotes a CSC phenotype in cancer cells. Since crosstalk between CSCs and MSCs promote drug resistance, mediates metastasis, and induces tumor growth by inducing CSC phenotype in cancer cells and restoration of CSCs, it is suggested that blockade of the crosstalk of MSCs with CSCs can provide a new avenue to improving the cancer therapeutic tools. Indeed, targeting the communication between MSCs with CSCs is more reasonable, possible, and clinically promising than the direct targeting of CSCs in the treatment of desmoplastic cancers.
Author Contributions
Conception and design: ZH, JT, and YJ. Collection and assembly of data: YJ, WL, and LZ. Manuscript writing: YJ, WL, LZ, and ZH. Made critical revisions: WL, ZH, and JT. All authors reviewed and approved of the final manuscript.
Funding
This study was supported by the Public Welfare Application Plan Project of Shaoxing (2018C30109); Project of Shaoxing Medical Key Discipline Construction Plan (2019SZD06); Health and Family Planning Commission of Zhejiang province (2018KY831). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Frank NY, Schatton T, Frank MH. The Therapeutic Promise of the Cancer Stem Cell Concept. J Clin Invest (2010) 120:41–50. doi: 10.1172/JCI41004
2. Ye J, Wu D, Wu P, Chen Z, Huang J. The Cancer Stemcell Niche: Cross Talk Between Cancer Stemcells and Their Microenvironment. Tumor Biol (2014) 35:3945–51. doi: 10.1007/s13277-013-1561-x
3. Bhowmick NA, Moses HL. Tumor-Stroma Interactions. Curr Opin Genet Dev (2005) 15:97–101. doi: 10.1016/j.gde.2004.12.003
4. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, et al. Mesenchymal Stem Cells Regulate Epithelial-Mesenchymal Transition and Tumor Progression of Pancreatic Cancer Cells. Cancer Sci (2013) 104:157–64. doi: 10.1111/cas.12059
5. Shibue T, Weinberg RA. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat Rev Clin Oncol (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
6. Jing Y, Han Z, Liu Y, Sun K, Zhang S, Jiang G, et al. Mesenchymal Stem Cells in Inflammation Microenvironment Accelerates Hepatocellular Carcinoma Metastasis by Inducing Epithelial-Mesenchymal Transition. PloS One (2012) 7:e43272. doi: 10.1371/journal.pone.0043272
7. Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, et al. Infiltrating Bone Marrow Mesenchymal Stem Cells Increase Prostate Cancer Stem Cell Population and Metastatic Ability via Secreting Cytokines to Suppress Androgen Receptor Signaling. Oncogene (2014) 33:2768–78. doi: 10.1038/onc.2013.233
8. Hossain A, Shinojima N, Gumin J, Feng G, Lang FF. Tumor-Associated Mesenchymal Stromal Cells Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the Il6/Stat3 Pathway. Neuro Oncol (2011) 13:19. doi: 10.1093/neuonc/nor148
9. Kong BH, Shin H, Kim SH, Mok HS, Shim JK, Lee JH, et al. Increased In Vivo Angiogenic Effect of Glioma Stromal Mesenchymal Stem-Like Cells on Glioma Cancer Stem Cells From Patients With Glioblastoma. Int J Oncol (2013) 42:1754–62. doi: 10.3892/ijo.2013.1856
10. McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, et al. Human Ovarian Carcinoma-Associated Mesenchymal Stem Cells Regulate Cancer Stem Cells and Tumorigenesis via Altered BMP Production. J Clin Invest (2011) 121:3206–19. doi: 10.1172/JCI45273
11. Vulcano F, Milazzo L, Ciccarelli C, Eramo A, Sette G, Mauro A, et al. Wharton’s Jelly Mesenchymal Stromal Cells Have Contrasting Effects on Proliferation and Phenotype of Cancer Stem Cells From Different Subtypes of Lung Cancer. Exp Cell Res (2016) 345:190–8. doi: 10.1016/j.yexcr.2016.06.003
12. Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, et al. Potential Role of Mesenchymal Stem Cells (MSCs) in the Breast Tumour Microenvironment: Stimulation of Epithelial to Mesenchymal Transition (EMT). Breast Cancer Res Treat (2010) 124:317–26. doi: 10.1007/s10549-010-0734-1
13. Xue Z, Wu X, Chen X, Liu Y, Wang X, Wu K, et al. Mesenchymal Stem Cells Promote Epithelial to Mesenchymal Transition and Metastasis in Gastric Cancer Though Paracrine Cues and Close Physical Contact. J Cell Biochem (2015) 116:618–27. doi: 10.1002/jcb.25013
14. Houghton JM, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, et al. Gastric Cancer Originating From Bone Marrow-Derived Cells. Science (2004) 306:1568–71. doi: 10.1126/science.1099513
15. Nishimura K, Semba S, Aoyagi K, Sasaki H, Yokozaki H. Mesenchymal Stem Cells Provide an Advantageous Tumor Microenvironment for the Restoration of Cancer Stem Cells. Pathobiology (2012) 79:290–306. doi: 10.1159/000337296
16. Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, et al. The Wnt5A/protein Kinase C Pathway Mediates Motility in Melanoma Cells via the Inhibition of Metastasis Suppressors and Initiation of an Epithelial to Mesenchymal Transition. J Biol Chem (2007) 282:17259–71. doi: 10.1074/jbc.M700075200
17. Vincent T, Neve EPA, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 Transcriptional Repressor Complex Promotes TGF-β Mediated Epithelial-Mesenchymal Transition. Nat Cell Biol (2009) 11:943–50. doi: 10.1038/ncb1905
18. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells With Properties of Stem Cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
19. Alcolea S, Antón R, Camacho M, Soler M, Alfranca A, Avilés-Jurado FX, et al. Interaction Between Head and Neck Squamous Cell Carcinoma Cells and Fibroblasts in the Biosynthesis of PGE 2. J Lipid Res (2012) 53:630–42. doi: 10.1194/jlr.M019695
20. Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-Stimulated Mesenchymal Stem Cells Create a Carcinoma Stem Cell Niche via Prostaglandin E2 Signaling. Cancer Discov (2012) 2:840–55. doi: 10.1158/2159-8290.CD-12-0101
21. Wang D, Dubois RN. Eicosanoids and Cancer. Nat Rev Cancer (2010) 10:181–93. doi: 10.1038/nrc2809
22. Rudnick JA, Arendt LM, Klebba I, Hinds JW, Iyer V, Gupta PB, et al. Functional Heterogeneity of Breast Fibroblasts Is Defined by a Prostaglandin Secretory Phenotype That Promotes Expansion of Cancer-Stem Like Cells. PloS One (2011) 6:e24605. doi: 10.1371/journal.pone.0024605
23. Ishimoto T, Oshima H, Oshima M, Kai K, Torii R, Masuko T, et al. CD44+ Slow-Cycling Tumor Cell Expansion Is Triggered by Cooperative Actions of Wnt and Prostaglandin E2 in Gastric Tumorigenesis. Cancer Sci (2010) 101:673–8. doi: 10.1111/j.1349-7006.2009.01430.x
24. Dinarello CA. Why Not Treat Human Cancer With Interleukin-1 Blockade? Cancer Metastasis Rev (2010) 29:317–29. doi: 10.1007/s10555-010-9229-0
25. Ravishankaran P, Karunanithi R. Clinical Significance of Preoperative Serum Interleukin-6 and C-Reactive Protein Level in Breast Cancer Patients. World J Surg Oncol (2011) 9:18. doi: 10.1186/1477-7819-9-18
26. Zhang G-J, Adachi I. Serum Interleukin-6 Levels Correlate to Tumor Progression and Prognosis in Metastatic Breast Carcinoma. Anticancer Res (1999) 19:1427–32.
27. Iliopoulos D, Hirsch HA, Struhl K. An Epigenetic Switch Involving NF-κb, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell (2009) 139:693–706. doi: 10.1016/j.cell.2009.10.014
28. Gunn WG, Conley A, Deininger L, Olson SD, Prockop DJ, Gregory CA. A Crosstalk Between Myeloma Cells and Marrow Stromal Cells Stimulates Production of DKK1 and Interleukin-6: A Potential Role in the Development of Lytic Bone Disease and Tumor Progression in Multiple Myeloma. Stem Cells (2006) 24:986–91. doi: 10.1634/stemcells.2005-0220
29. Arnulf B, Lecourt S, Soulier J, Ternaux B, Lacassagne MN, Crinquette A, et al. Phenotypic and Functional Characterization of Bone Marrow Mesenchymal Stem Cells Derived From Patients With Multiple Myeloma. Leukemia (2007) 21:158–63. doi: 10.1038/sj.leu.2404466
30. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PloS One (2009) 4:e4992. doi: 10.1371/journal.pone.0004992
31. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast Cancer Stem Cells are Regulated by Mesenchymal Stem Cells Through Cytokine Networks. Cancer Res (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
32. Kalwitz G, Endres M, Neumann K, Skriner K, Ringe J, Sezer O, et al. Gene Expression Profile of Adult Human Bone Marrow-Derived Mesenchymal Stem Cells Stimulated by the Chemokine CXCL7. Int J Biochem Cell Biol (2009) 41:649–58. doi: 10.1016/j.biocel.2008.07.011
33. Tang Z, Yu M, Miller F, Berk RS, Tromp G, Kosir MA. Increased Invasion Through Basement Membrane by CXCL7-Transfected Breast Cells. Am J Surg (2008) 196:690–6. doi: 10.1016/j.amjsurg.2008.08.001
34. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24-Breast Cancer Cells Exhibit Enhanced Invase Properties: An Early Step Necessary for Metastasis. Breast Cancer Res (2006) 8:R59. doi: 10.1186/bcr1610
35. Sethi N, Dai X, Winter CG, Kang Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell (2011) 19:192–205. doi: 10.1016/j.ccr.2010.12.022
36. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 Blockade Selectively Targets Human Breast Cancer Stem Cells In Vitro and in Xenografts. J Clin Invest (2010) 120:485–97. doi: 10.1172/JCI39397
37. Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast Cancer Cell Lines Contain Functional Cancer Stem Sells With Metastatic Capacity and a Distinct Molecular Signature. Cancer Res (2009) 69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741
38. Camussi G, C. Deregibus M, Tetta C. Tumor-Derived Microvesicles and the Cancer Microenvironment. Curr Mol Med (2012) 13:58–67. doi: 10.2174/156652413804486304
39. Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-Derived Microvesicles: Important and Underappreciated Mediators of Cell-to-Cell Communication. Leukemia (2006) 20:1487–95. doi: 10.1038/sj.leu.2404296
40. Vafaei S, Roudi R, Madjd Z, Aref AR, Ebrahimi M. Potential Theranostics of Circulating Tumor Cells and Tumor-Derived Exosomes Application in Colorectal Cancer. Cancer Cell Int (2020) 20:1–15. doi: 10.1186/s12935-020-01389-3
41. Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, et al. Microvesicles Released From Human Renal Cancer Stem Cells Stimulate Angiogenesis and Formation of Lung Premetastatic Niche. Cancer Res (2011) 71:5346–56. doi: 10.1158/0008-5472.CAN-11-0241
42. Al-Nedawi K, Meehan B, Rak J. Microvesicles: Messengers and Mediators of Tumor Progression. Cell Cycle (2009) 8:2014–8. doi: 10.4161/cc.8.13.8988
43. Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as Mediators of Intercellular Communication in Cancer–the Emerging Science of Cellular “Debris”. Semin Immunopathol (2011) 33:455–67. doi: 10.1007/s00281-011-0250-3
44. Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM, et al. Membrane Microvesicles as Actors in the Establishment of a Favorable Prostatic Tumoral Niche: A Role for Activated Fibroblasts and CX3CL1-CX3CR1 Axis. Cancer Res (2009) 69:785–93. doi: 10.1158/0008-5472.CAN-08-1946
45. Muturi HT, Dreesen JD, Nilewski E, Jastrow H, Giebel B, Ergun S, et al. Tumor and Endothelial Cell-Derived Microvesicles Carry Distinct CEACAMs and Influence T-Cell Behavior. PloS One (2013) 8:e74654. doi: 10.1371/journal.pone.0074654
46. Taylor DD, Gercel-Taylor C. MicroRNA Signatures of Tumor-Derived Exosomes as Diagnostic Biomarkers of Ovarian Cancer. Gynecol Oncol (2008) 110:13–21. doi: 10.1016/j.ygyno.2008.04.033
47. Salido-Guadarrama I, Romero-Cordoba S, Peralta-Zaragoza O, Hidalgo-Miranda A, Rodríguez-Dorantes M. MicroRNAs Transported by Exosomes in Body Fluids as Mediators of Intercellular Communication in Cancer. Onco Targets Ther (2014) 7:1327–38. doi: 10.2147/OTT.S61562
48. Li J, Sherman-Baust CA, Tsai-Turton M, Bristow RE, Roden RB, Morin PJ. Claudin-Containing Exosomes in the Peripheral Circulation of Women With Ovarian Cancer. BMC Cancer (2009) 9:244. doi: 10.1186/1471-2407-9-244
49. Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells Toward a Pro-Metastatic Phenotype Through MET. Nat Med (2012) 18:883–91. doi: 10.1038/nm.2753
50. Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB, et al. Transfer of microRNAs by Embryonic Stem Cell Microvesicles. PloS One (2009) 4:e4722. doi: 10.1371/journal.pone.0004722
51. Lindoso RS, Collino F, Camussi G. Extracellular Vesicles Derived From Renal Cancer Stem Cells Induce a Pro-Tumorigenic Phenotype in Mesenchymal Stromal Cells. Oncotarget (2015) 6:7959–69. doi: 10.18632/oncotarget.3503
52. Park SA, Ryu CH, Kim SM, Lim JY, Park SI, Jeong CH, et al. CXCR4-Transfected Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Exhibit Enhanced Migratory Capacity Toward Gliomas. Int J Oncol (2011) 38:97–103. doi: 10.3892/ijo-00000828
53. Gao H, Priebe W, Glod J, Banerjee D. Activation of Signal Transducers and Activators of Transcription 3 and Focal Adhesion Kinase by Stromal Cell-Derived Factor 1 Is Required for Migration of Human Mesenchymal Stem Cells in Response to Tumor Cell-Conditioned Medium. Stem Cells (2009) 27:857–65. doi: 10.1002/stem.23
54. Liu H, Liu S, Li Y, Wang X, Xue W, Ge G, et al. The Role of SDF-1-CXCR4/CXCR7 Axis in the Therapeutic Effects of Hypoxia-Preconditioned Mesenchymal Stem Cells for Renal Ischemia/Reperfusion Injury. PloS One (2012) 7:e34608. doi: 10.1371/journal.pone.0034608
55. Balabanian K, Lagane B, Infantino S, Chow KYC, Harriague J, Moepps B, et al. The Chemokine SDF-1/CXCL12 Binds to and Signals Through the Orphan Receptor RDC1 in T Lymphocytes. J Biol Chem (2005) 280:35760–6. doi: 10.1074/jbc.M508234200
56. Egeblad M, Werb Z. New Functions for the Matrix Metalloproteinases in Cancer Progression. Nat Rev Cancer (2002) 2:161–74. doi: 10.1038/nrc745
57. Nie XC, Wang JP, Zhu W, Xu XY, Xing YN, Yu M, et al. COL4A3 Expression Correlates With Pathogenesis, Pathologic Behaviors, and Prognosis of Gastric Carcinomas. Hum Pathol (2013) 44:77–86. doi: 10.1016/j.humpath.2011.10.028
58. Ando H, Aihara R, Ohno T, Ogata K, Mochiki E, Kuwano H. Prognostic Significance of the Expression of MUC1 and Collagen Type IV in Advanced Gastric Carcinoma. Br J Surg (2009) 96:901–9. doi: 10.1002/bjs.6635
59. Smrkolj Š, Eržen M, Rakar S. Prognostic Significance of Topoisomerase II Alpha and Collagen IV Immunoexpression in Cervical Cancer. Eur J Gynaecol Oncol (2010) 31:380–5.
60. Ortiz De Montellano PR. Catalytic Sites of Hemoprotein Peroxidases. Annu Rev Pharmacol Toxicol (1992) 32:89–107. doi: 10.1146/annurev.pa.32.040192.000513
61. Saed GM, Ali-Fehmi R, Jiang ZL, Fletcher NM, Diamond MP, Abu-Soud HM, et al. Myeloperoxidase Serves as a Redox Switch That Regulates Apoptosis in Epithelial Ovarian Cancer. Gynecol Oncol (2010) 116:276–81. doi: 10.1016/j.ygyno.2009.11.004
62. Matušan-Ilijaš K, Damante G, Fabbro D, Dordević G, Hadžisejdić I, Grahovac M, et al. Osteopontin Expression Correlates With Nuclear Factor-κb Activation and Apoptosis Downregulation in Clear Cell Renal Cell Carcinoma. Pathol Res Pract (2011) 207:104–10. doi: 10.1016/j.prp.2010.11.004
63. Zou C, Luo Q, Qin J, Shi Y, Yang L, Ju B, et al. Osteopontin Promotes Mesenchymal Stem Cell Migration and Lessens Cell Stiffness via Integrin β1, FAK, and ERK Pathways. Cell Biochem Biophys (2013) 65:455–62. doi: 10.1007/s12013-012-9449-8
64. Bruno S, Collino F, Iavello A, Camussi G. Effects of Mesenchymal Stromal Cell-Derived Extracellular Vesicles on Tumor Growth. Front Immunol (2014) 5:382. doi: 10.3389/fimmu.2014.00382
65. Bruno S, Collino F, Deregibus MC, Grange C, Tetta C, Camussi G. Microvesicles Derived From Human Bone Marrow Mesenchymal Stem Cells Inhibit Tumor Growth. Stem Cells Dev (2013) 22:758–71. doi: 10.1089/scd.2012.0304
66. Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, et al. Human Mesenchymal Stem Cells Exert Potent Antitumorigenic Effects in a Model of Kaposi’s Sarcoma. J Exp Med (2006) 203:1235–47. doi: 10.1084/jem.20051921
67. Wysoczynski M, Ratajczak MZ. Lung Cancer Secreted Microvesicles: Underappreciated Modulators of Microenvironment in Expanding Tumors. Int J Cancer (2009) 125:1595–603. doi: 10.1002/ijc.24479
68. Del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue-Factor-Bearing Microvesicles Arise From Lipid Rafts and Fuse With Activated Platelets to Initiate Coagulation. Blood (2005) 106:1604–11. doi: 10.1182/blood-2004-03-1095
69. Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. Stromal Fibroblasts Activated by Tumor Cells Promote Angiogenesis in Mouse Gastric Cancer. J Biol Chem (2008) 283:19864–71. doi: 10.1074/jbc.M800798200
70. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-Associated Fibroblast-Like Differentiation of Human Mesenchymal Stem Cells. Cancer Res (2008) 68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943
71. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J, et al. VEGF Expression by Mesenchymal Stem Cells Contributes to Angiogenesis in Pancreatic Carcinoma. Br J Cancer (2008) 99:622–31. doi: 10.1038/sj.bjc.6604508
72. Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, Takashi S, et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
73. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible Formation of Breast Cancer Stem Cells and Their Dynamic Equilibrium With Non-Stem Cancer Cells via IL6 Secretion. Proc Natl Acad Sci USA (2011) 108:1397–402. doi: 10.1073/pnas.1018898108
74. Korkaya H, Kim G, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol Cell (2012) 47:570–84. doi: 10.1016/j.molcel.2012.06.014
75. Korkaya H, Liu S, Wicha MS. Breast Cancer Stem Cells, Cytokine Networks, and the Tumor Microenvironment. J Clin Invest (2011) 121:3804–9. doi: 10.1172/JCI57099
76. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-Associated Fibroblasts Regulate the Plasticity of Lung Cancer Stemness via Paracrine Signalling. Nat Commun (2014) 5:1–17. doi: 10.1038/ncomms4472
77. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 Mediates Cross-Talk Between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem Cells. Cancer Res (2012) 72:2768–79. doi: 10.1158/0008-5472.CAN-11-3567
78. Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell (2011) 9:433–46. doi: 10.1016/j.stem.2011.10.001
79. Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-Mediated Paracrine Interaction for Pancreatic Cancer Therapy and Monitoring. Nature (2019) 569:131–5. doi: 10.1038/s41586-019-1130-6
80. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy Activates Cancer-Associated Fibroblasts to Maintain Colorectal Cancer-Initiating Cells by IL-17a. J Exp Med (2013) 210:2851–72. doi: 10.1084/jem.20131195
81. Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, et al. Metronomic Chemotherapy Prevents Therapy-Induced Stromal Activation and Induction of Tumor-Initiating Cells. J Exp Med (2016) 213:2967–88. doi: 10.1084/jem.20151665
82. Noubissi FK, Ogle BM. Cancer Cell Fusion: Mechanisms Slowly Unravel. Int J Mol Sci (2016) 17:1587. doi: 10.3390/ijms17091587
83. Ogle BM, Cascalho M, Platt JL. Biological Implications of Cell Fusion. Nat Rev Mol Cell Biol (2005) 6:567–75. doi: 10.1038/nrm1678
84. Li GC, Ye QH, Dong QZ, Ren N, Jia HL, Qin LX. Mesenchymal Stem Cells Seldomly Fuse With Hepatocellular Carcinoma Cells and Are Mainly Distributed in the Tumor Stroma in Mouse Models. Oncol Rep (2013) 29:713–9. doi: 10.3892/or.2012.2174
85. Pawelek JM. Cancer Cell Fusion With Migratory Bone Marrow-Derived Cells as an Explanation for Metastasis: New Therapeutic Paradigms. Futur Oncol (2008) 4:449–52. doi: 10.2217/14796694.4.4.449
86. Pawelek JM, Chakraborty AK. Fusion of Tumour Cells With Bone Marrow-Derived Cells: A Unifying Explanation for Metastasis. Nat Rev Cancer (2008) 8:377–86. doi: 10.1038/nrc2371
87. Rappa G, Mercapide J, Lorico A. Spontaneous Formation of Tumorigenic Hybrids Between Breast Cancer and Multipotent Stromal Cells is a Source of Tumor Heterogeneity. Am J Pathol (2012) 180:2504–15. doi: 10.1016/j.ajpath.2012.02.020
88. Dittmar T, Schwitalla S, Seidel J, Haverkampf S, Reith G, Meyer-Staeckling S, et al. Characterization of Hybrid Cells Derived From Spontaneous Fusion Events Between Breast Epithelial Cells Exhibiting Stem-Like Characteristics and Breast Cancer Cells. Clin Exp Metastasis (2011) 28:75–90. doi: 10.1007/s10585-010-9359-3
89. Xu MH, Gao X, Luo D, Zhou XD, Xiong W, Liu GX. EMT and Acquisition of Stem Cell-Like Properties Are Involved in Spontaneous Formation of Tumorigenic Hybrids Between Lung Cancer and Bone Marrow-Derived Mesenchymal Stem Cells. PloS One (2014) 9:e87893. doi: 10.1371/journal.pone.0087893
90. Powell AE, Anderson EC, Davies PS, Silk AD, Pelz C, Impey S, et al. Fusion Between Intestinal Epithelial Cells and Macrophages in a Cancer Context Results in Nuclear Reprogramming. Cancer Res (2011) 71:1497–505. doi: 10.1158/0008-5472.CAN-10-3223
91. He X, Li B, Shao Y, Zhao N, Hsu Y, Zhang Z, et al. Cell Fusion Between Gastric Epithelial Cells and Mesenchymal Stem Cells Results in Epithelial-to-Mesenchymal Transition and Malignant Transformation. BMC Cancer (2015) 15:1–11. doi: 10.1186/s12885-015-1027-1
92. Li H, Feng Z, Tsang TC, Tang T, Jia X, He X, et al. Fusion of HepG2 Cells With Mesenchymal Stem Cells Increases Cancer-Associated and Malignant Properties: An In Vivo Metastasis Model. Oncol Rep (2014) 32:539–47. doi: 10.3892/or.2014.3264
93. Wang Y, Fan H, Zhou B, Ju Z, Yu L, Guo L, et al. Fusion of Human Umbilical Cord Mesenchymal Stem Cells With Esophageal Carcinoma Cells Inhibits the Tumorigenicity of Esophageal Carcinoma Cells. Int J Oncol (2012) 40:370–7. doi: 10.3892/ijo.2011.1232
94. Harris H. The Analysis of Malignancy by Cell Fusion: The Position in 1988. Cancer Res (1986) 48:3302–6.
95. Schichor C, Albrecht V, Korte B, Buchner A, Riesenberg R, Mysliwietz J, et al. Mesenchymal Stem Cells and Glioma Cells Form a Structural as Well as a Functional Syncytium In Vitro. Exp Neurol (2012) 234:208–19. doi: 10.1016/j.expneurol.2011.12.033
96. Lu X, Kang Y. Cell Fusion Hypothesis of the Cancer Stem Cell. Adv Exp Med Biol (2011) 950:129–40. doi: 10.1007/978-94-007-0782-5_6
97. Zhang LN, Kong CF, Zhao D, Cong XL, Wang SS, Ma L, et al. Fusion With Mesenchymal Stem Cells Differentially Affects Tumorigenic and Metastatic Abilities of Lung Cancer Cells. J Cell Physiol (2019) 234:3570–82. doi: 10.1002/jcp.27011
98. Balch C, Nephew KP, Huang THM, Bapat SA. Epigenetic “Bivalently Marked” Process of Cancer Stem Cell-Driven Tumorigenesis. BioEssays (2007) 29:842–5. doi: 10.1002/bies.20619
99. Feinberg AP. Phenotypic Plasticity and the Epigenetics of Human Disease. Nature (2007) 447:433–40. doi: 10.1038/nature05919
100. Jones PA, Baylin SB. The Epigenomics of Cancer. Cell (2007) 128:683–92. doi: 10.1016/j.cell.2007.01.029
101. Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJA. The Origin of the Cancer Stem Cell: Current Controversies and New Insights. Nat Rev Cancer (2005) 5:899–904. doi: 10.1038/nrc1740
102. Gupta PB, Chaffer CL, Weinberg RA. Cancer Stem Cells: Mirage or Reality? Nat Med (2009) 15:1010–2. doi: 10.1038/nm0909-1010
103. Pardal R, Clarke MF, Morrison SJ. Applying the Principles of Stem-Cell Biology to Cancer. Nat Rev Cancer (2003) 3:895–902. doi: 10.1038/nrc1232
104. Wicha MS, Liu S, Dontu G. Cancer Stem Cells: An Old Idea - A Paradigm Shift. Cancer Res (2006) 66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153
105. Teng IW, Hou PC, Lee KD, Chu PY, Yeh KT, Jin VX, et al. Targeted Methylation of Two Tumor Suppressor Genes Is Sufficient to Transform Mesenchymal Stem Cells Into Cancer Stem/Initiating Cells. Cancer Res (2011) 71:4653–63. doi: 10.1158/0008-5472.CAN-10-3418
106. Teodoridis JM, Hall J, Marsh S, Kannall HD, Smyth C, Curto J, et al. CpG Island Methylation of DNA Damage Response Genes in Advanced Ovarian Cancer. Cancer Res (2005) 65:8961–7. doi: 10.1158/0008-5472.CAN-05-1187
107. Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter Hypermethylation of P15ink4b, HIC1, CDH1, and ER Is Frequent in Myelodysplastic Syndrome and Predicts Poor Prognosis in Early-Stage Patients. Eur J Haematol (2006) 76:23–32. doi: 10.1111/j.1600-0609.2005.00559.x
108. Chen WY, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, et al. Epigenetic and Genetic Loss of Hic1 Function Accentuates the Role of P53 in Tumorigenesis. Cancer Cell (2004) 6:387–98. doi: 10.1016/j.ccr.2004.08.030
109. Bao Q, Zhao Y, Niess H, Conrad C, Schwarz B, Jauch KW, et al. Mesenchymal Stem Cell-Based Tumor-Targeted Gene Therapy in Gastrointestinal Cancer. Stem Cells Dev (2012) 21:2355–63. doi: 10.1089/scd.2012.0060
110. Gargett CE, Masuda H. Adult Stem Cells in the Endometrium. Mol Hum Reprod (2010) 16:818–34. doi: 10.1093/molehr/gaq061
111. Torsvik A, Bjerkvig R. Mesenchymal Stem Cell Signaling in Cancer Progression. Cancer Treat Rev (2013) 39:180–8. doi: 10.1016/j.ctrv.2012.03.005
112. Riggi N, Suvà ML, De Vito C, Provero P, Stehle JC, Baumer K, et al. EWS-FLI-1 Modulates Mirna145 and SOX2 Expression to Initiate Mesenchymal Stem Cell Reprogramming Toward Ewing Sarcoma Cancer Stem Cells. Genes Dev (2010) 24:916–32. doi: 10.1101/gad.1899710
113. El-Badawy A, Ghoneim MA, Gabr MM, Salah RA, Mohamed IK, Amer M, et al. Cancer Cell-Soluble Factors Reprogram Mesenchymal Stromal Cells to Slow Cycling, Chemoresistant Cells With a More Stem-Like State. Stem Cell Res Ther (2017) 8:1–20. doi: 10.1186/s13287-017-0709-9
114. Timaner M, Letko-Khait N, Kotsofruk R, Benguigui M, Beyar-Katz O, Rachman-Tzemah C, et al. Therapy-Educated Mesenchymal Stem Cells Enrich for Tumor-Initiating Cells. Cancer Res (2018) 78:1253–65. doi: 10.1158/0008-5472.CAN-17-1547
115. Zhong H, Davis A, Ouzounova M, Carrasco RA, Chen C, Breen S, et al. A Novel IL6 Antibody Sensitizes Multiple Tumor Types to Chemotherapy Including Trastuzumab-Resistant Tumors. Cancer Res (2016) 76:480–90. doi: 10.1158/0008-5472.CAN-15-0883
116. Njatcha C, Farooqui M, Kornberg A, Johnson DE, Grandis JR, Siegfried JM. STAT3 Cyclic Decoy Demonstrates Robust Antitumor Effects in Non–Small Cell Lung Cancer. Mol Cancer Ther (2018) 17:1917–26. doi: 10.1158/1535-7163.MCT-17-1194
117. London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, et al. Calcium-Dependent Arrhythmias in Transgenic Mice With Heart Failure. Am J Physiol Heart Circ Physiol (2003) 284:H431–41. doi: 10.1152/ajpheart.00431.2002
118. Yen TWF, Aardal NP, Bronner MP, Thorning DR, Savard CE, Lee SP, et al. Myofibroblasts Are Responsible for the Desmoplastic Reaction Surrounding Human Pancreatic Carcinomas. Surgery (2002) 131:129–34. doi: 10.1067/msy.2002.119192
119. Waghray M, Yalamanchili M, Magliano MPd, Simeone DM. Deciphering the Role of Stroma in Pancreatic Cancer. Curr Opin Gastroenterol (2013) 29:537–43. doi: 10.1097/MOG.0b013e328363affe
120. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science (80- ) (2009) 324:1457–61. doi: 10.1126/science.1171362
121. Chan TS, Shaked Y, Tsai KK. Targeting the Interplay Between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front Oncol (2019) 9:688. doi: 10.3389/fonc.2019.00688
122. Polyak K, Weinberg RA. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat Rev Cancer (2009) 9:265–73. doi: 10.1038/nrc2620
123. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell (2018) 172:841–856.e16. doi: 10.1016/j.cell.2018.01.009
124. Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal Stem Cell Delivery of TRAIL can Eliminate Metastatic Cancer. Cancer Res (2009) 69:4134–42. doi: 10.1158/0008-5472.CAN-08-4698
125. Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, Van De Water JAJM, Mohapatra G, et al. Assessment of Therapeutic Efficacy and Fate of Engineered Human Mesenchymal Stem Cells for Cancer Therapy. Proc Natl Acad Sci USA (2009) 106:4822–7. doi: 10.1073/pnas.0806647106
126. Naseri Z, Oskuee RK, forouzandeh-moghadam M, Jaafari MR. Delivery of LNA-antimiR-142-3p by Mesenchymal Stem Cells-Derived Exosomes to Breast Cancer Stem Cells Reduces Tumorigenicity. Stem Cell Rev Rep (2020) 16:541–56. doi: 10.1007/s12015-019-09944-w
127. Scherzed A, Hackenberg S, Froelich K, Kessler M, Koehler C, Hagen R, et al. BMSC Enhance the Survival of Paclitaxel Treated Squamous Cell Carcinoma Cells In Vitro. Cancer Biol Ther (2011) 11:349–57. doi: 10.4161/cbt.11.3.14179
128. Lis R, Touboul C, Mirshahi P, Ali F, Mathew S, Nolan DJ, et al. Tumor Associated Mesenchymal Stem Cells Protects Ovarian Cancer Cells From Hyperthermia Through CXCL12. Int J Cancer (2011) 128:715–25. doi: 10.1002/ijc.25619
129. Roodhart JML, Daenen LGM, Stigter ECA, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal Stem Cells Induce Resistance to Chemotherapy Through the Release of Platinum-Induced Fatty Acids. Cancer Cell (2011) 20:370–83. doi: 10.1016/j.ccr.2011.08.010
130. Skolekova S, Matuskova M, Bohac M, Toro L, Demkova L, Gursky J, et al. Cisplatin-Induced Mesenchymal Stromal Cells-Mediated Mechanism Contributing to Decreased Antitumor Effect in Breast Cancer Cells. Cell Commun Signal (2016) 14:4. doi: 10.1186/s12964-016-0127-0
131. Poggi A, Varesano S, Zocchi MR. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front Immunol (2018) 9:262. doi: 10.3389/fimmu.2018.00262
132. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing CD44. Nat Med (2011) 17:211–6. doi: 10.1038/nm.2284
133. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of Cancer Relapse and Metastasis by Inhibiting Cancer Stemness. Proc Natl Acad Sci USA (2015) 112:1839–44. doi: 10.1073/pnas.1424171112
134. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast Cancer Stem Cells Transition Between Epithelial and Mesenchymal States Reflective of Their Normal Counterparts. Stem Cell Rep (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
135. Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-β-Id1 Signaling Opposes Twist1 and Promotes Metastatic Colonization via a Mesenchymal-to-Epithelial Transition. Cell Rep (2013) 5:1228–42. doi: 10.1016/j.celrep.2013.11.014
136. Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP, et al. Heterogeneity of Human Breast Stem and Progenitor Cells as Revealed by Transcriptional Profiling. Stem Cell Rep (2018) 10:1596–609. doi: 10.1016/j.stemcr.2018.03.001
137. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and Targeting of LGR5 + Human Colon Cancer Stem Cells. Nature (2017) 545:187–92. doi: 10.1038/nature22081
138. De Sousa E Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A Distinct Role for Lgr5 + Stem Cells in Primary and Metastatic Colon Cancer. Nature (2017) 543:676–80. doi: 10.1038/nature21713
139. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer With Reduced Survival. Cancer Cell (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
140. Miao L, Liu Q, Lin CM, Luo C, Wang Y, Liu L, et al. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res (2017) 77:719–31. doi: 10.1158/0008-5472.CAN-16-0866
141. Miao L, Lin CM, Huang L. Stromal Barriers and Strategies for the Delivery of Nanomedicine to Desmoplastic Tumors. J Control Release (2015) 219:192–204. doi: 10.1016/j.jconrel.2015.08.017
142. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine Plus Nab-Paclitaxel is an Active Regimen in Patients With Advanced Pancreatic Cancer: A Phase I/II Trial. J Clin Oncol (2011) 29:4548–54. doi: 10.1200/JCO.2011.36.5742
143. Wang S, El-Deiry WS. TRAIL and Apoptosis Induction by TNF-Family Death Receptors. Oncogene (2003) 22:8628–33. doi: 10.1038/sj.onc.1207232
144. Fakiruddin KS, Ghazalli N, Lim MN, Zakaria Z, Abdullah S. Mesenchymal Stem Cell Expressing TRAIL as Targeted Therapy Against Sensitised Tumour. Int J Mol Sci (2018) 19:2188. doi: 10.3390/ijms19082188
145. Cafforio P, Viggiano L, Mannavola F, Pellè E, Caporusso C, Maiorano E, et al. PIL6-TRAIL-Engineered Umbilical Cord Mesenchymal/Stromal Stem Cells are Highly Cytotoxic for Myeloma Cells Both In Vitro and In Vivo. Stem Cell Res Ther (2017) 8:1–13. doi: 10.1186/s13287-017-0655-6
146. Han J, Hwang HS, Na K. TRAIL-Secreting Human Mesenchymal Stem Cells Engineered by a Non-Viral Vector and Photochemical Internalization for Pancreatic Cancer Gene Therapy. Biomaterials (2018) 182:259–68. doi: 10.1016/j.biomaterials.2018.08.024
147. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and Characterization of a New Member of the TNF Family That Induces Apoptosis. Immunity (1995) 3:673–82. doi: 10.1016/1074-7613(95)90057-8
148. Franco AV, Zhang XD, Van Berkel E, Sanders JE, Zhang XY, Thomas WD, et al. The Role of NF-κb in TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Apoptosis of Melanoma Cells. J Immunol (2001) 166:5337–45. doi: 10.4049/jimmunol.166.9.5337
149. Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science (80- ) (1997) 277:818–21. doi: 10.1126/science.277.5327.818
150. Capper D, Gaiser T, Hartmann C, Habel A, Mueller W, Herold-Mende C, et al. Stem-Cell-Like Glioma Cells are Resistant to TRAIL/Apo2L and Exhibit Down-Regulation of Caspase-8 by Promoter Methylation. Acta Neuropathol (2009) 117:445–56. doi: 10.1007/s00401-009-0494-3
151. Loebinger MR, Sage EK, Davies D, Janes SM. TRAIL-Expressing Mesenchymal Stem Cells Kill the Putative Cancer Stem Cell Population. Br J Cancer (2010) 103:1692–7. doi: 10.1038/sj.bjc.6605952
152. Fakiruddin KS, Lim MN, Nordin N, Rosli R, Zakaria Z, Abdullah S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers (Basel) (2019) 11:1261. doi: 10.3390/cancers11091261
153. Lee SH, Hyun SK, Kim HB, Kang CD, Kim SH. Potential Role of CD133 Expression in the Susceptibility of Human Liver Cancer Stem-Like Cells to TRAIL. Oncol Res (2016) 24:495–509. doi: 10.3727/096504016X14685034103950
154. Signore M, Ricci-Vitiani L, De Maria R. Targeting Apoptosis Pathways in Cancer Stem Cells. Cancer Lett (2013) 332:374–82. doi: 10.1016/j.canlet.2011.01.013
155. Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O’Flaherty JD, et al. Generation and Characterisation of Cisplatin-Resistant Non-Small Cell Lung Cancer Cell Lines Displaying a Stem-Like Signature. PloS One (2013) 8:e54193. doi: 10.1371/journal.pone.0054193
156. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c Links Breast Cancer Stem Cells With Normal Stem Cells. Cell (2009) 138:592–603. doi: 10.1016/j.cell.2009.07.011
157. Isobe T, Hisamori S, Hogan DJ, Zabala M, Hendrickson DG, Dalerba P, et al. miR-142 Regulates the Tumorigenicity of Human Breast Cancer Stem Cells Through the Canonical WNT Signaling Pathway. Elife (2014) 3:e01977. doi: 10.7554/eLife.01977
158. Sundaram B, Herbert FJ, Kumar S. Human Mesenchymal Stem Cell (hMSC) - Derived Exosomes/Exosome Mimetics as a Potential Novel Therapeutic Tool for Regenerative Medicine. Springer, Singapore: Regenerative Medicine: Laboratory to Clinic (2017). p. 81–97. doi: 10.1007/978-981-10-3701-6_5
159. Mohsenzadegan M, Peng R, Roudi R. Dendritic Cell/Cytokine-Induced Killer Cell-Based Immunotherapy in Lung Cancer: What We Know and Future Landscape. J Cell Physiol (2020) 235:74–86. doi: 10.1002/jcp.28977
160. Hass R, Kasper C, Böhm S, Jacobs R. Different Populations and Sources of Human Mesenchymal Stem Cells (MSC): A Comparison of Adult and Neonatal Tissue-Derived MSC. Cell Commun Signal (2011) 9:1–14. doi: 10.1186/1478-811X-9-12
161. Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, et al. Immunoregulatory Mechanisms of Mesenchymal Stem and Stromal Cells in Inflammatory Diseases. Nat Rev Nephrol (2018) 14:493–507. doi: 10.1038/s41581-018-0023-5
162. Juneja SC, Viswanathan S, Ganguly M, Veillette C. A Simplified Method for the Aspiration of Bone Marrow From Patients Undergoing Hip and Knee Joint Replacement for Isolating Mesenchymal Stem Cells and In Vitro Chondrogenesis. Bone Marrow Res (2016) 2016:1–18. doi: 10.1155/2016/3152065
163. Yin JQ, Zhu J, Ankrum JA. Manufacturing of Primed Mesenchymal Stromal Cells for Therapy. Nat BioMed Eng (2019) 3:90–104. doi: 10.1038/s41551-018-0325-8
164. Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, et al. Mesenchymal Stem (Stromal) Cells for Treatment of ARDS: A Phase 1 Clinical Trial. Lancet Respir Med (2015) 3:24–32. doi: 10.1016/S2213-2600(14)70291-7
165. François M, Copland IB, Yuan S, Romieu-Mourez R, Waller EK, Galipeau J. Cryopreserved Mesenchymal Stromal Cells Display Impaired Immunosuppressive Properties as a Result of Heat-Shock Response and Impaired Interferon-γ Licensing. Cytotherapy (2012) 14:147–52. doi: 10.3109/14653249.2011.623691
166. Antunes MA, Abreu SC, Cruz FF, Teixeira AC, Lopes-Pacheco M, Bandeira E, et al. Effects of Different Mesenchymal Stromal Cell Sources and Delivery Routes in Experimental Emphysema. Respir Res (2014) 15:1–14. doi: 10.1186/s12931-014-0118-x
167. O’Cearbhaill ED, Ng KS, Karp JM. Emerging Medical Devices for Minimally Invasive Cell Therapy. Mayo Clin Proc (2014) 89:259–73. doi: 10.1016/j.mayocp.2013.10.020
168. Wang Y, Chen X, Cao W, Shi Y. Plasticity of Mesenchymal Stem Cells in Immunomodulation: Pathological and Therapeutic Implications. Nat Immunol (2014) 15:1009–16. doi: 10.1038/ni.3002
Keywords: mesenchymal stem cells, cancer stem cells, cytokines, exosomes, tumor microenvironment
Citation: Jing Y, Liang W, Zhang L, Tang J and Huang Z (2022) The Role of Mesenchymal Stem Cells in the Induction of Cancer-Stem Cell Phenotype. Front. Oncol. 12:817971. doi: 10.3389/fonc.2022.817971
Received: 18 November 2021; Accepted: 19 January 2022;
Published: 17 February 2022.
Edited by:
Hamid Morjani, Université de Reims Champagne-Ardenne, FranceReviewed by:
Raheleh Roudi, University of Minnesota, United StatesAntonella Zannetti, Institute of Biostructure and Bioimaging (CNR), Italy
Alexander Pedroza-Gonzalez, National Autonomous University of Mexico, Mexico
Copyright © 2022 Jing, Liang, Zhang, Tang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongliang Huang, em9uZ2xpYW5naHVhbmdAdG9uZ2ppLmVkdS5jbg==; Junjun Tang , c2h0amo0MDBAc2luYS5jb20=