 Yanhong Shang1†Jianming Mo2†Ran Huo1Xiaofang Li1Guotao Fang1Zichun Wei3Guomin Gu4Xiaodan Zhu4
Yanhong Shang1†Jianming Mo2†Ran Huo1Xiaofang Li1Guotao Fang1Zichun Wei3Guomin Gu4Xiaodan Zhu4 Chan Zhang5Chunling Liu6*
Chan Zhang5Chunling Liu6* Dong Yan5*
Dong Yan5*- 1Department of Medical Oncology, Affiliated Hospital of Hebei University, Hebei Key Laboratory of Cancer Radiotherapy and Chemotherapy, Baoding, China
- 2Department of Pulmonary and Critical Care Medicine, Peking University Shenzhen Hospital, Shenzhen, China
- 3Thoracic Surgery, Peking University Shenzhen Hospital, Shenzhen, China
- 4Second Department of Pulmonary Medicine, Affiliated Cancer Hospital of Xinjiang Medical University, Urumqi, China
- 5Department of Oncology, Beijing Luhe Hospital, Capital Medical University, Beijing, China
- 6Department of Pulmonary Medicine, Affiliated Cancer Hospital of Xinjiang Medical, Urumqi, China
Background: Although rare, ERBB2 exon 16 skipping mutations (ERBB2ΔEx16) have been implicated in resistance to anti-HER2 and anti-EGFR targeted agents. Our study investigated the prevalence and clinical significance of ERBB2ΔEx16 in Chinese pan-cancer patients.
Methods: We retrospectively screened 40996 patients, spanning 19 cancer types, who had available genomic profiles acquired with DNA-based next-generation sequencing (NGS). We characterized the clinical and molecular features of the ERBB2ΔEx16-positive patients. Furthermore, we also analyzed a pan-cancer dataset from the Cancer Genome Atlas (TCGA; n=8705).
Results: A total of 22 patients were detected with ERBB2ΔEx16, resulting in an overall prevalence rate of 0.054% (22/40996). Of them, 16 patients had lung cancer (LC; 0.05%, 16/30890), five patients had gastric cancer (GC; 0.35%, 5/1448), and one patient had ovarian cancer (0.12%, 1/826). Among the 16 LC patients, ERBB2ΔEx16 was detected in four treatment-naïve EGFR/ALK-negative patients and 12 EGFR-positive patients after the onset of resistance to EGFR tyrosine kinase inhibitors (TKIs). The treatment-naïve patients harbored no LC-associated oncogenic drivers except ERBB2 amplification, suggesting a potential oncogenic role for ERBB2ΔEx16. Consistently, ERBB2ΔEx16+ patients from TCGA data also carried no known drivers despite various concurrent alterations. In the 12 EGFR TKI-resistant LC patients, relative variant frequencies for ERBB2ΔEx16 were lower than in untreated patients, suggesting ERBB2ΔEx16 as secondary alterations following TKI treatment and thereby implicating ERBB2ΔEx16 in mediating therapeutic resistance.
Conclusions: Our study identified an overall ERBB2ΔEx16 prevalence rate of 0.054% and provided insights into the clinical implications of ERBB2ΔEx16 in Chinese pan-cancer patients.
Introduction
The Erb-B receptor tyrosine kinase 2 (HER2 or ERBB2) is a member of the Erb-B family and structurally related to the epidermal growth factor receptor (EGFR) (1). HER2 is a 185 kDa transmembrane receptor that lacks a ligand-binding structure and acts as a co-receptor by forming more potent heterodimers with HER1/EGFR and HER3/ErbB3 (1–5). Through its critical role in regulating cell growth and development, dysregulation of HER2 signaling, particularly HER2 overexpression, is one of the oncogenic drivers in various solid malignancies (1, 2, 4, 5). In addition to HER2 overexpression, genetic mutations affecting the extracellular, transmembrane, and kinase domains of ERBB2 have been reported as alternative mechanisms of HER2 activation in various solid tumors and affect tumor biology and treatment response (5–8). Genetic alterations affecting the exon 16 of ERBB2 (ERBB2ΔEx16) result in alternative splicing, lead to exon 16 skipping, and produce an altered HER2 protein that lacks 16 amino acids in the extracellular domain (amino acid positions 634-649) (5, 9, 10). ERBB2ΔEx16 is comprised of short in-frame deletions affecting exon 16 and missense mutations in splice donor or acceptor sites flanking exon 16. By altering gene splicing, the ERBB2ΔEx16 isoform could result in molecular conformational change by exposing cysteine residues crucial in intermolecular disulfide bond formation and lead to the constitutive activation and stable covalent binding of HER2 homodimers with more enhanced transformational activity (9–14).
ERBB2ΔEx16 was first reported in HER2-overexpressed breast cancer after prolonged targeted treatment with trastuzumab (5, 10–12). Preclinical studies in cell and mouse models have demonstrated the constitutive activation of ERBB2ΔEx16 and its critical role in tumorigenesis of breast and lung (9–15). Clinical studies have identified an ERBB2ΔEx16 prevalence of 0.01-0.19% in various solid tumor types (16, 17). Albeit rare, ERBB2ΔEx16 was implicated mostly in resistance to anti-HER2 agents in HER2-positive breast tumors and osimertinib in EGFR-mutant non-small-cell lung cancer (NSCLC) (5, 12, 16, 18). A deeper molecular understanding of the underlying pathogenesis of cancers could pave avenues for improving the treatment and survival outcomes of patients with ERBB2ΔEx16-positive (ERBB2ΔEx16+) tumors. In a large-scale analysis of Chinese cancer patients, Shi et al. identified 0.046% ERBB2ΔEx16+ cases (18/38680) spanning lung, colorectal, gastric, and ovarian cancers (19). Our study investigated the prevalence and clinical significance of ERBB2ΔEx16 in Chinese pan-cancer patients (n = 40996). To further characterize their mutational landscape and aberrant pathways, we also analyzed ERBB2ΔEx16-positive patients screened from pan-cancer datasets from the Cancer Genome Atlas (TCGA; n=8705) (20, 21).
Patients and methods
Patients
We retrospectively screened 40996 Chinese patients, spanning 19 cancer types, who voluntarily submitted either tissue or plasma samples for DNA-based next-generation sequencing (NGS) using either 168- or 520-gene panels between January 2018 to December 2020. We analyzed the clinical and molecular profile of the patients detected with ERBB2ΔEx16. For comparison, we obtained the molecular and survival data of a pan-cancer dataset from the TCGA (n=8705) (20, 21). This study was IRB approved and conducted in accordance with the ethical guidelines including Declaration of Helsinki and US Common Rule.
Targeted next-generation sequencing
DNA was isolated from blood and tumor samples and subjected to NGS in Burning Rock Biotech, a clinical laboratory accredited by the College of American Pathologists and certified by the Clinical Laboratory Improvement Amendments, according to optimized protocols as described previously (22, 23). NGS library construction required a minimum of 30 ng of DNA. Target capture was performed using commercial panels consisting of either 168 or 520 cancer-related genes, which respectively span 0.269 Megabases (Mb) and 1.003 Mb of the human genome (Burning Rock Biotech, Guangzhou, China) (24, 25). Indexed samples were sequenced on NovaSeq 6000 (Illumina, Inc., CA, USA) with 150-bp read lengths and a target sequencing depth of 1,000× for tissue samples and 10,000× for plasma samples. Maximum allelic frequency (MaxAF) was defined as the maximum allelic frequency detected from a sample, and relative allelic frequency (RAF) was calculated as the ratio of the allelic frequency of a certain variant to MaxAF.
Sequence data analysis
Sequence data were analyzed using the Burning Rock analysis system as previously described (22, 23, 26). Briefly, sequence data were mapped to the reference human genome (hg19). Variants with depth <100 or population frequency >0.1% in major databases were excluded from further analysis. Copy number was calculated based on the ratio between the depth of coverage in tumor samples and average coverage of an adequate number (n>50) of samples without CNV as references per capture interval. The cut-offs for copy number variations were 1.5 for copy number deletion and 2.5 for copy number amplifications.
Statistical analysis
Statistical analyses were performed using the Wilcoxon signed-rank test or Fisher’s exact test as appropriate in R software (version 4.0.2). A two-sided P value <0.05 was considered statistically significant.
Results
Pan-cancer prevalence of ERBB2ΔEx16 alterations
Figure 1A illustrates the study design. We started with screening 40996 Chinese patients spanning 19 cancer types, most of which were lung cancer (LC; 71.4%), for patients who harbored ERBB2ΔEx16 (Table S1). A total of 22 patients were detected with ERBB2ΔEx16, resulting in an overall prevalence rate of 0.054% (22/40996). Table 1 summarizes their clinical characteristics. The majority were female (54.5%) and had stage IV disease (60.9%). Most had LC (0.05%, 16/30,890), five patients had gastric cancer (GC; 0.35%, 5/1,448), and one patient had ovarian cancer (OC; 0.12%, 1/826). ERBB2ΔEx16 was not detected in other cancer types, including colorectal and breast.
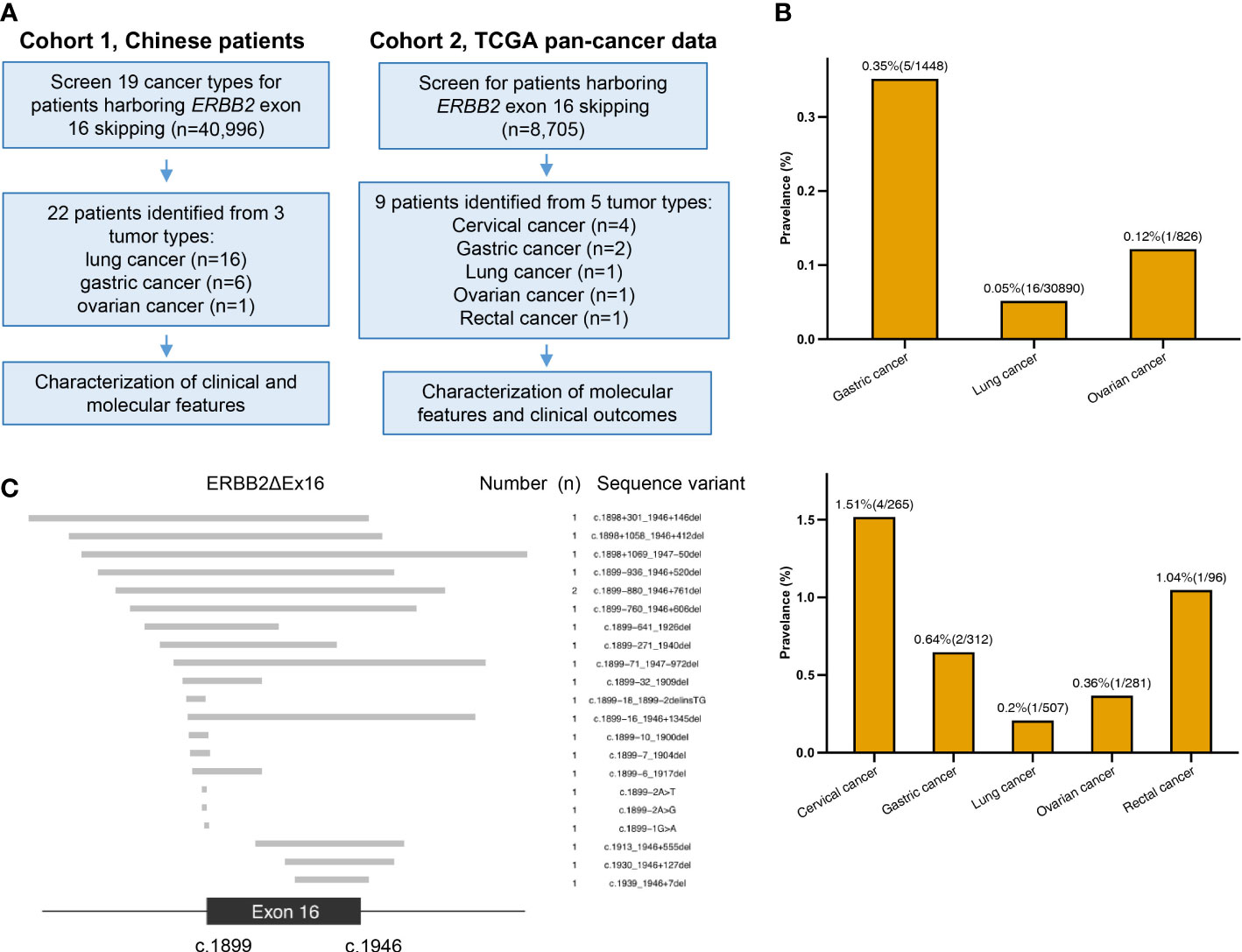
Figure 1 Study design and somatic ERBB2 exon 16 skipping (ERBB2ΔEx16) analyzed in this study. (A) A diagram of study workflow. (B) Number of ERBB2ΔEx16+ patients from Cohorts 1 (upper panel) and 2 (lower panel) per tumor type, shown above the bars are the tumor type-specific prevalence (number of ERBB2ΔEx16+ patients/number of screened patients). (C) Diagram of the 22 unique sequence variants detected in the Chinese cancer patients cohort. The exon 16 (between nucleotides 1899 to 1946) and the flanking regions are shown, in which the gray bars correspond to each unique ERBB2ΔEx16 isoform as shown by the sequence variant on the right column.
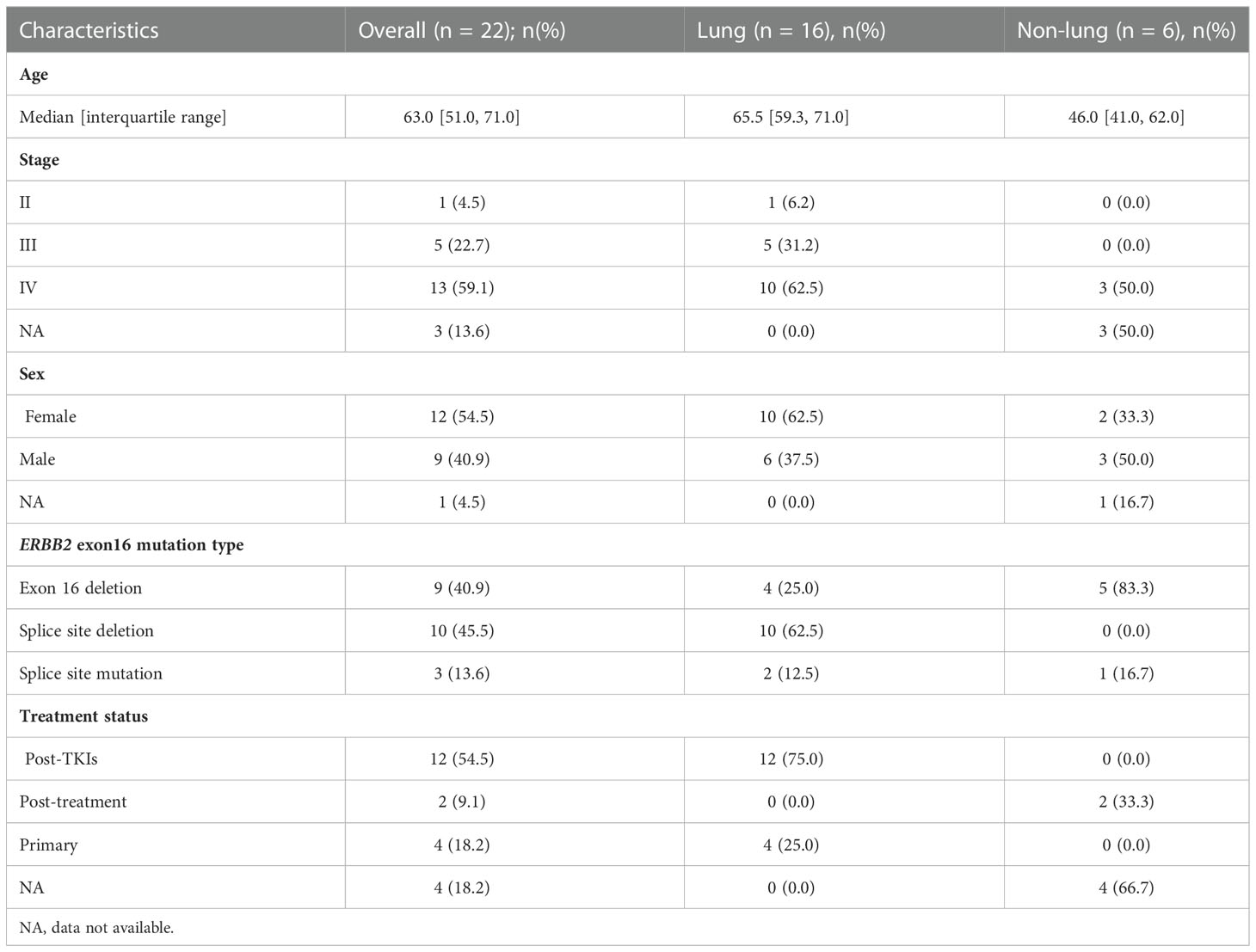
Table 1 Clinical characteristics of the 22 patients detected with ERBB2 exon 16 mutations in Cohort 1.
We also screened 34 TCGA pan-cancer datasets (n = 8705) and identified nine ERBB2ΔEx16+ patients (Figure 1B). Of them, four patients had cervical squamous cell carcinoma and endocervical adenocarcinoma, two patients had stomach adenocarcinoma, and a patient each had lung adenocarcinoma, OC, and rectum adenocarcinoma. Clinical characteristics of the group and each patient are respectively detailed in Tables S2 and S3.
Of the 21 unique ERBB2ΔEx16 variants detected from Chinese patients, 9 involved complete deletion of exon 16, 3 were deletions or point mutations involving splice donors, and 9 deletions or point mutations affecting the splice acceptors (Figure 1C). ERBB2 c.1899-880_1946+761del was detected from two patients. The other ERBB2ΔEx16 variants were unique and only detected in a patient each (Table 2). We also identified a novel variant ERBB2 c.1899-2A>G, which was the only mutation detected from the paired tissue and plasma samples of an OC patient after treatment (P17; Table 2; Figure 1C).
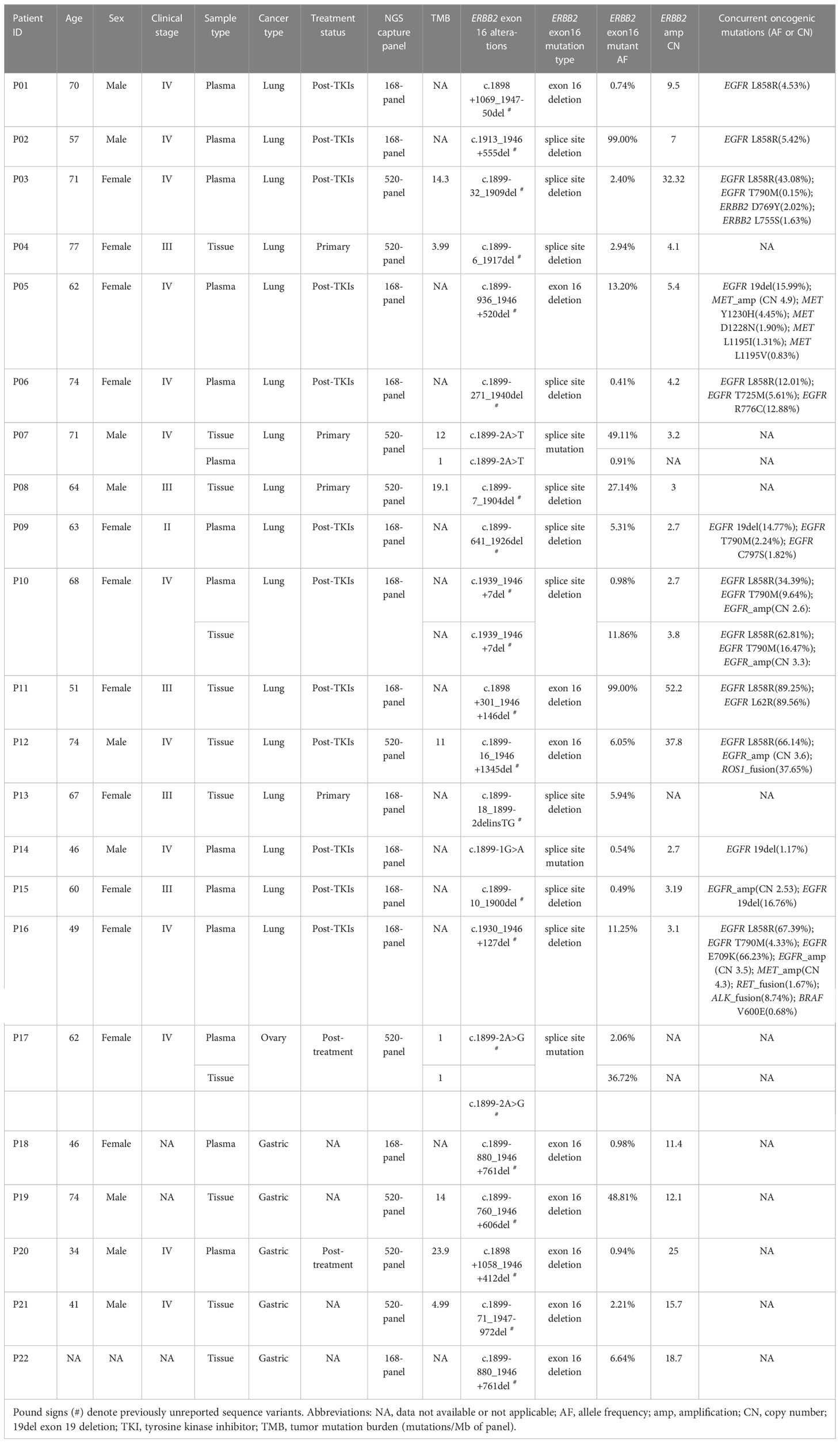
Table 2 Detailed clinical and mutational profile of the 22 patients detected with ERBB2 exon 16 alterations in Cohort 1.
Genomic profiles of ERBB2ΔEx16+ patients suggested potential roles in tumorigenesis and resistance to EGFR TKI in LC
Next, we characterized the clinical and molecular features of the 22 Chinese ERBB2ΔEx16+ patients. Among the 16 LC patients, 12 were EGFR-positive and had progressed on EGFR tyrosine kinase inhibitor (TKI) therapy (75.0%) and four were EGFR/ALK-negative and treatment-naïve (25.0%; Table 2). All ERBB2ΔEx16+ patients had concurrent ERBB2 gene amplification except P08 (94.0%, 15/16; Figure 2A). Among the four previously untreated patients, three also harbored ERBB2 amplification or STK11 point mutations, two harbored SMARCA4 or KEAP1 point mutations, and one carried mutated TP53, BRAF, or CTNNB1 (Figure 2A). No oncogenic driver gene mutations were detected from these four patients except ERBB2 amplification, suggesting mutual exclusivity between ERBB2ΔEx16 and established drivers, and therefore a potential oncogenic role of ERBB2ΔEx16 in LC. This potential tumor-promoting activity was also supported by higher relative variant frequencies (RAFs) of ERBB2ΔEx16 detected in untreated LC patients than in their EGFR TKI-resistant counterparts or in GC patients (Figure 2B). The higher RAFs suggested an increased likelihood for ERBB2ΔEx16 to be clonal in the untreated lung tumors and therefore more likely to function as an oncogenic driver in LC. Consistent with our observation in Chinese patients, ERBB2ΔEx16+ patients from TCGA datasets also harbored no oncogenic drivers despite various concurrent alterations (Figure S1).
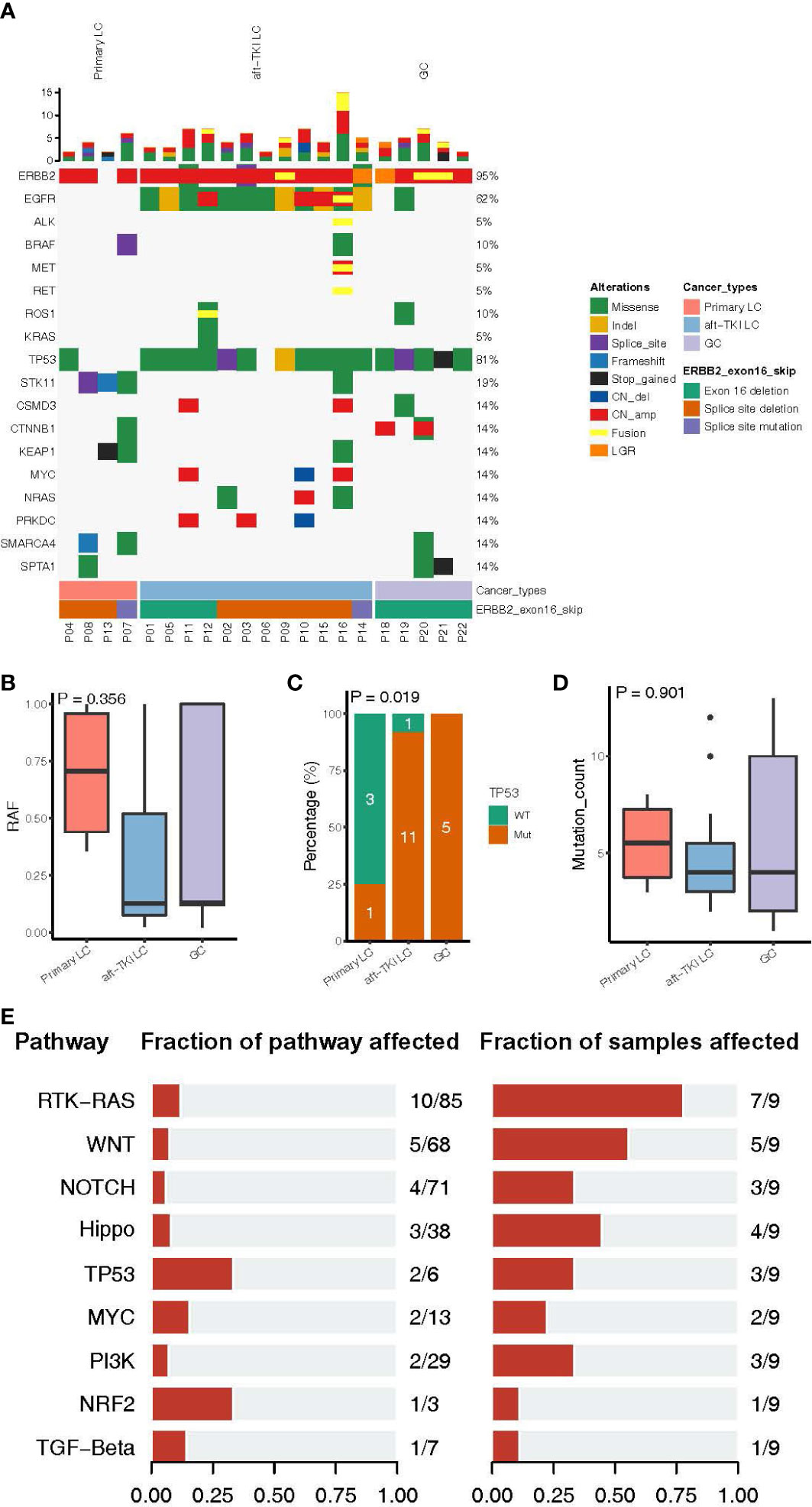
Figure 2 Molecular characteristics of patients harboring ERBB2 exon 16 skipping (ERBB2ΔEx16). (A) An oncoprint of somatic mutation landscape of ERBB2ΔEx16+ patients from Cohort 1. Each row represents a gene indicated on the left, with the mutation rate indicated on the right. Each column represents a patient. Different colors denote the mutation types. Bar plots on top of the oncoprint summarize the number of mutations each patient carries. The cancer type, ERBB2ΔEx16 variant type, and patient ID of each patient were indicated by various colors at the bottom. (B) Relative allele frequency (RAF), (C) mutation frequencies of TP53, and (D) number of somatic mutations in lung cancer (LC) patients before systemic therapy, LC patients after progression on EGFR tyrosine kinase inhibitors (TKIs), and in gastric cancer (GC) patients. (E) Genomic alterations harbored by ERBB2ΔEx16+ patients from TCGA were categorized by pathway and analyzed for the proportion of mutated among all pathway-related genes and the corresponding mutation frequency in patients. Number pairs in the middle (e.g. “10/85” for the RTK-RAS pathway), indicate the number of genes encoding for members of the indicated pathway that were found altered in ERBB2ΔEx16+ patients and number of genes encoding for members of the pathway, respectively. Number pairs at the rightmost (e.g. “7/9” for the RTK-RAS pathway), indicate the number of ERBB2ΔEx16+ patients carrying ≥1 alteration in genes in the indicated pathway and the number of ERBB2ΔEx16+ patients, respectively.
In addition to RAF, there were differences among the three patient subgroups in other molecular features. Treatment-naïve ERBB2ΔEx16+ LC patients were the least likely to carry concurrent TP53 alterations (Figure 2C) and had the highest number of somatic mutations (Figure 2D). Analysis of genomic profiles from the TCGA data revealed scarce concurrent oncogenic driver alterations (Figure S1). Consistently, grouping co-occurring aberrations by pathway revealed likely aberrant signaling cascades in ERBB2ΔEx16+ patients, the most frequently of which receptor tyrosine kinase (RTK)-Ras (7/9 patients), WNT (5/9 patients), and Hippo (4/9 patients) signaling pathways (Figure 2E). Although their significance in tumor biology is well documented, the roles of these cascades in promoting transformation or conferring therapeutic resistance still await more clinical evidence. Taken together, the landscape of concomitant genomic alterations implicated ERBB2ΔEx16 in mediating oncogenesis and EGFR-TKI resistance and warrants further clinical evidence for validation.
Among the Chinese ERBB2ΔEx16+ patients, 12 had EGFR-positive LC and had been previously treated with EGFR TKI(s), accounting for 0.068% of the patients carrying EGFR sensitizing mutations (n = 17753). The lower RAFs suggested ERBB2ΔEx16 as a subclonal and/or secondary aberration in these EGFR TKI-resistant patients (Figure 2B). All 12 harbored concurrent ERBB2 amplification and one had MET amplification and fusion (Figure 2A). The predominant majority (91.7%, 11/12) also harbored TP53 mutations, which was in stark contrast with the treatment-naïve LC patients (Figure 2C). While ERBB2 and MET amplification had been reported to confer resistance to EGFR TKI, their activities do not exclude the possibility of ERBB2ΔEx16 contributing to therapeutic resistance.
Case vignettes
P03 was a female patient with EGFR L858R-mutant advanced LUAD with bone metastasis. ERBB2ΔEx16 was detected after disease progression with osimertinib using her plasma samples but not in the paired tissue rebiopsy (Table 2; Figure 3A). Prior to fourth-line osimertinib, the patient had received first-line chemotherapy, followed by sequential EGFR TKIs, including second-line gefitinib and third-line afatinib monotherapies. In addition to EGFR L858R, her plasma sample collected after osimertinib progression was also detected with other concurrent ERBB2 alterations, including ERBB2ΔEx16 (c.1899-32_1909del), gene amplification (copy number: 32.32), and L755S and D769Y (27), suggesting one or more of these alterations as resistance mechanisms.
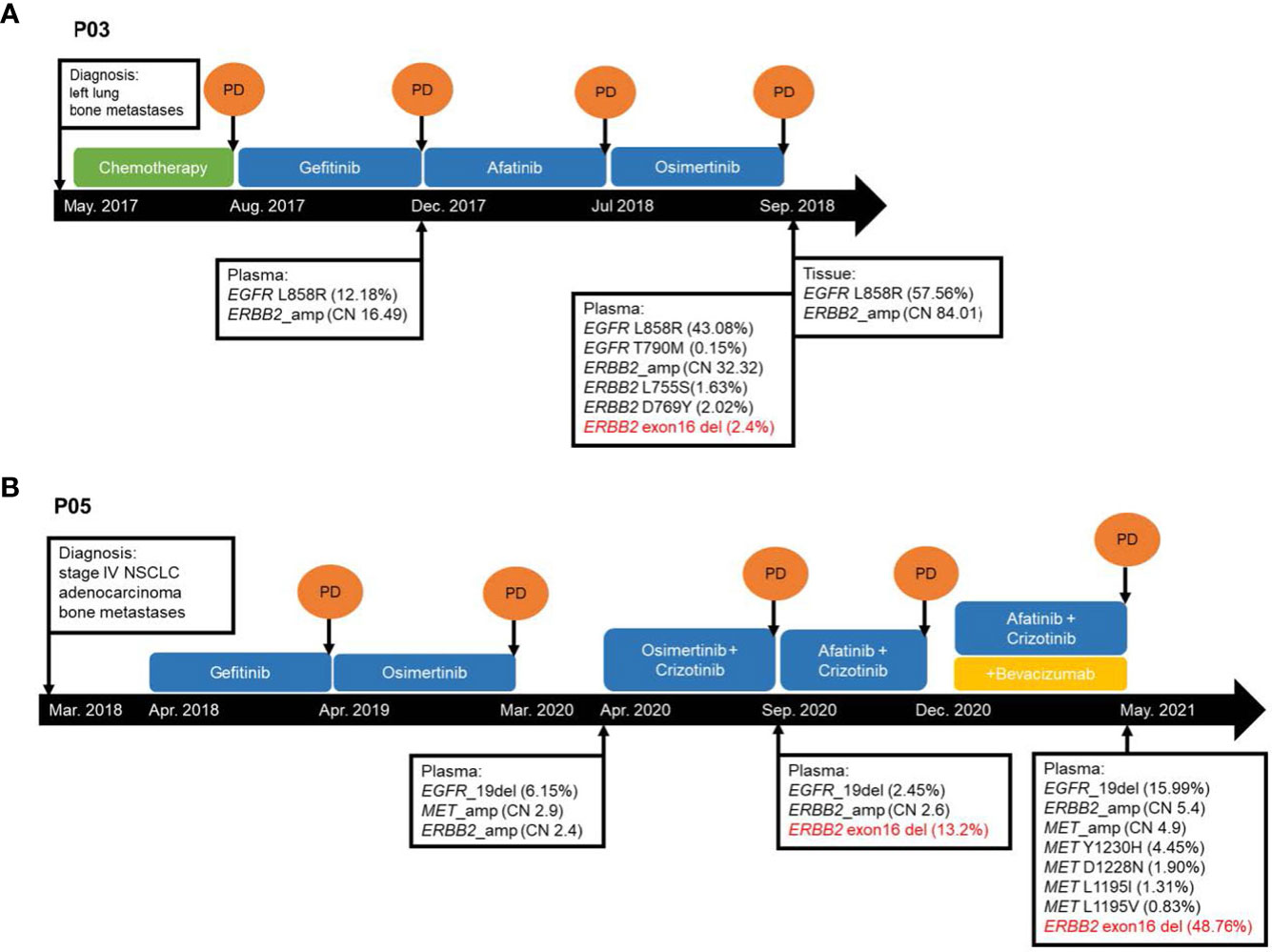
Figure 3 Schematic presentation of the courses of management for two ERBB2ΔEx16+ lung cancer patients (A) P03 and (B) P05 from Cohort 1. CN, copy number. NSCLC, non-small cell lung cancer. PD, progressive disease.
P05 was a female patient with EGFR exon 19 deletion-mutant stage IV LUAD with bone metastasis, and ERBB2ΔEx16 was identified from her plasma sample after progression on osimertinib plus crizotinib (Table 2; Figure 3B). She had received first-line gefitinib and second-line osimertinib and was subsequently detected with MET amplification after the second disease progression. After onset of resistance to third-line osimertinib plus crizotinib, her plasma sample was detected with EGFR exon 19 deletion, ERBB2ΔEx16 (c.1899-936_1946+520del), and ERBB2 amplification. She was then treated with afatinib and crizotinib but with no clinical benefit, and bevacizumab was subsequently added to the regimen. Upon progression, molecular testing revealed increased levels of ERBB2 amplification (copy number: 2.6 to 5.4) and ERBB2ΔEx16 (VAF from 13.2% to 48.8%) as well as various MET alterations, including amplification, Y1230H, D1288N L1195I, and L1195V, the latter four of which are known secondary mutations associated with MET TKI resistance (28, 29). Similar to patient P03, one or more of these genetic abnormalities may have conferred resistance to the targeted agents. Together, these cases illustrate the potential role of ERBB2ΔEx16 in mediating EGFR TKI resistance and disease progression in LC.
Discussion
Through screening a large real-world population, our findings demonstrated an overall ERBB2ΔEx16 prevalence of 0.054% (22/40996) among Chinese cancer patients. Our findings were consistent with another real-world study that reported a prevalence of 0.046% (18/38,680) (19). Importantly, almost all the in-frame deletions identified in our study were unique and were absent from the Catalogue of Somatic Mutations in Cancer (COSMIC) database. Of the 21 unique ERBB2ΔEx16 from our cohort, ERBB2 c.1899-1G>A was detected from a patient each with LC and GC. ERBB2 c.1899-1G>A was previously reported in breast, lung, and gastric cancer (16, 30). Moreover, ERBB2 c.1899-2A>T with concurrent ERBB2 gene amplification were the mutations detected from baseline tumor and plasma samples of a stage IV LC patient (P07). This variant was also identified in an LC patient in the TCGA datasets and a previous study (16). Contrastingly, ERBB2 c.1899-2A>G, a different transition located at the same splice site, is absent from the COSMIC database. We identified this variant from an OC patient (P17) as the only potentially oncogenic mutation in paired tumor (520-gene panel) and plasma (168-gene panel) samples, which suggested a tumorigenic role for this ERBB2ΔEx16 variant.
The oncogenic activity of ERBB2ΔEx16 was first suggested in breast tumors (10). Compared with ERBB2 wild-type tumors, ERBB2ΔEx16+ mammary tumors exhibit a higher degree of intratumoral heterogeneity as shown by distinct signaling and gene expression profiles associated with the activation of tumor initiation and progression (14, 31). Subsequent preclinical studies have established the critical role of ERBB2ΔEx16 in the increased aggressiveness of HER2-positive breast tumors (9–14, 31). Turpin et al. also demonstrated intrinsic resistance of ERBB2ΔEx16+ breast cancer cell lines to the antibody-drug conjugate ado-trastuzumab emtansine (T-DM1) due to the lack of internalization of the T-DM1-ERBB2ΔEx16 complex, which is crucial for T-DM1 efficacy (14). Although ERBB2ΔEx16 has been implicated in trastuzumab resistance in breast cancer, the role of ERBB2ΔEx16 in mediating inhibitor resistance remains controversial (32). Numerous studies have also reported trastuzumab sensitivity of cell samples from patients with HER2-positive breast and gastrointestinal cancer that express ERBB2ΔEx16 (9, 17, 31, 33). In our cohort, ERBB2ΔEx16 was not detected among the 778 women with breast cancer included in the screening population. We speculate that ERBB2ΔEx16 is rare in Chinese women with breast cancer and requires a larger cohort for its detection.
In LC, Smith et al. have also demonstrated the transforming activity of ERBB2ΔEx16 in vitro and in vivo (15). ERBB2ΔEx16 has been implicated in osimertinib resistance in vitro (18). Interestingly, we also observed that all but one ERBB2ΔEx16+ patients also had concurrent ERBB2 gene amplification (95.5%, 21/22). All 12 EGFR-positive LC patients detected with ERBB2ΔEx16 at progression on EGFR-targeted therapy also harbored concurrent ERBB2 amplification. As a well-documented resistance mechanism of EGFR inhibitors, ERBB2 amplification was found in 10-15% patients with acquired resistance to first- or second-generation EGFR-TKIs (34) and 2-5% of patients with acquired resistance to first- (35) or second-line osimertinib (36). In the study by Shi et al, 10 of the 12 ERBB2ΔEx16+ LC patients also harbored ERBB2 amplification, and although it was unknown whether 2 wild-type patients had been treated (19). Despite its presence, ERBB2 amplification (present in 2.9% patients with sensitizing EGFR mutations, 508/17753) did not exclude potential roles of the ΔEx16 mutant in mediating drug resistance. Moreover, the allele frequencies (AFs) of the ΔEx16 allele were both 99.0% in patients P02 and P11 (Table 2), both of whom were LC patients with acquired resistance to EGFR-TKI. These findings were consistent with a tumor-promoting role for the ERBB2ΔEx16 mutant. For the remaining patients, it is still possible that the ΔEx16 allele was the predominantly amplified allele, although the low AFs and amount of the wild-type allele from the non-malignant cells present in the sequenced samples did not allow for a definitive conclusion. Further research, including those using preclinical models that express abnormally high copies of ERBB2ΔEx16 and those analyzing the AFs for the ΔEx16 allele from circulating tumor cells, may unravel the significance of concurrent ERBB2 amplification and exon 16 skipping in conferring TKI resistance.
Additionally, ERBB2ΔEx16 was also detected without ERBB2 amplification in a treatment-naïve LC patient and an OC patient, neither of whom carrying LC-associated oncogenic driver alterations. ERBB2ΔEx16 has been previously found in a patient with low grade serous ovarian tumor (37) and one OC patient (19). In the latter patient, no mutations in known cancer driver genes were detected. Together, these and our findings supported tumor-promoting roles of ERBB2ΔEx16 in LC and OC.
Preclinical and clinical investigations have shown the differential activity of some irreversible pan-HER TKIs such as afatinib, dacomitinib, and neratinib in targeting various ERBB2 mutant alleles across cancer types (6–8, 38, 39). A subset of patients with breast, lung, or cervical cancer demonstrated promising clinical outcomes with pan-HER TKIs; however, the same inhibitors were inefficacious in other tumor types, including colorectal and bladder cancer (8). Various treatment strategies for HER2-mutant cancers are actively investigated in different phases of clinical trials (8). Since ERBB2ΔEx16 only affects the extracellular domain, resulting in a protein product with an intact tyrosine kinase domain, pan-HER TKIs could be efficacious. Tilio et al. reported that the ERBB2ΔEx16+ breast cancer cell lines were resistant to lapatinib but were sensitive to dacomitinib (40). Hsu et al. also reported a patient with EGFR L858R/T790M-positive advanced LC who acquired ERBB2ΔEx16 during osimertinib therapy (18). The authors also reported that afatinib was able to reverse ERBB2ΔEx16-mediated osimertinib resistance in EGFR L858R/T790M double mutant LC cell line (18). Our patient (P05) received afatinib combined with crizotinib after the emergence of ERBB2ΔEx16. However, no clinical benefit was observed, which may have been due to the intratumoral genetic heterogeneity. The clinical efficacy of pan-HER inhibitors and other novel treatment strategies in inhibiting ERBB2ΔEx16-mediated signaling in a certain subset of patients with ERBB2ΔEx16+ tumors deserves further investigation. It also remains to be explored whether other concurrent genomic alterations, such as TP53 mutations, could affect treatment response. TP53 is the most commonly co-mutated gene in ERBB2ΔEx16+ tumors in our cohort. The presence of co-occurring genetic alterations that affect treatment responses, such as ERBB2ΔEx16, ERBB2 gene amplification, or TP53 mutations, highlights the importance of elucidating the molecular profile of baseline and multiple rebiopsy samples to monitor treatment-related mutational changes and optimize treatment decisions.
Due to the retrospective nature of our study, clinical, treatment, and survival outcomes for some patients are not available, which severely limits our analysis. Our study did not use RNA-based analysis to investigate whether the ERBB2ΔEx16 variants detected in our cohort could result in alternative splicing.
Conclusion
Our study identified an overall ERBB2ΔEx16 prevalence rate of 0.054% and provided an insight into the rarity of ERBB2ΔEx16 in Chinese pan-cancer patients. Among patients with LC, ERBB2ΔEx16 was detected before receiving treatment and after developing resistance from EGFR TKIs, suggesting its potential role in inhibitor resistance. Our study also raises the need to develop novel drugs and implement novel therapeutic strategies to improve the survival outcomes of patients with ERBB2ΔEx16.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NODE (National Omics Data Encyclopedia) OEP003749.
Ethics statement
The studies involving human participants were reviewed and approved by IRB approved and conducted in accordance with the ethical guidelines including Declaration of Helsinki and US Common Rule. The patients/participants provided their written informed consent to participate in this study.
Author contributions
YS and JM conceived of the study and drafted the manuscript. YS, JM, CL, and YD collected data, analyzed and interpreted the data, and drafted the manuscript. CL and YD provided supervision and administrative support. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Beijing Municipal Education Commission Science and Technology Project (No. KM202010025005), Science and Technology Department of Xinjiang Uygur Autonomous Region (No. 2022D14010), and Government-funded Medical Talent Training Program of Hebei Province in 2020.
Acknowledgments
The authors thank the patient and his family. We also thank the investigators, study coordinators, operation staff, and the whole project team who worked on this case. We also appreciate the technical support from Jian Wang, Xiao Zou, and Wenjie Sun of Burning Rock Biotech. We also thank Dr. Analyn Lizaso for contributing to the draft.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1064598/full#supplementary-material
Supplementary Figure 1 | An oncoprint summarizing the somatic mutation landscape of the nine ERBB2ΔEx16+ patients arranged according to signaling pathway. The groups of genes within the gray box are genes related to the signaling pathway indicated at the bottom of the gray border (i.e. the first 5 rows of the oncoprint are genes related to Wnt signaling pathway as noted in the sixth row at the bottom of the gray border). Each row represents a gene or signaling pathway indicated on the left, with the mutation rate indicated on the right. Each column represents a patient. The bar plot on top of the oncoprint summarizes the number of mutations each patient carries. Different colors denote the alteration types. Dark gray denotes mutated signaling pathways. The tumor type was indicated by various colors at the bottom of the oncoprint.
Abbreviations
CESC, endocervical adenocarcinoma; CNV, copy number variants; EGFR, epidermal growth factor receptor; ERBB2 or HER2, Erb-B receptor tyrosine kinase 2; ERBB2ΔEx16, ERBB2 exon 16 in-frame deletion mutation; LUAD, lung adenocarcinoma; OV, ovarian cancer; READ, rectum adenocarcinoma; SNV, single nucleotide variant; STAD, stomach adenocarcinoma; TCGA, The Cancer Genome Atlas; TKI, tyrosine kinase inhibitor.
References
1. Rubin I, Yarden Y. The basic biology of HER2. Ann Oncol (2001) 12 Suppl 1:S3–8. doi: 10.1093/annonc/12.suppl_1.s3
2. Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, et al. The neu oncogene: an erb-b-related gene encoding a 185,000-Mr tumour antigen. Nature (1984) 312:513–6. doi: 10.1038/312513a0
3. Brennan PJ, Kumagai T, Berezov A, Murali R, Greene MI. HER2/Neu: mechanisms of dimerization/oligomerization. Oncogene (2002) 21:328. doi: 10.1038/sj.onc.1205119
4. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol (2006) 7:505–16. doi: 10.1038/nrm1962
5. Jackson C, Browell D, Gautrey H, Tyson-Capper A. Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol (2013) 2013:973584. doi: 10.1155/2013/973584
6. Pahuja KB, Nguyen TT, Jaiswal BS, Prabhash K, Thaker TM, Senger K, et al. Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell (2018) 34:792–806. doi: 10.1016/j.ccell.2018.09.010
7. Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature (2018) 554:189–94. doi: 10.1038/nature25475
8. Cocco E, Lopez S, Santin AD, Scaltriti M. Prevalence and role of HER2 mutations in cancer. Pharmacol Ther (2019) 199:188–96. doi: 10.1016/j.pharmthera.2019.03.010
9. Alajati A, Sausgruber N, Aceto N, Duss S, Sarret S, Voshol H, et al. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res (2013) 73:5320–7. doi: 10.1158/0008-5472.CAN-12-3186
10. Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog (1998) 23:62–8. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o
11. Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Ménard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer (2006) 13:221–32. doi: 10.1677/erc.1.01047
12. Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther (2009) 8:2152–62. doi: 10.1158/1535-7163.MCT-09-0295
13. Marchini C, Gabrielli F, Iezzi M, Zenobi S, Montani M, Pietrella L, et al. The human splice variant Δ16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLos One (2011) 6:e18727. doi: 10.1371/journal.pone.0018727
14. Turpin J, Ling C, Crosby EJ, Hartman ZC, Simond AM, Chodosh LA, et al. The ErbB2ΔEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene (2016) 35:6053–64. doi: 10.1038/onc.2016.129
15. Smith HW, Yang L, Ling C, Walsh A, Martinez VD, Boucher J, et al. An ErbB2 splice variant lacking exon 16 drives lung carcinoma. Proc Natl Acad Sci U.S.A. (2020) 117:20139–48. doi: 10.1073/pnas.2007474117
16. Cai Z, Wang Z, Liu C, Shi D, Li D, Zheng M, et al. Detection of microsatellite instability from circulating tumor DNA by targeted deep sequencing. J Mol Diagn (2020) 22:860–70. doi: 10.1016/j.jmoldx.2020.04.210
17. Volpi CC, Pietrantonio F, Gloghini A, Fucà G, Giordano S, Corso S, et al. The landscape of d16HER2 splice variant expression across HER2-positive cancers. Sci Rep (2019) 9:3545. doi: 10.1038/s41598-019-40310-5
18. Hsu C-C, Liao B-C, Liao W-Y, Markovets A, Stetson D, Thress K, et al. Exon 16-skipping HER2 as a novel mechanism of osimertinib resistance in EGFR L858R/T790M-positive non-small cell lung cancer. J Thorac Oncol (2020) 15:50–61. doi: 10.1016/j.jtho.2019.09.006
19. Shi L, Xu C, Ma Y, Ou Q, Wu X, Lu S, et al. Clinical significance of exon 16 skipping: analysis of a real-world retrospective observational cohort study. ESMO Open (2020) 5:e000985. doi: 10.1136/esmoopen-2020-000985
20. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell (2018) 173:400–16. doi: 10.1016/j.cell.2018.02.052
21. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst (2018) 6:271–81. doi: 10.1016/j.cels.2018.03.002
22. Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L, et al. Capture-based targeted ultradeep sequencing in paired tissue and plasma samples demonstrates differential subclonal ctDNA-releasing capability in advanced lung cancer. J Thorac Oncol (2017) 12:663–72. doi: 10.1016/j.jtho.2016.11.2235
23. Xie X, Wu J, Guo B, Wang L, Deng H, Lin X, et al. Comprehensive characterization reveals sputum supernatant as a valuable alternative liquid biopsy for genome profiling in advanced non-small cell lung cancer. Respir Res (2022) 23:175. doi: 10.1186/s12931-022-02097-4
24. Gong B, Li D, Kusko R, Novoradovskaya N, Zhang Y, Wang S, et al. Cross-oncopanel study reveals high sensitivity and accuracy with overall analytical performance depending on genomic regions. Genome Biol (2021) 22:109. doi: 10.1186/s13059-021-02315-0
25. Deveson IW, Gong B, Lai K, LoCoco JS, Richmond TA, Schageman J, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol (2021) 39:1115–28. doi: 10.1038/s41587-021-00857-z
26. Li YS, Jiang BY, Yang JJ, Zhang XC, Zhang Z, Ye JY, et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann Oncol (2018) 29:945–52. doi: 10.1093/annonc/mdy009
27. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery (2013) 3:224–37. doi: 10.1158/2159-8290.CD-12-0349
28. Kang J, Chen H-J, Wang Z, Liu J, Li B, Zhang T, et al. Osimertinib and cabozantinib combinatorial therapy in an EGFR-mutant lung adenocarcinoma patient with multiple MET secondary-site mutations after resistance to crizotinib. J Thorac Oncol (2018) 13:e49–53. doi: 10.1016/j.jtho.2017.10.028
29. Fujino T, Kobayashi Y, Suda K, Koga T, Nishino M, Ohara S, et al. Sensitivity and resistance of MET exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro. J Thorac Oncol (2019) 14:1753–65. doi: 10.1016/j.jtho.2019.06.023
30. Jiao X-D, Liu K, Wu Y, Zhou X-C, Qin B-D, Ling Y, et al. HER2 splice site mutation c.1899-1G>A as the potential acquired resistance to trastuzumab in a patient with HER2-positive gastric adenocarcinoma. Oncologist (2021) 26:717–21. doi: 10.1002/onco.13799
31. Castagnoli L, Ghedini GC, Koschorke A, Triulzi T, Dugo M, Gasparini P, et al. Pathobiological implications of the d16HER2 splice variant for stemness and aggressiveness of HER2-positive breast cancer. Oncogene (2017) 36:1721–32. doi: 10.1038/onc.2016.338
32. Castagnoli L, Ladomery M, Tagliabue E, Pupa SM. The d16HER2 splice variant: A friend or foe of HER2-positive cancers? Cancers (Basel) (2019) 11:902. doi: 10.3390/cancers11070902
33. Castagnoli L, Iezzi M, Ghedini GC, Ciravolo V, Marzano G, Lamolinara A, et al. Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res (2014) 74:6248–59. doi: 10.1158/0008-5472.CAN-14-0983
34. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol (2018) 29:i10–i9. doi: 10.1093/annonc/mdx703
35. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. LBA50 mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol (2018) 29:VIII740. doi: 10.1093/annonc/mdy424.063
36. Papadimitrakopoulou VA, Wu Y-L, Han J-Y, Ahn M-J, Ramalingam SS, John T, et al. LBA51 analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol (2018) 29:VIII741. doi: 10.1093/annonc/mdy424.064
37. Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew Y-E, Rowley SM, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget (2015) 6:37663–77. doi: 10.18632/oncotarget.5438
38. Ma CX, Bose R, Gao F, Freedman RA, Telli ML, Kimmick G, et al. Neratinib efficacy and circulating tumor DNA detection of mutations in nonamplified metastatic breast cancer. Clin Cancer Res (2017) 23:5687–95. doi: 10.1158/1078-0432.CCR-17-0900
39. Ou S-HI, Schrock AB, Bocharov EV, Klempner SJ, Haddad CK, Steinecker G, et al. HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol (2017) 12:446–57. doi: 10.1016/j.jtho.2016.11.2224
Keywords: deltaHER2, ERBB2, ERBB2delta16, exon 16 skipping, lung cancer
Citation: Shang Y, Mo J, Huo R, Li X, Fang G, Wei Z, Gu G, Zhu X, Zhang C, Liu C and Yan D (2023) Investigation of the prevalence and clinical implications of ERBB2 exon 16 skipping mutations in Chinese pan-cancer patients. Front. Oncol. 12:1064598. doi: 10.3389/fonc.2022.1064598
Received: 08 October 2022; Accepted: 13 December 2022;
Published: 06 January 2023.
Edited by:
Leonhard Müllauer, Medical University of Vienna, AustriaReviewed by:
Koichi Goto, National Cancer Center Hospital East, JapanHow-Wen Ko, Linkou Chang Gung Memorial Hospital, Taiwan
Copyright © 2023 Shang, Mo, Huo, Li, Fang, Wei, Gu, Zhu, Zhang, Liu and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Yan, eWQxNXl0ODhAMTYzLmNvbQ==; Chunling Liu, bGl1ZGV5b3V4aWFuZzY2QHNpbmEuY29t
†These authors have contributed equally to this work