
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 05 January 2023
Sec. Pediatric Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1106597
 Christina Fong1
Christina Fong1 Brian H. Kushner1
Brian H. Kushner1 Angela Di Giannatale2Gunes Gundem3Shanita Li1Stephen S. Roberts1Ellen M. Basu1Anita Price4
Angela Di Giannatale2Gunes Gundem3Shanita Li1Stephen S. Roberts1Ellen M. Basu1Anita Price4 Nai-Kong V. Cheung1
Nai-Kong V. Cheung1 Shakeel Modak1*
Shakeel Modak1*Introduction: While subcutaneous metastases are often observed with stage MS neuroblastoma, an entity that usually resolves spontaneously, skeletal muscle metastases (SMM) have been rarely described. The purpose of this retrospective study was to investigate the significance of SMM in neuroblastoma.
Patients and methods: Seventeen patients with neuroblastoma SMM were diagnosed at a median age of 4.3 (0.1-15.6) months. All had SMM at diagnosis and metastases at other sites. Fifteen (88%) had ≥ 2 SMM in disparate muscle groups. One, 14, and 2 patients had low, intermediate, and high-risk disease respectively. Fifteen tumors had favorable histology without MYCN amplification, and 2 were MYCN-amplified. Most SMM (80%; n=12/15 evaluated) were MIBG-avid.
Results: Only 1 patient (with MYCN-non-amplified neuroblastoma) had disease progression. All survive at median follow-up of 47.9 (16.9-318.9) months post-diagnosis. Biological markers (histology, chromosomal and genetic aberrations) were not prognostic. Whole genome sequencing of 3 matched primary and SMM lesions suggested that both primary and metastatic tumors arose from the same progenitor. SMM completely resolved in 10 patients by 12 months post-diagnosis. Of 4 patients managed with watchful observation alone without any cytotoxic therapy, 3 maintain complete remission with SMM resolving by 5, 13, and 21 months post-diagnosis respectively.
Conclusions: Children with neuroblastoma SMM have an excellent prognosis, with a clinical course suggestive of stage MS disease. Based on these results, the initial management of infants with non-MYCN-amplified NB with SMM could be watchful observation, which could eliminate or reduce exposure to genotoxic therapy.
Neuroblastoma, the most common extracranial solid tumor of childhood, typically develops along the path of migration of neural crest cells, usually in the abdomen or mediastinum (1). Common sites of metastases include bone, bone marrow (BM), lymph nodes, and liver. Rarely, neuroblastoma metastasizes to pleura, lung, central nervous system (CNS), or testis. Although preclinical studies indicate that neural crest cells can interact with skeletal muscle progenitors and influence myogenesis via BMP, WNT, and NOTCH signaling pathways (2), skeletal muscle metastases (SMM) are extraordinarily rare. Only a single case of SMM in a patient with neuroblastoma has been reported: an infant with skeletal and cardiac muscle involvement treated with intermediate-dose chemotherapy who maintained remission >12 months from diagnosis (3). Two additional cases of primary muscle neuroblastoma without metastases have been described (4, 5). Among adult cancers, SMMs are rare, partly because of mechanical stress, increased lactic acid and decreased pH (6). When present in adults, they are usually symptomatic of grave prognosis, and treatment is primarily palliative (7).
While metastatic neuroblastoma has been a therapeutic challenge, a unique group of patients diagnosed in infancy with metastases to liver, skin, and BM have a favorable prognosis, often with spontaneous resolution of all sites of disease. Initially described in 1971, this special, Evans stage IV-S of neuroblastoma was defined by the presence of a small adrenal primary tumor and metastases to the liver, skin and BM without cortical bone involvement and with limited BM involvement among infants <12 months of age (8). However, computed tomography (CT), magnetic resonance imaging (MRI), and scintigraphy with meta-iodobenzylguanidine (MIBG) and FDG positron emission tomography (PET) were unavailable for routine staging in the 1970s with unusual sites of metastases likely going undetected. IV-S neuroblastoma was later incorporated into the International Neuroblastoma Staging System (termed stage 4S) and in 2009 into the International Neuroblastoma Risk Group (INRG) risk-stratification system and renamed stage MS, without materially changing its definition, except that diagnosis be made at <18 months of age (9, 10). SMM were not included in stages IV-S, 4S or MS definitions. Therefore, the clinical significance of SMM and their impact on risk assessment is unclear.
Several studies have recently highlighted the importance of better understanding spatial and temporal heterogeneity of primary tumor and distant metastasis, specifically pertaining to segmental chromosomal aberrations, MYCN, and various mutations including ALK (11, 12). The current literature comparing matched primary-relapse samples using whole exome or whole genome sequencing (WGS) suggests that branched evolution and clonal evolution of specific mutations leads to the clonal heterogeneity seen in relapsed neuroblastoma (13, 14). This clonal relationship between the primary tumor and SMM and other metastases noted in stage MS disease is unclear. We reviewed cases seen at our center and analyzed paired diagnostic and SMM samples in order to develop a better understanding of outcomes and clonality for this rare entity.
Inclusion criteria for this retrospective review included patients diagnosed with neuroblastoma and SMM treated between 1995 and 2021 at our institution. Clinical and biological data were analyzed. Staging was recorded using the INRG staging system and response using International Neuroblastoma Response Criteria (10, 15). Progression-free survival (PFS) was defined as time from diagnosis to relapse or progression. Overall survival (OS) time was censored at date of last contact. Survival was analyzed by the Kaplan-Meier method. Approval from the Institutional Review Board of Memorial Sloan Kettering Cancer Center (MSKCC) was obtained prior to initiating this retrospective review of patient data.
Targeted tumor sequencing was performed using previously described methods, i.e., MSK-IMPACT (16) or Foundation One platforms (17). For WGS, tumor DNA was extracted from fresh frozen or OCT-embedded tissue biopsies and matched normal DNA from buffy coat using the DNeasy Blood & Tissue Kit (Qaigen). FFPE tissues were deparaffinized and DNA eluted as previously described (17). Briefly, genomic DNA was sheared, and sequencing libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with modifications. Libraries were subjected to a 0.5X size selection using AMPure XP beads (Beckman Coulter) after post-ligation cleanup. Libraries were not amplified by PCR and were pooled equivolume for sequencing. Samples were run on a NovaSeq 6000 (Illumina). Tumors were covered to an average of 80-90X, and normals covered at 42-52X.
Analysis of WGS data was executed using Isabl platform (18). Briefly, upon completion of each sequencing run, Isabl imports paired tumor-normal FASTQ files, executes alignment and quality control algorithms, extracts signatures (i.e., mutation signatures, microsatellite instability score, gene expression), and generates tumor purity and ploidy estimates. For samples with sufficient coverage (>60x) and tumor purity (>20%) Ensembl Variant calling is performed. High confidence somatic mutations were classified with regards to their putative role in cancer pathogenesis and microsatellite instability scores, and mutation signatures were statistically derived. Clinical relevance of mutations in common cancer genes was annotated using OncoKb, COSMIC, Ensembl Variant Effect Predictor, VAGrENT, gnomAD and ClinVar databases. Signals across data modalities (germline, somatic mutations, somatic signatures, copy number segments and gene expression profiles) were integrated for analysis.
Clonal structure was analyzed using the union of high confidence SNVs from all biopsies for a patient. DPClust (v0.2.2, https://github.com/Wedge-Oxford/dpclust) was used for calculation of cancer cell fraction corrected for purity and local copy number as well as clustering and assignment of mutations across samples with the exception of the Gibbs Sampling Dirichlet Process step which has been rewritten internally for performance (19).
Seventeen patients (10 male; 7 female) with SMM detected at diagnosis were identified. Median age was 4.3 (range 0.1-15.6) months. Clinical and biologic features, involved muscles, and therapies administered are described in Table 1. Most patients presented with abdominal distension or cutaneous nodules. One patient each also had poor feeding, orbital swelling and widespread bruising. In general, disease-related symptoms at diagnosis were not severe, and no patient required ventilator support. All had stage M neuroblastoma, stratified as high (n=2), intermediate (n=14), or low-risk (n=1) (20) Most patients (15/17; 88%) had multiple SMM (median 3, range 1-8) in widely disparate muscle groups detected by CT or MRI. Frequently involved muscle groups included the gluteus, paraspinal, psoas, abdominal wall, quadriceps, hamstring, adductor, and abductor muscles. The maximum diameter ranged from 0.8-5.3 (median 2) cm. The majority (80%; n=12/15 tested) of SMM were MIBG-avid. The three MIBG-non avid SMM had maximum diameters of >1cm, and their corresponding primary tumors were MIBG-avid. Two patients did not undergo MIBG scans at diagnosis. Additional metastases were noted in bone (n=12), lymph node (n=10), liver (n=11), skin (n=11), BM (n=4), lung/pleural (n=4), dura (n=4), spleen (n=1), and CNS parenchyma (n=1).
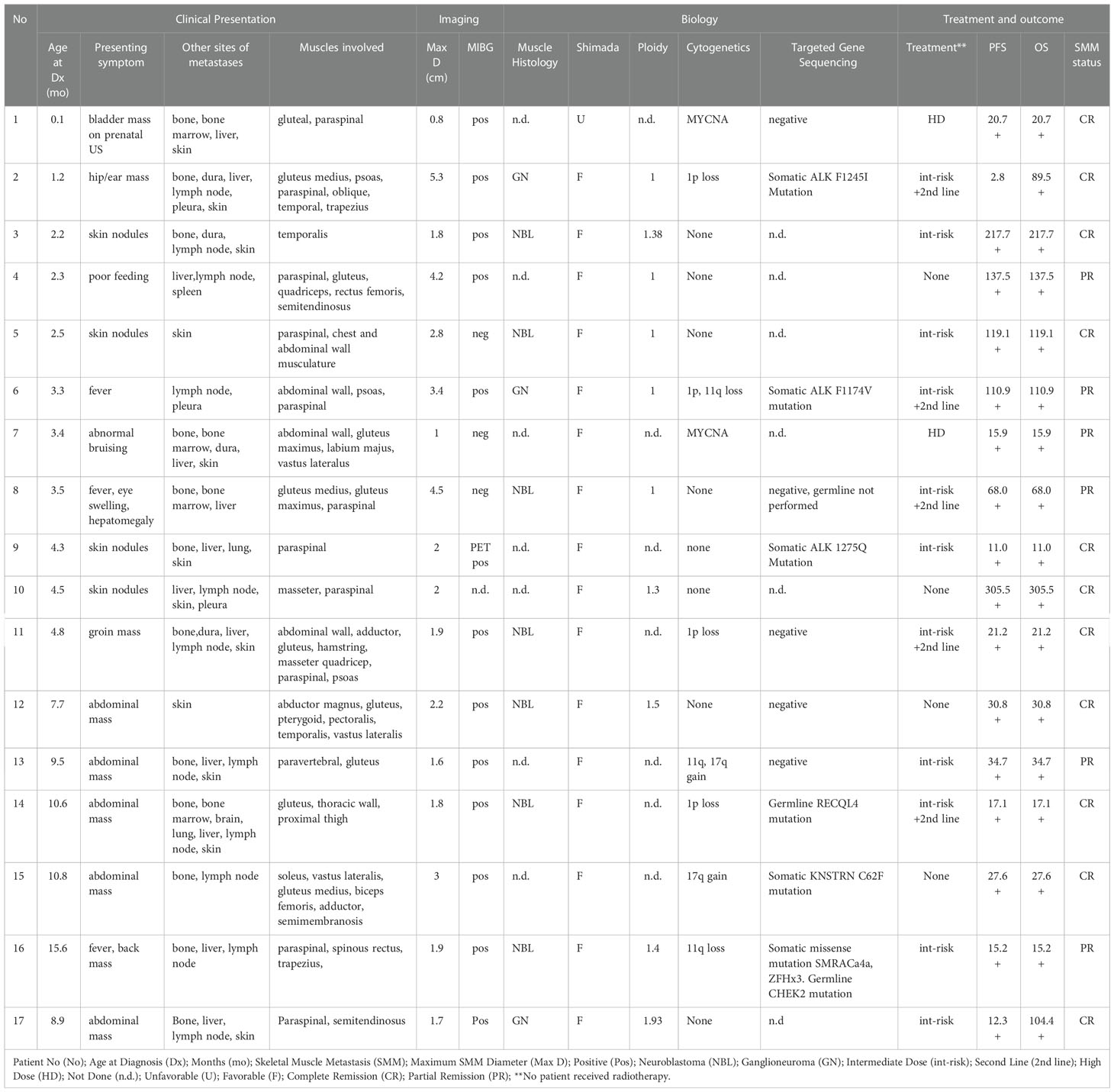
Table 1 Clinical Presentation, Diagnostic Imaging, Pathology, and Outcomes of Patients with SMM.
Ten patients underwent biopsies of SMM. Seven biopsies directly correlated with the histopathology of the primary tumor, i.e., favorable histology neuroblastoma. Three biopsies (from patients #2, #6, and #17) revealed ganglioneuroma without immature elements while the matched primary tumor showed partially differentiated neuroblastoma. Fifteen (88%) primary tumors had favorable histology without MYCN-amplification. Two tumors were MYCN-amplified. Half (5/10 tested) of the tumors were hyperdiploid, and 41% (7/17) had aberrations of chromosome 1p, 11q and/or 17q. Targeted exome sequencing on primary tumors detected somatic ALK mutations in 3/10 tested (F1174V, F1245I, 1275Q). One patient each had germline RECQL4 and CHEK2 mutation.
Three patients (Table 1, patients #3, #12, #14) had both a primary diagnostic sample and muscle metastasis sample available for WGS analysis. By comparing the matched tumors in each patient, we identified a set of substitutions found on the trunk that were fully clonal in both the primary and metastatic tumor (Figure 1). Overall, truncal substitutions as well as the shared copy number aberrations (CNAs) designate the most recent common ancestor shared between tumor cells in the primary sample and cells leading to muscle metastasis. In two patients (#3, #12) with numeric CNAs only, the genome-wide CNA profile was the same in both primary and metastatic tumor. In another patient (#14), we observed segmental CNAs including losses on 1p and 4p as well as potential whole genome duplication. Of note, the clone leading to the muscle metastasis had a complex structural rearrangement in the form of a chromothripsis on chromosome 5q. SMM resolved in all three patients, one of whom was managed with observation only without cytotoxic therapy (patient #12).
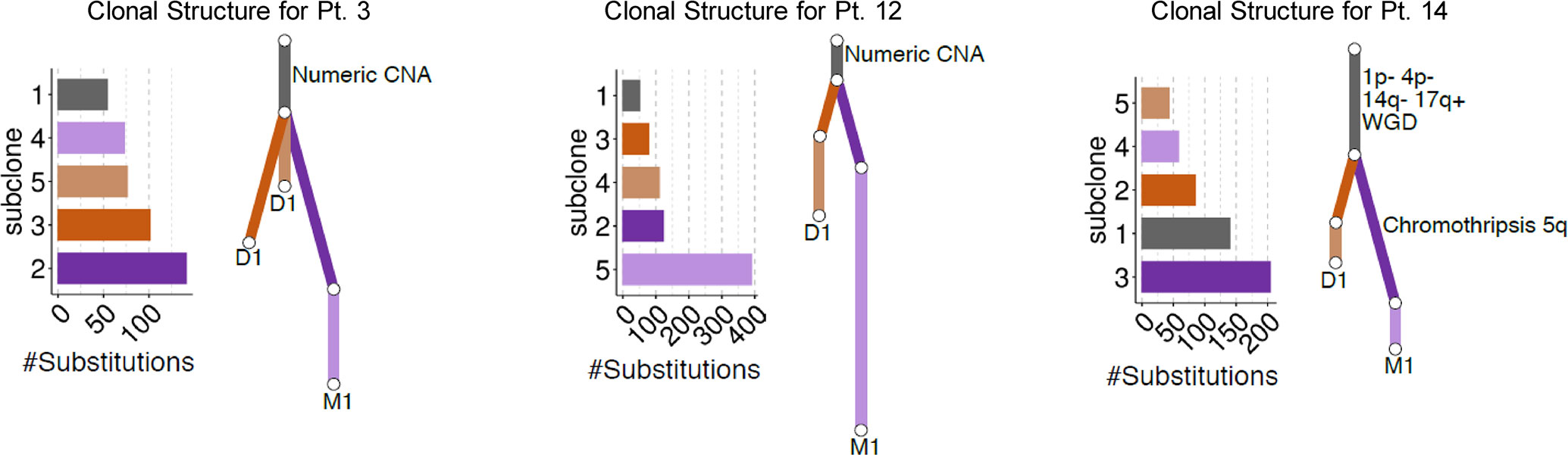
Figure 1 Summary of the clonal relationship between the diagnostic (D1) and muscle metastatic (M1) tumors for each patient. Mutation clusters identified in each patient are shown bar plots. Inferred clonal relationship between clusters is summarized in the form of a phylogenetic tree where branch length is proportional to the number of mutations in the cluster. Mutations shared fully clonally in both tumors are shown in dark grey followed by tumor-specific subclones.
At diagnosis, patients with MYCN-non-amplified (MYCN-NA) neuroblastoma received no cytotoxic chemotherapy (n=4) or intermediate-risk chemotherapy (n=11) (Table 1). Whereas the standard initial approach at MSKCC for patients without severe symptoms would be watchful observation without cytotoxic therapy, most (n=11) patients were referred after starting treatment at other centers where initial treatment decisions were made based on local protocols. One patient (#2), who was initially managed with observation alone, relapsed 2.4 months post-diagnosis (but not in muscle), subsequently received intermediate-dose chemotherapy and has maintained remission 87+ months later. Of the 15 patients with MYCN-NA neuroblastoma, 10 and 5 demonstrated complete and partial resolution of SMM respectively. No patient received dose-intensive chemotherapy, radiotherapy or immunotherapy.
Both patients with MYCN-amplified disease had multiple sites of metastases in addition to SMM (Patients #1 and #7). Both achieved remission with high-dose induction chemotherapy and surgery. One (#1) was consolidated with naxitamab-based immunotherapy (21) and a bivalent gangliosides vaccine (22), without autologous transplant or radiotherapy, and the other (#7) with myeloablative chemotherapy, radiotherapy and dinutuximab-based immunotherapy. Both remain in remission 27+ and 15+ months post-diagnosis.
We noted 3-year PFS and OS of 87% and 100% respectively. Median PFS for the entire group is 40.1 (12.3-318.2) months. All patients survive at a median follow up of 47.9 (16.9-318.9) months after diagnosis. Eleven patients are in complete remission including complete resolution of SMM at 12 months post-diagnosis (three patients managed with observation alone, #10, #12, and #15) (Figure 2 [patient #9]). Median time to resolution of SMM was 10.5 (range 5 – 21) months. The remaining 6 patients have completed therapy but have persistent, non-progressive disease with decreasing size of SMM. Of the 4 patients managed with watchful observation alone without any cytotoxic therapy, one is in partial remission and 3 maintain complete remission with skeletal muscle metastases resolving by 5, 13, and 21 months post-diagnosis respectively.
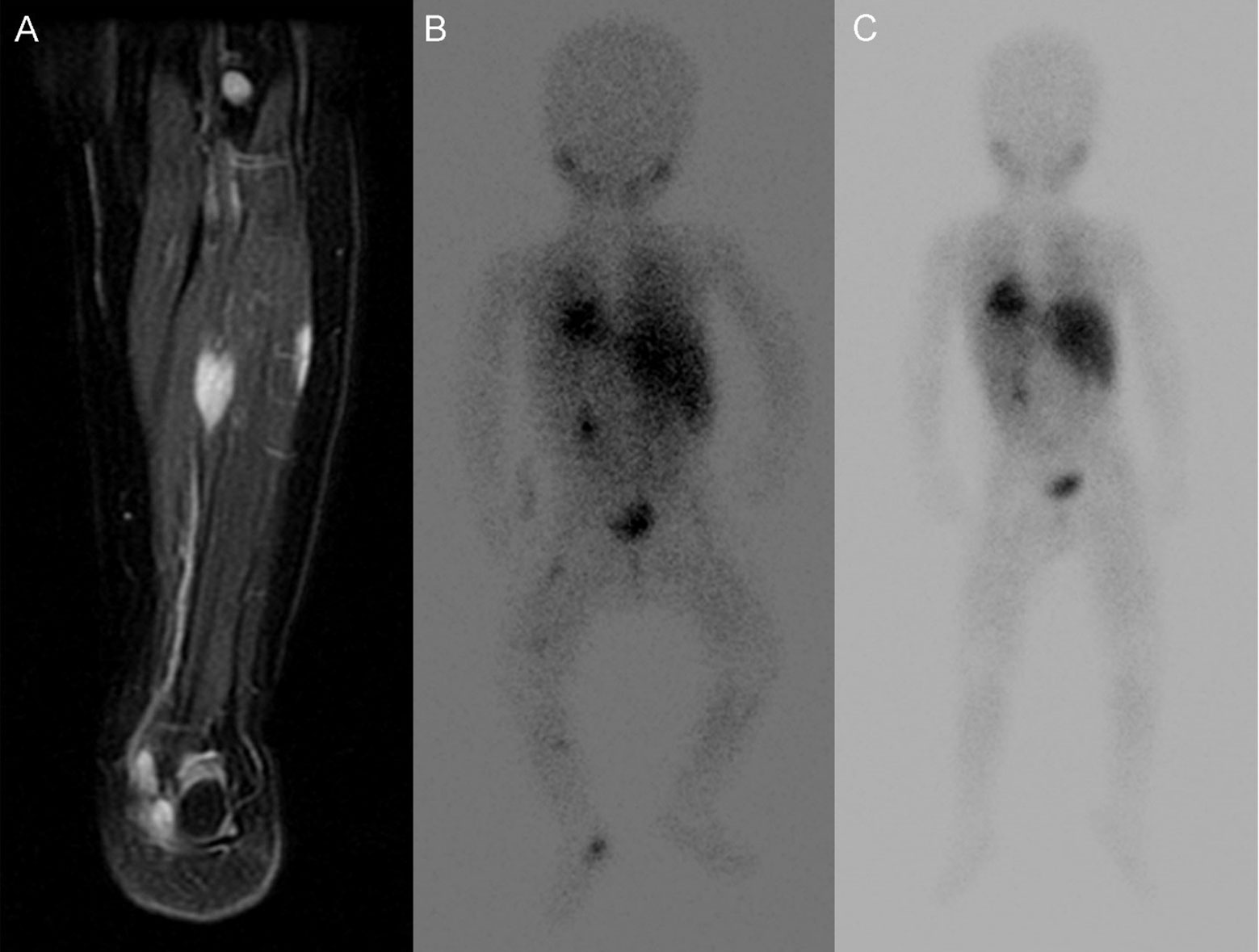
Figure 2 Images of lower extremity SMM of Patient No. 15. (A) T2 MR of SMM at diagnosis (B) corresponding MIBG avid SMM at diagnosis. (C) MIBG show resolution of SMM without treatment after 21 months.
The understanding of SMM in neuroblastoma remains limited due to their rarity. Our data substantially improves this understanding by identifying skeletal muscle as unique sites of metastasis. SMM share a close clonal relationship to primary tumors and are associated with an excellent prognosis. SMM were diagnosed at <18 months in all patients analyzed and were accompanied by metastases at sites typical for high-risk neuroblastoma (bone or BM) or at atypical sites (dura, pleura, or CNS). Therefore, our patients did not meet the criteria for classical stage MS disease. Nevertheless, the clinical course of our patients resembled that of stage MS disease with an excellent survival outcome despite receiving no chemotherapy or truncated intermediate-risk chemotherapy regimens (for those with MYCN-NA disease) (23). Furthermore, biological markers including chromosomal aberrations, ploidy, or ALK mutations did not impact outcome. The 2 patients with MYCN-A disease remain progression-free, one despite not having undergone myeloablative transplant or radiotherapy (patient #1). Of the 4 patients managed without any cytotoxic therapy, 3 achieved and maintain remission in all disease sites and the fourth, treated with intermediate-dose chemotherapy after relapse, has not had further disease progression for 87+ months. Of note, among the 4 patients not treated at diagnosis, the SMMs continued to regress, similar to regressions of disease seen in patients with classical MS disease.
The young age at diagnosis of these synchronous lesions could lead to speculation that they represent multiclonal disease as has been suggested for synchronous bilateral neuroblastoma (24). These were considered to be independent neoplasms that separated early in embryogenesis and developed in parallel. In contrast, using whole exome and whole genome sequencing, several studies have identified common mutations and rearrangements between primary and metastatic biopsy samples in high-risk stage M neuroblastoma suggesting that the two are clonally related (11, 13, 14). The limited availability of both diagnostic and metastatic disease specimens for patients with stage MS disease has hindered the investigation of clonality in patients with stage MS disease. Our genomic analysis of paired primary tumor biopsies and SMM biopsies demonstrated a close clonal relationship between the primary tumor and the SMM. Based on identification of shared truncal substitutions and CNAs in primary and SMM biopsies, both the primary tumor and SMM appeared to have arisen from a common tumor cell progenitor – from a monoclonal cell of origin. Interestingly, 2 patients had SMM with ganglioneuroma without immature elements despite their matched primary tumor biopsy showing partially differentiated neuroblastoma. These patients each had involvement of >3 muscle groups, 1p chromosomal aberrations and ALK mutations and are alive progression free at 89+ and 110+ months post-diagnosis with remission of SMM. The discrepancy in histological findings between primary and metastatic tumor could be attributed to different rates of tumor-cell maturation in different sites. Further genomic, epigenomic, transcriptomic or proteomic studies comparing primary versus SMM lesions in a larger sample size might shed light on a signature for diagnosing MS disease, possibly uncovering candidate genes or pathways to study spontaneous regression of human cancer.
We identified a subgroup of patients all diagnosed at a favorable age with a rare clinical presentation of SMM who did not meet criteria for stratification as stage MS. However, we observed a favorable outcome both for survival and resolution of disease that more closely parallel that of stage MS disease, even when no cytotoxic therapy was administered. This was a retrospective study of patients treated at a single tertiary center and our cohort included several patients who received initial therapy at other institutions. Although a larger cohort would help confirm our findings, SMM in neuroblastoma are extraordinarily rare events. The excellent outcome in all patients, and the spontaneous regression of many SMM and other metastases in some of our patients suggest that SMM constitute a forme fruste of classical stage MS disease associated with spontaneous resolution. Reducing cytotoxic therapy without compromising cure is an unmet need in treating metastatic pediatric cancers. At our institution we have a long-standing practice of avoiding cytotoxic therapy for non-high-risk neuroblastoma. While withholding all chemotherapy may not be feasible for all patients with SMM, the results presented here suggest that the presence of metastases in skeletal muscles should not lead to upstaging patients, rather it should strengthen the MS staging where a wait-and-watch approach could be justified.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
The retrospective analysis of patient data was approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center (MSKCC). Written informed consent was not required.
CF drafted the original manuscript. BK, AD, SM contributed to the conception of the work and the acquisition of data. All authors provided critical review and revisions for important intellectual content; the work reported in the paper has been performed by the authors, unless clearly specified in the text. All authors contributed to the article and approved the submitted version.
The authors acknowledge NIH Cancer Center Support Grant P30 CA008748 for financial support.
We thank Joseph Olechnowicz (editor, Memorial Sloan Kettering Cancer Center) for assistance.
SM is a consultant to Ymabs Therapeutics Inc, Illumina RP, US World Meds, and Recordati Pharmaceuticals. NKC reports receiving commercial research grants from Y-mAbs Therapeutics and Abpro-Labs Inc.; holding ownership interest/equity in Y-mAbs Therapeutics Inc., holding ownership interest/equity in Abpro-Labs, and owning stock options in Eureka Therapeutics. NKC is the inventor and owner of issued patents licensed by MSK to Y-mAbs Therapeutics, Biotec Pharmacon, and Abpro-labs. Hu3F8 and 8H9 were licensed by MSKCC to Y-mAbs therapeutics. Both MSK and NKC have financial interest in Y-mAbs. NKC is an advisory board member for Abpro-Labs and Eureka Therapeutics. G.G. is a consultant for Isabl Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cheung NK, Dyer MA. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer (2013) 13(6):397–411. doi: 10.1038/nrc3526
2. Van Ho AT, Hayashi S, Bröhl D, Auradé F, Rattenbach R, Relaix F. Neural crest cell lineage restricts skeletal muscle progenitor cell differentiation through Neuregulin1-ErbB3 signaling. Dev Cell (2011) 21(2):273–87. doi: 10.1016/j.devcel.2011.06.019
3. Faingold R, Babyn PS, Yoo SJ, Dipchand AI, Weitzman S. Neuroblastoma with atypical metastases to cardiac and skeletal muscles: MRI features. Pediatr Radiol (2003) 33(8):584–6. doi: 10.1007/s00247-002-0858-5
4. Giuliani S, Marachelian A, Franklin A, Shimada H, Grikscheit T. Forearm skeletal muscle neuroblastoma in a child: a rare primary location. J Pediatr Hematol Oncol (2013) 35(1):61–3. doi: 10.1097/MPH.0b013e31827b0cf1
5. Kang T, Dormans J, Maris J, Carpentieri D, Pawel BR, Adamson PC. Congenital neuroblastoma arising in the deltoid muscle. J Pediatr Hematol Oncol (2004) 26(2):101–3. doi: 10.1097/00043426-200402000-00006
6. LaBan MM, Nagarajan R, Riutta JC. Paucity of muscle metastasis in otherwise widely disseminated cancer: a conundrum. Am J Phys Med Rehabil (2010) 89(11):931–5. doi: 10.1097/PHM.0b013e3181f713c3
7. Molina-Garrido MJ, Guillén-Ponce C. Muscle metastasis of carcinoma. Clin Trans Oncol (2011) 13(2):98–101. doi: 10.1007/s12094-011-0625-x
8. Evans AE, D'Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. children's cancer study group a. Cancer (1971) 27(2):374–8. doi: 10.1002/1097-0142(197102)27:2<374::aid-cncr2820270221>3.0.co;2-g
9. Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol (1993) 11(8):1466–77. doi: 10.1200/JCO.1993.11.8.1466
10. Park JR, Bagatell R, Cohn SL, Pearson AD, Villablanca JG, Berthold F, et al. Revisions to the international neuroblastoma response criteria: A consensus statement from the national cancer institute clinical trials planning meeting. J Clin Oncol (2017) 35(22):2580–7. doi: 10.1200/JCO.2016.72.0177
11. Chicard M, Colmet-Daage L, Clement N, Danzon A, Bohec M, Bernard V, et al. Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin Cancer Res (2018) 24(4):939–49. doi: 10.1158/1078-0432.CCR-17-1586
12. Chicard M, Boyault S, Colmet Daage L, Richer W, Gentien D, Pierron G, et al. Genomic copy number profiling using circulating free tumor DNA highlights heterogeneity in neuroblastoma. Clin Cancer Res (2016) 22(22):5564–73. doi: 10.1158/1078-0432.CCR-16-0500
13. Schramm A, Köster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet (2015) 47(8):872–7. doi: 10.1038/ng.3349
14. Fransson S, Martinez-Monleon A, Johansson M, Sjöberg RM, Björklund C, Ljungman G, et al. Whole-genome sequencing of recurrent neuroblastoma reveals somatic mutations that affect key players in cancer progression and telomere maintenance. Sci Rep (2020) 10(1):22432. doi: 10.1038/s41598-020-78370-7
15. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The international neuroblastoma risk group (INRG) classification system: an INRG task force report. J Clin Oncol (2009) 27(2):289–97. doi: 10.1200/JCO.2008.16.6785
16. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn (2015) 17(3):251–64. doi: 10.1016/j.jmoldx.2014.12.006
17. Padovan-Merhar OM, Raman P, Ostrovnaya I, Kalletla K, Rubnitz KR, Sanford EM, et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PloS Genet (2016) 12(12):e1006501. doi: 10.1371/journal.pgen.1006501
18. Medina-Martínez JS, Arango-Ossa JE, Levine MF, Zhou Y, Gundem G, Kung AL, et al. Isabl platform, a digital biobank for processing multimodal patient data. BMC Bioinf (2020) 21(1):549. doi: 10.1186/s12859-020-03879-7
19. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell (2012) 149(5):994–1007. doi: 10.1016/j.cell.2012.04.023
20. Twist CJ, Schmidt ML, Naranjo A, London WB, Tenney SC, Marachelian A, et al. Maintaining outstanding outcomes using response- and biology-based therapy for intermediate-risk neuroblastoma: A report from the children's oncology group study ANBL0531. J Clin Oncol (2019) 37(34):3243–55. doi: 10.1200/JCO.19.00919
21. Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3F8 anti-GD2 monoclonal antibody dosing with granulocyte-macrophage colony-stimulating factor in patients with resistant neuroblastoma: A phase 1 clinical trial. JAMA Oncol (2018) 4(12):1729–35. doi: 10.1001/jamaoncol.2018.4005
22. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res (2014) 20(5):1375–82. doi: 10.1158/1078-0432.CCR-13-1012
23. Twist CJ, Naranjo A, Schmidt ML, Tenney SC, Cohn SL, Meany HJ, et al. Defining risk factors for chemotherapeutic intervention in infants with stage 4S neuroblastoma: A report from children's oncology group study ANBL0531. J Clin Oncol (2019) 37(2):115–24. doi: 10.1200/JCO.18.00419
Keywords: neuroblastoma, metastasis, skeletal muscle metastasis, pediatric oncology, chemotherapy, tumor evolution
Citation: Fong C, Kushner BH, Di Giannatale A, Gundem G, Li S, Roberts SS, Basu EM, Price A, Cheung N-KV and Modak S (2023) Skeletal muscle metastases in neuroblastoma share common progenitors with primary tumor and biologically resemble stage MS disease. Front. Oncol. 12:1106597. doi: 10.3389/fonc.2022.1106597
Received: 23 November 2022; Accepted: 15 December 2022;
Published: 05 January 2023.
Edited by:
Chi-kong Li, The Chinese University of Hong Kong, ChinaReviewed by:
Cai Jiaoyang, Shanghai Children’s Medical Center, ChinaCopyright © 2023 Fong, Kushner, Di Giannatale, Gundem, Li, Roberts, Basu, Price, Cheung and Modak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shakeel Modak, bW9kYWtzQG1za2NjLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.