 Ian Burns1†
Ian Burns1† William D. Gwynne2†
William D. Gwynne2† Yujin Suk1,3
Yujin Suk1,3 Stefan Custers3
Stefan Custers3 Iqra Chaudhry3
Iqra Chaudhry3 Chitra Venugopal2
Chitra Venugopal2 Sheila K. Singh2*
Sheila K. Singh2*- 1Michael G. DeGroote School of Medicine, McMaster University, Hamilton, ON, Canada
- 2Department of Surgery, McMaster University, Hamilton, ON, Canada
- 3Department of Biochemistry and Biomedical Sciences, Faculty of Health Sciences, McMaster University, Hamilton, ON, Canada
Pediatric central nervous system (CNS) tumors are the most common solid tumors diagnosed in children and are the leading cause of pediatric cancer-related death. Those who do survive are faced with the long-term adverse effects of the current standard of care treatments of chemotherapy, radiation, and surgery. There is a pressing need for novel therapeutic strategies to treat pediatric CNS tumors more effectively while reducing toxicity – one of these novel modalities is chimeric antigen receptor (CAR) T-cell therapy. Currently approved for use in several hematological malignancies, there are promising pre-clinical and early clinical data that suggest CAR-T cells could transform the treatment of pediatric CNS tumors. There are, however, several challenges that must be overcome to develop safe and effective CAR T-cell therapies for CNS tumors. Herein, we detail these challenges, focusing on those unique to pediatric patients including antigen selection, tumor immunogenicity and toxicity. We also discuss our perspective on future avenues for CAR T-cell therapies and potential combinatorial treatment approaches.
Introduction
Pediatric central nervous system (CNS) tumors are the most common solid tumors diagnosed in children (1). Despite advances in the molecular characterization of these tumors and the fine-tuning of multimodal therapies, numerous patients experience high rates of tumor recurrence and mortality (2, 3). In fact, CNS tumors are the leading cause of pediatric cancer-related death, recently surpassing leukemia (1, 4). Those who survive face lifelong challenges associated with the standard of care (SoC) treatment, which usually consists of surgery, chemotherapy and/or local or craniospinal irradiation. Chemotherapy leaves patients with off-target organ damage and often neurocognitive deficits (5), and radiation causes debilitating damage to the developing brain (6). Given this persistent mortality and morbidity, there is an urgent need for novel therapies that effectively eradicate CNS tumors in children, providing durable remissions while minimizing treatment-related toxicity.
Recent developments in cancer immunotherapy have unveiled targeted treatment strategies that can prevent tumor recurrence and negate long-term neurotoxic sequalae caused by cytotoxic therapies. Immune checkpoint inhibition with antibodies targeting programmed cell death protein 1 (PD-1) and CTLA4 demonstrates superior efficacy in comparison to the SoC in several cancers (7, 8). In many children with relapsed and treatment-refractory leukemia, treatment with Chimeric Antigen Receptor (CAR) T-cells has led to durable remission (9, 10). CAR T-cells are generated by engineering patient T-cells to express the hybrid CAR protein, which contains an extracellular antibody-like domain that recognizes a cancer-specific antigen and intracellular signalling components that trigger an immune response (11). Each new generation of CAR T-cell has comprised more sophisticated co-stimulatory signalling domains, including CD28 and 4-1BB, and other genetic modifications, such as transgenes for cytokine secretion, to optimize anti-tumor activity (12). Currently, CAR T-cells are approved for use in hematological malignancies including relapsed/refractory non-Hodgkin lymphoma, multiple myeloma, and pediatric relapsed acute lymphoblastic leukemia (13). Solid tumors have emerged as the next frontier for CAR T-cell therapies.
Pre-clinical and early clinical trial data have suggested that CAR T-cells could play an important role in the treatment of pediatric CNS tumors, including medulloblastomas (MB), atypical rhabdoid teratoid tumors (ATRT), high grade gliomas (HGG) and ependymomas (14–16). Many obstacles remain, however, to the successful development of CAR T-cell therapies in pediatric CNS tumors. The paucity of targetable antigens and the unfavourable immunological characteristics of these tumors present unique challenges, and children have unique and poorly understood vulnerabilities to treatment-related toxicities. Herein, we review the major challenges associated with developing CAR T-cell therapies specifically for pediatric CNS tumors and present our perspective on possible avenues for the future development of more effective CAR T-cell and combinatorial immunotherapies.
Challenges
Antigen Selection
Whereas adult CNS tumors display an abundance of neoantigens that arise from high mutational burden, there is a marked paucity of neoantigens on pediatric CNS tumors (17, 18). Children are exposed to fewer environmental factors that contribute to DNA damage and the resultant lack of neoantigens presents a unique challenge for pediatric immunotherapy target selection. Target antigens should have tumor-specific (little to no expression in normal cells) or tumor-associated (overexpressed in tumor tissue) expression to spare the developing brain from off-tumor toxicity (19). One promising strategy to overcome the paucity of true neoantigens is to target oncofetal antigens, a class of cell surface markers normally expressed exclusively during prenatal tissue development that can become re-expressed during neoplastic transformation (20). For example, CAR T-cells have been developed to target tumor-specific exons of the oncofetal antigen cerebroglycan GPC2 (21, 22). Alternatively, they can be made to target tumor-specific antigen epitopes. CAR T-cells targeting the epidermal growth factor receptor (EGFR) 806 epitope that is uniquely expressed on the surface of tumor cells can effectively eradicate glioblastoma (GBM) cells while sparing EGFR-expressing human fetal astrocytes (23).
In addition to a reduced neoantigen abundance, there is extensive intratumoral phenotypic heterogeneity among pediatric CNS tumor cells (24). Brain tumor initiating cells (BTICs) are an infrequent subpopulation of tumuor cells CAR T-cell that share properties with normal stem cells, including the capacity for limitless self-renewal and proliferation. BTICs are resistant to chemotherapy (25) and radiation (26) and seed pediatric CNS tumor recurrence and leptomeningeal metastasis (27, 28). The identification of a target that selectively marks BTICs may provide an effective means to eradicate therapy refractory tumor cells, thus delaying or preventing recurrence. Unfortunately, existing BTIC markers amenable to immunotherapy in adult gliomas, such as prominin 1 (PROM1; CD133), are also expressed by human neural stem and progenitor cells (29).
Selection of tumor cells with reduced target antigen expression throughout the course of treatment will also induce temporal heterogeneity. This antigen escape is an impediment to effective CAR T-cell treatment (24, 30). Multivalent CARs are a potential way to improve targeting of tumors with heterogenous antigen expression. Bielamowicz et al. demonstrated improved anti-tumor efficacy in GBM models using trivalent CAR T-cells targeting ephrin A receptor 2, human epidermal growth factor receptor 2 (HER2) and interleukin-13 receptor alpha-2 (IL13Rα2) (30). With the same trivalent design, a significant survival benefit was observed in patient-derived xenograft (PDX) models of MB and ependymoma. Notably, modest expression of HER2 and IL13Rα2 on patient samples in this study suggests additional, more highly expressed targets are needed (15).
Currently, there are a limited number of CAR T-cell clinical trials for children with CNS tumors, all at phase I. Targets include HER2, B7 homolog 3 (B7H3), EGFR806, the disialoganglioside GD2 and IL13Rα2 (Table 1).

Table 1 Current clinical trials investigating CAR T-cells for pediatric CNS tumors.
Delivery
In comparison to hematological malignancies, solid tumors and especially CNS tumors situated behind the blood brain barrier (BBB) present unique physical challenges that hinder effective delivery of CAR T-cells. While peripherally infused CAR T-cells have been found to modestly cross the BBB (31–33), numerous pre-clinical studies evaluating the comparative efficacy of intravenous (IV), intratumoral (IT) and intraventricular (ICV) delivery of CAR T-cells targeting CNS tumors have produced compelling evidence favoring locoregional administration via surgically-inserted catheter (IT or ICV). Locoregional delivery is associated with more effective tumor infiltration, improved anti-tumor efficacy, and reduced systemic toxicity (16, 34–36). For example, Theruvath et al. tested B7H3 CAR T-cells against ATRT patient-derived xenografts in mice and showed dramatically more rapid tumor homing and expansion with locoregional delivery, in comparison to the far higher doses of CAR T-cells delivered via IV. Additionally, significantly higher levels of systemic inflammatory cytokines were detected upon IV delivery (16). Notably, ICV delivery may be superior to IT in cases of leptomeningeal spread, as CAR T-cells are able to more freely traffic throughout the CNS (34). In current pediatric clinical trials, locoregional delivery is the preferred method (Table 1).
Homing and Persistence
Other important challenges impeding the development of effective CAR T-cells for pediatric CNS tumors include CAR T-cell homing and persistence. To improve homing to tumor sites, CAR T-cells expressing chemokine receptors have been developed (37, 38). Once CAR T-cells reach target sites, they must be capable of exerting an antitumoral response prior to exhaustion. Should exhaustion occur prior to tumor clearance, CAR T-cell efficacy drops dramatically. A recent study found that co-expression of AP1 transcription factor, c-Jun, in CAR T-cells led to an increased capacity for expansion, and diminished terminal differentiation. These exhaustion-resistant CAR T-cells also exhibit a dramatic increase in antitumoral efficacy (39).
Additional strategies to improve CAR T-cell persistence and reduce exhaustion include optimizing T-cell activation and co-stimulation signalling and interfering with molecules that impair T-cell activation (40). For example, CAR T-cells engineered to express pro-inflammatory cytokines such as IL-12 and IL-18 and those with constitutively active IL-15 and IL-7 have increased anti-tumor efficacy and improved persistence in solid tumors (41–44). Particularly in immunologically “cold” pediatric CNS tumors, additional inflammatory cytokine secretion by CAR T-cells could also augment local immune cell activation. This benefit must be balanced with local and systemic toxicity associated with increased cytokine production (40). Finally, issues of CAR-T cell persistence can be addressed by optimizing the timing of their delivery. For example, the use of small, frequent (usually weekly) dosing regimens may help maximize the therapeutic window while minimizing infusion-associated toxicity (37). It is unclear, however, whether frequent CAR T-cell dosing translates to improved anti-tumor efficacy in comparison to infrequent or one-time dosing.
Toxicity
Cytokine release syndrome (CRS), a systemic inflammatory response following excess cytokine production by endogenous immune cells and/or CAR T-cells, and the toxic encephalopathy known as immune effector cell-associated neurotoxicity syndrome (ICANS) that often follows, are major systemic side effects of CAR T-cell therapies targeting hematological malignancies (45). Relatively little is known regarding these toxicities in the context of CAR T-cells for CNS tumors, especially in pediatrics. Nevertheless, the locoregional delivery strategies currently employed with many CNS-targeting CAR T-cell therapies reduce much of the concern for systemic toxicity, which is known to be a dose-dependent (46) manifestation of the systemic administration and peripheral activation of CAR T-cells (16, 45). This is in keeping with the CRS reported by Goff et al. after IV infusion of only the highest dose of EGFRvIII-targeting CAR T-cells in a GBM patient (47), and that most trials with CAR T-cells targeting CNS tumors have shown few adverse events (48). There is, however, reasonable concern for excess cytokine production leading to local CNS toxicity following locoregional delivery. Promisingly, 3 pediatric patients recently treated with locally-infused CAR T-cells targeting HER2 experienced no dose limiting toxicity while still showing local CNS immune activation (49). Interestingly, CRS and ICANS were not predicted by pre-clinical studies of CD19-targeting CAR T-cells (45) – perhaps similarly unexpected toxicities will emerge through the development of CAR T-cells for CNS tumors.
Given that CAR T-cell dosing, antigen affinity and other design factors remain largely empiric, off-target and particularly on-target/off-tumor toxicity are major concerns. Illustrating this concern, Richman et al. showed that high-affinity CAR T-cells targeting GD2 caused fatal encephalitis after acting on normal brain tissues expressing GD2 in a neuroblastoma mouse model (50). It has also been observed that ICV-administered CAR T-cells migrate effectively into the periphery (16), suggesting that even with locoregional delivery strategies, off-tumor toxicity within the periphery must be considered.
In creating CAR T-cells for the pediatric population, attention must be drawn to the fact that the childhood brain and other tissues are still developing and also have different antigen expression in comparison to adults. This is particularly relevant with CAR T-cells targeting known or potential stem cell antigens. For example, CD133 is expressed on neural stem cells (51) and hematopoietic stem cells (52). Hence, while treatment with CD133-targeting CAR T-cells may be tolerated in adults with GBM, this target may not be appropriate in pediatric patients. Preclinical development of novel targets must ensure proper examination of appropriate control tissues, such as human neural stem cells and fetal tissue arrays, to get insights into potential toxicities. Building inducible control into CAR T-cells provides clinicians with the ability to rapidly regulate CAR T-cell activity during treatment and in case of anticipated or unanticipated toxicities. These include suicide genes such as inducible Caspase 9 and herpes simplex virus tyrosine kinase, and cell surface elimination markers that allow for antibody-mediated control (53).
Tumor Immune Microenvironment
Tumors comprise a distinct network of tumor cells, immune cells, stromal cells, and extracellular matrix proteins, a spectrum collectively termed the tumor immune microenvironment (TIME). Immunologically “hot” tumors comprise high numbers of tumor-infiltrating lymphocytes (TILs) and increased PD-1 ligand expression, whereas immunologically “cold” tumors have low numbers of TILs and reduced PD-1 expression. Pediatric CNS tumors are immunologically cold due to their low mutational burden and a lack of neoantigen expression (54, 55). Cold tumors respond poorly to immune checkpoint inhibition (56) and are associated with poor clinical outcomes (18, 57). Colder tumors are also less responsive to adoptive T-cell and CAR T-cell therapies (58, 59). In such cases, administered CAR T-cells must be capable of activation and infiltration, where endogenous T-cells are unable to do the same. To overcome the cold TIMEs of pediatric CNS tumors, novel CAR T-cell engineering approaches can be applied to optimize their function in these environments. Potential tools include cytokine switch receptors, which transform an inhibitory signal into a growth-inducing signal, and optimization of CAR T-cell metabolism in the hypoxic and reactive oxygen species-filled microenvironment (40).
In addition to being immunologically cold, there is substantial heterogeneity in the TIME between and among pediatric CNS tumor types. To develop effective immunotherapies, this heterogeneity must be understood and exploited. Grabovska et al. analyzed genome-wide DNA methylation data from >6,000 pediatric CNS tumors – interestingly, the immune infiltrate subgroups that they identified exist independent of molecular subgroup and are predictive of outcomes in multiple pediatric tumor types. They also showed that specific molecular drivers like H3.3G34 mutations in HGG are associated with characteristic immune infiltrates independent of tumor subtype (18). In MB, several studies have shown that Sonic Hedgehog tumors have an increased proportion of T-cells in comparison to other subgroups, rendering them promising candidates for immunotherapy (18, 60). Notably, pediatric midline gliomas are exceptionally immunologically cold and have very low inflammatory cytokine expression (61). In comparison to normal brain tissue, Diffuse Intrinsic Pontine Glioma (DIPG) tumors do not display increased macrophage or T-cell infiltration, or PD1L expression (62).
Looking forward, a deeper understanding of the heterogenous and cold TIMEs of pediatric CNS tumors will allow for the development of novel treatment approaches that help overcome these unfavorable environments. In addition to novel CAR T-cell design, combining CAR T-cells with other immunotherapies or small molecules may allow for the induction of a potent inflammatory response and improve outcomes.
Combinatorial Therapies
Agents, including small-molecule drugs and other immunotherapies, that can prime CAR T-cells to overcome immunosuppressive effects of tumor cells or those that can convert a cold TIME into a hot TIME may act in combination with CAR T-cell therapies to elicit a more powerful antitumoral response in the pediatric CNS (63). Inhibition of the PD-1/PD-1 ligand immune checkpoint axis, which tumor cells exploit to avoid detection from host immune cells, is a strategy that may enhance the activity of CAR T-cells through increased target engagement (63). The development of small molecules capable of targeting PD-1 have been hindered, however, in part due to the hydrophobic PD-1/PDL-1 interface. The use of cytotoxic/cytolytic agents like cisplatin chemotherapy (64, 65), or oncolytic viruses such as HSV-1 G207 (66), can also enhance the effectiveness of immunotherapy by releasing tumor-associated antigens and cytosolic DNA that promote the conversion of a typically cold pediatric TIME into a hot TIME. The latter presents a potential treatment window of opportunity in pediatric CNS brain tumor patients that are treated with chemoradiotherapy. Researchers have exploited a metabolic vulnerability of immunosuppressive regulatory T-cells (T-reg) to overcome their immunosuppressive nature. Small molecule inhibitors of Indoleamine-pyrrole 2,3-dioxygenase (IDO1) reduce T-reg activity in the TIME and increase immunotherapy efficacy (67).
The capacity for small molecules to be administered systemically, penetrate the BBB, and modulate intracellular targets provides combinatorial immunotherapeutic opportunities for small-molecule agents that monoclonal antibodies and other larger molecules cannot fulfill. Cytotoxic and cytolytic agents also have the potential to greatly enhance the efficacy of CAR T-cell therapies. These combinatorial treatment approaches may be the key to overcoming the challenges presented by solid pediatric CNS tumors.
Discussion
CAR T-cell therapies for hematological malignancies represent major breakthroughs in cancer research and adapting CAR T-cells to target solid tumors represents the next frontier. Here we have reviewed the unique physical and biological challenges associated with developing CAR T-cells for pediatric CNS tumors, and highlighted promising avenues of current and future research (Figure 1). The paucity of targetable antigens, intratumoral heterogeneity, and the co-expression of many potential antigens in normal and developing tissues are all fundamental challenges. Potential solutions include using appropriate preclinical controls, exploring BTIC-specific antigens and novel CAR T-cell engineering strategies such as multivalent CARs. In terms of CAR T-cell administration, IT and ICV methods improve delivery and reduce systemic toxicity. There are also many unknowns regarding the local and systemic toxicity of CAR T-cell therapies for pediatric brain tumors and therefore, a cautious approach guided by an awareness of the potential unique susceptibilities of the pediatric brain is called for. It is unclear how treatment of CNS tumors with CAR T-cells may impact brain development. Other novel approaches are also necessary to improve the homing and persistence of administered cells. Finally, the cold and heterogeneous TIMEs of some pediatric CNS tumors necessitate the development and application of novel combinatorial therapies to support CAR T-cells in generating an immune response sufficient to eradicate tumor cells. With creative use of existing and novel therapies and continued innovation in CAR T-cell design, there is potential for a new era of improved outcomes and reduced toxicity for children with CNS tumors.
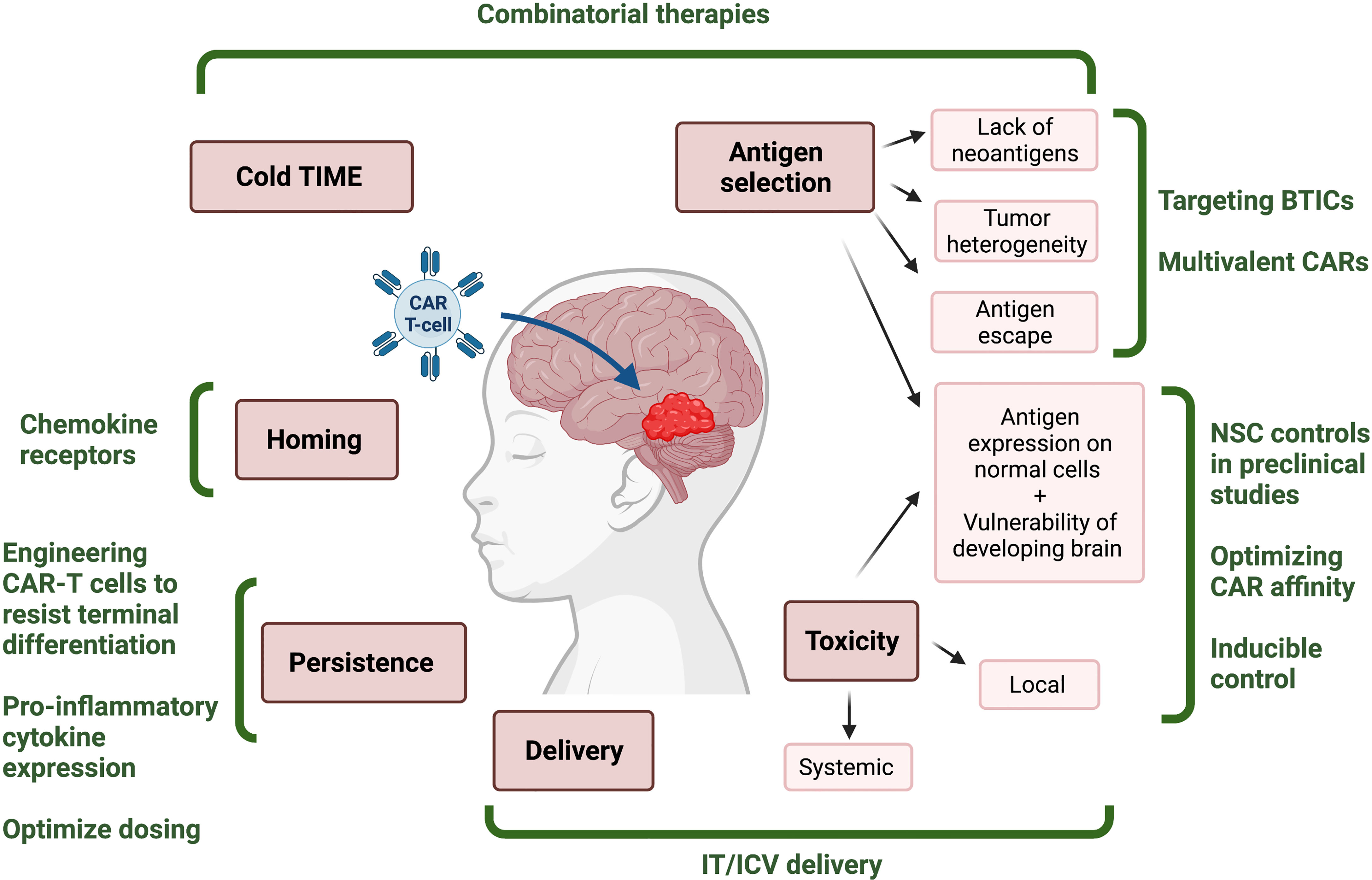
Figure 1 Challenges and potential solutions for development of effective CAR T-cell therapies for pediatric CNS tumors. Infographic depicting the challenges (red) associated with immunotherapies for malignant pediatric CNS tumors, and the proposed solutions (green) that might mitigate them. TIME, tumor immune microenvironment; BTIC, brain tumor initiating cell; NSC, neural stem cell; IT, intrathecal; ICV, intraventricular. Figured created with BioRender.com.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
IB, WG, YS, SC, and IC contributed to the conception and drafting of the manuscript. All authors reviewed and approved the final version.
Conflict of Interest
SS is a shareholder and a scientific advisory board member of Century Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol (2020) 22(12 Suppl 2):iv1–iv96. doi: 10.1093/neuonc/noaa200
2. Girardi F, Allemani C, Coleman MP. Worldwide Trends in Survival From Common Childhood Brain Tumors: A Systematic Review. J Global Oncol (2019) 5):1–25. doi: 10.1200/jgo.19.00140
3. Hill RM, Richardson S, Schwalbe EC, Hicks D, Lindsey JC, Crosier S, et al. Time, Pattern, and Outcome of Medulloblastoma Relapse and Their Association With Tumour Biology at Diagnosis and Therapy: A Multicentre Cohort Study. Lancet Child Adolesc Health (2020) 4(12):865–74. doi: 10.1016/S2352-4642(20)30246-7
4. Withrow DR, Berrington de Gonzalez A, Lam CJK, Warren KE, Shiels MS. Trends in Pediatric Central Nervous System Tumor Incidence in the United States 1998-2013. Cancer Epidemiol Biomark Prev (2019) 28(3):522–30. doi: 10.1158/1055-9965.Epi-18-0784
5. Askins MA, Moore BD. Preventing Neurocognitive Late Effects in Childhood Cancer Survivors. J Child Neurol (2008) 23(10):1160–71. doi: 10.1177/0883073808321065
6. Radcliffe J, Packer RJ, Atkins TE, Bunin GR, Schut L, Goldwein JW, et al. Three- and Four-Year Cognitive Outcome in Children With Noncortical Brain Tumors Treated With Whole-Brain Radiotherapy. Ann Neurol (1992) 32(4):551–4. doi: 10.1002/ana.410320411
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
8. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab Plus Ipilimumab Versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
9. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
10. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
11. Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov (2013) 3(4):388–98. doi: 10.1158/2159-8290.Cd-12-0548
12. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus Med Hemother (2019) 46(1):15–24. doi: 10.1159/000496870
13. Goldsmith SR, Ghobadi A, DiPersio JF. Hematopoeitic Cell Transplantation and CAR T-Cell Therapy: Complements or Competitors? Front Oncol (2020) 10:608916–608916. doi: 10.3389/fonc.2020.608916
14. Wang SS, Bandopadhayay P, Jenkins MR. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol (2019) 40(8):748–61. doi: 10.1016/j.it.2019.05.009
15. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional Delivery of CAR T Cells to the Cerebrospinal Fluid for Treatment of Metastatic Medulloblastoma and Ependymoma. Nat Med (2020) 26(5):720–31. doi: 10.1038/s41591-020-0827-2
16. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally Administered B7-H3-Targeted CAR T Cells for Treatment of Atypical Teratoid/Rhabdoid Tumors. Nat Med (2020) 26(5):712–9. doi: 10.1038/s41591-020-0821-8
17. Hutzen B, Ghonime M, Lee J, Mardis ER, Wang R, Lee DA, et al. Immunotherapeutic Challenges for Pediatric Cancers. Mol Ther Oncolyt (2019) 15:38–48. doi: 10.1016/j.omto.2019.08.005
18. Grabovska Y, Mackay A, O’Hare P, Crosier S, Finetti M, Schwalbe EC, et al. Pediatric Pan-Central Nervous System Tumor Analysis of Immune-Cell Infiltration Identifies Correlates of Antitumor Immunity. Nat Commun (2020) 11(1): 4324. doi: 10.1038/s41467-020-18070-y
19. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol (2015) 33(24):2646–54. doi: 10.1200/jco.2014.60.1591
21. Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell (2017) 32(3):295–309.e212. doi: 10.1016/j.ccell.2017.08.003
22. Li N, Torres MB, Spetz MR, Wang R, Peng L, Tian M, et al. CAR T Cells Targeting Tumor-Associated Exons of Glypican 2 Regress Neuroblastoma in Mice. Cell Rep Med (2021) 2(6):100297. doi: 10.1016/j.xcrm.2021.100297
23. Ravanpay AC, Gust J, Johnson AJ, Rolczynski LS, Cecchini M, Chang CA, et al. EGFR806-CAR T Cells Selectively Target a Tumor-Restricted EGFR Epitope in Glioblastoma. Oncotarget (2019) 10:7080–95. doi: 10.18632/oncotarget.27389
24. Kabir TF, Kunos CA, Villano JL, Chauhan A. Immunotherapy for Medulloblastoma: Current Perspectives. Immunotargets Ther (2020) 9:57–77. doi: 10.2147/ITT.S198162
25. Beier D, Schriefer B, Brawanski K, Hau P, Weis J, Schulz JB, et al. Efficacy of Clinically Relevant Temozolomide Dosing Schemes in Glioblastoma Cancer Stem Cell Lines. J Neurooncol (2012) 109(1):45–52. doi: 10.1007/s11060-012-0878-4
26. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444(7120):756–60. doi: 10.1038/nature05236
27. Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, et al. Quiescent Sox2(+) Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell (2014) 26(1):33–47. doi: 10.1016/j.ccr.2014.05.005
28. Kahn SA, Wang X, Nitta RT, Gholamin S, Theruvath J, Hutter G, et al. Notch1 Regulates the Initiation of Metastasis and Self-Renewal of Group 3 Medulloblastoma. Nat Commun (2018) 9(1):4121. doi: 10.1038/s41467-018-06564-9
29. Vora P, Venugopal C, Salim SK, Tatari N, Bakhshinyan D, Singh M, et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell (2020) 26(6):832–44.e836. doi: 10.1016/j.stem.2020.04.008
30. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro Oncol (2018) 20(4):506–18. doi: 10.1093/neuonc/nox182
31. Davila ML, Brentjens RJ. CD19-Targeted CAR T Cells as Novel Cancer Immunotherapy for Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia. Clin Adv Hematol Oncol (2016) 14(10):802–8.
32. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity After Adoptive Immunotherapy With CD19 CAR-T Cells. Cancer Discov (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
33. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Trans Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
34. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-Tumor Efficacy Against Glioblastoma. Mol Ther (2018) 26(1):31–44. doi: 10.1016/j.ymthe.2017.10.002
35. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res An Off J Am Assoc Cancer Res (2018) 24(1):95–105. doi: 10.1158/1078-0432.CCR-17-2041
36. Mulazzani M, Fräßle SP, von Mücke-Heim I, Langer S, Zhou X, Ishikawa-Ankerhold H, et al. Long-Term In Vivo Microscopy of CAR T Cell Dynamics During Eradication of CNS Lymphoma in Mice. Proc Natl Acad Sci USA (2019) 116(48):24275–84. doi: 10.1073/pnas.1903854116
37. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol Rev (2019) 290(1):60–84. doi: 10.1111/imr.12773
38. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10(1):4016. doi: 10.1038/s41467-019-11869-4
39. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
40. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
41. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors. Vivo Oncoimmunol (2015) 4(3):e994446. doi: 10.4161/2162402x.2014.994446
42. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc Natl Acad Sci USA (2016) 113(48):E7788–e7797. doi: 10.1073/pnas.1610544113
43. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
44. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov (2017) 7(11):1238–47. doi: 10.1158/2159-8290.Cd-17-0538
45. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat Rev Immunol (2021). doi. doi: 10.1038/s41577-021-00547-6
46. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome After CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
47. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother (Hagerstown Md 1997) (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
48. Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front Neurosci (2021) 15:662064(535). doi: 10.3389/fnins.2021.662064
49. Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Künkele A, et al. Locoregional Infusion of HER2-Specific CAR T Cells in Children and Young Adults With Recurrent or Refractory CNS Tumors: An Interim Analysis. Nat Med (2021) 27:1544–52. doi: 10.1038/s41591-021-01404-8
50. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol Res (2018) 6(1):36–46. doi: 10.1158/2326-6066.Cir-17-0211
51. Schwartz PH, Bryant PJ, Fuja TJ, Su H, O'Dowd DK, Klassen H. Isolation and Characterization of Neural Progenitor Cells From Post-Mortem Human Cortex. J Neurosci Res (2003) 74(6):838–51. doi: 10.1002/jnr.10854
52. Handgretinger R, Kuçi S. CD133-Positive Hematopoietic Stem Cells: From Biology to Medicine. Adv Exp Med Biol (2013) 777:99–111. doi: 10.1007/978-1-4614-5894-4_7
53. Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front Immunol (2020) 11:326(326). doi: 10.3389/fimmu.2020.00326
54. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The Landscape of Genomic Alterations Across Childhood Cancers. Nature (2018) 555(7696):321–7. doi: 10.1038/nature25480
55. Abedalthagafi M, Mobark N, Al-Rashed M, AlHarbi M. Epigenomics and Immunotherapeutic Advances in Pediatric Brain Tumors. NPJ Precis Oncol (2021) 5(1):34. doi: 10.1038/s41698-021-00173-4
56. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J Immunother Cancer (2018) 6(1):157. doi: 10.1186/s40425-018-0479-7
57. Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME, et al. CD8+ T-Cell Infiltrate in Newly Diagnosed Glioblastoma Is Associated With Long-Term Survival. J Clin Neurosci (2010) 17(11):1381–5. doi: 10.1016/j.jocn.2010.03.031
58. Galon J, Rossi J, Turcan S, Danan C, Locke FL, Neelapu SS, et al. Characterization of Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell-Mediated Tumor Microenvironment Immune Gene Profile in a Multicenter Trial (ZUMA-1) With Axicabtagene Ciloleucel (Axi-Cel, KTE-C19). J Clin Oncol (2017) 35(15_suppl):3025–5. doi: 10.1200/JCO.2017.35.15_suppl.3025
59. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31(5):711–23.e714. doi: 10.1016/j.ccell.2017.04.003
60. Bockmayr M, Mohme M, Klauschen F, Winkler B, Budczies J, Rutkowski S, et al. Subgroup-Specific Immune and Stromal Microenvironment in Medulloblastoma. Oncoimmunology (2018) 7(9):e1462430. doi: 10.1080/2162402x.2018.1462430
61. Bailey CP, Wang R, Figueroa M, Zhang S, Wang L, Chandra J. Computational Immune Infiltration Analysis of Pediatric High-Grade Gliomas (pHGGs) Reveals Differences in Immunosuppression and Prognosis by Tumor Location. Comput Syst Oncol (2020) n/a(n/a):e1016. doi: 10.1002/cso2.1016
62. Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, et al. Characterization of the Immune Microenvironment of Diffuse Intrinsic Pontine Glioma: Implications for Development of Immunotherapy. Neuro Oncol (2019) 21(1):83–94. doi: 10.1093/neuonc/noy145
63. Yang J, Hu L. Immunomodulators Targeting the PD-1/PD-L1 Protein-Protein Interaction: From Antibodies to Small Molecules. Med Res Rev (2019) 39(1):265–301. doi: 10.1002/med.21530
64. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell (2015) 28(6):690–714. doi: 10.1016/j.ccell.2015.10.012
65. Krombach J, Hennel R, Brix N, Orth M, Schoetz U, Ernst A, et al. Priming Anti-Tumor Immunity by Radiotherapy: Dying Tumor Cell-Derived DAMPs Trigger Endothelial Cell Activation and Recruitment of Myeloid Cells. Oncoimmunology (2019) 8(1):e1523097. doi: 10.1080/2162402x.2018.1523097
66. Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N Engl J Med (2021) 384(17):1613–22. doi: 10.1056/NEJMoa2024947
Keywords: chimeric antigen receptor T-cell, pediatric brain tumor, immunotherapy, CNS tumor, combinatorial immunotherapy
Citation: Burns I, Gwynne WD, Suk Y, Custers S, Chaudhry I, Venugopal C and Singh SK (2022) The Road to CAR T-Cell Therapies for Pediatric CNS Tumors: Obstacles and New Avenues. Front. Oncol. 12:815726. doi: 10.3389/fonc.2022.815726
Received: 15 November 2021; Accepted: 07 January 2022;
Published: 27 January 2022.
Edited by:
David D. Eisenstat, Royal Children’s Hospital, AustraliaReviewed by:
Nicholas Vitanza, Seattle Children’s Hospital, United StatesStacie Wang, Royal Children’s Hospital, Australia
Copyright © 2022 Burns, Gwynne, Suk, Custers, Chaudhry, Venugopal and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheila K. Singh, c3NpbmdoQG1jbWFzdGVyLmNh
†These authors have contributed equally to this work and share first authorship