 Ran Huang
Ran Huang Qiong Dai2
Qiong Dai2 Johannes Haybaeck
Johannes Haybaeck Zhihui Yang
Zhihui Yang- 1Department of Pathology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 2Department of Human Anatomy, School of Basic Medical Sciences, Southwest Medical University, Luzhou, China
- 3Department of Cardiology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 4Institute of Pathology, Neuropathology and Molecular Pathology, Medical University of Innsbruck, Innsbruck, Austria
- 5Diagnostic & Research Center for Molecular BioMedicine, Institute of Pathology, Medical University of Graz, Graz, Austria
Esophageal squamous cell carcinoma (ESCC) is a malignant tumor developing from the esophageal squamous epithelium, and is the most common histological subtype of esophageal cancer (EC). EC ranks 10th in morbidity and sixth in mortality worldwide. The morbidity and mortality rates in China are both higher than the world average. Current treatments of ESCC are surgical treatment, radiotherapy, and chemotherapy. Neoadjuvant chemoradiotherapy plus surgical resection is recommended for advanced patients. However, it does not work in the significant promotion of overall survival (OS) after such therapy. Research on targeted therapy in ESCC mainly focus on EGFR and PD-1, but neither of the targeted drugs can significantly improve the 3-year and 5-year survival rates of disease. Phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway is an important survival pathway in tumor cells, associated with its aggressive growth and malignant progression. Specifically, proliferation, apoptosis, autophagy, and so on. Related genetic alterations of this pathway have been investigated in ESCC, such as PI3K, AKT and mTOR-rpS6K. Therefore, the PI3K/AKT/mTOR pathway seems to have the capability to serve as research hotspot in the future. Currently, various inhibitors are being tested in cells, animals, and clinical trials, which targeting at different parts of this pathway. In this work, we reviewed the research progress on the PI3K/AKT/mTOR pathway how to influence biological behaviors in ESCC, and discussed the interaction between signals downstream of this pathway, especially eukaryotic translation initiation factors (eIFs) and the development and progression of ESCC, to provide reference for the identification of new therapeutic targets in ESCC.
Introduction
The General Status of Esophageal Carcinoma
In 2020, the incidence and mortality of esophageal carcinoma (EC) ranked tenth and sixth in the world, with about 70% of the cases affection males. According to statistics, in 2020, 544,076 people died due to EC worldwide1]. The incidence in eastern Asia is the highest in the world (1). EC mainly includes ESCC and esophageal adenocarcinoma (EAC). The incidence of ESCC is higher than EAC in Asia (2). Currently, the incidence of ESCC has decreased significantly in Asia (e.g., in China), probably due to the decline in poverty (3), but the mortality rate is still not optimistic.
With the improvement of various examination methods, the detection rate of esophageal squamous dysplasia is increasing, especially in the areas with high incidence of ESCC. Esophageal squamous dysplasia has the potential to develop into ESCC. The two can be regarded as a continuous pathological process (4). The symptoms of EC are very insidious. Early EC is often detected by gastroscopy, CT, or MRI (5). These examinations may reveal ulceration or protuberance on the mucosal esophageal surface, or thickening of the esophageal wall (5). As the lesion progresses, patients might have difficulty swallowing. They can only take half liquid diet or even liquid diet, until they are completely unable to eat and thus significantly lose weight. Some patients even feel chest pain (6).
According to the degree of differentiation of ESCC, three grades can be differentiated: highly, moderately, and poorly (7) (Figures 1A–C). Many keratins and intercellular bridges can be seen in a highly differentiated ESCC. The poorly differentiated ESCC does not have keratin pearls and intercellular bridges. These cells are disorderly arranged hierarchy, with higher cellular atypia and nuclear pleomorphism.
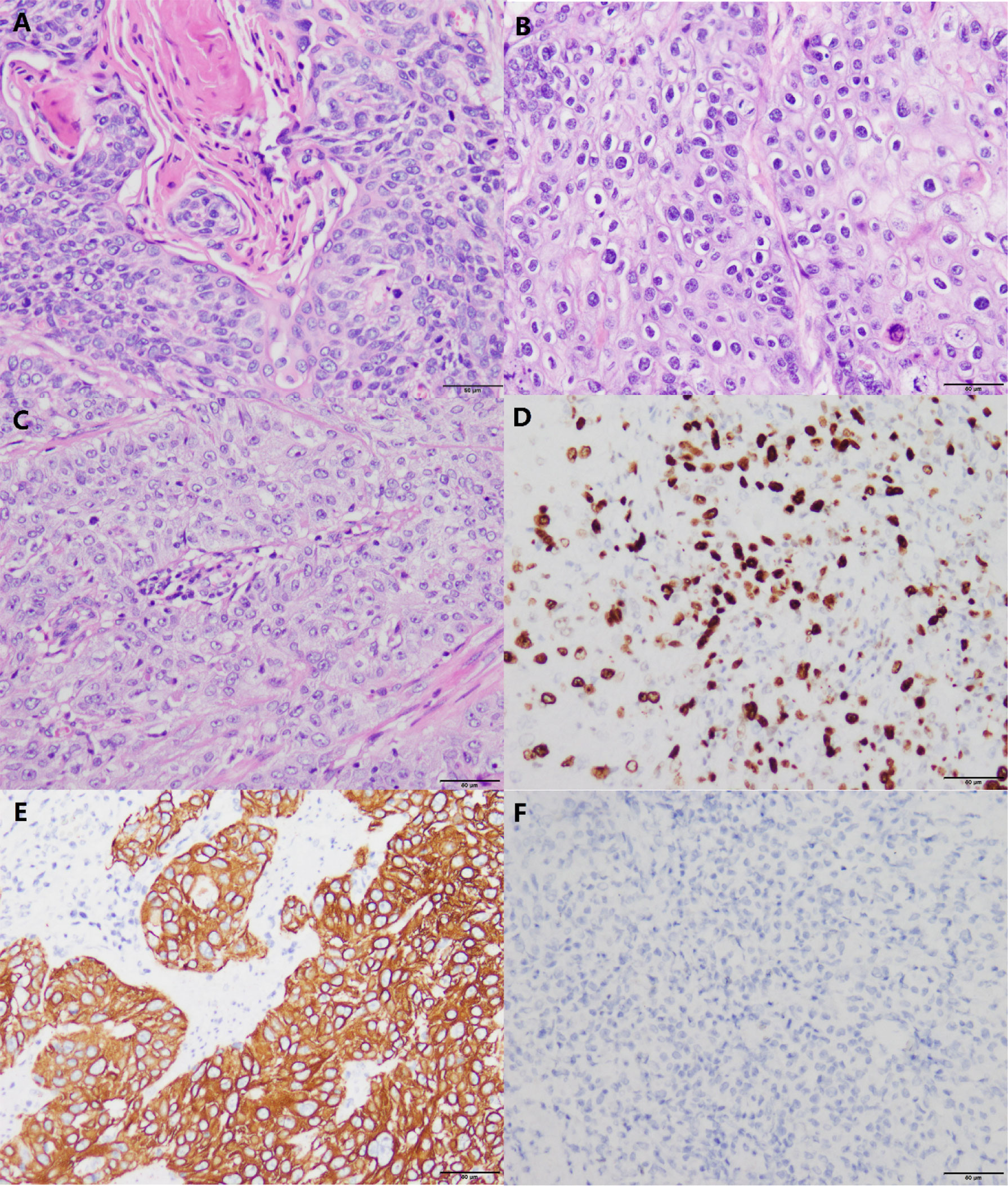
Figure 1 Histomorphology and immunohistochemistry in variably differentiated ESCC (own images, have not been published in elsewhere, the scale bar is 50 µm). (A) Highly differentiated ESCC (H&E stain, ×200). (B) Moderately differentiated ESCC (H&E stain, ×200). (C) Poorly differentiated ESCC (H&E stain, × 200). (D) In this case of poorly differentiated ESCC, the positive index of Ki-67 reaches 50% (Envision stain, ×200). (E) Poorly differentiated ESCC cells are positive for CK5/6 (Envision stain, ×200). (F) Poorly differentiated ESCC cells are negative for CK7 (Envision stain, ×200).
Poorly differentiated ESCC and EAC are difficult to distinguish by HE staining alone (7). Immunohistochemistry (IHC) can distinguish them. In poorly differentiated ESCC, markers indicating squamous epithelial differentiation (e.g., CK5/6) (Figures 1D, E) are positive and markers indicating glandular epithelial differentiation are negative (e.g., CK7, CK20) (7) (Figure 1F). P63 is an indicator that has been shown to be positively expressed in ESCC (8). Some patients can have components of both squamous cell carcinoma and adenocarcinoma, which is termed adenosquamous carcinoma (9).
The Methods and Efficacy of Current Treatments for ESCC
Most ESCC patients are diagnosed at advanced stage. Thus, more attention should be paid to prevention and early diagnosis. If this disease is diagnosed early, endoscopic therapy is possible. This can save the patient’s organs and improve the well-being (10, 11). Some scholars suggest that endoscopic ultrasound (EUS) should be performed before treatment to accurately evaluate the condition (it is not recommended for some patients with extreme esophageal stenosis) and guide the therapy. T staging and regional lymph node status are important prognostic factors for ESCC. EUS is a test comparable to PET, accurate for T staging and inexpensive. More importantly, EUS is superior to CT and MRI in the detection of lymph node involvement, with higher sensitivity (10).
In a randomized controlled trial conducted by Joel Shapiro and colleagues, the addition of neoadjuvant chemoradiotherapy (preoperative chemoradiotherapy) during surgery for patients with resectable EC was found to have an overall survival benefit (12). It was confirmed during the follow-up for a long term. In a phase III clinical trial conducted by Chinese scholars, it was also found that neoadjuvant chemoradiotherapy plus surgery (NCT01216527) improved the survival rate of locally advanced ESCC patients compared with surgery alone, with acceptable and controllable adverse events (13).
Currently, neoadjuvant chemoradiotherapy and esophagectomy are the mainly therapy for ESCC. In a guide to the management of EC, it is suggested that patients with locally advanced ESCC should receive neoadjuvant chemoradiotherapy (14). Multimodal therapy has advantages over performing surgical resection alone. Similarly, patients without metastatic disease should receive esophageal resection after neoadjuvant therapy, if the evaluation of surgery is safe (14).
Japanese scholar Masayuki Watanabe and colleagues found that patients with ESCC received neoadjuvant chemoradiotherapy plus surgical resection, the 3-year survival rate was 29.8% and the 5-year survival rate was 15.0% (15). This data indicates that there is still a large proportion of patients who do not achieve better outcomes. Researchers are still exploring other ways to treat ESCC.
Currently, many researchers are focusing on the treatment of ESCC by molecular targeting. EGFR and PD1 are the hottest targets. It has been demonstrated that EGFR is one of the cancer genes responsible for the common somatic copy number variations (SCNV) in ESCC (16). At the same time, some researchers found abnormally high expression of EGFR in ESCC (17). In a phase II, single-group, multicenter trial conducted in several Chinese hospitals, researchers found that icotinib (an EGFR tyrosine kinase inhibitor, TKI, NCT01855854) exhibited promising activity in advanced ESCC patients whose EGFR was overexpressed or amplified (18). These trial results showed that only a small number of patients respond well to icotinib. So, it can be speculated that ESCC patients may have developed resistance to EGFR-targeted therapy (18). Curtis R Chong and colleagues proposed that the resisting to EGFR-targeted therapy in tumor cells could be relevant to the abnormally activating of PI3K/AKT/mTOR pathway (19).
In the phase III AIO/EORTC clinical trial conducted by M. Moehler and colleagues, they found that the use of panitumumab (an anti-EGFR antibody) combined with cisplatin and 5-fluorouracil did not improve survival compared to unselected advanced ESCC patients who received 5-fluorouracil alone (20). This result supports further studies of serum and tumor biomarkers (20).
Programmed cell death protein 1 (PD-1) is an inhibitory receptor expressed on activated lymphocytes, can connect with the ligands of PD-L1 and PD-L2. And they are favorable to regulating the balance of T cell activation, immune tolerance, and immune-mediated tissue damage (21, 22). Blockading the immune checkpoint has fundamentally improved the treatment of melanoma patients (23). At the same time, many researchers are exploring its efficacy in other cancers (24). A monoclonal antibody targeting PD-1, Nivolumab, could increase tumor antigen-specific T cell proliferation and cytokine secretion in vitro (25, 26). It has been approved for the treatment of many other diseases, for example, advanced non-small cell lung cancer and Hodgkin’s lymphoma (27, 28). Nivolumab has been approved for ESCC patients who have progressed after chemotherapy in Japan since February 2020 (29). Toshihiro Kudo and colleagues found that Nivolumab is safe and effective in advanced EC patients who are refractory to standard chemotherapy (28). Jiyun Lee and colleagues found that Nivolumab showed some efficacy as second-line therapy for ESCC in a phase III trial, but the improvement of OS was not significant (29). Through analysis, Qu and colleagues found that overexpression of PD-L1 in ESCC might relate to short OS. However, the difference was not statistical significant (P=0.07) (30). Some scholars have also found that the overexpression of PD-L1 in ESCC is related to its disease-free survival (DFS), but it has no correlation with its prognosis (31).
Therefore, the current targeted therapies for EGFR and PD-1 respectively have encountered bottlenecks. The solution is to dig deeper into the molecular mechanisms of ESCC and find other sensitive targets. In the future, combination therapy with multiple molecular-specific targeted drugs may be a good option for the treatment of ESCC.
Environmental Factors and Probably Genetic Mechanisms of ESCC Development
The causes of ESCC are complex. Environmental and genetic factors are contributors for ESCC formation. It is believed that pathogenic genes have an important influence on it.
Previous studies have found that ESCC has a variety of related environmental predisposing factors (32). Such as tobacco (33, 34), alcoholic beverages (35, 36), little or no-intake vegetables and fruits (37, 38), pickled vegetables (39), hot foods (40) and so on. If the body is exposed to these factors for a long time, it may increase the susceptibility of ESCC. Some studies found a genetic link with these exposures for developing ESCC. Chen Wu and colleagues found that there was a gene-environment interaction between alcohol abuse and genetic variation in alcohol metabolic pathways that leads to the development of ESCC. It has been reported that drinkers who carried both the risk alleles of ADH1B and ALDH2 had the highest risk to develop cancer (41).
In ESCC, Yongmei Song and colleagues found genomic alterations in several important pathways (e.g., the RTK-RAS and AKT pathways) and genes (e.g., PIK3CA) (42).
De-Chen Lin and colleagues found that the MAPK and PI3K pathways were activated through a variety of mechanisms in ESCC (16). At the same time, several potentially altered genes have been identified in ESCC. Such as ERBB, HDAC, PI3K family, XPO1, FGFR1, TP53, JAK-STAT3, and mTOR-rpS6K were defined to be recurrent candidate druggable targets (16). These genes have implications for future molecular studies.
The results of a whole-exome sequencing conducted by Genta Sawada and colleagues are consistent with those of De-Chen Lin and colleagues. Their sequencing included 144 Japanese patients with ESCC, including neoplastic and non-neoplastic esophageal tissues (43). In addition, they also found some other gene mutations in many tumor tissues (43). For example, some mutations in genes that regulate cell cycle (TP53, CCND1, CDKN2A, and FBXW7) and epigenetic process (MLL2, EP300, CREBBP, and TET2). It should be noted that TP53 plays a role in inhibiting carcinogenesis in organisms (44). The most common genetic change in a variety of human cancers is the mutation of TP53 (45). According to reports, the mutation rate of TP53 in ESCC ranges from 60% to 93.1% (16, 43). Many researchers have found that TP53 is closely related to PI3K/AKT/mTOR pathway, which may be involved in the occurrence and development of tumors (Figure 2). Such as, it has been found that TP53 can negatively regulate PI3K/AKT/mTOR pathway by upregulating related proteins (45). Besides, it was also found that PI3K/AKT/mTOR pathway could negatively affect the expression of TP53 by upregulating MDM2, which promotes the degradation of p53 (A protein encoded by TP53) (44). Mutations were also found in some key genes of signaling pathways, such as NOTCH, WNT (FAT1, YAP1, and AJUBA) and RTK-PI3K (PIK3CA, EGFR, and ERBB2) (43).
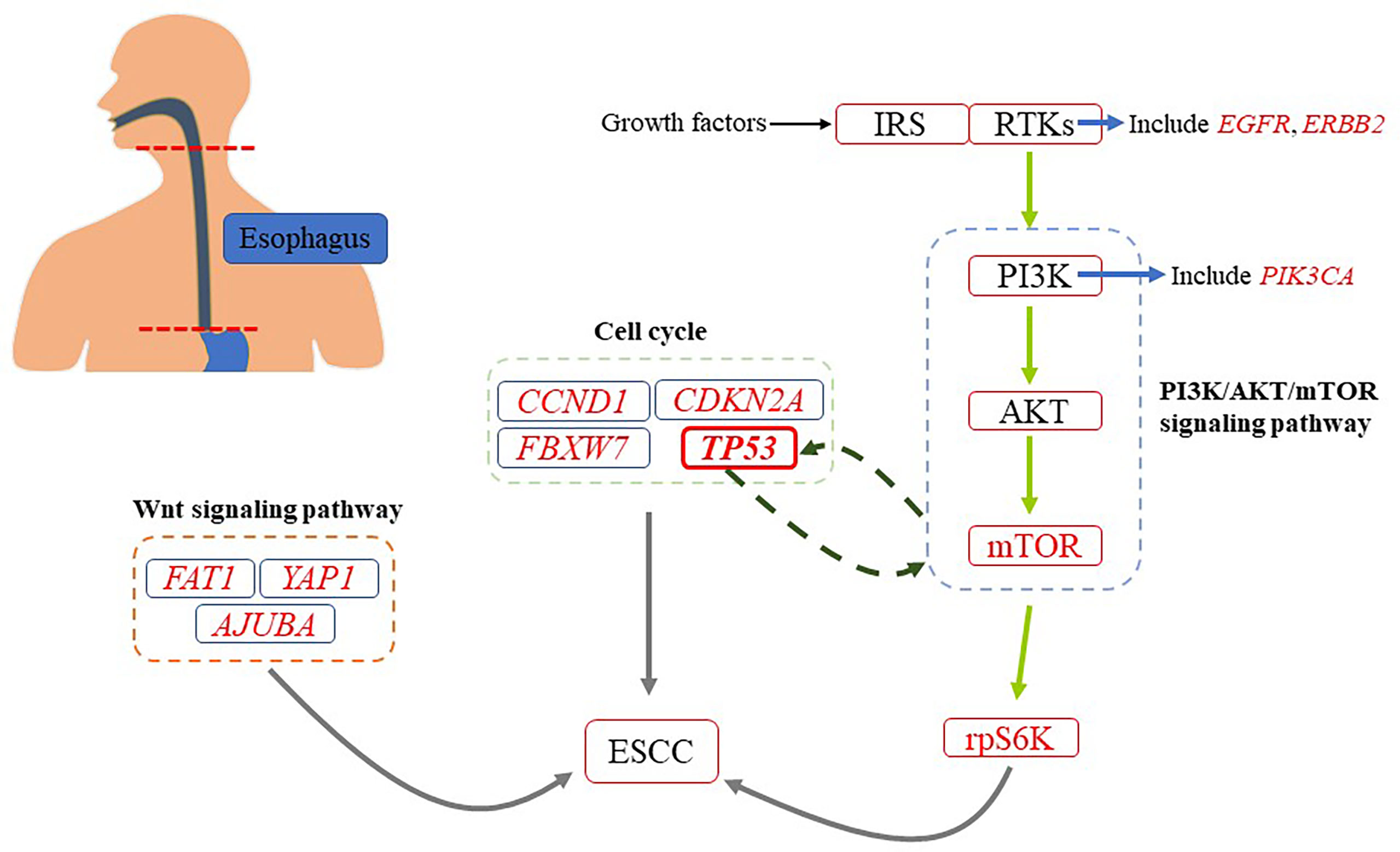
Figure 2 A simplified diagram of genetic changes about ESCC. There are many genetic changes associated with ESCC. It has been found that PI3K/AKT/mTOR pathway is closely related to the occurrence and development of ESCC.
At present, abnormal activation of PI3K signaling has been found in ESCC, and genetic mutations of PI3K, AKT and mTOR-rpS6K have been found. The researchers found that EGFR, ERBB2 and FGFR1 genes were mutated, and their downstream key pathways were PI3K/AKT/mTOR pathway (16, 43). To some extent, the functional realization of the three components depends on PI3K/AKT/mTOR pathway. Therefore, the occurrence and development of ESCC are closely related to the PI3K/AKT/mTOR pathway (Figure 3).
At the same time, PI3K/AKT/mTOR pathway has been proved to be an important pathway controlling growth and metabolism in cells, which is an important guarantee for the survival and normal function. Abnormal activation of PI3K/AKT/mTOR pathway has been found in many tumors, and inhibition of this pathway has also achieved certain therapeutic effects for tumors.
PI3K/AKT/mTOR Signaling Pathway
The PI3K/AKT/mTOR signaling pathway plays an important role in basic functions of cell growth, apoptosis, translation, and cell metabolism (46). There were found abnormal expressions of PI3K/AKT/mTOR signaling pathway in many tumors (47). These evidences suggested that this pathway acting as an essential part in the development of tumors and suggested their potential as new therapeutic targets.
PI3Ks constitute an important enzyme family namely the lipid kinase family. It can be divided into three categories. The class I PI3Ks are heterodimers composed of catalytic subunits and regulatory subunits (48). Class I PI3Ks could be subdivided into class IA and IB enzymes. The class IA consists of three catalytic subunits (p110α, p110β and p110δ) encoded by PIK3CA, PIK3CB and PIK3CD genes, which can be activated by receptor tyrosine kinases (RTKs). While the class IB is composed of p110γ (a catalytic subunit) encoded by PIK3CG and activated by G protein-coupled receptors (GPCRs) (49, 50). The regulatory subunits of class IA and IB are also different in structure (51–54). Class II PI3Ks consist of three different subtypes (PI3K-C2α, PI3K-C2β and PI3K-C2γ) (55). Class III PI3Ks are composed of two subunits (Vps34 and Vps15), and could play an important role in the autophagy and phagocytosis pathway of lysosomes (56).
Class I PI3Ks are the research hotspot of PI3K signal transduction. In addition, class IA PI3Ks are widely found in carcinogenic processes. RTK or GPCR activation enrolls class I PI3Ks into the plasma membrane, where p85 (regulatory subunit) -mediated inhibition of p110 is released and p110 directly phosphorylates PIP2 (phosphatidylinositol 4,5-bisphosphate) into PIP3 (phosphatidylinositol 3,4,5-triphosphate) (57). This lipid is similar to the model of second messenger, which activates downstream proteins and participates in cell growth and survival (58). Phosphatase and tensin homolog (PTEN) can dephosphorylate the third site of the PIP3 inositol ring, result in the conversion to PIP2. It is a negative regulatory factor that inhibits the transduction of PI3K signal to pyruvate dehydrogenase kinase 1 (PDK1) (59) (Figure 3).
AKT is a serine/threonine kinase and a key downstream signal of PI3K (60). AKT has three subtypes: AKT1, AKT2 and AKT3. Overexpression and phosphorylation of AKT can be found in a variety of cancers (61). Sundaramoorthy Revathidevi and colleagues searched the TCGA data and found that compared with other activation methods such as amplification, overexpression and phosphorylation, the activation of AKT by mutation was rare (60). In many cancers, methylation of its upstream regulators, including PTEN, has been shown to activate AKT (62). Also the activation mutation of PI3K, RAS can potentially activate AKT (63). It is established that PI3K can directly activate mTORC2, and the activated mTORC2 can activate AKT (59) (Figure 3).
mTOR signal is one of the key genetic variation targets in cancers, which is often associated with tumor occurrence and progression. The mTOR protein is a serine-threonine kinase of PI3K related family, which is a part of mTORC1 and mTORC2 complexes. These two complexes have different structures and functions (64). The mTORC1 contains regulatory associated protein of mTOR (Raptor), while the mTORC2 contains rapamycin insensitive companion of mTOR (Rictor) (64). Which can explain that mTORC1 is sensitive to rapamycin while mTORC2 is not sensitive to rapamycin treatment. The two both contain mammalian lethal sec-13 protein 8 (mLST8). In addition, mTORC2 contains mammalian stress-activated MAPK-interacting protein 1 (mSIN1; also known as MAPKAP1) (64) (Figure 3). The tuberous sclerosis complex (TSC) is a factor that can regulate mTORC1, and it is also one of the convergence points of multiple pathways in vivo. TSC is a heterotrimer consisted of TSC1, TSC2 and TBC1D7. At the same time, TSC functions as a GTPase activation protein (GAP) could activate the ras homolog enriched in brain (RHEB), a small GTP enzyme. When AKT is activated, it can mediate TSC2 phosphorylation, inhibit TSC1/2 complex and activate mTORC1 signal (65).
Growth factors, amino acids, and oxygen can activate mTORC1. When it was activated, it can participate in protein, lipid and nucleotide synthesis and autophagy (59). For example, after mTORC1 activation, the phosphorylation of its downstream signal molecule 4E-binding protein 1 (4E-BP1) could be inhibited, thus the eukaryotic translation initiation factor 4E (eIF4E) will be released to participate in protein synthesis (59). Autophagy-related protein 1(Atg-1) is a node in several different signaling pathways regulating autophagy in vivo. mTORC1 is one of the upstream signals of Atg-1, and the activation of mTOR signal can inhibit the autophagy induction ability of Atg-1 (66) (Figure 3).
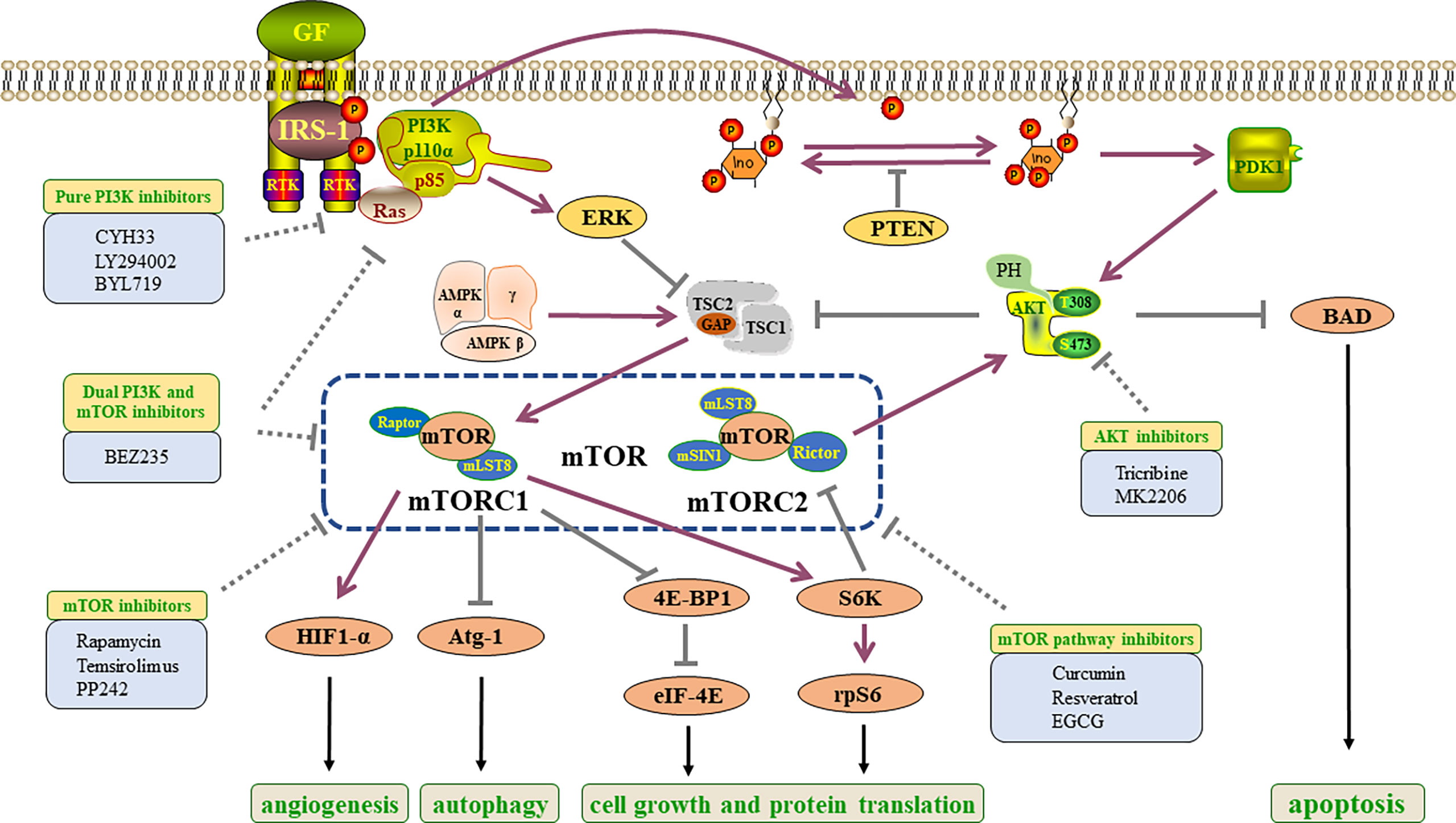
Figure 3 Constituent elements and inhibitors of PI3K/AKT/mTOR pathway in ESCC. Growth factors bind to RTKs to activate the PI3K/AKT/mTOR pathway, which directly and indirectly results in tumorigenesis, the activation of protein translation and angiogenesis, the inhibition of apoptosis and autophagy. GF, growth factors; PI3K, phosphatidylinositol 3-kinase; IRS1, insulin receptor substrate 1; RTK, receptor tyrosine kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; ERK, extracellular signal-related kinase; PIP3, phosphatidylinositol 3,4,5-trisphosphate; TSC, tuberous sclerosis protein; PTEN, phosphatase and tensin homolog; PDK1, pyruvate dehydrogenase lipoamide kinase isozyme 1; AMP,: AMP-activated protein kinase; AKT, protein kinase B; mTORC, mammalian target of rapamycin complex; BAD, Bcl2-related death protein; Raptor, regulatory associated protein of mTOR; HIF1-α, Hypoxia-inducible factor 1-α; mLST8, mammalian lethal with sec-13 protein 8; eIF4E, eukaryotic translation initiation factor 4E; mSIN1, mammalian stress-activated MAPK- interacting protein 1; Rictor, rapamycin insensitive companion of mTOR; Atg-1, autophagy-related protein 1; 4E-BP1, 4E-binding protein 1; S6K, ribosomal S6 kinase; rpS6, ribosomal protein S6.
mTORC2 is often over-activated in cancer cells, and can promote cell survival and migration through phosphorylation of Akt Ser 47 (67, 68). In addition, mTORC2 can regulate additional physiological functions by phosphorylating different substrates such as glycolytic enzyme pyruvate dehydrogenase kinase 1 (PDHK1), serum and glucocorticoid induced kinase (SGK), protein kinase C ζ (PKC ζ) and so on (59, 69, 70).
The Influence of PI3K/AKT/mTOR Pathway on ESCC Development
Many studies have found that the PI3K/AKT/mTOR pathway is associated with cell proliferation, apoptosis, autophagy, and drug resistance of ESCC. Therefore, therapy targeting PI3K/AKT/mTOR pathway should be a promising therapeutic strategy.
Influence on Cell Proliferation and Apoptosis
The proliferation is inextricably linked with apoptosis, whether in normal cells or in tumor cells (71). Numerous studies have found that mTOR regulates cell growth and division. mTORC1 directly activates the ribosomal protein S6 kinase (p70S6K) and inhibits 4E-BP1, thereby increasing translation. At the same time, to a certain extent, it can regulate cell proliferation by controlling cell cycle. And mTORC2 promotes metabolism mainly by activating AKT2.
Shau-Hsuan Li and colleagues demonstrated overexpression of phosphorylated mTOR, p70S6K, and 4EBP1 in 56% tumor tissues of ESCC patients (72). Survival analysis also found that p-mTOR and p-p70S6K overexpression, Ki-67 index >50% were associated with poor OS. Among them, the overexpression of p-p70S6K can be considered as an independent prognostic indicator of ESCC. It is confirmed that everolimus (an inhibitor of mTOR) can inhibit the growth of ESCC in both cell lines and transplanted tumor models (72). Before this, Guiqin Hou and colleagues found that rapamycin and siRNA against mTOR can rapidly inhibit the expression of mTOR and phosphorylation of p70S6K and 4EBP1. In addition, inhibition of mTOR can also make cell cycle arrest at G0/G1 phase and induce apoptosis of ESCC cells (73).
In an experiment conducted by Jiarui Yu and colleagues, these scientists explored the effects of Gambogic acid (GA), the mainly active component secreted by Garcinia hanburryi tree, on the ESCC cells (74, 75). It was found that GA could inhibit the proliferation, migration, and invasion of ESCC cells. Meanwhile, GA induced dose-dependent apoptosis in ESCC cells by inhibiting the expression of Bcl2 and up-regulating the expression of apoptosis-related proteins such as Bax and cleaved-caspase3/9 (75). The probably mechanism was that GA can down-regulate the levels of PI3K, p-AKT and p-mTOR, and promote the expression of PTEN in ESCC cells (75).
These experimental data suggest that proliferation and apoptosis are closely related to PI3K/AKT/mTOR pathway in ESCC.
Influence on Cell Autophagy
Autophagy restricts malignant transformation, balances cell metabolism, and maintains cell survival, but autophagy can promote the cells growth and progression of the cancer (66). Many studies have confirmed that autophagy can protect the cancer cells from anticancer therapy by blocking the apoptotic pathway (also called protective autophagy), to keep the cancer cells alive, allowing them to grow and metastasize (76). Some studies have confirmed that autophagy is mainly induced through PI3K/AKT/mTOR signaling pathway (77). If this pathway is blocked, the autophagy will be inhibited, and the apoptosis will be activated. They together enhance the sensitivity of tumor cells to treatment (77). Beclin-1 synergistic with PI3K pathway enhances autophagy vacuole and activates autophagy cascade reaction (78). Microtubule-associated protein light chain 3 (LC3), now widely used as a monitoring autophagy body formed by specific molecular markers (79). Yu and colleagues found that enhanced autophagy was associated with cisplatin resistance in ESCC cell lines (80). O’Donovan and colleagues found that in drug-resistant ESCC cells, LC3-II levels were significantly increased after treatment with 5-fluorouracil (81). However, inhibition of autophagy induction by siRNA targeting Beclin1 and ATG7 significantly enhanced the effect of 5-fluorouracil (81). These studies suggest that in ESCC cells, autophagy acts as a protective mechanism to promote cell survival during antitumor therapy, leading to therapeutic resistance. In addition, Le Yu and colleagues found that autophagy inhibition can enhance the sensitivity of ESCC cells to cisplatin in vivo (80). Chi Lu and colleagues reported that ionizing radiation activates autophagy in ESCC cell lines. In addition, they found that inhibiting autophagy can enhance apoptosis and cell cycle arrest in vitro induced by radiation (82). Yan Cai and colleagues found that chloroquine inhibited the growth and proliferation of ESCC cell EC109, and this was mediated by regulating autophagy (83).
The Related Inhibitors to PI3K/AKT/mTOR Pathway of ESCC
Pure PI3K Inhibitors
According to selectivity, pure PI3K inhibitors can be divided into two types: pan PI3K inhibitors and selective isoform PI3K inhibitors (84). CYH33 is a novel selective inhibitor of PI3Kα with a unique structure. Jia-jie Shi and colleagues reported that CYH33 combined with radiotherapy can synergistically inhibit the proliferation of ESCC (85). Clinical trial of CYH33 in the treatment of advanced ESCC(NCT03544905) is currently under way (Table 1). LY294002 was identified as a generic PI3K inhibitor. Guiqin Hou and colleagues found that LY294002 could inhibit proliferation of ESCC cells through PI3K/AKT/mTOR/p70S6K signaling pathway. However, LY294002 triggered AKT (Ser473)/PRAS40 (Thr246) feedback activation mediated by mTORC2 in Eca109 and Ec9706 cells. This may lead to limited therapeutic effect of LY294002 on ESCC (86). Therefore, the role of a single PI3K inhibitor is limited. Further experiments by Guiqin Hou and colleagues highlighted that shRNA inhibition of Rictor could reduce phosphorylation of AKTSer473 and Thr308 sites, and counteract activation of AKT (Ser473)/PRAS40 (Thr246) induced by LY294002, which significantly improved the sensitivity of ESCC cells to LY294002 in vitro and in vivo (86). BYL719 is a PI3Kα inhibitor. Moshe Elkabets and colleagues found that AXL is involved in ESCC resistance to BYL719 (87). The mechanism of drug resistance may be that AP-1 transcription factors c-JUN and c-FOS regulate the overexpression of AXL (88). The combination of BYL719-SP600125 (blocking JNK signaling pathway) has achieved certain results in vitro and in vivo (88). For PI3K inhibitor, the application of PI3K inhibitor combined with other drugs should be a hot spot in the future. However, the result of a completed clinical trial of combined drug use was not satisfactory. Combined application of LJM716(HER3 targeting antibody) and BYL719 (NCT01822613) in ESCC patients, the tumor did not shrink as expected.
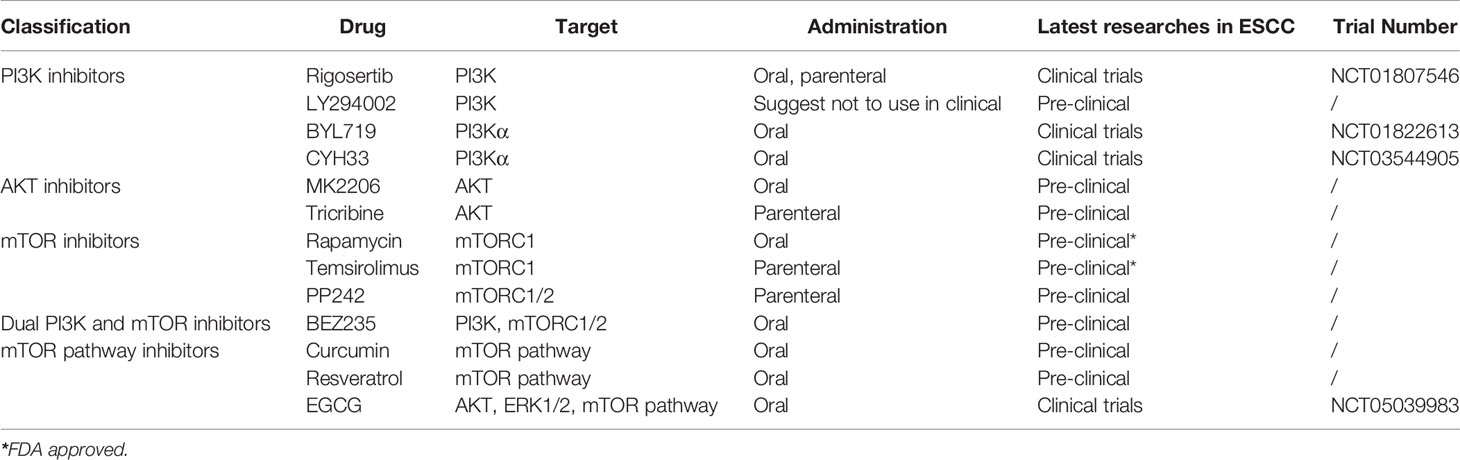
Table 1 The researches about inhibitors of PI3K/AKT/mTOR pathway in ESCC.
AKT Inhibitors
Few Akt inhibitors are currently used in clinical trials. Tricribine (TCN), an Akt inhibitor, significantly inhibited p-Akt, HIF-1α, and VEGF expression in vitro and in vivo, enhancing the radiosensitivity of ESCC in vitro and in vivo (89). ESCC has been proved to have a close relationship with PI3K/AKT/mTOR. The phosphorylation at Thr308 and at Ser473 is both necessary for full AKT activation. MK-2206 is an oral inhibitor targeting on all three AKT subtypes. Ni Shi and colleagues found that in ESCC cells, the phosphorylation level of AKT at Ser473 only slightly decreased upon treatment with MK2206 (Table 1) (90). The effect of MK2206 alone in the mouse model of ESCC was also not ideal. However, in a clinical trial conducted by Timothy A. Yap and colleagues, it was found that MK-2206 has a certain therapeutic effect on solid tumors such as lung and colorectal cancer (91). The underlying molecular changes in ESCC may be more complex. For example, when MK2206 is used alone, the up or downstream targets of AKT may be activated to affect drug action. Subsequent experiments by Nishi and colleagues found that MK2206 combined with BEZ235 (a co-inhibitor of PI3K and mTOR) enhanced the inhibiting of proliferation in ESCC cells, both in vivo and in vitro.
mTOR Inhibitors
The mTOR inhibitors are divided into three generations (92). Current researches are mainly focused on the first and second generations.
The First Generation of mTOR Inhibitors
Rapamycin and its analogues are the first generation of mTOR inhibitors. Through allosteric mechanism, they can partially inhibit the activity of mTORC1 and slow down the proliferation of cancer cells (93). Guiqin Hou and colleagues reported that rapamycin could induce apoptosis in ESCC cells. In addition, rapamycin was found to inhibit tumor growth in human ESCC cell line EC9706 in nude mice. Its inhibitory effect was stronger than that of cisplatin used alone. But the combination of rapamycin and cisplatin was the strongest (94). Temsirolimus (CCI-779, TriceITM) is one of these analogues. Toshio Nishikawa and colleagues found that in some ESCC cell lines (such as TE-1, TE-8, and TE-10), the level of mTOR phosphorylation was increased, accompanied by the upregulation of hypoxia-inducible factor-1α (HIF-1α). Temsirolimus significantly inhibited the activation of mTOR and its downstream effector proteins, resulting in decreased proliferation of ESCC cells. Finally, in vitro, temsirolimus significantly reduced the size of subcutaneous tumors in nude mice and effectively extended the survival of mice with esophageal carcinoma in situ (the cell used for this experiment was TE-8) (95).
At the same time, the first generation of mTOR inhibitors only had little effect on the phosphorylation of 4E-BP1 (96).These inhibitors do not inhibit the activity of mTORC2, so the direct activation of AKT by mTORC2 is not affected. And the negative feedback loop formed after suppressing mTORC1 can activate the PI3K/AKT signal (93). This may be the cause of inhibitor resistance. Now some studies have found that some tumors are resistant to these inhibitors. For example, T Fujishita and colleagues found that everolimus (a rapamycin analogue) had little effect on blocking tumor invasion when used in the later phase of locally aggressive intestinal adenocarcinoma (cis-Apc/Smad4 mice model). But inhibiting mTOR and EGFR or MEK at the same time may be more effective in treating colon cancer (97). As such, the molecular in tumors are more complex than expected, and combination of drugs seems to be a more meaningful route.
Clinical trials with rapamycin and its analogues related to ESCC have not been carried out. Still some researchers have also had limited success in treating a small number of rare cancers with monotherapy, including mantle cell lymphoma and pancreatic neuroendocrine tumors (98, 99).
The Second Generation of mTOR Inhibitors
The second generation mTOR inhibitors are some small molecule ATP competitive inhibitors. They can target mTOR or both mTOR and PI3K. Several categories according to the chemical structure exist (92). The pyrazolopyrimidine class is one of them. PP242 is a typical example of a pyrazolopyrimidine. The inhibitory effect of PP242 was stronger than that of rapamycin, and PP242 could inhibit the activities of mTORC1 and mTORC2.
Yu Huang and colleagues examined the antitumor effect of PP242 in ESCC cell lines include Eca-109 and TE-1 (100). As expected, they found that PP242 can weaken the activities of both mTORC1 and mTORC2 signaling in ESCC, stronger than rapamycin. PP242 could inhibit 4E-BP1 phosphorylation and abrogate PI3K/AKT feedback activation relying on mTORC1 (100). It seems that the anti-tumor effect of the second generation of inhibitors should be far more obvious than that of the first generation. In ESCC cells, Yu Huang and colleagues found PP242 can effectively inhibit the proliferation, induce apoptosis, and arrest cell cycle (100).
PP242 is not tested in currently ongoing clinical trials. TAK-228 (derived from PP242) was well tolerated as a single agent and showed initial therapeutic activity in hematological malignancies (NCT01118689) (101).
Dual PI3K and mTOR Inhibitors
Regarding second generation mTOR inhibitors, some compounds were found to target both PI3K and mTOR (92). BEZ235 is one of them. Ning Wu and colleagues found the activity of p-AKT, p-mTOR, and p-p70S6K can be reduced significantly by BEZ235 in ESCC cells include Eca-109 and TE-1. This inhibitory effect can induce autophagy and apoptosis of human ESCC cells (102). At the same time, they found that BEZ235 combined with Trichostatin A(histone deacetylase inhibitor) had better tumor inhibition effect than single drug (102).Clinical trials of dual PI3K and mTOR inhibitors have not been conducted in the ESCC. But the clinical trials of BEZ235 in other tumors are ongoing. In a clinical trial, the dual PI3K and mTOR inhibitor, PF-05212384 was found to have manageable safety and antitumor activity. This trial provides support for further clinical studies in patients with advanced solid malignancies (NCT 00940498) (103).
mTOR Pathway Inhibitors
Some natural polyphenols extracted from plants, such as curcumin and resveratrol, have been confirmed to inhibit mTOR signaling pathway directly or indirectly in certain tumors (92). Researchers have also explored the role of these extracts in ESCC (104, 105).
Curcumin is a polyphenolic compound extracted from turmeric roots. It is safe, non-toxic, and has anti-tumor effects in the human body (106). Many studies have shown that curcumin and PI3K/AKT/mTOR signaling pathway are closely related. Lian Deng and colleagues found that curcumin combined with docetaxel can induce apoptosis and autophagy in ESCC cells, which may be based on the PI3K/AKT/mTOR signaling pathway (104).
Resveratrol is rich in grapes, red wine, and peanuts (107). It is a plant defensin that has specific cytotoxicity for multiple carcinoma cells (such as melanoma and breast cancer), with certain treatment potential (108, 109). Qishan Tang and colleagues has confirmed that resveratrol can induce cell cycle arrest at the sub-G1 phase and result in subsequent apoptosis, in a dose-dependent manner (110). They also confirmed that resveratrol can inactivate the mTOR signal (110).
Epigallocatechin-3-gallate (EGCG), a primary tea polyphenol, has been shown to inhibit the growth of certain human cancer cells (111). The mechanisms of inhibiting tumor are antioxidation, inhibiting cell proliferation and angiogenesis, as well as increasing cancer apoptosis (111). Yao-Kuang Wang and colleagues has confirmed that EGCG can inhibit the proliferation and colony formation of arecoline-induced ESCC cells by inhibiting AKT and ERK1/2 pathway (105). Exactly, an important downstream signal of AKT and ERK1/2 is mTOR. A clinical trial on EGCG (NCT05039983) in ESCC is currently ongoing in China.
These natural compounds can inhibit the growth of ESCC cells and are inseparable from mTOR signal.
eIFs and PI3K/AKT/mTOR Pathway
PI3K/AKT/mTOR Pathway Can Regulate eIFs to Influence the Translation
Translation is an important and complicated process of gene expression in eukaryotes. Translation mainly includes four processes: initiation, elongation, termination, and ribosome recycling (112). The regulation of translation mainly takes place at initiation phase and which is the rate limiting phase of protein synthesis (113). The regulators of translation initiation are the eIFs. The activation of RTKs, MAPK and PI3K/AKT signaling pathways could be stimulated by some signals which promoting tumorigenesis (114). These pathways play an important role in the regulating of eIF functions (115). Both MAPK and PI3K/AKT pathways regulate the functions of eIFs via mTOR. Therefore, mTOR plays a leading role in the regulation of eIF functions (115, 116). Their mis-regulation usually causes abnormal translation, synthesizes aberrant proteins, finally leading to tumorigenesis (117–120).
Overview on the Role of eIFs in Translation Initiation
The initiation process of translation begins with the formation of the 43S pre-initiation complex (121). The 43S initiation complex consists of 40S ribosomal subunit, eIF2–GTP–Met-tRNAiMet, eIF1, eIF1a and eIF3 (121, 122). The 43S initiation complex can then be guided by the eIF4F complex to bind with mRNA. The eIF4F complex is a heterotrimer composed of eIF4A, eIF4E and eIF4G subunits (123). The mRNA was scanned by 43S initiation complex. With the assistance of eIF1, the tRNA anticodon ring correctly binds to the start codon AUG on the mRNA (121). eIF4A has helicase activity. eIF4E binds to the mRNA’s cap structure. It is usually bonded with 4E-BP1. Once 4E-BP1 was phosphorylated, the eIF4E could be released. The role of eIF4G is to link mRNA with ribosomes (123, 124). eIF3 acts more like a scaffold, links other eIFs with 40S ribosomal subunit (125). When the complex encounters the correct AUG start codon, eIF2 will be hydrolyzed, and other eIFs will be released. At the last stage of translation initiation, eIF2 is in an inactive GDP binding state, the GTP bound to eIF5B is hydrolyzed, and these translation factors are separated from the ribosome (122, 126). In addition, eIF6, which has not yet been mentioned, is the first eIF associated with the 60S subunit that modulates translation in response to extracellular signals (127, 128).
eIFs-Potential Therapeutic Targets in Tumors
Moreover, some researchers found that changes in the expression of certain translation promoters (primarily increased expression) were associated with the development of specific tumors (129–131). For example, eIF1A is a small 17kDa promoter and highly conserved in all eukaryotes. Somatic mutations in the N-terminal tail (NTT) of eIF1A have been found to be associated with uveal melanoma, thyroid cancer, and ovarian cancer (132–134). Urmila Sehrawat and colleagues found that eIF1A could regulate different mRNAs differently in mammalian cells (135). The eIF1A NTT mutants enhanced the scanning of the 5 ‘UTR-containing cell cycle genes, possibly affecting the cell cycle and promoting cell proliferation (135). eIF3H is one of the central subunits of eIF3 complex. It has been observed that eIF3H is often amplified in breast and prostate cancer together with proto-oncogene Myc (136, 137). Researchers have found the amplification of eIF3H gene in colorectal cancer and non-small cell lung cancer (NSCLC) through genome-wide analyses and fluorescent in situ hybridization (FISH) (117, 138, 139). In addition, the expression level of eIF3H is positively correlated with the poor differentiation and invasive growth of prostate cancer (117, 137).
In ESCC, there are also some limited studies on eIFs. For example, Ting Liu and colleagues found that eIF4E increased significantly in clinical ESCC tissues and ESCC cell lines, and its expression level was associated with lymph node metastasis, TNM period, and ESCC’s overall and disease-free survival (140). After using the shRNA knockout eIF4E, it was found that the induced cytotoxicity by cisplatin has increased in the ESCC cell lines, and the chemosensitivity to cisplatin in xenograft tumor models also has increased (140). Hong Yang and colleagues found that excessive expression of eIF5A2 in ESCC cells resulted in increased chemoresistance to 5-fluorouracil, docetaxel, and taxol. Conversely, the shRNAs of eIF5A2 could increase the sensitivity of tumors to these chemotherapeutic drugs. It was found that in patients who underwent taxane-based chemotherapy after esophagectomy, eIF5A2 overexpression was associated with poor total survival rate (P <0.05) (141). Therefore, targeting eIF4E or eIF5A2 may be a feasible method of improving ESCC chemotherapy sensitivity. However, eIFs in ESCC still require more researches.
These previous examples reveal eIFs may be a promising target for future tumor treatment. At present, there are some inhibitors of eIFs in preclinical and clinical trials (Table 2). Ribavirin is an antiviral drug approved by the Food and Drug Administration (FDA) for hepatitis C, which can also treat syncytial virus infection and viral hemorrhagic fever (144–146). Importantly, ribavirin has been extensively documented as an inhibitor of eIF4E (147–149).
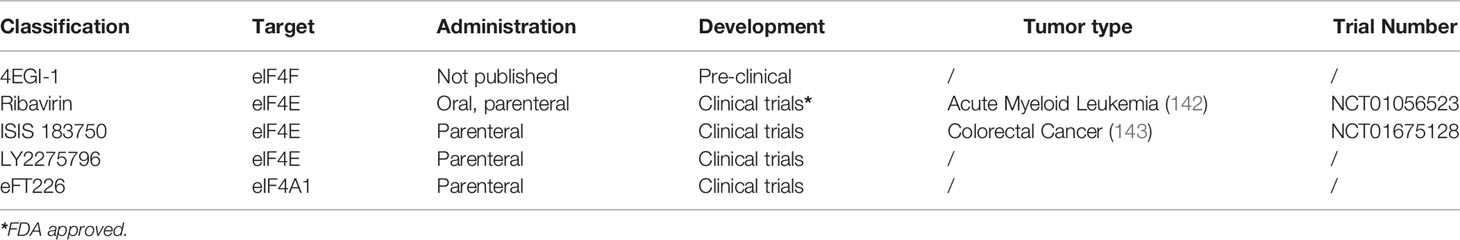
Table 2 The inhibitors of eIFs in tumor and the specific tumor types that can be inhibited in clinical trials.
JingJin and colleagues found an overexpression of eIF4E in most ovarian cancer patients. In addition, the eIF4E function is critical for the growth and survival of tumors (150). eIF4E inhibition was found to be achieved at clinically achievable doses of ribavirin. Inhibition of eIF4E by ribavirin may be a potential therapeutic approach to improve clinical management of ovarian cancer (150). Sakibul Huq and colleagues found that ribavirin enhanced radiosensitivity in nasopharyngeal carcinoma (NPC). At the same time, it can inhibit the expression of various proteins which are all overexpressed in NPC and correlated with poor prognosis, also can inhibit the mTOR/eIF4E axis (151). These studies indicate that ribavirin is a potential targeted drug for tumor therapy.
Several clinical trials related to ribavirin are underway, such as oropharynx squamous cell cancer (NCT01721525), acute myeloid leukemia (NCT01056523), melanoma (NCT00897312).
Conclusions and Discussions
ESCC is a complex disease, the external predisposing factors and genetic mutations both have an important impact on oncogenesis and tumor progression. So, we should focus on the prevention, warning high-risk individuals away from alcohol, cigarettes, and so on. At the same time, we should pay attention to the screening of disease and improve the early diagnostic rate. Once ESCC patients could be diagnosed in the early stage, their prognosis and living quality will be improved significantly.
From the global cancer report, the incidence and mortality rate of ESCC are currently ranked tenth and sixth (1). This data shows that the current situation of ESCC is not optimistic. At present, neoadjuvant chemoradiotherapy and esophagectomy are the mainstream treatment methods of ESCC. Many researchers focus on treating ESCC by targeted therapy (EGFR or PD-1). There are currently a variety of related drugs used for clinical, and some ESCC patients respond particularly well to them. Still the increase of patients’ overall five-year survival rate has no statistical significance (18, 31).
In all results of ESCC gene testing conducted by several research groups, the abnormal expression of PI3K/AKT/mTOR and its related pathways have been found. In this article, we discussed how the PI3K/AKT/mTOR pathway affects the growth, proliferation, and autophagy of ESCC. It is also discussed that inhibitors in different parts of PI3K/AKT/mTOR pathway can affect the growth and biological behavior of ESCC (Table 1). Additionally, the eIFs regulated by PI3K/AKT/mTOR pathway, also has an important influence on the occurrence and development of tumors. Through the discussion, it was found that the PI3K/AKT/mTOR pathway and eIFs could be the future therapeutic target of ESCC.
Still, it should not be ignored that there are only few relevant studies on the application of PI3K/AKT/mTOR pathway inhibitors in ESCC. Most studies have used a single inhibitor and with limited efficacy. At present, there are few studies on the combination of multiple inhibitors with different targets in ESCC ongoing. It may be that researchers have not yet paid attention to the potential therapeutic effects of PI3K/AKT/mTOR pathway and its regulated eIFs in ESCC. It also could be that new drugs are being developed slowly and in fewer varieties. But why the effectiveness of a single inhibitor is limited?
The researchers found that the activation of multiple pathways is the main cause of drug resistance in tumor cells (115). Such as, Jessie Villanueva and colleagues found that melanoma cells show enhanced activation of PI3K signaling after treatment with BRAF inhibitors, leading to drug resistance of tumor cells (152). In ESCC, some researchers found the resistance of tumor cells to EGFR-targeted therapy might be related to the abnormal activation of PI3K/AKT/mTOR pathway (19). Thus, the growth of tumors carrying oncogenes that activate multiple pathways does not depend on a single signaling pathway.
The PI3K/AKT/mTOR pathway and its regulated eIFs have been proved to be a key pathway involved in growth. And many other pathways in the body are inseparable from it. Therefore, the future treatment of ESCC must be related tightly with the PI3K/AKT/mTOR pathway and its regulated eIFs. Simultaneous suppression of multiple targets of this pathway may be a future research focus. One hypothesis: it may be focused more on the co-inhibition of PI3K/AKT/mTOR pathway and eIFs.
In this context, multiple components of several oncogenic signaling pathways and eIFs participated in mRNA translation have been identified as biomarkers with potential diagnostic, therapeutic and prognostic value. Therefore, anti-tumor agents targeting the core elements of protein synthesis and related signaling pathways can get over intratumor heterogeneity and represent as novel promising anticancer drugs.
Author Contributions
Conceptualization, RH, JH, and YD. Writing—original draft preparation, RH, QD, RY, and QZ. Writing—review, revising and editing, RH, JH, and ZY. All authors have read and agreed to the published version of the manuscript.
Funding
The project of Department of Science and Technology, Sichuan province, Grant/Award, Number: 2018JY0398.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Yang HX, Hou X, Liu QW, Zhang LJ, Liu JG, Lin P, et al. Tumor Location Does Not Impact Long-Term Survival in Patients With Operable Thoracic Esophageal Squamous Cell Carcinoma in China. Ann Thorac Surg (2012) 93:1861–6. doi: 10.1016/j.athoracsur.2012.03.002
3. Akhtar S, Sheikh AA, Qureshi HU. Chewing Areca Nut, Betel Quid, Oral Snuff, Cigarette Smoking and the Risk of Oesophageal Squamous-Cell Carcinoma in South Asians: A Multicentre Case-Control Study. Eur J Cancer (2012) 48:655–61. doi: 10.1016/j.ejca.2011.06.008
4. Taylor PR, Abnet CC, Dawsey SM. Squamous Dysplasia–The Precursor Lesion for Esophageal Squamous Cell Carcinoma. Cancer Epidemiol Biomark Prev (2013) 22:540–52. doi: 10.1158/1055-9965.Epi-12-1347
5. Oda I, Abe S, Kusano C, Suzuki H, Nonaka S, Yoshinaga S, et al. Correlation Between Endoscopic Macroscopic Type and Invasion Depth for Early Esophagogastric Junction Adenocarcinomas. Gastric Cancer (2011) 14:22–7. doi: 10.1007/s10120-011-0001-0
6. Takubo K, Sasajima K, Yamashita K, Tanaka Y, Fujita K. Prognostic Significance of Intramural Metastasis in Patients With Esophageal Carcinoma. Cancer (1990) 65:1816–9. doi: 10.1002/1097-0142(19900415)65:8<1816::aid-cncr2820650825>3.0.co;2-l
7. DiMaio MA, Kwok S, Montgomery KD, Lowe AW, Pai RK. Immunohistochemical Panel for Distinguishing Esophageal Adenocarcinoma From Squamous Cell Carcinoma: A Combination of P63, Cytokeratin 5/6, MUC5AC, and Anterior Gradient Homolog 2 Allows Optimal Subtyping. Hum Pathol (2012) 43:1799–807. doi: 10.1016/j.humpath.2012.03.019
8. Hara T, Kijima H, Yamamoto S, Kenmochi T, Kise Y, Tanaka H, et al. Ubiquitous P63 Expression in Human Esophageal Squamous Cell Carcinoma. Int J Mol Med (2004) 14:169–73. doi: 10.3892/ijmm.14.2.169
9. Zhang HD, Chen CG, Gao YY, Ma Z, Tang P, Duan XF, et al. Primary Esophageal Adenosquamous Carcinoma: A Retrospective Analysis of 24 Cases. Dis Esophagus (2014) 27:783–9. doi: 10.1111/dote.12153
10. Luo LN, He LJ, Gao XY, Huang XX, Shan HB, Luo GY, et al. Endoscopic Ultrasound for Preoperative Esophageal Squamous Cell Carcinoma: A Meta-Analysis. PloS One (2016) 11:e0158373. doi: 10.1371/journal.pone.0158373
11. Probst A, Aust D, Märkl B, Anthuber M, Messmann H. Early Esophageal Cancer in Europe: Endoscopic Treatment by Endoscopic Submucosal Dissection. Endoscopy (2015) 47:113–21. doi: 10.1055/s-0034-1391086
12. Shapiro J, van Lanschot JJB, Hulshof MCCM, van Hagen P, van Berge Henegouwen MI, Wijnhoven BPL, et al. Neoadjuvant Chemoradiotherapy Plus Surgery Versus Surgery Alone for Oesophageal or Junctional Cancer (CROSS): Long-Term Results of a Randomised Controlled Trial. Lancet Oncol (2015) 16:1090–8. doi: 10.1016/s1470-2045(15)00040-6
13. Yang H, Liu H, Chen Y, Zhu C, Fang W, Yu Z, et al. Neoadjuvant Chemoradiotherapy Followed by Surgery Versus Surgery Alone for Locally Advanced Squamous Cell Carcinoma of the Esophagus (NEOCRTEC5010): A Phase III Multicenter, Randomized, Open-Label Clinical Trial. J Clin Oncol (2018) 36:2796–803. doi: 10.1200/jco.2018.79.1483
14. Little AG, Lerut AE, Harpole DH, Hofstetter WL, Mitchell JD, Altorki NK, et al. The Society of Thoracic Surgeons Practice Guidelines on the Role of Multimodality Treatment for Cancer of the Esophagus and Gastroesophageal Junction. Ann Thorac Surg (2014) 98:1880–5. doi: 10.1016/j.athoracsur.2014.07.069
15. Watanabe M, Mine S, Nishida K, Yamada K, Shigaki H, Matsumoto A, et al. Salvage Esophagectomy After Definitive Chemoradiotherapy for Patients With Esophageal Squamous Cell Carcinoma: Who Really Benefits From This High-Risk Surgery? Ann Surg Oncol (2015) 22:4438–44. doi: 10.1245/s10434-015-4556-6
16. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, et al. Genomic and Molecular Characterization of Esophageal Squamous Cell Carcinoma. Nat Genet (2014) 46:467–73. doi: 10.1038/ng.2935
17. Jiang D, Li X, Wang H, Shi Y, Xu C, Lu S, et al. The Prognostic Value of EGFR Overexpression and Amplification in Esophageal Squamous Cell Carcinoma. BMC Cancer (2015) 15:377. doi: 10.1186/s12885-015-1393-8
18. Huang J, Fan Q, Lu P, Ying J, Ma C, Liu W, et al. Icotinib in Patients With Pretreated Advanced Esophageal Squamous Cell Carcinoma With EGFR Overexpression or EGFR Gene Amplification: A Single-Arm, Multicenter Phase 2 Study. J Thorac Oncol (2016) 11:910–7. doi: 10.1016/j.jtho.2016.02.020
19. Chong CR, Janne PA. The Quest to Overcome Resistance to EGFR-Targeted Therapies in Cancer. Nat Med (2013) 19:1389–400. doi: 10.1038/nm.3388
20. Moehler M, Maderer A, Thuss-Patience PC, Brenner B, Meiler J, Ettrich TJ, et al. Cisplatin and 5-Fluorouracil With or Without Epidermal Growth Factor Receptor Inhibition Panitumumab for Patients With Non-Resectable, Advanced or Metastatic Oesophageal Squamous Cell Cancer: A Prospective, Open-Label, Randomised Phase III AIO/EORTC Trial (POWER). Ann Oncol (2020) 31:228–35. doi: 10.1016/j.annonc.2019.10.018
21. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The Function of Programmed Cell Death 1 and Its Ligands in Regulating Autoimmunity and Infection. Nat Immunol (2007) 8:239–45. doi: 10.1038/ni1443
22. Okazaki T, Honjo T. PD-1 and PD-1 Ligands: From Discovery to Clinical Application. Int Immunol (2007) 19:813–24. doi: 10.1093/intimm/dxm057
23. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
24. Raufi AG, Klempner SJ. Immunotherapy for Advanced Gastric and Esophageal Cancer: Preclinical Rationale and Ongoing Clinical Investigations. J Gastrointest Oncol (2015) 6:561–9. doi: 10.3978/j.issn.2078-6891.2015.037
25. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
26. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol (2015) 33:4015–22. doi: 10.1200/jco.2015.62.3397
27. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol (2010) 28:3167–75. doi: 10.1200/jco.2009.26.7609
28. Kudo T, Hamamoto Y, Kato K, Ura T, Kojima T, Tsushima T, et al. Nivolumab Treatment for Oesophageal Squamous-Cell Carcinoma: An Open-Label, Multicentre, Phase 2 Trial. Lancet Oncol (2017) 18:631–9. doi: 10.1016/s1470-2045(17)30181-x
29. Lee J, Kim B, Jung HA, La Choi Y, Sun JM. Nivolumab for Esophageal Squamous Cell Carcinoma and the Predictive Role of PD-L1 or CD8 Expression in Its Therapeutic Effect. Cancer Immunol Immunother (2021) 70:1203–11. doi: 10.1007/s00262-020-02766-7
30. Qu HX, Zhao LP, Zhan SH, Geng CX, Xu L, Xin YN, et al. Clinicopathological and Prognostic Significance of Programmed Cell Death Ligand 1 (PD-L1) Expression in Patients With Esophageal Squamous Cell Carcinoma: A Meta-Analysis. J Thorac Dis (2016) 8:3197–204. doi: 10.21037/jtd.2016.11.01
31. Rong L, Liu Y, Hui Z, Zhao Z, Zhang Y, Wang B, et al. PD-L1 Expression and its Clinicopathological Correlation in Advanced Esophageal Squamous Cell Carcinoma in a Chinese Population. Diagn Pathol (2019) 14:6. doi: 10.1186/s13000-019-0778-4
32. Abnet CC, Arnold M, Wei WQ. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology (2018) 154:360–73. doi: 10.1053/j.gastro.2017.08.023
33. Zendehdel K, Nyrén O, Luo J, Dickman PW, Boffetta P, Englund A, et al. Risk of Gastroesophageal Cancer Among Smokers and Users of Scandinavian Moist Snuff. Int J Cancer (2008) 122:1095–9. doi: 10.1002/ijc.23076
34. Ishiguro S, Sasazuki S, Inoue M, Kurahashi N, Iwasaki M, Tsugane S. Effect of Alcohol Consumption, Cigarette Smoking and Flushing Response on Esophageal Cancer Risk: A Population-Based Cohort Study (JPHC Study). Cancer Lett (2009) 275:240–6. doi: 10.1016/j.canlet.2008.10.020
35. Liu Y, Chen H, Sun Z, Chen X. Molecular Mechanisms of Ethanol-Associated Oro-Esophageal Squamous Cell Carcinoma. Cancer Lett (2015) 361:164–73. doi: 10.1016/j.canlet.2015.03.006
36. Nieminen MT, Novak-Frazer L, Collins R, Dawsey SP, Dawsey SM, Abnet CC, et al. Alcohol and Acetaldehyde in African Fermented Milk Mursik–a Possible Etiologic Factor for High Incidence of Esophageal Cancer in Western Kenya. Cancer Epidemiol Biomark Prev (2013) 22:69–75. doi: 10.1158/1055-9965.EPI-12-0908
37. Jeurnink SM, Büchner FL, Bueno-de-Mesquita HB, Siersema PD, Boshuizen HC, Numans ME, et al. Variety in Vegetable and Fruit Consumption and the Risk of Gastric and Esophageal Cancer in the European Prospective Investigation Into Cancer and Nutrition. Int J Cancer (2012) 131:E963–73. doi: 10.1002/ijc.27517
38. Engel LS, Chow WH, Vaughan TL, Gammon MD, Risch HA, Stanford JL, et al. Population Attributable Risks of Esophageal and Gastric Cancers. JNCI J Natl Cancer Inst (2003) 95:1404–13. doi: 10.1093/jnci/djg047
39. Islami F, Ren J-S, Taylor PR, Kamangar F. Pickled Vegetables and the Risk of Oesophageal Cancer: A Meta-Analysis. Br J Cancer (2009) 101:1641–7. doi: 10.1038/sj.bjc.6605372
40. Munishi MO, Hanisch R, Mapunda O, Ndyetabura T, Ndaro A, Schüz J, et al. Africa’s Oesophageal Cancer Corridor: Do Hot Beverages Contribute? Cancer Causes Control (2015) 26:1477–86. doi: 10.1007/s10552-015-0646-9
41. Wu C, Kraft P, Zhai K, Chang J, Wang Z, Li Y, et al. Genome-Wide Association Analyses of Esophageal Squamous Cell Carcinoma in Chinese Identify Multiple Susceptibility Loci and Gene-Environment Interactions. Nat Genet (2012) 44:1090–7. doi: 10.1038/ng.2411
42. Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of Genomic Alterations in Oesophageal Squamous Cell Cancer. Nature (2014) 509:91–5. doi: 10.1038/nature13176
43. Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology (2016) 150:1171–82. doi: 10.1053/j.gastro.2016.01.035
44. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of P53. Cell (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
45. Ekshyyan O, Anandharaj A, Nathan CA. Dual PI3K/mTOR Inhibitors: Does P53 Modulate Response? Clin Cancer Res (2013) 19:3719–21. doi: 10.1158/1078-0432.Ccr-13-1291
46. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt Signalling Pathway and Cancer. Cancer Treat Rev (2004) 30:193–204. doi: 10.1016/j.ctrv.2003.07.007
47. Fruman DA, Rommel C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat Rev Drug Discov (2014) 13:140–56. doi: 10.1038/nrd4204
48. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol (2014) 4:64. doi: 10.3389/fonc.2014.00064
49. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide Kinases. Annu Rev Biochem (1998) 67:481–507. doi: 10.1146/annurev.biochem.67.1.481
50. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat Rev Cancer (2014) 15:7–24. doi: 10.1038/nrc3860
51. Ciraolo E, Gulluni F, Hirsch E. Methods to Measure the Enzymatic Activity of PI3Ks. Methods Enzymol (2014) 543:115–40. doi: 10.1016/B978-0-12-801329-8.00006-4
52. Suire S, Coadwell J, Ferguson GJ, Davidson K, Hawkins P, Stephens L. P84, a New Gbetagamma-Activated Regulatory Subunit of the Type IB Phosphoinositide 3-Kinase P110gamma. Curr Biol (2005) 15:566–70. doi: 10.1016/j.cub.2005.02.020
53. Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural Basis for Activation and Inhibition of Class I Phosphoinositide 3-Kinases. Sci Signaling (2011) 4:re2. doi: 10.1126/scisignal.2002165
54. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat Rev Mol Cell Biol (2010) 11:329–41. doi: 10.1038/nrm2882
55. Wang H, Lo WT, Vujicic Zagar A, Gulluni F, Lehmann M, Scapozza L, et al. Autoregulation of Class II Alpha PI3K Activity by Its Lipid-Binding PX-C2 Domain Module. Mol Cell (2018) 71:343–51.e344. doi: 10.1016/j.molcel.2018.06.042
56. Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, et al. Structure and Flexibility of the Endosomal Vps34 Complex Reveals the Basis of Its Function on Membranes. Science (2015) 350:aac7365. doi: 10.1126/science.aac7365
57. Alzahrani AS. PI3K/Akt/mTOR Inhibitors in Cancer: At the Bench and Bedside. Semin Cancer Biol (2019) 59:125–32. doi: 10.1016/j.semcancer.2019.07.009
58. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
59. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
60. Revathidevi S, Munirajan AK. Akt in Cancer: Mediator and More. Semin Cancer Biol (2019) 59:80–91. doi: 10.1016/j.semcancer.2019.06.002
61. Hay N. The Akt-mTOR Tango and its Relevance to Cancer. Cancer Cell (2005) 8:179–83. doi: 10.1016/j.ccr.2005.08.008
62. Nakakido M, Deng Z, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R. Dysregulation of AKT Pathway by SMYD2-Mediated Lysine Methylation on PTEN. Neoplasia (2015) 17:367–73. doi: 10.1016/j.neo.2015.03.002
63. Downward J. Targeting RAS Signalling Pathways in Cancer Therapy. Nat Rev Cancer (2003) 3:11–22. doi: 10.1038/nrc969
64. Mossmann D, Park S, Hall MN. mTOR Signalling and Cellular Metabolism are Mutual Determinants in Cancer. Nat Rev Cancer (2018) 18:744–57. doi: 10.1038/s41568-018-0074-8
65. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the rictor-mTOR Complex. Science (2005) 307:1098–101. doi: 10.1126/science.1106148
66. Scott RC, Juhász G, Neufeld TP. Direct Induction of Autophagy by Atg1 Inhibits Cell Growth and Induces Apoptotic Cell Death. Curr Biol (2007) 17:1–11. doi: 10.1016/j.cub.2006.10.053
67. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, et al. Oncogenic EGFR Signaling Activates an Mtorc2-NF-κb Pathway That Promotes Chemotherapy Resistance. Cancer Discov (2011) 1:524–38. doi: 10.1158/2159-8290.Cd-11-0124
68. Xu K, Chen G, Li X, Wu X, Chang Z, Xu J, et al. MFN2 Suppresses Cancer Progression Through Inhibition of Mtorc2/Akt Signaling. Sci Rep (2017) 7:41718. doi: 10.1038/srep41718
69. Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H, et al. mTOR Complex Component Rictor Interacts With PKCzeta and Regulates Cancer Cell Metastasis. Cancer Res (2010) 70:9360–70. doi: 10.1158/0008-5472.Can-10-0207
70. Zhang J, Jia L, Liu T, Yip YL, Tang WC, Lin W, et al. Mtorc2-Mediated PDHE1α Nuclear Translocation Links EBV-LMP1 Reprogrammed Glucose Metabolism to Cancer Metastasis in Nasopharyngeal Carcinoma. Oncogene (2019) 38:4669–84. doi: 10.1038/s41388-019-0749-y
71. Guo M, Hay BA. Cell Proliferation and Apoptosis. Curr Opin Cell Biol (1999) 11:745–52. doi: 10.1016/S0955-0674(99)00046-0
72. Li SH, Chen CH, Lu HI, Huang WT, Tien WY, Lan YC, et al. Phosphorylated P70s6k Expression Is an Independent Prognosticator for Patients With Esophageal Squamous Cell Carcinoma. Surgery (2015) 157:570–80. doi: 10.1016/j.surg.2014.10.014
73. Hou G, Xue L, Lu Z, Fan T, Tian F, Xue Y. An Activated mTOR/P70s6k Signaling Pathway in Esophageal Squamous Cell Carcinoma Cell Lines and Inhibition of the Pathway by Rapamycin and siRNA Against mTOR. Cancer Lett (2007) 253:236–48. doi: 10.1016/j.canlet.2007.01.026
74. Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, et al. Discovery, Characterization and SAR of Gambogic Acid as a Potent Apoptosis Inducer by a HTS Assay. Bioorg Med Chem (2004) 12:309–17. doi: 10.1016/j.bmc.2003.11.013
75. Yu J, Wang W, Yao W, Yang Z, Gao P, Liu M, et al. Gambogic Acid Affects ESCC Progression Through Regulation of PI3K/AKT/mTOR Signal Pathway. J Cancer (2020) 11:5568–77. doi: 10.7150/jca.41115
76. Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G, et al. Targeting PI3K/AKT/mTOR-Mediated Autophagy for Tumor Therapy. Appl Microbiol Biotechnol (2020) 104:575–87. doi: 10.1007/s00253-019-10257-8
77. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The Role of Autophagy in Cancer Development and Response to Therapy. Nat Rev Cancer (2005) 5:726–34. doi: 10.1038/nrc1692
78. Han T, Guo M, Gan M, Yu B, Tian X, Wang JB. TRIM59 Regulates Autophagy Through Modulating Both the Transcription and the Ubiquitination of BECN1. Autophagy (2018) 14:2035–48. doi: 10.1080/15548627.2018.1491493
79. Yoshioka A, Miyata H, Doki Y, Yamasaki M, Sohma I, Gotoh K, et al. LC3, an Autophagosome Marker, Is Highly Expressed in Gastrointestinal Cancers. Int J Oncol (2008) 33:461–8. doi: 10.3892/ijo_00000028
80. Yu L, Gu C, Zhong D, Shi L, Kong Y, Zhou Z, et al. Induction of Autophagy Counteracts the Anticancer Effect of Cisplatin in Human Esophageal Cancer Cells With Acquired Drug Resistance. Cancer Lett (2014) 355:34–45. doi: 10.1016/j.canlet.2014.09.020
81. O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of Autophagy by Drug-Resistant Esophageal Cancer Cells Promotes Their Survival and Recovery Following Treatment With Chemotherapeutics. Autophagy (2011) 7:509–24. doi: 10.4161/auto.7.6.15066
82. Lu C, Xie C. Radiation-Induced Autophagy Promotes Esophageal Squamous Cell Carcinoma Cell Survival Via the LKB1 Pathway. Oncol Rep (2016) 35:3559–65. doi: 10.3892/or.2016.4753
83. Cai Y, Cai J, Ma Q, Xu Y, Zou J, Xu L, et al. Chloroquine Affects Autophagy to Achieve an Anticancer Effect in EC109 Esophageal Carcinoma Cells. Vitro Oncol Lett (2018) 15:1143–8. doi: 10.3892/ol.2017.7415
84. Elmenier FM, Lasheen DS, Abouzid KAM. Phosphatidylinositol 3 Kinase (PI3K) Inhibitors as New Weapon to Combat Cancer. Eur J Med Chem (2019) 183:111718. doi: 10.1016/j.ejmech.2019.111718
85. Shi JJ, Xing H, Wang YX, Zhang X, Zhan QM, Geng MY, et al. PI3Kalpha Inhibitors Sensitize Esophageal Squamous Cell Carcinoma to Radiation by Abrogating Survival Signals in Tumor Cells and Tumor Microenvironment. Cancer Lett (2019) 459:145–55. doi: 10.1016/j.canlet.2019.05.040
86. Hou G, Zhao Q, Zhang M, Fan T, Liu M, Shi X, et al. Down-Regulation of Rictor Enhances Cell Sensitivity to PI3K Inhibitor LY294002 by Blocking Mtorc2-Medicated Phosphorylation of Akt/PRAS40 in Esophageal Squamous Cell Carcinoma. BioMed Pharmacother (2018) 106:1348–56. doi: 10.1016/j.biopha.2018.07.075
87. Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL Mediates Resistance to PI3Kalpha Inhibition by Activating the EGFR/PKC/mTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell (2015) 27:533–46. doi: 10.1016/j.ccell.2015.03.010
88. Badarni M, Prasad M, Balaban N, Zorea J, Yegodayev KM, Joshua BZ, et al. Repression of AXL Expression by AP-1/JNK Blockage Overcomes Resistance to PI3Ka Therapy. JCI Insight (2019). doi: 10.1172/jci.insight.125341
89. Guo Q, He J, Shen F, Zhang W, Yang X, Zhang C, et al. TCN, an AKT Inhibitor, Exhibits Potent Antitumor Activity and Enhances Radiosensitivity in Hypoxic Esophageal Squamous Cell Carcinoma. Vitro Vivo Oncol Lett (2017) 13:949–54. doi: 10.3892/ol.2016.5515
90. Shi N, Yu H, Chen T. Inhibition of Esophageal Cancer Growth Through the Suppression of PI3K/AKT/mTOR Signaling Pathway. Onco Targets Ther (2019) 12:7637–47. doi: 10.2147/OTT.S205457
91. Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-In-Man Clinical Trial of the Oral Pan-AKT Inhibitor MK-2206 in Patients With Advanced Solid Tumors. J Clin Oncol (2011) 29:4688–95. doi: 10.1200/JCO.2011.35.5263
92. Xu T, Sun D, Chen Y, Ouyang L. Targeting mTOR for Fighting Diseases: A Revisited Review of mTOR Inhibitors. Eur J Med Chem (2020) 199:112391. doi: 10.1016/j.ejmech.2020.112391
93. Guertin DA, Sabatini DM. The Pharmacology of mTOR Inhibition. Sci Signal (2009) 2:pe24. doi: 10.1126/scisignal.267pe24
94. Hou G, Zhang Q, Wang L, Liu M, Wang J, Xue L. mTOR Inhibitor Rapamycin Alone or Combined With Cisplatin Inhibits Growth of Esophageal Squamous Cell Carcinoma in Nude Mice. Cancer Lett (2010) 290:248–54. doi: 10.1016/j.canlet.2009.09.015
95. Nishikawa T, Takaoka M, Ohara T, Tomono Y, Hao H, Bao X, et al. Antiproliferative Effect of a Novel mTOR Inhibitor Temsirolimus Contributes to the Prolonged Survival of Orthotopic Esophageal Cancer-Bearing Mice. Cancer Biol Ther (2013) 14:230–6. doi: 10.4161/cbt.23294
96. Choo AY, Yoon S-O, Kim SG, Roux PP, Blenis J. Rapamycin Differentially Inhibits S6Ks and 4E-BP1 to Mediate Cell-Type-Specific Repression of mRNA Translation. Proc Natl Acad Sci USA (2008) 105:17414–9. doi: 10.1073/pnas.0809136105
97. Fujishita T, Kojima Y, Kajino-Sakamoto R, Taketo MM, Aoki M. Tumor Microenvironment Confers mTOR Inhibitor Resistance in Invasive Intestinal Adenocarcinoma. Oncogene (2017) 36:6480–9. doi: 10.1038/onc.2017.242
98. Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III Study to Evaluate Temsirolimus Compared With Investigator’s Choice Therapy for the Treatment of Relapsed or Refractory Mantle Cell Lymphoma. J Clin Oncol (2009) 27:3822–9. doi: 10.1200/JCO.2008.20.7977
99. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Cutsem EV, et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med (2011) 364:514–23. doi: 10.1056/NEJMoa1009290
100. Huang Y, Xi Q, Chen Y, Wang J, Peng P, Xia S, et al. A Dual Mtorc1 and Mtorc2 Inhibitor Shows Antitumor Activity in Esophageal Squamous Cell Carcinoma Cells and Sensitizes Them to Cisplatin. Anti-Cancer Drugs (2013) 24:889–98. doi: 10.1097/CAD.0b013e328363c64e
101. Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, et al. TAK-228 (Formerly MLN0128), an Investigational Oral Dual TORC1/2 Inhibitor: A Phase I Dose Escalation Study in Patients With Relapsed or Refractory Multiple Myeloma, non-Hodgkin Lymphoma, or Waldenstrom’s Macroglobulinemia. Am J Hematol (2016) 91:400–5. doi: 10.1002/ajh.24300
102. Wu N, Zhu Y, Xu X, Zhu Y, Song Y, Pang L, et al. The Anti-Tumor Effects of Dual PI3K/mTOR Inhibitor BEZ235 and Histone Deacetylase Inhibitor Trichostatin A on Inducing Autophagy in Esophageal Squamous Cell Carcinoma. J Cancer (2018) 9:987–97. doi: 10.7150/jca.22861
103. Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, et al. First-In-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients With Advanced Cancer. Clin Cancer Res (2015) 21:1888–95. doi: 10.1158/1078-0432.CCR-14-1306
104. Deng L, Wu X, Zhu X, Yu Z, Liu Z, Wang J, et al. Combination Effect of Curcumin With Docetaxel on the PI3K/AKT/mTOR Pathway to Induce Autophagy and Apoptosis in Esophageal Squamous Cell Carcinoma. Am J Transl Res (2021) 13:57–72.
105. Wang YK, Chen WC, Lai YH, Chen YH, Wu MT, Kuo CT, et al. Influence of Tea Consumption on the Development of Second Esophageal Neoplasm in Patients With Head and Neck Cancer. Cancers (Basel) (2019). doi: 10.3390/cancers11030387
106. Shen CY, Jiang JG, Yang L, Wang DW, Zhu W. Anti-Ageing Active Ingredients From Herbs and Nutraceuticals Used in Traditional Chinese Medicine: Pharmacological Mechanisms and Implications for Drug Discovery. Br J Pharmacol (2017) 174:1395–425. doi: 10.1111/bph.13631
107. Foroozesh M, Primrose G, Guo Z, Bell LC, Alworth WL, Guengerich FP. Aryl Acetylenes as Mechanism-Based Inhibitors of Cytochrome P450-Dependent Monooxygenase Enzymes. Chem Res Toxicol (1997) 10:91–102. doi: 10.1021/tx960064g
108. Fuggetta MP, D’Atri S, Lanzilli G, Tricarico M, Cannavo E, Zambruno G, et al. In Vitro Antitumour Activity of Resveratrol in Human Melanoma Cells Sensitive or Resistant to Temozolomide. Melanoma Res (2004) 14:189–96. doi: 10.1097/01.cmr.0000130007.54508.b2
109. Osman AM, Bayoumi HM, Al-Harthi SE, Damanhouri ZA, Elshal MF. Modulation of Doxorubicin Cytotoxicity by Resveratrol in a Human Breast Cancer Cell Line. Cancer Cell Int (2012) 12:47. doi: 10.1186/1475-2867-12-47
110. Tang Q, Li G, Wei X, Zhang J, Chiu JF, Hasenmayer D, et al. Resveratrol-Induced Apoptosis is Enhanced by Inhibition of Autophagy in Esophageal Squamous Cell Carcinoma. Cancer Lett (2013) 336:325–37. doi: 10.1016/j.canlet.2013.03.023
111. Yang CS, Wang H. Cancer Preventive Activities of Tea Catechins. Molecules (2016). doi: 10.3390/molecules21121679
112. Hershey JW, Sonenberg N, Mathews MB. Principles of Translational Control: An Overview. Cold Spring Harb Perspect Biol (2012). doi: 10.1101/cshperspect.a011528
113. Verma M, Choi J, Cottrell KA, Lavagnino Z, Thomas EN, Pavlovic-Djuranovic S, et al. A Short Translational Ramp Determines the Efficiency of Protein Synthesis. Nat Commun (2019) 10:5774. doi: 10.1038/s41467-019-13810-1
114. Ali MU, Ur Rahman MS, Jia Z, Jiang C. Eukaryotic Translation Initiation Factors and Cancer. Tumour Biol (2017) 39:1010428317709805. doi: 10.1177/1010428317709805
115. Chen K, Yang J, Li J, Wang X, Chen Y, Huang S, et al. Eif4b is a Convergent Target and Critical Effector of Oncogenic Pim and PI3K/Akt/mTOR Signaling Pathways in Abl Transformants. Oncotarget (2016) 7:10073–89. doi: 10.18632/oncotarget.7164
116. Golob-Schwarzl N, Krassnig S, Toeglhofer AM, Park YN, Gogg-Kamerer M, Vierlinger K, et al. New Liver Cancer Biomarkers: PI3K/AKT/mTOR Pathway Members and Eukaryotic Translation Initiation Factors. Eur J Cancer (2017) 83:56–70. doi: 10.1016/j.ejca.2017.06.003
117. Hao P, Yu J, Ward R, Liu Y, Hao Q, An S, et al. Eukaryotic Translation Initiation Factors as Promising Targets in Cancer Therapy. Cell Commun Signal (2020) 18:175. doi: 10.1186/s12964-020-00607-9
118. Silvera D, Formenti SC, Schneider RJ. Translational Control in Cancer. Nat Rev Cancer (2010) 10:254–66. doi: 10.1038/nrc2824
119. Smolle E, Taucher V, Pichler M, Petru E, Lax S, Haybaeck J. Targeting Signaling Pathways in Epithelial Ovarian Cancer. Int J Mol Sci (2013) 14:9536–55. doi: 10.3390/ijms14059536
120. Golob-Schwarzl N, Wodlej C, Kleinegger F, Gogg-Kamerer M, Birkl-Toeglhofer AM, Petzold J, et al. Eukaryotic Translation Initiation Factor 6 Overexpression Plays a Major Role in the Translational Control of Gallbladder Cancer. J Cancer Res Clin Oncol (2019) 145:2699–711. doi: 10.1007/s00432-019-03030-x
121. Jackson RJ, Hellen CU, Pestova TV. The Mechanism of Eukaryotic Translation Initiation and Principles of its Regulation. Nat Rev Mol Cell Biol (2010) 11:113–27. doi: 10.1038/nrm2838
122. Spilka R, Ernst C, Mehta AK, Haybaeck J. Eukaryotic Translation Initiation Factors in Cancer Development and Progression. Cancer Lett (2013) 340:9–21. doi: 10.1016/j.canlet.2013.06.019
123. Walsh D, Perez C, Notary J, Mohr I. Regulation of the Translation Initiation Factor Eif4f by Multiple Mechanisms in Human Cytomegalovirus-Infected Cells. J Virol (2005) 79:8057–64. doi: 10.1128/jvi.79.13.8057-8064.2005
124. Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, et al. Topology and Regulation of the Human Eif4a/4G/4H Helicase Complex in Translation Initiation. Cell (2009) 136:447–60. doi: 10.1016/j.cell.2009.01.014
125. Masutani M, Sonenberg N, Yokoyama S, Imataka H. Reconstitution Reveals the Functional Core of Mammalian Eif3. EMBO J (2007) 26:3373–83. doi: 10.1038/sj.emboj.7601765
126. Pöyry TA, Kaminski A, Jackson RJ. What Determines Whether Mammalian Ribosomes Resume Scanning After Translation of a Short Upstream Open Reading Frame? Genes Dev (2004) 18:62–75. doi: 10.1101/gad.276504
127. Gandin V, Miluzio A, Barbieri AM, Beugnet A, Kiyokawa H, Marchisio PC, et al. Eukaryotic Initiation Factor 6 is Rate-Limiting in Translation, Growth and Transformation. Nature (2008) 455:684–8. doi: 10.1038/nature07267
128. Golob-Schwarzl N, Schweiger C, Koller C, Krassnig S, Gogg-Kamerer M, Gantenbein N, et al. Separation of Low and High Grade Colon and Rectum Carcinoma by Eukaryotic Translation Initiation Factors 1, 5 and 6. Oncotarget (2017) 8:101224–43. doi: 10.18632/oncotarget.20642
129. Smolle MA, Czapiewski P, Lapinska-Szumczyk S, Majewska H, Supernat A, Zaczek A, et al. The Prognostic Significance of Eukaryotic Translation Initiation Factors (eIFs) in Endometrial Cancer. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20246169
130. Skofler C, Kleinegger F, Krassnig S, Birkl-Toeglhofer AM, Singer G, Till H, et al. Eukaryotic Translation Initiation Factor 4ai: A Potential Novel Target in Neuroblastoma. Cells (2021) 10. doi: 10.3390/cells10020301
131. Krassnig S, Wohlrab C, Golob-Schwarzl N, Raicht A, Schatz C, Birkl-Toeglhofer AM, et al. A Profound Basic Characterization of eIFs in Gliomas: Identifying Eif3i and 4H as Potential Novel Target Candidates in Glioma Therapy. Cancers (2021) 13. doi: 10.3390/cancers13061482
132. Ewens KG, Kanetsky PA, Richards-Yutz J, Purrazzella J, Shields CL, Ganguly T, et al. Chromosome 3 Status Combined With BAP1 and EIF1AX Mutation Profiles Are Associated With Metastasis in Uveal Melanoma. Invest Ophthalmol Vis Sci (2014) 55:5160–7. doi: 10.1167/iovs.14-14550
133. Karunamurthy A, Panebianco F, J.Hsiao S, Vorhauer J, Nikiforova MN, Chiosea S, et al. Prevalence and Phenotypic Correlations of EIF1AX Mutations in Thyroid Nodules. Endocr Relat Cancer (2016) 23:295–301. doi: 10.1530/erc-16-0043
134. Etemadmoghadam D, Azar WJ, Lei Y, Moujaber T, Garsed DW, Kennedy CJ, et al. EIF1AX and NRAS Mutations Co-Occur and Cooperate in Low-Grade Serous Ovarian Carcinomas. Cancer Res (2017) 77:4268–78. doi: 10.1158/0008-5472.Can-16-2224
135. Sehrawat U, Koning F, Ashkenazi S, Stelzer G, Leshkowitz D, Dikstein R. Cancer-Associated Eukaryotic Translation Initiation Factor 1a Mutants Impair Rps3 and Rps10 Binding and Enhance Scanning of Cell Cycle Genes. Mol Cell Biol (2019). doi: 10.1128/MCB.00441-18
136. Zhang L, Smit-McBride Z, Pan X, Rheinhardt J, Hershey JW. An Oncogenic Role for the Phosphorylated H-Subunit of Human Translation Initiation Factor Eif3. J Biol Chem (2008) 283:24047–60. doi: 10.1074/jbc.M800956200
137. Nupponen NN, Porkka K, Kakkola L, Tanner M, Persson K, Borg A, et al. Amplification and Overexpression of P40 Subunit of Eukaryotic Translation Initiation Factor 3 in Breast and Prostate Cancer. Am J Pathol (1999) 154:1777–83. doi: 10.1016/s0002-9440(10)65433-8
138. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA Circularization by METTL3-Eif3h Enhances Translation and Promotes Oncogenesis. Nature (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
139. Golob-Schwarzl N, Puchas P, Gogg-Kamerer M, Weichert W, GÖPpert B, Haybaeck J. New Pancreatic Cancer Biomarkers Eif1, Eif2d, Eif3c and Eif6 Play a Major Role in Translational Control in Ductal Adenocarcinoma. Anticancer Res (2020) 40:3109–18. doi: 10.21873/anticanres.14292
140. Liu T, Li R, Zhao H, Deng J, Long Y, Shuai MT, et al. Eif4e Promotes Tumorigenesis and Modulates Chemosensitivity to Cisplatin in Esophageal Squamous Cell Carcinoma. Oncotarget (2016) 7:66851–64. doi: 10.18632/oncotarget.11694
141. Yang H, Li XD, Zhou Y, Ban X, Zeng TT, Li L, et al. Stemness and Chemotherapeutic Drug Resistance Induced by EIF5A2 Overexpression in Esophageal Squamous Cell Carcinoma. Oncotarget (2015) 6:26079–89. doi: 10.18632/oncotarget.4581
142. Assouline S, Culjkovic-Kraljacic B, Bergeron J, Caplan S, Cocolakis E, Lambert C, et al. A Phase I Trial of Ribavirin and Low-Dose Cytarabine for the Treatment of Relapsed and Refractory Acute Myeloid Leukemia With Elevated Eif4e. Haematologica (2015) 100:e7–9. doi: 10.3324/haematol.2014.111245
143. Duffy AG, Makarova-Rusher OV, Ulahannan SV, Rahma OE, Fioravanti S, Walker M, et al. Modulation of Tumor Eif4e by Antisense Inhibition: A Phase I/II Translational Clinical Trial of ISIS 183750-an Antisense Oligonucleotide Against Eif4e-in Combination With Irinotecan in Solid Tumors and Irinotecan-Refractory Colorectal Cancer. Int J Cancer (2016) 139:1648–57. doi: 10.1002/ijc.30199
144. Casaos J, Gorelick NL, Huq S, Choi J, Xia Y, Serra R, et al. The Use of Ribavirin as an Anticancer Therapeutic: Will It Go Viral? Mol Cancer Ther (2019) 18:1185–94. doi: 10.1158/1535-7163.Mct-18-0666
145. Liu WL, Su WC, Cheng CW, Hwang LH, Wang CC, Chen HL, et al. Ribavirin Up-Regulates the Activity of Double-Stranded RNA-Activated Protein Kinase and Enhances the Action of Interferon-Alpha Against Hepatitis C Virus. J Infect Dis (2007) 196:425–34. doi: 10.1086/518894
146. Crotty S, Cameron C, Andino R. Ribavirin’s Antiviral Mechanism of Action: Lethal Mutagenesis? J Mol Med (Berl) (2002) 80:86–95. doi: 10.1007/s00109-001-0308-0
147. Loustaud-Ratti V, Debette-Gratien M, Jacques J, Alain S, Marquet P, Sautereau D, et al. Ribavirin: Past, Present and Future. World J Hepatol (2016) 8:123–30. doi: 10.4254/wjh.v8.i2.123
148. Tan K, Culjkovic B, Amri A, Borden KL. Ribavirin Targets Eif4e Dependent Akt Survival Signaling. Biochem Biophys Res Commun (2008) 375:341–5. doi: 10.1016/j.bbrc.2008.07.163
149. Chen J, Xu X, Chen J. Clinically Relevant Concentration of Anti-Viral Drug Ribavirin Selectively Targets Pediatric Osteosarcoma and Increases Chemosensitivity. Biochem Biophys Res Commun (2018) 506:604–10. doi: 10.1016/j.bbrc.2018.10.124
150. Jin J, Xiang W, Wu S, Wang M, Xiao M, Deng A. Targeting Eif4e Signaling With Ribavirin as a Sensitizing Strategy for Ovarian Cancer. Biochem Biophys Res Commun (2019) 510:580–6. doi: 10.1016/j.bbrc.2019.01.117
151. Huq S, Casaos J, Serra R, Peters M, Xia Y, Ding AS, et al. Repurposing the FDA-Approved Antiviral Drug Ribavirin as Targeted Therapy for Nasopharyngeal Carcinoma. Mol Cancer Ther (2020) 19:1797–808. doi: 10.1158/1535-7163.MCT-19-0572
Keywords: esophageal squamous cell carcinoma (ESCC), PI3K/AKT/mTOR signaling pathway, inhibitors, eukaryotic translation initiation factors (eIFs), therapeutic target
Citation: Huang R, Dai Q, Yang R, Duan Y, Zhao Q, Haybaeck J and Yang Z (2022) A Review: PI3K/AKT/mTOR Signaling Pathway and Its Regulated Eukaryotic Translation Initiation Factors May Be a Potential Therapeutic Target in Esophageal Squamous Cell Carcinoma. Front. Oncol. 12:817916. doi: 10.3389/fonc.2022.817916
Received: 18 November 2021; Accepted: 01 April 2022;
Published: 28 April 2022.
Edited by:
Jiang Chen, Zhejiang University, ChinaCopyright © 2022 Huang, Dai, Yang, Duan, Zhao, Haybaeck and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Haybaeck, am9oYW5uZXMuaGF5YmFlY2tAaS1tZWQuYWMuYXQ=; Zhihui Yang, eXpoaWg3M0Bzd211LmVkdS5jbg==