Corrigendum: Comprehensive analysis of the transcriptome-wide m6A methylome in lung adenocarcinoma by MeRIP sequencing
 Wenli Mao1
Wenli Mao1 Qingzhen Yu1,2Kefeng Wang1Qiang Ma1Yuxin Zheng1Guojun Zhang3
Qingzhen Yu1,2Kefeng Wang1Qiang Ma1Yuxin Zheng1Guojun Zhang3 Wei Luo4Nianwu Wang1
Wei Luo4Nianwu Wang1 Yukun Wang1,5*
Yukun Wang1,5*- 1Department of Pharmacology, School of Medicine, Southern University of Science and Technology, Shenzhen, China
- 2Medical Research Center, Southern University of Science and Technology Hospital, Shenzhen, China
- 3Nutrition Department, Southern University of Science and Technology Hospital, Shenzhen, China
- 4Department of Clinical Laboratory, Southern University of Science and Technology Hospital, Shenzhen, China
- 5Department of Pharmacy, Southern University of Science and Technology Hospital, Shenzhen, China
N6-methyladenosine (m6A) is the most abundant internal modification on eukaryotic mRNAs. There is increasing evidence that m6A plays a key role in tumor progression, so it is important to analyze m6A modifications within the transcriptome-wide in lung adenocarcinoma (LUAD). Three pairs of LUAD samples and tumor-adjacent normal tissues were obtained from the South University of Science and Technology Hospital. And then methylated RNA immunoprecipitation sequencing (MeRIP-seq) and RNA sequencing (RNA-seq) were used to identify differential m6A modifications between tumor and tumor-adjacent normal tissues. We identified 4041 aberrant m6A peaks, of which 1192 m6A peaks were upregulated and 2849 m6A peaks downregulated. It was found that genes with the dysregulated m6A peaks were enriched in the pathways in cancer, Rap1 signaling pathway, and insulin resistance. Additionally, 612 genes with abnormal regulation of m6A peaks and RNA expression were identified by combining MeRIP-seq and RNA-seq data. Through KEGG analysis, the 612 genes were enriched in cancer-related signaling pathways, such as the cGMP-PKG signaling pathway, and the Rap1 signaling pathway. What’s more, GSEA enrichment analysis showed these genes were enriched in cell cycle phase transition, cell division, cellular response to DNA damage stimulus, and chromosome organization. To further explore the relationship between differential m6A modified genes and clinical parameters of LUAD patients, we searched The Cancer Genome Atlas (TCGA) and identified 2 genes (FCRL5 and GPRIN1) that were associated with the prognosis and diagnosis of LUAD patients. Furthermore, we found a positive correlation between GPRIN1 and m6A reader YTHDF1 in the GEPIA2 database. It was verified that YTHDF1 binds to GPRIN1 mRNA and regulates its expression. Our study results suggest that m6A modification plays important role in the progression and prognosis of LUAD and maybe a potential new therapeutic target for LUAD patients in the future.
Introduction
RNA methylation mostly regulates gene expression at the posttranscriptional level, which is considered to be another layer of epigenetic regulation similar to DNA methylation and histone modification. Recently, a variety of molecular modifications of RNA (including rRNA, tRNA, snRNA, mRNA, and long non-coding RNA) have been discovered (1). The ways of RNA methylation modification include N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), 5-hydroxymethylcytosine (5hmC), N6, 2′-Odimethyladenosine (m6Am), 7-methylguanine (m7G) (2), of which m6A modification is the most abundant type in eukaryotic (3). The m6A modification is a methylation modification on the RNA level that was first discovered in the 1970s (4). The m6A high-throughput sequencing study revealed that m6A is a selective modification based on the recruitment of specific mRNAs. The m6A modification mainly takes place on the RRACH (R = G or A, H = A, C or U) consensus sequence (5–7), of which G(m6A)C (70%) or A(m6A)C (30%) (8), and is enriched near the 3’UTR, and stop codon (9). m6A modification is a dynamic and reversible methylation modification regulated by the m6A regulators, which is mainly composed of “writers” (METTL3, METTL14), and “erasers” (ALKBH5, FTO), and “readers” (YTHDFs, YTHDCs, IGF2BPs) (10). Increasing studies have confirmed that m6A modification plays an important role in malignant tumors (11, 12). For instance, m6A reader YTHDF1 promotes the proliferation, migration, invasion, and cell cycle of hepatocellular carcinoma cells by activating the PI3K/AKT/mTOR signaling pathway (13). Additionally, METTL3 mediates m6A modification of circCUX1 and stabilizes its expression, which binds caspase1 to reduce the release of inflammatory factors, leading to radiotherapy tolerance in hypopharyngeal squamous cell carcinoma (HPSCC) (14).
Lung cancer is the most common malignant tumor, with the highest mortality rate in the world, and only 21% of the 5-year survival rate (15). LUAD is the most common histological type of lung cancer, accounting for about 40% (16). In recent years, some studies have reported that m6A modification plays an important role in the progression of lung adenocarcinoma (17). Li et al. (16) revealed that YTHDF2 was involved in the development of LUAD via promoting the attenuation of AXIN1 mRNA to activate the Wnt/β-catenin signaling pathway, which may be a potential therapeutic target for LUAD. In addition, miR-600 induces degradation of METTL3 mRNA by targeting the 3’UTR of METTL3 mRNA and leads to inactivation of the PI3K/AKT pathway related to cell growth and survival (18). What’s more, METTL3 promotes the translation of YAP mRNA and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis, inducing drug resistance and metastasis of A549 cells (19). Many studies have described the important role of m6A modification in LUAD. However, the transcriptome-wide m6A methylome of LUAD has not been determined.
In this study, we identified differential methylated peaks and expressed genes through MeRIP-seq and RNA-seq. Furthermore, 2 genes with significant changes in both the m6A modification and expression associated with prognosis and diagnosis of LUAD were discovered by additional TCGA clinical data analysis.
Materials and Methods
Patients and Samples
Three pairs of LUAD samples and tumor-adjacent normal tissues were obtained from the South University of Science and Technology Hospital. The LUAD samples and tumor-adjacent normal tissues were immediately collected and separated into centrifuge tubes after surgery. These samples were examined by experienced pathologists who confirmed the diagnosis of disease samples. All tissues were stored at −80°C until RNA isolation. This study was approved by the Ethics Committee of the South University of Science and Technology Hospital.
MeRIP Sequencing and RNA Sequencing
Total RNA was isolated and purified using TRIzol™ Reagent (Invitrogen™, cat. no15596018) following the instructions of the manufacturer. Total RNA was chemically broken into 200nt fragments. The product was added to anti-N6-Methyladenosine (m6A) antibody (Sigma-Aldrich, cat. no ABE572), protein A-magnetic beads (Invitrogen™, cat. no 10002D), protein G-magnetic beads (Invitrogen™, cat. no 10004D), which was then mixed and incubated overnight. RNA was extracted by phenol-chloroform lysate to afford the purified product. A portion of the initial fragmented RNA was used as the input RNA-seq library for MeRIP-seq. Ribosomal RNA was removed from the products. The first-strand cDNA was synthesized by the SMART principle, and enriched library fragments were amplified by PCR. Ultrafine RNA methylation m6A detection library fragments were obtained by DNA purification of magnetic bead library fragments. Bioptic Qsep100 Analyzer was used to perform a quality inspection on the library and detect whether the size distribution of the library is consistent with the theoretical size. NovaSeq high-throughput sequencing platform and PE150 sequencing mode were used.
Cell Culture and Transfection
Lung adenocarcinoma cell line A549 cells were cultivated in 1640 medium containing 10% fetal bovine serum (FBS) (ExCell Bio Inc., Shanghai, China) and 100 U/mL penicillin-streptomycin mixture (Gibco, Grand Island, NY, USA). The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. YTHDF1 siRNA was designed and constructed by Genechem Bio (Shanghai, China). In transfection, siRNAs were transfected by LipoRNAiMAX Transfection Reagent (Invitrogen) according to product instructions. Briefly, siRNAs were diluted in the 100 μL Opti-MEM (Gibco) at 10 nmol/L and mixed with 2.5 μL Lipofectamine RNAi Max. After incubation at room temperature for 18 min, the mixture was added to the cell culture medium, and the cells were collected after 48 h. The sequences of the targets were as follows: YTHDF1-Homo-370 sense 5’- GGAUACAGUUCAUGACAAUTT-3’, antisense 5’-AUUGUCAUGAACUGUAUCCTT-3’; YTHDF1-Homo-1076 sense 5’-CCUCCACCCAUAAAGCAUATT-3’, antisense UAUGCUUUAUGGGUGGAGGTT-3’; YTHDF1-Homo-1503 sense 5’-GCUCCAUUAAGUACUCCAUTT-3’, antisense 5’-AUGGAGUACUUAAUGGAGCTT-3’.
RNA Isolation and RT-qPCR
Total RNA was extracted from A549 cells using Trizol (Takara, Japan) according to the manufacturer’s instructions. For RT-qPCR, RNA was reverse transcribed to cDNA by using a reverse transcription kit (Takara, Japan). The levels of RNA transcripts were analyzed by the Jena qTOWER3 RT-qPCR system (Jena, Germany). The sequences of the primers were as follows: GAPDH forward primer, 5’-CTCCTCCTGTTCGACAGTCAGC-3’, reverse primer 5’-CCCAATACGACCAAATCCGTT-3’; YTHDF1 forward primer, 5’-GCACACAACCTCCATCTTCG-3’, reverse primer 5’-AACTGGTTCGCCCTCATTGT-3’; GPRIN1 forward primer 5’-AAAGCAGGCCGATTCCACTTC-3’, reverse primer 5’-TCCTTCCTCGGTGACACTGTA-3’.
RIP-qPCR
The collected cell precipitates were coprecipitated by EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore 17-701) following the manufacturer’s procedure. Cells were lysed in a moderate volume RIP lysis buffer on ice for 5 min, and then the lysates were moved to -80°C overnight. Lysates were centrifuged at 4°C, and 12,000 rpm for 30 min to obtain the supernatant. 5μg YTHDF1 antibody or rabbit IgG was conjugated to protein A/G magnetic beads on a shaker for 30 min at room temperature. Magnetic beads containing antibodies were added to the supernatant, followed by rotating overnight at 4°C. TRIzol LS reagent (Invitrogen) was used to extract the RNA according to the manufacturer’s instructions. RT-qPCR follows the above method.
Public Databases and Analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) database (https://david.ncifc rf.gov/) was used in Gene Oncology (GO) functional enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. 594 LUAD patients’ RNA-seq data were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). The expression levels of genes were analyzed using the R package DESeq2 [version 1.26.0]. Then, 526 LUAD patients including primary tumor, lymph node metastasis, distant metastasis, and clinical stage were selected for the further study. A univariate Cox regression model was adopted to calculate the hazard ratios (HRs) of the genes’ overall survival. Kaplan-Meier analysis was used to construct survival curves, and the Cox regression model was utilized to estimate the significance of the differences. ROC curves were constructed to estimate the accuracy of genes in predicting outcomes. YTHDF1-RIP-seq data was downloaded from the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE136433.
Results
Overview of the m6A Methylation Map in LUAD
MeRIP-seq analysis of tumor tissue and tumor-adjacent normal tissue from three LUAD patients was performed. The sequence logo showed the top three m6A motifs enriched from altered m6A peaks and the “GGAC” sequence was ranked in the first place (Figure 1A). The distribution of m6A peaks in normal and tumor tissues was further investigated. Figures 1B–D shows that the distribution of m6A signals around mRNA and lncRNA were comparable in the two classes of tissue samples. Also, m6A peaks in mRNA both in tumor and normal tissues were mainly enriched in the coding sequence near the stop codon. It was found that their m6A mRNA methylation was reduced globally in the tumor compartment compared with adjacent, normal control tissues (Figure 1E). In Figure 1F, by comparing with normal tissues, tumor tissues had 1192 significantly upregulated m6A peaks, corresponding to transcripts of 1092 genes, as well as 2849 significantly downregulated m6A peaks, representing transcripts of 2415 genes. The top 20 altered m6A peaks are listed in Table 1. By analyzing the distribution of m6A peaks per mRNA, one m6A peak in the majority of mRNAs was found (Figure 1G). All altered m6A peaks were mapped to human chromosomes. Dysregulated m6A peaks were also observed in all chromosomes, particularly in chr1, chr2, chr16, chr17, and chr19 (Figure 1H).
FIGURE 1
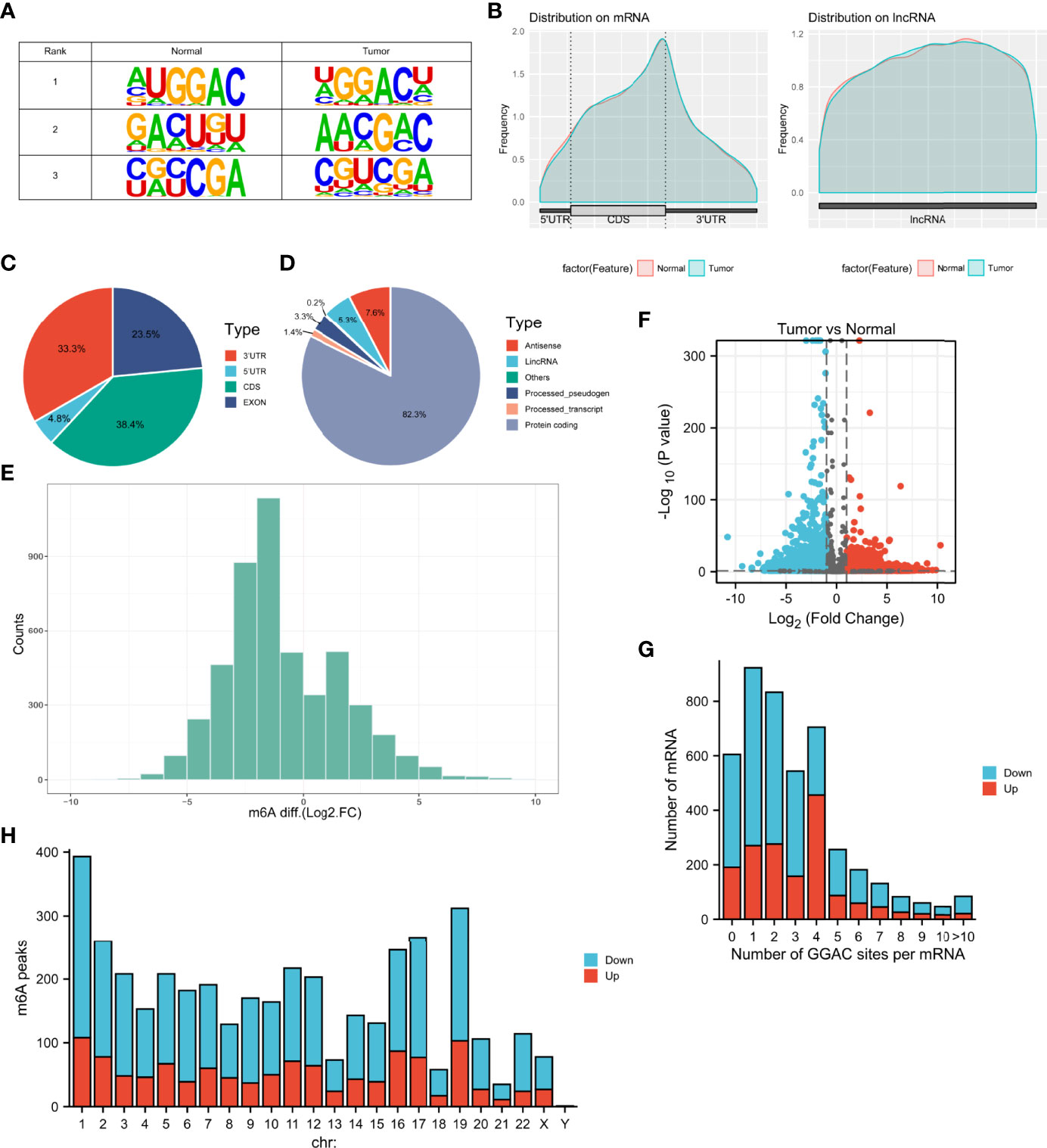
Figure 1 Characteristics of m6A methylation in LUAD. (A) The sequence logo showed the top three m6A motifs. (B) The distribution of m6A peaks in normal and tumor tissues. (C, D) Pie charts showing the distribution of differently methylated m6A peaks. (E) Histogram showing the changes in m6A enrichment between normal and tumor samples. (F) Volcano plots show significantly different m6A peaks. (G) The distribution of altered GGAC sequence per mRNA. (H) The distribution of differentially methylated m6A peaks in human chromosomes. Fold change ≥ 2 and P < 0.05.
TABLE 1

Table 1 Top 20 differently methylated m6A peaks in Tumor vs Normal.
m6A-Containing Genes are Involved in Important Biological Processes and Pathways
To investigate the biological significance of m6A modification in LUAD, GO terms and KEGG pathway analyses of differential methylated genes were performed. For GO analyses, biological processes (BP), cellular components (CC), and molecular functions (MF) were taken into consideration. Figure 2A demonstrates the top 10 significantly enriched BP, CC, and MF of genes with downregulated m6A peaks, while GO analysis of genes goes along with upregulated m6A peaks as shown in Figure 2B. KEGG pathway analysis showed that genes with downregulated m6A peaks in LUAD were significantly associated with the pathways in cancer, Rap1 signaling pathway, and insulin resistance (Figure 2C). Genes with upregulated m6A peaks were significantly associated with the ascorbate and alternate metabolism, pentose and glucuronate interconversions, and sphingolipid signaling pathway (Figure 2D). Additionally, in terms of the fold change, differential methylated genes between tumor and normal were associated with the negative regulation of protein phosphorylation, glycoprotein biosynthetic process, and regulation of GTPase activity (Figure 3).
FIGURE 2
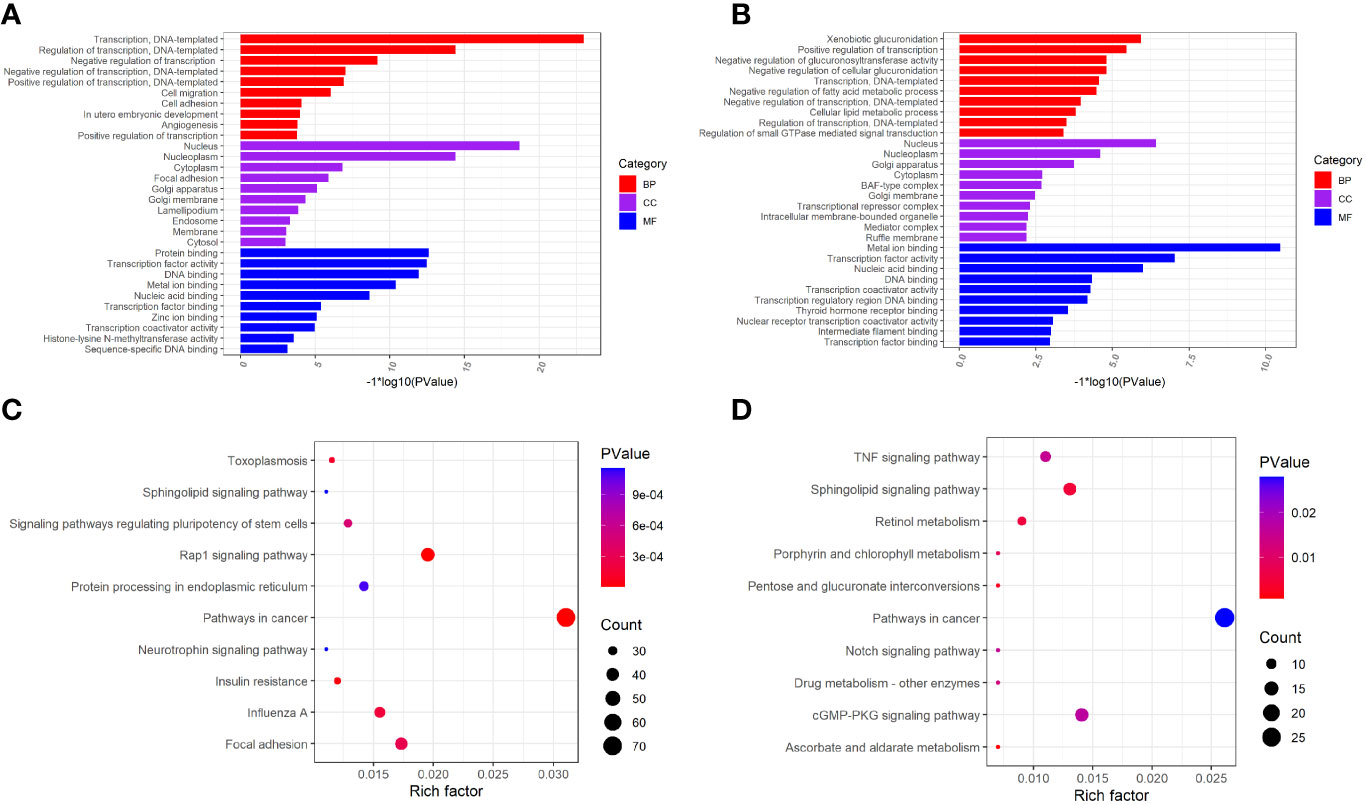
Figure 2 GO function and KEGG pathway enrichment of differently methylated m6A genes. (A) The top 10 GO terms of genes with down-regulated m6A peaks. (B) The top 10 GO terms of genes with up-regulated m6A peaks. (C) The top 10 KEGG pathways of genes with down-regulated m6A peaks. (D) The top 10 KEGG pathways of genes with up-regulated m6A peaks.
FIGURE 3
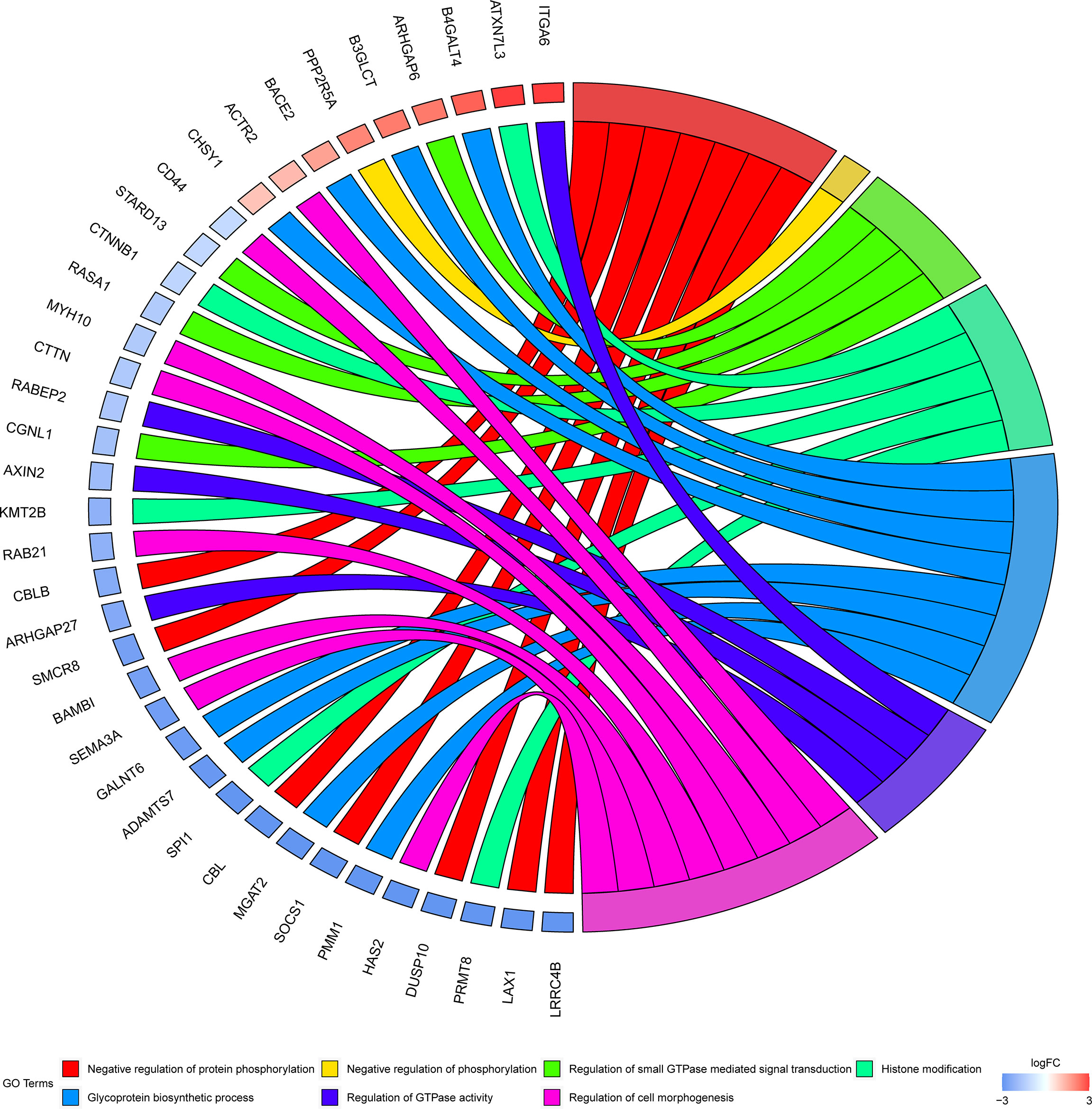
Figure 3 GO terms enrichment analyses of differently methylated genes between in tumor and normal. Squares in the left semicircle refer to the differently methylated mRNAs, and the squares in the right semicircle refer to the GO terms. Fold change ≥ 2 and P-value < 0.05, genes compared to GO terms.
Overview of Transcriptome Profiles and Conjoint Analyses of MeRIP-Seq and RNA-Seq Data
The transcriptome profiles of tumor tissues versus normal tissues from three LUAD patients were detected using RNA-seq (MeRIP-seq input library). Compared to normal tissues, tumor tissues had 1726 up-regulated genes and 2011 down-regulated genes (Figures 4A, B). The top 20 differently expressed genes are listed in Table 2. Then, all differential methylated m6A peaks with differential RNA levels (612) were divided into four groups by conjoint analysis for the MeRIP-seq and RNA-seq data. It was found that 177 hypermethylated m6A peaks in RNAs were significantly upregulated (128; hyper-up) or downregulated (49; hyper-down), while 435 hypomethylated m6A peaks in RNAs that were significantly upregulated (88; hypo-up) or downregulated (347; hypo-down) (Figure 4C). GO and KEGG pathway analyses for the biological significance of those genes (612) with differential methylated m6A peaks and synchronously differential expression were performed. GO analysis indicated that these genes were mainly enriched in the vasculogenesis (BP), an integral component of the plasma membrane (CC), and GTPase activator activity (MF) (Figure 4D). KEGG pathway analysis revealed that these genes were predominately enriched in the cGMP-PKG signaling pathway, focal adhesion, and Rap1 signaling pathway (Figure 4E). By contrast, GSEA enrichment of genes with significant changes in both the m6A modification and RNA levels showed that these genes were enriched in cell cycle phase transition, cell division, cellular response to DNA damage stimulus, and chromosome organization (Figures 4F–I).
FIGURE 4
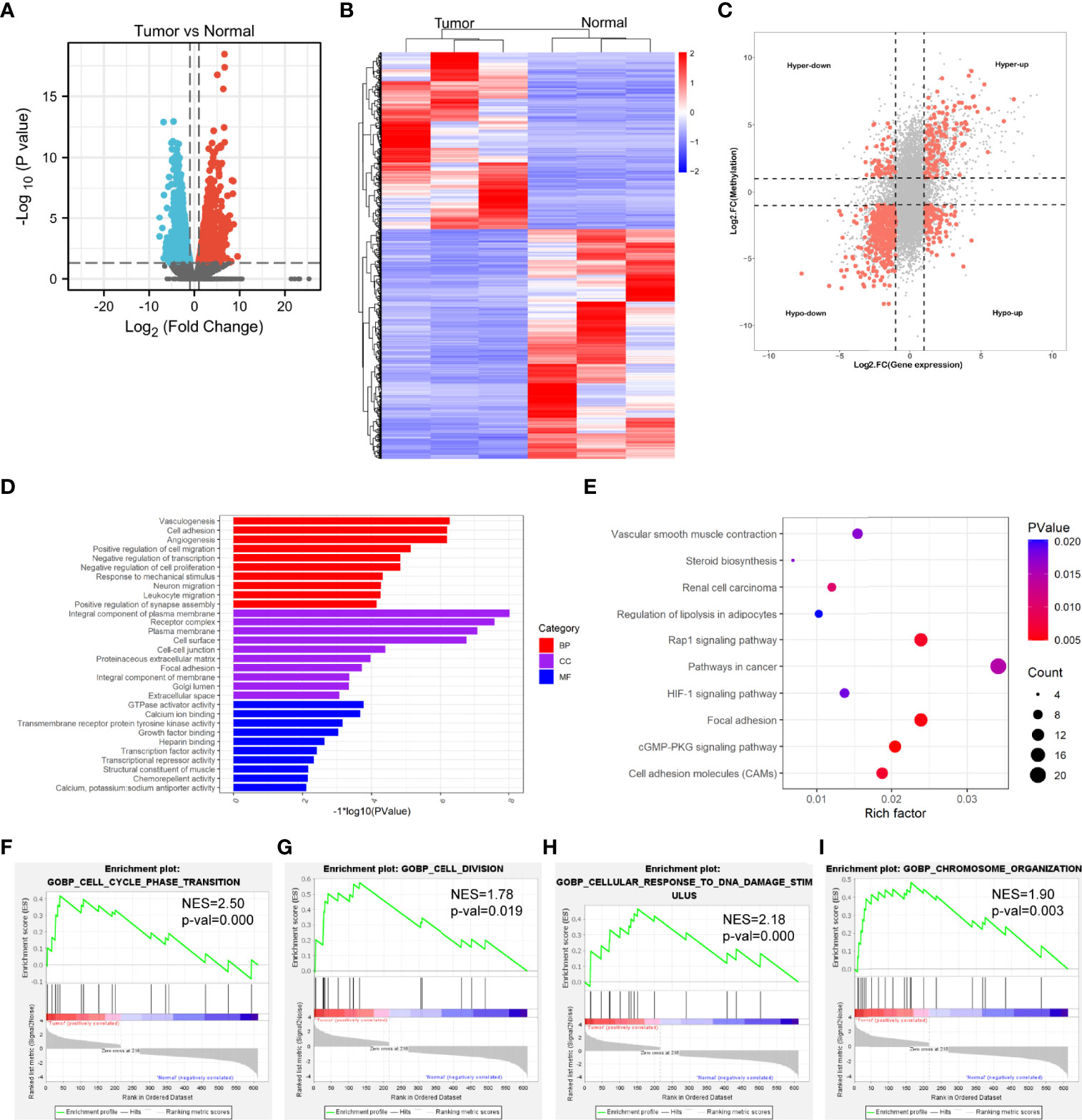
Figure 4 Conjoint analysis of MeRIP-seq and RNA-seq data. (A) Volcano plots showing the differentially expressed genes in tumor vs normal. (B) Heatmap plots showing the differentially expressed genes in tumor vs normal. (C) Four-quadrant plots showing the distribution of genes with significant changes in both the m6A modification and RNA levels. (D) The top 10 GO terms of genes with significant changes in both the m6A modification and mRNA levels. (E) The top 10 KEGG pathways of genes with significant changes in both the m6A modification and RNA levels. (F, G, H, I) GSEA enrichment of genes with significant changes in both the m6A modification and RNA levels.
TABLE 2
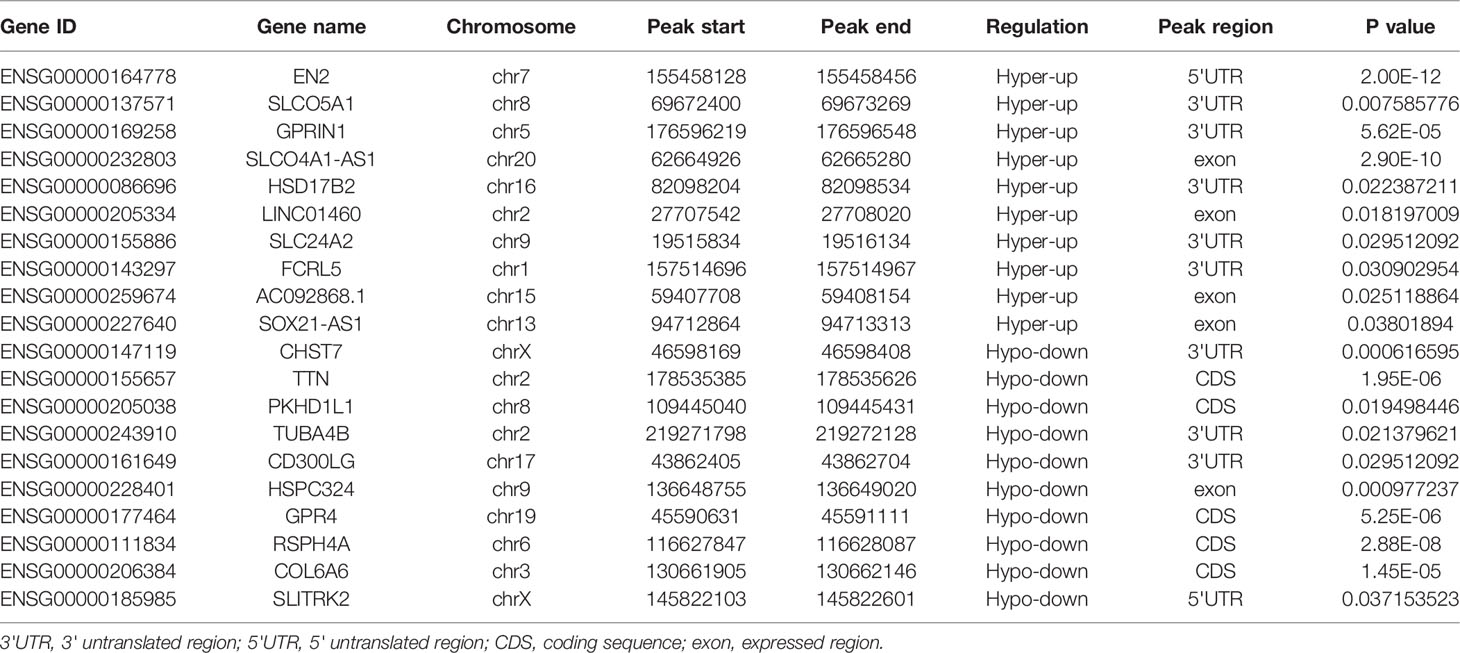
Table 2 Top 20 differently genes in m6A-seq&RNA-seq (Tumor/Normal).
Identifying the 2 genes Containing m6A Modification Correlates with Clinical Parameters of LUAD Patients
To evaluate the clinical significance of m6A modification regulating gene expression, the TCGA database was explored. 428 differential expressed genes commonly expressed in this study and the TCGA database were identified (Figure 5A). The top 20 differently expressed genes are listed in Table 3. The univariate Cox regression analysis of the expression of the top 20 differential expressed genes was performed to identify prognostic genes. The results demonstrated that the expression of 7 candidate genes was significantly related to the prognosis (p < 0.05) of LUAD patients (Figure 5B). Furthermore, we identified 2 genes (FCRL5 and GPRIN1) correlated with the prognosis of patients with LUAD. The overall survival curves of these 2 genes are shown in Figures 5C, D. This result showed that high expression of FCRL5 was associated with a better prognosis for LUAD patients, while high expression of GPRIN1 was associated with a worse prognosis for LUAD patients. The relationship between these 2 genes and clinical features was also analyzed in this study. GPRIN1 was found to be significantly high in patients with lymph node metastasis (Figure 5E), while FCRL5 was low expressed in patients with the clinical stage (Figure 5F), lymph node metastasis (Figure 5G), and distant metastasis (Figure 5H). In addition, the relationship between these 2 genes and clinical diagnosis showed that GPRIN1 had high accuracy in predicting the outcome, while FCRL5 had certain accuracy (Figures 5I, J). Previously, Li et al. (20) also reported GPRIN1 as a factor for poor prognosis of non-small cell lung cancer by analyzing RNA-seq data in TCGA and GEO databases. This indicates that GPRIN1 is a vital oncogene in LUAD, and m6A modification plays an important role in the expression level of GPRIN1.
TABLE 3
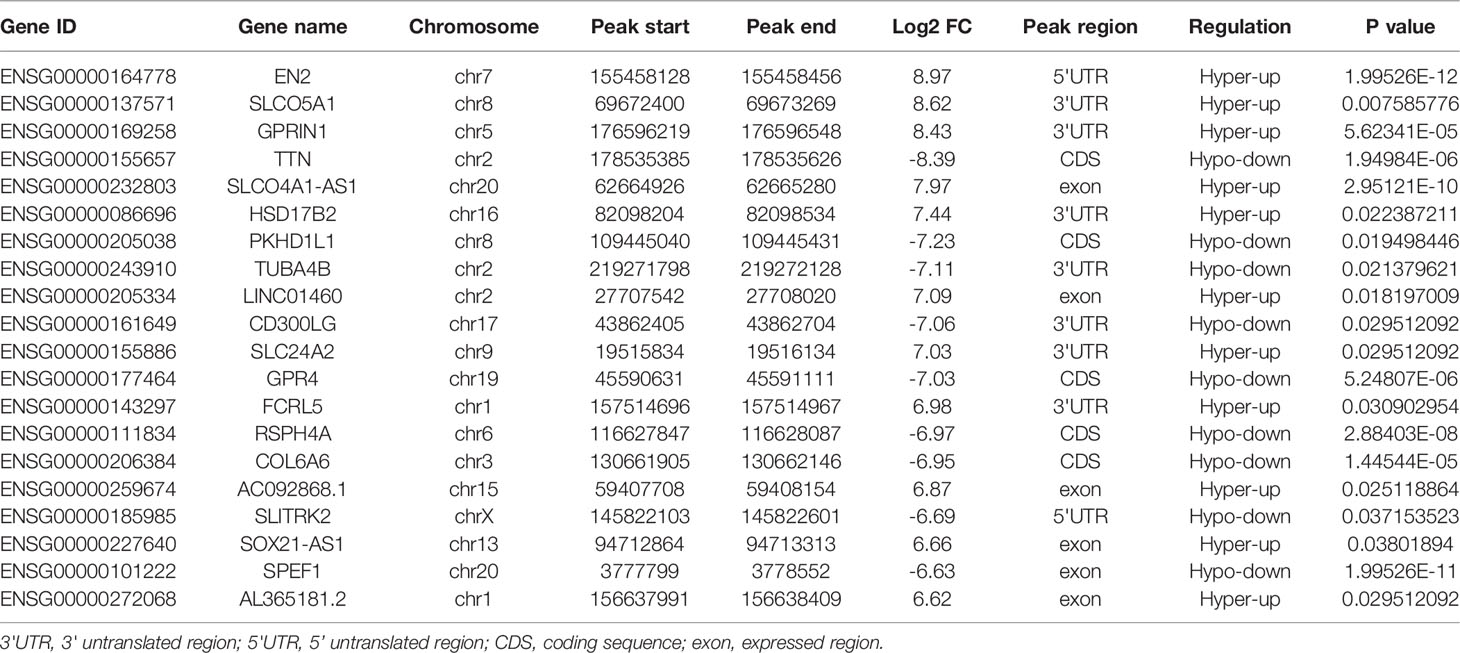
Table 3 Top 20 differently genes in m6A-seq&RNA-seq&TCGA (Tumor/Normal).
FIGURE 5
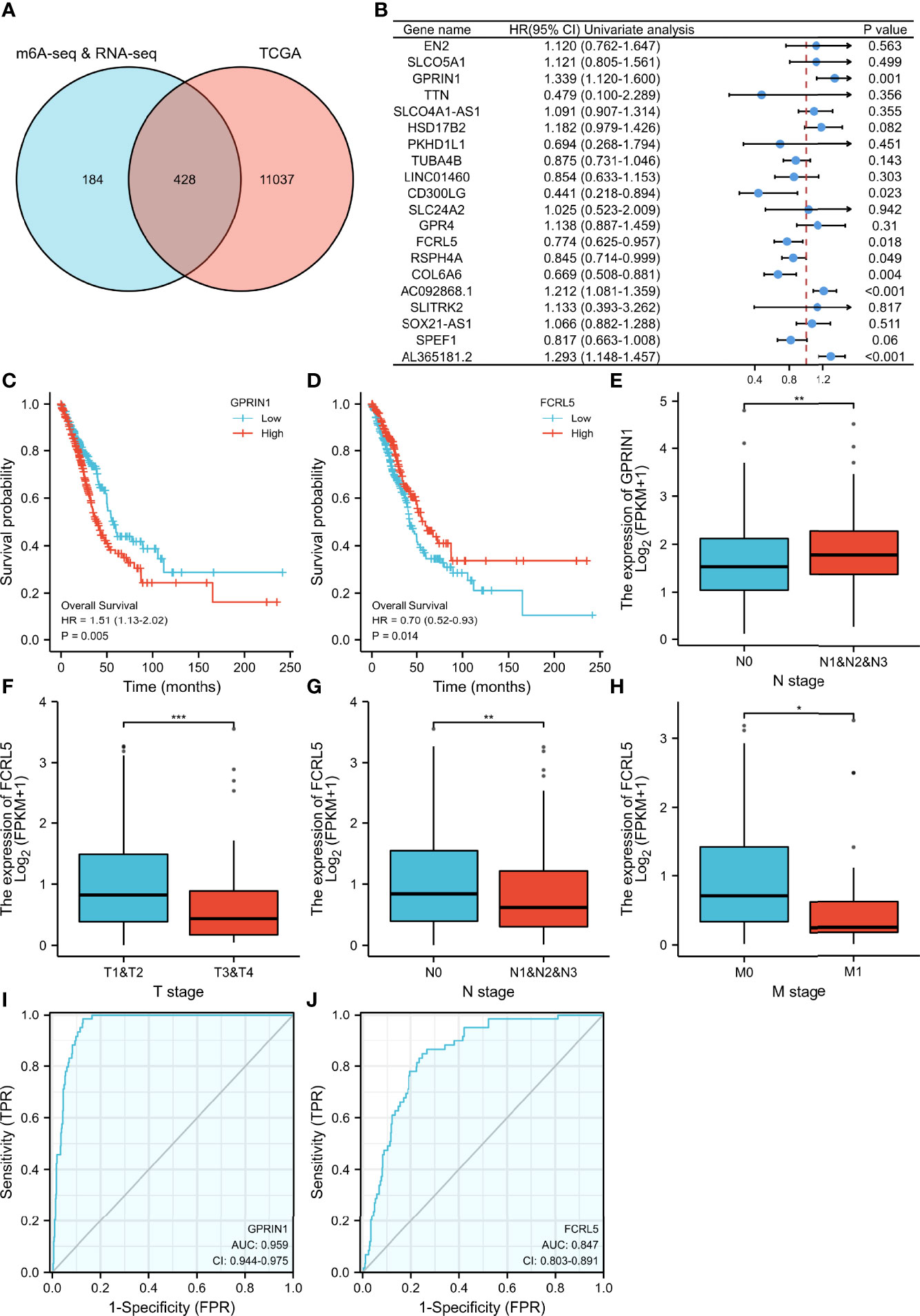
Figure 5 The relationship between gene expression regulated by m6A modification and clinical parameters in the TCGA database. (A) The intersection of genes in our study and TCGA database. (B) 20 prognostic genes were identified via univariate Cox regression analysis. (C, D) Overall survival curves of GPRIN1 and FCRL5, respectively. (E)The relationships between GPRIN1 expression and N. (F, G, H) The relationships between FCRL5 expression and M, N, and clinical stage. (I, J) The ROC curves of GPRIN1 and FCRL5, respectively. P*<0.5, P**<0.01, P***<0.001. (N: lymph node metastasis; M: distant metastasis.).
To explore the potential roles of m6A modification in GPRIN1 expression, we conducted a bioinformatics analysis using the GEPIA2 and CPTAC databases. It was found that GPRIN1 was strongly correlated with m6A reader YTHDF1 (r = 0.41) (Figure 6A). Also, both GPRIN1 and YTHDF1 proteins were highly expressed in LUAD (Figures 6B, C). Further, the m6A RIP-seq data showed that the m6A peaks were enriched in the 3’UTRs of GPRIN1 transcript in LUAD tissues. Compared to the normal tissues, m6A methylation was up-regulated in the vicinity of the 3’UTR of GPRIN1. The region with a gain of m6A in LUAD is the same that gains enrichment of YTHDF1 in A549 cells (Figure 6D). It was reported that YTHDF1 regulated gene expression via control of mRNA translation efficiency in an m6A-dependent manner (21). To evaluate whether YTHDF1 involved the expression of GPRIN1, siRNAs targeting YTHDF1 were transfected efficiently into A549 cells. As shown in Figure 6E, knockdown YTHDF1 significantly down-regulated the expression level of GPRIN1. Furthermore, we verified that YTHDF1 could bind GPRIN1 mRNA by DF1-RIP-qPCR assay, in which the fold enrichment of the YTHDF1-IP group was significantly higher than that of the IgG-IP group (Figure 6F). Therefore, we hypothesized that YTHDF1 regulates the expression of GPRIN1 by recognizing the m6A modification on GPRIN1 mRNA.
FIGURE 6
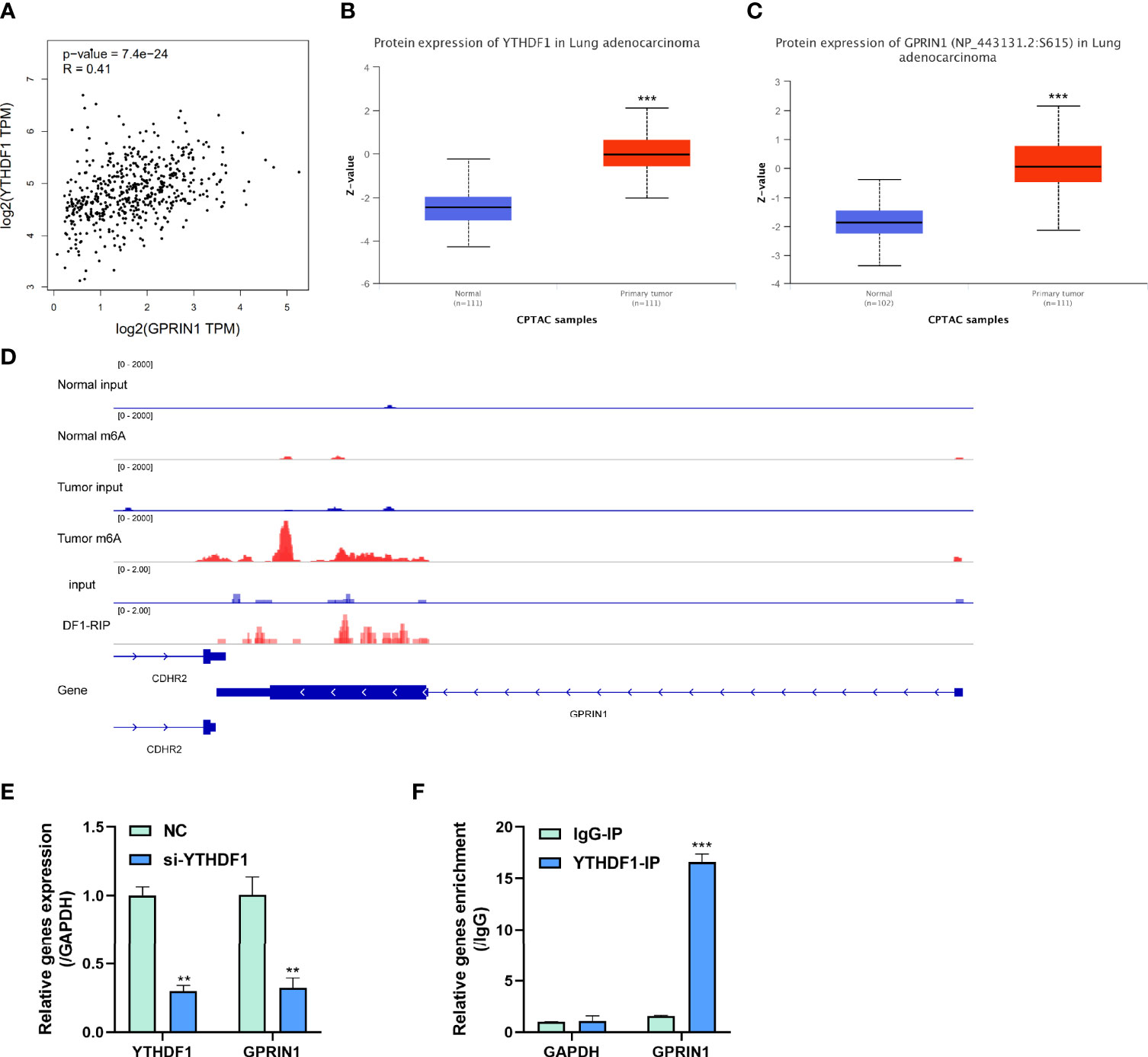
Figure 6 m6A reader YTHDF1 regulated GPRIN1 expression. (A) A strong positive correlation between YTHDF1 and GPRIN1 in the GEPIA2 database. (B, C) The protein expression of YTHDF1 and GPRIN1 in the CPTAC database, respectively. (D) Tumor m6A signal was increased at specific positions 3’UTR of GPRIN1. The region with a gain of m6A in LUAD is the same that gains enrichment of YTHDF1 in A549 cells. (E) Knockdown of YTHDF1 downregulated the RNA levels of GPRIN1 in LUAD A549 cells. (F) RIP-derived RNA in A549 cells was measured by RT-qPCR. P*<0.5, P**<0.01, P***<0.001.
Discussion
m6A is the most abundant internal modification on eukaryotic mRNAs. Recently, the significance of m6A modification is revealed that it involves almost all aspects of mRNA metabolism, including pre-mRNA processing, splicing, nuclear export, translation, and degradation (22). m6A modification also plays critical roles in physiology and pathology such as embryonic development (23), neurodevelopment (24), immunoregulation (25), and tumorigenesis (26). It is also reported that m6A modification plays a crucial role in the development and progression of LUAD (16, 27). However, the transcriptome-wide m6A methylome of LUAD has not been characterized. In this study, MeRIP-seq and RNA-seq were performed to reveal the m6A mapping in LUAD. We identified 4041 anomalously regulated m6A peaks in tumor tissues, of which 1192 m6A peaks were upregulated and 2849 m6A peaks were downregulated. Moreover, GO and KEGG pathway analyses revealed the potential functions of differential methylated transcripts. Additionally, by combining MeRIP-seq and RNA-seq data, genes with differential methylated m6A peaks and synchronously differential expression were enriched in cancer-related pathways. Finally, 2 genes were further proven to be associated with the prognosis and diagnosis of LUAD patients in the TCGA database.
In this study, it was detected that differential methylated RNAs related to important biological pathways via KEGG pathway analysis. It was found that upregulated m6A modification genes were associated with the ascorbate and alternate metabolism, pentose, and glucuronate interconversions, and sphingolipid signaling pathway. Moreover, downregulated m6A modification genes were significantly associated with the pathways in cancer, Rap1 signaling pathway, and insulin resistance.
Combining MeRIP-seq and RNA-seq data, 612 genes with differential methylated m6A peaks and synchronously differential expressed in RNA were identified. We performed GO and KEGG pathway analyses to investigate the biological significance of those genes. KEGG pathway analysis revealed that these genes were primarily enriched in the cGMP-PKG signaling pathway, focal adhesion, and Rap1 signaling pathway. It was revealed that these biological pathways are closely implicated in the onset and development of tumors, including tumor growth, and metastasis (28, 29). Rap1 is activated in response to upstream signalings, such as growth factors, cytokines, and chemokines that act on receptor tyrosine kinases and G-protein coupled receptors (30). Rap1 plays an important role in cell-matrix adhesion and supports the key role of the RAP1/RAC1 signaling axis in head and neck squamous cell carcinoma (HNSCC) cell migration via inducing α5β1 integrin via the extracellular matrix molecule fibronectin (31). Rap1 activity is increased in progressively metastatic prostate cancer cell lines and promotes metastasis in in vivo models through the integrins (32). Xiang et al. found that H. pylori infection enhances PRTG expression by promoting the stabilization of transcription factor ZEB1 and recruitment of PRTG promoters, and subsequently activates the cGMP/PKG signaling pathway to further promote proliferation, metastasis, and chemotherapy resistance of gastric cancer cells (33). The effects of EMX2OS and FUS on proliferation, invasion, and migration of human cell lines DU145 and PC3 mediated by the cGMP-PKG pathway were investigated by functional gain and functional loss experiments (34). The attenuation of cellular β-catenin accumulation through the activated cGMP-PKG pathway and the increased phosphorylation of β-catenin were observed in exisulind-treated colonic tumor cells (35).
In further investigation, 2 genes (FCRL5 and GPRIN1) were identified as most correlated with the development of LUAD using TCGA databases. Overexpression of GPRIN1 served as a factor for poor prognosis, while FCRL5 favors prognosis. Besides, high expression of GPRIN1 is highly involved in the lymph node metastasis, and FCRL5 with not only lymph node metastasis but also distant metastasis and the clinical stage of LUAD patients. Further analysis revealed that the region with a gain of m6A in LUAD is the same that gains enrichment of YTHDF1 in A549 cells. What’s more, YTHDF1 binds GPRIN1 mRNA by YTHDF1-RIP-qPCR assay, in which the fold enrichment of the YTHDF1-IP group was significantly higher than that of the IgG-IP group.
However, our research still has some shortcomings. Firstly, LUAD is a kind of highly heterogeneous cancer, and the tissue samples of clinical patients vary greatly. Although the three pairs of clinical samples met the statistical requirements, they were too small a sample size to reflect the differential m6A modification and gene expression between LUAD and adjacent tissue in the LUAD population, and the number of cases still must be further increased. Secondly, although we have performed some experiments to verify that YTHDF1 regulates GPRIN1 expression, more experiments are needed to strengthen the credibility, which is also the direction of our future in-depth research.
In summary, the different m6A methylome in LUAD relative to the corresponding normal controls was analyzed, which demonstrated a strong association between m6A modifcation and LUAD progression. 2 genes (FCRL5 and GPRIN1) with differential methylated m6A peaks and synchronously differential expression were associated with the prognosis and diagnosis of LUAD patients. Further one step research, GPRIN1 was found to act as an downstream target of YTHDF1. Targeting the YTHDF1-m6A-GPRIN1 axes may offer a promising therapeutic approach in curing LUAD.
Data Availability Statement
The data presented in the study are deposited in the GEO repository, accession number GSE198288.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the South University of Science and Technology Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
WM was a major contributor to all of the experimental work, data analysis, and manuscript writing. KW, YZ, and WL were involved in the experimental work. QY, QM, NW, and GZ were involved in the data analysis. YW acquired funding and assisted with the manuscript development. The final manuscript was reviewed and approved by all of the authors.
Funding
This work was supported by the Stability Support Plan for Higher Education Institutions in Shenzhen (20200809124551001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The author thanks Fengxin Gao from Guangzhou Epibiotek Co., Ltd. for technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.791332/full#supplementary-material
References
1. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res (2018) 46(D1):D303–7. doi: 10.1093/nar/gkx1030
2. Li J, Yang X, Qi Z, Sang Y, Liu Y, Xu B, et al. The Role of mRNA M(6)A Methylation in the Nervous System. Cell Biosci (2019) 9:66. doi: 10.1186/s13578-019-0330-y
3. He L, Li J, Wang X, Ying Y, Xie H, Yan H, et al. The Dual Role of N6-Methyladenosine Modification of RNAs is Involved in Human Cancers. J Cell Mol Med (2018) 22(10):4630–9. doi: 10.1111/jcmm.13804
4. Desrosiers R, Friderici K, Rottman F. Identification of Methylated Nucleosides in Messenger RNA From Novikoff Hepatoma Cells. Proc Natl Acad Sci U.S.A. (1974) 71(10):3971–5. doi: 10.1073/pnas.71.10.3971
5. Narayan P, Rottman FM. An In Vitro System for Accurate Methylation of Internal Adenosine Residues in Messenger RNA. Sci (New York NY) (1988) 242(4882):1159–62. doi: 10.1126/science.3187541
6. Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM. Context Effects on N6-Adenosine Methylation Sites in Prolactin mRNA. Nucleic Acids Res (1994) 22(3):419–26. doi: 10.1093/nar/22.3.419
7. Csepany T, Lin A, Baldick CJ, Beemon K. Sequence Specificity of mRNA N6-Adenosine Methyltransferase. J Biol Chem (1990) 265(33):201117–22. doi: 10.1016/S0021-9258(17)30477-5
8. Wei CM, Moss B. Nucleotide Sequences at the N6-Methyladenosine Sites of HeLa Cell Messenger Ribonucleic Acid. Biochemistry (1977) 16(8):1672–6. doi: 10.1021/bi00627a023
9. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR M(6)A Promotes Cap-Independent Translation. Cell (2015) 163(4):999–1010. doi: 10.1016/j.cell.2015.10.012
10. Uddin MB, Wang Z, Yang C. The M(6)A RNA Methylation Regulates Oncogenic Signaling Pathways Driving Cell Malignant Transformation and Carcinogenesis. Mol Cancer (2021) 20(1):61. doi: 10.1186/s12943-021-01356-0
11. Du A, Li S, Zhou Y, Disoma C, Liao Y, Zhang Y, et al. M6A-Mediated Upregulation of circMDK Promotes Tumorigenesis and Acts as a Nanotherapeutic Target in Hepatocellular Carcinoma. Mol Cancer (2022) 21(1):109. doi: 10.1186/s12943-022-01575-z
12. Raj N, Wang M, Seoane JA, Zhao RL, Kaiser AM, Moonie NA, et al. The Mettl3 Epitranscriptomic Writer Amplifies P53 Stress Responses. Mol Cell (2022) 27:S1097-2765(22)00321-5. doi: 10.1016/j.molcel.2022.04.010
13. Luo X, Cao M, Gao F, He X. YTHDF1 Promotes Hepatocellular Carcinoma Progression via Activating PI3K/AKT/mTOR Signaling Pathway and Inducing Epithelial-Mesenchymal Transition. Exp Hematol Oncol (2021) 10(1):35. doi: 10.1186/s40164-021-00227-0
14. Wu P, Fang X, Liu Y, Tang Y, Wang W, Li X, et al. N6-Methyladenosine Modification of Circcux1 Confers Radioresistance of Hypopharyngeal Squamous Cell Carcinoma Through Caspase1 Pathway. Cell Death Dis (2021) 12(4):298. doi: 10.1038/s41419-021-03558-2
15. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
16. Li Y, Sheng H, Ma F, Wu Q, Huang J, Chen Q, et al. RNA M(6)A Reader YTHDF2 Facilitates Lung Adenocarcinoma Cell Proliferation and Metastasis by Targeting the AXIN1/Wnt/beta-Catenin Signaling. Cell Death Dis (2021) 12(5):479. doi: 10.1038/s41419-021-03763-z
17. Khan RIN, Malla WA. M(6)A Modification of RNA and its Role in Cancer, With a Special Focus on Lung Cancer. Genomics (2021) 113(4):2860–9. doi: 10.1016/j.ygeno.2021.06.013
18. Wei W, Huo B, Shi X. miR-600 Inhibits Lung Cancer via Downregulating the Expression of METTL3. Cancer Manag Res (2019) 11:1177–87. doi: 10.2147/CMAR.S181058
19. Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. M(6)A mRNA Methylation Initiated by METTL3 Directly Promotes YAP Translation and Increases YAP Activity by Regulating the MALAT1-miR-1914-3p-YAP Axis to Induce NSCLC Drug Resistance and Metastasis. J Hematol Oncol (2019) 12(1):135. doi: 10.1186/s13045-019-0830-6
20. Li J, Gong Z, Jiang H, Gao J, Liang J, Chang P, et al. Comprehensive Analysis Reveals GPRIN1 is a Potential Biomarker for Non-Sm All Cell Lung Cancer. #N/A (2021) 16(1):130–8. doi: 10.2174/1574893615999200530201333
21. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
22. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic Transcriptomic M(6)A Decoration: Writers, Erasers, Readers and Functions in RNA Metabolism. Cell Res (2018) 28(6):616–24. doi: 10.1038/s41422-018-0040-8
23. Kwon J, Jo YJ, Namgoong S, Kim NH. Functional Roles of Hnrnpa2/B1 Regulated by METTL3 in Mammalian Embryonic Development. Sci Rep (2019) 9(1):8640. doi: 10.1038/s41598-019-44714-1
24. Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Wu M. Epigenetics in Neurodevelopment: Emerging Role of Circular RNA. Front Cell Neurosci (2019) 13:327. doi: 10.3389/fncel.2019.00327
25. Zhang C, Fu J, Zhou Y. A Review in Research Progress Concerning M6a Methylation and Immunoregulation. Front Immunol (2019) 10:922. doi: 10.3389/fimmu.2019.00922
26. Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D, et al. Mechanism of RNA Modification N6-Methyladenosine in Human Cancer. Mol Cancer (2020) 19(1):104. doi: 10.1186/s12943-020-01216-3
27. Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 Subunit of System XC(-) is Essential for M(6)A Reader YTHDC2 to be an Endogenous Ferroptosis Inducer in Lung Adenocarcinoma. Free Radical Biol Med (2021) 168:25–43. doi: 10.1016/j.freeradbiomed.2021.03.023
28. Li H, Liang J, Wang J, Han J, Li S, Huang K, et al. Mex3a Promotes Oncogenesis Through the RAP1/MAPK Signaling Pathway in Colorectal Cancer and is Inhibited by hsa-miR-6887-3p. Cancer Commun (Lond) (2021) 41(6):472–91. doi: 10.1002/cac2.12149
29. Piazza GA, Ward A, Chen X, Maxuitenko Y, Coley A, Aboelella NS, et al. PDE5 and PDE10 Inhibition Activates cGMP/PKG Signaling to Block Wnt/beta-Catenin Transcription, Cancer Cell Growth, and Tumor Immunity. Drug Discovery Today (2020) 25(8):1521–7. doi: 10.1016/j.drudis.2020.06.008
30. Shah S, Brock EJ, Ji K, Mattingly RR. Ras and Rap1: A Tale of Two GTPases. Semin Cancer Biol (2019) 54:29–39. doi: 10.1016/j.semcancer.2018.03.005
31. Liu M, Banerjee R, Rossa C Jr., D'Silva NJ. RAP1-RAC1 Signaling Has an Important Role in Adhesion and Migration in HNSCC. J Dent Res (2020) 99(8):959–68. doi: 10.1177/0022034520917058
32. Bailey CL, Kelly P, Casey PJ. Activation of Rap1 Promotes Prostate Cancer Metastasis. Cancer Res (2009) 69(12):4962–8. doi: 10.1158/0008-5472.CAN-08-4269
33. Xiang T, Yuan C, Guo X, Wang H, Cai Q, Xiang Y, et al. The Novel ZEB1-Upregulated Protein PRTG Induced by Helicobacter Pylori Infection Promotes Gastric Carcinogenesis Through the cGMP/PKG Signaling Pathway. Cell Death Dis (2021) 12(2):150. doi: 10.1038/s41419-021-03440-1
34. Wang Z, Zhang C, Chang J, Tian X, Zhu C, Xu W. LncRNA EMX2OS, Regulated by TCF12, Interacts With FUS to Regulate the Proliferation, Migration and Invasion of Prostate Cancer Cells Through the cGMP-PKG Signaling Pathway. Onco Targets Ther (2020) 13:7045–56. doi: 10.2147/OTT.S243552
Keywords: lung adenocarcinoma, m6A, MeRIP-seq, GPRIN1, prognosis
Citation: Mao W, Yu Q, Wang K, Ma Q, Zheng Y, Zhang G, Luo W, Wang N and Wang Y (2022) Comprehensive Analysis of the Transcriptome-wide m6A Methylome in Lung Adenocarcinoma by MeRIP Sequencing. Front. Oncol. 12:791332. doi: 10.3389/fonc.2022.791332
Received: 08 October 2021; Accepted: 21 June 2022;
Published: 11 July 2022.
Edited by:
Luis Del Valle, Louisiana State University, United StatesReviewed by:
Daniel Hernández Sotelo, Autonomous University of Guerrero, MexicoHua Xiao, Michigan State University, United States
Copyright © 2022 Mao, Yu, Wang, Ma, Zheng, Zhang, Luo, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukun Wang, wangyk_sustech@126.com