 Bin Jia
Bin Jia Peiyi Xia2†
Peiyi Xia2† Wenjia Wang
Wenjia Wang Enjie Liu
Enjie Liu Guozhong Jiang
Guozhong Jiang Yanru Qin
Yanru Qin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 17 January 2023
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1086908
This article is part of the Research TopicImplementation of Genomic and Epigenomic Innovation in Clinical Cancer DiagnosticsView all 8 articles
Background: Sarcomatoid hepatocellular carcinoma (SHC) is a rare epithelial malignancy with high invasiveness and poor prognosis. However, the molecular characteristics and main driver genes for SHC have not been determined. The aim of this study is to explore the potentially actionable mutations of driver genes, which may provide more therapeutic options for SHC.
Methods: In this study, DNA extraction and library preparation were performed using tumor tissues from 28 SHC patients. Then we used Miseq platform (Illumina) to sequence the target-enriched library, and we aligned and processed the sequencing data. The gene groups were tested for SNVs/Indels/CNVs. Tumor mutation burden (TMB) was assessed by the 425-cancer-relevant gene panel. Multivariate analysis of COX’s model was used for survival analysis (OS) of patients’ clinical characteristics.
Result: The median overall survival (OS) of the patients was only 4.4 months. TP53, TERT, and KRAS were the top three frequently mutated genes, with frequencies of 89.3%, 64.3%, and 21.4%, respectively. A considerable number of patients carried mutations in genes involved in the TP53 pathway (96%) and DNA Damage Repair (DDR) pathway (21%). Multiple potentially actionable mutations, such as NTRK1 fusions and BRCA1/2 mutations, were identified in SHCs.
Conclusions: This study shows a landscape of gene mutations in SHC. SHC has high mutation rates in TP53 pathway and DDR pathway. The potentially actionable mutations of driver genes may provide more therapeutic options for SHC. Survival analysis found that age, smoking, drinking, and tumor diameter may be independent prognostic predictors of SHC.
Sarcomatoid hepatocellular carcinoma (SHC) is a rare epithelial malignancy with high invasiveness and poor prognosis and accounts for no more than 4% of primary malignant hepatic tumors (1, 2). Studies have reported that its occurrence may be related to viral infection, cirrhosis, radiotherapy, chemotherapy, and interventional therapy (3). Typical SHC contains definite malignant epithelial components and spindle cells and other sarcomatoid components in the same tissue, and most scholars believe that the latter is transformed from the former rather than the real mesenchymal tissue (4, 5). Sarcomatoid carcinomas(SC) and carcinosarcomas(CS) are different in pathology and immunohistochemistry. CSalso contain malignant epithelial and sarcomatoid components, but sarcomatoid regions may be composed of different malignant mesenchymal components, such as malignant fibrous histiocytic carcinoma, fibrosarcoma, leiomyosarcoma, osteosarcoma, etc (6). Sarcomatoid areas of SC consist of a single malignant spindle cell and express epithelioid features that can only be detected by immunohistochemistry and electron microscopy. Therefore, different names, including spindle cell carcinoma, pleomorphic carcinoma, and metaplastic carcinoma, can be observed (2).
The pathogenesis of SHC has not been fully clarified yet. The key is the tissue source of sarcomatoid components in the tumor. Histologically, the sarcomatoid cells of SC can exhibit immunoreactivity for cytokeratin, suggesting that SC may originate from the sarcomatoid change of epithelial carcinoma. Many studies suggest a common origin instead of a collision tumor composed of sarcoma and carcinoma (4, 5), however, differential diagnosis between them is still difficult for pathologists. One of the most significant histopathological characteristics that supports SC is the identification of transitional zones between epithelial and mesenchymal cells, but such a “zone” is not always observed (7). Compared with traditional liver cancer, SHC has a high degree of malignancy, fast growth, easy distant metastasis, low surgical resection rate, and recurrence even after radical resection and liver transplantation, eventually, the prognosis is not satisfied (2, 8, 9). The median postoperative survival of SHC patients has been reported as about 8 months, and the 1-year postoperative survival rate of patients is around 22% (1, 8). Due to its rarity, the current literature on these tumors is limited to either case reports or small case series (3, 5, 10).
SHC is insensitive to most chemotherapeutic agents, including conventional platinum-based chemotherapy, leading to an extremely poor prognosis. Meanwhile, little is known about molecular characteristics and main driver genes for SHC, which results in a lack of targeted drugs. In this study, we screened a panel of 425 genes on dissected tumor tissues from 28 SHC patients to show the mutational landscape of SHC, and explore the potentially actionable mutations of driver genes, which may provide more therapeutic options for SHC.
A total of 28 patients with sarcomatoid liver cancer diagnosed by pathology and treated in the First Affiliated Hospital of Zhengzhou University from May 2015 to September 2019 were retrospectively collected in this study. The study was approved by the Review Broad of the First Affiliated Hospital of Zhengzhou University. For each patient, Formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks/sections from biopsies or surgically removed liver lesions were obtained. All specimens of these patients were examined by two experienced pathologists under the guidance of 2019 WHO classification of SHC.
DNA extraction, library preparation, and targeted-capture enrichment were performed as previously described (11). Briefly, tissue genomic DNA(gDNA)was extracted from tumor tissue as described using DNeasy Tissue Kits (Qiagen Inc., Germantown, MD, USA). Genomic DNA from the white blood cells(WBCs)was extracted using DNeasy Blood & Tissue Kit (Qiagen Inc., Germantown, MD, USA)and used as the normal control to distinguish germline mutations. The KAPA Hyper Prep kit (Kapa Biosystems, Wilmington, MA, USA) was used for DNA library preparation adapted to an Illumina MiSeq®. Customized xGen lockdown probes panel (containing 425 predefined cancer-related genes, Integrated DNA Technologies Inc., Coralville, IA, USA) were used in hybridization enrichment. The capture reaction was performed with Dynabeads M-270 (Life Technologies, Waltham, MA, USA) and the NimbleGen SeqCap EZ Hybridization & Wash Kit (Roche Inc., Indianapolis, IN, USA). Captured libraries were PCR-amplified with KAPA HiFi HotStart ReadyMix (KAPA Biosystems). The purified library was quantified using the KAPA Library Quantification kit (KAPA Biosystems). The target-enriched library was sequenced using Miseq platform (Illumina) with a mean coverage depth of 1000X and 100X for the tumor tissue samples and WBCs, respectively.
Sequencing data were demultiplexed by bcl2fastq v2.16.0.10 (Illumina, Inc.), and analyzed by Trimmomatic (12) to remove low-quality (quality<15) or N bases. Then the data were aligned to the hg19 reference human genome with the Burrows-Wheeler Aligner (v0.7.12) (13) with the BWA-MEM algorithm and default parameters to create SAM files. Picard 1.119 (http://picard.sourceforge.net/) was used to convert SAM files to compressed BAM files which were then sorted according to chromosome coordinates. The Genome Analysis Toolkit (14)(GATK, version 3.4-0) was modified and used to locally realign the BAMs files at intervals with indel mismatches and recalibrate base quality scores of reads in BAM files (15).
VarScan2 was employed for the detection of single-nucleotide variations (SNVs) and insertion/deletion mutations, followed by annotation using ANNOVAR (16, 17). SNVs were filtered out if the mutant allele frequency (MAF) was less than 0.5% for tumor tissue. Common SNVs were excluded if they were present in >1% of the population in the 1000 Genomes Project or dbSNP database. The resulting mutation list was further filtered by an in-house list of recurrent artifacts based on a normal pool of whole blood samples. Parallel sequencing of matched white blood cells from each patient was performed to further remove sequencing artifacts, germline variants, and clonal hematopoiesis. Copy number variations (CNVs) were detected using ADTEx (http://adtex.sourceforge.net) with default parameters (18). Genomic fusions were identified by FACTERA with default parameters. The fusion reads were further manually reviewed and confifirmed on Integrative Genomics Viewer (IGV).
Tumor mutation burden (TMB), as assessed by the 425-cancer-relevant gene panel (Nanjing Geneseeq Technology). TMB was defined as the total number of base substitutions and indels in the coding regions of the targeted genes, including synonymous alterations to reduce sampling noise and excluding known driver alterations, as previously described (19).
Continuous variables were expressed as mean ± standard deviation (SD) or median (range). The OS rates were estimated by the Kaplan–Meier method, and comparisons were made using the log-rank test. Multivariate analysis was done with the Cox proportional hazard regression model. Statistical analysis was performed using SPSS 21.0 for Windows (SPSS Inc., Chicago, Il, USA). TMB analysis in different gene groups was ranked by Mann–Whitney U test. Overall survival (OS) was calculated from the date of first referral to the date of death (uncensored) or last contact (censored). Multivariate analysis of COX’s model was used for survival analysis (OS) of patients’ clinical characteristics. A p-value ≤0.05 was considered statistically significant.
In the patient cohort, there were 20 males and 8 females, with a male-to-female ratio of 2.5:1. The onset age was 44 ~ 79 years old, and the median age was 55 years old. Eighteen patients were infected with the hepatitis B virus, one with the hepatitis C virus and nine without the hepatitis virus. The tumor site was located in the left lobe of the liver in 7 cases, the right lobe of the liver in 17 cases, multiple livers in 3 cases, and caudate lobe in 1 case. According to the International Union of Cancer (International Union Against Cancer, UICC)/(American Committee on Cancer, AJCC) released the eighth edition of TNM staging of liver Cancer, including 2 stages I cases, 1 stage II case, 9 stage III cases, and 16 stage IV cases. The mean maximum tumor diameter was 6.27cm (1.94cm ~ 14.9cm), as shown in Table 1.
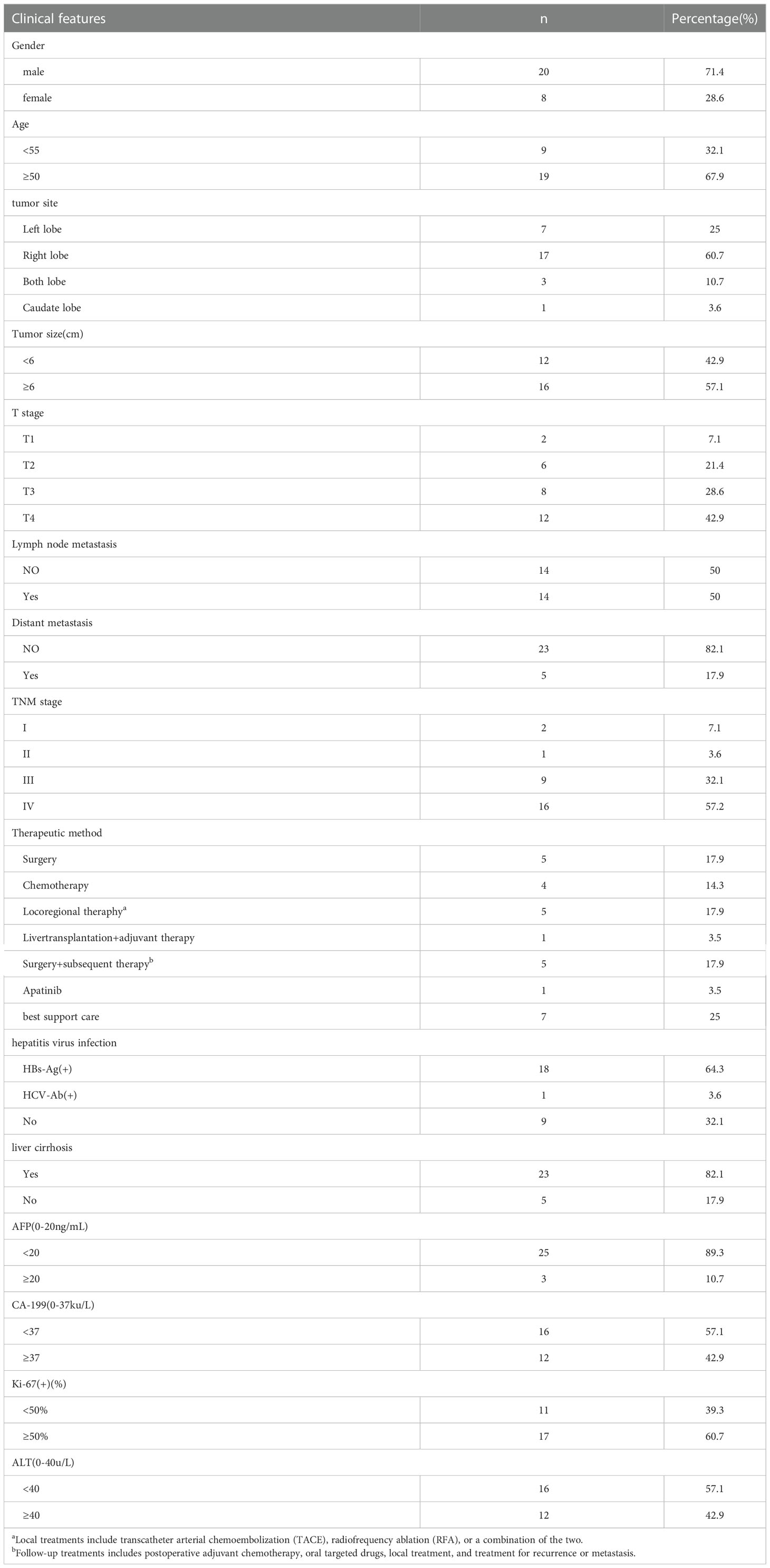
Table 1 Clinical features of 28 sarcomatoid hepatocellular carcinoma patients.
A total of 28 SHC tumor samples were analyzed using a 425 gene NGS panel (Supplemental Table S1). Overall, 239 genomic alterations of 138 distinct cancer-relevant genes were detected in the SHC tumor samples, including 127 missenses (53%), 24 nonsense (10%), 17 frameshifts (7%), 35 copy number variances (15%), 3 indels (1%), 14 splicing site mutations (6%) and 19 other alterations (8%) (Supplemental Figure 1).
TP53, TERT, and KRAS were the top three frequently mutated genes, with frequencies of 89.3%, 64.3%, and 21.4%, respectively (Figure 1). Alterations of the TP53 tumor suppressor gene were detected in 25 out of 28 samples in the cohort, indicating that inactivation of the TP53 pathway was a common genetic event in the SHCs (Figure 1). Majority of TP53 mutated cases were rare mutations and seven nonsense mutations were included, hot spot mutation R249S was found in five cases, R175H and R273L were only found in one case, respectively. Especially, TP53-ARID1A co-mutation was found in one case. Seventeen cases showed alterations in TERT, among which sixteen cases carried missense mutations in promoter regions, including c.-124C>T in fourteen cases, c.-146C>T, and c.-57A>C in one case, respectively. One case occurred CNV events, which is an amplification of TERT. We also found 15 cases with TERT mutations harboring TP53 mutations (15/28, 53.6%). KRAS was altered in five patients, including copy number variation (CNV) in one case, G12D in three cases, and G13D in one case.
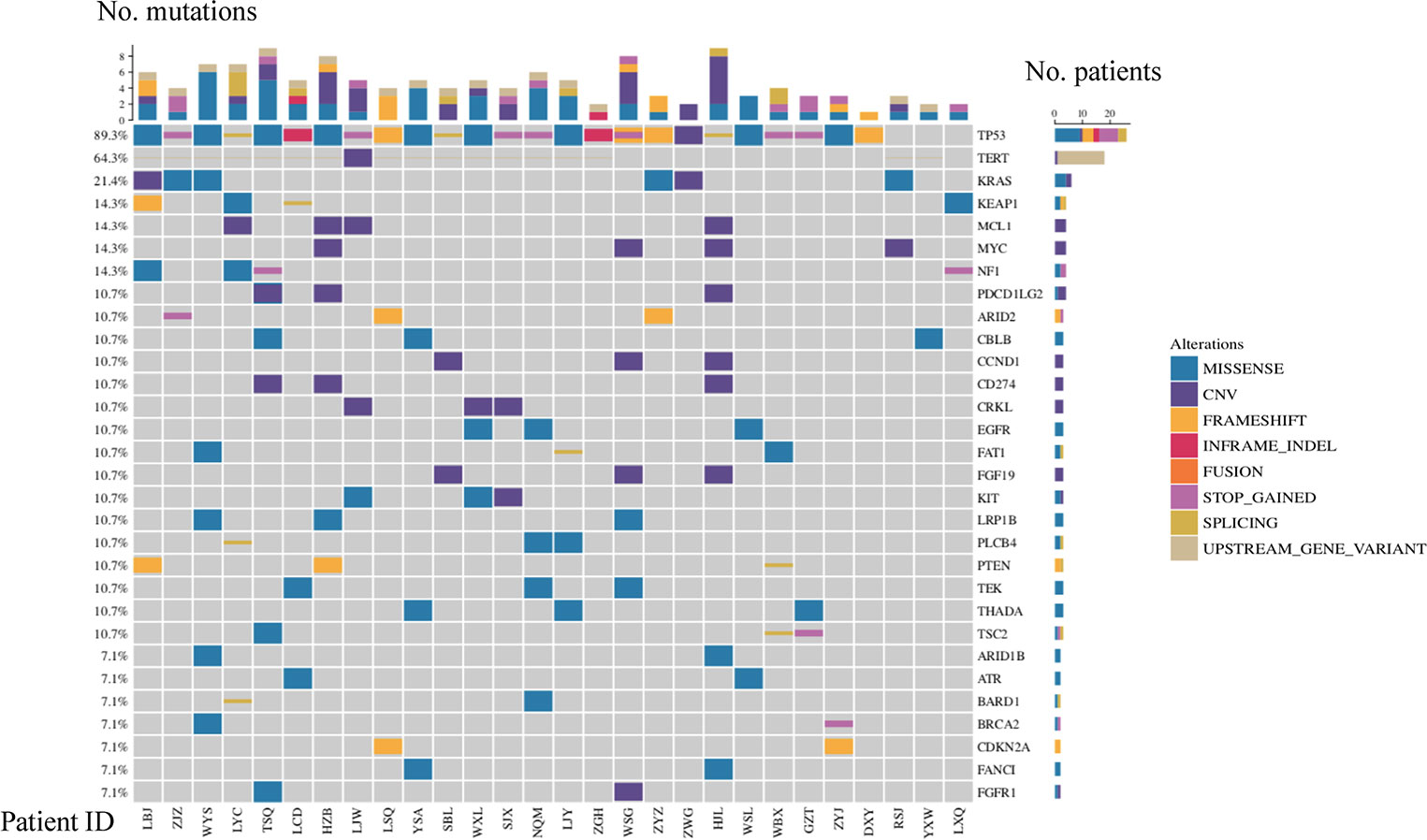
Figure 1 Mutational landscape of SHC.
Other frequently mutated genes included KEAP1, NF1, MCL1, and MYC with the same frequency of 14.3% (Figure 1). KEAP1 and NF1 are tumor suppressor genes, while MCL1 and MYC are oncogenes. The type of mutation in MCL1 and MYC were only copy number variants, and MCL1 and MYC appeared in two cases at the same time. EGFR mutations were also detected in three cases (11%, two females, and one male), which were all rare mutations including P265T, A647T, and P596L. Of note, we found a considerable number of patients carrying mutations in genes involved in the TP53 pathway (96%) and DDR pathway (21%). Based on all gene mutations In patients, TP53, TERT, CCND1, RB1, CDKN2A, CTNNB1 and CDK6 were enriched in the TP53 pathway. It was also found that ATR, POLE, BRCA1 and BRCA2 were enriched in the DDR pathway, respectively (Supplemental Figure 2).
Additional actionable driver gene alterations were also identified, including BRCA1/2 mutations(n=2) and NTRK1 fusions(n=1). Two cases with BRCA1/2 pathogenic mutations (BRCA1 c.5467+1G>A and BRCA2 p.Q1095*) were likely sensitive to PARP inhibitors (Figure 1, Supplemental Table S2). Downstream CRABP2 was jointed to intron 9 of NTRK1, and was identified in one patient with NTRK1 fusion, indicating that treatment of NTRK1 inhibitors may be a potential therapy.(Supplemental Table S2)
The tumor mutation burden (TMB) in 28 SHC tumors was ranging from 1.15 to 20.69 mutations/MB with a median number of 6.9 mutations/MB (Figures 2A, B, Supplemental Table S3). Six (21.4%) patients had high TMB using a clinically validated cut-off of 10 mutations/Mb (20). We also analyzed the association between TMB and OS by stratifying the patients into high TMB (≥10 mutations/Mb) and low TMB (<10 mutations/Mb), but there was no significant difference in OS between these two groups (Figure 2C) To further analyze the relationship between genetic alterations and TMB in SHC, the TMB distribution in SHC samples was compared between the wild type gene group and mutated gene group. SHCs with FAT1 or EGFR mutations showed a trend toward higher TMB, but there was no significant difference between the wild-type and mutated groups (Figure 3).
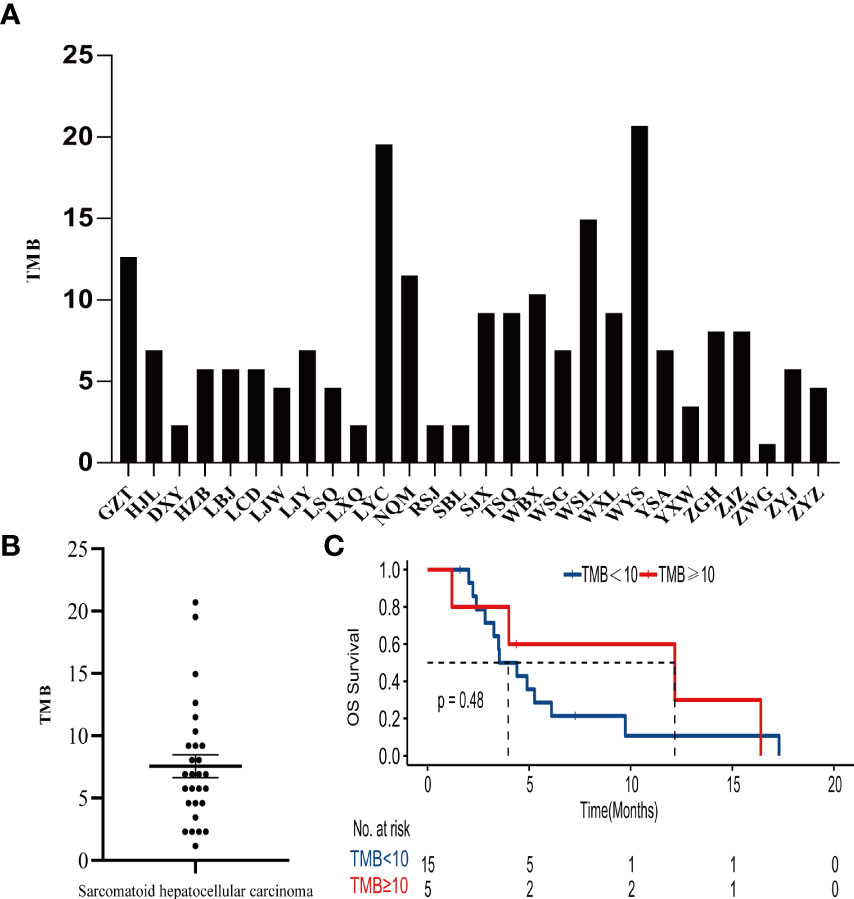
Figure 2 (A)Distribution of TMB in each patient, (B) the median number of TMB in the SHC patients, (C) Kaplan- Meier estimates of overall survival in SHC comparing patients with high TMB and low TMB.
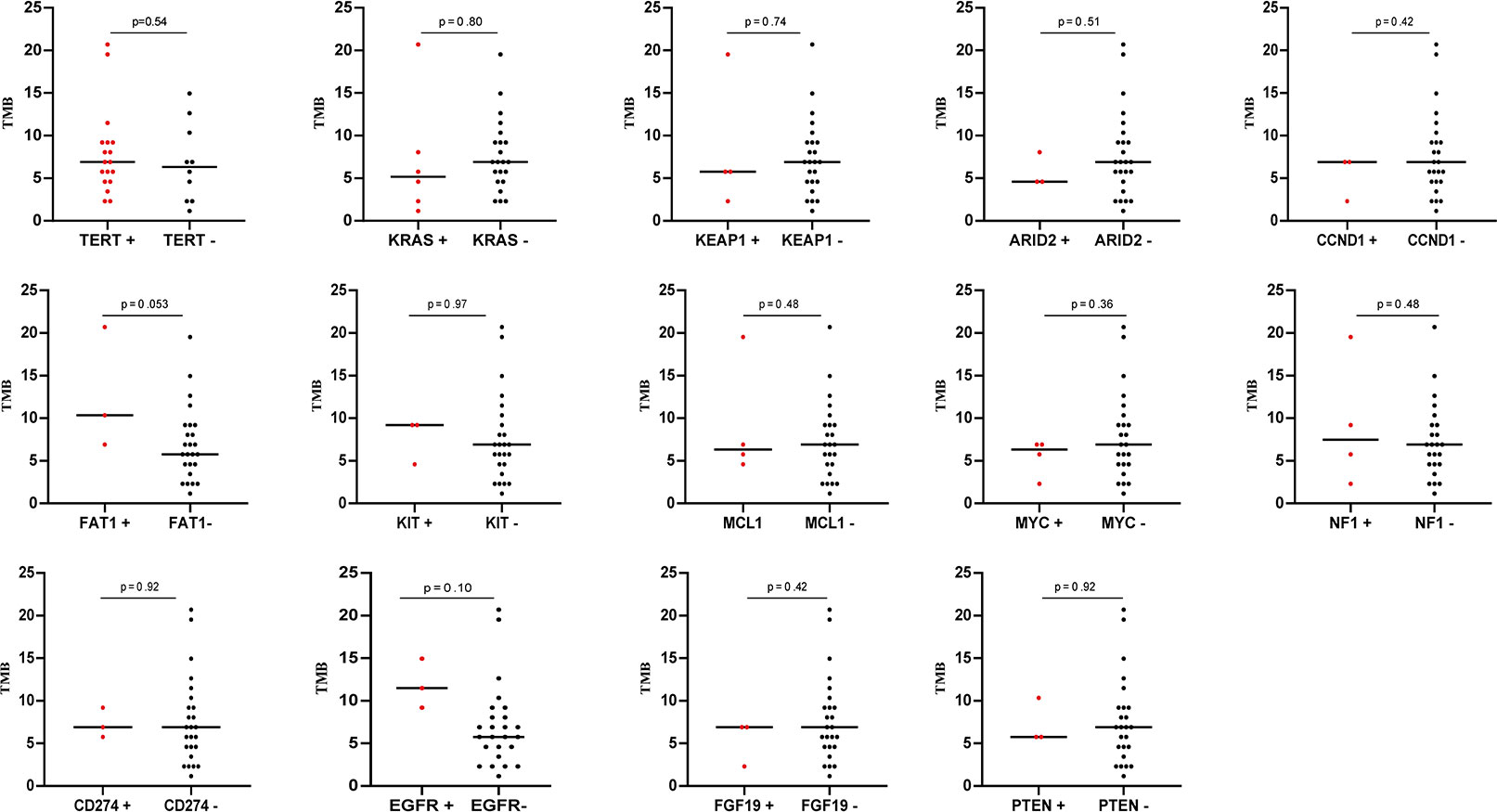
Figure 3 The correlation of TMB with mutation status of different genes.
We obtained the survival data of 20 patients during the follow-up, 17 patients died and 3 survived by the end of follow-up. The median overall survival (OS) of the patients was only 4.4 months (95%CI: 2.5-6.3 months). The 6-month survival rate was 30.0%, and the 1-year survival rate was 15.0%. Furthermore, we analyzed the correlation of patients’ clinical characteristics with OS using a multivariate COX regression model and found that the clinical variables, including age, smoking, drinking, and tumor diameter, may be independent prognostic predictors of SHC (Table 2). Age ≤56 years old was associated with poor prognostic(mOS=12.2 vs 3.5 months, HR 0.18, 95% CI 0.06 to 0.57; log-rank p=0.004; Figure 4A), drinking(mOS=2.8 vs 6.1 months, HR 26.54, 95% CI 4.72 to 149.3; log-rank p<0.001), smoking(mOS=3.3 vs 6.1 months, HR 6.81, 95% CI 1.56 to 29.7; log-rank p=0.011), tumor diameter >8 centimeters (mOS=2.6 vs 6.1 months, HR 16.16, 95% CI 3.17 to 82.37; log-rank p<0.001) were also associated with a poor prognostic(Figures 4B–D). Then we analyzed the correlation of patients’ genetic alterations with OS and found that mutations in genes in the RAS/RAF pathway or KRAS, TERT, ARID2, EGFR, KEAP1, NF1, and TEK showed no significant correlation with the survival of SHC patients (Figure 5). It’s worth noting that the group with wild-type KRAS had a longer mean survival time than those with KRAS mutations (6.24 vs 4.18 months). Three patients (21.4%) in the wild-type KRAS lived longer than a year, and all patients (0%) in the KRAS mutations group died within a year. We compared the treatment between mutated KRAS and wild-type KRAS groups, finding no statistical difference (Supplemental Table S4).
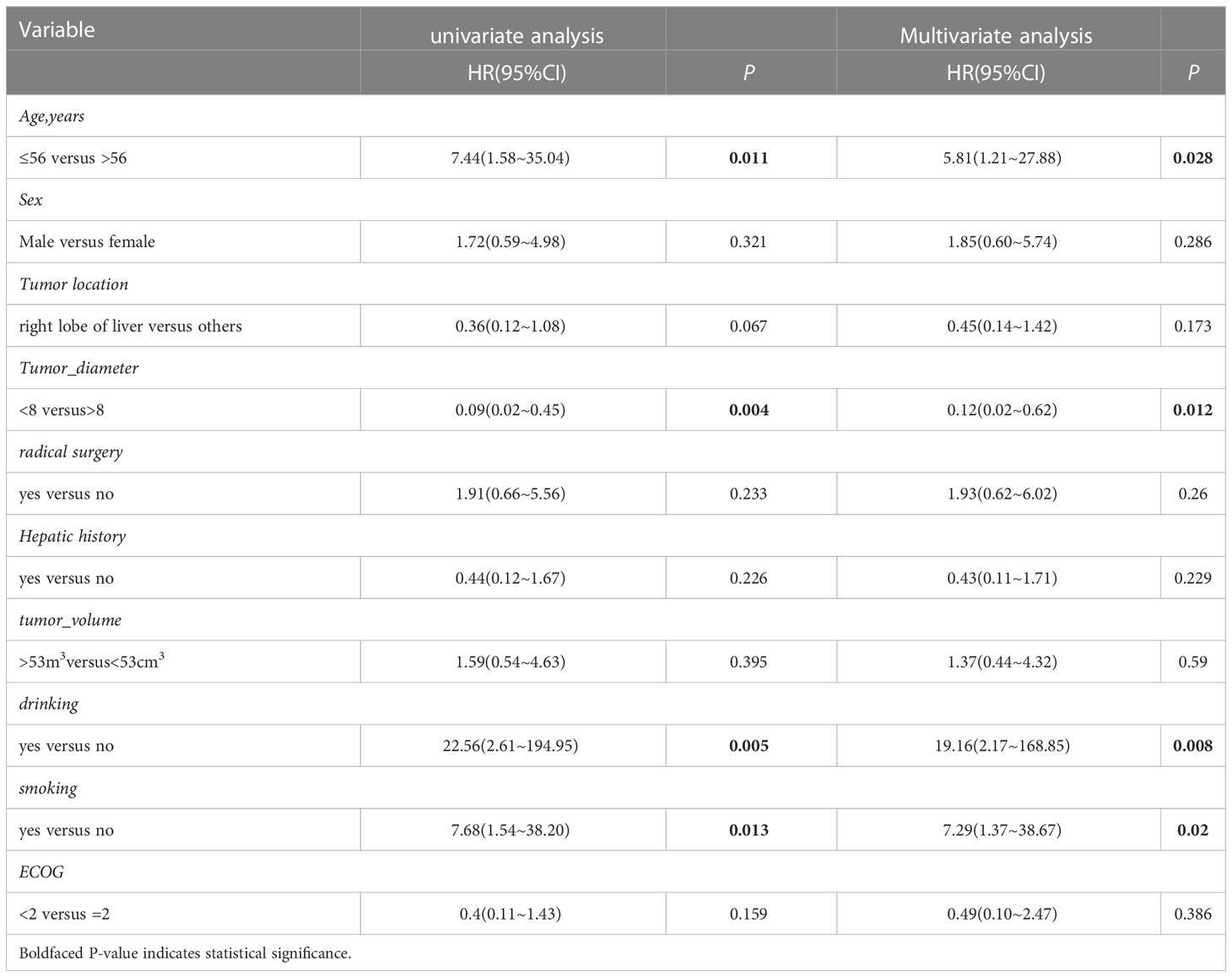
Table 2 Univariate and multivariate cox hazards analysis for OS in patients with SHC.
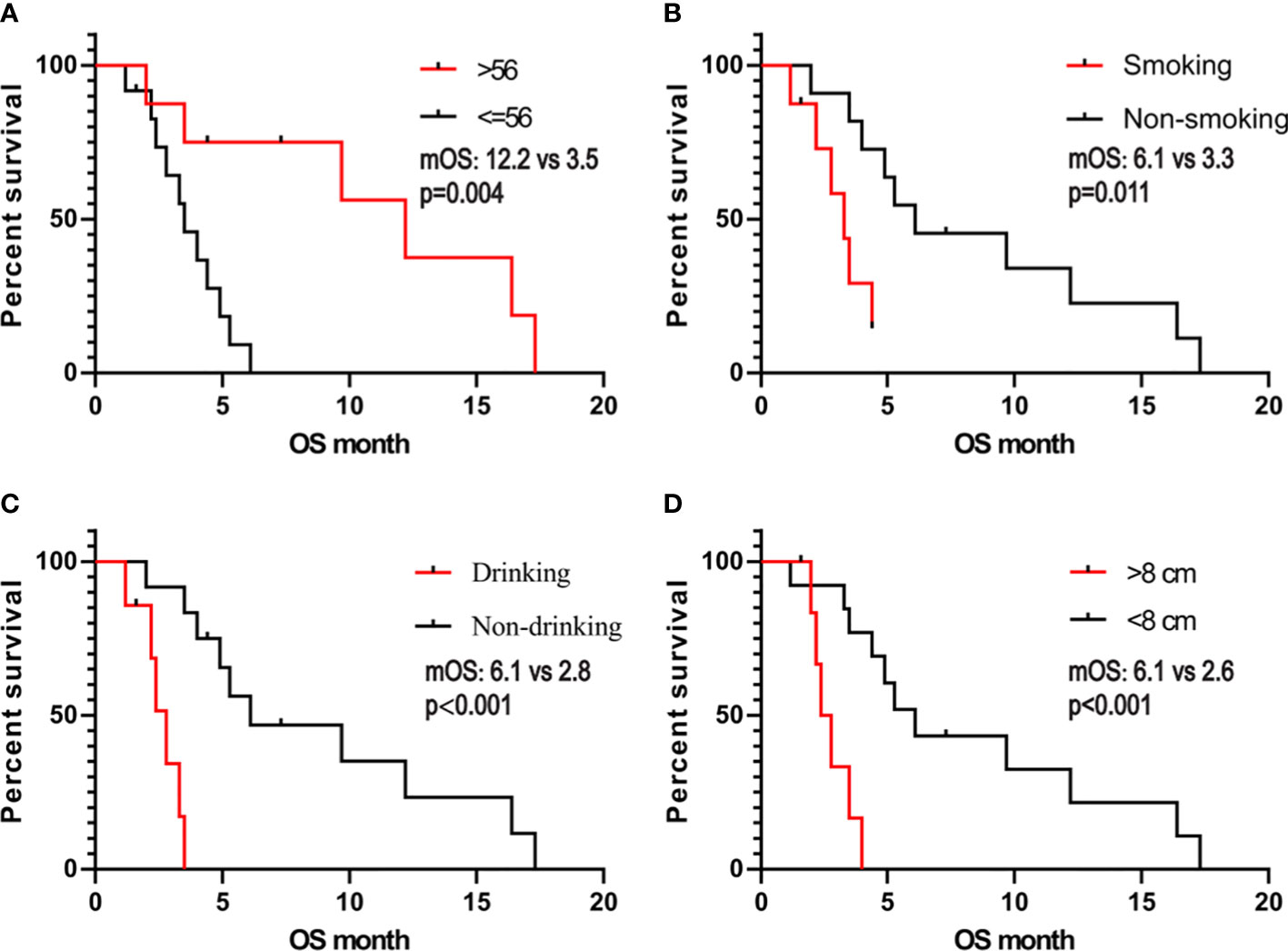
Figure 4 Kaplan- Meier estimates of overall survival in SHC comparing patients with (A) age, (B) smoking, (C) alcohol consumption, and (D) tumor diameter.
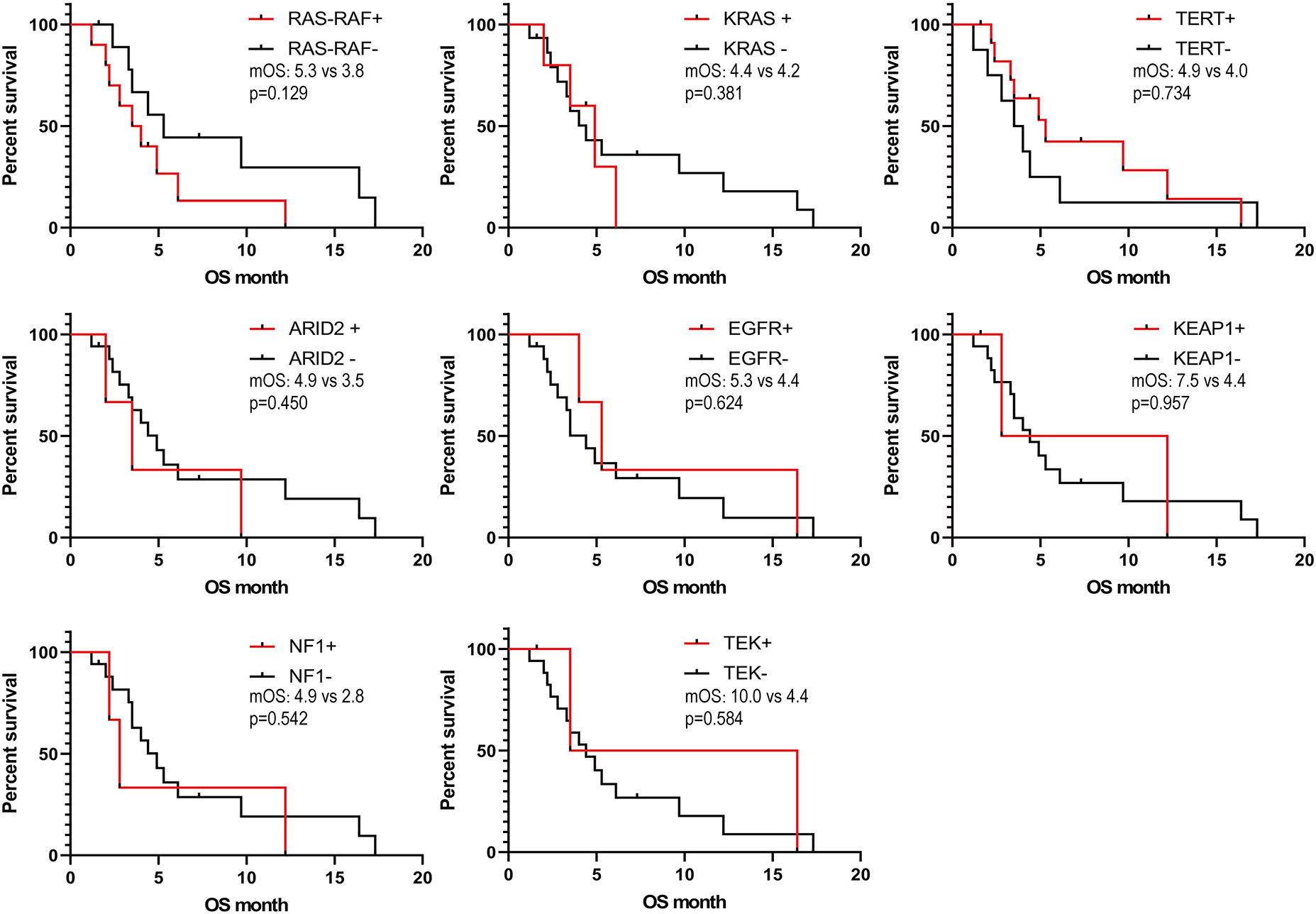
Figure 5 Kaplan- Meier estimates of overall survival in SHC comparing patients with different gene mutations.
In this current study, we described the genetic profiles of SHC using 28 FFPE samples and identified TP53, TERT, and KRAS as the most frequent mutations that give new insight into the understanding of the etiology of SHC. In addition, we also indicated the multiple prognostic factors for the progression of SHC. Genetic alterations are key determinants of tumors’ sensitivity to targeted therapies. For example, mutations of KRAS and NRAS predict resistance to anti-EGFR antibodies in colorectal cancers, and patients with lung adenocarcinoma that harbor EGFR mutations or ALK rearrangements have shown remarkable efficacy in relevant targeted drug therapy (21, 22). Thus, we focus on the effects of genetic mutations in SHC and possible targeted therapies.
TP53-TERT co-mutations (53.6%), are much higher than in previous reports (10). Several studies including small samples revealed that the frequency of TP53 mutations were highest among all gene detected in SHC (10, 23), consistent with our study. Since hepatocellular carcinoma phenotypes are closely associated with gene mutations, TP53 and CTNNB1 mutations could define two mutually exclusive groups of distinct phenotypes (24). SHC is more invasive accompanying increased cell proliferation and epithelial-to-mesenchymal transition, corresponding to the characteristics of the “TP53 mutation group” (24). KEAP1 is a key factor controlling the endogenous antioxidant response, functioning as a negative regulator of the transcription factor nuclear factor erythroid-2 like 2 (NFE2L2/NRF2) (25). Mutations in KEAP1 are associated with a worse prognosis in cancer (26). It has been suggested that NF1 was involved in the activation of the RAS/MAPK pathway directly; other studies showed NF1 mutation played an important role in drug resistance to BRAF, EGFR inhibitors, and tamoxifen, and was associated with shorter survival (27, 28).
Our study showed that the mutation rate of KRAS is the highest among oncogenes, followed by MCL1 and MYC. KRAS mutations were also detected in another study including 10 cases of liver sarcomatoid carcinomas (23). While the association of KRAS mutations with poor prognosis may not be unique to SHCs (29), the occurrence of KRAS mutation may induce sarcomatoid phenotype (30) and indicate poor prognosis in cancers with sarcomatoid component (23). KRAS is one of front-line sensors that allow the transmission of transducing signals from the cell surface to the nucleus, and affecting a series of essential cellular processes such as cell differentiation, growth, chemotaxis and apoptosis. KRAS mutation causes aberrant activation, which is associated spontaneous tumor development in KRAS-driven cancer. Meanwhile, KRAS mutation regulates tumor microenvironment via secreting molecules in a paracrine manner and inducing various chemokines, cytokines and growth factors, which contributes to the promotion and maintenance of malignancy (31). In our study, we found no statistical significance of Kaplan- Meier curves between groups with KRAS mutations and wild-type KRAS, and it may be due to the different KRAS mutation subtypes compared with other studies. G12D (60%) is the dominant mutant subtype of KRAS in this study, and we still found that the numbers of patients who survived over one year in wild- type KRAS group were greater than those in the KRAS mutation (n=3 vs n=0). Evidence exists that KRAS- mutant cancers with the G12C subtype, instead of the G12D subtype, was associated with higher TMB and PD-L1 positivity rate compared with wild-type KRAS (32). Novel inhibitors targeting KRAS (G12C), such as AMG510 (sotorasib) and MRTX849 (adagrasib), have displayed promising results in preclinical and clinical trials, due to the covalent inhibition of cysteine and high GTPase activity of KRAS (G12C) (33). KRAS itself apart, to target the KRAS-driven malignant phenotypes, such as the metabolic vulnerabilities of KRAS mutant cancer mentioned in recent review (31), might represent another effective strategy. MYC encodes transcription factors associated with cancer cell-cycle progression, proliferation, and biosynthesis (34). Myeloid leukemia 1 (MCL-1) is an antiapoptotic protein of the BCL-2 family that prevents apoptosis by binding to the pro-apoptotic BCL-2 proteins (35). A previous study showed MYC and MCL1 conferred resistance to chemotherapy by expanding CSCs via mtOXPHOS (36).
In line with our findings, TERT mutations have been reported in 53.3% of SHC in a previous study (10). TERT is the catalytic subunit of telomerase, which activates telomerase to maintain the integrity of telomerase and enables tumor cells to obtain infinite proliferation (37). TERT promoter mutations generate novel transcription factor binding sites, contributing to increased TERT expression in cancer cells. The presence of TERT mutation was associated with worse prognosis in breast cancer, thyroid carcinoma, and lung adenocarcinoma (37), meanwhile, TERT promoter mutations are highly associated with sarcomatoid histology in patients with metastatic pleural mesothelioma (38) and urothelial carcinomas (39). Although vaccines (e.g. GV1001) and oligonucleotide inhibitors (e.g. imetelstat) of telomerase have advanced to early stage clinical trials, neither approach has yet demonstrated clinical efficacy, raising questions over their failure to translate (40). Several studies are attempting to specifically target cancer cells harbouring TERT promoter mutations, for example, by suppressing GABPβ1L-driven transcription at these de novo ETS binding sites (41). It is worth noting that TERT mutations had little effect on patients’ survival in our study, which may be associated with small samples and poor prognosis of SHC.
We also found a considerable number of patients carrying mutations in genes involved in the TP53 pathway (96%) and DDR pathway (28%). Alterations in DDR genes are associated with genomic instability and increased somatic TMB, which may enhance immunogenicity through increased tumor-specific neoantigen load (42). ARID1A, encoding a subunit of the SWI/SNF chromatin-remodeling complex, is the most frequently mutated epigenetic regulator in cancers (43). In the endometrial epithelium, the p53 pathway is activated following ARID1A loss, and ARID1A normally directly represses p53 pathway genes, so ARID1A and TP53 mutations are typically mutually exclusive (44). However, in our study, co-mutations of TP53-ARID1A existed in one patient, which may lead to invasive phenotypes including sarcomatoid component (44).
Combination therapy with immune checkpoint inhibitors (ICIs) and an antivascular endothelial growth factor antibody has shown remarkable effects for advanced HCCs in several clinical trials. Unfortunately, SHC patients have often been excluded from key clinical trials with ICIs (45). Luckily, a case report showed a patient with advanced PSHC achieved a complete response to nivolumab after 2 cycles of treatment and the duration of complete remission was longer than 8 months (46). In a large retrospective study of hepatocellular carcinoma(HCC), comprehensive genomic profiling (CGP) of cancer-related genes was performed on 755 consecutive cases of HCC using NGS, finding that the median TMB for the entire cohort was 4 mutations/Mb, with 95% of cases having a TMB of < 10 mutations/Mb (47). Interestingly, the median of TMB was 6.9 mutations/MB in our study, suggesting that SHC may be more likely to have a higher TMB than conventional HCC. Of note, KRAS mutant tumor showed prominently increased TMB and remarkable clinical benefit to PD-1 inhibitors in TP53 or KRAS mutant patients, especially those with co-occurring TP53/KRAS mutations in cohorts of lung adenocarcinoma immunotherapeutic patients (48). In addition, we observed mutations in EGFR or FAT1 have significantly higher TMB. Three EGFR mutations in our study were rare mutations, in contrast to previous studies that reported lower TMB in EGFR-mutant lung cancer compared with EGFR wild-type (49), we found TMB in EGFR-mutant patients was higher in our study, which might be associated with mutation type and the signature of SHC. FAT1 is a Drosophila tumor suppressor, which has important functions in regulating the Wnt pathway and the Hippo pathway (50). Mutations in FAT1 are associated with a higher TMB and lower multiple lymphocyte infiltration (51), which is consistent with our results. Compared with wild-type, patients with FAT1 mutations could have higher durable clinical benefits during immunotherapy (20). Consequently, such patients might have chance to benefit from immune checkpoint blockade.
Previous studies have reported that the median OS after surgery is about 8 months and the 1-year survival rate is only 22% (1, 8). In our study, the median OS of patients was only 4 months, and the 1-year survival rate was 10%. The prognosis of patients was worse than reported before, which may be related to the high proportion(89.3%) of stage III~IV patients. Survival analysis found that age, smoking, drinking and tumor diameter, may be independent prognostic predictors of SHC. However, the link between genetic alterations and prognosis was not statistically significant. Fortunately, we described some potentially actionable mutations in SHC, including NTRK1 fusions(n=1) and BRCA1/2 mutations(n=2), which may provide more treatment options in SHC. BRCA1 and BRCA2 are key regulators of DNA maintenance through homologous recombination (HR) (52). Additionally, they function in DNA crosslink repair as part of the Fanconi anemia (FA) complex and play important roles in the protection of stalled replication forks, transcription regulation, chromatin modulation, cell cycle regulation, checkpoint enforcement and telomere maintenance (52, 53). Meanwhile, DNA repair defects due to BRCA1/2 mutation instigate immune signaling through the cGAS/STING pathway, and the inflammatory signaling provides both tumor-suppressive as well as tumor-promoting traits (52, 54). Mutations in BRCA1/2 confer high-penetrance susceptibility to breast and ovarian cancers, increasing the risk of developing breast cancer by 49–57% and ovarian cancer by 18–40% (55). Poly-adenosine diphosphate ribose polymerase (PARP) inhibitors (PARPi) are effective against tumors with an impaired ability to repair double-strand DNA breaks, and several FDA-approved PARPi are available for treatment of BRCA1/2 carriers with tumors originating at various sites including breast, ovaries, pancreas and prostate (53, 56). However, PARPis used in the clinic remain vulnerable to acquired drug resistance currently, many ongoing clinical trials will evaluate the combination therapy of PARPi and other treatments in breast cancers (56). NTRK fusions, encoding TRK fusion proteins, are oncogenic drivers of a wide variety of adult and paediatric tumors. NTRK gene fusions occur at a low frequency (<1%) in common solid tumors but tend to be high in rare cancers (such as infantile fibrosarcoma and mammary secretory cancer) (57). Luckily, the solid tumors with NTRK fusion can be treated with targeted therapies, such as larotrectinib and entrectinib, the first two TRK inhibitors approved in the United States (58). Many clinical trials have shown good therapeutic results and safety of TRK inhibitors (59, 60). Thus we should encourage broader screening for these fusions in patients with rare tumors as they may benefit from TRK inhibitors, though NTRK fusions are rare.
There are several limitations to our study. First, we did not detect the gene mutation of non-sarcomatoid hepatocellular carcinoma, therefore we could not obtain more specific gene information on SHC through comparison. Second, the cohort size was small, resulting in limited statistical significance between genetic alterations and prognosis.
In conclusion, our study showed a landscape of gene mutations in SHC. TP53, TERT, and KRAS were the top three most frequently mutated genes. Meanwhile, SHC had high mutation rates in the TP53 pathway and DDR pathway. Multiple potentially actionable mutations, such as NTRK1 fusions and BRCA1/2 mutations, might provide additional therapeutic options. More samples for genetic variants analysis are still needed for further investigation.
The data presented in the study are deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human, accession number: HRA003771.
The studies involving human participants were reviewed and approved by The Review Broad of the First Affiliated Hospital of Zhengzhou University. The patients/participants provided their written informed consent to participate in this study.
YQ and GJ conceptualized this work. YQ and GJ supervised the study. BJ, PX, JD, WF, WW, and EL acquisition of data, and BJ, PX, and JD performed the statistical analysis and interpreted data. BJ, PX, and JD prepared the manuscript. YQ, GJ, BJ, PX, JD, WF, WW, and EL revised the manuscript. All authors approved the protocol. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Key Research and Development Project (No. 222102310069), the Key Research Project of Colleges and Universities (No. 20A320053) of Henan, grants from National Natural Science Foundation of China (No. 82172941) and Henan Province Medical Science and Technology Research Project (No. 212102310134).
We express our gratitude to Dr. Feng Zhang for her invaluable help with data collection for this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1086908/full#supplementary-material
1. Liao SH, Su TH, Jeng YM, Liang PC, Chen DS, Chen CH, et al. Clinical manifestations and outcomes of patients with sarcomatoid hepatocellular carcinoma. Hepatology (2019) 69(1):209–21. doi: 10.1002/hep.30162
2. Wu L, Tsilimigras DI, Farooq A, Hyer JM, Merath K, Paredes AZ, et al. Management and outcomes among patients with sarcomatoid hepatocellular carcinoma: A population-based analysis. Cancer (2019) 125(21):3767–75. doi: 10.1002/cncr.32396
3. Koo HR, Park MS, Kim MJ, Lim JS, Yu JS, Jin H, et al. Radiological and clinical features of sarcomatoid hepatocellular carcinoma in 11 cases. J Comput Assist Tomogr (2008) 32(5):745–9. doi: 10.1097/RCT.0b013e3181591ccd
4. Dahm HH. Immunohistochemical evaluation of a sarcomatoid hepatocellular carcinoma with osteoclastlike giant cells. Diagn Pathol (2015) 10:40. doi: 10.1186/s13000-015-0274-4
5. Yoshida N, Midorikawa Y, Kajiwara T, Yoshida N, Nakayama H, Sugitani M, et al. Hepatocellular carcinoma with sarcomatoid change without anticancer therapies. Case Rep Gastroenterol (2013) 7(1):169–74. doi: 10.1159/000350558
6. Wang QB, Cui BK, Weng JM, Wu QL, Qiu JL, Lin XJ. Clinicopathological characteristics and outcome of primary sarcomatoid carcinoma and carcinosarcoma of the liver. J Gastrointest Surg (2012) 16(9):1715–26. doi: 10.1007/s11605-012-1946-y
7. Kan A, Guo RP. The prognosis of subsequent surgical treatment in patients with sarcomatoid carcinoma in the liver: A retrospective study. Int J Surg (2018) 55:145–51. doi: 10.1016/j.ijsu.2018.05.736
8. Li Z, Wu X, Bi X, Zhang Y, Huang Z, Lu H, et al. Clinicopathological features and surgical outcomes of four rare subtypes of primary liver carcinoma. Chin J Cancer Res (2018) 30(3):364–72. doi: 10.21147/j.issn.1000-9604.2018.03.08
9. Lin CC, Chen CL. Living donor liver transplantation for hepatocellular carcinoma achieves better outcomes. Hepatobiliary Surg Nutr (2016) 5(5):415–21. doi: 10.21037/hbsn.2016.08.02
10. Zhang C, Feng S, Tu Z, Sun J, Rui T, Zhang X, et al. Sarcomatoid hepatocellular carcinoma: From clinical features to cancer genome. Cancer Med (2021) 10(18):6227–38. doi: 10.1002/cam4.4162
11. Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating novel resistance mechanisms to third-generation egfr tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res (2018) 24(13):3097–107. doi: 10.1158/1078-0432.Ccr-17-2310
12. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
13. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
14. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
15. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From fastq data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinf (2013) 43(1110):11.0.1–.0.33. doi: 10.1002/0471250953.bi1110s43
16. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res (2012) 22(3):568–76. doi: 10.1101/gr.129684.111
17. Wang K, Li M, Hakonarson H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) 38(16):e164. doi: 10.1093/nar/gkq603
18. Amarasinghe KC, Li J, Hunter SM, Ryland GL, Cowin PA, Campbell IG, et al. Inferring copy number and genotype in tumour exome data. BMC Genomics (2014) 15(1):732. doi: 10.1186/1471-2164-15-732
19. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(1):34. doi: 10.1186/s13073-017-0424-2
20. Fang W, Ma Y, Yin JC, Hong S, Zhou H, Wang A, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-Pd-(L)1 therapies in non-small cell lung cancer. Clin Cancer Res (2019) 25(16):5015–26. doi: 10.1158/1078-0432.Ccr-19-0585
21. Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with alk-positive non-Small-Cell lung cancer: Results from a global phase 2 study. Lancet Oncol (2018) 19(12):1654–67. doi: 10.1016/S1470-2045(18)30649-1
22. Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic kras in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer (2021) 20(1):143. doi: 10.1186/s12943-021-01441-4
23. Ding Y, Shao Y, Na C, Yin JC, Hua H, Tao R, et al. Genetic characterisation of sarcomatoid carcinomas reveals multiple novel actionable mutations and identifies kras mutation as a biomarker of poor prognosis. J Med Genet (2022) 59(1):10–7. doi: 10.1136/jmedgenet-2020-107083
24. Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouze E, Blanc JF, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol (2017) 67(4):727–38. doi: 10.1016/j.jhep.2017.05.014
25. Rojo de la Vega M, Chapman E, Zhang DD. Nrf2 and the hallmarks of cancer. Cancer Cell (2018) 34(1):21–43. doi: 10.1016/j.ccell.2018.03.022
26. Zhu G, Ren D, Lei X, Shi R, Zhu S, Zhou N, et al. Mutations associated with no durable clinical benefit to immune checkpoint blockade in non-S-Cell lung cancer. Cancers (Basel) (2021) 13(6):1397. doi: 10.3390/cancers13061397
27. Bowman L, Tiu R, Smyth EN, Willard MD, Li L, Beyrer J, et al. Clinical characteristics, treatments, and concurrent mutations in non-small cell lung cancer patients with Nf1 mutations. Clin Lung Cancer (2021) 22(1):32–41 e1. doi: 10.1016/j.cllc.2020.09.011
28. Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The Nf1 somatic mutational landscape in sporadic human cancers. Hum Genomics (2017) 11(1):13. doi: 10.1186/s40246-017-0109-3
29. Timar J, Kashofer K. Molecular epidemiology and diagnostics of kras mutations in human cancer. Cancer Metastasis Rev (2020) 39(4):1029–38. doi: 10.1007/s10555-020-09915-5
30. Fu Y, Cruz-Monserrate Z, Helen Lin H, Chung Y, Ji B, Lin SM, et al. Ductal activation of oncogenic kras alone induces sarcomatoid phenotype. Sci Rep (2015) 5:13347. doi: 10.1038/srep13347
31. Liu P, Wang Y, Li X. Targeting the untargetable kras in cancer therapy. Acta Pharm Sin B (2019) 9(5):871–9. doi: 10.1016/j.apsb.2019.03.002
32. Wang S, Li Q, Ma P, Fang Y, Yu Y, Jiang N, et al. Kras mutation in rare tumors: A landscape analysis of 3453 Chinese patients. Front Mol Biosci (2022) 9:831382. doi: 10.3389/fmolb.2022.831382
33. Huang L, Guo Z, Wang F, Fu L. Kras mutation: From undruggable to druggable in cancer. Signal Transduct Target Ther (2021) 6(1):386. doi: 10.1038/s41392-021-00780-4
35. Wang H, Guo M, Wei H, Chen Y. Targeting mcl-1 in cancer: Current status and perspectives. J Hematol Oncol (2021) 14(1):67. doi: 10.1186/s13045-021-01079-1
36. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. Myc and Mcl1 cooperatively promote chemotherapy-resistant breast cancer stem cells Via regulation of mitochondrial oxidative phosphorylation. Cell Metab (2017) 26(4):633–47 e7. doi: 10.1016/j.cmet.2017.09.009
37. Dratwa M, Wysoczańska B, Łacina P, Kubik T, Bogunia-Kubik K. Tert-regulation and roles in cancer formation. Front Immunol (2020) 11:589929. doi: 10.3389/fimmu.2020.589929
38. Tallet A, Nault JC, Renier A, Hysi I, Galateau-Salle F, Cazes A, et al. Overexpression and promoter mutation of the tert gene in malignant pleural mesothelioma. Oncogene (2014) 33(28):3748–52. doi: 10.1038/onc.2013.351
39. Wang X, Lopez-Beltran A, Osunkoya AO, Wang M, Zhang S, Davidson DD, et al. Tert promoter mutation status in sarcomatoid urothelial carcinomas of the upper urinary tract. Future Oncol (2017) 13(8):705–14. doi: 10.2217/fon-2016-0414
40. Guterres AN, Villanueva J. Targeting telomerase for cancer therapy. Oncogene (2020) 39(36):5811–24. doi: 10.1038/s41388-020-01405-w
41. Mancini A, Xavier-Magalhaes A, Woods WS, Nguyen KT, Amen AM, Hayes JL, et al. Disruption of the Beta1l isoform of gabp reverses glioblastoma replicative immortality in a tert promoter mutation-dependent manner. Cancer Cell (2018) 34(3):513–28 e8. doi: 10.1016/j.ccell.2018.08.003
42. Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA damage response and repair (Ddr) gene mutations on efficacy of pd-(L)1 immune checkpoint inhibition in non-small cell lung cancer. Clin Cancer Res (2020) 26(15):4135–42. doi: 10.1158/1078-0432.CCR-19-3529
43. Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, et al. Arid1a-mutated ovarian cancers depend on Hdac6 activity. Nat Cell Biol (2017) 19(8):962–73. doi: 10.1038/ncb3582
44. Reske JJ, Wilson MR, Holladay J, Siwicki RA, Skalski H, Harkins S, et al. Co-Existing Tp53 and Arid1a mutations promote aggressive endometrial tumorigenesis. PloS Genet (2021) 17(12):e1009986. doi: 10.1371/journal.pgen.1009986
45. Morisue R, Kojima M, Suzuki T, Nakatsura T, Ojima H, Watanabe R, et al. Sarcomatoid hepatocellular carcinoma is distinct from ordinary hepatocellular carcinoma: Clinicopathologic, transcriptomic and immunologic analyses. Int J Cancer (2021) 149(3):546–60. doi: 10.1002/ijc.33545
46. Zhu SG, Li HB, Yuan ZN, Liu W, Yang Q, Cheng Y, et al. Achievement of complete response to nivolumab in a patient with advanced sarcomatoid hepatocellular carcinoma: A case report. World J Gastrointest Oncol (2020) 12(10):1209–15. doi: 10.4251/wjgo.v12.i10.1209
47. Ang C, Klempner SJ, Ali SM, Madison R, Ross JS, Severson EA, et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget (2019) 10(40):4018–25. doi: 10.18632/oncotarget.26998
48. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of Tp53 and kras mutation status for response to pd-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res (2017) 23(12):3012–24. doi: 10.1158/1078-0432.CCR-16-2554
49. Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor mutation burden and efficacy of egfr-tyrosine kinase inhibitors in patients with egfr-mutant lung cancers. Clin Cancer Res (2019) 25(3):1063–9. doi: 10.1158/1078-0432.CCR-18-1102
50. Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, et al. Recurrent somatic mutation of Fat1 in multiple human cancers leads to aberrant wnt activation. Nat Genet (2013) 45(3):253–61. doi: 10.1038/ng.2538
51. Martin D, Degese MS, Vitale-Cross L, Iglesias-Bartolome R, Valera JLC, Wang Z, et al. Assembly and activation of the hippo signalome by Fat1 tumor suppressor. Nat Commun (2018) 9(1):2372. doi: 10.1038/s41467-018-04590-1
52. van Vugt M, Parkes EE. When breaks get hot: Inflammatory signaling in Brca1/2-mutant cancers. Trends Cancer (2022) 8(3):174–89. doi: 10.1016/j.trecan.2021.12.003
53. Patel PS, Algouneh A, Hakem R. Exploiting synthetic lethality to target Brca1/2-deficient tumors: Where we stand. Oncogene (2021) 40(17):3001–14. doi: 10.1038/s41388-021-01744-2
54. Groelly FJ, Porru M, Zimmer J, Benainous H, De Visser Y, Kosova AA, et al. Anti-tumoural activity of the G-quadruplex ligand pyridostatin against Brca1/2-deficient tumours. EMBO Mol Med (2022) 14(3):e14501. doi: 10.15252/emmm.202114501
55. Chen S, Parmigiani G. Meta-analysis of Brca1 and Brca2 penetrance. J Clin Oncol (2007) 25(11):1329–33. doi: 10.1200/jco.2006.09.1066
56. Menezes MCS, Raheem F, Mina L, Ernst B, Batalini F. Parp inhibitors for breast cancer: Germline Brca1/2 and beyond. Cancers (Basel) (2022) 14(17):4332. doi: 10.3390/cancers14174332
57. Cocco E, Scaltriti M, Drilon A. Ntrk fusion-positive cancers and trk inhibitor therapy. Nat Rev Clin Oncol (2018) 15(12):731–47. doi: 10.1038/s41571-018-0113-0
58. Rolfo C. Ntrk gene fusions: A rough diamond ready to sparkle. Lancet Oncol (2020) 21(4):472–4. doi: 10.1016/s1470-2045(20)30026-7
59. Haratake N, Seto T. Ntrk fusion-positive non-Small-Cell lung cancer: The diagnosis and targeted therapy. Clin Lung Cancer (2021) 22(1):1–5. doi: 10.1016/j.cllc.2020.10.013
Keywords: genetic testing, prognosis, gene mutation, hepatic tumors, sarcomatoid carcinoma
Citation: Jia B, Xia P, Dong J, Feng W, Wang W, Liu E, Jiang G and Qin Y (2023) Genetic testing and prognosis of sarcomatoid hepatocellular carcinoma patients. Front. Oncol. 12:1086908. doi: 10.3389/fonc.2022.1086908
Received: 01 November 2022; Accepted: 20 December 2022;
Published: 17 January 2023.
Edited by:
Shamini Selvarajah, University Health Network (UHN), CanadaReviewed by:
Yang Xu, Geneseeq Technology Inc., CanadaCopyright © 2023 Jia, Xia, Dong, Feng, Wang, Liu, Jiang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guozhong Jiang, Z3VvemhvbmdqaWFuZ0B6enUuZWR1LmNu; Yanru Qin, eWFucnVxaW5AMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.