
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 09 January 2023
Sec. Pediatric Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1082525
This article is part of the Research TopicMolecular Diagnostics of Pediatric Cancer, volume IIView all 10 articles
 Xueliang Wang1
Xueliang Wang1 Decheng Deng1Yaping Yan1Mansi Cai1Xiaodan Liu2Ailing Luo1Shanshan Liu1Xiaohong Zhang1
Decheng Deng1Yaping Yan1Mansi Cai1Xiaodan Liu2Ailing Luo1Shanshan Liu1Xiaohong Zhang1 Hua Jiang1
Hua Jiang1 Xiaoping Liu1*
Xiaoping Liu1*Objective: To explore the functions of the polymorphisms in 5-methylcytosine (m5C) modification-related coding genes on the susceptibility of pediatric acute lymphoblastic leukemia (ALL).
Methods: Case–control study and multinomial logistic regression analysis were performed to construct models to evaluate the susceptibility of pediatric ALL. The relationship between five functional SNPs in m5C modification-coding genes and pediatric ALL risk was analyzed. Genotyping of 808 cases and 1,340 healthy samples from South China was identified using a TaqMan assay; odds ratios (ORs) and 95% confidence intervals (CIs) were calculated to estimate the relationship between the five selected SNPs and pediatric ALL susceptibility.
Results: Among the five analyzed SNPs, NOL1 rs3764909 and NSUN4 rs10252 variants significantly increased the susceptibility of pediatric ALL, while NSUN3 rs7653521, NSUN5 rs1880948, and NSUN6 rs3740102 variants were not associated with the risk of ALL. Stratification analyses demonstrated that NOL1 rs3764909 C>A exhibited a significant association with increased pediatric ALL risk in subgroups of common B ALL, pre-B ALL, T-cell ALL, low and middle risk, other gene fusion types, non-gene fusion, hypodiploid, normal diploid, primitive lymphocytes in marrow < 5% on week 12, and minimal residual disease (MRD) <0.01% on week 12 after induced therapy; NSUN4 rs10252 G>A was related to increased risk of ALL children in subgroups of age ≥ 120 months, normal white blood cell (WBC) number, middle risk, non-gene fusion, MRD ≥ 0.01 on days 15–19, and primitive lymphocytes in marrow < 5% on day 33 after induced therapy. Compared with the reference haplotype CAGTA, children who harbored haplotypes CCGTG and ACATA were remarkably related to increased ALL susceptibility. rs3764909 and rs10252 varieties of alleles were not associated with MRD levels after the selected chemotherapeutics.
Conclusions: In conclusion, NOL1 rs3764909 and NSUN4 rs10252 variants were enhanced by pediatric ALL risk and were suggested to be potential biomarkers for pediatric ALL.
Acute lymphoblastic leukemia (ALL) is the most common malignant tumor in children and adolescents (1–3). Despite the heterogeneity, the clinical cure rate of childhood ALL is over 85%. Combination chemotherapy and allogeneic hematopoietic cell transplantation is the main treatment of ALL. In order to obtain the best treatment effect, different chemotherapy regimens are formulated according to the risk of recurrence. Even though these efforts were made, there are still a significant proportion of children with ALL having a high risk of relapse (4). Several genetic factors are verified to enhance the risk of pediatric ALL, but a certain percentage of children were not recognized to inherit risk genetic factors (5). Plenty of studies have discovered polymorphic variants in genes that are connected with an elevated risk of ALL (6–8).
In recent years, abundant studies have revealed that epigenetic regulation participates in the initiation and procession of tumors. The role and regulatory mechanism of RNA methylation in tumors have attracted the close attention of researchers. RNA methylation refers to the chemical modification of RNA methyladenine by the selective addition of methyl groups under the catalysis of methyltransferase. Common RNA methylations include several sites (9). A number of m6A-methylated genes take part in the carcinogenesis of leukemia. m6A methyltransferase METTL14 was demonstrated to promote leukemogenesis via mRNA m6A modification (10). m6A demethylase FTO attenuates aerobic glycolysis and accelerates leukemia. Our previous studies identified that genetic variants in m6A methyltransferase METTL3 and METTL14 were associated with the increased risk of pediatric ALL (11, 12).
As we know, N6-methyladenosine is the most common modification of RNA methylation, and 5-methylcytosine (m5C) is another common and conserved modification in RNA, including mRNAs and non-coding RNAs. m5C regulates RNA stability assembly and translation as well as m6A (13). The enzymes modulating m5C of RNAs can be functionally classified into writers, erasers, and readers. Methyltransferases (writers) can install m5C on RNA. The reported m5C writers include NSUN1–7 and DNMT2 (14). Ten-eleven translocation family proteins (TETs) can oxidize 5-methylcytosine to cytosine-5-hydroxymethylation (hm5C), so these are regarded as “erasers” for m5C (15). As for “reader”, only YBX1 and ALYREF have been identified as recognition proteins for m5C modification sites at present (16). NOL1/NOP2/SUN family is also documented as m5C methyltransferase to regulate RNA stability and functions (14). These RNA modifiers can regulate the expression of various oncogenes and promote tumorigenesis and development. In addition, methyltransferases are abnormally expressed in a variety of tumors and have been used to predict the prognosis of patients. There is only one available study on the epidemiological assessment of single-nucleotide polymorphisms (SNPs) in the m5C modification core gene. Chen and Cao et al. performed a case–control study and verified that m5C modification genes were related to survival and chemotherapy efficacy of colorectal cancer. Two SNPs of YBX1 gene, rs10890208 and rs3862218, may predict a reduction by using the Cox regression model to analyze the association between 13 candidate SNPs of m5C modifier gene and overall survival (OS) of colorectal cancer (CRC) after chemotherapy (17). However, the role of SNPs in the m5C methyltransferase gene in ALL risk has not been reported. Because of the evidence that cells regulated by the m5C methyltransferase gene promote tumorigenesis, we conducted a case–control study to explore the association of genetic variations in m5C modification-coding genes with the risk to pediatric ALL in China.
A total of 808 pediatric ALL cases and 1,340 age-matched, gender-matched, and ethnicity-matched control samples from South China were enrolled from January 2017 to May 2019 in this study, as summarized in our previous studies (11, 12). All children were diagnosed with ALL by at least two hematologists. The control samples were free from hematological diseases, malignancy, or any type of autoimmune disorder.
The potentially functional SNPs in five m5C methyltransferases were selected as previously described, and the protocol was as follows; the National Center for Biotechnology Information (NCBI) dbSNP database and SNP info (https://snpinfo.niehs.nih.gov/) were used. The selected SNPs should fulfill the following criteria: (1) the minor allele frequency (MAF) was >5% of Chinese Han subjects in HapMap and (2) located in the exon, 5′ untranslated regions (5′ UTR), and 3′ UTR of genes, which were predicted to be potential functional; (3) each SNP should be in low linkage disequilibrium (R2 < 0.8). Five SNPs were selected (NOL1 rs3764909, NSUN3 rs7653521, NSUN4 rs10252, NSUN5 rs1880948, and NSUN6 rs3740102). rs3764909 is located in the exonic region of NOL1 and might be a transcriptional factor binding site. rs7653521 is located in the exonic region of NSUN3 and was predicted to have the potential to bind transcriptional factors. rs10252 is located in the exon of NSUN4 and was predicted as a miRNA binding site. rs1880948 is located upstream of NSUN5 transcriptional start site and may be a transcriptional factor binding site. rs3740102 is located in an exon of NSUN6 and was predicted to be a transcriptional factor binding site.
Peripheral blood genomic DNA was extracted using the QIAamp DNA blood mini kit (QIAGEN, Valencia, CA, USA). For genotyping, assay probes were purchased from Thermo Fisher (Waltham, MA, USA; TaqMan SNP Assays, 4351379). The detailed information on these assays is presented in Table S1. The genotype was identified by TaqMan PCR on an ABI 7900 (Applied Biosystems, Foster City, CA, USA). The conditions of reactions were described previously (11). To ensure the accuracy of these genotyping results, 10% of the samples were randomly selected to be genotyped by a DNA sequencing method. A concordance rate of 100% for the quality control samples was obtained (11).
The compliance of genotypes with the Hardy–Weinberg equilibrium (HWE) in the control group and differences in clinical characteristics between ALL children and healthy children were evaluated using the χ2 test. The age- and gender-adjusted odds ratios (ORs) and 95% confidence intervals (CIs) for the association between the SNPs and ALL susceptibility were calculated by multivariate logistic regression analysis. All these analyses were performed using the software SAS v10.0 (SAS Institute, Cary, NC, USA).
Five m5C modification core gene SNPs (NOL1 rs3764909 C>A, NSUN3 rs7653521 T>C, NSUN4 rs10252 G>A, NSUN5 rs1880948 G>A, and NSUN6 rs3740102 C>A) were genotyped in 808 pediatric ALL samples and 1,340 age- and gender-matched healthy controls. The five SNPs comply with the HWE in control populations. Single-locus analysis was used to analyze the relationship between the five SNPs and pediatric ALL risk. The NOL1 rs3764909 (AC/CC versus AA: adjusted OR = 1.684, 95% CI = 1.408–2.014, p < 0.001) and NSUN4 rs10252 (adjusted OR = 1.140, 95% CI = 1.001–1.298, p = 0.049) variant alleles were associated with an increased risk of ALL. However, there was no association between the remaining polymorphisms, NSUN3 rs7653521 (OR = 1.067, 95% CI = 0.946–1.203, p = 0.291), NSUN5 rs1880948 (OR = 0.980, 95% CI = 0.862–1.114, p = 0.759), NSUN6 rs3740102 (OR = 0.959, 95% CI = 0.825–1.114, p = 0.581), and pediatric ALL risk (Table 1).
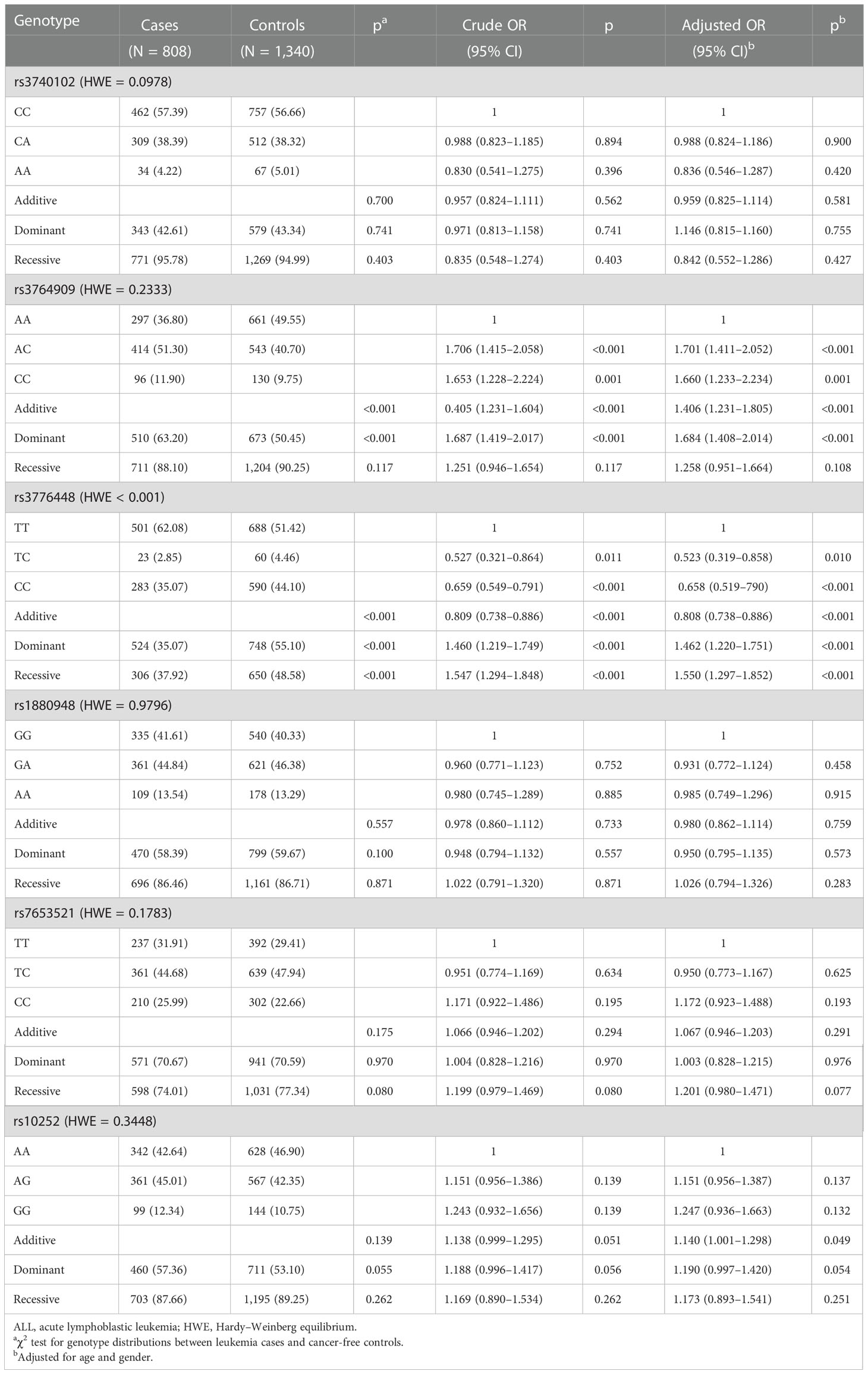
Table 1 Logistic regression analysis of associations between m5C modification key gene polymorphisms and ALL susceptibility.
SNPs rs3764909 C>A and rs10252 T>C with statistically significant differences were stratified according to age, gender, white blood cell (WBC), immunophenotype, gene infusion, karyotype, primitive lymphocytes in the marrow, minimal residual disease (MRD), and relapse (Table 2). NOL1 rs3764909 AC/CC increased ALL risk in children aged <120 months (adjusted OR = 1.592, 95% CI = 1.319–1.923, p < 0.001), children aged ≥120 months (adjusted OR = 2.759, 95% CI = 1.568–4.855, p < 0.001), female (adjusted OR = 1.785, 95% CI = 1.408–2.265, p < 0.001), male (adjusted OR = 1.860, 95% CI = 1.381–2.505, p < 0.001), number of WBC > 10 × 107 (adjusted OR = 1.811, 95% CI = 1.417–2.315, p < 0.001), normal WBC number (adjusted OR = 1.668, 95% CI = 1.322–2.106, p < 0.001), common B ALL (adjusted OR = 3.323, 95% CI = 2.495–4.426, p < 0.001), pre-B ALL (adjusted OR = 1.566, 95% CI = 1.125–2.181, p = 0.008), T-ALL (adjusted OR = 1.890 95% CI = 1.132–3.155, p = 0.015), low-risk ALL (adjusted OR = 1.705, 95% CI = 1.278–2.276, p < 0.001), middle-risk ALL (adjusted OR = 2.074, 95% CI = 1.630–2.640, p < 0.001), other gene fusion types (adjusted OR = 4.806, 95% CI = 1.623–14.242, p = 0.005), non-gene fusion (adjusted OR = 4.524, 95% CI = 2.896–7.065, p < 0.001), hypodiploid (adjusted OR = 3.330, 95% CI = 1.327–8.539, p = 0.010), normal diploid (OR = 3.403, 95% CI = 2.364–4.898, p < 0.001), primitive/naïve lymphocytes in marrow ≥ 5% on days 15–19 (adjusted OR = 1.718, 95% CI = 1.395–2.116, p < 0.001) and day 33 (adjusted OR = 1.466, 95% CI = 1.185–1.813, p < 0.001), and <5% (adjusted OR = 1.623, 95% CI = 1.247–2.112, p < 0.001) on days 15–19, day 33 (adjusted OR = 2.082, 95% CI = 1.603–2.688, p < 0.001), and week 12 (adjusted OR = 2.704, 95% CI = 2.153–3.397, p < 0.001) after induced therapy with MRD ≥ 0.01% on days 15–19 (adjusted OR = 1.952, 95% CI = 1.537–2.479, p < 0.001) and day 33 (adjusted OR = 1.729, 95% CI = 1.144–2.613, p = 0.009), with MRD < 0.01% on days 15–19 (adjusted OR = 1.486, 95% CI = 1.190–1.854, p < 0.001), day 33 (adjusted OR = 1.677, 95% CI = 1.391–2.023, p < 0.001), and week 12 (adjusted OR = 1.710, 95% CI = 1.428–3.397, p < 0.001) after induced chemotherapy, with relapse (adjusted OR = 2.332, 95% CI = 1.058–5.138, p = 0.036) and no relapse (adjusted OR = 1.262, 95% CI = 1.034–1.541, p = 0.022).
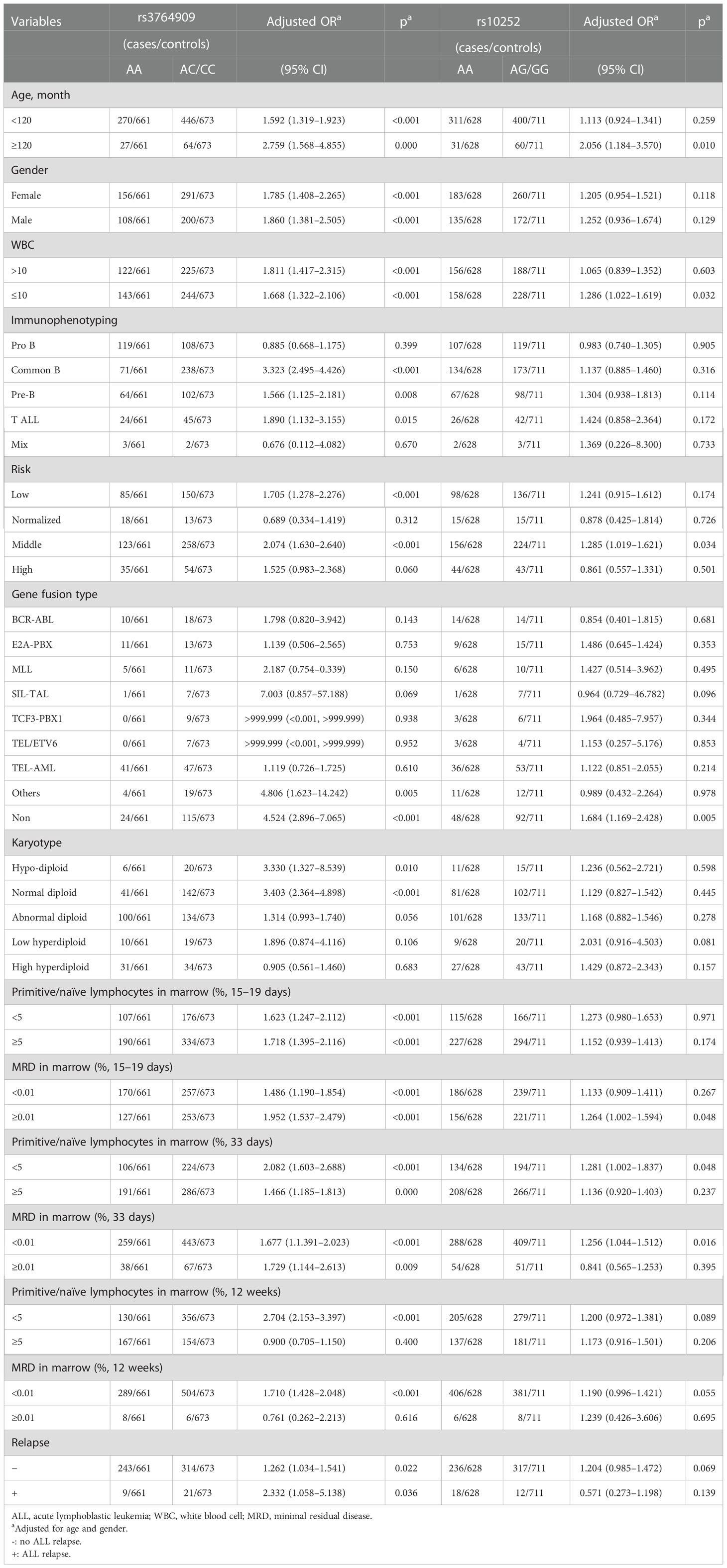
Table 2 Stratification analysis of m5C-related gene polymorphisms with ALL susceptibility.
NSUN4 rs10252 AG/GG also increased ALL risk in children of age ≥ 120 months (adjusted OR = 2.056, 95% CI = 1.184–3.570, p = 0.010), normal WBC number (adjusted OR = 1.286, 95% CI = 1.022–1.619, p = 0.032), middle risk (adjusted OR = 1.285, 95% CI = 1.019–1.621, p = 0.034), non-gene fusion (adjusted OR = 1.684, 95% CI = 1.169–2.428, p = 0.005), primitive/naïve lymphocyte <5% on days 15–19 (adjusted OR = 1.281, 95% CI = 1.002–1.837, p = 0.048), MRD ≥ 0.01% on days 15–19 (adjusted OR = 1.264 95% CI = 1.002–1.594 p = 0.048), and MRD < 0.01% (adjusted OR = 1.256, 95% CI = 1.044–1.512, p = 0.016) after induced chemotherapy.
Furtherly, whether the haplotypes of NOL1 rs3764909, NSUN3 rs7653521, NSUN4 rs10252, NSUN5 rs1880948, and NSUN6 rs3740102 are linked to pediatric ALL susceptibility were evaluated. The wild-type allele CAGTA was considered as the reference group The results showed that children with haplotypes CCGTG (adjusted OR = 2.035, 95% CI = 1.095–3.782, p = 0.025) and ACATA (adjusted OR = 3.169, 95% CI = 1.455–6.899, p = 0.004) would have enhanced ALL susceptibility (Table 3).
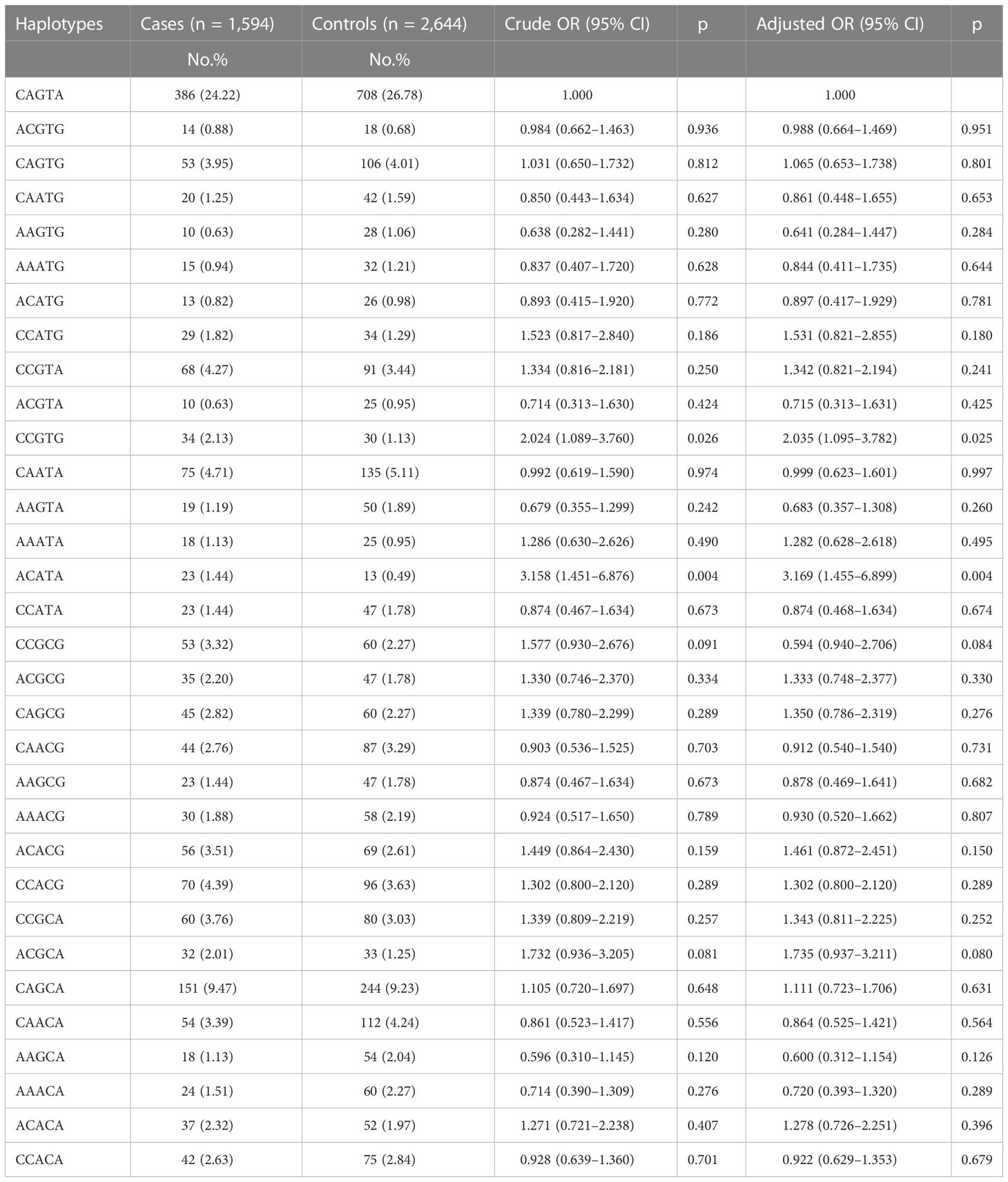
Table 3 Association between inferred haplotypes of the m5C-related genes and pediatric ALL risk.
The MRD in the marrow of pediatric ALL samples with different NOL1 rs3764909 and NSUN4 rs10252 alleles after treatment with Chinese Children Cancer Group chemotherapeutics (CCCGs) or South China Children Leukemia Group chemotherapeutics (SCCLGs) was detected. The differences between varieties of alleles were estimated. Unfortunately, we did not identify the association between rs3764909 or rs10252 varieties of alleles and the sensitivity to CCCG treatment or SCCLG treatment in ALL children (Table 4).
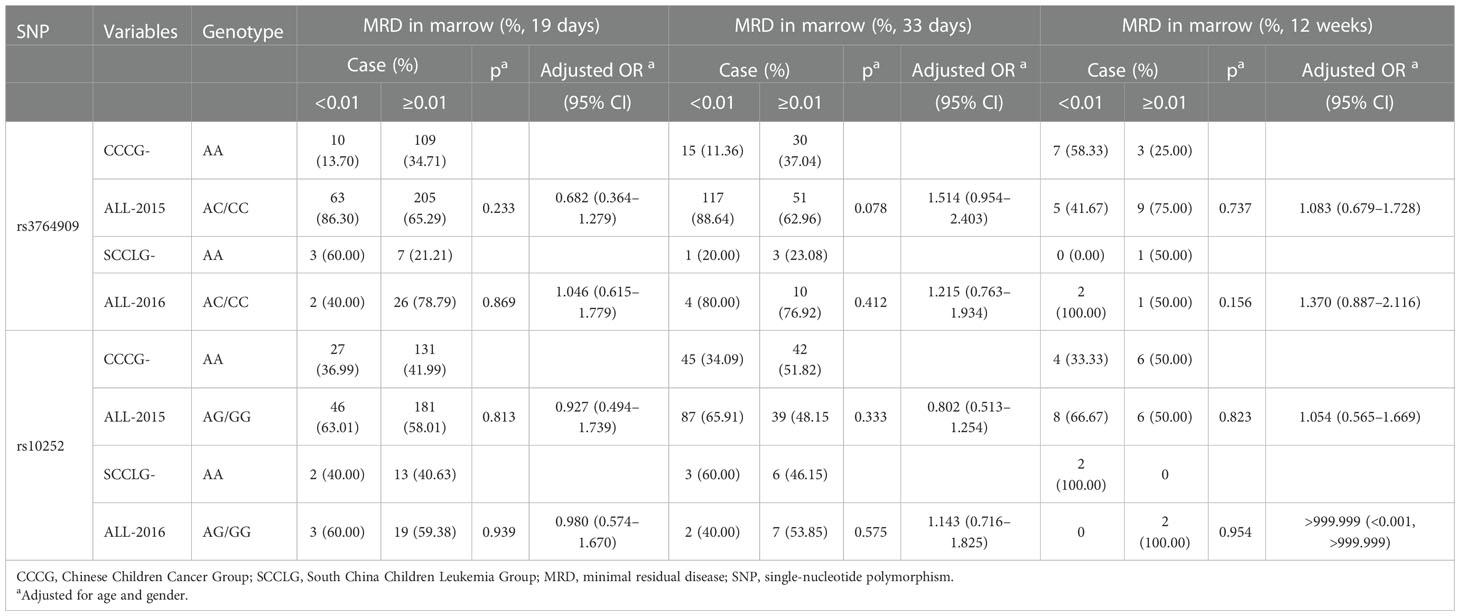
Table 4 The influence of m5C-related gene polymorphisms on sensitivity to different treatment strategies based on MRD levels.
In this case–control study, the possible relationship of m5C methyltransferase coding gene polymorphisms with pediatric ALL risk from a population in southern China was explored. The results discovered that two of the five selected SNPs, NOL1 rs3764909 G>A and NSUN4 rs10252 G>A, were associated with increased pediatric ALL, and the other m5C methyltransferase coding genes SNPs were not related to pediatric ALL risk. This is the first study on the association between m5C methyltransferase coding gene polymorphisms and pediatric ALL susceptibility.
In recent years, it has been reported that epigenetic changes, including DNA methylation, RNA methylation, histone modification, and non-coding RNAs, can promote the progression of ALL (18). Several lines of data have introduced m5C modification as an important regulator in post-transcription. In the study of carcinogenesis, m5C-modified genes have been reported to be associated with a variety of cancers, including bladder cancer (19), hepatocellular carcinoma (20), glioblastoma multiforme (21), and leukemia (22). However, limited pieces of evidence have focused on the function of polymorphisms of m5C genes on disease susceptibility. There is only one available study on the epidemiological assessment of SNPs in the m5C modification core gene and cancer. In July of this year, a case–control study was performed and revealed that two SNPs of YBX1 gene, rs10890208 and rs3862218, may predict a reduction by using the Cox regression model to analyze the association between 13 candidate SNPs of the m5C modifier gene and OS of CRC after chemotherapy (17).
Our study identified that NOL1 rs3764909 and NSUN4 rs10252 variants could contribute to the increased pediatric ALL risk. NOL1 and NSUN4 are important methyltransferases involved in m5C RNA modification (14). Similar to m6A methylation modification, m5C RNA modification can participate in other biological processes including cell growth, proliferation, apoptosis, and differentiation by affecting RNA translation, nuclear export, and stability (23). NOL1, one of the m5C methyltransferases, can generate m5C at C72 of tRNA (24, 25). Hong et al. demonstrated that NOL1 could bind to the T-cell factor binding element of the cyclin D1 gene promoter and enhance transcriptional expression (26). Interestingly, telomerase can also interact with NOL1 to affect the transcription of the cyclin D1 gene. NOL1 can promote tumor proliferation by activating cyclins (26). NOL1-E2A fusion was regarded as the pathogenesis of acute leukemia in a case report (27). NOL1 was also reported to promote hepatocellular carcinoma cell proliferation by TGF-β1/hPVT1/NOP2 pathway (28). NSUN4 is located at chr1 1p33, as the “writer” of m5C, involved in rRNA methylation. It can mediate mitochondrial protein synthesis by regulating the assembly processing and maturation of mt-ribosome (24, 29). It has been shown to be involved in tumor effects. NSUN4 promotes the malignant progression of hepatocellular carcinoma (30). NOL1 and NSUN4 polymorphisms may be involved in tumor risk-related biological functions by affecting m5C modification of coding and non-coding RNAs (24, 31).
In this study, we found that NOL1 rs3764909 C>A and NSUN4 rs10252 G>A were associated with increased ALL susceptibility. NOL1 has been found to be associated with the pathogenesis of leukemia (27), so NOL1 SNP variants may contribute to the development of ALL. NSUN4 can affect tumorigenesis by affecting mitochondrial protein synthesis (24). NSUN4 rs10252 variants may regulate mitochondrial protein synthesis in ALL cells and increase the risk of ALL.
However, there are some limitations to this study. On the one hand, we did not perform independent experimental studies to verify the relationship between selected SNPs and specific risk factors in children with leukemia. On the other hand, for rs3764909 and rs10252, deeply functional verifications are needed to explain the mechanisms of NOL1 and NSUN4 in ALL.
In conclusion, NOL1 rs3764909 and NSUN4 rs10252 variants are associated with increased ALL tumor susceptibility. The specific mechanisms by which NOL1 and NSUN4 polymorphisms are involved in pediatric ALL susceptibility require further study. Genetic variants in m5C modification coding genes were associated with enhanced pediatric ALL susceptibility and suggested that NOL1 and NSUN4 gene polymorphisms might be a potential liquid biopsy biomarker for pediatric ALL.
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Guangzhou Women and Children’s Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
XPL and HJ contributed to the conception and design of the study. XW and DD wrote the first draft of the manuscript. DD and MC extracted genomic DNA. XW, YY and SL conducted Taqman PCR. AL and XDL collected samples. XZ performed the statistical analysis. XPL revised the manuscript. All authors contributed to the manuscript revision and read and approved the submitted version.
Guangzhou Municipal Science and Technology Project (202201020603 and 202102010262) and Guangzhou Municipal Clinical Featured Technology Project (2019TS56).
We thank the Clinical Biological Resource Bank of Guangzhou Women and Children’s Medical Center for providing part of the clinical samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1082525/full#supplementary-material
1. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood (2015) 125:3977–87. doi: 10.1182/blood-2015-02-580043
2. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35:975–83. doi: 10.1200/JCO.2016.70.7836
3. Pui C, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33:2938–48. doi: 10.1200/JCO.2014.59.1636
4. Vrooman LM, Silverman LB. Treatment of childhood acute lymphoblastic leukemia: Prognostic factors and clinical advances. Curr Hematol Malig Rep (2016) 11:385–94. doi: 10.1007/s11899-016-0337-y
5. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
6. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet (2009) 41:1006–10. doi: 10.1038/ng.430
7. Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, et al. Inherited GATA3 variants are associated with ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet (2013) 45:1494–98. doi: 10.1038/ng.2803
8. Treviño LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet (2009) 41:1001–05. doi: 10.1038/ng.432
9. You C, Dai X, Wang Y. Position-dependent effects of regioisomeric methylated adenine and guanine ribonucleosides on translation. Nucleic Acids Res (2017) 45:9059–67. doi: 10.1093/nar/gkx515
10. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic Stem/Progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell (2018) 22:191–205. doi: 10.1016/j.stem.2017.11.016
11. Luo A, Yang L, Li M, Cai M, Huang A, Liu X, et al. Genetic variants in METTL14 are associated with the risk of acute lymphoblastic leukemia in southern Chinese children: A five-center case-control study. Cancer Manag Res (2021) 13:9189–200. doi: 10.2147/CMAR.S335925
12. Liu X, Huang L, Huang K, Yang L, Yang X, Luo A, et al. Novel associations between METTL3 gene polymorphisms and pediatric acute lymphoblastic leukemia: A five-center case-control study. Front Oncol (2021) 11:635251. doi: 10.3389/fonc.2021.635251
13. Gao Y, Fang J. RNA 5-methylcytosine modification and its emerging role as an epitranscriptomic mark. RNA Biol (2021) 18:117–27. doi: 10.1080/15476286.2021.1950993
14. Li M, Tao Z, Zhao Y, Li L, Zheng J, Li Z, et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J Transl Med (2022) 20:214. doi: 10.1186/s12967-022-03427-2
15. Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J Am Chem Soc (2014) 136:11582–85. doi: 10.1021/ja505305z
16. Yang X, Yang Y, Sun B, Chen Y, Xu J, Lai W, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res (2017) 27:606–25. doi: 10.1038/cr.2017.55
17. Chen S, Cao X, Ben S, Zhu L, Gu D, Wu Y, et al. Genetic variants in RNA m(5) c modification genes associated with survival and chemotherapy efficacy of colorectal cancer. Cancer Med (2022). doi: 10.1002/cam4.5018
18. Cruz-Rodriguez N, Combita AL, Zabaleta J. Epigenetics in hematological malignancies. Methods Mol Biol (Clifton NJ) (2018) 1856:87–101. doi: 10.1007/978-1-4939-8751-1_5
19. Chen X, Li A, Sun B, Yang Y, Han Y, Yuan X, et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol (2019) 21:978–90. doi: 10.1038/s41556-019-0361-y
20. Yang X, Yang F, Lan L, Wen N, Li H, Sun X. Diagnostic and prognostic value of m5C regulatory genes in hepatocellular carcinoma. Front Genet (2022) 13:972043. doi: 10.3389/fgene.2022.972043
21. Zhou H, Meng M, Wang Z, Zhang H, Yang L, Li C, et al. The role of m5C-related lncRNAs in predicting overall prognosis and regulating the lower grade glioma microenvironment. Front Oncol (2022) 12:814742. doi: 10.3389/fonc.2022.814742
22. Xue C, Zhao Y, Li L. Advances in RNA cytosine-5 methylation: detection, regulatory mechanisms, biological functions and links to cancer. Biomark Res (2020) 8:43. doi: 10.1186/s40364-020-00225-0
23. Dominissini D, Rechavi G. 5-methylcytosine mediates nuclear export of mRNA. Cell Res (2017) 27:717–19. doi: 10.1038/cr.2017.73
24. Lenarčič T, Jaskolowski M, Leibundgut M, Scaiola A, Schönhut T, Saurer M, et al. Stepwise maturation of the peptidyl transferase region of human mitoribosomes. Nat Commun (2021) 12:3671. doi: 10.1038/s41467-021-23811-8
25. Li J, Li H, Long T, Dong H, Wang E, Liu R. Archaeal NSUN6 catalyzes m5C72 modification on a wide-range of specific tRNAs. Nucleic Acids Res (2019) 47:2041–55. doi: 10.1093/nar/gky1236
26. Hong J, Lee JH, Chung IK. Telomerase activates transcription of cyclin D1 gene through an interaction with NOL1. J Cell Sci (2016) 129:1566–79. doi: 10.1242/jcs.181040
27. Zhong C, Prima V, Liang X, Frye C, McGavran L, Meltesen L, et al. E2A-ZNF384 and NOL1-E2A fusion created by a cryptic t(12;19)(p13.3; p13.3) in acute leukemia. Leukemia (2008) 22:723–29. doi: 10.1038/sj.leu.2405084
28. Wang F, Yuan J, Wang S, Yang F, Yuan S, Ye C, et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatol (Baltimore Md) (2014) 60:1278–90. doi: 10.1002/hep.27239
29. Cámara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab (2011) 13:527–39. doi: 10.1016/j.cmet.2011.04.002
30. Cui M, Qu F, Wang L, Liu X, Yu J, Tang Z, et al. m5C RNA methyltransferase-related gene NSUN4 stimulates malignant progression of hepatocellular carcinoma and can be a prognostic marker. Cancer Biomark A Dis Markers (2022) 33:389–400. doi: 10.3233/CBM-210154
Keywords: ALL, pediatric, 5-methylcytosine, susceptibility, polymorphism
Citation: Wang X, Deng D, Yan Y, Cai M, Liu X, Luo A, Liu S, Zhang X, Jiang H and Liu X (2023) Genetic variants in m5C modification core genes are associated with the risk of Chinese pediatric acute lymphoblastic leukemia: A five-center case–control study. Front. Oncol. 12:1082525. doi: 10.3389/fonc.2022.1082525
Received: 28 October 2022; Accepted: 12 December 2022;
Published: 09 January 2023.
Edited by:
Jinhong Zhu, Harbin Medical University Cancer Hospital, ChinaReviewed by:
Adolfo Martinez, General Hospital of Mexico, MexicoCopyright © 2023 Wang, Deng, Yan, Cai, Liu, Luo, Liu, Zhang, Jiang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoping Liu, bGl1X3hpYW9waW5nQGd3Y21jLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.