 Zakia Akter
Zakia Akter Narges Salamat
Narges Salamat Md. Yousuf Ali
Md. Yousuf Ali Li Zhang
Li Zhang- Department of Biological Science, The University of Texas at Dallas, Richardson, TX, United States
Cancer immunotherapy shows durable treatment responses and therapeutic benefits compared to other cancer treatment modalities, but many cancer patients display primary and acquired resistance to immunotherapeutics. Immunosuppressive tumor microenvironment (TME) is a major barrier to cancer immunotherapy. Notably, cancer cells depend on high mitochondrial bioenergetics accompanied with the supply of heme for their growth, proliferation, progression, and metastasis. This excessive mitochondrial respiration increases tumor cells oxygen consumption, which triggers hypoxia and irregular blood vessels formation in various regions of TME, resulting in an immunosuppressive TME, evasion of anti-tumor immunity, and resistance to immunotherapeutic agents. In this review, we discuss the role of heme, heme catabolism, and mitochondrial respiration on mediating immunosuppressive TME by promoting hypoxia, angiogenesis, and leaky tumor vasculature. Moreover, we discuss the therapeutic prospects of targeting heme and mitochondrial respiration in alleviating tumor hypoxia, normalizing tumor vasculature, and TME to restore anti-tumor immunity and resensitize cancer cells to immunotherapy.
1 Introduction
Cancer is one of the leading causes of death worldwide with nearly 2 million new cases in 2022 (1). It is accounted for an approximately 10 million deaths in 2020 (2) and lung cancer alone contributed to 350 deaths per day in 2022 (1). This demands the development of sustainable treatment options to cure and manage cancer. Cancer immunotherapy either in the form of immune checkpoint blockades (ICBs), also known as immune checkpoint inhibitors (ICIs), or chimeric antigen receptor (CAR) T-cell therapy (3) has improved cancer treatment over traditional therapies including chemotherapy, surgery, radiation therapy, and so on. It shows stable clinical responses, progression free survival, and long survival benefits with a greater aim to reactivate the immune system to kill cancer cells (4).
Unfortunately, many cancer patients show resistance to immunotherapies either as primary or acquired resistance. Some patients are unresponsive to cancer immunotherapy treatment at early stages, known as primary resistance, while some patients manifest therapeutic resistance after 8 to 10 months, known as acquired resistance (5, 6). Different types of factors including immunosuppressive TME, high mitochondrial bioenergetics or oxidative metabolism, hypoxia, and tumor vasculature are mechanistically involved in inducing and promoting resistance to immunotherapy (7–9).
Various components of TME such as tumor cells, blood cells, and stromal cells stimulate cancer cells proliferation, migration, and metastasis, affect the infiltration of cytotoxic T cells into cancer cells, and consequently help cancer cells evade the immune surveillance mechanism (3, 10). These cells promote CD8+ T cell exhaustion, immunosuppressive environment, and immunoresistance by releasing proinflammatory cytokines, triggering angiogenesis and hypoxia and up-regulating immune checkpoint proteins (ICPs) such as programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), cytotoxic T-lymphocyte antigen-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin mucin -3 (TIM-3) (11–13). Thus, an interest has grown in the better understanding of the mechanism that modulates TME into immunosuppressive TME.
Cancer cells need high energy for their unregulated cell growth, progression, and metastasis, which cause them to use high mitochondrial bioenergetics and excessive oxygen compared to normal cells (14). Heme plays a central role in mitochondrial respiration by supplying the prosthetic groups of oxidative phosphorylation (OXPHOS) complexes (15). It had been found that cancer cells upregulate mitochondrial respiration, hence uptake more heme compared to normal cells. High ATP demand as well as enhanced oxygen consumption by tumor cells create hypoxia in TME areas. This hypoxic condition makes immunosuppressive TME by upregulating vascular endothelial growth factor (VEGF), resulting in neo-vasculature and activating hypoxia-inducible factor-1 (HIF-1) signaling pathways involved in therapeutic resistance (16, 17).
Hypoxia caused by rapidly proliferating tumor cells triggers the formation of new blood vessels, neo-vascularization, from existing blood vessels into TME, which is functionally and structurally abnormal compared to normal vessels (18). This disorganized tumor vasculature also inhibits the effective delivery of immunotherapeutic agents and other drugs to tumor areas and prevents the infiltration of cytotoxic CD8+ T cells, hence supporting cancer cells survival and progression with limited sensitivity to therapeutics (19).
Therefore, an investigation of the mechanism of an immunosuppressive TME amplified by heme and dysregulated mitochondrial respiration are required to improve the recruitment of effector immune cells into tumor areas, enhance anti-tumor immunity, and resensitize cancer cells to immunotherapy. This review will focus on the role of heme and mitochondrial respiration on mediating immunosuppressive TME and present the promising therapeutic prospects of targeting heme and mitochondrial respiration to normalize TME and potentiate immunotherapy.
2 Background of cancer immunotherapy
Immunotherapy has emerged in the last decades as an exciting and promising new modality of cancer treatment and is considered as the “fifth pillar” of cancer therapy. It has a long history, dating back to William Bradley Coley’s use of Streptococcus pyogenes and Serratia marcescens, known as “Coley’s toxins”, which were injected into tumors to treat a variety of malignancies including sarcoma and lymphomas, and this cemented his place as the “father of immunotherapy” (20, 21). However, the modern use of immunotherapies in cancer can be traced back to Dr. James Allison and Dr. Tasuku Honjo’s work on immune checkpoint molecules, culminating in the first ICIs, ipilimumab and nivolumab, and earning the researchers the 2018 Nobel Prize in Medicine or Physiology (22). Immune checkpoint molecules are important proteins that modulate the immune response and allow cancer cells to evade anti-tumor immunity. ICIs are antibodies that target these ICPs to block interaction between ICPs and allow the body to resume an immune response capable of targeting the cancer. The US Food and Drug Administration (FDA) has approved three categories of ICIs such as anti-PD-1 (Nivolumab, Pembrolizumab, and Cemiplimab), anti-PD-L1 (Atezolimumab, Durvalumab and Avelum), and anti-CTLA-4 (Ipilimumab) (4). ICIs targeting CTLA-4, PD-1, and PD-L1 are used for different cancers, including melanomas, breast cancers, and lung cancers (23). The use of ICIs has helped thousands of patients to achieve a stable remission of their cancers.
Other forms of cancer immunotherapy include cytokines that can regulate the host immune response, such as interleukin -2 (IL-2) and interferons (IFNs). IL-2, which was first discovered in 1976, has been approved by FDA for kidney cancer and melanoma (24). IL-2 enhances the effects of T-cell and natural killer (NK) cell activity, allowing the host immune response to target cancers. It is also combined with other immunotherapies, such as ICIs, in order to boost the host immune response and allow targeting of “cold” tumors. Cold tumors are tumors that are poorly responsive to immunotherapy due to an immunosuppressive TME. IFNs work via a similar mechanism like interleukins and are also used in cancer immunotherapy (25). Another emerging immunotherapy treatment modality is CAR T-cell therapy enabling the use of the patients' own T-cells which are adapted to target specific cancers. T-cells are isolated from patients’ blood, and then T cells are edited to express a CAR that allows them to bind to specific tumor antigens. These modified CAR T-cells are then infused back into patient (26). There are currently 6 different CAR T-cell therapies approved by FDA to target a variety of blood cancers including acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma (NHL), and multiple myeloma (26). Cancer vaccines are another form of immunotherapy which can target a host immune response against antigens expressed by different cancers (27). They are generally divided into autologous, made from the patient’s own cells, and allogenic, made from artificial cells. These cancer vaccines hold promise for durable cancer response, including prevention of cancer (28). Immunotherapies have significantly improved the overall survival rate of patients, especially in case of melanoma cancers (29). In non-small cell lung cancer (NSCLC), pembrolizumab increased median progression-free survival to 10.3 months versus 6.0 months in chemotherapy group (30).
Unfortunately, many cancer patients show limited efficacy and adverse effects to immunotherapies due to different factors such as an immunosuppressive TME or mutations limiting the T cell infiltration to tumor cells (31). An adverse effects could be a cytokine storm, a sudden inflammatory response because of activating the immune system that can be dangerous to the host (32). Such adverse events have limited the possibilities of combining different immunotherapies as well as efficacy but significantly increased patients’ health risk. Additionally, not all cancers are responsive to immunotherapies, due to the presence of different immune evasion mechanisms and tumor heterogeneity phenotype in TME that allow TME to be immunosuppressive and resistance to immunotherapeutics. This is an area of major interest for researchers to target TME following combinatorial approaches such as combining immunotherapies with other agents and molecules to turn “cold” tumors into “hot” tumors favoring therapeutic responses to immunotherapy (32).
3 The crosstalk between TME and cancer immunotherapy
TME consists of cancer cells, tumor-antagonizing immune cells, such as effector T cells, dendritic cells (DC), NK cells, and tumor-promoting immune suppressive cells such as cancer associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), regulatory B cells (Bregs), and extracellular matrix (ECM) (33, 34). These cells and components of TME work together and their interaction mostly favor the growth of cancer, an immune evasion of cancer cells, and the insensitivity of cancer cells to immunotherapy (35). CAFs stimulate the release of cytokines and growth factors including platelet derived growth factor (PDGF), VEGF, hepatocyte growth factor (HGF), stromal cell-derived factor 1 (SDF1), and so on, which activates endothelial cells, resulting in an enhancement of tumor growth, angiogenesis process, and tumor vasculature. Immunosuppressive cells upregulate immunosuppressive cytokines such as IL-4, IL-10, IL-13 and transforming growth factor beta (TGF-β), stimulate angiogenesis by expressing VEGF and HIF-1α, and express ICPs, which are involved in tumor development, immunosuppressive TME formation, and anti-tumor immunity evasion (11–13, 36). Macrophage stimulates the release of tumor cells in the circulatory and lymphatic system and works as immunosuppressive agents through suppressing anti-tumor response and mechanism (37–39). TAMs are another prominent cancer promoting cells in TME. TAMs are present in two heterogenous form. One is pro-inflammatory M1 macrophage and another one is anti-inflammatory M2 macrophages, which are controlled and regulated by prostaglandin E2 (PE2), TGF-β, IL-4, IL-10, and IL-13, and involved in tumor-promoting activities by secreting anti-inflammatory cytokines such as TGF-β and IL-10 (40). To promote tumor growth and angiogenesis in TME, most of the macrophages in immune system are polarized to M2 macrophage, which suppress anti-tumor immune responses, subsequently releasing tumorigenic factors and promoting the remodulation of ECM, motility of tumor cells, and intravasation (41).
Cytotoxic T-lymphocytes (CTLs), also known as CD8+T cells, and helper T cells, also known as CD4+ T cells (TH1, TH2, and TH17), are the most prominent components of anti-cancer immunity (42, 43). Helper CD4+ T cells and cytotoxic CD8+ T cells mediate anti-tumor immunity through recognizing and killing cancer cells, but immunosuppressive activities within TME makes CTLs dysfunctional as well as causes T cells exhaustion (44, 45). Interaction between PD-1 and PD-L1, mainly expressed by T-cells and cancer cells, respectively, suppresses T-cells and stimulates the survival of cancer cells (46). Cancer immunotherapies mainly works by targeting T-cells and immune cells to restore anti-tumor immunity, promoting T-cells activation and T cell infiltration within the TME (47). Some constituents of TME have the potential to prevent tumor development, hence they are targeted for cancer immunotherapy. These cells in TME prevent tumor generation by releasing cytokine molecules as well as activating cytokines signaling pathways and direct cell contact (48). Many preclinical studies suggested that the inhibition of fibroblasts activating proteins in CAFs stimulate immunosuppression (49–51). Activation of CAFs by inhibiting different signaling pathways including nuclear factor kappa B (NF-kβ), C-X-C chemokine receptor 4 (CXCR4), fibroblast growth factor receptor (FGFR), and Hedgehog using selective inhibitors showed tumor suppressive or antitumor activity (52). CAFs also participate in an immunosuppressive function through suppressing cytotoxic T cell infiltration and function by releasing TGF-β (53). Targeting MDSCs in TME is another potential strategy for cancer immunotherapeutics. Preventing the immunosuppressive activities of MDSCs involves in blockage of MDSCs proliferation, recruitment, and differentiation of MDSCs into mature cells (54). Several studies showed that using inhibitors or neutralizing antibodies against tumor-derived factors like as IL-6, colony stimulating factor 1 (CSF1), and granulocyte-macrophage-colony stimulating factor (GM-CSF) and chemokines such as CXCR2, CXCR4, CC chemokine ligand 2 (CCL2) suppress MDSCs expansion and recruitment (55–59). Many synthetic inhibitors such as COX-2 inhibitors (celecoxib and acetylsalicylic acid), phosphodiesterase-5 inhibitors (tadalafil, sildenafil, and vardenafil) as well as bardoxolone are used to inhibit the immunosuppressive activities of MDSCs, eventually leading to the differentiation of MDSCs into non-suppressive mature cells (60–62).
However, tumor cells in TME remain in hypoxic state due to high oxidative metabolism and comparatively slow growth of blood vessels than the rapid growth of tumor cells. The simultaneous hypoxia and reoxygenation occurring in the TME areas causes the excessive production of reactive oxygen species (ROS) such as superoxide radical (O2.-), hydroxyl radical (OH), singlet oxygen (O2), and hydrogen peroxide (H2O2) which induce oxidative stress in the cells of TME that ultimately break both single and double DNA stand, initiate abnormal DNA synthesis, cause genetic instability, and finally generate aggressive tumor phenotype (63, 64). Although ROS plays a significant role in cancer progression, there are numerous studies suggesting that ROS also has negative effect on tumor growth. One study suggested that an excess level of ROS is produced during T cell activation by T cell receptor (TCR) signaling which regulates cell proliferation and clonal expansion and finally prevent progression of cancer and cancerous infection (65–68). ROS is also responsible for the development of cell-mediated and humoral immunity through the activation of B cell and T cell. H2O2 is a key element of ROS which has a significant role in B cell maturation and activation and enhance B cell receptor signaling (65, 69). ROS also controls tumor growth through creating balance between T cells and modulate T cell apoptosis (70). ROS from TME can spread up the phagocytosis process through recognizing and engulfing cancer cells (71). Moreover, phagocytic cells such as macrophage, monocytes, and neutrophils generate ROS, eventually leading to killing and clearance of damaged cells (72). ROS degrade pathogens through the formation of neutrophils extracellular traps by signaling cascade (73). In TME, excess ROS are also produced during the differentiation of macrophage (M1 and M2) (74, 75).
Hypoxia contributes to the generation of immunosuppressive TME and immunotherapeutic resistance by upregulating HIF-1α signaling pathways and triggering the expression of VEGF, PDGF, and other cytokines, resulting in a modification of tumor metabolism, angiogenesis, and lymphangiogenesis (76) (Figure 1). Tumor vasculature is structurally and functionally abnormal which leads to the uncontrolled proliferation of endothelial cells and the insufficient oxygen and nutrient supply to immune cells eventually blocking the function of T cells. Moreover, it interferes with the infiltration and delivery of functional T cells towards the tumor, downregulates the expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), and develops the loss of antitumor immunity and immunotherapeutic resistance (77). Therefore, it is necessary to regulate TME to enhance the sensitivity of cancer cells to immunotherapy by understanding key mechanisms triggering hypoxia, tumor angiogenesis, and other processes mediating immunosuppression.
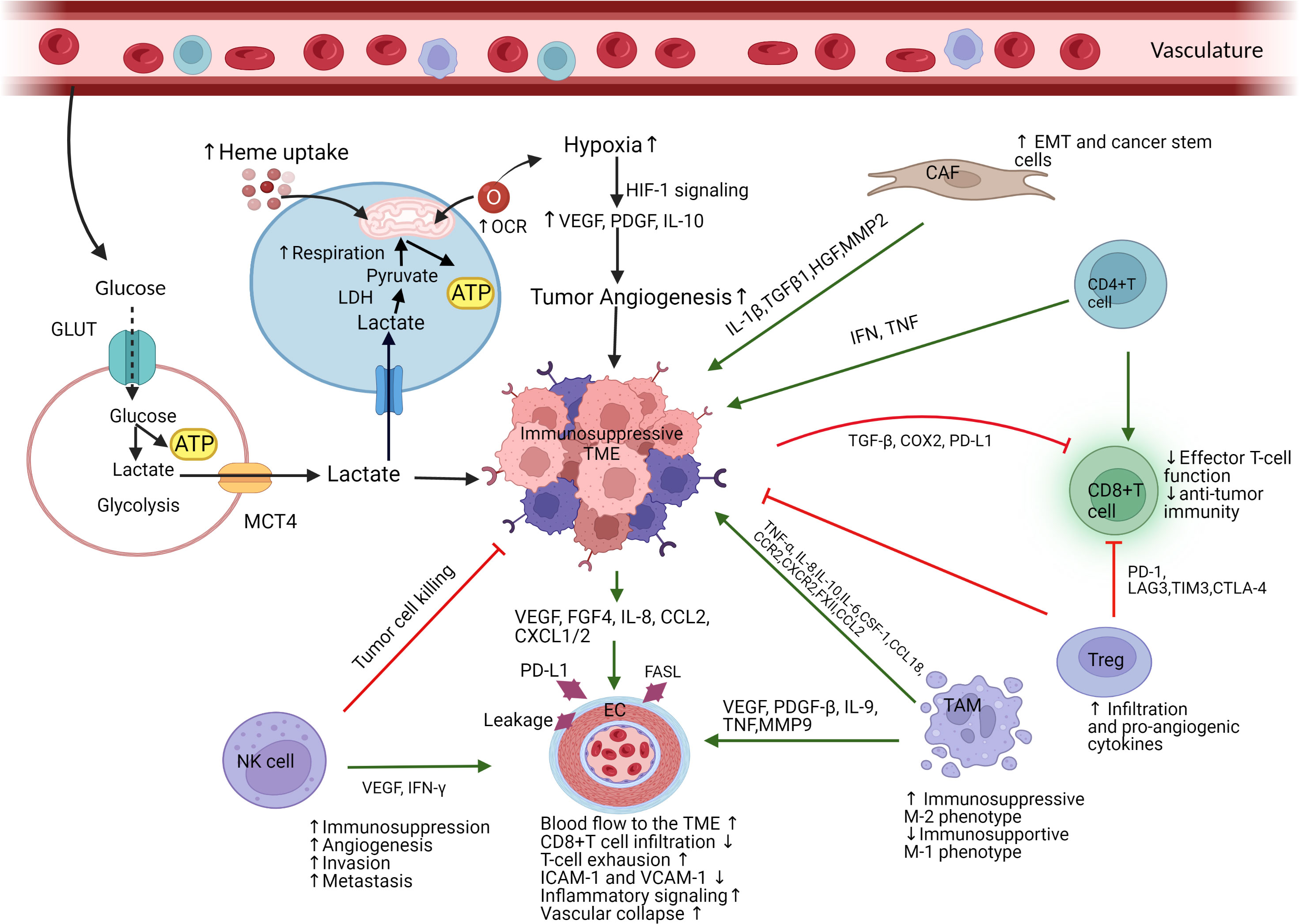
Figure 1 Schematic representation of immunosuppressive TME mainly mediated by tumor cells’ high heme uptake and mitochondrial respiration. (Figure created with BioRender.com accessed on 19 November 2022). Immune suppressive cells and components infiltrate into the TME, interact with tumor cells, causes immune resistance (immunosuppressive), and promote tumorigenesis. Due to excessive consumption of nutrients, tumor cells release lactate which causes acidosis and recruits immunosuppressive cells into TME. Tumor cells’ high heme uptake and mitochondrial respiration associated with an elevated consumption of oxygen promote hypoxia and angiogenesis, which drive an immunosuppressive TME. Hypoxia activates HIF-1 and VEGF signaling cascade which causes dysfunctional vascularization and generates tumor invasion and metastasis. TAMs enhance an immunosuppressive TME function via the polarization of immunosupportive M1-like phenotype to immunosuppressive M2-like phenotype. TAMs also promote tumor EC function via activating VEGF, PDGF-β, TNF, and IL-9. NK-cells stimulate EC function by targeting VEFG and IFN- γ. Receiving signal from TAMs and NK-cells, tumor endothelial cell become immunosuppressive by increasing blood flow into TME and stimulating the expression of PD-L1 which ultimately increase T cells exhaustion and apoptosis. Immunosuppressive TME causes cytotoxic CD8+ T cells dysfunction and T cells exhaustion. CAFs are responsible for EMT and cancer stem cell generation by activating ECs and releasing cytokines and growth factors which promote tumor growth, angiogenesis, and tumor vasculature. GLUT, glucose transporter; ATP, adenosine triphosphate; MCT4, monocarboxylate transporter 4; LDH, lactate dehydrogenase; OCR, oxygen consumption rate; HIF, hypoxia-inducible factor; VEGF, vascular endothelial growth factor; CAF, cancer associated fibroblast; EMT, epithelial mesenchymal transition; Treg, regulatory T cells; TAM, tumor associated macrophage; EC, endothelial cells; NK, natural killer cells; IL, interleukin; TGF, transforming growth factor; HGF, hepatocyte growth factor; MMP2, matrix metalloproteinase 2; IFN, interferon; TNF, tumor necrosis factor; COX, cyclooxygenase; PD-L1, programmed death-ligand 1; PD-1, programmed death -1; LAG3, lymphocyte activating 3; TIM, T-cell immunoglobulin mucin-3; CTLA, cytotoxic T-lymphocyte associated protein; CSF, colony stimulating factor; CCR, C-C chemokine receptor; CCL, CC chemokine ligand; CXCR, C-X-C chemokine receptor; PDGF, platelet derived growth factor; FGF, fibroblast growth factor; CXCL, C-X-C motif chemokine ligand.
4 Modulation of TME by heme and mitochondrial respiration
Various studies reported that cancer cells import elevated levels of heme and upregulate mitochondrial oxidative metabolism. These cause the modulation of TME into an immunosuppressive state that prevents antitumor immunity and supports cancer cells growth and proliferation, which are discussed into following sections and represented in Figure 1.
4.1 The immunosuppressive role of heme on TME
Heme is an essential signaling and metabolic molecules that are involved in many biological processes, from metabolism to the regulation of transcription (78) in host. Heme is composed of four pyrrole rings, chelated by a central iron ion. This iron-containing porphyrin has been shown in epidemiological studies to be associated with various diseases such as cancers, Alzheimer’s dementia, vascular diseases, and metabolic diseases (79). Damaged red blood cells release heme, which is known as labile or free heme, into the body that drives immune response and inflammation (80). Labile heme acts as a danger associated molecular pattern (DAMP) that provokes immune responses by binding to toll-like receptor 4 (TLR4) (80). The induction of TLR4 signaling pathways by labile heme causes the release of tumor necrosis factor α (TNF-α) and IL-6 (81), which are associated with immunosuppressive TME and immune evasion (82, 83). Heme also causes endothelial cells dysfunction as well as the induction of endothelial cells or vascular permeability (81). Free heme produces ROS by Fenton reaction, resulting in an induction of oxidative stress and DNA damage (80).
Labile heme also causes the expression of oncogenes and cancer driver genes such as c-MYC, bcl-2, TGF-β, and Wnt by working on guanine quadruplex (G4) DNA structure as a heme-G4 DNA complex, resulting in a stimulation of cancer cell growth and proliferation (84). It is known that TGF-β promotes immunosuppressive TME and the loss of anti-tumor immunity by triggering the clonal expansion of Treg cells and inhibiting the function of effector T cells and DCs (85). Wnt/β-catenin signaling pathway also influences TME and cancer immunotherapy by inhibiting the infiltration, activation, and proliferation of CD8+ T cells and increasing Treg cells recruitment in TME (86). Therefore, targeting free or labile heme could possibly regulate vascular integrity and the expression of these genes and subsequently these immunosuppressive activities.
Cancer cells display intensified heme uptake and synthesis (79, 87) (Figure 1), which is manifested by the overexpression of heme carrier protein 1 (HCP-1) in lung cancer and gastric cancer cells (88). This elevated heme uptake of cancer cells also enhances mitochondrial oxidative metabolism. Heme does not only help coordinate the assembly of several OXPHOS complexes, but it is itself incorporated into complexes II-IV of the electron transport chain (87). This intensified heme uptake and synthesis by cancer cells helps to fuel an elevated ATP production that allows cancer cells to proliferate, and further fuels hallmarks of cancer such as metastasis and angiogenesis (89). Various studies reported the involvement of angiogenesis with immunosuppressive TME and poor immunotherapy responses.
Furthermore, studies in the authors’ lab have shown that heme plays an important role in TME modulation (90). Heme can modulate TME by working on tumor-associated endothelial cells (TECs) and TAMs, that promote angiogenesis and tumor immunosuppression (88) respectively, besides supporting cancer cell growth, survival, and metastasis. There is a metabolic interaction between heme, TECs, and cancer cells (CCs) within TME (91). TECs upregulate angiogenesis through secreting VEGF, PDGF, and FGF molecules and Notch signaling pathways (92). They also promote neovascularization based on CXCR4 signaling pathways and inhibit anti-tumor immunity by preventing cytotoxic reactions of the immune cells (92). Thus, the action of heme on TECs could lead to immunosuppressive activities in TME. Heme causes macrophage polarization, a process that converts macrophages into M1-like and M2-like phenotype macrophage favoring the enhancement of pro-tumor functions and the tumor escape from immune surveillance (88). M2-like macrophages, comprising of TAMs, produce different growth factors, matrix metalloproteinase (MMP), angiogenic factors including VEGF, PDGF, and FGF, and cytokines (93, 94). Taken together, these secreted molecules stimulate tumor cell growth, invasion, metastasis, tissue remodeling, angiogenesis, and immunosuppression within the TME.
Indoleamine 2,3-dioxygenase 1 (IDO1) is upregulated in different types of cancers including lung cancers, ovarian cancers, prostate cancers, colon cancers, breast cancers, and kidney cancers (95). It is a heme-binding enzyme that catalyzes the conversion of tryptophan into kynurenine, an immunosuppressive metabolite. Cancer cells depend on IDO1 for exerting immunosuppressive effects. For instance, IDO1 stimulates Treg cells and prevents the activity and proliferation of effector T cells by producing kynurenine, which leads to an immune evasion and immunosuppression (96). Hence, targeting heme could be another strategy to prevent the binding between heme and IDO1, which may enhance anti-tumor immune responses by preventing kynurenine production and restoring the function of effector T cells.
Heme catabolism also acts as an important factor to modulate TME and immune responses by inducing the expression of heme oxygenase -1 (HO-1) (84). The expression of HO-1 is predominantly triggered by heme which is observed from the immunoblot analysis of tumor lysates collected from a mice treated with heme compared to untreated mice in prostate cancer (84). HO-1 also triggers macrophage polarization, which in turn causes immune suppression in TME areas and immune evasion of cancer cells found in nasopharyngeal carcinoma (97). HO-1 upregulation is associated with oncological events such as tumorigenesis, metastasis, angiogenesis, and hypoxia as well as with poor survival outcome and limited immunotherapy efficacy (98, 99).
HO-1 induction has been shown to be implicated in prostate cancer progression by inducing metastasis (100). HO-1 may also have a direct impact on immune cells. One study found that HO-1 may prevent NK cell function by inhibiting CD48 expression, resulting in an immunosuppression and immune invasion of cancer as shown in both in vitro and in vivo studies (101). Yet, studies have also shown a dual role of HO-1, demonstrating its mediation of ferroptosis induction, while it may have a protective role in cases of elevated ROS (102). The upregulation of HO-1 showed anti-PD-1 resistance in melanoma and breast cancer mouse models (103), suggesting the role of heme catabolism in limiting immunotherapy response.
4.2 The immunosuppressive role of mitochondrial respiration on TME
Dysfunctional mitochondrial metabolism is one of the hallmarks in cancer, leading to an amplification of mitochondrial respiration and many biosynthetic pathways. Alterations of mitochondria, both in the cancer and the immune cells of the TME, leads to an immunosuppressive condition (Figure 1) and limited sensitivity to immunotherapies by overexpressing PD-1 and PD-L1 signaling pathways (104). According to various studies, mitochondrial metabolic reprogramming is implicated in triggering resistance to multiple cancer therapeutics, such as radiotherapy, chemotherapy, targeted therapy, and immunotherapy. Song et al., showed that the reprogramming of mitochondria by an epigenetic alternation led to the loss of ATP synthase subunit that secrete ROS and stabilize HIF-1α under hypoxic conditions, perpetuating a hypoxic phenotype (105). This loss of ATP synthase was associated with resistance to several cancer therapies. Therefore, it could be essential to understand the immunosuppressive mechanism of upregulated mitochondrial oxidative metabolism for targeting mitochondrial respiration in order to normalize TME.
4.2.1 Mitochondria modulate T cell response
During cellular stress, mitochondria can be disrupted and release ROS, mitochondrial DNA (mtDNA), and other mitochondrial DAMPs (mtDAMPs) into the extracellular environment (106). These mtDAMPs released mostly in response to severe cellular damage activate an immune signaling pathway that mounts an immune response against the cancer cells (107). In the immediate aftermath of cellular damage and mtDAMPs release, the immune-primed TME leads to a T cell response that allows for targeting cancer cells (108). Unfortunately, a chronic inflammatory state arises from prolonged mtDAMPs signaling that leads to T cells exhaustion and immunosuppressive condition in TME due to the hyperactivation (104). Therefore, targeting mitochondrial mtDAMPs release within a certain threshold to elicit an immune response, yet not cause an immune suppression by chronic mtDAMPs release, can be a therapeutic strategy in targeting “cold” tumors.
4.2.2 High mitochondrial OXPHOS triggers an immunosuppressive TME
Elevated mitochondrial OXPHOS is a hallmark of various cancers, including breast cancer, gastric cancer, hepatocellular carcinoma, and NSCLC (87, 109). Increased oxidative metabolism does not conflict with the fact that most cancer cells exhibit enhanced glycolytic rates, which were observed by Warburg (110). In fact, elevated cancer cells’ glycolysis is linked with an intensified OXPHOS as pyruvate and lactate, products of glycolysis, enter and amplify TCA cycle in nearly all NSCLC tumors (111, 112). Additionally, components of OXPHOS complexes and markers of mitochondrial biogenesis are found to be highly predictive of reduced overall survival rate in NSCLC patients (113).
Studies have shown that elevated tumoral mitochondrial OXPHOS can lead to an enhanced tumor hypoxia (114) associated with an immunosuppressive TME due to the inadequate supply of oxygen to effector T cells. In case of melanoma, tumor oxidative metabolism was found to be associated with limited anti-PD-1 therapeutic responses (115). They demonstrated that the excess consumption of oxygen by tumor cells causes hypoxia and T-cell dysfunction. Interestingly, the suppression of tumor oxidative metabolism decreases hypoxia in tumor areas and consequently improves therapeutic responses to anti-PD-1 treatment (115).
Tumor hypoxia is a common feature among a variety of solid tumors, and many studies have been done aimed at attempting to relieve tumor hypoxia (107) to normalize TME. Hypoxia suppresses the activation of immune cells including helper T cells and cytotoxic T cells, enhances the activity of the immune suppressed cells, and provokes multidrug resistance (MDR) (16). It can not only lead to the expression of T cell inhibitory molecules, but also can cause the recruitment of immunosuppressive cells such as MDSCs, Treg cells, CAFs, and TAMs (114) into TME.
As mentioned earlier, hypoxia induces angiogenesis by stimulating the expression of angiogenic factors (VEGF) (116). Angiogenesis is the creation of new blood vessels surrounding the tumor in order to satisfy its demand for increased oxygen and nutrients. However, the creation of these new blood vessels is not always successful, and in fact, many of these newly formed blood vessels are improperly created which are structurally leaky and impair oxygen delivery to the tumor which in turn mediates increased angiogenesis (104). Angiogenesis leads to immunosuppression by this enhanced cycle of hypoxia signaling and buildup of toxic metabolites in the TME, resulting in an effector T cell exhaustion.
In addition, toxic metabolic waste products from the elevated tumor OXPHOS can exacerbate the immunosuppressive TME. These toxic metabolites can affect T cells, leading to the decreased function of T cells. Further, glucose depletion in the TME, mainly by the energy intensive tumor cells, can lead to the decreased access of glucose to T cells, which make a switch to glycolytic metabolism upon activation and thus have an increased need for readily available glucose. This can decrease T cell activity and lead to the tumor cells to maintain an immunosuppressive TME (114). Enhanced mitochondrial respiration also produces ROS that support cancer cell proliferation, progression, metastasis, and the modulation of various cells residing in TME, leading to immunosuppressive characteristics (117).
5 Therapeutic prospects of targeting heme and mitochondrial respiration
Cancer immunotherapy has been showing promising results and durable responses for lung cancer, breast cancer, head and neck cancer, and melanoma in various clinical trials studying the effects of blocking PD-1/PD-L1 interaction (118). However, resistance to immunotherapy is very common phenomenon to majority of cancer patients due to cancer heterogeneity and dynamic TME (35, 119). Different types of cells in TME interact with each other and create a hostile condition to anti-tumor immunity and cancer immunotherapy (35). In addition to making immunosuppressive TME, these mechanisms reduce the supply of oxygen and nutrients to immune cells, prevent the infiltration of effector T cells in tumor areas, and induce immunotherapeutic resistance (11). Therefore, it could be a promising approach to target heme and mitochondrial respiration for overcoming immunosuppressive mechanisms in TME and restoring anti-tumor immunity, which are discussed in the following section and summarized in Tables 1, 2.
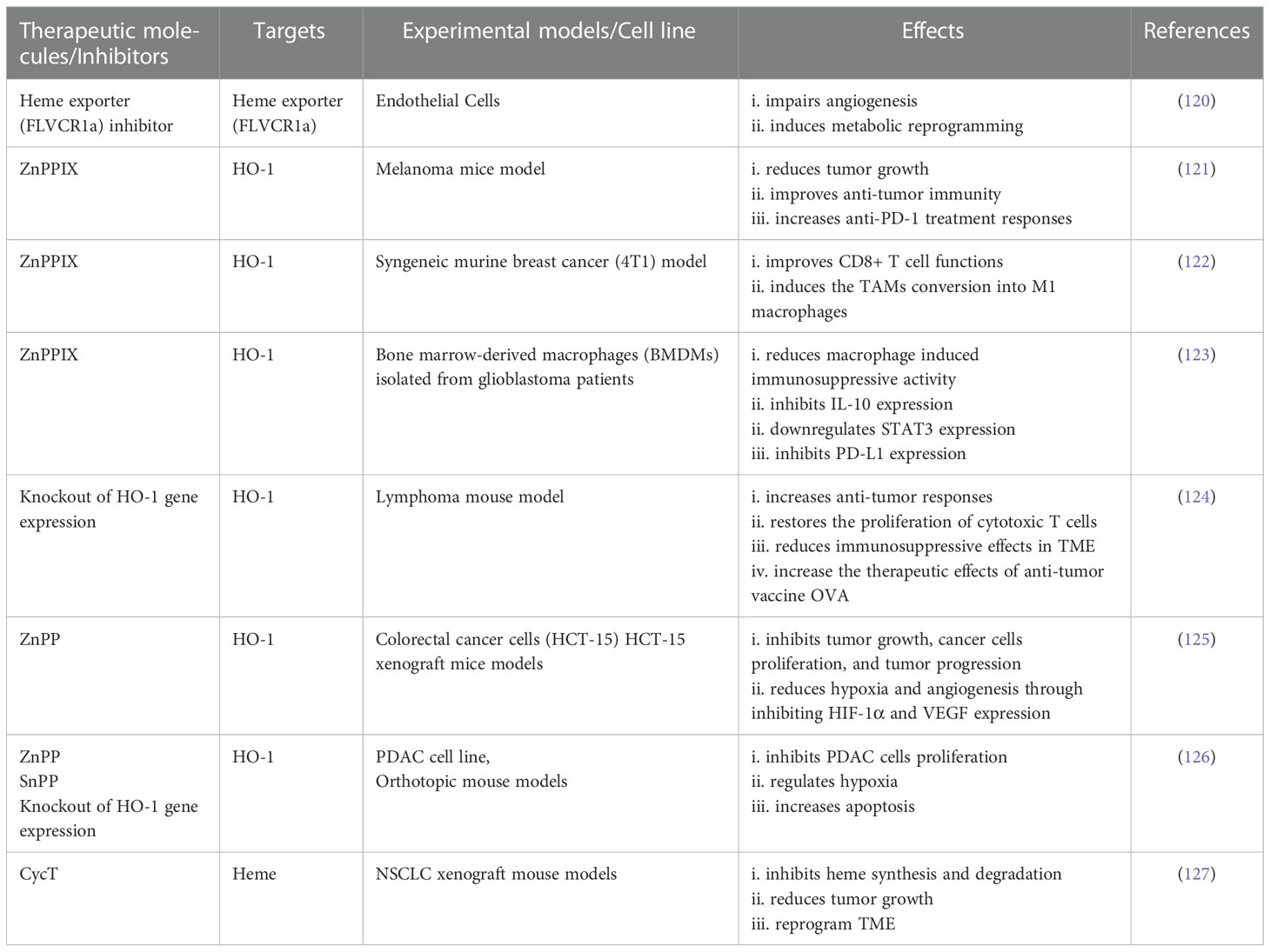
Table 1 Studies targeting heme and heme catabolism and their effects in TME and anti-tumor immunity.
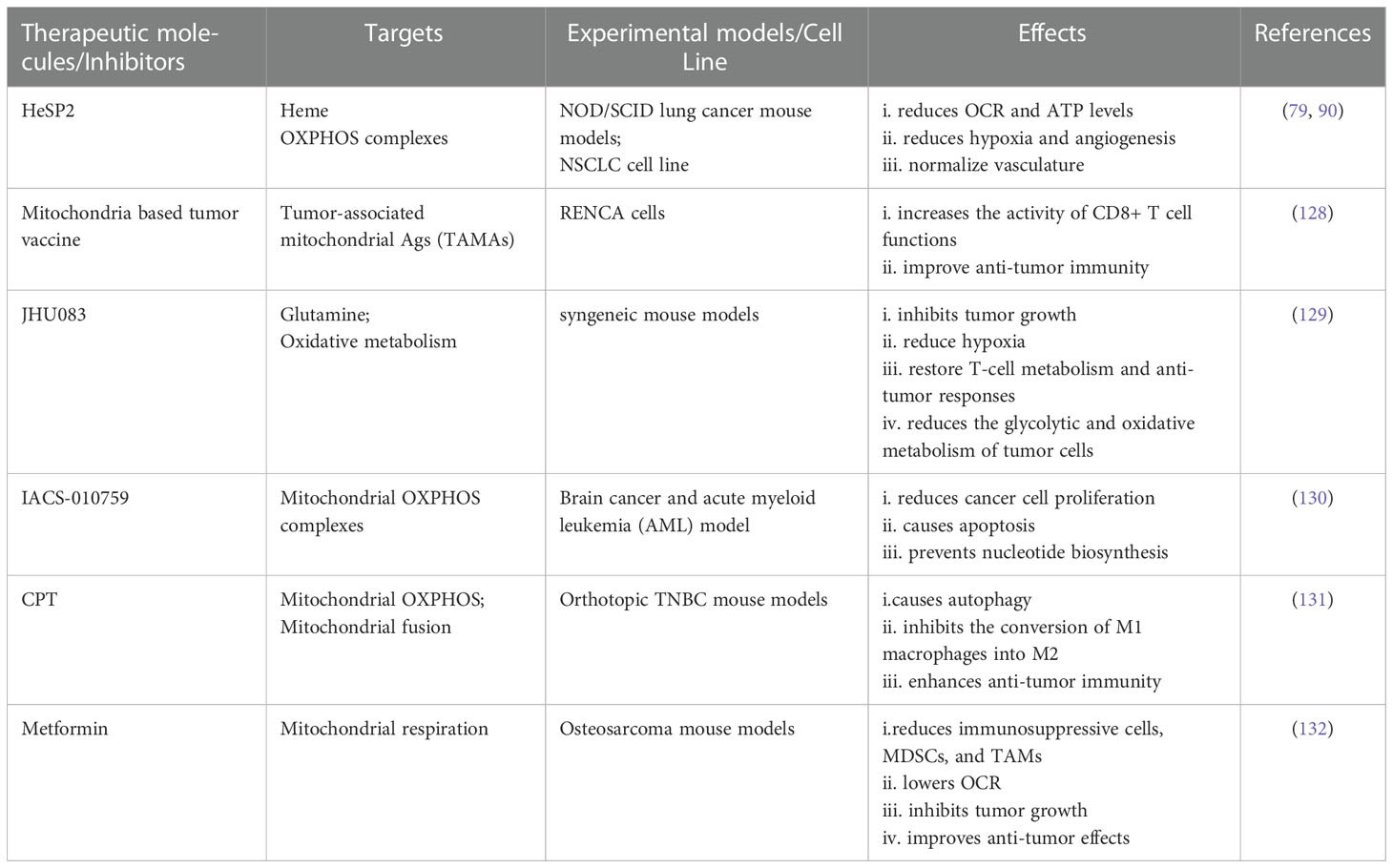
Table 2 Studies targeting mitochondrial respiration and OXPHOS complexes and their effects in TME and anti-tumor immunity.
5.1 Targeting heme and normalizing TME
As mentioned earlier, heme modulates TME, so it might be useful to target heme to normalize TME and improve immunotherapy responses. Multispectral optoacoustic tomography (MSOT) analysis showed the improvement of TME by targeting heme in NSCLC xenograft models (89). Kalainayakan et al. studied the effects of cyclopamine tartrate (CycT), which inhibits heme synthesis and degradation, resulting in a reduction of tumor growth and an improvement of TME areas, in NSCLC xenograft mouse models (127). It had been found that the inhibition of heme exporter, FLVCR1a, in TECs perturbs the metabolic interaction among heme, TECs, and CCs and impairs angiogenesis, resulting in a metabolic rewiring within TME by triggering fatty acid oxidation and ketone bodies accumulation (91, 120).
Various studies reported HO-1 as a potential therapeutic target to improve immunosuppressive TME and restore anti-tumor responses and found promising results both in vivo and in vitro studies. For instance, TAM-induced HO-1 inhibition or removal of myeloid specific HO-1 led to an enhancement of anti-tumor immunity and anti-PD-1 treatment responses in melanoma cancer model (121).
Kim et al. found that targeting HO-1 using HO-1 inhibitor, zinc protophorphyrin IX (ZnPP), facilitates the conversion of TAMs into M1-like phenotype, resulting in an improvement of cytotoxic T cell functions (122) in syngeneic murine breast cancer (4T1) model. The inhibition of HO-1 prevented immunosuppressive mechanism within TME by inhibiting the expression of IL-10, the upregulation of signal transducer and activator of transcription 3 (STAT3), and expression of PD-L1 in case of gliobastoma found by Magri et al. (123). The downregulation of PD-L1 ultimately could enhance the activity of effector T cells by blocking the interaction between PD-L1 and PD-1.
The pharmacological inhibition of HO-1 from myeloid areas improved anti-tumor responses and anti-tumor efficacy of OVA vaccine by enhancing the proliferation of CD8+ T cells and preventing the immunosuppressive effects of TME (124). ZnPP treatment in colorectal cancer cells and xenograft mouse models inhibits HO-1 that consequently reduces the expression of VEGF and HIF1-α (125). The downregulation of HO-1 by tin protoporphyrin IX (SnPP) was found to control hypoxia and pancreatic ductal adenocarcinoma (PDAC) cell proliferation (126). Bauer et al. showed that the silencing of HO-1 using small interfering RNA (siRNA) causes the inhibition of VEGF driven endothelial cells proliferation by regulating the downstream targets of HO-1, vimentin and calpain (133). As a results, this caused angiogenesis impairment, hypoxia improvement, and tumor growth reduction (125). Taken together, all these studies suggest that targeting heme and HO-1 could overcome the immunosuppressive effects of TME and enhance immunotherapeutic efficacy.
5.2 Targeting mitochondrial respiration and normalizing TME
Dysfunctional mitochondrial bioenergetics play a potential role in mediating the loss of anti-tumor immunity and immunotherapy. As stated above, upregulated mitochondrial respiration by cancer cells causes hypoxia within TME areas because of the cancer cells’ elevated oxygen and energy consumption (134). It implies the necessity of increasing the supply of oxygen to immune cells to enhance anti-tumor immunity by targeting the OXPHOS complexes and mitochondrial respiration. Nowadays, the supply of oxygen using nanomaterials are getting attention to mitigate the effects of immunosuppressive TME (135).
Moreover, hypoxia upregulates the expression of VEGF and pro-inflammatory cytokines based on HIF-1α signaling pathways, which orchestrates angiogenesis and the accumulation of immunosuppressive cells within TME (134, 136). These angiogenesis processes represent the loss of anti-tumor immunity and resistance to immunotherapeutics including ICIs and CAR T-cells (134, 136). Different clinical trials are ongoing based on combinatorial approaches following anti-VEGF and ICIs targeting angiogenesis and immune system, respectively (137). However, anti-angiogenics treatment sometimes can prune tumor vessels and other vessels based on dose and time duration, which results in an induction of hypoxia, immunosuppression, and upregulation of PD-L1 (138). Therefore, targeting mitochondrial respiration could be a potential approach to reduce hypoxia and consequently angiogenesis process and immunosuppressive condition within TME areas. It had been found that the use of heme-sequestering peptide (HeSP2) inhibits heme uptake to lung cancer cells, resulting in a decrease of OXPHOS complexes, a reduction of oxygen consumption rate (OCR) and ATP production, and a reduced expression of angiogenic factors (VEGF and VEGFR) (79). Dey et al. showed that the use of HeSP2 reduced angiogenesis, improved hypoxia, and normalized vasculature in NOD/SCID lung cancer mouse models (90). It is worth mentioning that targeting mitochondrial respiration can prevent resistance to antiangiogenics, TKIs (139).
As mentioned earlier, when becoming dysfunctional and damaged, mitochondria release mtDAMP, mtDNA, and so on. Interestingly, one study targeted mitochondrial released abnormal protein, known as tumor-associated mitochondrial Ags (TAMAs), to produce anti-tumor response. Pierini et al. produced a mitochondria based tumor vaccine that showed effective CD8+ T-cell responses and sustainable protection against RENCA cells (128). The inhibition of glutamine using JHU083 lowers the tumor cells oxidative and glycolytic metabolism in syngeneic mouse models, and these effects lead to the reduction of hypoxia, suppression of tumor growth, and the normalization of T- cell metabolism with anti-tumor responses (129).
The use of natural products, cryptotanshinone (CPT), isolated from the herb Salvia miltiorrhiza (Danshen) showed the improvement of anti-tumor immunity through targeting mitochondrial respiration and normalizing TME in orthotopic TNBC mouse models (131). Through targeting mitochondrial respiration, metformin reduced immunosuppressive cells (MDSCs and TAMs) in TME, which results in an improvement of anti-tumor immunity and inhibition of tumor growth (132). Therapeutic benefits of targeting mitochondrial OXPHOS are currently being studied in phase I clinical trials in advanced cancer patients using IACS-010759, an inhibitor of mitochondrial complex I (130, 140).
Interestingly, tumors showed increased sensitivity to therapy when antioxidants were administered that reversed the effects of mitochondria secreted ROS (105). Zhang et al. sensitized MDR breast cancer to chemotherapy doxorubicin by targeting mitochondria using photochemotherapeutic nanoparticles, resulting in an enhancement of anti-tumor immune response and immunogenic cell death (141).
6 Future prospects and limitations
Cancer immunotherapy targets the immune system to restore anti-tumor immunity and enable T cells to kill cancer cells; however, immunosuppressive TME limits the effects of cancer immunotherapy and immune responses (142). Hence, combinatorial approaches following the improvement of immunosuppressive TME and anti-tumor immunity could be useful to inhibit the progression of cancer, prevent immunotherapeutic resistance, and ensure sustainable therapeutic benefits with long-term prognosis. As immunotherapy agents mainly work on immune cells, combination of regimens that target different immunosuppressive mechanisms in TME might be useful to enhance the function of effector T cells, prevent the recruitment of immunosuppressive cells, and improve anti-tumor immunity. Many studies are currently ongoing that are aimed at targeting heme and mitochondrial respiration for overcoming therapeutic resistance and improving treatment outcomes. For instance, the use of IDO1 inhibitor is currently under therapeutic intervention as a cancer immunotherapy. This IDO1 inhibitor specifically targets IDO1, which is involved in cancer cells proliferation and immune suppression, and displaces heme from IDO1 by forming a complex (143, 144). It would be possible to improve the efficacy of cancer immunotherapy by regulating heme catabolism because of its role in inducing immunosuppressive characteristics. For example, Schillingmann et al. showed the potentiality of inhibiting HO-1 expression to improve adoptive T-cell responses against cancer patients producing wilms tumor protein -1 (WT1) (145). Synergistic effects of targeting heme catabolism and ICIs were observed in another study. Blockage of HO-1 expression and anti-PD-1 treatment together enhanced anti-tumor effects and anti-PD-1 treatment responses in melanoma mice models (121). However, the dependency of tumor cells and immune cells on oxidative metabolism is very complicated and immune cells need energy for their function, suggesting a judicious strategy for targeting mitochondrial OXPHOS (142). It had been found that the reprogramming of T cells metabolism by targeting mitochondrial OXPHOS increase tumor infiltrating cytotoxic T cells function, anti-tumor immunity, and cancer immunotherapy responses (146, 147). Combination of bezafibrate, which is an agonist of peroxisome proliferator-activated receptor-gamma coactivator 1-alpha/peroxisome proliferator–activated receptors (PGC-1α/PPAR) complexes and selectively targets cytotoxic T cells oxidative metabolism, and anti-PD-L1 amplifies the number of T cells, effector T cells function, and anti-tumor immunity (148). It is also possible to epigenetically target T cells mitochondrial biogenesis, improve effector T cells function, and increase anti-tumor immunity found in an in vivo study (149). A number of OXPHOS inhibitors, such as metformin and canagliflozin, are actively being used in clinical trials examining their efficacy in combination with current cancer therapies (16). Notably, metformin combination therapy with anti-PD-1 immunotherapies, nivolumab and pembrolizumab, is being studied in several in-progress phase I and II clinical trials (16). Metformin acts by targeting mitochondrial complex I and thereby reducing OXPHOS, leading to reduced tumor hypoxia, which allows greater T-cell infiltration. Synergistic effects of metformin and IR-780 improved hypoxia by inhibiting mitochondrial respiration and delivering oxygen, respectively. This combination ultimately reduced immunosuppressive MDSC cells, cleared immunosuppressive TME, and enhanced anti-tumor responses (150). Scharping et al. showed that the treatment of metformin with anti-PD-1 reduces oxygen supply to tumor cells and increases the function of T cells (151). Hypoxia-activated prodrug TH-302 was studied in combination with anti-PD-1 or anti-CTLA-4 in prostate cancer mouse model. This combination was shown to reduce 80% tumor, hypoxia and the recruitment of immunosuppressive cells (152). Combinational treatments of IACS-010759, radiotherapy, and anti-PD-1 prevented resistance to anti-PD-1 and improved anti-tumor immunity in NSCLC adenocarcinoma xenograft mice models (153). Therefore, combinational treatments that target heme and mitochondrial OXPHOS along with the immune system could be a novel therapeutic approach by normalizing TME and improving anti-tumor immunity and responses to cancer immunotherapy. It would be desirable to study further in clinical settings to define mechanism and optimal doses, timing, and duration to which targeting heme, heme catabolism, and mitochondrial respiration could potentiate cancer immunotherapy. The heterogeneity of TME and individuals metabolic profiling, which affects therapeutic responses, should be considered before designing any drug regimens for better treatment outcomes and clinical responses. Moreover, some challenges are associated with developing metabolism based therapeutics, for instance, the dependency of both normal and cancer cells on metabolism, the rapid metabolic adaptation of cancer cells, drug delivery issues to mitochondria (154, 155), and so on, which should be addressed before developing anticancer therapies based on metabolic targets.
Conclusion
Immunosuppressive TME limits the success of cancer immunotherapy. Numerous studies have shown that heme, HO-1, and upregulated mitochondrial respiration play an important role in mediating immunosuppressive TME by promoting the recruitment of immunosuppressive cells, hypoxia, angiogenesis, and immune-evasion in TME areas. Various in vitro and in vivo studies show that targeting heme and mitochondrial respiration using heme sequestration molecules, HO-1 inhibitor, mitochondrial respiration inhibitor, and inhibitor of OXPHOS complexes could improve hypoxia, normalize vasculature, reduce oxygen consumption of cancer cells, and ultimately restore immunosupportive TME and anti-tumor immunity. However, much more attention is necessary to study the therapeutic effects of targeting heme and mitochondrial respiration and their combination with immunotherapeutic molecules, especially in clinical trials, which can help to develop effective immunotherapeutic drug combinations.
Author contributions
Conceptualization: ZA and LZ. Literature survey: ZA, N.S, and MA. Writing—original draft preparation: ZA, NS, and MA. Review and editing: ZA, NS, and LZ. Visualization: ZA, NS, and M.Y.A. Supervision: LZ. Funding acquisition: LZ. All authors contributed to the article and approved the submitted version.
Funding
Cancer Prevention and Research Institute of Texas grant PR200021.
Conflict of interest
LZ reports a patent US 11,358,991 B2. LZ has financial interests in HemePro, which has proprietary interests in materials discussed in this manuscript. This conflict of interest is managed with UTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Organization WH. Cancer (Fact sheet) (2022). Available at: https://www.who.int/news-room/fact-sheets/detail/cancer.
3. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol (2021) 14(1):1–33. doi: 10.1186/s13045-021-01164-5
4. Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China cancer immunotherapy workshop in Beijing. J Hematol Oncol (2018) 11(1):1–15. doi: 10.1186/s13045-018-0684-3
5. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book (2019) 39:147–64. doi: 10.1200/EDBK_240837
6. Wang F, Wang S, Zhou Q. The resistance mechanisms of lung cancer immunotherapy. Front Oncol (2020) 10:568059. doi: 10.3389/fonc.2020.568059
7. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
8. Syn NL, Teng MW, Mok TS, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol (2017) 18(12):e731–e41. doi: 10.1016/S1470-2045(17)30607-1
9. Pérez-Ruiz E, Melero I, Kopecka J, Sarmento-Ribeiro AB, García-Aranda M, De Las Rivas J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist Updates (2020) 53:100718. doi: 10.1016/j.drup.2020.100718
10. Wu Z, Zhang C, Najafi M. Targeting of the tumor immune microenvironment by metformin. J Cell Communication Signaling (2021) 16:1–16. doi: 10.1007/s12079-021-00648-w
11. Saleh R, Elkord E. Acquired resistance to cancer immunotherapy: role of tumor-mediated immunosuppression. Seminars in Cancer Biology ( Elsevier) (2020) 65.
12. Horvath L, Thienpont B, Zhao L, Wolf D, Pircher A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)-novel approaches and future outlook. Mol Cancer (2020) 19(1):1–15. doi: 10.1186/s12943-020-01260-z
13. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7(1):1–9. doi: 10.1038/ncomms10501
14. Sica V, Bravo-San Pedro JM, Stoll G, Kroemer G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int J Cancer (2020) 146(1):10–7. doi: 10.1002/ijc.32616
15. Kalainayakan SP, FitzGerald KE, Konduri PC, Vidal C, Zhang L. Essential roles of mitochondrial and heme function in lung cancer bioenergetics and tumorigenesis. Cell Biosci (2018) 8(1):1–12. doi: 10.1186/s13578-018-0257-8
16. Boreel DF, Span PN, Heskamp S, Adema GJ, Bussink J. Targeting oxidative phosphorylation to increase the efficacy of radio-and immune-combination TherapyTargeting OXPHOS to increase radio-immunotherapy efficacy. Clin Cancer Res (2021) 27(11):2970–8. doi: 10.1158/1078-0432.CCR-20-3913
17. Shi S, Chen L, Huang G. Antiangiogenic therapy improves the antitumor effect of adoptive cell immunotherapy by normalizing tumor vasculature. Med Oncol (2013) 30(4):1–7. doi: 10.1007/s12032-013-0698-1
18. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis (2018) 9(2):1–14. doi: 10.1038/s41419-017-0061-0
19. Sun Y, Chen W, Torphy RJ, Yao S, Zhu G, Lin R, et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci Trans Med (2021) 13(604):eabc8922. doi: 10.1126/scitranslmed.abc8922
20. McCarthy EF. The toxins of William b. coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthopaedic J (2006) 26:154.
21. Oncology T. A brief history of immunotherapy (2014). Available at: https://www.targetedonc.com/view/a-brief-history-of-immunotherapy.
22. Dobosz P, Dzieciątkowski T. The intriguing history of cancer immunotherapy. Front Immunol (2019) 10:2965. doi: 10.3389/fimmu.2019.02965
23. Webb ES, Liu P, Baleeiro R, Lemoine NR, Yuan M, Wang Y. Immune checkpoint inhibitors in cancer therapy. J Biomed Res (2018) 32(5):317. doi: 10.7555/JBR.31.20160168
24. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology (2016) 5(6):e1163462. doi: 10.1080/2162402X.2016.1163462
25. Abdolvahab MH, Darvishi B, Zarei M, Majidzadeh-A K, Farahmand L. Interferons: Role in cancer therapy. Immunotherapy (2020) 12(11):833–55. doi: 10.2217/imt-2019-0217
26. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):1–11. doi: 10.1038/s41408-021-00459-7
27. Saxena M, van der Burg SH, Melief CJ, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer (2021) 21(6):360–78. doi: 10.1038/s41568-021-00346-0
28. Apostolopoulos V. Cancer vaccines: research and applications. MDPI; (2019) 11:1041. doi: 10.3390/cancers11081041
29. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AM, Redmond WL, et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer (2017) 81:116–29. doi: 10.1016/j.ejca.2017.01.035
30. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
31. Halima A, Vuong W, Chan TA. Next-generation sequencing: unraveling genetic mechanisms that shape cancer immunotherapy efficacy. J Clin Invest (2022) 132(12). doi: 10.1172/JCI154945
32. Esfahani K, Roudaia L, Buhlaiga NA, Del Rincon S, Papneja N, Miller W. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol (2020) 27(s2):87–97. doi: 10.3747/co.27.5223
34. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–R5. doi: 10.1016/j.cub.2020.06.081
35. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res (2020) 30(6):507–19. doi: 10.1038/s41422-020-0337-2
36. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
37. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
38. Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol (2016) 40:41–8. doi: 10.1016/j.copbio.2016.02.007
39. Del Prete A, Schioppa T, Tiberio L, Stabile H, Sozzani S. Leukocyte trafficking in tumor microenvironment. Curr Opin Pharmacol (2017) 35:40–7. doi: 10.1016/j.coph.2017.05.004
40. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch'ng ES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol (2020) 9:1512. doi: 10.3389/fonc.2019.01512
41. Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol (2019) 10:1799. doi: 10.3389/fimmu.2019.01799
42. Oh DY, Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity (2021) 54(12):2701–11. doi: 10.1016/j.immuni.2021.11.015
43. Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T Cell dysfunction in cancer immunity and immunotherapy. Front Immunol (2019) 10:1719. doi: 10.3389/fimmu.2019.01719
44. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
45. Liudahl SM, Coussens LM. To help or to harm: dynamic roles of CD4+ T helper cells in solid tumor microenvironments. Immunol: Elsevier (2018) 1:97–116. doi: 10.1016/B978-0-12-809819-6.00008-3
46. Lu D, Ni Z, Liu X, Feng S, Dong X, Shi X, et al. Beyond T cells: understanding the role of PD-1/PD-L1 in tumor-associated macrophages. J Immunol Res (2019) 2019. doi: 10.1155/2019/1919082
47. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res (2014) 74(24):7168–74. doi: 10.1158/0008-5472.CAN-14-2458
48. Lin W-W, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117(5):1175–83. doi: 10.1172/JCI31537
49. Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res (2014) 2(3):187–93. doi: 10.1158/2326-6066.CIR-14-0002
50. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3–CCL2 SignalingFAP via STAT3–CCL2 promote tumor immunosuppression. Cancer Res (2016) 76(14):4124–35. doi: 10.1158/0008-5472.CAN-15-2973
51. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in CancerFAP+ CAF diversity and immunotherapy response. Cancer Discov (2020) 10(9):1330–51. doi: 10.1158/2159-8290.CD-19-1384
52. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
53. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol Cancer (2021) 20(1):1–30. doi: 10.1186/s12943-021-01428-1
54. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
55. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell (2012) 21(6):822–35. doi: 10.1016/j.ccr.2012.04.025
56. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood J Am Soc Hematol (2010) 115(7):1461–71. doi: 10.1182/blood-2009-08-237412
57. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, et al. Anti-IL-6 receptor m a b eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol (2012) 42(8):2060–72. doi: 10.1002/eji.201142335
58. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Trans Med (2014) 6(237):237ra67–ra67. doi: 10.1126/scitranslmed.3007974
59. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE2-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer EnvironmentPGE2 controls CXCR4-driven accumulation of MDSCs. Cancer Res (2011) 71(24):7463–70. doi: 10.1158/0008-5472.CAN-11-2449
60. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor CellsCOX-2 blockade and gliomagenesis. Cancer Res (2011) 71(7):2664–74. doi: 10.1158/0008-5472.CAN-10-3055
61. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203(12):2691–702. doi: 10.1084/jem.20061104
62. Nagaraj S, Youn J-I, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res (2010) 16(6):1812–23. doi: 10.1158/1078-0432.CCR-09-3272
63. Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis. Mutation Research/Fundamental and molecular mechanisms of mutagenesis. Mutation Research (2005) 569(1-2):75–85. doi: 10.1016/j.mrfmmm.2004.03.013
64. Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev (2010) 29(2):285–93. doi: 10.1007/s10555-010-9224-5
65. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
66. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol (2018) 39(6):489–502. doi: 10.1016/j.it.2018.01.005
67. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T Cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol (2004) 5(8):818–27. doi: 10.1038/ni1096
68. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195(1):59–70. doi: 10.1084/jem.20010659
69. Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol (2002) 3(12):1129–34. doi: 10.1038/ni1202-1129
70. Yarosz EL, Chang C-H. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw (2018) 18(1). doi: 10.4110/in.2018.18.e14
71. Tyurin V, Balasubramanian K, Winnica D, Tyurina Y, Vikulina A, He R, et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’signals: cleavage and inhibition of phagocytosis by lp-PLA2. Cell Death Differ (2014) 21(5):825–35. doi: 10.1038/cdd.2014.1
72. Fokam D, Hoskin D. Instrumental role for reactive oxygen species in the inflammatory response. Front Biosci Landmark (2020) 25(6):1110–9. doi: 10.2741/4848
73. Araźna M, Pruchniak MP, Demkow U. Reactive oxygen species, granulocytes, and NETosis. Respir Virol Immunogenicity (2014) 836:1–7. doi: 10.1007/5584_2014_12
74. Tan H-Y, Wang N, Li S, Hong M, Wang X, Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longevity (2016) 2016. doi: 10.1155/2016/2795090
75. Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, et al. NADPH oxidases are essential for macrophage differentiation. J Biol Chem (2016) 291(38):20030–41. doi: 10.1074/jbc.M116.731216
76. Roy S, Kumaravel S, Sharma A, Duran CL, Bayless KJ, Chakraborty S. Hypoxic tumor microenvironment: implications for cancer therapy. Exp Biol Med (2020) 245(13):1073–86. doi: 10.1177/1535370220934038
77. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
78. Zhang L. Heme biology: heme acts as a versatile signaling molecule regulating diverse biological processes. In: World scientific (2020).
79. Wang T, Ashrafi A, Konduri PC, Ghosh P, Dey S, Modareszadeh P, et al. Heme sequestration as an effective strategy for the suppression of tumor growth and ProgressionHeme sequestration for suppressing tumor growth. Mol Cancer Ther. World Scientific Publishing Co (2021) 20(12):2506–18. doi: 10.1158/1535-7163.MCT-21-0033
80. Canesin G, Muralidharan AM, Swanson KD, Wegiel B. HO-1 and heme: G-quadruplex interaction choreograph DNA damage responses and cancer growth. Cells (2021) 10(7):1801. doi: 10.3390/cells10071801
81. Ryter SW. Significance of heme and heme degradation in the pathogenesis of acute lung and inflammatory disorders. Int J Mol Sci (2021) 22(11):5509. doi: 10.3390/ijms22115509
82. Bent EH, Millán-Barea LR, Zhuang I, Goulet DR, Fröse J, Hemann MT. Microenvironmental IL-6 inhibits anti-cancer immune responses generated by cytotoxic chemotherapy. Nat Commun (2021) 12(1):1–13. doi: 10.1038/s41467-021-26407-4
83. Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Ségui B. The TNF paradox in cancer progression and immunotherapy. Front Immunol (2019) 10:1818. doi: 10.3389/fimmu.2019.01818
84. Canesin G, Di Ruscio A, Li M, Ummarino S, Hedblom A, Choudhury R, et al. Scavenging of labile heme by hemopexin is a key checkpoint in cancer growth and metastases. Cell Rep (2020) 32(12):108181. doi: 10.1016/j.celrep.2020.108181
85. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
86. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
87. Sohoni S, Ghosh P, Wang T, Kalainayakan SP, Vidal C, Dey S, et al. Elevated heme synthesis and uptake underpin intensified oxidative metabolism and tumorigenic functions in non–small cell lung cancer CellsAltered heme homeostasis in non–small cell lung cancer. Cancer Res (2019) 79(10):2511–25. doi: 10.1158/0008-5472.CAN-18-2156
88. Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The multifaceted role of heme in cancer. Front Oncol (2020) 9:1540. doi: 10.3389/fonc.2019.01540
89. Ghosh P, Guo Y, Ashrafi A, Chen J, Dey S, Zhong S, et al. Oxygen-enhanced optoacoustic tomography reveals the effectiveness of targeting heme and oxidative phosphorylation at normalizing tumor vascular OxygenationNormalizing tumor vascular function via heme targeting. Cancer Res (2020) 80(17):3542–55. doi: 10.1158/0008-5472.CAN-19-3247
90. Dey S, Ashrafi A, Vidal C, Jain N, Kalainayakan SP, Ghosh P, et al. Heme sequestration effectively suppresses the development and progression of both lung adenocarcinoma and squamous cell CarcinomaHeme sequestration for suppressing both ADC and SCC. Mol Cancer Res (2022) 20(1):139–49. doi: 10.1158/1541-7786.MCR-21-0385
91. Petrillo S, De Giorgio F, Kopecka J, Genova T, Fiorito V, Allocco AL, et al. Endothelial heme dynamics drive cancer cell metabolism by shaping the tumor microenvironment. Biomedicines (2021) 9(11):1557. doi: 10.3390/biomedicines9111557
92. Yang D, Guo P, He T, Powell CA. Role of endothelial cells in tumor microenvironment. Clin Trans Med (2021) 11(6). doi: 10.1002/ctm2.450
93. Salmaninejad A, Valilou SF, Soltani A, Ahmadi S, Abarghan YJ, Rosengren RJ, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (2019) 42(5):591–608. doi: 10.1007/s13402-019-00453-z
94. Cheng H, Wang Z, Fu L, Xu T. Macrophage polarization in the development and progression of ovarian cancers: an overview. Front Oncol (2019) 9:421. doi: 10.3389/fonc.2019.00421
95. Théate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld J-C, et al. Extensive profiling of the expression of the indoleamine 2, 3-dioxygenase 1 protein in normal and tumoral human TissuesIDO1 expression profiling in normal and tumoral tissues. Cancer Immunol Res (2015) 3(2):161–72. doi: 10.1158/2326-6066.CIR-14-0137
96. Balog A, T-a L, Maley D, Gullo-Brown J, Kandoussi EH, Zeng J, et al. Preclinical characterization of linrodostat mesylate, a novel, potent, and selective oral indoleamine 2, 3-dioxygenase 1 inhibitor. Mol Cancer Ther (2021) 20(3):467–76. doi: 10.1158/1535-7163.MCT-20-0251
97. Huang J, Wan B, Li S, Liu G, Pang Q, Wu J, et al. High expression of heme oxygenase-1 in tumor-associated macrophages characterizes a poor-prognosis subtype in nasopharyngeal carcinoma. Aging (Albany NY) (2021) 13(4):5674. doi: 10.18632/aging.202492
98. Ye W, Liu Z, Liu F, Luo C. Heme oxygenase-1 predicts risk stratification and immunotherapy efficacy in lower grade gliomas. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.760800
99. Tauber S, Jais A, Jeitler M, Haider S, Husa J, Lindroos J, et al. Transcriptome analysis of human cancer reveals a functional role of heme oxygenase-1 in tumor cell adhesion. Mol Cancer (2010) 9(1):1–16. doi: 10.1186/1476-4598-9-200
100. Nemeth Z, Li M, Csizmadia E, Döme B, Johansson M, Persson JL, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget (2015) 6(32):33675. doi: 10.18632/oncotarget.5284
101. Zhang T, Fang Q, Liu P, Wang P, Feng C, Wang J. Heme oxygenase 1 overexpression induces immune evasion of acute myeloid leukemia against natural killer cells by inhibiting CD48. J Trans Med (2022) 20(1):1–13. doi: 10.1186/s12967-022-03589-z
102. Chiang S-K, Chen S-E, Chang L-C. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci (2018) 20(1):39. doi: 10.3390/ijms20010039
103. Khojandi N, Kuehm LM, Piening A, Donlin MJ, Hsueh EC, Schwartz TL, et al. Oxidized lipoproteins promote resistance to cancer immunotherapy independent of patient ObesityOxidized lipids promote resistance to cancer immunotherapy. Cancer Immunol Res (2021) 9(2):214–26. doi: 10.1158/2326-6066.CIR-20-0358
104. Kopecka J, Gazzano E, Castella B, Salaroglio IC, Mungo E, Massaia M, et al. Mitochondrial metabolism. In: Inducer or therapeutic target in tumor immuneresistance? Seminars in Cancer Biology (Elsevier) (2020). doi: 10.1016/j.semcdb.2019.05.008
105. Song K-H, Kim J-H, Lee Y-H, Bae HC, Lee H-J, Woo SR, et al. Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance. J Clin Invest (2018) 128(9):4098–114. doi: 10.1172/JCI96804
106. Jiang Q, Zhang C, Wang H, Peng T, Zhang L, Wang Y, et al. Mitochondria-targeting immunogenic cell death inducer improves the adoptive T-cell therapy against solid tumor. Front Oncol (2019) 9:1196. doi: 10.3389/fonc.2019.01196
107. Klein K, He K, Younes AI, Barsoumian HB, Chen D, Ozgen T, et al. Role of mitochondria in cancer immune evasion and potential therapeutic approaches. Front Immunol (2020) 11:573326. doi: 10.3389/fimmu.2020.573326
108. Li W, Zhang L. Rewiring mitochondrial metabolism for CD8+ T cell memory formation and effective cancer immunotherapy. Front Immunol (2020) 11:1834. doi: 10.3389/fimmu.2020.01834
109. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res (2018) 24(11):2482–90. doi: 10.1158/1078-0432.CCR-17-3070
110. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519. doi: 10.1085/jgp.8.6.519
111. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell (2016) 164(4):681–94. doi: 10.1016/j.cell.2015.12.034
112. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell (2017) 171(2):358–71.e9. doi: 10.1016/j.cell.2017.09.019
113. Sotgia F, Lisanti MP. Mitochondrial markers predict survival and progression in non-small cell lung cancer (NSCLC) patients: Use as companion diagnostics. Oncotarget (2017) 8(40):68095. doi: 10.18632/oncotarget.19677
114. Liu A, Curran MA. Tumor hypermetabolism confers resistance to immunotherapy. Seminars in Cancer Biology (Elsevier) (2020). doi: 10.1016/j.semcancer.2020.01.009
115. Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight (2019) 4(5). doi: 10.1172/jci.insight.124989
116. Taghizadeh-Hesary F, Akbari H, Bahadori M, Behnam B. Targeted anti-mitochondrial therapy: the future of oncology. Genes (2022) 13(10):1728. doi: 10.3390/genes13101728
117. Aboelella NS, Brandle C, Kim T, Ding Z-C, Zhou G. Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers (2021) 13(5):986. doi: 10.3390/cancers13050986
118. Tang J, Yu JX, Hubbard-Lucey VM, Neftelinov ST, Hodge JP, Lin Y. Trial watch: the clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat Rev Drug Discov (2018) 17(12):854–6. doi: 10.1038/nrd.2018.210
119. Merlano M, Abbona A, Denaro N, Garrone O. Knowing the tumour microenvironment to optimise immunotherapy. Acta Otorhinolaryngol Italica (2019) 39(1):2. doi: 10.14639/0392-100X-2481
120. Petrillo S, Chiabrando D, Genova T, Fiorito V, Ingoglia G, Vinchi F, et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ (2018) 25(3):573–88. doi: 10.1038/s41418-017-0001-7
121. Consonni FM, Bleve A, Totaro MG, Storto M, Kunderfranco P, Termanini A, et al. Heme catabolism by tumor-associated macrophages controls metastasis formation. Nat Immunol (2021) 22(5):595–606. doi: 10.1038/s41590-021-00921-5
122. Kim SH, Kim S-J, Park J, Joe Y, Lee SE, Saeidi S, et al. Reprograming of tumor-associated macrophages in breast tumor-bearing mice under chemotherapy by targeting heme oxygenase-1. Antioxidants (2021) 10(3):470. doi: 10.3390/antiox10030470
123. Magri S, Musca B, Pinton L, Orecchini E, Belladonna L, Orabona C, et al. The immunosuppression pathway of tumor-associated macrophages is controlled by heme oxygenase-1 in glioblastoma patients. Int J Cancer (2022) 51:2265–77. doi: 10.1002/ijc.34270
124. Alaluf E, Vokaer B, Detavernier A, Azouz A, Splittgerber M, Carrette A, et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight (2020) 5(11). doi: 10.1172/jci.insight.133929
125. Cheng C-C, Guan S-S, Yang H-J, Chang C-C, Luo T-Y, Chang J, et al. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J Biomed Sci (2016) 23(1):1–10. doi: 10.1186/s12929-016-0219-6
126. Abdalla MY, Ahmad IM, Rachagani S, Banerjee K, Thompson CM, Maurer HC, et al. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Trans Res (2019) 207:56–69. doi: 10.1016/j.trsl.2018.12.008
127. Kalainayakan SP, Ghosh P, Dey S, Fitzgerald KE, Sohoni S, Konduri PC, et al. Cyclopamine tartrate, a modulator of hedgehog signaling and mitochondrial respiration, effectively arrests lung tumor growth and progression. Sci Rep (2019) 9(1):1–15. doi: 10.1038/s41598-018-38345-1
128. Pierini S, Fang C, Rafail S, Facciponte JG, Huang J, De Sanctis F, et al. A tumor mitochondria vaccine protects against experimental renal cell carcinoma. J Immunol (2015) 195(8):4020–7. doi: 10.4049/jimmunol.1500281
129. Oh M-H, Sun I-H, Zhao L, Leone RD, Sun I-M, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/JCI131859
130. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24(7):1036–46. doi: 10.1038/s41591-018-0052-4
131. Yen J-H, Huang W-C, Lin S-C, Huang Y-W, Chio W-T, Tsay GJ, et al. Metabolic remodeling in tumor-associated macrophages contributing to antitumor activity of cryptotanshinone by regulating TRAF6-ASK1 axis. Mol Therapy Oncolytics (2022) 26:158–74. doi: 10.1016/j.omto.2022.06.008
132. Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, et al. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int Immunol (2019) 31(4):187–98. doi: 10.1093/intimm/dxy079
133. Bauer A, Mylroie H, Thornton CC, Calay D, Birdsey GM, Kiprianos AP, et al. Identification of cyclins A1, E1 and vimentin as downstream targets of heme oxygenase-1 in vascular endothelial growth factor-mediated angiogenesis. Sci Rep (2016) 6(1):1–16. doi: 10.1038/srep29417
134. Jayaprakash P, Vignali PDA, Delgoffe GM, Curran MA. Hypoxia reduction sensitizes refractory cancers to immunotherapy. Annu Rev Med (2022) 73:251–65. doi: 10.1146/annurev-med-060619-022830
135. Zhang C, Yan Q, Li J, Zhu Y, Zhang Y. Nanoenabled tumor oxygenation strategies for overcoming hypoxia-associated immunosuppression. ACS Appl Bio Mater (2020) 4(1):277–94. doi: 10.1021/acsabm.0c01328
136. Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updates (2021) 59:100787. doi: 10.1016/j.drup.2021.100787
137. Ileiwat ZE, Tabish TA, Zinovkin DA, Yuzugulen J, Arghiani N, Pranjol MZI. The mechanistic immunosuppressive role of the tumour vasculature and potential nanoparticle-mediated therapeutic strategies. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.976677
138. Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis (2017) 20(2):185–204. doi: 10.1007/s10456-017-9552-y
139. Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mourón S, et al. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Rep (2016) 15(12):2705–18. doi: 10.1016/j.celrep.2016.05.052
140. Yap TA, Ahnert JR, Piha-Paul SA, Fu S, Janku F, Karp DD, et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J Clin Oncol (2019) 37(15_suppl):3014. doi: 10.1200/JCO.2019.37.15_suppl.3014
141. Zhang J, Zhang D, Li Q, Jiang Y, Song A, Li Z, et al. Task-specific design of immune-augmented nanoplatform to enable high-efficiency tumor immunotherapy. ACS Appl Mater Interf (2019) 11(46):42904–16. doi: 10.1021/acsami.9b13556
142. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer ImmunotherapyTailoring cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi: 10.1158/1078-0432.CCR-14-1860
143. Kassab SE, Mowafy S. Structural basis of selective human indoleamine-2, 3-dioxygenase 1 (hIDO1) inhibition. Chem Med Chem (2021) 16(20):3149–64. doi: 10.1002/cmdc.202100253
144. Sun L. Advances in the discovery and development of selective heme-displacing IDO1 inhibitors. Expert Opin Drug Discov (2020) 15(10):1223–32. doi: 10.1080/17460441.2020.1781811
145. Schillingmann DA, Riese SB, Vijayan V, Tischer-Zimmermann S, Schmetzer H, Maecker-Kolhoff B, et al. Inhibition of heme oxygenase-1 activity enhances wilms tumor-1-Specific T-cell responses in cancer immunotherapy. Int J Mol Sci (2019) 20(3):482. doi: 10.3390/ijms20030482
146. Guo Y, Xie Y-Q, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat Immunol (2021) 22(6):746–56. doi: 10.1038/s41590-021-00940-2
147. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
148. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti–PD-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
149. Malinee M, Pandian GN, Sugiyama H. Targeted epigenetic induction of mitochondrial biogenesis enhances antitumor immunity in mouse model. Cell Chem Biol (2022) 29(3):463–75.e6. doi: 10.1016/j.chembiol.2021.08.001
150. Zuo H, Hou Y, Yu Y, Li Z, Liu H, Liu C, et al. Circumventing myeloid-derived suppressor cell-mediated immunosuppression using an oxygen-generated and-economized nanoplatform. ACS Appl Mater Interf (2020) 12(50):55723–36. doi: 10.1021/acsami.0c18180
151. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor HypoxiaMetformin improves PD-1 blockade immunotherapy. Cancer Immunol Res (2017) 5(1):9–16. doi: 10.1158/2326-6066.CIR-16-0103
152. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest (2018) 128(11):5137–49. doi: 10.1172/JCI96268
153. Chen D, Barsoumian HB, Fischer G, Yang L, Verma V, Younes AI, et al. Combination treatment with radiotherapy and a novel oxidative phosphorylation inhibitor overcomes PD-1 resistance and enhances antitumor immunity. J Immunother Cancer (2020) 8(1). doi: 10.1136/jitc-2019-000289
154. Sainero-Alcolado L, Liaño-Pons J, Ruiz-Pérez MV, Arsenian-Henriksson M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ (2022) 29(7):1304–17. doi: 10.1038/s41418-022-01022-y
Keywords: tumor micoenvironment, heme, mitochondrial respiration, hypoxia, angiogenesis, cancer immunotherapy
Citation: Akter Z, Salamat N, Ali MY and Zhang L (2023) The promise of targeting heme and mitochondrial respiration in normalizing tumor microenvironment and potentiating immunotherapy. Front. Oncol. 12:1072739. doi: 10.3389/fonc.2022.1072739
Received: 17 October 2022; Accepted: 12 December 2022;
Published: 04 January 2023.
Edited by:
Tong Xiang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Sergio Dias, Universidade de Lisboa, PortugalXiaopin Duan, Southern Medical University, China
Copyright © 2023 Akter, Salamat, Ali and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, bGkuemhhbmdAdXRkYWxsYXMuZWR1
†These authors have contributed equally to this work and share first authorship