 Dima El-Sharkawi
Dima El-Sharkawi Ayoma Attygalle
Ayoma Attygalle Claire Dearden
Claire Dearden
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 09 March 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.777066
This article is part of the Research TopicPathogenesis, Treatment, and Future Directions for Rare T-Cell LeukemiasView all 12 articles
T-cell clones can frequently be identified in peripheral blood. It can be difficult to appreciate whether these are benign and transient or whether they signify a clonal disorder. We review factors that aid in understanding the relevance of T-cell clones. Conversely, obvious pathological T-cell clones can be detected in blood, but there is uncertainty in how to categorize this clonal T cell population, thus, we adopt a multidisciplinary review of the clinical features, diagnostic material and radiology before making the diagnosis. In this review we shall discuss some of these challenges faced when diagnosing mature T-cell leukemias.
Mature T-cell neoplasms with leukemic involvement are rare and while many can present with archetypal features that allow for easy diagnostic categorization, other cases can be more difficult to sub-classify. Accurate and precise diagnosis requires integration of all the clinical findings along with morphological assessment, immunophenotyping, cytogenetic and molecular analysis of the peripheral blood, bone marrow and lymph node and radiology (1). A multi-disciplinary review of these cases is paramount to avoid incorrect diagnosis, for example, a histopathologist reviewing a lymph node biopsy may suggest a patient has nodal peripheral T-cell lymphoma in the absence of information regarding the white cell count and clinical picture (2).
In this review, we shall discuss some of the features that aid in subclassifying the mature T-cell leukemias and differentiating them from nodal peripheral T-cell lymphoma with leukemic involvement. We shall highlight rare examples of these diseases in order to avoid potential diagnostic pitfalls.
Lymphocytosis due to an increase in T-lymphocytes can easily be distinguished from clonal B-cell populations by basic flow cytometry methods. Once it has been established that the increase in lymphocytes are T cells, and further characterizing the T-lymphocyte population by morphology, immunophenotyping, cytogenetic and molecular analysis, an attempt to establish clonality is recommended, particularly in cases where the white cell count is low. This can be performed by several methods, namely, PCR-based methods, next generation sequencing (NGS) and flow cytometry of the TCR-Vβ repertoire (albeit limited use in everyday practice) or more recently TRBC1 (3–7).
Persistent T-cell lymphocytosis and expansion of T-cell populations can be seen in many cases of chronic infection, for example HIV and CMV and indeed reactive to other malignancies (8–10). Similarly immune dysregulation due to primary immunodeficiencies or autoimmune conditions such as autoimmune lymphoproliferative disorders (ALPS) can lead to significant lymphoid proliferation and peripheral blood involvement. Thus, even in the absence of clonal T-cell expansion a persistent T-cell lymphocytosis may indicate significant pathology that requires multi-disciplinary team input both for both diagnosis and management. This discussion is beyond the scope of this paper. One important point to note is that many of these conditions in themselves increase the risk of lymphoma.
The identification of a T-cell clone is not synonymous with a neoplasm. T-cell clones can be detected due to reactive causes, infection and senescence and can be persistent in these cases (11, 12). There is a spectrum of disorders both infective and autoimmune that can be associated with polyclonal expansion of T cells through to monoclonal expansion through to neoplastic proliferations of T cells. This is especially the case with large granular lymphocytic proliferations which can be seen in autoimmune conditions, and Felty’s syndrome, but similarly it is known that there is a strong link between Rheumatoid arthritis and LGLL. Furthermore, the pathogenesis of lymphoproliferative disorders, such as LGLL has been linked to chronic T cell activation with viruses such as HTLV or EBV implicated (13). This boundary and how we define these clones can be complex and can change with time (14). Furthermore, with improvements in diagnostics and also availability, there will be an increase in individuals identified with persistent T-cell clones with normal or even low lymphocyte counts. Interestingly, there is an association between clonal hematopoiesis (CHIP), MDS and clonal T-cell disorders. Not only do they share many of the same recurrent mutations seen predominantly in epigenetic regulators such as DNMT3A and TET2, suggestive that this may be early mutations in common progenitors, but they also often co-exist and there are numerous reports of co-existing MDS with LGLL or angioimmunoblastic T-cell lymphoma (15, 16).
T-cell clones of uncertain significance may be detected by molecular analysis solely, or there may be a small T-cell population identified by flow cytometry often with a large granular lymphocyte phenotype (as described below) (17, 18). While there is no equivalent to monoclonal B-lymphocytosis, many of these incidental clones can be considered as ‘T-cell clones of uncertain significance’ if the criteria for diagnosis of large granular lymphocytic leukemia (LGLL) or other mature T-cell leukemia are not met. However, the significance of the T-cell clones in the context of cytopenias and therefore how to manage them is not clear (19). It should also be noted that large granular lymphocytic proliferations can be seen with other hematological and non-hematological conditions, namely, myelodysplasia, plasma cell dyscrasias, aplastic anaemia, post- stem cell transplant, HIV infection and treatment with dasatinib (20–24). The presence of mutations in STAT3 and STAT5b does not immediately define a diagnosis of LGLL as these mutations are not specific to this disease (14). Thus, if patients do not have sufficient evidence for a positive diagnosis of a defined T-cell leukemia, then we prefer to consider them as having T-cell clones of uncertain significance. Akin to MGUS and monoclonal B cell lymphocytosis of uncertain significance, these should however be followed up as these clones may acquire secondary events that drive progression and develop into malignancy (25). Our preference depending on clinical situation is to monitor patients every 6 months in the first instance and then annually if no progression occurs.
While distinguishing between T-lymphoblastic leukemia (T-ALL) and mature T-cell neoplasms is usually very straightforward with the presence of immature markers such as CD1a, CD34, and TdT in the former, there have been unusual cases of T-prolymphocytic leukemia, T-PLL seen with lack of surface CD3 and CD45 that can make the diagnosis more difficult (26, 27). Similarly, there have been reports of mature T-cell neoplasms aberrantly expressing immature markers, such as CD1a (28, 29).
The mature T-cell leukemias are sufficiently diverse from one another that they are usually readily discernible; however distinguishing them from nodal or cutaneous lymphomas with leukemic involvement can be challenging and thus requires integration of all results before reaching a diagnosis, occasionally this can require multiple biopsies and can take time before a conclusion can be made (Table 1).
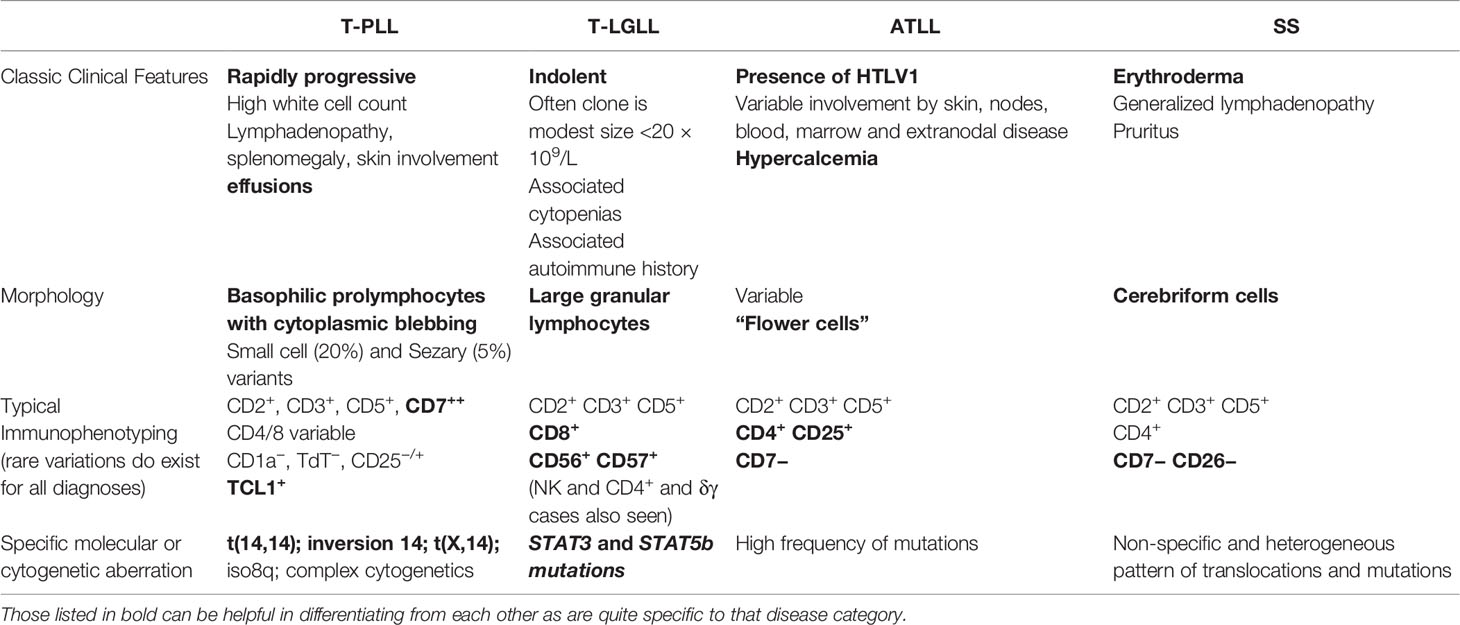
Table 1 Summary of the defining features of the mature T cell leukemias.
Large granular lymphocytic leukemia (LGLL) typically presents with cytopenias, most commonly neutropenia. Median age at presentation is 66 years (30–32). There is a strong association with autoimmune conditions, and approximately 15% of patients with LGLL will also have rheumatoid arthritis.
Typically there are no dysplastic features on the peripheral blood unless there are co-existing conditions and the cytopenias are evident. The large granular lymphocytes tend to be infrequent but are characteristic large lymphocytes with abundant cytoplasm and azurophilic granules. The distribution of the lymphocytes is intrasinusoidal in the bone marrow trephine that is otherwise normo- or hypercellular.
The characteristic immunophenotypic profile of LGLL is that of mature cytotoxic T cells most commonly αβTCR, CD2, CD3, CD8, CD56, and CD57 positive. Less commonly, LGLL can be comprised of CD4 positive T-cells, NK cells (classified as chronic lymphoproliferative disorder of NK cells in the most recent WHO classification), or have a γδTCR (33). While these can all be readily differentiated from T-LGLL by flow cytometry, it is important to consider their differential diagnoses such as aggressive NK cell leukemia or hepatosplenic T cell lymphoma, especially if patents have a more aggressive clinical picture.
Clonality may be assessed by flow cytometry using TRBC1 or more commonly by assessing for TCR gene rearrangements. Molecular analysis of STAT3 and STAT5b can be helpful as recurrent mutations in these genes have been identified in LGLL, but are not specific.
The WHO diagnostic criteria for LGLL are defined as a persistent (>6 months) increase in the number of peripheral blood large granular lymphocytes, usually 2–20 × 109/L without a clearly defined cause (33). However, it is stated that LGL counts of less than 2 × 109/L that otherwise meet the diagnosis are still consistent.
Hence, this diagnosis can only be made once persistence of the clone has been demonstrated. Often the authors are asked to review cases where patients have been investigated for cytopenias and while there are persistent T-cell rearrangements identified by molecular analysis, there is no associated lymphocyte population with LGLL phenotype identified by flow cytometry or infiltrate seen on bone marrow trephine. In these cases we would suggest continued infrequent monitoring and reassessment if the clinical situation changes but that this does not meet the diagnostic criteria for LGLL (34).
We have seen cases with very high white count with lymphocytes >100 × 109/L and so while low level clones are more common, they are not a defining feature.
Similarly, rare patients have presented with a predominantly nodal distribution of disease and this must not be assumed to be PTCL NOS based on distribution alone.
Cases of LGLL with more unusual immunophenotypic profiles such as γδTCR can lead to other differential diagnoses such as gamma delta hepatosplenic T-cell lymphoma (35, 36). However, by combining the clinical features such as generalized symptoms, rapidity of onset of symptoms, presence of hepatosplenomegaly, and bone marrow sinusoidal expansion by lymphoma cells the two can be readily distinguished, emphasizing that the pathologist cannot make the diagnosis in isolation, without knowing the clinical picture.
T-prolymphocytic leukemia (T-PLL) characteristically presents at a median age of 65 years. Patients with ataxia telangiectasia have an increased risk of T-PLL and in these cases, the presentation can be in their 20s. Often the illness presents rapidly, with a rapidly rising white cell count, with generalized symptoms and also effusions, ascites, edema, and peri-orbital edema and skin infiltration. However, T-PLL can have an indolent pre-phase that is detected incidentally, when patients will not have these symptoms and have smaller more stable clones but with the characteristic phenotype as described below.
The morphology can be variable with three characteristic appearances described. These include the more typical prolymphocytes with blebbing of the cytoplasm and single nucleolus; small cell variant with cells displaying condensed chromatin and nucleoli invisible by light microscopy; and cerebriform variant with an irregular nuclear outline similar to the lymphocytes seen in Sézary syndrome.
The lymphocytes are post-thymic and express mature markers positive for CD2, CD3, CD5 and CD7 and CD52. CD4+ CD8− is most commonly seen, with rarer cases expressing only CD8 or double positive. The latter is quite specific to T-PLL compared to other mature T cell leukemias, and so can be helpful for making the diagnosis. Similarly, TCL1 expression can be assessed by flow cytometry and is specific to T-PLL.
Changes involving chromosome 14 are the most common genetic alteration, seen in over 90% of cases. Inv(14)(q11q32) and t(14;14)(q11;q32) causes juxtaposition of TCRα and TCL1 or TCL1B leading to activation (37). This rearrangement can be identified by FISH (karyotype has a lower sensitivity), and the aberrant TCL1 protein expression can also be detected by flow cytometry or immunohistochemistry (38). The translocation t(X;14) is present in approximately 10–20% cases and involves the rearrangement of the TCRα locus with the proto-oncogene MTCP1 (39–41).
Other cytogenetic abnormalities are also commonly found, namely, abnormalities of chromosome 8 which often results in increased expression of MYC, deletions in 11q23, 12p, 22q, and 17 or abnormalities in chromosome 6 have also been identified with the majority of patients exhibiting a complex karyotype (37, 40, 42–47). While molecular analysis is also performed, the recurrent mutations in genes such as ATM, JAK3, and STAT5b are not specific (40).
As well as assessing for TCL1 expression, in our center we will also perform FISH to look for the characteristic inversion 14(q11q32) or t(14;14)(q11q32), but importantly also perform cytogenetics to look for other aberrations that are frequently seen in T-PLL.
International consensus criteria have been recently published to guide the diagnosis (48). When specific cytogenetic aberrations or protein expression are detected, the diagnosis is certain; however, there is a small subset of cases which have been diagnosed elsewhere to have T-PLL on the basis of a leukemic clonal T-cell population “compatible” with T-PLL by flow cytometry and with involvement by a “T-PLL specific site” that on further investigation with a wider T-cell panel, has been reclassified as PTCL-NOS due to lack of cytogenetic aberrations, and features that would be more unusual for T-PLL such as weak CD7.
Identification of CD4+ CD8+ double positive T-cell populations, can be very suggestive of the diagnosis of T-PLL. While ATLL is characteristically CD4+ CD25+, CD25 expression can also be seen in T-PLL and so does not differentiate between the two, HTLV analysis aids in differentiation of these cases. Typically Sézary cells do not express CD7, which can help diagnostically. A recent case that had been referred to our center as possible T-PLL was in a patient with marked erythroderma and a relatively modest lymphocytosis, in this case the weak CD7 positivity pushed the referring center to this diagnosis, however, the clinical history of the erythroderma being significant for many years and the discrepancy of the extent of cutaneous involvement and progression with lack of progression of the leukemic component made the diagnosis of T-PLL very unlikely. Skin biopsy showed evidence of a CD4 positive T-cell lymphoma infiltrate with small to intermediate sized T-cell infiltrate with focal epidermotropism. This in conjunction with the lack of any specific cytogenetic aberration by FISH analysis for TCRAD break-apart allowed the regional skin lymphoma unit and our center to conclude that this was most in keeping with Sézary syndrome. This highlights the importance of multidisciplinary involvement in difficult cases such as these.
Similarly, cases can be seen where nodal lymphomas are incorrectly diagnosed as T-PLL due to the leukemic involvement or the converse when initially the patient presents with lymphadenopathy but the lymphocytosis is not such a feature or perhaps in a more indolent phase (49, 50).
Despite the marked heterogeneity of this disease, given the knowledge of the etiological infectious agent, HTLV1, differentiating this from other mature T cell neoplasms is generally easier than other mature T cell leukemias. However, HTLV1 serology should be undertaken in any case of cutaneous, nodal or leukemic mature T-cell neoplasm in order not to miss this diagnosis (51).
Several morphological variants have been described with the archetypal variant being medium to large sized “flower cells” with nuclear indentations (33, 52).
Mature T-cell markers are expressed, namely, CD2, CD3, and CD5, but usually lack CD7. The majority are CD4 positive and CD8 negative but CD8 positive and double positive cases have been described (33, 53). CD25 is strongly expressed in nearly all cases. CD30 expression is variable.
The genomic landscape of ATLL is complex with a high frequency of mutations with regional variations and variations dependent on subtype of ATLL (54–56). Most frequently mutated genes are PLCG1, PRKCB, VAV1, IRF4, FYN, CARD11, and STAT3.
Sézary syndrome usually presents in patients in the older (60 years plus) age group. Symptoms most frequently include erythroderma and generalized lymphadenopathy. The classical triad of erythrodermic pruritic rash covering >80% of the body surface area, lymphadenopathy and circulating Sézary cells can aid in diagnosis (33, 57). The history is usually quite short, due to the rapid progression, however a secondary Sézary syndrome occurring following a more prolonged history with documented preceding mycosis fungoides has also been defined (57).
Sézary cells in the peripheral blood typically show cerebreiform nuclei. Skin changes histologically are similar to mycosis fungoides with less epidermotropism. Effacement of the lymph nodes with dense monotonous infiltrates can be seen (33).
The immunophenotype of the cells usually are CD3 and CD4 positive and lack CD7 and CD26. Rarer phenotypes have been seen such as loss of other T cell antigens, CD4 negative CD8 positive disease or double positive (57)
The cytogenetic rearrangement seen are non-specific with complex cytogenetics and numerous mutations identified in studies exploring the molecular landscape (58, 59).
It is important to analyze all compartments (skin, peripheral blood, lymph nodes) for possible involvement. Histological changes in the skin can be non-specific and thus numerous skin biopsies are frequently taken before a diagnosis is made. Furthermore, many inflammatory skin conditions may lead to reactive T cell clones in the peripheral blood which can complicate diagnosing the skin condition. This emphasizes the importance of peripheral blood analysis which can help confirm the diagnosis. Skin biopsy and peripheral blood show the same TR gene rearrangements. The total Sezary count is greater than 1 ×10°/L with an expanded CD4 positive population resulting in CD4:CD8 ratio of greater than 10 with loss of CD7 being quite characteristic (2, 33).
All nodal lymphomas have been reported to have leukemic involvement in rare cases (60–62). The diagnosis of these would not be performed on peripheral blood alone and would need to be correlated with the bone marrow, lymph node histology and any clinical information. We have had a number of cases referred for second opinion on diagnosis with quite marked T-lymphocytosis, who are ultimately classified as PTCL NOS due to lack of defining markers to suggest an alternative diagnosis.
Not all patients who present with mature T-cell leukamias have easily classifiable disease, and in these cases, if they do not fulfil the currently recognized diagnostic categories, by definition they need to be considered as peripheral T cell lymphoma, not otherwise specified. As our use of next generation sequencing, and gene expression and methylation profiling increases, how we define these neoplasms is likely to change and improve. In the meantime, the integration of clinical, morphological, genetic and histopathological features is paramount to ensure that optimal management is employed to avoid under- or over-treatment of the patient.
DE, AA, and CD devised concept of article. DE wrote the first draft. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A Systematic Approach to Diagnosis of Mature T-Cell Leukemias Reveals Heterogeneity Among WHO Categories. Blood (2004) 104:328–35. doi: 10.1182/blood-2004-01-0002
2. Foucar K. Mature T-Cell Leukemias Including T-Prolymphocytic Leukemia, Adult T-Cell Leukemia/Lymphoma, and Sézary Syndrome. Am J Clin Pathol (2007) 127:496–510. doi: 10.1309/KWJYBCCGTB90B6AE
3. Mahe E, Pugh T, Kamel-Reid S. T Cell Clonality Assessment: Past, Present and Future. J Clin Pathol (2018) 71:195–200. doi: 10.1136/jclinpath-2017-204761
4. Tembhare P, Yuan CM, Xi L, Morris JC, Liewehr D, Venzon D, et al. Flow Cytometric Immunophenotypic Assessment of T-Cell Clonality by Vβrepertoire Analysis. Am J Clin Pathol (2011) 135:890–900. doi: 10.1309/AJCPV2D1DDSGJDBW
5. Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T Cell Receptor β-Chain Constant Region for Immunotherapy of T Cell Malignancies. Nat Med (2017) 23:1416–23. doi: 10.1038/nm.4444
6. Horna P, Shi M, Olteanu H, Johansson U. Emerging Role of T-Cell Receptor Constant β Chain-1 (TRBC1) Expression in the Flow Cytometric Diagnosis of T-Cell Malignancies. Int J Mol Sci (2021) 22:1817. doi: 10.3390/ijms22041817
7. Bruggemann M, White H, Gaulard P, Garcia-Sanz R, Gameiro P, Oeschger S, et al. Powerful Strategy for Polymerase Chain Reaction-Based Clonality Assessment in T-Cell Malignancies Report of the BIOMED-2 Concerted Action BHM4 CT98-3936. Leukemia (2007) 21:215–21. doi: 10.1038/sj.leu.2404481
8. Mudd JC, Lederman MM. CD8 T Cell Persistence in Treated HIV Infection. Curr Opin HIV AIDS (2014) 9:500–5. doi: 10.1097/COH.0000000000000086
9. Morales M, Trujillo M, Del Carmen Maeso M, Piris MA. Thymoma and Progressive T-Cell Lymphocytosis. Ann Oncol (2007) 18:603–4. doi: 10.1093/annonc/mdl406
10. Kronenberg A, Seebach JD, Bossart W, Weber R. Polyclonal Proliferation of Large Granular Lymphocytes During Cytomegalovirus Primary Infection in a Human Immunodeficiency Virus-Infected Patient Receiving Antiretroviral Therapy. Clin Infect Dis (2001) 33:E34–6. doi: 10.1086/322652
11. Maini MK, Gudgeon N, Wedderburn LR, Rickinson AB, Beverley PCL. Clonal Expansions in Acute EBV Infection Are Detectable in the CD8 and Not the CD4 Subset and Persist With a Variable CD45 Phenotype. J Immunol (2000) 165:5729–37. doi: 10.4049/jimmunol.165.10.5729
12. Chou JP, Effros RB. T Cell Replicative Senescence in Human Aging. Curr Pharm Des (2013) 19:1680–98. doi: 10.2174/1381612811319090016
13. Loughran TP Jr, Hadlock KG, Yang Q, Perzova R, Zambello R, Semenzato G, et al. Seroreactivity to an Envelope Protein of Human T-Cell Leukemia/Lymphoma Virus in Patients With CD3- (Natural Killer) Lymphoproliferative Disease of Granular Lymphocytes. Blood (1997) 90:1977–81. doi: 10.1182/blood.V90.5.1977
14. Mustjoki S, Young NS. Somatic Mutations in “Benign” Disease. New Engl J Med (2021) 384:2039–52. doi: 10.1056/NEJMra2101920
15. Durrani J, Awada H, Kishtagari A, Visconte V, Kerr C, Adema V, et al. Large Granular Lymphocytic Leukemia Coexists With Myeloid Clones and Myelodysplastic Syndrome. Leukemia (2020) 34:957–62. doi: 10.1038/s41375-019-0601-y
16. Lewis NE, Petrova-Drus K, Huet S, Epstein-Peterson ZD, Gao Q, Sigler AE, et al. Clonal Hematopoiesis in Angioimmunoblastic T-Cell Lymphoma With Divergent Evolution to Myeloid Neoplasms. Blood Adv (2020) 4:2261–71. doi: 10.1182/bloodadvances.2020001636
17. Shi M, Olteanu H, Jevremovic D, He R, Viswanatha D, Corley H, et al. T-Cell Clones of Uncertain Significance are Highly Prevalent and Show Close Resemblance to T-Cell Large Granular Lymphocytic Leukemia. Implications for Laboratory Diagnostics. Modern Pathol (2020) 33:2046–57. doi: 10.1038/s41379-020-0568-2
18. Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical Spectrum of Clonal Proliferations of T-Large Granular Lymphocytes: A T-Cell Clonopathy of Undetermined Significance? Blood (1994) 84:1620–7. doi: 10.1182/blood.V84.5.1620.bloodjournal8451620
19. Wlodarski MW, Nearman Z, Jiang Y, Lichtin A, Maciejewski JP. Clonal Predominance of CD8(+) T Cells in Patients With Unexplained Neutropenia. Exp Hematol (2008) 36:293–300. doi: 10.1016/j.exphem.2007.11.011
20. Zhang X, Sokol L, Bennett JM, Moscinski LC, List A, Zhang L. T-Cell Large Granular Lymphocyte Proliferation in Myelodysplastic Syndromes: Clinicopathological Features and Prognostic Significance. Leuk Res (2016) 43:18–23. doi: 10.1016/j.leukres.2016.02.006
21. Sidiqi MH, Aljama MA, Viswanatha DS, Dingli D. T-Cell Large Granular Lymphocytic Leukemia and Plasma Cell Disorders. Haematologica (2019) 104:e108–e10. doi: 10.3324/haematol.2018.204099
22. Go RS, Lust JA, Phyliky RL. Aplastic Anemia and Pure Red Cell Aplasia Associated With Large Granular Lymphocyte Leukemia. Semin Hematol (2003) 40:196–200. doi: 10.1016/S0037-1963(03)00140-9
23. Nann-Rütti S, Tzankov A, Cantoni N, Halter J, Heim D, Tsakiris D, et al. Large Granular Lymphocyte Expansion After Allogeneic Hematopoietic Stem Cell Transplant Is Associated With a Cytomegalovirus Reactivation and Shows an Indolent Outcome. Biol Blood Marrow Transplant (2012) 18:1765–70. doi: 10.1016/j.bbmt.2012.07.007
24. Smith PR. Benign Monoclonal Expansion of CD8+ Lymphocytes in HIV Infection. J Clin Pathol (2000) 53:177–81. doi: 10.1136/jcp.53.3.177
25. Stern M, Theodorou I, Aurias A, Maier-Redelsperger M, Debre M, Debre P, et al. T-Cell Nonmalignant Clonal Proliferation in Ataxia Telangiectasia: A Cytological, Immunological, and Molecular Characterization. Blood (1989) 73:1285–90. doi: 10.1182/blood.V73.5.1285.bloodjournal7351285
26. Thakral B, Wang SA. T-Cell Prolymphocytic Leukemia Negative for Surface CD3 and CD45. Blood (2018) 132:111–. doi: 10.1182/blood-2018-03-840611
27. Chen X, Cherian S. Immunophenotypic Characterization of T-Cell Prolymphocytic Leukemia. Am J Clin Pathol (2013) 140:727–35. doi: 10.1309/AJCPG71KYOXTKLQW
28. Mendonca Bryne S, Hanaganahalli Basavaiah S, Christina Pinto A, Bhat G, Upadyaya K, Thahir M. An Unusual Presentation of HTLV-Associated Adult T-Cell Lymphoma/Leukemia - An Eye That Said It "A (T) Ll". Cancer Invest (2020) 38:209–13. doi: 10.1080/07357907.2020.1728298
29. Juncà J, Botín T, Vila J, Navarro J-T, Millá F. Adult T-Cell Leukemia/Lymphoma With an Unusual CD1a Positive Phenotype. Cytometry Part B: Clin Cytometry (2014) 86:292–6. doi: 10.1002/cytob.21130
30. Shah MV, Hook CC, Call TG, Go RS. A Population-Based Study of Large Granular Lymphocyte Leukemia. Blood Cancer J (2016) 6:e455. doi: 10.1038/bcj.2016.59
31. Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, et al. Analysis of a French Cohort of Patients With Large Granular Lymphocyte Leukemia: A Report on 229 Cases. Haematologica (2010) 95:1534–41. doi: 10.3324/haematol.2009.018481
32. Sanikommu SR, Clemente MJ, Chomczynski P, Afable MG 2nd, Jerez A, Thota S, et al. Clinical Features and Treatment Outcomes in Large Granular Lymphocytic Leukemia (LGLL). Leuk Lymphoma (2018) 59:416–22. doi: 10.1080/10428194.2017.1339880
33. Swerdlow SH, World Health O and International Agency for Research on C. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Vol. 585. Lyon: International Agency for Research on Cancer (2017). col. ill. ; 27 cm.
34. Singleton TP, Yin B, Teferra A, Mao JZ. Spectrum of Clonal Large Granular Lymphocytes (LGLs) of Alphabeta T Cells: T-Cell Clones of Undetermined Significance, T-Cell LGL Leukemias, and T-Cell Immunoclones. Am J Clin Pathol (2015) 144:137–44. doi: 10.1309/AJCPJ57YTEGLIUOI
35. Yabe M, Medeiros LJ, Wang SA, Konoplev S, Ok CY, Loghavi S, et al. Clinicopathologic, Immunophenotypic, Cytogenetic, and Molecular Features of Gammadelta T-Cell Large Granular Lymphocytic Leukemia: An Analysis of 14 Patients Suggests Biologic Differences With Alphabeta T-Cell Large Granular Lymphocytic Leukemia. [Corrected]. Am J Clin Pathol (2015) 144:607–19. doi: 10.1309/AJCPJSA1E1YWSZEY
36. Yabe M, Medeiros LJ, Wang SA, Tang G, Bueso-Ramos CE, Jorgensen JL, et al. Distinguishing Between Hepatosplenic T-Cell Lymphoma and Gammadelta T-Cell Large Granular Lymphocytic Leukemia: A Clinicopathologic, Immunophenotypic, and Molecular Analysis. Am J Surg Pathol (2017) 41:82–93. doi: 10.1097/PAS.0000000000000743
37. Brito-Babapulle V, Pomfret M, Matutes E, Catovsky D. Cytogenetic Studies on Prolymphocytic Leukemia. II. T Cell Prolymphocytic Leukemia. Blood (1987) 70:926–31. doi: 10.1182/blood.V70.4.926.bloodjournal704926
38. Sun Y, Tang G, Hu Z, Thakral B, Miranda RN, Medeiros LJ, et al. Comparison of Karyotyping, TCL1 Fluorescence in Situ Hybridisation and TCL1 Immunohistochemistry in T Cell Prolymphocytic Leukaemia. J Clin Pathol (2018) 71:309–15. doi: 10.1136/jclinpath-2017-204616
39. Stern MH, Soulier J, Rosenzwajg M, Nakahara K, Canki-Klain N, Aurias A, et al. MTCP-1: A Novel Gene on the Human Chromosome Xq28 Translocated to the T Cell Receptor Alpha/Delta Locus in Mature T Cell Proliferations. Oncogene (1993) 8:2475–83.
40. Stengel A, Kern W, Zenger M, Perglerova K, Schnittger S, Haferlach T, et al. Genetic Characterization of T-PLL Reveals Two Major Biologic Subgroups and JAK3 Mutations as Prognostic Marker. Genes Chromosomes Cancer (2016) 55:82–94. doi: 10.1002/gcc.22313
41. Madani A, Choukroun V, Soulier J, Cacheux V, Claisse JF, Valensi F, et al. Expression of P13mtcp1 Is Restricted to Mature T-Cell Proliferations With T(X;14) Translocations. Blood (1996) 87:1923–7. doi: 10.1182/blood.V87.5.1923.1923
42. Maljaei SH, Brito-Babapulle V, Hiorns LR, Catovsky D. Abnormalities of Chromosomes 8, 11, 14, and X in T-Prolymphocytic Leukemia Studied by Fluorescence In Situ Hybridization. Cancer Genet Cytogenet (1998) 103:110–6. doi: 10.1016/S0165-4608(97)00410-X
43. Brito-Babapulle V, Hamoudi R, Matutes E, Watson S, Kaczmarek P, Maljaie H, et al. P53 Allele Deletion and Protein Accumulation Occurs in the Absence of P53 Gene Mutation in T-Prolymphocytic Leukaemia and Sezary Syndrome. Br J Haematol (2000) 110:180–7. doi: 10.1046/j.1365-2141.2000.02174.x
44. Costa D, Queralt R, Aymerich M, Carrio A, Rozman M, Vallespi T, et al. High Levels of Chromosomal Imbalances in Typical and Small-Cell Variants of T-Cell Prolymphocytic Leukemia. Cancer Genet Cytogenet (2003) 147:36–43. doi: 10.1016/S0165-4608(03)00161-4
45. Nowak D, Le Toriellec E, Stern MH, Kawamata N, Akagi T, Dyer MJ, et al. Molecular Allelokaryotyping of T-Cell Prolymphocytic Leukemia Cells With High Density Single Nucleotide Polymorphism Arrays Identifies Novel Common Genomic Lesions and Acquired Uniparental Disomy. Haematologica (2009) 94:518–27. doi: 10.3324/haematol.2008.001347
46. Hu Z, Medeiros LJ, Fang L, Sun Y, Tang Z, Tang G, et al. Prognostic Significance of Cytogenetic Abnormalities in T-Cell Prolymphocytic Leukemia. Am J Hematol (2017) 92:441–7. doi: 10.1002/ajh.24679
47. Soulier J, Pierron GL, Vecchione D, Garand R, Brizard FO, Sigaux FO, et al. A Complex Pattern of Recurrent Chromosomal Losses and Gains in T-Cell Prolymphocytic Leukemia. Genes Chromosomes Cancer (2001) 31:248–54. doi: 10.1002/gcc.1141
48. Staber PB, Herling M, Bellido M, Jacobsen ED, Davids MS, Kadia TM, et al. Consensus Criteria for Diagnosis, Staging, and Treatment Response Assessment of T-Cell Prolymphocytic Leukemia. Blood (2019) 134:1132–43. doi: 10.1182/blood.2019000402
49. Kawamoto K, Miyoshi H, Yanagida E, Yoshida N, Kiyasu J, Kozai Y, et al. Comparison of Clinicopathological Characteristics Between T-Cell Prolymphocytic Leukemia and Peripheral T-Cell Lymphoma, Not Otherwise Specified. Eur J Haematol (2017) 98:459–66. doi: 10.1111/ejh.12856
50. Dearden C. How I Treat Prolymphocytic Leukemia. Blood (2012) 120:538–51. doi: 10.1182/blood-2012-01-380139
51. Cook LB, Fuji S, Hermine O, Bazarbachi A, Ramos JC, Ratner L, et al. Revised Adult T-Cell Leukemia-Lymphoma International Consensus Meeting Report. J Clin Oncol (2019) 37:677–87. doi: 10.1200/JCO.18.00501
52. Ohshima K. Pathological Features of Diseases Associated With Human T-Cell Leukemia Virus Type I. Cancer Sci (2007) 98:772–8. doi: 10.1111/j.1349-7006.2007.00456.x
53. Cook LB, Phillips AA. How I Treat Adult T-Cell Leukemia/Lymphoma. Blood (2021) 137:459–70. doi: 10.1182/blood.2019004045
54. Kogure Y, Kataoka K. Genetic Alterations in Adult T-Cell Leukemia/Lymphoma. Cancer Sci (2017) 108:1719–25. doi: 10.1111/cas.13303
55. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated Molecular Analysis of Adult T Cell Leukemia/Lymphoma. Nat Genet (2015) 47:1304–15. doi: 10.1038/ng.3415
56. Kataoka K, Iwanaga M, Yasunaga JI, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic Relevance of Integrated Genetic Profiling in Adult T-Cell Leukemia/Lymphoma. Blood (2018) 131:215–25. doi: 10.1182/blood-2017-01-761874
57. Vonderheid EC, Bernengo MG, Burg G, Duvic M, Heald P, Laroche L, et al. Update on Erythrodermic Cutaneous T-Cell Lymphoma: Report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol (2002) 46:95–106. doi: 10.1067/mjd.2002.118538
58. Vermeer MH, van Doorn R, Dijkman R, Mao X, Whittaker S, van Voorst Vader PC, et al. Novel and Highly Recurrent Chromosomal Alterations in Sezary Syndrome. Cancer Res (2008) 68:2689–98. doi: 10.1158/0008-5472.CAN-07-6398
59. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic Analyses Reveal Recurrent Mutations in Epigenetic Modifiers and the JAK-STAT Pathway in Sezary Syndrome. Nat Commun (2015) 6:8470. doi: 10.1038/ncomms9470
60. Nguyen JT, Condron MR, Nguyen ND, De J, Medeiros LJ, Padula A. Anaplastic Large Cell Lymphoma in Leukemic Phase: Extraordinarily High White Blood Cell Count. Pathol Int (2009) 59:345–53. doi: 10.1111/j.1440-1827.2009.02376.x
61. Jain G, Kumar C, Malhotra A, Mallick SR, Bakhshi S, Chopra A. Peripheral Blood Involvement in Angioimmunoblastic T-Cell Lymphoma: A Case Report and Review of the Literature. Am J Blood Res (2020) 10:257–65.
Keywords: diagnostics, mature T and NK-cell neoplasms, T-PLL, T-cell prolymphocytic leukemia, large granular lymphocyte (LGL) leukemia, adult T-cell leukemia/lymphoma (ATL), T cell lymphoma
Citation: El-Sharkawi D, Attygalle A and Dearden C (2022) Mature T-Cell leukemias: Challenges in Diagnosis. Front. Oncol. 12:777066. doi: 10.3389/fonc.2022.777066
Received: 14 September 2021; Accepted: 11 January 2022;
Published: 09 March 2022.
Edited by:
Jonathan Brammer, The Ohio State University, United StatesReviewed by:
Giorgio Alberto Croci, University of Milan, ItalyCopyright © 2022 El-Sharkawi, Attygalle and Dearden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dima El-Sharkawi, ZGltYS5lbC1zaGFya2F3aUBybWgubmhzLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.