
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 09 January 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1074726
This article is part of the Research Topic The Role of BRAF Signaling in Pediatric & Adult Neuro-Oncology View all 10 articles
 Monica Capogiri1
Monica Capogiri1 Andrea J. De Micheli1
Andrea J. De Micheli1 Alvaro Lassaletta2
Alvaro Lassaletta2 Denise P. Muñoz3
Denise P. Muñoz3 Jean-Philippe Coppé3
Jean-Philippe Coppé3 Sabine Mueller1,4
Sabine Mueller1,4 Ana S. Guerreiro Stucklin1*
Ana S. Guerreiro Stucklin1*BRAFV600E represents the most common BRAF mutation in all human cancers. Among central nervous system (CNS) tumors, BRAFV600E is mostly found in pediatric low-grade gliomas (pLGG, ~20%) and, less frequently, in pediatric high-grade gliomas (pHGG, 5-15%) and adult glioblastomas (GBM, ~5%). The integration of BRAF inhibitors (BRAFi) in the treatment of patients with gliomas brought a paradigm shift to clinical care. However, not all patients benefit from treatment due to intrinsic or acquired resistance to BRAF inhibition. Defining predictors of response, as well as developing strategies to prevent resistance to BRAFi and overcome post-BRAFi tumor progression/rebound growth are some of the main challenges at present in the field. In this review, we outline current achievements and limitations of BRAF inhibition in gliomas, with a special focus on potential mechanisms of resistance. We discuss future directions of targeted therapy for BRAFV600E mutated gliomas, highlighting how insights into resistance to BRAFi could be leveraged to improve outcomes.
Precision medicine brings new therapeutic avenues to clinical practice, tailoring therapies to tumor-specific vulnerabilities. Yet, with the increasing use of targeted agents, new patterns of tumor response and drug resistance are emerging. Tumor development under therapeutic stress is an evolutionary process. Treatment interventions impose selective pressures on tumor cells and their microenvironment that in turn impact response to therapy. A comprehensive understanding of a tumor’s biology and its evolution under treatment has thus become crucial for improving clinical outcomes, as well as successfully implementing novel cancer therapeutics.
Several targeted therapies are directed at activated kinases. BRAFV600E is the most frequent mutation in BRAF (serine/threonine-protein kinase B-raf), a proto-oncogene and the most commonly mutated kinase in human cancers. The biological and clinical relevance of BRAFV600E has been thoroughly described in melanoma, colorectal cancer, thyroid cancer, non-small-cell lung cancer, and hairy cell leukemia (1–4).
BRAFV600E is also detected in glioma, the most common primary brain tumor across all ages (5, 6). Among gliomas, the highest incidence of BRAFV600E is in low-grade gliomas, including pleomorphic xanthoastrocytoma (PXA, 56%), ganglioglioma (GG, 40%) and pilocytic astrocytoma (PA, 3%). While rarer in high-grade gliomas overall (< 5%), its incidence is higher in the epithelioid glioblastoma subtype (69%) (7).
Targeting BRAFV600E with specific BRAF inhibitors (BRAFi) in monotherapy (8, 9) and in combination with downstream MEK inhibitors (MEKi) (10–12) led to improved patient outcomes in different cancer entities, including gliomas (13–17). Despite these encouraging results, patients may experience therapy failure at different stages of treatment, with some tumors being refractory to BRAFi upfront (intrinsic resistance), and others developing resistance, after an initial response, while on therapy (acquired resistance). Moreover, rapid tumor progression is often observed after BRAFi discontinuation (rebound growth).
Drug resistance is a multifaceted phenomenon involving genetic, epigenetic, metabolic and (micro)environmental changes (18, 19). Direct target reactivation, presence or gain of additional genetic alterations, activation of compensatory oncogenic pathways, and adaptive survival mechanisms are some of the features of cancer cells driving drug resistance. Additionally, environmental features such as hypoxia, blood-brain barrier (BBB) and tumor microenvironment (TME) may also play a critical role in response to treatment.
To improve outcomes and mitigate treatment failure, it is essential to understand, predict, prevent, and overcome resistance to BRAFi. In this review, we discuss the role of BRAFV600E in glioma, with a focus on lessons learned from molecularly informed clinical trials as well as our current understanding of the mechanisms involved in resistance to targeted therapy. We highlight upcoming strategies to surpass potential roadblocks ahead.
The B-raf proto-oncogene (BRAF) is located on chromosome 7 (7q34) and encodes the BRAF protein of the RAF kinases family, a core component of the proliferation and survival RAS-RAF-MEK-MAPK cascade (Figure 1). In response to growth factor binding to receptor tyrosine kinases, RAF dimerizes and activates MEK1/2, which in turn phosphorylates ERK1/2. These events lead to the activation of multiple substrates that can promote cell growth and proliferation (20–22).
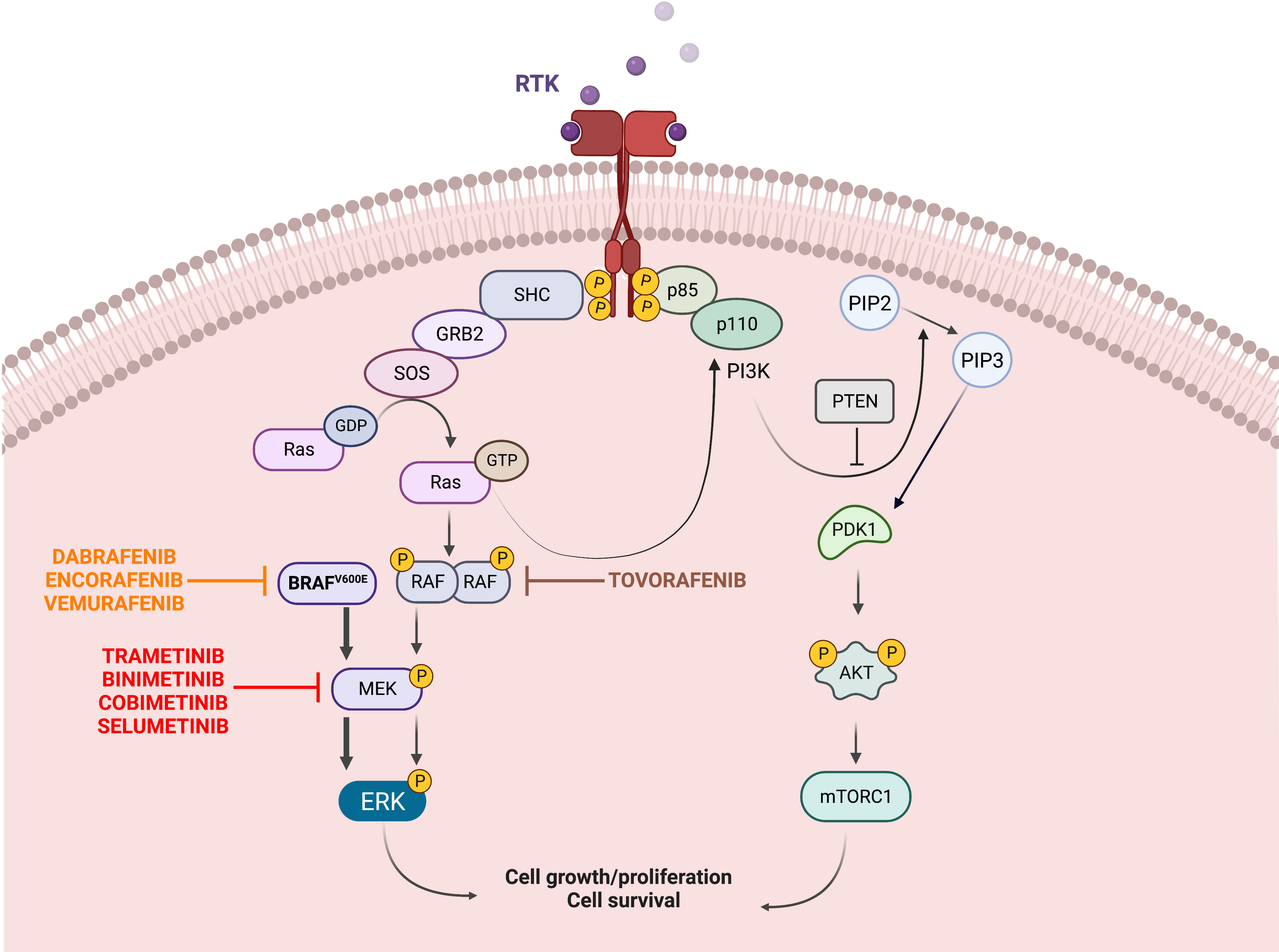
Figure 1 Oncogenic BRAF/MAPK signaling. The mitogen-activated protein kinase (MAPK) pathway is essential to the regulation of cellular growth, proliferation, and survival. Upstream of BRAF, growth factors binding to receptor tyrosine kinases (RTKs) at the cell surface lead to phosphorylation of RAS proteins, which then activate BRAF. Signal transduction continues downstream from BRAF to MAPK kinase (MEK) 1 and MEK2, and finally to ERK, which phosphorylates multiple targets, leading to increased cell survival, proliferation and differentiation. The V600E mutation results in constitutive activation of BRAF and downstream activation of MEK and ERK, in a RAS-independent manner. Specific inhibitors have been synthetized to target the mutated BRAF (Dabrafenib, Encorafenib, Vemurafenib) and the downstream MEK (Trametinib, Binimetinib, Cobimetinib, Selumetinib); more recently pan-raf inhibitors such as Tovorafenib have been developed. The PI3K/AKT/mTOR pathway is another survival pathway and its overactivation has been associated with BRAFi resistance (Created with BioRender.com).
A missense mutation located in exon 15, nucleotide 1799, results in substitution of valine (V) by glutamic acid (E) at codon 600 (V600E) and causes a 500-fold increase in constitutive activity, without the need for dimerization (23). BRAF is mutated in nearly 7% of all human cancers, and V600E point-mutation represents more than 90% of observed alterations (24). In the same exon, other less frequent alterations have also been detected (V600K, V600D, V600R) (25).
The first generation of RAF inhibitors were small-molecule ATP-competitive multikinase inhibitors. Most notably Sorafenib (23, 26, 27), blocking CRAF, BRAF (wildtype and mutant), KIT, VEGFR1/2, FLT1 and PDGFR, was tested in several clinical trials. Unexpectedly, Sorafenib led to accelerated tumor growth in children with BRAF-fused and NF1-driven low-grade gliomas, most likely due to ERK/MAPK paradoxical activation (27). Selective BRAF inhibitors, includingVemurafenib (28–30), Dabrafenib (31, 32) and Encorafenib (33, 34), efficiently inhibit the catalytic activity (and MAPK signaling) of the mutant BRAFV600E, which can function as active monomers, but have limited efficacy against RAF in dimeric forms. Moreover, they can induce increase in functional RAF kinase dimers and paradoxical activation of MAPK signaling in wild-type BRAF or BRAF-fusion expressing cells (35).
A new generation of RAF inhibitors, developed after the characterization of BRAF dimerization, are now in clinical trials. Among these are “paradox breakers” (compounds such as PLX7904 and PLX8394) designed to avoid the paradoxical activation of MAPK. Another subclass includes compounds that inhibit both the monomeric and dimeric forms of BRAF, thus limiting the dimerization of RAF, a known mechanism of resistance (DAY101/Tovorafenib) (36).
After several case studies (37–41), a growing number of clinical trials have established the clinical benefit of BRAFV600E-directed therapies in pediatric and adult gliomas (15, 16, 42). Reports of successful tumor responses and improved survival outcomes with favorable tolerability have been shown with Dabrafenib (42) and Vemurafenib (15, 17) as single agents or in combinations with MEK inhibitors (MEKi) (14, 16).
As part of the VE-BASKET multi-cohort study for non-melanoma cancers (15), BRAF inhibition with Vemurafenib led to an objective response rate (ORR) of 25% in a cohort of 24 adult patients with relapsed/progressive BRAFV600E mutant gliomas. Reponses varied but were detected in all histological glioma subsets, with greater effect detected in low-grade gliomas and PXAs. Meaningful responses were also seen upon treatment with Dabrafenib in pediatric patients with low-grade gliomas [n=32, ORR 44% (95% CI 26 – 62)], with 1-year progression-free survival (PFS) of 85% (95% CI 64 – 94) (43). Supported by preclinical data showing a correlation of extent of MAPK inhibition with response to BRAFi treatment (44) and data emerging from trials evaluating MEK inhibitors (Selumetinib, Trametinib and Binimetinib) in pediatric glioma patients (45–49), subsequent studies assessed the effect of combinatorial BRAFV600E and MEK inhibition.
Treatment with dabrafenib plus trametinib was evaluated in the phase 2 Rare Oncology Agnostic Research (ROAR) trial (16). An interim analysis showed an ORR of 69% (95% CI 39 – 91) in adult patients with low-grade gliomas (9 of a total of 13 patients). For the high-grade glioma cohort (n = 45, 31/45 with glioblastoma) a lower but meaningful ORR was reported of 33% (95% CI 39 – 91, 15 of 45 patients) at a median follow-up time of 12.7 months (IQR 5.4 – 32.3 months). In pediatric patients with low-grade gliomas, when compared to conventional chemotherapy with vincristine and carboplatin (VC), dabrafenib plus trametinib significantly increased ORR (47%, 95% CI 35-59 vs 11%, 95% CI 3-25 for the VC cohort; odds ratio 7.2) (14). Comparison with previous single-agent trials is limited by differences in design and heterogeneity in patient inclusion, however current data suggest an advantage of combinatorial BRAF and MEK inhibition, when compared to BRAFi monotherapy. Beyond progression-free and overall survival, time to response is another important aspect of therapy. Rapid tumor shrinkage is especially relevant in CNS locations where tumors can lead to potentially irreversible neurological damage (e.g. patients with optic pathway gliomas, for whom preservation of vision is critical). Future studies should provide further insights regarding extent/timing of response using targeted treatments, compared to conventional chemotherapy.
The therapeutic benefit of BRAFi and concurrent BRAFi/MEKi in gliomas is indisputable (14, 16). Whether complete and sustained tumor responses can be achieved remains unclear and several key questions remain to be addressed. A large subset of patients may experience rebound growth upon discontinuation of treatment (13). However, most data so far were collected for patients with progressive gliomas, treated with BRAFi after one or several lines of conventional cytotoxic therapies. The optimal timing for introduction of targeted agents (upfront vs at progression), duration of treatment, schedule for treatment discontinuation and best approach to post-BRAFi progression remain under debate. Also, long-term side effects of these targeted therapies in children remain unknown.
The tumor’s ability to escape therapeutic pressure can precede (intrinsic resistance) or emerge during treatment (acquired resistance). As a result, patients can be refractory to treatment or experience an initial response followed by tumor growth while on treatment. Tumor progression after BRAFi discontinuation (rebound growth) is another form of treatment failure but not strictly driven by resistance to BRAFi, since it occurs also in patients with good response to treatment, in the absence of intrinsic or acquired resistance.
In a general context, intrinsic resistance to targeted therapy is usually caused by pre-existing concomitant genetic alterations. Resistance may also derive from a non-genetic rewiring of signaling pathways, epigenetic modulation and/or changes in the tumor microenvironment, leading to the activation of compensatory signals and/or reactivation of the targeted pathway (50–52) (Figure 2).
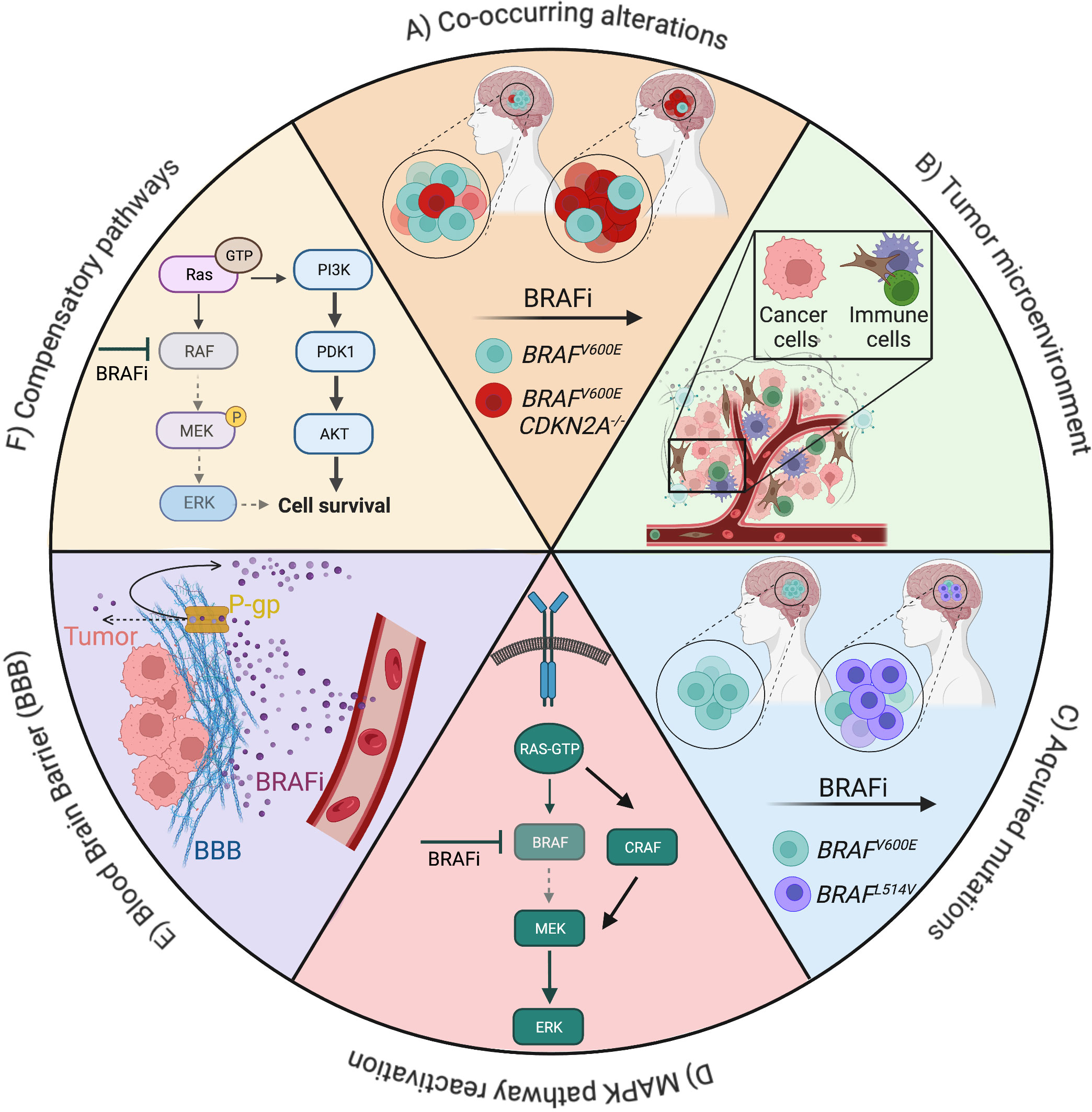
Figure 2 Overview of mechanisms of resistance to BRAF inhibitors (BRAFi) in gliomas. Drug resistance can be driven by cell-intrinsic factors, such as mutations (A, C), pathway activation (D, F), cell-extrinsic factors, such as the tumor microenvironment (B), the presence of physical barriers like the blood brain barrier and its P-glycoprotein (E). Examples of alterations and signaling pathways related to resistance to BRAFis in low- and high-grade gliomas are depicted (Created with BioRender.com).
A common reason for BRAFi treatment failure is the presence of concomitant response-modifying alterations (53, 54). CDKN2A deletion can co-occur with BRAFV600E in different cancer entities, including gliomas, where it is associated with a higher risk of progression, malignant transformation, and overall poorer outcome (55, 56). Intrinsic resistance to BRAFi has been associated with PI3K-mTOR pathway aberrations in several BRAFV600E mutated non-melanoma tumors, including glioblastoma (57). All patients presenting PI3K-mTOR pathway mutations (such as PTENP339fs* 2, PIK3CAI391M, AKTD46E) and BRAFV600E progressed within less than 3 months of targeted monotherapy. Later clinical trials further corroborated this observation: five pediatric and young adult patients with BRAFV600E-mutated brain tumors and PI3K pathway alterations (58) were treated with a combination of Vemurafenib and Everolimus. Overall, two patients (40%) had a partial response and one (20%) had stable disease as best response. Similarly, H3.3 K27M mutation is a poor prognostic marker in gliomas overall and has been detected with concomitant BRAFV600E mutation in rare cases (13, 59).
BRAFV600E has been detected in a tumor harboring also a BRAF-KIAA1549 fusion. In a phase 1 trial of the MEK inhibitor Selumetinib (45), 1 out of 38 pLGG patients carried both alterations, and rapidly progressed on treatment. Though thought to be generally mutually exclusive, it is unclear how patients with BRAFV600E gliomas harboring additional genetic alterations at the level of BRAF (mutations and/or fusions) respond to BRAFi.
About 80% of drug resistant cases in BRAFV600E mutated tumors overall are thought to arise from the reactivation of MAPK pathway (60). Despite positive initial response to inhibitors such as Dabrafenib or Vemurafenib, it is well documented that cells can find alternative ways to activate the pro-survival MAPK pathway in many cancer subtypes (61, 62), including gliomas (63). Secondary mutations in the MAPK pathway, BRAF copy number gains, BRAF alternative splicing as well as increased expression of receptor tyrosine kinases are some of the diverse mechanisms that can cause MAPK pathway reactivation (64).
Binding of BRAFi to one monomer induces allosteric transactivation of the second monomer, while at the same time the binding of the drug at the second monomer is sterically prevented (65). In tumor cells bearing BRAFV600E mutants, the mutated form can dimerize with BRAFV600E, wild-type BRAF and wild-type CRAF, leading to MAPK pathway reactivation (35).
Mutations or hyperactivation in the PI3K/AKT pathway can also cause the reactivation of MAPK (66). In patient-derived BRAFV600E HGG xenograft models chronic exposure to Dabrafenib led to an up-regulation of RAS, phospho-c-Raf, phospho-p90RSK, and phospho-AKT, with paradoxical activation of the MAPK pathway and AKT/mTOR pathway causing resistance to BRAFi (67).
Signaling redundancies and interconnections through pathway crosstalk have been identified as contributors to drug resistance (68). Negative feedback loops, cross inhibition, cross activation, and pathway convergence are hallmarks of cell signaling regulation. Activation of compensatory pathways is more likely to occur in HGG when compared to LGG, typically driven by a single alteration resulting in up-regulation of the RAS/MAPK pathway (69, 70).
Feedback activation of EGFR signaling is one way by which BRAFV600E gliomas adjust and escape BRAFi treatment. BRAFV600E inhibition can suppress MAPK signaling, which in turn downregulates the EGFR phosphatase PTPN9, resulting in sustained EGFR phosphorylation and enhanced EGFR activity (71). One case report supports this observation, in which a glioblastoma patient carrying both BRAFV600E and an activating mutation of EGFR experienced no clinical benefit from RAF and MEK inhibition (72).
Additional pathways have been implicated in resistance to BRAFi. For example, Vemurafenib-resistant glioma cells up-regulate pro-survival mediators such as Wnt and increase Axl receptor tyrosine kinase activity (73).
In melanoma, around 50% of patients treated with BRAFi alone or in combination with MEKi experience an initial significant shrinking of the tumor followed by tumor re-growth due to the acquisition of a new mutation (74). Loss-of-function mutations in STAG2 (75), BOP1 downregulation (76) and COT alteration (77) are just some of the pro-survival mutations that bypass the inhibition of MAPK pathway in melanoma. A case report described an acquired BRAFL514V mutation at time of relapse in a BRAFV600E pLGG, leading to dimerization of BRAFV600E and rendering the monomer-specific inhibitor inefficient (78). In a larger case series, paired pre- and post-BRAFi pediatric and adult glioma samples (n = 15 patients) were analyzed, and 9 cases displayed a novel mutation acquired after BRAFi monotherapy or in combination with MEKi (79). Among these alterations, loss of NF1, PTEN, CBL and CRAF activation were functionally validated using patient-derived cell lines. Further alterations included mutations in PIK3C2G, ERRFI1, BAP1, ANKHD1, and MAP2K1. This study also revealed the heterogeneity of mechanisms of resistance to BRAFi, with all post-treatment samples with acquired mutations presenting unique novel alterations.
The BBB and blood-tumor barrier constitute two sequential barriers that drugs need to cross to reach the tumor. Most commercially available drugs have a reduced capability of crossing the BBB, thus representing an important limitation to therapy effectiveness in brain tumors. The presence of tight junctions and efflux proteins on the BBB impacts distribution of BRAFi and MEKi within the central nervous system. P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), expressed on the luminal side of the BBB, can actively efflux various therapeutic agents (80–82) including Vemurafenib, Dabrafenib, Trametinib, and Cobimetinib (83, 84). Furthermore, many kinase inhibitors such as Dasatinib, Gefitinib, Sorafenib and Erlotinib (85–87) are substrates for important transporters at the BBB and this significantly limits their concentrations in the brain.
Although it is not clear whether these findings have a direct link with therapy failure in glioma patients, it is likely that intrinsic resistance to targeted therapy is partially due to the inability of some drugs to reach therapeutic concentrations in the brain.
Like the BBB, the tumor microenvironment (TME) can impose additional roadblocks to targeted therapies. While most studies have been performed on melanoma, some of the conclusions could be applicable to gliomas. The TME contains various non-cancerous cells such as astrocytes, endothelial cells, microglia, and macrophages, which by their own distinct mechanisms can limit the efficacy of targeted inhibitors. For example, TME stromal cells secrete high levels of ligands capable of activating the MAPK pathway through receptor tyrosine kinases, thereby activating the same pathways that BRAFi are designed to block (83). In BRAFV600E-mutant melanoma, a positive correlation between intrinsic resistance to BRAFi and HGF expression by stromal cells has been described (88).
BRAFi also affects the immune phenotype of the TME in ways it can either benefit treatment or explain mechanisms of resistance. For instance, BRAFi have been associated with an increased number of T-cells in the TME and up-regulation of MHC proteins on cancer cells (89, 90). In a study on metastatic melanoma, patients treated with Dabrafenib and Trametinib displayed an increase in T-cell infiltration and an increase in melanocytic differentiation agents (91). While these changes in the immune composition could facilitate BRAFi treatment, other changes observed in the TME were also associated with resistance. For examples, BRAF inhibition have also been linked to an increase in expression of the immunosuppressive ligand PD-L1 (91). While the mechanisms by which the immune phenotype of the TME evolves to adapt to treatment are yet to be fully characterized, these studies suggest that better therapeutic benefit could be derived from combining BRAFi with immune checkpoint inhibitors or other immunomodulating drugs.
BRAF inhibitors have shown promising results against a wide variety of BRAFV600E mutated cancers. For children,adolescents, and adults with low-grade gliomas, responses to BRAFi are superior to conventional chemotherapy and a growing body of evidence supports upfront targeted therapy in these patients. Though to a lesser extent, encouraging responses further support the use of BRAFi in the treatment of pediatric and adult patients with HGG.
With BRAFi now part of routine clinical care, new challenges emerge. The most pressing concern at present is resistance to targeted therapy as a cause of treatment failure, morbidity, and mortality. In many human cancers, important progress has been made using genomic and transcriptomic profiling of paired clinical samples to track the tumor evolution during and after targeted treatment (92, 93). Yet, re-biopsy at time of relapse is not a standard of care in patients with relapsed gliomas and data are limited.
Several ongoing preclinical and clinical efforts are expected to further elucidate the molecular causes underlying BRAFi treatment failure. Recent reports highlight the evolution of gliomas under BRAF inhibition and reveal a high diversity of acquired alterations at relapse (79). This heterogeneity also suggests that the mechanisms driving drug resistance are patient-/tumor-specific, further supporting the need for tumor biopsies at time of relapse. Though challenging, such an approach may reveal tumor-specific vulnerabilities as well as inform the design of new drug interventions and personalized treatment options for patients with otherwise limited treatment options.
One of the main mechanisms of resistance to BRAFi in gliomas is reprogramming of kinase and cell signaling activity. Several proteomic approaches such as Reverse-Phase Protein Array (RPPA), Mass Spectrometry (MS; total or phospho), or Inhibitor-Bead Mass Spectrometry (IB-MS) can reveal how tumors rewire signaling cascades upon therapeutic pressure but require relatively large amounts of sample. Another recently developed method is the High Throughput-Kinase Activity Mapping (HT-KAM), which measures the activity of hundreds of kinases simultaneously, in cells or in tumor tissues’ extracts (94, 95). The readout of this assay provides a rank of kinases that are overactive/underactive upon treatment, thus uncovering parallel/orthogonal mechanisms that can be targeted to overcome therapeutic resistance. With such understanding of the molecular mechanisms treatment failure, BRAFi could then be rationally combined with inhibitors of resistance mechanisms, to increase treatment efficacy.
Lastly, very little is known about the long-term side effects of these therapies, especially in pediatric patients (96). Their potential impact in the development of young children and adolescents must be carefully assessed in comprehensive longitudinal prospective studies. Advanced imaging- and/or liquid biopsy-based strategies for disease monitoring are further areas of research, expected to improve how we predict response, monitor and tailor treatment with BRAFi. Translation of new technologies and innovation into clinical care will thus be key to understand and overcome BRAFi resistance.
MC, AD and AS wrote the first draft of the manuscript. MC generated the figures. All authors contributed to writing and editing of the final manuscript. All authors listed in the paper have made a substantial, direct, and intellectual contribution to the work and approved it for publication. All authors contributed to the article and approved the submitted version.
This work is supported by grants from the Krebsliga Zürich, Promedica Stiftung, Botha-Chan Low Grade Glioma Consortium and Swiss National Science Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer (2014) 14(7):455–67. doi: 10.1038/nrc3760
2. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417(6892):949–54. doi: 10.1038/nature00766
3. Fiskus W, Mitsiades N. B-raf inhibition in the clinic: present and future. Annu Rev Med (2016) 67:29–43. doi: 10.1146/annurev-med-090514-030732
4. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer (2017) 17:676–91. doi: 10.1038/nrc.2017.79
5. Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer (2010) 46(4):765–81. doi: 10.1016/j.ejca.2009.12.014
6. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2013–2017. Neuro Oncol (2020) 22(Supplement_1):iv1–96. doi: 10.1093/neuonc/noaa200
7. Andrews LJ, Thornton ZA, Saincher SS, Yao IY, Dawson S, McGuinness LA, et al. Prevalence of BRAFV600 in glioma and use of BRAF inhibitors in patients with BRAFV600 mutation-positive glioma: Systematic review. Neuro-Oncology (2022) 24:528–40. doi: 10.1093/neuonc/noab247
8. Brose MS, Cabanillas ME, Cohen EEW, Wirth LJ, Riehl T, Yue H, et al. Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol (2016) 17(9):1272–82. doi: 10.1016/S1470-2045(16)30166-8
9. Hauschild A, Ascierto PA, Schadendorf D, Grob JJ, Ribas A, Kiecker F, et al. Long-term outcomes in patients with BRAFV600-mutant metastatic melanoma receiving dabrafenib monotherapy: Analysis from phase 2 and 3 clinical trials. Eur J Cancer (2020) 125:114–20. doi: 10.1016/j.ejca.2019.10.033
10. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol (2017) 36:7–13. doi: 10.1200/JCO.2017
11. Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol (2016) 17(7):984–93. doi: 10.1016/S1470-2045(16)30146-2
12. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. New Engl J Med (2019) 381(7):626–36. doi: 10.1056/NEJMoa1904059
13. Nobre L, Zapotocky M, Ramaswamy V, Ryall S, Bennett J, Alderete D, et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis Oncol (2020) 4:561–571. doi: 10.1200/PO.19.00298
14. Bouffet E, Hansford J, Garré ML, Hara J, Plant-Fox A, Aerts I, et al. Primary analysis of a phase II trial of dabrafenib plus trametinib (dab + tram) in BRAF V600–mutant pediatric low-grade glioma (pLGG). J Clin Oncol (2022) 40(17_suppl):LBA2002–LBA2002. doi: 10.1200/JCO.2022.40.17_suppl.LBA2002
15. Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF inhibition in BRAFV600-mutant gliomas: Results from the VE-BASKET study. J Clin Oncol (2018) 36(35):3477–84. doi: 10.1200/JCO.2018.78.9990
16. Wen PY, Stein A, van den Bent M, de Greve J, Wick A, de Vos FYFL, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol (2022) 23(1):53–64. doi: 10.1016/S1470-2045(21)00578-7
17. Nicolaides T, Nazemi KJ, Crawford J, Kilburn L, Minturn J, Gajjar A, et al. Phase I study of vemurafenib in children with recurrent or progressive BRAF V600E mutant brain tumors: Pacific pediatric neuro-oncology consortium study (PNOC-002). (2020). doi: 10.18632/oncotarget.27600
18. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature (2019) 575:299–309. doi: 10.1038/s41586-019-1730-1
19. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: A brief review. Advanced Pharm Bull (2017) 7:339–48. doi: 10.15171/apb.2017.041
20. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol (2015) 16:281–98. doi: 10.1038/nrm3979
21. Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, et al. Raf family kinases: Old dogs have learned new tricks. Genes Cancer (2011) 2:232–60. doi: 10.1177/1947601911407323
22. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
23. Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of b-RAF. Cell (2004) 116(6):855–67. doi: 10.1016/S0092-8674(04)00215-6
24. Cantwell-Dorris ER, O’Leary JJ, Sheils OM. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol Cancer Ther (2011) 10:385–94. doi: 10.1158/1535-7163.MCT-10-0799
25. Martin-Algarra S, Labiano T, Echeveste JI, Gomez N, Montañana M, Aguirre M, et al. Use of cobas 4800 BRAF mutation test for the analysis of BRAF V600 mutations in cytological samples (CS) from metastatic melanoma (MM). Am Soc Clin Oncol (2012). doi: 10.1200/jco.2012.30.15_suppl.8572
26. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res (2004) 64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443
27. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol (2014) 16(10):1408–16. doi: 10.1093/neuonc/nou059
28. Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic b-raf kinase with potent antimelanoma activity. Proc Natl Acad Sci (2008) 105(8):3041–6. doi: 10.1073/pnas.0711741105
29. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl J Med (2011) 364(26):2507–16. doi: 10.1056/NEJMoa1103782
30. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature (2010) 467(7315):596–9. doi: 10.1038/nature09454
31. Falchook GS, Long G v, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet (2012) 379(9829):1893–901. doi: 10.1016/S0140-6736(12)60398-5
32. Hauschild A, Grob JJ, Demidov L v, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X
33. van Geel RMJM, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, et al. A phase ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal CancerEncorafenib+ cetuximab ± alpelisib in BRAF mCRC. Cancer Discovery (2017) 7(6):610–9. doi: 10.1158/2159-8290.CD-16-0795
34. Huang T, Karsy M, Zhuge J, Zhong M, Liu D. B-raf and the inhibitors: from bench to bedside. J Hematol Oncol (2013) 6(1):1–9. doi: 10.1186/1756-8722-6-30
35. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF Inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature (2010) 464(7287):427–30. doi: 10.1038/nature08902
36. Landi DB, Ziegler DS, Franson AF, Baxter PA, Leary S, Larouche V, et al. FIREFLY-1 (PNOC 026): A phase 2 study to evaluate the safety and efficacy of tovorafenib (DAY101) in pediatric patients with RAF-altered recurrent or progressive low-grade glioma or advanced solid tumors. J Clin Oncol (2022) 40(16_suppl):TPS10062–TPS10062. doi: 10.1200/JCO.2022.40.16_suppl.TPS10062
37. Lassaletta A, Guerreiro Stucklin A, Ramaswamy V, Zapotocky M, McKeown T, Hawkins C, et al. Profound clinical and radiological response to BRAF inhibition in a 2-month-old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr Blood Cancer (2016) 63(11):2038–41. doi: 10.1002/pbc.26086
38. Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L, et al. Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer (2014) 61(6):1101–3. doi: 10.1002/pbc.24891
39. Drobysheva A, Klesse LJ, Bowers DC, Rajaram V, Rakheja D, Timmons CF, et al. Targeted MAPK pathway inhibitors in patients with disseminated pilocytic astrocytomas. JNCCN J Natl Compr Cancer Network (2017) 15(8):978–82. doi: 10.6004/jnccn.2017.0139
40. Rush S, Foreman N, Liu A. Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol (2013) 31(10):e159–60. doi: 10.1200/JCO.2012.44.1568
41. Leaver KE, Zhang N, Ziskin JL, Vogel H, Recht L, Thomas RP. Response of metastatic glioma to vemurafenib. Neurooncol Pract (2016) 3(4):268–71. doi: 10.1093/nop/npv054
42. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation–positive relapsed or refractory low-grade glioma: Results from a phase I/IIa study. Clin Cancer Res (2019) 25(24):7303–11. doi: 10.1158/1078-0432.CCR-19-2177
43. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation–positive relapsed or refractory low-grade glioma: Results from a phase I/IIa study. Clin Cancer Res (2019) 25(24):7303–11. doi: 10.1158/1078-0432.CCR-19-2177
44. Long G v., Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. New Engl J Med (2014) 371(20):1877–88. doi: 10.1056/NEJMoa1406037
45. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase i trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A pediatric brain tumor consortium (PBTC) study. Neuro Oncol (2017) 19(8):1135–44. doi: 10.1093/neuonc/now282
46. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol (2019) 20(7):1011–22. doi: 10.1016/S1470-2045(19)30277-3
47. Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, Ecker J, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neurooncol (2020) 149(3):499–510. doi: 10.1007/s11060-020-03640-3
48. Manoharan N, Choi J, Chordas C, Zimmerman MA, Scully J, Clymer J, et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J Neurooncol (2020) 149(2):253–62. doi: 10.1007/s11060-020-03592-8
49. Lavrador JP, Robinson C, Hurwitz V, Giamouriadis A, Vergani F, Ashkan K, et al. Low grade glioma clinic - establishing an innovative service based on a multidisciplinary approach. Neuro Oncol (2018) 20(suppl_5):v352–2. doi: 10.1093/neuonc/noy129.036
50. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer (2009) 9(1):28–39. doi: 10.1038/nrc2559
51. Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol (2008) 15(10):1109–18. doi: 10.1038/nsmb.1486
52. Sun C, Bernards R. Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies. Trends Biochem Sci (2014) 39(10):465–74. doi: 10.1016/j.tibs.2014.08.010
53. Wagenaar TR, Ma L, Roscoe B, Park SM, Bolon DN, Green MR. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res (2014) 27(1):124–33. doi: 10.1111/pcmr.12171
54. Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep (2013) 4(6):1090–9. doi: 10.1016/j.celrep.2013.08.023
55. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun (2020) 8. doi: 10.1186/s40478-020-00902-z
56. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol (2015) 33(9):1015–22. doi: 10.1200/JCO.2014.58.3922
57. Sen S, Meric-Bernstam F, Hong DS, Hess KR, Subbiah V. Co-Occurring genomic alterations and association with progression-free survival in BRAFV600-mutated nonmelanoma tumors. JNCI: J Natl Cancer Institute (2017) 109(10):djx094. doi: 10.1093/jnci/djx094
58. Sen S, Tanaka R, Khatua S, Zaky W, Janku F, Penas-Prado M, et al. Dual inhibition of BRAF and mTOR in BRAFV600E-mutant pediatric, adolescent, and young adult brain tumors. Cold Spring Harb Mol Case Stud (2020) 6(4). doi: 10.1101/mcs.a005041
59. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol (2017) 35:2934–41. doi: 10.1200/JCO.2016
60. Degirmenci U, Yap J, Sim YRM, Qin S, Hu J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resistance (2021) 4:665–83. doi: 10.20517/cdr.2021.36
61. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med (2013) 19(11):1401–9. doi: 10.1038/nm.3392
62. Koumaki K, Kontogianni G, Kosmidou V, Pahitsa F, Kritsi E, Zervou M, et al. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600E CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim Biophys Acta (BBA) - Mol Basis Dis (2021) 1867(4):166061. doi: 10.1016/j.bbadis.2020.166061
63. Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U.S.A. (2013) 110(15):5957–62. doi: 10.1073/pnas.1219232110
64. Luebker SA, Koepsell SA. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front Oncol (2019) 9. doi: 10.3389/fonc.2019.00268
65. Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell (2016) 30(3):485–98. doi: 10.1016/j.ccell.2016.06.024
66. Villanueva J, Vultur A, Herlyn M. Resistance to BRAF inhibitors: Unraveling mechanisms and future treatment options. Cancer Res (2011) 71:7137–40. doi: 10.1158/0008-5472.CAN-11-1243
67. Sasame J, Ikegaya N, Kawazu M, Natsumeda M, Hayashi T, Isoda M, et al. HSP90 inhibition overcomes resistance to molecular targeted therapy in BRAFV600E-mutant high-grade glioma. Clin Cancer Res (2022) 28(11):2425–39. doi: 10.1158/1078-0432.CCR-21-3622
68. Jänne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discovery (2009) 8(9):709–23. doi: 10.1038/nrd2871
69. Northcott PA, Pfister SM, Jones DTW. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol (2015) 16(6):e293–302. doi: 10.1016/S1470-2045(14)71206-9
70. Jones DTW, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci (2012) 69:1799–811. doi: 10.1007/s00018-011-0898-9
71. Yao TW, Zhang J, Prados M, Weiss WA, David James C, Nicolaides T. EGFR blockade prevents glioma escape from BRAFV600E targeted therapy. Oncotarget (2015) 6(26):21993–2005. doi: 10.18632/oncotarget.4014
72. Smith-Cohn M, Davidson C, Colman H, Cohen AL. Challenges of targeting BRAFV600E mutations in adult primary brain tumor patients: a report of two cases. CNS Oncol (2019) 8(4):CNS48. doi: 10.2217/cns-2019-0018
73. Yao TW, Zhang J, Prados M, Weiss WA, David James C, Nicolaides T. Oncotarget 583 www.impactjournals.com/oncotarget acquired resistance to BRAF inhibition in BRAF V600E mutant gliomas. Oncotarget (2017) 8. doi: 10.18632/oncotarget.11882
74. Tangella LP, Clark ME, Gray ES. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma - a mini review. Biochim Biophys Acta - Gen Subj (2021) 1865:583–595. doi: 10.1016/j.bbagen.2020.129736
75. Shen CH, Kim SH, Trousil S, Frederick DT, Piris A, Yuan P, et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat Med (2016) 22(9):1056–61. doi: 10.1038/nm.4155
76. Gupta R, Bugide S, Wang B, Green MR, Johnson DB, Wajapeyee N. Loss of BOP1 confers resistance to BRAF kinase inhibitors in melanoma by activating MAP kinase pathway. Proc Natl Acad Sci (2019) 116(10):4583–91. doi: 10.1073/pnas.1821889116
77. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature (2010) 468(7326):968–72. doi: 10.1038/nature09627
78. Wang J, Yao Z, Jonsson P, Allen AN, Qin ACR, Uddin S, et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAFV600E-mutant brain tumor. Cancer Discovery (2018) 8(9):1130–41. doi: 10.1158/2159-8290.CD-17-1263
79. Schreck KC, Morin A, Zhao G, Allen AN, Flannery P, Glantz M, et al. Deconvoluting mechanisms of acquired resistance to RAF inhibitors in BRAFV600E-mutant human glioma. Clin Cancer Res (2021) 27(22):6197–208. doi: 10.1158/1078-0432.CCR-21-2660
80. Gampa G, Vaidhyanathan S, Wilken-Resman B, Parrish KE, Markovic SN, Sarkaria JN, et al. Challenges in the delivery of therapies to melanoma brain metastases. Curr Pharmacol Rep (2016) 2(6):309–25. doi: 10.1007/s40495-016-0072-z
81. Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. Impact of p-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther (2012) 342(1):33–40. doi: 10.1124/jpet.112.192195
82. Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci (2001) 24(12):719–25. doi: 10.1016/S0166-2236(00)02004-X
83. Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. Mechanisms limiting distribution of the threonine-protein kinase b-RaFV600E inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther (2013) 344(3):655–64. doi: 10.1124/jpet.112.201475
84. Choo EF, Ly J, Chan J, Shahidi-Latham SK, Messick K, Plise E, et al. Role of p-glycoprotein on the brain penetration and brain pharmacodynamic activity of the MEK inhibitor cobimetinib. Mol Pharm (2014) 11(11):4199–207. doi: 10.1021/mp500435s
85. Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by p-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther (2010) 334(1):147. doi: 10.1124/jpet.110.167601
86. Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther (2009) 330(3):956. doi: 10.1124/jpet.109.154781
87. de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JHM, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters p-gp and BCRP. Invest New Drugs (2012) 30(2):443–9. doi: 10.1007/s10637-010-9569-1
88. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature (2012) 487(7408):500–4. doi: 10.1038/nature11183
89. Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology (2013) 2(1):e22890. doi: 10.4161/onci.22890
90. Wilmott JS, Long G v, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic MelanomaBRAF inhibitors induce T-cell infiltration in melanoma. Clin Cancer Res (2012) 18(5):1386–94. doi: 10.1158/1078-0432.CCR-11-2479
91. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res (2013) 19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630
92. Lee JW, Park YS, Choi JY, Chang WJ, Lee S, Sung JS, et al. Genetic characteristics associated with drug resistance in lung cancer and colorectal cancer using whole exome sequencing of cell-free DNA. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.843561
93. Cohen YC, Zada M, Wang SY, Bornstein C, David E, Moshe A, et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat Med (2021) 27(3):491–503. doi: 10.1038/s41591-021-01232-w
94. Coppé JP, Mori M, Pan B, Yau C, Wolf DM, Ruiz-Saenz A, et al. Mapping phospho-catalytic dependencies of therapy-resistant tumours reveals actionable vulnerabilities. Nat Cell Biol (2019) 21(6):778–90. doi: 10.1038/s41556-019-0328-z
95. Olow A, Chen Z, Niedner RH, Wolf DM, Yau C, Pankov A, et al. An atlas of the human kinome reveals the mutational landscape underlying dysregulated phosphorylation cascades in cancer. Cancer Res (2016) 76(7):1733–45. doi: 10.1158/0008-5472.CAN-15-2325-T
Keywords: glioma, BRAF inhibitors, drug resistance, BRAFV600E mutation, high-grade glioma (HGG), low-grade glioma (LGG)
Citation: Capogiri M, De Micheli AJ, Lassaletta A, Muñoz DP, Coppé J-P, Mueller S and Guerreiro Stucklin AS (2022) Response and resistance to BRAFV600E inhibition in gliomas: Roadblocks ahead? Front. Oncol. 12:1074726. doi: 10.3389/fonc.2022.1074726
Received: 19 October 2022; Accepted: 12 December 2022;
Published: 09 January 2023.
Edited by:
Claudia Katharina Petritsch, Stanford University, United StatesReviewed by:
David S. Ziegler, Sydney Children’s Hospital, AustraliaCopyright © 2023 Capogiri, De Micheli, Lassaletta, Muñoz, Coppé, Mueller and Guerreiro Stucklin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana S. Guerreiro Stucklin, YW5hLnN0dWVja2xpbkBraXNwaS51emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.