 Yuan Wang1,2†
Yuan Wang1,2† Manyu Xu
Manyu Xu Wenyu Shi
Wenyu Shi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 February 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.815654
This article is part of the Research Topic Epidemiology, Biology, and Treatment Strategy of Peripheral T Cell Lymphoma View all 6 articles
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase expressed at early stages of normal development and in various cancers including ALK-positive anaplastic large cell lymphoma (ALK+ ALCL), in which it is the main therapeutic target. ALK tyrosine kinase inhibitors (ALK TKIs) have greatly improved the prognosis of ALK+ALCL patients, but the emergence of drug resistance is inevitable and limits the applicability of these drugs. Although various mechanisms of resistance have been elucidated, the problem persists and there have been relatively few relevant clinical studies. This review describes research progress on ALK+ ALCL including the application and development of new therapies, especially in relation to drug resistance. We also propose potential treatment strategies based on current knowledge to inform the design of future clinical trials.
Anaplastic large cell lymphoma (ALCL) is an aggressive cluster of differentiation (CD)30+ peripheral T-cell lymphoma (PTCL) that accounts for approximately 10%–15% of pediatric and 1%–2% of adult non-Hodgkin lymphoma (NHL) cases (1). Over 90% of children and adolescents with ALCL are ALK positive (ALK+) while the rate among adult patients is 40%–50% (2). The main feature of ALK+ ALCL is the expression of ALK fusion proteins such as nucleophosmin (NPM)–ALK, TNF receptor-associated factor (TRAF)1–ALK, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase ATIC–ALK, ring finger protein (RNF)213–ALK, and tropomyosin (TPM)3–ALK (3–6). NPM–ALK is the most prevalent of these fusions and is detected in 75%–80% of adults and 90% of children with ALK+ ALCL (7, 8). NPM–ALK arises from the fusion of the ALK gene on chromosome 2p23 with the NPM gene on chromosome 5q35. ALK is normally expressed in cells of the small intestine, testis, and colon but not in lymphocytes (9). In ALK+ ALCL, NPM–ALK is highly expressed as a result of a high copy number of the NPM promoter and constitutively activates NPM–ALK and downstream signaling including signal transducer and activator of transcription (STAT)3, phospholipase (PLC)γ, phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT), and mitogen-activated protein kinase (MAPK)–extracellular signal-regulated kinase (ERK) pathways that are important for cell survival and proliferation, metabolic transformation, and immune evasion via the oligomerization of NPM (10). Therefore, NPM–ALK is a target of therapeutic strategies in ALK+ ALCL. Alectinib was approved in Japan for the treatment of children and adults with relapsed/refractory (R/R) NPM–ALK+ ALCL, while crizotinib has been approved for this indication by the US Food and Drug Administration. However, a subset of patients responds poorly to ALK TKIs due to mutation/amplification of the ALK gene, which reduces sensitivity to these drugs. Tumors also survive by other mechanisms such as autophagy, anti-apoptosis, and ALK bypass substitution, among others.
STAT3 is a downstream effector of ALK that plays an important role in promoting cell survival, proliferation, and immune evasion (11). STAT3 exerts anti-apoptotic effects in ALK+ ALCL mainly by upregulating the anti-apoptotic protein B cell lymphoma extra-large (Bcl-xL) and antagonizing the tumor suppressor P53 (12). STAT3-induced expression of transforming growth factor (TGF)-β, interleukin (IL)-10, and programmed death ligand (PD-L1,CD274, B7-H1) creates a tumor-suppressive microenvironment (13–15). STAT3 directly binds to the promoter of hypoxia-inducible factor (HIF)-1α to promote gene expression; HIF-1 α in turn induces the expression of vascular endothelial growth factor (VEGF) and promotes tumor angiogenesis (16, 17). STAT3 also plays an important role in epigenetic regulation of gene expression. Under normal conditions, NPM–ALK phosphorylates the Y405 residue of STAT3, which causes the dimerization of phosphorylated STAT3 and its translocation into the nucleus where it modulates gene transcription in a methylation-dependent manner (18, 19). This leads to the silencing of oncogenes such as STAT5A, IL-2 receptor gamma (IL-2Rγ), Bcl-2-like protein 11 (BIM), protein tyrosine phosphatase non-receptor type 6 (SHP1), and CD48 (20–25) and suppresses the expression of micro (mi)RNAs with oncogenic effects such as miR-150, miR-497, miR-21, miR-29a, miR-939, miR-96, miR-155, and miR-146a (26–31). The consequent silencing of T-cell receptor-related genes including CD3ϵ, zeta-chain-associated protein kinase (ZAP)70, linker for activation of T cells (LAT), and SH2 domain-containing leukocyte protein of 76 kDa (SLP76) results in the loss of T cell identity (32). It was recently reported that NPM–ALK mediates STAT3 acetylation to inhibit the expression of tumor suppressor genes; inhibiting STAT3 acetylation resulted in their re-expression and ALCL cell apoptosis (33) (Figure 1).
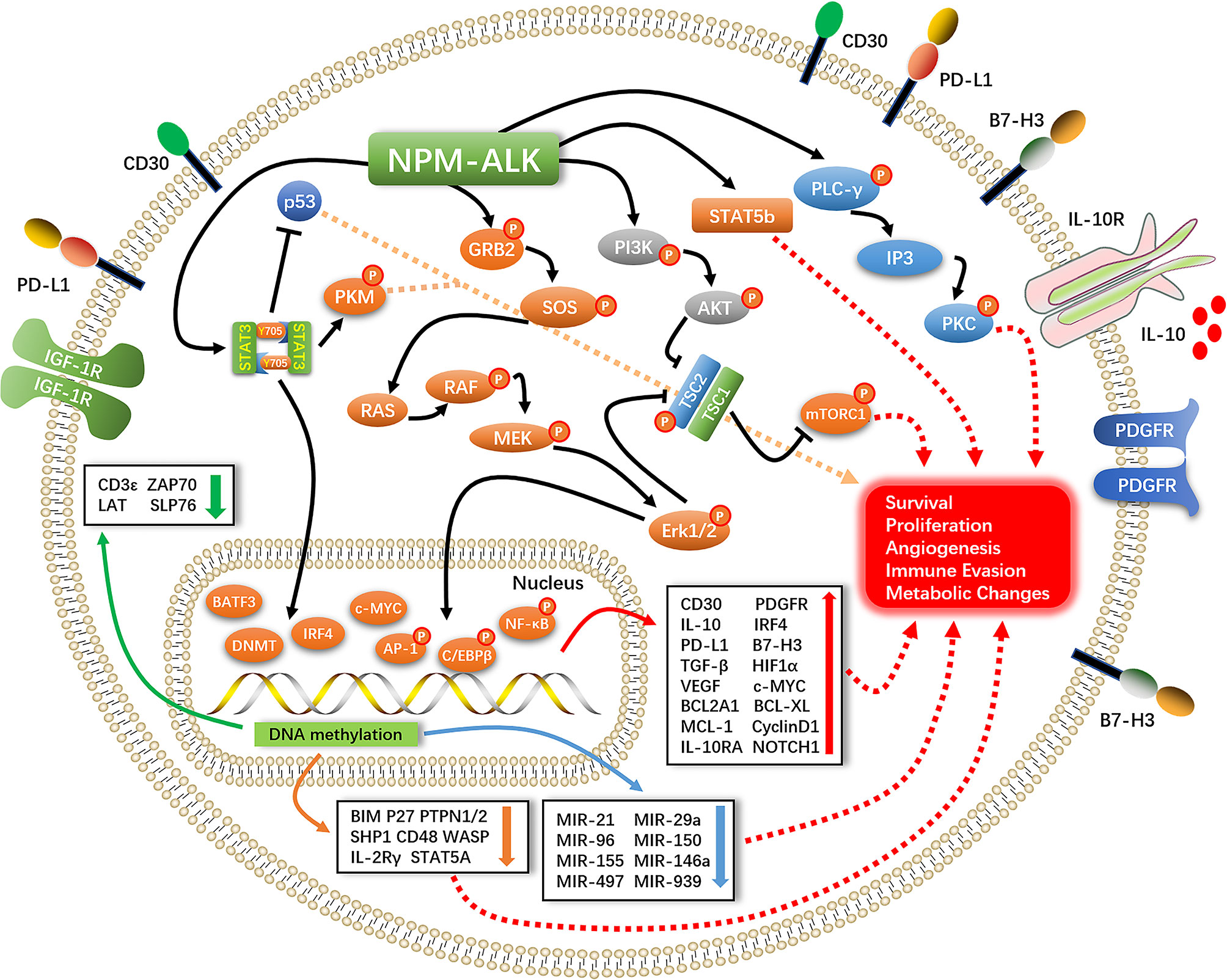
Figure 1 Pathogenesis of ALK+ALCL.
Activation of NPM–ALK leads to PLC-γ tyrosine phosphorylation (Y783, Y1254) and induction of its catalytic activity, leading to the breakdown of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol trisphosphate (IP3) and activation of protein kinase (PK)C; this process plays an important role in NPM–ALK-mediated mitogenic signaling (34–36) (Figure 1).
NPM–ALK interacts with the SH2/SH3 domain of the PI3K regulatory subunit (p85) (37). P85 binds to the catalytic subunit (p110) and converts PIP2 to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which binds to the pleckstrin homology domain of AKT and facilitates its translocation from the cytoplasm to the cell membrane. T308 and S473 phosphorylation by phosphatidylinositol-dependent protein kinase (PDK)1 and PDK2 activates AKT (38–40), mammalian target of rapamycin (mTOR) is then phosphorylated and activated by AKT to promote the survival and proliferation of ALK+ ALCL cells (41) (Figure 1).
MEK–ERK1/2 signaling stimulates cell proliferation by promoting cell cycle progression via the protein-dependent kinase (CDK)4 and retinoblastoma protein (RB) pathways. CDK4 binds to cyclin D1 to phosphorylate Rb and release the transcription factor E2F, which induces the expression of CDK4 as well as cyclin D1, leading to the entry of cells into S phase; it also promotes cell survival and proliferation via ERK1/2–mTOR signaling (42, 43). ERK1/2 phosphorylates JUNB, a member of the activator protein (AP)-1 family and an important transcription factor in ALK+ ALCL. JUNB transcriptional targets have been shown to be involved in cell proliferation, anti-apoptosis, immune evasion, and immune phenotype transition (44, 45) (Figure 1).
ALK+ ALCL treated with the chemotherapy regimen consisting of cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate, and prednisone (CHOP) has a favorable clinical outcome, although the rate of recurrence is high. Patients who relapse after first-line chemotherapy usually have a poor prognosis, while some patients are inherently resistant to chemotherapy regimens such as CHOP. ALK TKIs can improve the prognosis of patients with R/R ALK+ ALCL. In a clinical trial of children with R/R ALK+ ALCL treated with crizotinib (NCT00939770), the objective response rate (ORR) was 90% and the complete response (CR) rate was 80% (46). In a phase 2 clinical trial of alectinib for the treatment of R/R ALK+ ALCL(Range, 6-70 years), the ORR after alectinib treatment was 80% and the CR rate was 60%, while the 1-year PFS, event-free survival (EFS), and overall survival (OS) rates were 58.3%, 70.0%, and 70.0% respectively (47). But a new problem has arisen: Among patients who have a high response to crizotinib monotherapy, about 30-40% of patients have developed further resistance to the drug (48). ALK mutation/amplification reduces sensitivity to ALK TKIs, necessitating a switch to another drug. Furthermore, tumors mitigate the cytotoxicity of ALK TKIs through a variety of mechanisms (ALK bypass substitution, autophagy, anti-apoptosis, etc), thereby reducing/eliminating their dependence on ALK signaling and promoting tumor cell survival. In this situation, even complete inhibition of ALK cannot prevent tumor progression, and combination therapy or targeting of proteins other than ALK (eg, CD30, B7-H3, heat shock protein [HSP]90, etc) may be necessary (Figure 2).
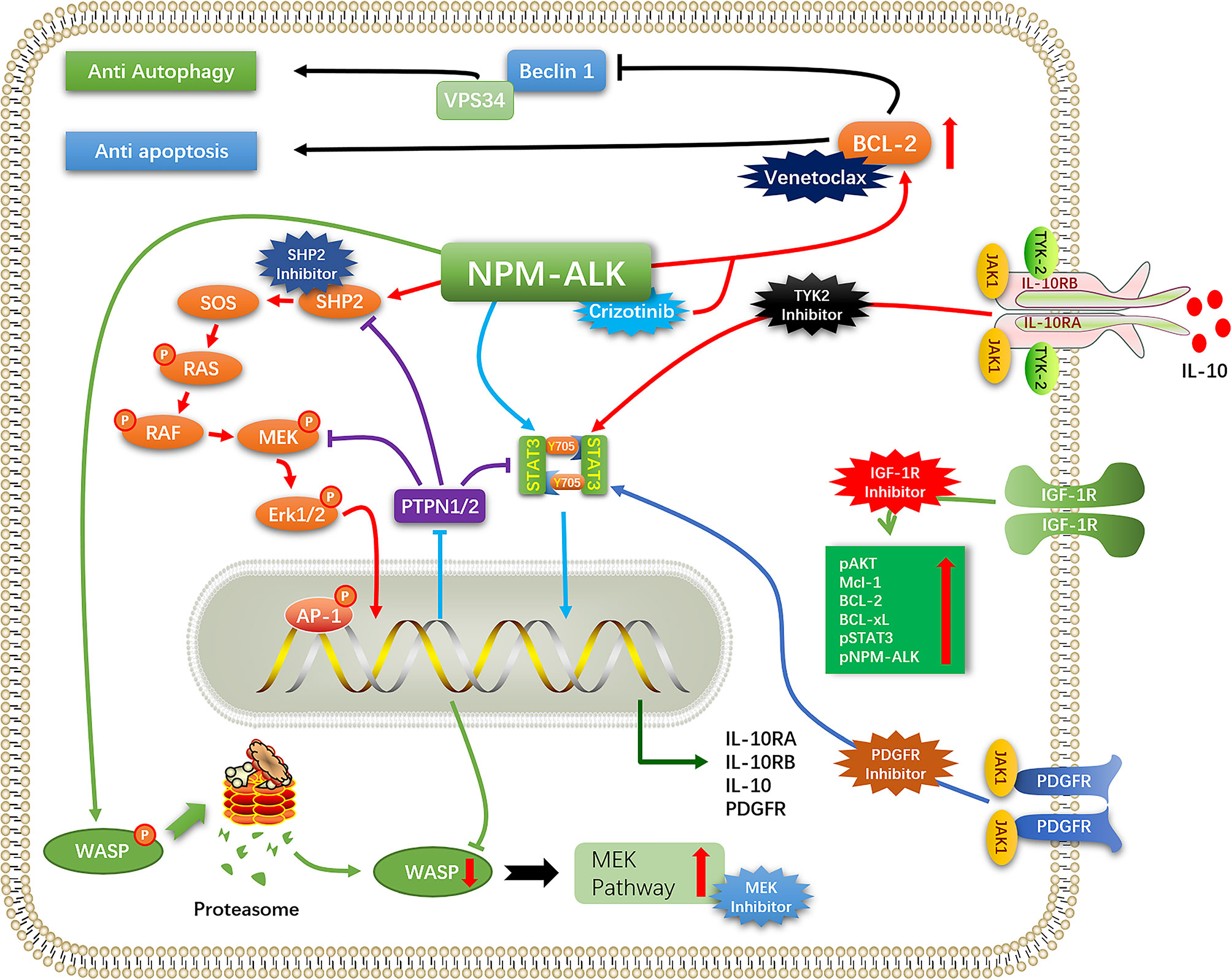
Figure 2 Mechanisms of resistance to ALK-TKI.
ALK mutations can lead to the development of ALK resistance through a variety of mechanisms such as by blocking the binding of ALK TKI to the ATP-binding site of ALK (L1196M, I1171T/N/S, G1269A, G1202R/del, G1202N, S1206Y/C, etc) (47, 49–54). Several point mutations in the structural domain of NPM–ALK kinase have been identified in ALK+ ALCL (L1122V, L1196M, F1174V, L1198F, P1139S, and G1202R) that have been implicated in the development of resistance to ALK TKIs (55). For example, the F1174V/L1198F and L1196M/D1203N double mutations were shown to confer higher resistance to the inhibitors (56).
ALK+ ALCL tumor cells also develop ALK TKI resistance through amplification of NPM–ALK, which was shown to be overexpressed in ALK+ALCL-resistant cell lines (48, 55, 57, 58).
IL-10 signaling bypass leads to crizotinib resistance (59). In ALK+ ALCL, IL-10 promotes the proliferation of ALK+ ALCL and reduces the sensitivity of ALCL cells to ALK TKI. A screen of tumors from ALK+ ALCL patients who progressed within 3 months of crizotinib treatment found that IL-10RA was overexpressed in tumor cells, leading to ALK TKI resistance. In resistant ALK+ ALCL cell lines, autocrine IL-10/IL-10RA signaling activated tyrosine kinase (TYK)2/Janus kinase (JAK)1 signaling instead of the ALK–STAT3 pathway to activate STAT3, which stimulated the expression of IL-10, IL-10RA, and IL-10RB. Thus, the strength of IL-10RA signaling in ALK+ ALCL affects tumor sensitivity to crizotinib. IL-10 is a known immunosuppressive factor that directly inhibits effector T cells, promotes T cell exhaustion, and inhibits T cell activation through myeloid-derived suppressor cells and induction of regulatory T cells (60, 61). This could allow ALK+ ALCL to achieve immune evasion and may result in the development of drug resistance, although there have been no studies investigating this possibility.
Activation of insulin-like growth factor 1 receptor (IGF-1R) signaling is another mechanism of ALK resistance. Type 1 IGF-1R is commonly expressed in ALK+ ALCL. In a crizotinib resistance model, IGF-1R signaling was shown to be increase with drug concentration and reduced the cytotoxicity of crizotinib; conversely, crizotinib combined with IGF-1R inhibitors partly restored the sensitivity of resistant cells (62). ALK phosphorylates IGF-1R at the C-terminal Y338 residue; in turn, IGF-1R increases the phosphorylation of ALK and its downstream effectors and promotes the survival of ALK+ ALCL cells by increasing the expression of anti-apoptotic proteins (myeloid cell leukemia [Mcl]-1, Bcl-2, Bcl-xl, etc) (63). The combined use of IGF-1R inhibitors and ALK TKIs can suppress ALK and downstream signaling, thereby enhancing the sensitivity of ALK+ ALCL to ALK TKIs and reducing crizotinib resistance caused by activation of the IGF-1R pathway, making combination therapy a potential treatment regimen. (64).
PDGFR expression and activation is a key driver of ALCL proliferation, survival and spread. NPM-ALK promoted the expression of PDGFRB through NPM-ALK/AP-1 (JUN/JUNB) (65). High expression of PDGFRB could also be seen in most ALK+ALCL. PDGFR promoted cell survival via JAK1/STAT3 and AKT(PKB)/mTOR pathways (66, 67). The level of PDGFR-mRNA decreased after application of PDGFR inhibitors, indicating that PDGFR is promoting its own expression, ALK-TKI in combination with PDGFR inhibitors reduces lymphoma growth and decreases relapse rates, and is a potential treatment option for patients with drug-resistant lymphoma. (65).
Activation of the MEK pathway also contributes to ALK TKI resistance, mainly through deficiency/low expression of Wiskott–Aldrich syndrome protein (WASP) (68), a tumor suppressor whose loss leads to tumor development and invasion (69). NPM–ALK negatively regulates the expression of WASP by directly phosphorylating Y102, leading to proteasome-mediated protein degradation and preventing protein binding to WASP-interacting protein (WIP) (70). NPM–ALK was also shown to repress WASP protein expression via STAT3–CCAAT/enhancer-binding protein (C/EBP)-β signaling. Downregulation of WASP was shown to enhance MEK pathway activation in ALK+ALCL. NPM–ALK can form a complex with the guanine exchange factor VAV1 to enhance the activity of cell division cycle (CDC)42, which promotes the progression of ALK+ ALCL; meanwhile, WASP inhibits the binding of CDC42 to GTP (71). Protein tyrosine phosphatase (PTPN)1/2 expression was found to be downregulated in ALK+ ALCL, leading to the overactivation of MEK, SHP2, and Janus kinase (JAK)/STAT and resistance to ALK TKIs (72).
Although ALK inhibition undermines the survival and proliferation of ALCL cells, not all cellular changes caused by ALK expression are reversed. For example, IL-10R, WASP, and MEK signaling was not downregulated by application of crizotinib; instead, tumor cells developed resistance to the drug through autocrine IL-10 secretion by ALK+ ALCL cells and activation of IL-10R signaling, which replaced the role of ALK in promoting IL-10R expression, suggesting that cytologic changes caused by ALK led to the emergence of ALK-independent mechanisms of tumor cell survival (59, 68). Some oncogenes that were downregulated via epigenetic modifications were not upregulated by crizotinib, but were instead involved in crizotinib resistance, suggesting that the decrease in their expression was not reversed (68, 72) (Figure 2).
Autophagy activation and inhibition play different roles according to the tumor type. In ALK+ ALCL, inhibition of autophagy facilitated tumor survival in the presence of ALK TKIs (73). Crizotinib was shown to decrease autophagic flux in ALK+ ALCL, which was important for tumor survival of NPM–ALK+ ALCL; this cytoprotective response reduced the cytotoxicity of crizotinib (56, 74). Rapamycin, an mTOR inhibitor that activates autophagy, reduces the survival of ALCL cells (75). The expression level of miR-7-5P was found to be downregulated in ALK+ ALCL by the application of crizotinib; it was later shown that miR-7-5P directly targets the 3′ untranslated region of RAF1 transcript, thereby negatively regulating RAF1 expression and reducing the inhibitory phosphorylation of Unc-51–like autophagy-activating kinase (ULK)1 (S757) to promote autophagy. Conversely, downregulation of miR-7-5P following crizotinib application inhibited autophagy. Potentiating the effect of miR-7-5P using an miRNA mimic enhanced crizotinib-induced autophagic flux and cytotoxicity (76). Interestingly, in ALK+ ALCL stem cells, crizotinib more potently induced autophagy and enhanced tumor cell resistance to the drug, an effect mediated by MYC (77).
Elevated levels of Bcl-2 have been observed upon treatment with crizotinib. Bcl2 is anti-apoptotic protein that inhibits autophagy, which can lead to drug resistance in tumors. Inhibition of Bcl-2 significantly increased crizotinib-induced autophagy (78, 79).
Decreased autophagic flux in ALK+ ALCL plays a role in drug resistance, and tumor cells are regulated in various ways that lead to inhibition of autophagy and crizotinib resistance including regulation of miRNAs and Bcl-2 expression. Autophagy inhibition can reverse the crizotinib-induced decrease in cell viability, thus limiting the cytotoxicity of crizotinib (Figure 2).
For patients with ALK TKI resistance, tissue biopsy and pathologic examination can reveal the mechanism of resistance and inform clinical decisions, especially drug selection. For example, ALK TKIs can be selected that target specific ALK mutations. On the other hand, the absence of ALK mutation can indicate the development of resistance by the tumor, requiring ALK TKI combination therapy. For highly resistant (especially compound) mutations, ALK amplification may not be detected or the cause of resistance may not be clear, in which case switching ALK TKIs or using combination therapy may not be effective and therapeutic targets other than ALK should be considered. The development of ALK sequencing has aided the precision treatment of ALK+ ALCL and has revealed novel drug resistance mutations; the elucidation of ALK TKI resistance mechanisms will facilitate the development of new treatment strategies.
For drug resistance caused by NPM–ALK overexpression, crizotinib discontinuation can lead to overactivation of ALK oncogenic signaling and enhanced mitochondrial activity, reactive oxygen species (ROS) production, and activation of the MEK–ERK1/2 pathway, causing DNA damage. This may result in apoptosis of ALK TKI-resistant cells and the resensitization of tumor cells to crizotinib (56, 57, 80–83). NPM–ALK overexpression promotes STAT1 phosphorylation, and high levels of phosphorylated (p)STAT1 antagonize STAT3 and activate tumor suppressor genes, promoting cell death. Enhanced NPM–ALK expression following withdrawal of ALK TKI leads to the upregulation of pSTAT1, which antagonizes STAT3 and induces apoptosis (84, 85). Given the damaging effects of excessive tumorigenic signals on DNA, agents that Enhance DNA damage and inhibit DNA repair can be used during drug withdrawal to enhance the effects NPM–ALK (Figure 3).
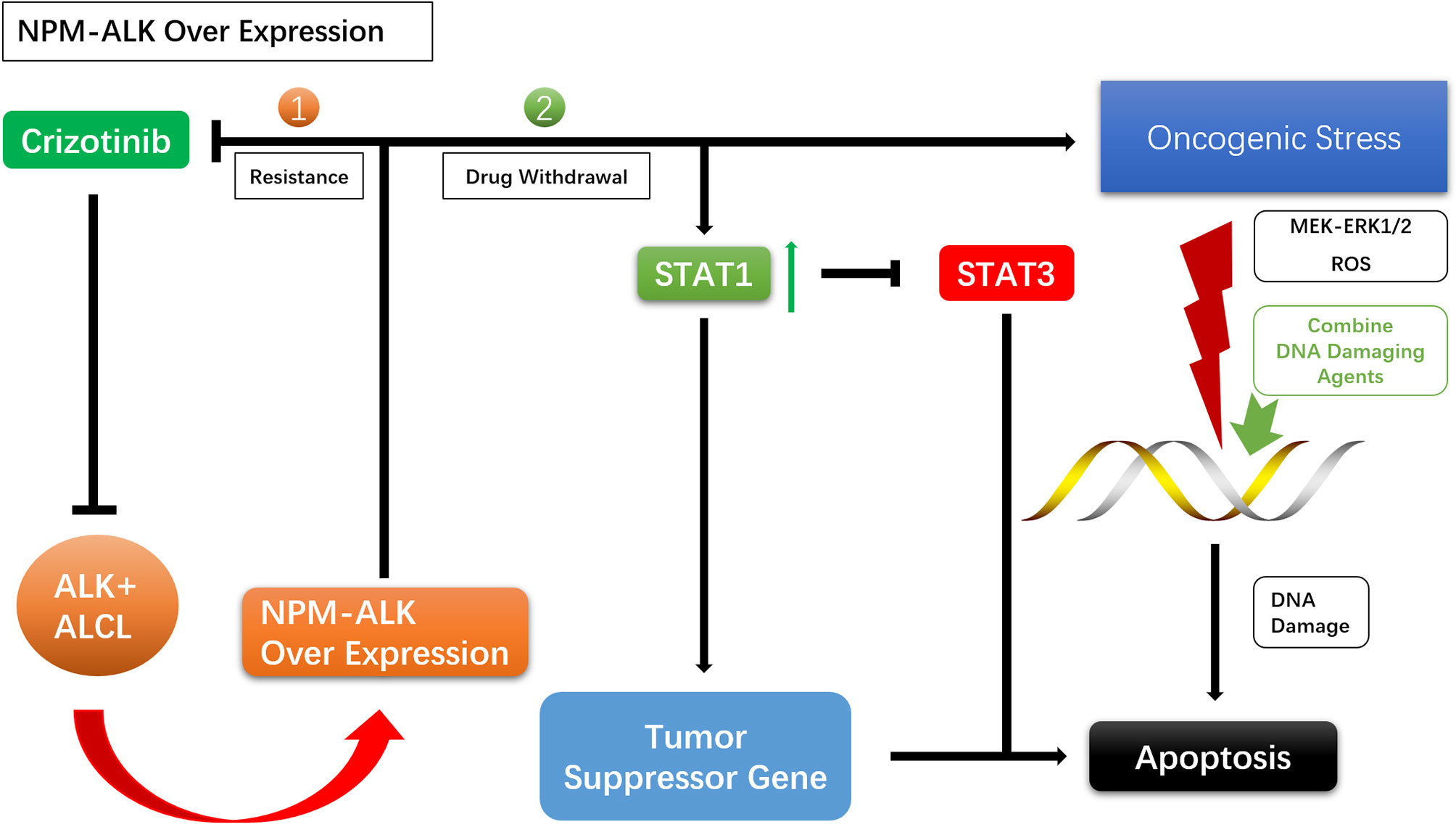
Figure 3 Mechanisms of intermittent treatment.
Second-generation ALK TKIs such as alactinib, ceritinib, brigatinib, lorlatinib, and ZX-29 have more potent activity in the central nervous system than crizotinib and can overcome the effects of most crizotinib resistance mutations (86) (Table 1). Alectinib is an orally administered drug with greater potency than crizotinib that was shown to be effective in crizotinib-resistant tumors (100). including ALK+ non-small cell lung cancer (NSCLC), with neutropenia and elevated levels of creatine kinase as the most serious adverse effects (101, 102). Ceritinib is a small molecule ALK TKI that has demonstrated efficacy in ALK-positive tumors including ALCL, inflammatory myofibroblastic tumor, neuroblastoma, and rhabdomyosarcoma; in ALK+ ALCL, the most commonly reported complication was elevated transaminases (103). Brigatinib is an ALK/ROS1 inhibitor with higher selectivity than alectinib or ceritinib that can overcome most crozotinib resistance mutations including G1202R, with an ORR of 74%, median PFS of 14.5 months, and 1-year OS rate of 83% in crizotinib-resistant patients (96, 104). Brigatinib has shown superior efficacy to crizotinib in the first-line setting, and an ongoing study is investigating brigatinib in ALCL (105). However, second-generation ALK TKIs are more likely to lead to the development of resistance than crizotinib (54). The third-generation ALK TKI loratinib overcomes nearly all single resistance mutations to second-generation inhibitors and is more potent than brigatinib against the G1202R mutation. However, G1202R was shown to reduce the sensitivity of tumor cells to brigatinib and loratinib, and tumor cells harboring double mutations (D1203N+E1210K and F1174C+D1203N) had lower sensitivity to loratinib, whereas none of the second-generation ALK TKIs were effective (54). ZX-29 is a novel ALK TKI that induces apoptosis by stimulating the production of ROS, overcoming the drug resistance conferred by the ALK G1202R mutation and exhibiting greater cytotoxicity than ceritinib (106, 107). The Fms-related receptor tyrosine kinase (FLT)3/AXL inhibitor gilteritinib is currently mainly used for R/R acute myeloid leukemia (108). Gilteritinib inhibits not only wild-type and mutant ALK but also overcomes highly resistant double mutations (I1171N+F1174I, I1171N+L1198H, and others containing I1171N) as well as single mutations by forming hydrogen bonds with the ALK E1197, M1199, and E1210 residues (99, 109). Mutations such as L1196M, I1171T/N/S, G1269A, G1202R/del, G1202N, and S1206Y/C that sterically hinder the binding of ALK TKIs to ALK are a common resistance mechanism (47, 49–54). Gilteritinib inserts into the ATP-binding site of ALK and therefore has an advantage over the first 3 generations of ALK TKI in terms of drug resistance, providing an additional treatment option for patients. Gilteritinib has also demonstrated efficacy against ALK+ ALCL in preclinical studies (110).

Table 1 Resistant and sensitive mutations of ALK-TKIs.
ALK kinase structural domain mutations common to NSCLC and ALCL have been identified. Table 1 summarizes mutations known to confer resistance/sensitivity to ALK TKIs that have potential application in the treatment of ALK+ ALCL (Table 1).
PROTACs are used to promote proteasome-mediated protein degradation (111). PROTAC is composed of 2 ligands that connect the target protein and E3 ubiquitin ligase, forming a ternary complex. The ubiquitin-binding enzyme E2 binds to the E2 binding site on E3 ligase and transfers ubiquitin to the target protein, leading to its degradation (111–113). ALK TKIs (alectinib, brigatinib, ceritinib, etc.) are often used as PROTAC ligands to promote the degradation of ALK protein and thereby inhibit tumor growth driven by ALK (114). This allows a small dose of PROTAC to achieve a strong inhibitory effect, which is especially advantageous for overcoming drug resistance due to ALK amplification. PROTAC directly targets and degrades proteins and is unaffected by ALK point mutations; this explains how PROTAC designed with alectinib/brigatinib can degrade the ALK G1202R mutant, which is resistant to the drugs themselves. PROTAC designed with alectinib as the ligand was more effective than alectinib in ALK+ ALCL patients and enhanced ALK degradation (115–117). ARV-110, the first PROTAC drug targeting the androgen receptor, has been used in patients with metastatic debulking-resistant prostate cancer with promising results (118). A second PROTAC drug, ARV-471 (targeting the estrogen receptor [ER]), is being evaluated in clinical trials for the treatment of patients with locally advanced or metastatic ER+/human epidermal growth factor receptor (HER)2− breast cancer. Interim results have shown that ARV-471 reduced ER expression levels by 62% and up to 90% and was effective against both wild-type and mutant ER (119). PROTACs targeting ALK are not yet in clinical use but several ALK-RROTACs are being developed such as SIAIS001, SIAIS117, and SIAIS164018 that can potentially overcome resistance to ALK TKIs (115, 116, 120) (Figure 4A).
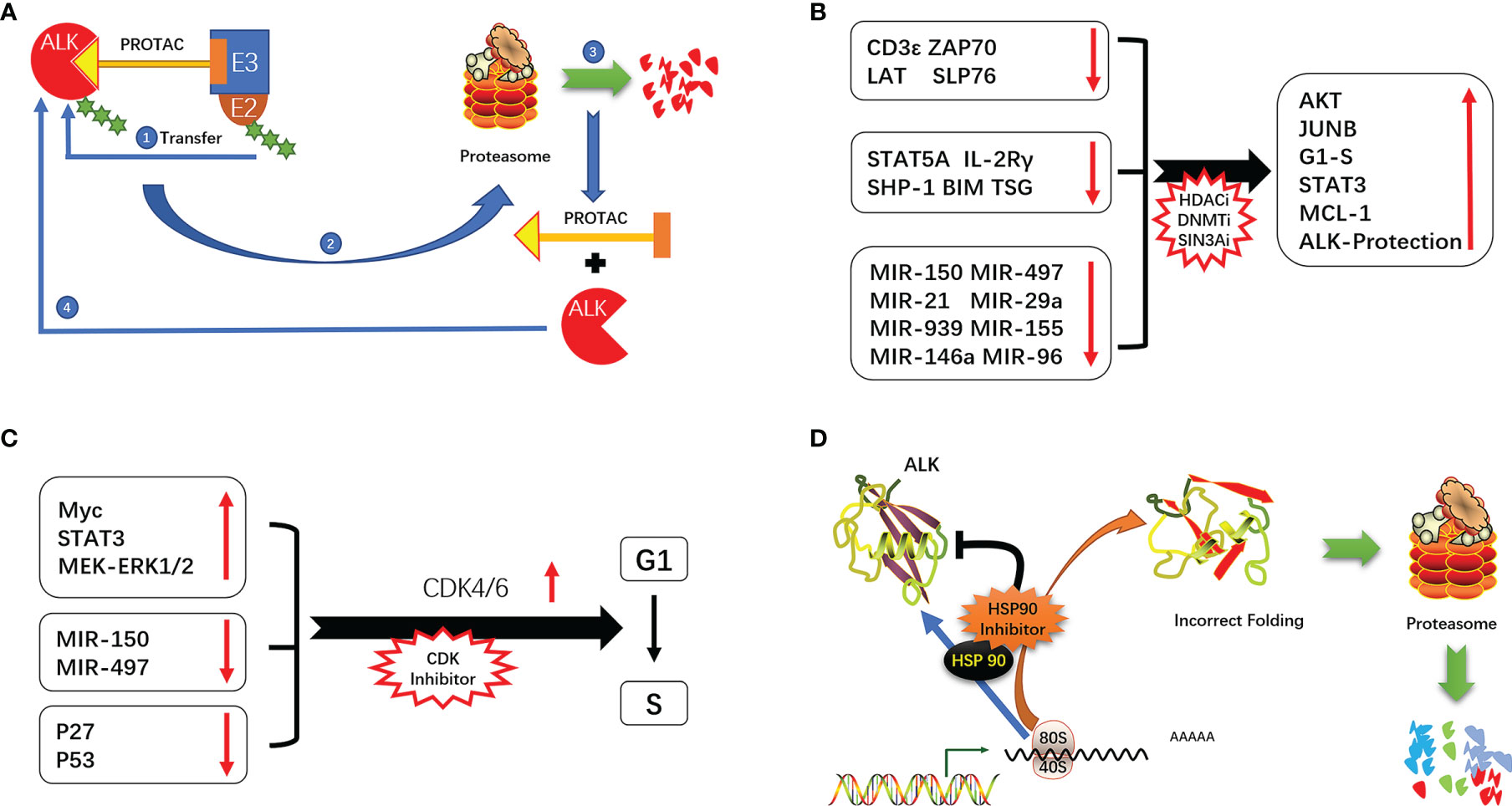
Figure 4 (A) Mechanism of action of PROTAC. (B) ALK signaling-mediated cycle operation. (C) Epigenetic alterations promote the development of ALK-positive ALCL. (D) Mechanism of ALK inhibition by HSP90.
ALK-independent drug resistance is normally treated by a combination of drugs that inhibit ALK along with other pathways involved in cell survival, thereby enhancing the cytotoxicity of ALK TKIs. The use of ALK TKI combinations can delay the development of ALK resistance. Crizotinib combined with everolimus, CHOP, decitabine, and trametinib prolonged the emergence of resistance, possibly because of a reduction in the dose of individual drugs and the effects of dual targeting (48).
Elevated expression of IL-10RA not only confers resistance to crizotinib but also to second- or third-generation inhibitors. Accordingly, ALK TKI combined with IL-10 pathway inhibitors (e.g., STAT3/pan-JAK/TYK2 inhibitors) may be effective (48) in patients who are resistant to ALK TKIs and have high IL-10RA expression. TYK2 acts upstream of STAT3; it was reported that the survival of ALK+ ALCL cells depended on TYK2/STAT1/MCL1. Regardless of the presence of ALK fusion protein, TYK2 inhibitors can induce tumor cell apoptosis. Inhibiting TY2 blocks the activation of STAT3 by IL-10 and other pathways via T2, suppressing tumor cell survival via a bypass mechanism (121). The combination of IGF-1R and NPM–ALK inhibited tumor growth in a mouse model of ALCL cell lymphoma. Inhibition of IGF-1R promoted cell apoptosis and blocked phosphorylation of NPM–ALK and its downstream effectors (122). Thus, IGF-1R inhibition combined with crizotinib can have a synergistic effect, allowing dose reduction of crizotinib and delaying the emergence of drug resistance (123). The expression level of platelet-derived growth factor receptor (PDGFR)B in ALK+ ALCL was found to be positively correlated with the cytotoxicity of PDGFR inhibitors, which may be effective in the treatment of PDGFRB+ ALK+ ALCL. Findings from in vitro experiments and mouse models have demonstrated that imatinib treatment suppresses the proliferation of tumor cells and promotes apoptosis. Notably, a rapid and durable antitumor response was achieved with imatinib in a patient with advanced refractory ALCL (65). Activation of the MEK pathway was observed in WASP− ALK+ ALCL and was associated with reduced efficacy of ALK TKIs. The combination of the MEK inhibitor trametinib with crizotinib has achieved better clinical outcomes than crizotinib by delaying the emergence of drug resistance (65); thus, targeting MEK can reduce the survival of lymphoma with low or absent WASP expression via the MAPK pathway (68). Additionally, γ-secretase inhibitors suppressed the proliferation of ALK+ ALCL cells with crizotinib resistance, suggesting NOTCH1 as a therapeutic target in crizotinib-resistant ALK+ ALCL (124, 125).
Crizotinib combined with everolimus more potently induced cell cycle arrest, DNA damage, and apoptosis than monotherapy in the Karpas299 transplantation model (126, 127). Although everolimus can inhibit mTOR, it can cause the activation of AKT and RAS–ERK, an effect that is blocked by the addition of crizotinib. The combination treatment also prevented the occurrence of selective drug resistance after long-term use of monotherapy (126). In neuroblastoma, crizotinib combined with mTOR inhibitor was shown to overcome crizotinib resistance and promote tumor cell apoptosis (128). In ALK+ ALCL cells, the mTOR inhibitor rapamycin combined with crizotinib increased autophagic flux and promoted cell death (75) (Figure 2).
Given the reversibility and importance of epigenetic regulation of gene expression in the development of ALK+ ALCL, drugs targeting epigenetic modifications and allowing “re-expression of tumor suppressor proteins” or “reduced expression of oncogenic proteins”, such as DNA methylation, HDAC and SIN3A inhibitors, are a promising therapeutic approach. (20, 22, 23, 30, 31, 33). In a multicenter clinical study of cidapenem in R/R PTCL, patients with ALK+ ALCL treated with cidapenem had a higher ORR (66.67%) and disease control rate (83.33%) and better prognosis compared to those with other PTCL subtypes (129). The use of decitabine (a DNA methyltransferase inhibitor) not only enhanced the efficacy of ALK TKIs but also prolonged the time to emergence of drug resistance by more than 3 fold compared to monotherapy (48) (Figure 4B).
ALK promotes cell cycle progression through a variety of mechanisms (eg, activation of MEK/ERK and STAT3 signaling, induction of cell cycle-related gene expression, and downregulation of P27 and P53) (12, 42, 130, 131). NPM–ALK also promotes cell cycle progression via regulation of miRNAs. In ALK+ ALCL, the combination therapy of cell cycle inhibitors has shown the potential of resistance to ALK (132). In a mouse xenograft model of neuroblastoma with ALK F1174L and F1245C mutations, the CDK4/6 inhibitor ribociclib blocks the binding of CDK4/6 to CyclinD1, thereby inhibiting the operation of the cell cycle. In combination with Ceritinib exerts synergistic cytotoxicity, inhibits tumor growth, enhances cycle arrest, and promotes cell death (133) (Figure 4C).
ALK+ ALCL expresses CD30, PD-L1, and B7-H3, which are potential therapeutic targets for promoting tumor cell death through pathways independent of ALK, thereby overcoming ALK TKI resistance.
ALK promotes CD30 expression through the MEK–ERK–AP1–JUNB pathway (134, 135). Brentuximab vedotin (BV) is an antibody–drug conjugate (ADC) that targets CD30 in which anti-CD30 antibody is linked to the microtubule destroyer monomethyl auristatin E (MMAE) via a protease-cleavable linker. After binding to CD30, BV forms phagosomes through receptor-mediated endocytosis before it is hydrolyzed by lysosomal proteases; this releases MMAE, leading to cell cycle arrest and cell apoptosis (136–139). At the 5-year follow-up of a clinical trial of BV in R/R ALCL, 66% of patients continued to show a CR (140). In another study of BV in the treatment of R/R ALCL, 86% of patients achieved ORR including 57% with CR and 29% with partial remission; the duration of response was 12.6 months and the duration of CR was 13.2 months (141). In a clinical trial of BV combined with chemotherapy in the treatment of pediatric patients with first-onset ALK+ ALCL (NCT01979536), the 2-year EFS rate was 79.1% and 2-year OS rate was 97.0% (142). Only one patient (1.5%) relapsed during treatment. These results demonstrate that BV is superior to conventional chemotherapy in preventing recurrence.
In a clinical trial of chimeric antigen receptor (CAR) T-cell (CAR-T) therapy targeting CD30 in ALCL patients (NCT01316146), the duration of CR was up to 9 months after 4 infusions. Importantly, CD30 CAR-T was still detectable after 6 weeks of treatment, indicating that CAR-T had sustained antitumor effects. ADCs targeting CD30 are associated with a number of adverse effects including gastrointestinal reactions that reduce the tolerability of these drugs and can lead to treatment discontinuation. CD30 CAR-T therapy circumvents this problem and under standardized care and execution, is a safe and effective treatment for CD30+ lymphoma (143).
HSP90 is highly expressed in tumors including ALK+ ALCL. HSP90 inhibitors not only block the binding of HSP90 to ATP but also promote proteasome-mediated degradation of HSP90 target proteins (144). Inhibition of HSP90 resulted in the downregulation of NPM–ALK. The combined use of the HSP90 inhibitor onalespib and ALK TKI in ALK+ NSCLC delayed the emergence of ALK resistance and preserved sensitivity to onalespib (145). A dual-target inhibitor designed based on the active structure of HSP90 and ALK TKIs (resorcinol and 2,4-diaminopyrimidine motifs) has been proposed that is expected to overcome ALK TKI resistance (146) (Figure 4D).
In ALK+ ALCL, whether NPM-ALK promotes PD-L1 expression through downstream signals or in view of the role of PD-1/PD-L1 in TME, PD1/PD-L1 is expected to become a potential therapeutic target for ALK+ ALCL. Recently, studies have found that the expression of PD-L1 and the number of tumor-infiltrating T cells are related to the prognosis of ALK+ ALCL (147). Geptanolimab (GB226) is a PD-1 monoclonal antibody. In an open study (NCT03502629), it was found that the higher the level of PD-L1 in R/R PTCL, the effect of PD-1 blockers is better. In R/R PTCL, the ORR was 40.4% and the 12-month Duration of Response (DOR) was 48.5%. In PTCL with PD-L1 expression>50%, the ORR (53.3%) and median PFS (6.2 months) were higher, especially in ENKTL, ALK-ALCL, ALK+ALCL, Geptanolimab has a better curative effect (148). Similar to this, in an ALK-resistant ALK+ ALCL patient whose tumor tissue highly expresses PD-L1, the tumor tissue completely disappeared after 5 months of Navumab treatment and the complete remission was maintained for up to 18 months (149). In addition, when B7-H3 CAR-T is used to treat ALK+ ALCL, it shows strong cytokine secretion and cytotoxicity both in in vivo and in vitro experiments. It has obvious proliferative activity and memory phenotype after receiving B7-H3 stimulation. Under the premise of strict control of CRS, B7-H3 CAR-T is expected to become another important therapeutic target for ALK+ ALCL after ALK and CD30 (150).
ALK TKI resistance is a major challenge in the treatment of ALK+ ALCL; therefore, therapeutic strategies to overcome this resistance are a key research direction for this malignancy. Many aspects of the resistance mechanisms remain to be elucidated including the upregulation of Bcl-2, ALK amplification after the application of crizotinib, bypass signaling, autophagy, and apoptosis. There have been few studies of single drugs/combination therapies that can improve/reverse ALK resistance, which may be related to the low prevalence of ALK+ ALCL and do not provide sufficient impetus for clinical trials. There are also considerable disparities in the treatment of ALK TKI resistance, especially in terms of therapeutic options for patients. Strategies that target resistance mutations are expected to greatly improve the clinical outcome of ALK+ ALCL.
Selection of appropriate ALK TKIs by sequencing ALK mutation sites can alleviate ALK TKI resistance, although highly resistant mutants and multiple mutations are problematic. The mechanisms by which mutations lead to ALK resistance have been systematically investigated in studies of ALK protein structure and binding to TKI and ATP, and they have served as the basis for the evaluation of candidate drugs such as ZX-29 and ginitinib. The latter in particular—whose binding to ALK is largely unaffected by ALK mutations—can overcome the effects of most ALK single mutations as well as double mutations conferring loratanib resistance (99, 106, 107).
ALK amplification also underlies resistance to ALK TKIs (57) although it may not benefit tumor cells as it can trigger oncogenic stress and induce DNA damage especially upon discontinuation of an ALK TKI, resulting in tumor cell apoptosis and restoration of sensitivity to the inhibitor (85). PROTAC technology is a new treatment strategy that targets ALK amplification and is effective against drug resistance mutations as it induces mutant ALK proteins to undergo ubiquitin-mediated degradation (114).
ALK-independent drug resistance usually necessitates treatment with a combination of drugs, and many studies have shown that although ALK inhibition decreases tumor cell survival and proliferation, many cellular changes caused by ALK overexpression are not reversed such as IL-10RA and IGF-1R dysregulation (59, 62–64, 68). When the inhibition of ALK fusion proteins is alleviated, these changes allow tumor cells to survive and proliferate, leading to the development of ALK TKI resistance. Pro-survival pathways other than ALK can be blocked by a combination of drugs that synergistically enhance the cytotoxicity of ALK TKIs. Additionally, drug combinations can also delay the emergence of resistance through dose reduction of single agents and dual targeting. Although there have been only a few clinical studies examining ALK TKI combinations in ALK+ ALCL, clinical trials are currently underway (Table 2). ALK TKIs also cause adaptive changes in tumor cells that favor their survival such as the upregulation of Bcl-2 and inhibition of autophagy; therapeutic strategies that target these changes may be effective in the treatment of ALK+ ALCL (56, 77, 79).
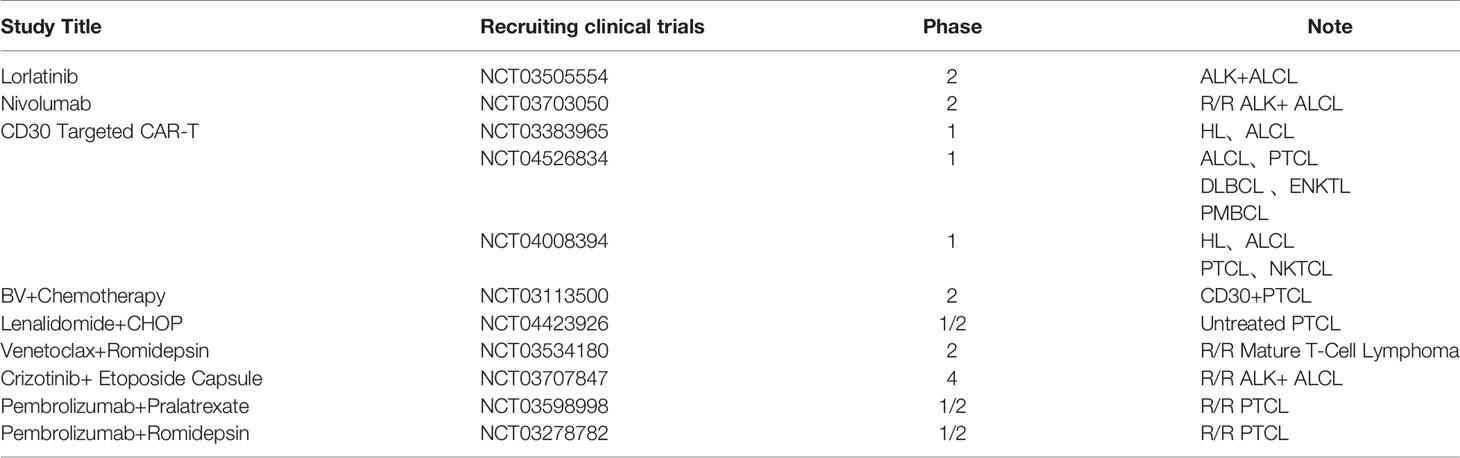
Table 2 Recruiting ALCL clinical trials.
In addition to ALK fusion proteins and their associated signaling pathways, activation of ALK and its downstream effectors in ALK+ ALCL results in the expression of CD30, PD-L1, and B7-H3, which are potential drug targets in patients who are highly resistant to ALK TKIs. However, it is unclear whether such targeted therapies can restore tumor cell sensitivity to the inhibitors.
In this review, we summarized research progress on ALK resistance to provide a reference for the design of clinical studies and development of new drugs for the treatment of ALK+ ALCL. ALK is an important therapeutic target in ALK+ ALCL. ALK TKIs have broadened the therapeutic options for ALK+ALCL patients who are resistant to or relapse on chemotherapy, but the emergence of drug resistance is an outstanding problem. Considerable progress has been made in the elucidation of ALK resistance mechanisms including those associated with and independent of ALK, and novel TKIs are being developed that can bring lasting remission to ALK TKI-resistant patients.
YW and JH performed the analysis and wrote the manuscript. MX and QX checked and embellished the language. CZ searched the relevant literature and drew the figures. WS and YZ designed the ideas for the article. All authors contributed to the article and unanimously agreed to submit the review.
This study was supported by the National Natural Science Foundation international cooperation (81570184), the Science and Technology Project of Nantong City (MS22018008), the Science and Technology Project of Nantong City (MS12017003-2), China Postdoctoral Science Foundation (2019M660127), Jiangsu Province Postdoctoral Science Foundation (2019K062), Jiangsu Province Postdoctoral Foundation (2019Z146), the Municipal Natural Science Foundation of Nantong (Nos. JCZ20207).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We sincerely appreciate all members. We thank Charlesworth Author Services (www.cwauthors.com) for its linguistic assistance during the preparation of this manuscript.
1. Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, et al. The Expression of the Hodgkin’s Disease Associated Antigen Ki-1 in Reactive and Neoplastic Lymphoid Tissue: Evidence That Reed-Sternberg Cells and Histiocytic Malignancies Are Derived From Activated Lymphoid Cells. Blood (1985) 66(4):848–58. doi: 10.1182/blood.V66.4.848.848
2. Turner SD, Lamant L, Kenner L, Brugières L. Anaplastic Large Cell Lymphoma in Paediatric and Young Adult Patients. Br J Haematol (2016) 173(4):560–72. doi: 10.1111/bjh.13958
3. Lamant L, Dastugue N, Pulford K, Delsol G, Mariamé B. A New Fusion Gene TPM3-ALK in Anaplastic Large Cell Lymphoma Created by a (1;2)(Q25;P23) Translocation. Blood (1999) 93(9):3088–95. doi: 10.1182/blood.V93.9.3088.409k30_3088_3095
4. Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, et al. Inv(2)(P23q35) in Anaplastic Large-Cell Lymphoma Induces Constitutive Anaplastic Lymphoma Kinase (ALK) Tyrosine Kinase Activation by Fusionan Enzyme Involved in Purine Nucleotide Biosynthesis. Blood (2000) 95(6):2144–9. doi: 10.1182/blood.V95.6.2144
5. Feldman AL, Vasmatzis G, Asmann YW, Davila J, Middha S, Eckloff BW, et al. Novel TRAF1-ALK Fusion Identified by Deep RNA Sequencing of Anaplastic Large Cell Lymphoma. Genes Chromosomes Cancer (2013) 52(11):1097–102. doi: 10.1002/gcc.22104
6. van der Krogt JA, Bempt MV, Ferreiro JF, Mentens N, Jacobs K, Pluys U, et al. Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma With the Variant RNF213-, ATIC- and TPM3-ALK Fusions Is Characterized by Copy Number Gain of the Rearranged ALK Gene. Haematologica (2017) 102(9):1605–16. doi: 10.3324/haematol.2016.146571
7. Pulford K, Morris SW, Turturro F. Anaplastic Lymphoma Kinase Proteins in Growth Control and Cancer. J Cell Physiol (2004) 199(3):330–58. doi: 10.1002/jcp.10472
8. Damm-Welk C, Klapper W, Oschlies I, Gesk S, Röttgers S, Bradtke J, et al. Distribution of NPM1-ALK and X-ALK Fusion Transcripts in Paediatric Anaplastic Large Cell Lymphoma: A Molecular-Histological Correlation. Br J Haematol (2009) 146(3):306–9. doi: 10.1111/j.1365-2141.2009.07754.x
9. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science (1994) 263(5151):1281–4. doi: 10.1126/science.8122112
10. Wasik MA, Zhang Q, Marzec M, Kasprzycka M, Wang HY, Liu X. Anaplastic Lymphoma Kinase (ALK)-Induced Malignancies: Novel Mechanisms of Cell Transformation and Potential Therapeutic Approaches. Semin Oncol (2009) 36(2 Suppl 1):S27–35. doi: 10.1053/j.seminoncol.2009.02.007
11. Al Zaid Siddiquee K, Turkson J. STAT3 as a Target for Inducing Apoptosis in Solid and Hematological Tumors. Cell Res (2008) 18(2):254–67. doi: 10.1038/cr.2008.18
12. Pham TH, Park HM, Kim J, Hong JT, Yoon DY. STAT3 and P53: Dual Target for Cancer Therapy. Biomedicines (2020) 8(12):637. doi: 10.3390/biomedicines8120637
13. Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/Anaplastic Lymphoma Kinase (NPM/ALK) Oncoprotein Induces the T Regulatory Cell Phenotype by Activating STAT3. Proc Natl Acad Sci USA (2006) 103(26):9964–9. doi: 10.1073/pnas.0603507103
14. Kasprzycka M, Zhang Q, Witkiewicz A, Marzec M, Potoczek M, Liu X, et al. Gamma C-Signaling Cytokines Induce a Regulatory T Cell Phenotype in Malignant CD4+ T Lymphocytes. J Immunol (2008) 181(4):2506–12. doi: 10.4049/jimmunol.181.4.2506
15. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic Kinase NPM/ALK Induces Through STAT3 Expression of Immunosuppressive Protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA (2008) 105(52):20852–7. doi: 10.1073/pnas.0810958105
16. Marzec M, Liu X, Wong W, Yang Y, Pasha T, Kantekure K, et al. Oncogenic Kinase NPM/ALK Induces Expression of HIF1α Mrna. Oncogene (2011) 30(11):1372–8. doi: 10.1038/onc.2010.505
17. Martinengo C, Poggio T, Menotti M, Scalzo MS, Mastini C, Ambrogio C, et al. ALK-Dependent Control of Hypoxia-Inducible Factors Mediates Tumor Growth and Metastasis. Cancer Res (2014) 74(21):6094–106. doi: 10.1158/0008-5472.Can-14-0268
18. Kim KW, Mutter RW, Cao C, Albert JM, Shinohara ET, Sekhar KR, et al. Inhibition of Signal Transducer and Activator of Transcription 3 Activity Results in Down-Regulation of Survivin Following Irradiation. Mol Cancer Ther (2006) 5(11):2659–65. doi: 10.1158/1535-7163.Mct-06-0261
19. Hoareau-Aveilla C, Meggetto F. Crosstalk Between Microrna and DNA Methylation Offers Potential Biomarkers and Targeted Therapies in ALK-Positive Lymphomas. Cancers (Basel) (2017) 9(8):100. doi: 10.3390/cancers9080100
20. Zhang Q, Wang HY, Liu X, Wasik MA. STAT5A Is Epigenetically Silenced by the Tyrosine Kinase NPM1-ALK and Acts as a Tumor Suppressor by Reciprocally Inhibiting NPM1-ALK Expression. Nat Med (2007) 13(11):1341–8. doi: 10.1038/nm1659
21. Hegazy SA, Wang P, Anand M, Ingham RJ, Gelebart P, Lai R. The Tyrosine 343 Residue of Nucleophosmin (NPM)-Anaplastic Lymphoma Kinase (ALK) Is Important for Its Interaction With SHP1, a Cytoplasmic Tyrosine Phosphatase With Tumor Suppressor Functions. J Biol Chem (2010) 285(26):19813–20. doi: 10.1074/jbc.M110.121988
22. Zhang Q, Wang HY, Liu X, Bhutani G, Kantekure K, Wasik M. IL-2R Common Gamma-Chain Is Epigenetically Silenced by Nucleophosphin-Anaplastic Lymphoma Kinase (NPM-ALK) and Acts as a Tumor Suppressor by Targeting NPM-ALK. Proc Natl Acad Sci USA (2011) 108(29):11977–82. doi: 10.1073/pnas.1100319108
23. Piazza R, Magistroni V, Mogavero A, Andreoni F, Ambrogio C, Chiarle R, et al. Epigenetic Silencing of the Proapoptotic Gene BIM in Anaplastic Large Cell Lymphoma Through an Mecp2/SIN3a Deacetylating Complex. Neoplasia (2013) 15(5):511–22. doi: 10.1593/neo.121784
24. Hassler MR, Pulverer W, Lakshminarasimhan R, Redl E, Hacker J, Garland GD, et al. Insights Into the Pathogenesis of Anaplastic Large-Cell Lymphoma Through Genome-Wide DNA Methylation Profiling. Cell Rep (2016) 17(2):596–608. doi: 10.1016/j.celrep.2016.09.018
25. Wu R, Ivan E, Sahasrabuddhe AA, Shaw T, Mullighan CG, Leventaki V, et al. Epigenetic Modulation of CD48 by NPM-ALK Promotes Immune Evasion in ALK+ ALCL. Blood (2019) 134(Supplement_1):1510–0. doi: 10.1182/blood-2019-127453
26. Desjobert C, Renalier MH, Bergalet J, Dejean E, Joseph N, Kruczynski A, et al. Mir-29a Down-Regulation in ALK-Positive Anaplastic Large Cell Lymphomas Contributes to Apoptosis Blockade Through MCL-1 Overexpression. Blood (2011) 117(24):6627–37. doi: 10.1182/blood-2010-09-301994
27. Vishwamitra D, Li Y, Wilson D, Manshouri R, Curry CV, Shi B, et al. MicroRNA 96 Is a Post-Transcriptional Suppressor of Anaplastic Lymphoma Kinase Expression. Am J Pathol (2012) 180(5):1772–80. doi: 10.1016/j.ajpath.2012.01.008
28. Liu C, Iqbal J, Teruya-Feldstein J, Shen Y, Dabrowska MJ, Dybkaer K, et al. Microrna Expression Profiling Identifies Molecular Signatures Associated With Anaplastic Large Cell Lymphoma. Blood (2013) 122(12):2083–92. doi: 10.1182/blood-2012-08-447375
29. Hoareau-Aveilla C, Valentin T, Daugrois C, Quelen C, Mitou G, Quentin S, et al. Reversal of Microrna-150 Silencing Disadvantages Crizotinib-Resistant NPM-ALK(+) Cell Growth. J Clin Invest (2015) 125(9):3505–18. doi: 10.1172/jci78488
30. Hoareau-Aveilla C, Quelen C, Congras A, Caillet N, Labourdette D, Dozier C, et al. Mir-497 Suppresses Cycle Progression Through an Axis Involving CDK6 in ALK-Positive Cells. Haematologica (2019) 104(2):347–59. doi: 10.3324/haematol.2018.195131
31. Garbin A, Lovisa F, Holmes AB, Damanti CC, Gallingani I, Carraro E, et al. Mir-939 Acts as Tumor Suppressor by Modulating JUNB Transcriptional Activity in Pediatric Anaplastic Large Cell Lymphoma. Haematologica (2021) 106(2):610–3. doi: 10.3324/haematol.2019.241307
32. Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, et al. NPM-ALK Oncogenic Tyrosine Kinase Controls T-Cell Identity by Transcriptional Regulation and Epigenetic Silencing in Lymphoma Cells. Cancer Res (2009) 69(22):8611–9. doi: 10.1158/0008-5472.Can-09-2655
33. Gambi G, Di Simone E, Basso V, Ricci L, Wang R, Verma A, et al. The Transcriptional Regulator Sin3A Contributes to the Oncogenic Potential of STAT3. Cancer Res (2019) 79(12):3076–87. doi: 10.1158/0008-5472.Can-18-0359
34. Kim JW, Sim SS, Kim UH, Nishibe S, Wahl MI, Carpenter G, et al. Tyrosine Residues in Bovine Phospholipase C-Gamma Phosphorylated by the Epidermal Growth Factor Receptor In Vitro. J Biol Chem (1990) 265(7):3940–3. doi: 10.1016/S0021-9258(19)39684-X
35. Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-Anaplastic Lymphoma Kinase of Large-Cell Anaplastic Lymphoma Is a Constitutively Active Tyrosine Kinase That Utilizes Phospholipase C-Gamma to Mediate Its Mitogenicity. Mol Cell Biol (1998) 18(12):6951–61. doi: 10.1128/mcb.18.12.6951
36. Rhee SG. Regulation of Phosphoinositide-Specific Phospholipase C. Annu Rev Biochem (2001) 70:281–312. doi: 10.1146/annurev.biochem.70.1.281
37. Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A Retroviral Oncogene, Akt, Encoding a Serine-Threonine Kinase Containing an SH2-Like Region. Science (1991) 254(5029):274–7. doi: 10.1126/science.254.5029.274
38. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Balpha. Curr Biol (1997) 7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9
39. Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, et al. Role of Translocation in the Activation and Function of Protein Kinase B. J Biol Chem (1997) 272(50):31515–24. doi: 10.1074/jbc.272.50.31515
40. Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The Akt Kinase: Molecular Determinants of Oncogenicity. Proc Natl Acad Sci USA (1998) 95(25):14950–5. doi: 10.1073/pnas.95.25.14950
41. Vega F, Medeiros LJ, Leventaki V, Atwell C, Cho-Vega JH, Tian L, et al. Activation of Mammalian Target of Rapamycin Signaling Pathway Contributes to Tumor Cell Survival in Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma. Cancer Res (2006) 66(13):6589–97. doi: 10.1158/0008-5472.Can-05-3018
42. Marzec M, Kasprzycka M, Liu X, Raghunath PN, Wlodarski P, Wasik MA. Oncogenic Tyrosine Kinase NPM/ALK Induces Activation of the MEK/ERK Signaling Pathway Independently of C-Raf. Oncogene (2007) 26(6):813–21. doi: 10.1038/sj.onc.1209843
43. Kent LN, Leone G. The Broken Cycle: E2F Dysfunction in Cancer. Nat Rev Cancer (2019) 19(6):326–38. doi: 10.1038/s41568-019-0143-7
44. Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, et al. The Oncoprotein NPM-ALK of Anaplastic Large-Cell Lymphoma Induces JUNB Transcription via ERK1/2 and Junb Translation via Mtor Signaling. Blood (2007) 110(9):3374–83. doi: 10.1182/blood-2007-02-071258
45. Wu Z, Nicoll M, Ingham RJ. AP-1 Family Transcription Factors: A Diverse Family of Proteins That Regulate Varied Cellular Activities in Classical Hodgkin Lymphoma and ALK+ ALCL. Exp Hematol Oncol (2021) 10(1):4. doi: 10.1186/s40164-020-00197-9
46. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and Activity of Crizotinib for Paediatric Patients With Refractory Solid Tumours or Anaplastic Large-Cell Lymphoma: A Children’s Oncology Group Phase 1 Consortium Study. Lancet Oncol (2013) 14(6):472–80. doi: 10.1016/s1470-2045(13)70095-0
47. Fukano R, Mori T, Sekimizu M, Choi I, Kada A, Saito AM, et al. Alectinib for Relapsed or Refractory Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma: An Open-Label Phase II Trial. Cancer Sci (2020) 111(12):4540–7. doi: 10.1111/cas.14671
48. Arosio G, Sharma GG, Villa M, Mauri M, Crespiatico I, Fontana D, et al. Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma. Cancers (Basel) (2021) 13(17):4422. doi: 10.3390/cancers13174422
49. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK Mutations in Lung Cancer That Confer Resistance to ALK Inhibitors. N Engl J Med (2010) 363(18):1734–9. doi: 10.1056/NEJMoa1007478
50. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of Resistance to Crizotinib in Patients With ALK Gene Rearranged Non-Small Cell Lung Cancer. Clin Cancer Res (2012) 18(5):1472–82. doi: 10.1158/1078-0432.Ccr-11-2906
51. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Sci Transl Med (2012) 4(120):120ra117. doi: 10.1126/scitranslmed.3003316
52. Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, et al. Next-Generation Sequencing Reveals a Novel NSCLC ALK F1174V Mutation and Confirms ALK G1202R Mutation Confers High-Level Resistance to Alectinib (CH5424802/RO5424802) in ALK-Rearranged NSCLC Patients Who Progressed on Crizotinib. J Thorac Oncol (2014) 9(4):549–53. doi: 10.1097/jto.0000000000000094
53. Toyokawa G, Hirai F, Inamasu E, Yoshida T, Nosaki K, Takenaka T, et al. Secondary Mutations at I1171 in the ALK Gene Confer Resistance to Both Crizotinib and Alectinib. J Thorac Oncol (2014) 9(12):e86–7. doi: 10.1097/jto.0000000000000358
54. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov (2016) 6(10):1118–33. doi: 10.1158/2159-8290.Cd-16-0596
55. Ceccon M, Mologni L, Giudici G, Piazza R, Pirola A, Fontana D, et al. Treatment Efficacy and Resistance Mechanisms Using the Second-Generation ALK Inhibitor AP26113 in Human NPM-ALK-Positive Anaplastic Large Cell Lymphoma. Mol Cancer Res (2015) 13(4):775–83. doi: 10.1158/1541-7786.Mcr-14-0157
56. Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, et al. Lorlatinib Treatment Elicits Multiple on- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res (2018) 78(24):6866–80. doi: 10.1158/0008-5472.Can-18-1867
57. Ceccon M, Merlo MEB, Mologni L, Poggio T, Varesio LM, Menotti M, et al. Excess of NPM-ALK Oncogenic Signaling Promotes Cellular Apoptosis and Drug Dependency. Oncogene (2016) 35(29):3854–65. doi: 10.1038/onc.2015.456
58. Rajan SS, Amin AD, Li L, Rolland DC, Li H, Kwon D, et al. The Mechanism of Cancer Drug Addiction in ALK-Positive T-Cell Lymphoma. Oncogene (2020) 39(10):2103–17. doi: 10.1038/s41388-019-1136-4
59. Prokoph N, Probst NA, Lee LC, Monahan JM, Matthews JD, Liang HC, et al. IL10RA Modulates Crizotinib Sensitivity in NPM1-ALK+ Anaplastic Large Cell Lymphoma. Blood (2020) 136(14):1657–69. doi: 10.1182/blood.2019003793
60. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv Cancer Res (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
61. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive Plasticity of IL-10(+) and IL-35(+) T(Reg) Cells Cooperatively Promotes Tumor T Cell Exhaustion. Nat Immunol (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
62. Li Y, Wang K, Song N, Hou K, Che X, Zhou Y, et al. Activation of IGF-1R Pathway and NPM-ALK G1269A Mutation Confer Resistance to Crizotinib Treatment in NPM-ALK Positive Lymphoma. Invest New Drugs (2020) 38(3):599–609. doi: 10.1007/s10637-019-00802-7
63. Shi P, Lai R, Lin Q, Iqbal AS, Young LC, Kwak LW, et al. IGF-IR Tyrosine Kinase Interacts With NPM-ALK Oncogene to Induce Survival of T-Cell ALK+ Anaplastic Large-Cell Lymphoma Cells. Blood (2009) 114(2):360–70. doi: 10.1182/blood-2007-11-125658
64. Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for Co-Targeting IGF-1R and ALK in ALK Fusion-Positive Lung Cancer. Nat Med (2014) 20(9):1027–34. doi: 10.1038/nm.3667
65. Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O, et al. PDGFR Blockade Is a Rational and Effective Therapy for NPM-ALK-Driven Lymphomas. Nat Med (2012) 18(11):1699–704. doi: 10.1038/nm.2966
66. Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M. Platelet-Derived Growth Factor Induces Phosphorylation of Multiple JAK Family Kinases and STAT Proteins. Mol Cell Biol (1996) 16(4):1759–69. doi: 10.1128/mcb.16.4.1759
67. Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. Pdgfrs Are Critical for PI3K/Akt Activation and Negatively Regulated by Mtor. J Clin Invest (2007) 117(3):730–8. doi: 10.1172/jci28984
68. Menotti M, Ambrogio C, Cheong TC, Pighi C, Mota I, Cassel SH, et al. Wiskott-Aldrich Syndrome Protein (WASP) Is a Tumor Suppressor in T Cell Lymphoma. Nat Med (2019) 25(1):130–40. doi: 10.1038/s41591-018-0262-9
69. Han SS, Wen KK, Vyas YM. Deficiency of Wiskott-Aldrich Syndrome Protein has Opposing Effect on the Pro-Oncogenic Pathway Activation in Nonmalignant Versus Malignant Lymphocytes. Oncogene (2021) 40(2):345–54. doi: 10.1038/s41388-020-01533-3
70. Murga-Zamalloa CA, Mendoza-Reinoso V, Sahasrabuddhe AA, Rolland D, Hwang SR, McDonnell SR, et al. NPM-ALK Phosphorylates Wasp Y102 and Contributes to Oncogenesis of Anaplastic Large Cell Lymphoma. Oncogene (2017) 36(15):2085–94. doi: 10.1038/onc.2016.366
71. Ambrogio C, Voena C, Manazza AD, Martinengo C, Costa C, Kirchhausen T, et al. The Anaplastic Lymphoma Kinase Controls Cell Shape and Growth of Anaplastic Large Cell Lymphoma Through Cdc42 Activation. Cancer Res (2008) 68(21):8899–907. doi: 10.1158/0008-5472.Can-08-2568
72. Karaca Atabay E, Mecca C, Wang Q, Ambrogio C, Mota I, Prokoph N, et al. Tyrosine Phosphatases Regulate Resistance to ALK Inhibitors in ALK+ Anaplastic Large Cell Lymphoma. Blood (2021) blood.2020008136. doi: 10.1182/blood.2020008136
73. Mitou G, Frentzel J, Desquesnes A, Le Gonidec S, AlSaati T, Beau I, et al. Targeting Autophagy Enhances the Anti-Tumoral Action of Crizotinib in ALK-Positive Anaplastic Large Cell Lymphoma. Oncotarget (2015) 6(30):30149–64. doi: 10.18632/oncotarget.4999
74. Gao J, Yin M, Zhu Y, Gu L, Zhang Y, Li Q, et al. Prognostic Significance and Therapeutic Potential of the Activation of Anaplastic Lymphoma Kinase/Protein Kinase B/Mammalian Target of Rapamycin Signaling Pathway in Anaplastic Large Cell Lymphoma. BMC Cancer (2013) 13:471. doi: 10.1186/1471-2407-13-471
75. Lim MS, Carlson ML, Crockett DK, Fillmore GC, Abbott DR, Elenitoba-Johnson OF, et al. The Proteomic Signature of NPM/ALK Reveals Deregulation of Multiple Cellular Pathways. Blood (2009) 114(8):1585–95. doi: 10.1182/blood-2009-02-204735
76. Sorrentino D, Frentzel J, Mitou G, Blasco RB, Torossian A, Hoareau-Aveilla C, et al. High Levels of Mir-7-5p Potentiate Crizotinib-Induced Cytokilling and Autophagic Flux by Targeting RAF1 in NPM-ALK Positive Lymphoma Cells. Cancers (Basel) (2020) 12(10):2951. doi: 10.3390/cancers12102951
77. Shang C, Hassan B, Haque M, Song Y, Li J, Liu D, et al. Crizotinib Resistance Mediated by Autophagy Is Higher in the Stem-Like Cell Subset in ALK-Positive Anaplastic Large Cell Lymphoma, and This Effect Is MYC-Dependent. Cancers (Basel) (2021) 13(2):181. doi: 10.3390/cancers13020181
78. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell (2005) 122(6):927–39. doi: 10.1016/j.cell.2005.07.002
79. Torossian A, Broin N, Frentzel J, Daugrois C, Gandarillas S, Saati TA, et al. Blockade of Crizotinib-Induced BCL2 Elevation in ALK-Positive Anaplastic Large Cell Lymphoma Triggers Autophagy Associated With Cell Death. Haematologica (2019) 104(7):1428–39. doi: 10.3324/haematol.2017.181966
80. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-Induced Senescence Is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature (2006) 444(7119):633–7. doi: 10.1038/nature05268
81. Halazonetis TD, Gorgoulis VG, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science (2008) 319(5868):1352–5. doi: 10.1126/science.1140735
82. Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol Oncol (2015) 9(3):601–16. doi: 10.1016/j.molonc.2014.11.001
83. Ceccon M, Mauri M, Massimino L, Giudici G, Piazza R, Gambacorti-Passerini C, et al. Mitochondrial Hyperactivation and Enhanced ROS Production Are Involved in Toxicity Induced by Oncogenic Kinases Over-Signaling. Cancers (Basel) (2018) 10(12):509. doi: 10.3390/cancers10120509
84. Kim HS, Lee MS. STAT1 as a Key Modulator of Cell Death. Cell Signal (2007) 19(3):454–65. doi: 10.1016/j.cellsig.2006.09.003
85. Wu C, Molavi O, Zhang H, Gupta N, Alshareef A, Bone KM, et al. STAT1 Is Phosphorylated and Downregulated by the Oncogenic Tyrosine Kinase NPM-ALK in ALK-Positive Anaplastic Large-Cell Lymphoma. Blood (2015) 126(3):336–45. doi: 10.1182/blood-2014-10-603738
86. Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and Activity of Alectinib Against Systemic Disease and Brain Metastases in Patients With Crizotinib-Resistant ALK-Rearranged Non-Small-Cell Lung Cancer (AF-002JG): Results From the Dose-Finding Portion of a Phase 1/2 Study. Lancet Oncol (2014) 15(10):1119–28. doi: 10.1016/s1470-2045(14)70362-6
87. Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N Engl J Med (2016) 374(1):54–61. doi: 10.1056/NEJMoa1508887
88. Okada K, Araki M, Sakashita T, Ma B, Kanada R, Yanagitani N, et al. Prediction of ALK Mutations Mediating ALK-Tkis Resistance and Drug Re-Purposing to Overcome the Resistance. EBioMedicine (2019) 41:105–19. doi: 10.1016/j.ebiom.2019.01.019
89. Yanagitani N, Uchibori K, Koike S, Tsukahara M, Kitazono S, Yoshizawa T, et al. Drug Resistance Mechanisms in Japanese Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer and the Clinical Responses Based on the Resistant Mechanisms. Cancer Sci (2020) 111(3):932–9. doi: 10.1111/cas.14314
90. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non-Small Cell Lung Cancer. Cancer Discov (2014) 4(6):662–73. doi: 10.1158/2159-8290.Cd-13-0846
91. Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK Inhibitors can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov (2018) 8(6):714–29. doi: 10.1158/2159-8290.Cd-17-1256
92. Ou SH, Greenbowe J, Khan ZU, Azada MC, Ross JS, Stevens PJ, et al. I1171 Missense Mutation (Particularly I1171N) Is a Common Resistance Mutation in ALK-Positive NSCLC Patients Who Have Progressive Disease While on Alectinib and Is Sensitive to Ceritinib. Lung Cancer (2015) 88(2):231–4. doi: 10.1016/j.lungcan.2015.02.005
93. Ou SH, Milliken JC, Azada MC, Miller VA, Ali SM, Klempner SJ. ALK F1174V Mutation Confers Sensitivity While ALK I1171 Mutation Confers Resistance to Alectinib. The Importance of Serial Biopsy Post Progression. Lung Cancer (2016) 91:70–2. doi: 10.1016/j.lungcan.2015.09.006
94. Gettinger SN, Zhang S, Hodgson JG, Bazhenova L, Burgers S, Kim D-W, et al. Activity of Brigatinib (BRG) in Crizotinib (CRZ) Resistant Patients (Pts) According to ALK Mutation Status. J Clin Oncol (2016) 34(15_suppl):9060–0. doi: 10.1200/JCO.2016.34.15_suppl.9060
95. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, Hansen KH, Kim S-W, et al. Brigatinib (BRG) in Patients (Pts) With Crizotinib (CRZ)-Refractory ALK+ Non-Small Cell Lung Cancer (NSCLC): First Report of Efficacy and Safety From a Pivotal Randomized Phase (Ph) 2 Trial (ALTA). J Clin Oncol (2016) 34(15_suppl):9007–7. doi: 10.1200/JCO.2016.34.15_suppl.9007
96. Zhang S, Anjum R, Squillace R, Nadworny S, Zhou T, Keats J, et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin Cancer Res (2016) 22(22):5527–38. doi: 10.1158/1078-0432.Ccr-16-0569
97. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell (2015) 28(1):70–81. doi: 10.1016/j.ccell.2015.05.010
98. Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin Cancer Res (2020) 26(1):242–55. doi: 10.1158/1078-0432.Ccr-19-1104
99. Mizuta H, Okada K, Araki M, Adachi J, Takemoto A, Kutkowska J, et al. Gilteritinib Overcomes Lorlatinib Resistance in ALK-Rearranged Cancer. Nat Commun (2021) 12(1):1261. doi: 10.1038/s41467-021-21396-w
100. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-Rearranged Non-Small-Cell Lung Cancer. N Engl J Med (2014) 370(13):1189–97. doi: 10.1056/NEJMoa1311107
101. Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, et al. Synthesis, Structure-Activity Relationships, and In Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-Isopropoxy-5-Methyl-4-(Piperidin-4-Yl)Phenyl)-N4-(2-(Isopropylsulfonyl)Phenyl)Pyrimidine-2,4-Diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J Med Chem (2013) 56(14):5675–90. doi: 10.1021/jm400402q
102. Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, et al. Novel ALK Inhibitors in Clinical Use and Development. J Hematol Oncol (2015) 8:17. doi: 10.1186/s13045-015-0122-8
103. Fischer M, Moreno L, Ziegler DS, Marshall LV, Zwaan CM, Irwin MS, et al. Ceritinib in Paediatric Patients With Anaplastic Lymphoma Kinase-Positive Malignancies: An Open-Label, Multicentre, Phase 1, Dose-Escalation and Dose-Expansion Study. Lancet Oncol (2021) 22(12):1764–76. doi: 10.1016/s1470-2045(21)00536-2
104. Huang WS, Liu S, Zou D, Thomas M, Wang Y, Zhou T, et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J Med Chem (2016) 59(10):4948–64. doi: 10.1021/acs.jmedchem.6b00306
105. Pearson ADJ, Barry E, Mossé YP, Ligas F, Bird N, de Rojas T, et al. Second Paediatric Strategy Forum for Anaplastic Lymphoma Kinase (ALK) Inhibition in Paediatric Malignancies: ACCELERATE in Collaboration With the European Medicines Agency With the Participation of the Food and Drug Administration. Eur J Cancer (2021) 157:198–213. doi: 10.1016/j.ejca.2021.08.022
106. Gou W, Li Z, Xu X, Shen J, Guo M, Zhou X, et al. ZX-29, a Novel ALK Inhibitor, Induces Apoptosis via ER Stress in ALK Rearrangement NSCLC Cells and Overcomes Cell Resistance Caused by an ALK Mutation. Biochim Biophys Acta Mol Cell Res (2020) 1867(7):118712. doi: 10.1016/j.bbamcr.2020.118712
107. Zhou X, Zhang X, Wu Z, Xu X, Guo M, Zhai X, et al. The Novel ALK Inhibitor ZX-29 Induces Apoptosis Through Inhibiting ALK and Inducing ROS-Mediated Endoplasmic Reticulum Stress in Karpas299 Cells. J Biochem Mol Toxicol (2021) 35(3):e22666. doi: 10.1002/jbt.22666
108. Pulte ED, Norsworthy KJ, Wang Y, Xu Q, Qosa H, Gudi R, et al. FDA Approval Summary: Gilteritinib for Relapsed or Refractory Acute Myeloid Leukemia With a FLT3 Mutation. Clin Cancer Res (2021) 27(13):3515–21. doi: 10.1158/1078-0432.Ccr-20-4271
109. Dhillon S. Gilteritinib: First Global Approval. Drugs (2019) 79(3):331–9. doi: 10.1007/s40265-019-1062-3
110. Kuravi S, Cheng J, Fangman G, Polireddy K, McCormick S, Lin TL, et al. Preclinical Evaluation of Gilteritinib on NPM1-ALK-Driven Anaplastic Large Cell Lymphoma Cells. Mol Cancer Res (2021) 19(5):913–20. doi: 10.1158/1541-7786.Mcr-20-0738
111. Dale B, Cheng M, Park KS, Kaniskan H, Xiong Y, Jin J. Advancing Targeted Protein Degradation for Cancer Therapy. Nat Rev Cancer (2021) 21(10):638–54. doi: 10.1038/s41568-021-00365-x
112. Kleiger G, Mayor T. Perilous Journey: A Tour of the Ubiquitin-Proteasome System. Trends Cell Biol (2014) 24(6):352–9. doi: 10.1016/j.tcb.2013.12.003
113. Kang CH, Lee DH, Lee CO, Du Ha J, Park CH, Hwang JY. Induced Protein Degradation of Anaplastic Lymphoma Kinase (ALK) by Proteolysis Targeting Chimera (PROTAC). Biochem Biophys Res Commun (2018) 505(2):542–7. doi: 10.1016/j.bbrc.2018.09.169
114. Zhou X, Dong R, Zhang JY, Zheng X. PROTAC: A Promising Technology for Cancer Treatment. Eur J Med Chem (2020) 203:112539. doi: 10.1016/j.ejmech.2020.112539
115. Sun N, Ren C, Kong Y, Zhong H, Chen J, Li Y, et al. Development of a Brigatinib Degrader (SIAIS117) as a Potential Treatment for ALK Positive Cancer Resistance. Eur J Med Chem (2020) 193:112190. doi: 10.1016/j.ejmech.2020.112190
116. Ren C, Sun N, Liu H, Kong Y, Sun R, Qiu X, et al. Discovery of a Brigatinib Degrader SIAIS164018 With Destroying Metastasis-Related Oncoproteins and a Reshuffling Kinome Profile. J Med Chem (2021) 64(13):9152–65. doi: 10.1021/acs.jmedchem.1c00373
117. Xie S, Sun Y, Liu Y, Li X, Li X, Zhong W, et al. Development of Alectinib-Based Protacs as Novel Potent Degraders of Anaplastic Lymphoma Kinase (ALK). J Med Chem (2021) 64(13):9120–40. doi: 10.1021/acs.jmedchem.1c00270
118. Petrylak D, Crews C, Yeh E, Ciulli A, Poh A. Proof-of-Concept With Protacs in Prostate Cancer. Cancer Discov (2020) 10(8):1084. doi: 10.1158/2159-8290.Cd-nb2020-054
119. Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front Pharmacol (2021) 12:692574. doi: 10.3389/fphar.2021.692574
120. Ren C, Sun N, Kong Y, Qu X, Liu H, Zhong H, et al. Structure-Based Discovery of SIAIS001 as an Oral Bioavailability ALK Degrader Constructed From Alectinib. Eur J Med Chem (2021) 217:113335. doi: 10.1016/j.ejmech.2021.113335
121. Prutsch N, Gurnhofer E, Suske T, Liang HC, Schlederer M, Roos S, et al. Dependency on the TYK2/STAT1/MCL1 Axis in Anaplastic Large Cell Lymphoma. Leukemia (2019) 33(3):696–709. doi: 10.1038/s41375-018-0239-1
122. George B, George SK, Shi W, Haque A, Shi P, Eskandari G, et al. Dual Inhibition of IGF-IR and ALK as an Effective Strategy to Eradicate NPM-ALK(+) T-Cell Lymphoma. J Hematol Oncol (2019) 12(1):80. doi: 10.1186/s13045-019-0768-8
123. Wilson C, Nimick M, Nehoff H. ALK and IGF-1R as Independent Targets in Crizotinib Resistant Lung Cancer. Sci Rep (2017) 7(1):13955. doi: 10.1038/s41598-017-14289-w
124. Kamstrup MR, Biskup E, Gjerdrum LM, Ralfkiaer E, Niazi O, Gniadecki R. The Importance of Notch Signaling in Peripheral T-Cell Lymphomas. Leuk Lymphoma (2014) 55(3):639–44. doi: 10.3109/10428194.2013.807510
125. Larose H, Prokoph N, Matthews JD, Schlederer M, Högler S, Alsulami AF, et al. Whole Exome Sequencing Reveals NOTCH1 Mutations in Anaplastic Large Cell Lymphoma and Points to Notch Both as a Key Pathway and a Potential Therapeutic Target. Haematologica (2021) 106(6):1693–704. doi: 10.3324/haematol.2019.238766
126. Redaelli S, Ceccon M, Antolini L, Rigolio R, Pirola A, Peronaci M, et al. Synergistic Activity of ALK and Mtor Inhibitors for the Treatment of NPM-ALK Positive Lymphoma. Oncotarget (2016) 7(45):72886–97. doi: 10.18632/oncotarget.12128
127. Xu W, Kim JW, Jung WJ, Koh Y, Yoon SS. Crizotinib in Combination With Everolimus Synergistically Inhibits Proliferation of Anaplastic Lymphoma Kinase–Positive Anaplastic Large Cell Lymphoma. Cancer Res Treat (2018) 50(2):599–613. doi: 10.4143/crt.2016.357
128. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell (2012) 22(1):117–30. doi: 10.1016/j.ccr.2012.06.001
129. Shi Y, Jia B, Xu W, Li W, Liu T, Liu P, et al. Chidamide in Relapsed or Refractory Peripheral T Cell Lymphoma: A Multicenter Real-World Study in China. J Hematol Oncol (2017) 10(1):69. doi: 10.1186/s13045-017-0439-6
130. Slupianek A, Skorski T. NPM/ALK Downregulates p27Kip1 in a PI-3K-Dependent Manner. Exp Hematol (2004) 32(12):1265–71. doi: 10.1016/j.exphem.2004.11.002
131. Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, et al. Inhibition of Akt Increases p27Kip1 Levels and Induces Cell Cycle Arrest in Anaplastic Large Cell Lymphoma. Blood (2005) 105(2):827–9. doi: 10.1182/blood-2004-06-2125
132. Boi M, Todaro M, Vurchio V, Yang SN, Moon J, Kwee I, et al. Therapeutic Efficacy of the Bromodomain Inhibitor OTX015/MK-8628 in ALK-Positive Anaplastic Large Cell Lymphoma: An Alternative Modality to Overcome Resistant Phenotypes. Oncotarget (2016) 7(48):79637–53. doi: 10.18632/oncotarget.12876
133. Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy Against Neuroblastoma. Clin Cancer Res (2017) 23(11):2856–68. doi: 10.1158/1078-0432.Ccr-16-1114
134. Watanabe M, Sasaki M, Itoh K, Higashihara M, Umezawa K, Kadin ME, et al. Junb Induced by Constitutive CD30-Extracellular Signal-Regulated Kinase 1/2 Mitogen-Activated Protein Kinase Signaling Activates the CD30 Promoter in Anaplastic Large Cell Lymphoma and Reed-Sternberg Cells of Hodgkin Lymphoma. Cancer Res (2005) 65(17):7628–34. doi: 10.1158/0008-5472.Can-05-0925
135. Watanabe M, Ogawa Y, Itoh K, Koiwa T, Kadin ME, Watanabe T, et al. Hypomethylation of CD30 Cpg Islands With Aberrant Junb Expression Drives CD30 Induction in Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma. Lab Invest (2008) 88(1):48–57. doi: 10.1038/labinvest.3700696
136. Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat Biotechnol (2003) 21(7):778–84. doi: 10.1038/nbt832
137. Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. Cac10-Vcmmae, an Anti-CD30-Monomethyl Auristatin E Conjugate With Potent and Selective Antitumor Activity. Blood (2003) 102(4):1458–65. doi: 10.1182/blood-2003-01-0039
138. Sutherland MS, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, et al. Lysosomal Trafficking and Cysteine Protease Metabolism Confer Target-Specific Cytotoxicity by Peptide-Linked Anti-CD30-Auristatin Conjugates. J Biol Chem (2006) 281(15):10540–7. doi: 10.1074/jbc.M510026200
139. Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, et al. Intracellular Activation of SGN-35, a Potent Anti-CD30 Antibody-Drug Conjugate. Clin Cancer Res (2010) 16(3):888–97. doi: 10.1158/1078-0432.Ccr-09-2069
140. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Five-Year Results of Brentuximab Vedotin in Patients With Relapsed or Refractory Systemic Anaplastic Large Cell Lymphoma. Blood (2017) 130(25):2709–17. doi: 10.1182/blood-2017-05-780049
141. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Brentuximab Vedotin (SGN-35) in Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. J Clin Oncol (2012) 30(18):2190–6. doi: 10.1200/jco.2011.38.0402
142. Lowe EJ, Reilly AF, Lim MS, Gross TG, Saguilig L, Barkauskas DA, et al. Brentuximab Vedotin in Combination With Chemotherapy for Pediatric Patients With ALK+ ALCL: Results of COG Trial ANHL12P1. Blood (2021) 137(26):3595–603. doi: 10.1182/blood.2020009806
143. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and Immunological Responses After CD30-Specific Chimeric Antigen Receptor-Redirected Lymphocytes. J Clin Invest (2017) 127(9):3462–71. doi: 10.1172/jci94306
144. Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM, Workman P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol Cancer Res (2015) 13(11):1445–51. doi: 10.1158/1541-7786.Mcr-15-0234
145. Courtin A, Smyth T, Hearn K, Saini HK, Thompson NT, Lyons JF, et al. Emergence of Resistance to Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer can be Delayed by an Upfront Combination With the HSP90 Inhibitor Onalespib. Br J Cancer (2016) 115(9):1069–77. doi: 10.1038/bjc.2016.294
146. Geng K, Liu H, Song Z, Zhang C, Zhang M, Yang H, et al. Design, Synthesis and Pharmacological Evaluation of ALK and Hsp90 Dual Inhibitors Bearing Resorcinol and 2,4-Diaminopyrimidine Motifs. Eur J Med Chem (2018) 152:76–86. doi: 10.1016/j.ejmech.2018.04.019
147. Iwafuchi H, Nakazawa A, Sekimizu M, Mori T, Osumi T, Iijima-Yamashita Y, et al. Clinicopathological Features and Prognostic Significance of Programmed Death Ligand 1 in Pediatric ALK-Positive Anaplastic Large Cell Lymphoma: Results of the ALCL99 Treatment in Japan. Hum Pathol (2021) 116:112–21. doi: 10.1016/j.humpath.2021.07.011
148. Shi Y, Wu J, Wang Z, Zhang L, Wang Z, Zhang M, et al. Efficacy and Safety of Geptanolimab (GB226) for Relapsed or Refractory Peripheral T Cell Lymphoma: An Open-Label Phase 2 Study (Gxplore-002). J Hematol Oncol (2021) 14(1):12. doi: 10.1186/s13045-021-01033-1
149. Rigaud C, Abbou S, Minard-Colin V, Geoerger B, Scoazec JY, Vassal G, et al. Efficacy of Nivolumab in a Patient With Systemic Refractory ALK+ Anaplastic Large Cell Lymphoma. Pediatr Blood Cancer (2018) 65(4):e26902. doi: 10.1002/pbc.26902
Keywords: ALCL, ALK, ALK-TKI, lymphoma, drug resistance, therapy
Citation: Wang Y, He J, Xu M, Xue Q, Zhu C, Liu J, Zhang Y and Shi W (2022) Holistic View of ALK TKI Resistance in ALK-Positive Anaplastic Large Cell Lymphoma. Front. Oncol. 12:815654. doi: 10.3389/fonc.2022.815654
Received: 15 November 2021; Accepted: 04 January 2022;
Published: 08 February 2022.
Edited by:
Ning Ding, Peking University Cancer Hospital, ChinaReviewed by:
Mariusz Wasik, Fox Chase Cancer Center, United StatesCopyright © 2022 Wang, He, Xu, Xue, Zhu, Liu, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenyu Shi, c2hpd2VueXVAaG90bWFpbC5jb20=; Yaping Zhang, enp5YXBpbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.