 Shaojun Liu
Shaojun Liu Yuxuan Li
Yuxuan Li Qing Song
Qing Song
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 January 2023
Sec. Breast Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1060495
Triple-negative breast cancer (TNBC) is ineligible for hormonal therapy and Her-2-targeted therapy due to the negative expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor-2. Although targeted therapy and immunotherapy have been shown to attenuate the aggressiveness of TNBC partially, few patients have benefited from them. The conventional treatment for TNBC remains chemotherapy. Chemoresistance, however, impedes therapeutic progress over time, and chemotherapy toxicity increases the burden of cancer on patients. Therefore, introducing more advantageous TNBC treatment options is a necessity. Metabolic reprogramming centered on glucose metabolism is considered a hallmark of tumors. It is described as tumor cells tend to convert glucose to lactate even under normoxic conditions, a phenomenon known as the Warburg effect. Similar to Darwinian evolution, its emergence is attributed to the selective pressures formed by the hypoxic microenvironment of pre-malignant lesions. Of note, the Warburg effect does not disappear with changes in the microenvironment after the formation of malignant tumor phenotypes. Instead, it forms a constitutive expression mediated by mutations or epigenetic modifications, providing a robust selective survival advantage for primary and metastatic lesions. Expanding evidence has demonstrated that the Warburg effect mediates multiple invasive behaviors in TNBC, including proliferation, metastasis, recurrence, immune escape, and multidrug resistance. Moreover, the Warburg effect-targeted therapy has been testified to be feasible in inhibiting TNBC progression. However, not all TNBCs are sensitive to glycolysis inhibitors because TNBC cells flexibly switch their metabolic patterns to cope with different survival pressures, namely metabolic plasticity. Between the Warburg effect-targeted medicines and the actual curative effect, metabolic plasticity creates a divide that must be continuously researched and bridged.
Global cancer statistics reported that by the end of 2021, female breast cancer incidence had surpassed that of lung cancer, ranking first in the world and being the leading cause of female cancer death (1). Breast cancer is classified into three subtypes based on the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (Her-2). The luminal A subtype (ER positive, PR positive, Her-2 negative), the luminal B subtype (ER positive, PR positive, Her-2 negative or positive), the Her-2 subtype (ER negative, PR negative, Her-2 positive), and the basal-like subtype (ER negative, PR negative, Her-2 negative) (2, 3). The basal-like subtype overlaps with triple-negative breast cancer (TNBC) (4) and is the most aggressive subtype of breast cancer, with a higher recurrence rate, tremendous metastatic potential, and shorter overall survival, and is generally associated with a higher cancer burden and poorer disease outcomes (5–7). The lack of relevant receptor expression means that TNBC has lost its candidate status for both hormone therapy and Her-2-targeted therapy (8, 9). Despite significant advances in developing emerging targets in recent years, few patients have benefited from them, and chemotherapy remains the current standard of care (10–12). However, evolving chemoresistance has hampered treatment progress, and chemotherapy toxicity has significantly increased the patient burden (13, 14). The Warburg effect may be a promising therapeutic entry point for the immediate introduction of more effective treatment procedures for the clinical management of TNBC.
As is common knowledge, cancer cells carry out metabolic reprogramming to deal with the survival pressures derived from several aspects, such as the immune system, chemotherapeutics, and anoikis. Glucose metabolism rewiring is a significant part of this process, manifested as tumor cells maintaining a high-flux glycolysis phenotype rather than oxidative phosphorylation (OXPHOS) even under normoxia conditions, known as the Warburg effect (or aerobic glycolysis) (15, 16). The Warburg effect was postulated by the team of Otto Warburg, who found that tumor cells allocate 66% of glucose under oxygen availability conditions for fermentation and the remainder for respiration (17). Impaired mitochondrial structure and function in tumor cells is the initial explanation for this phenomenon (18). A recent study demonstrated a high frequency of mitochondrial defects in TNBC cells, resulting in a metabolic bias towards glycolysis and the promotion of cancer metastasis (19). On the contrary, Reda A. et al. suggest that the mitochondria of tumor cells function normally and serve as a protective mechanism against glycolysis inhibition (20). Current opinion prefers the latter, where the metabolic phenotypes of tumor cells are plastic. However, its regulatory agencies have not been entirely clarified (21).
The emergence of the Warburg phenotype is in line with an evolutionary model. The localized hypoxic microenvironment in which the pre-malignant lesion is located creates a selective pressure that drives its cellular metabolism to glycolysis. Mutations and epigenetic modifications allow glycolysis to break through hypoxic constraints, leading to its constitutive expression (22, 23). Furthermore, hypoxia-induced remodeling of the TNBC extracellular matrix (ECM) positively affects the development of the Warburg phenotype (24). Evidence suggests that the Warburg effect participates in TNBC invasive behaviors, including proliferation, metastasis, recurrence, drug resistance, and immune escape (25–28). The up-regulation of TNBC aerobic glycolysis is a high-probability event in breast cancer subtypes (29, 30). The enhanced reliance of TNBC stem cells on the Warburg effect supports their aggressive phenotype (31). However, there are always two sides to the coin. While the Warburg effect confers TNBC with a higher degree of malignancy, it simultaneously creates opportunities for metabolic-targeted therapeutic strategies (32). In addition, the Warburg effect flux and its product abundance have been used to evaluate the prognosis of TNBC patients receiving glycolysis inhibitors (33). Interestingly, not all TNBC cells are sensitive to glycolysis inhibitors (34), indicating that different states of TNBC cells differ from their choice of glycolytic pathways (35), which is consistent with the metabolic plasticity theory. This theory is strongly supported by the fact that mitochondrial copper depletion drives the MDA-MB-231 cells to reorient their metabolism from mitochondrial respiration to glycolysis to suppress TNBC translocation (36, 37). Consequently, precision medicine requires the customized and stratified management of TNBC patients based on their metabolic directivity.
Hypoxia-mediated Warburg-like metabolic changes are analogous to the Darwinian evolutionary model (23). Epithelial cells acquiring oncogenic transformation is the first step in breast carcinogenesis, whether under the stochastic or stem cell hypothesis (38). Upon reciprocal touch, non-oncogenic breast epithelial cells abide by contact inhibition and halt growing and reproducing (39). However, cells with oncogenic transformation evade contact inhibition (40), continuing to grow and proliferate in a compacted epithelial environment until apical extrusion occurs, completing the transition from monolayer to multilayer epithelium under the coordination of the CD44/COL17A1 pathway and progressively developing into pre-malignant lesions (41) which are separated from the surrounding matrix by a nearly continuous layer of myoepithelial and basement membrane proteins (42). Therefore, oxygen in the vascular matrix must traverse the basement membrane and tumor cell layer. Studies have indicated a limiting distance for oxygen diffusion (approximately 150µm), pre-malignant lesions with uncontrolled proliferation are bound to exceed this diffusion limit, resulting in a hypoxic region (23). Under normoxic conditions, hypoxia-inducible factors (HIFs) are rapidly degraded by prolyl hydroxylation of EglN family members, whereas hypoxia inhibits EglN1 activity, resulting in a massive accumulation of HIFs. In addition, glutamate secretion from TNBC causes EglN1 to undergo oxidative self-inactivation, which can also prevent HIFs from being degraded (43). Studies have shown that HIF directly or indirectly up-regulates the expression of critical glycolysis regulatory elements, resulting in metabolic adaptive transformation (OXPHOS to glycolysis), and it simultaneously stimulates angiogenesis to prepare for eventual vascular normalization. However, angiogenesis invariably lags behind, exposing tumor cells to persistent hypoxic stress (44, 45). Only glycolytic tumor cells, according to the evolutionary models, can survive in a constant hypoxic environment. Mutations or epigenetic modifications during selection lead to a glycolytic constitutive expression (manifested as the removal of oxygen concentration limitation) that is up-regulated in response to specific survival pressures, thereby conferring a survival advantage to tumor cells (Figure 1) (23). The “specific survival pressures” argument is based on the metabolic plasticity doctrine, which holds that the dominant metabolism maintained by tumor cells must be elastic in response to variations in survival pressures (15). For example, TNBC, which keeps a hybrid metabolic phenotype, activates the AMPK pathway to enhance OXPHOS when exposed to glycolytic inhibitor toxicity and vice versa (21). In addition, the 3D model of TNBC developed by Liu C. et al. revealed that the tumor tissue exhibits escalating matrix stiffness from the core to the periphery, and the dominant metabolism also manifests a distribution from the core glycolysis to the peripheral OXPHOS and fatty acid metabolism (46). Microenvironmental acidification and glucose limitation as consequences of up-regulation of glycolysis will further select tumor cells with survival advantages (47–50).
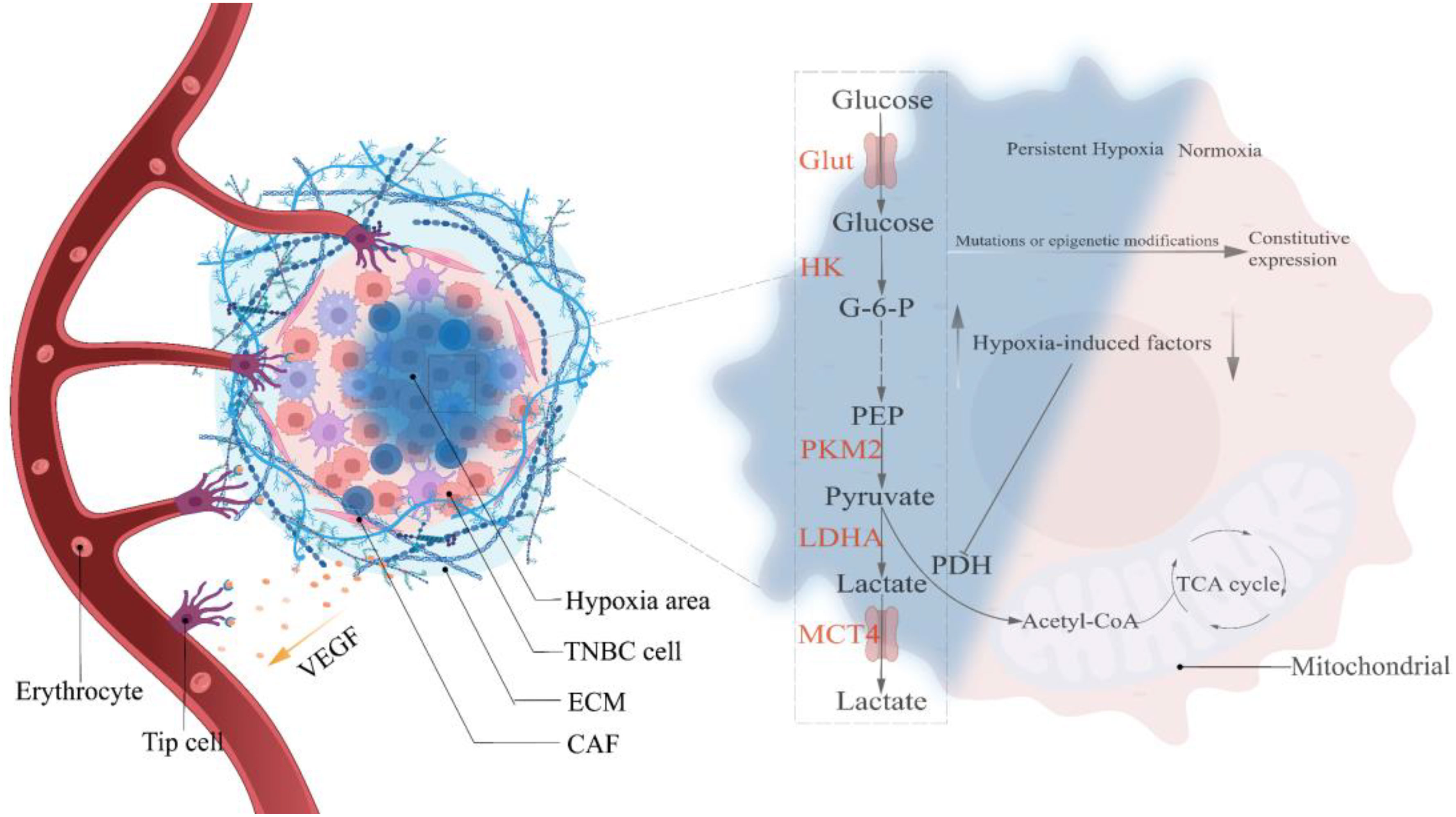
Figure 1 Hypoxia selects the Warburg phenotype. Hypoxia-mediated angiogenesis is gradual in precancerous lesions, and the creation of persistent hypoxia induces the Warburg phenotype to become the predominant metabolic pathway in TNBC cells. Continuous mutations and epigenetic modifications throughout the growth of cancers lead glycolysis to exceed the oxygen concentration restriction, resulting in the constitutive expression of the Warburg phenotype. HIF, Hypoxia-induced factor; VEGF, Vascular endothelial growth factor; Glut, Glucose transporter; HK, Hexokinase; PKM2, pyruvate kinase isozyme typeM2; LDHA, Lactate dehydrogenase; MCT4, Monocarboxylate transporter 4; G-6-P, Glucose-6-phosphate; PEP, Phosphoenolpyruvate; Acetyl-CoA, Acetyl-coenzyme A.
ECM, a substantial 3D supramolecular entity composed of hundreds of different building blocks, is an essential component of TNBC, and its changes profoundly affect the fate of TNBC cells (51, 52). Divergent matrix-remodeling strategies mediated by Mmp14/MT1-MMP define the branching morphology of developing and neoplastic epithelial cells early in breast carcinogenesis (53). This process may involve the effect of matrix remodeling on the chromatin state of mammary epithelial cells (54). On the other hand, ECM stimulates tumor growth in various ways throughout the development of TNBC. For instance, ECM compliance controls the cellular response to contact guidance by generating population transfer between round and elongated cells, hence affecting the migratory dynamics of tumor cells (55). Consequently, ECM remodeling plays a crucial role in carcinogenesis and progression. The preceding section identified hypoxia as an early event in TNBC. Interestingly, current research indicates hypoxia as a potential trigger for ECM remodeling (56, 57). Increased matrix proteins due to disruption of ECM deposition and degradation homeostasis (58), enhanced matrix cross-linking driven by lysyl oxidase (LOX) (59), and local compaction and distant stretching of matrix fibers derived from CAFs-mediated contractile forces (60, 61) are direct behaviors that cause ECM remodeling. Studies have demonstrated that hypoxia affects the aforementioned behaviors in a HIF-dependent manner (57), such as HIF-1α/miR-142-3p/LOX pathway (62) and the HIF-1α/CAF pathway (63), ultimately leading to changes in ECM biophysical and biological properties. Notably, a recent study in which ECM remodeling activated the glycolytic pathway in murine 4T1 cells suggests that ECM is involved in regulating the metabolism of TBNC cells (64). Concerning its regulation mechanism, Sullivan WJ et al. determined that ECM remodeling boosted lactate generation in TNBC cells via the Zfp36/TXNIP/Glut1 signaling pathway (65). Additionally, connective tissue growth factor (CTGF) in conjunction with ECM deposition accelerated TNBC glycolysis via integrin β3/FAK/Src/NF-B P65/Glut3 signaling (66). Moreover, ROCK isoforms differentially regulate the RhoA/ROCK1/p-MLC and RhoA/ROCK2/p-cofilin pathways through integrin β1-activated FAK signaling in an ECM stiffness-dependent manner to regulate MDA-MB-231 cell motility (67), and Rho/ROCK signaling can stimulate Glut1 translocation to the PM and enhance extra-membrane glucose uptake TNBC cells (68). In summary, hypoxia promotes matrix remodeling in various ways that further influence the metabolic pattern of TNBC cells (Figure 2), which simultaneously provides suggestions for developing innovative ECM-targeting treatment approaches (69).
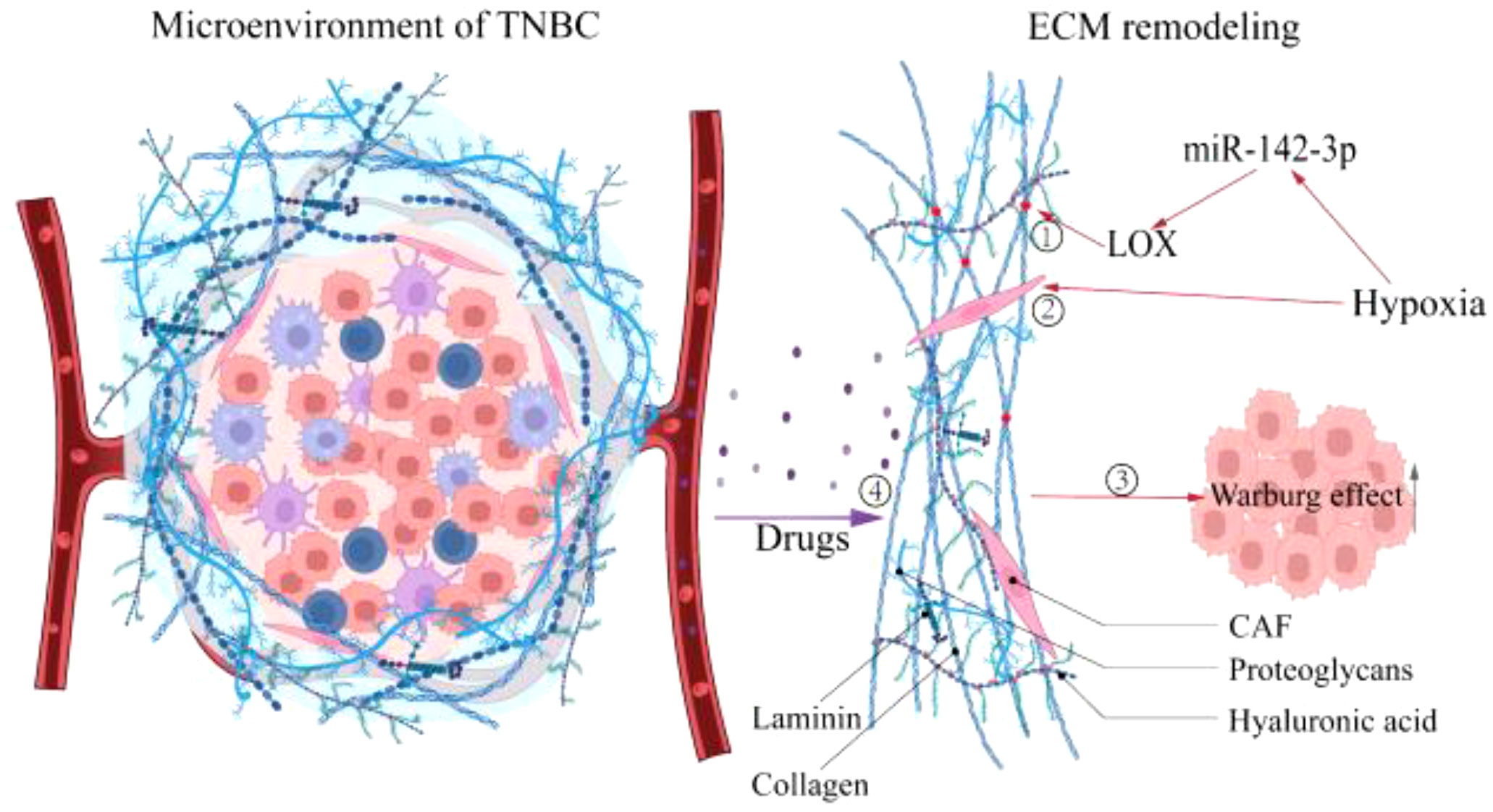
Figure 2 Hypoxia promotes the ECM remodeling. Hypoxia causes ECM remodeling, which affects tumor progression further. ①Enhanced matrix cross-linking. ②Local compaction and distant stretching of matrix fibers derived from CAFs-mediated contractile forces. ③ECM remodeling promotes the Warburg effect. ④ECM stiffness prevents drug penetration. LOX, Lysyl oxidase; CAF, Cancer-associated fibroblast.
Growth factor stimulation of TNBC cells commences the proliferation program (70), and cell proliferation undergoes three periods: I. interphase; II. mitosis; and III. cytokinesis which is called the cell cycle. The interphase accounts for about 90% of the time required for the entire cell cycle and is divided into three phases, namely G1, S, and G2. The cell cycle is precisely modulated by checkpoints such as cyclin-cyclin-dependent kinase complex (cyclin-CDK) and CDK inhibitors (examples include P15, P21, P27), inactivation or mutation of anti-oncogenes, and overexpression or amplification of proto-oncogenes leading to cell cycle checkpoint disorders and ultimately to uncontrolled cancer cell proliferation (71, 72). Positive cell cycle regulation of the Warburg effect is generally achieved through cell cycle checkpoints (73), such as critical glycolytic kinase 6-phosphoglucose-2-kinase (PFKFB3), pyruvate kinase (PKM2) was shown to promote cell cycle (72). Mechanistically, PFKFB3 is transported to the nucleus, and its products F2 and 6BP activate CDKs. Moreover, F2 and 6BP induce degradation of the CDK-mediated G1/S transition inhibitor P27 (74, 75). PKM2 binds to β-linked proteins to form the PKM2-β-linked protein complex to up-regulate cell cycle protein D1 to drive the G1/S transition (72). A study targeting PKM2 to regulate cell cycle progression and inhibit TNBC invasion suggests that further study of the interaction between the Warburg effect and cell cycle is a concern in TNBC management (76).
Tumor metastasis starts from the early stage of the tumor and continues until the cancer is removed. It can be divided into three periods: dissemination, metastatic dormancy, and host organ colonization (77). This process is not a simple linear cascade; concurrency and overlap are more prevalent (78). During dissemination, metastasis-initiating cells (MICs), which are assumed to be derived from cancer stem cells (CSCs) or CSC-like cells, lose their epithelial features and acquire mesenchymal traits through epithelial-mesenchymal transition (EMT) (79, 80), primarily manifested by downregulation of E-calmodulin and reduced cell adhesion, in which the Warburg effect plays an instrumental role (81). For example, high expression of Y-box binding protein 1 (YBX1) was detected in TNBC, which up-regulates aerobic glycolytic flux to accelerate EMT progression (82). Immediately afterward, MICs with mesenchymal properties detach from the tumor bed as single cells or cell clusters and become circulating tumor cells (CTCs) (79). In fact, CTC clusters are more conducive to tumor metastasis (83), so primary tumors significantly up-regulate HIF-1α-mediated Desmoglein2 (DSG2) to promote the formation of CTC clusters (84).
Subsequently, CTCs enter the TME, and the acidic TME shaped by the accumulation of glycolytic acid metabolites intensifies CTCs migration (85, 86). In addition, Glut3 overexpression in TNBC induces activation of M1 tumor-associated macrophages (M1-TAM) via lactate/C-X-C motif Chemokine ligand 8 (CXCL8), consequently creating an inflammatory TME that promotes CTCs metastasis (87). Notably, TNBC delivers Integrin beta 4 (ITGB4) to CAFs located in TME through exosomes and initiates the glycolytic phenotype of CAFs via BNIP3L-dependent mitophagy (known as the reverse Warburg effect) (88). Its product lactate shuttles between CAFs and CTCs via MCT4/MCT1 as a metabolic coupling link and activates the TGFβ1/P38 MAPK/MMP2/9 signaling axis to up-regulate CTCs’ mitochondrial activity and promote metastasis (89).Even more to the point, glycolysis is not the sole up-regulated metabolic phenotype in cancer cell migration. Other metabolic patterns, such as OXPHOS, fatty acid metabolism, and glutamine metabolism, are similarly up-regulated under certain conditions (90). And the metabolic phenotype of CTCs is dynamically remodeled at different stages of tumor metastasis to help them manage the changing microenvironment of the metastatic cascade and ensure a successful transition (Figure 3) (91). Next, CTCs migrate towards the vasculature, and the Glut1/integrin β1/Src/focal adhesion kinase (FAK) signaling pathway activated by PCB29-P exposure may be involved in this process (92). CTCs entering the blood circulation are challenged by ROS, and high concentrations of ROS are cytotoxic. At this time, the advantages of CTCs clusters gradually emerge, which induce microenvironmental hypoxia and activate HIF-1α-mediated glycolysis in response to oxidative stress (93). Moreover, when CTC reaches the bloodstream and is threatened by Anoikis, only a few Anoikis-resistant tumor cells survive (94). And the phosphorylation and activation of lactate dehydrogenase (LDHA), a glycolytic critical kinase, has been discovered to boost Anoikis resistance (95).
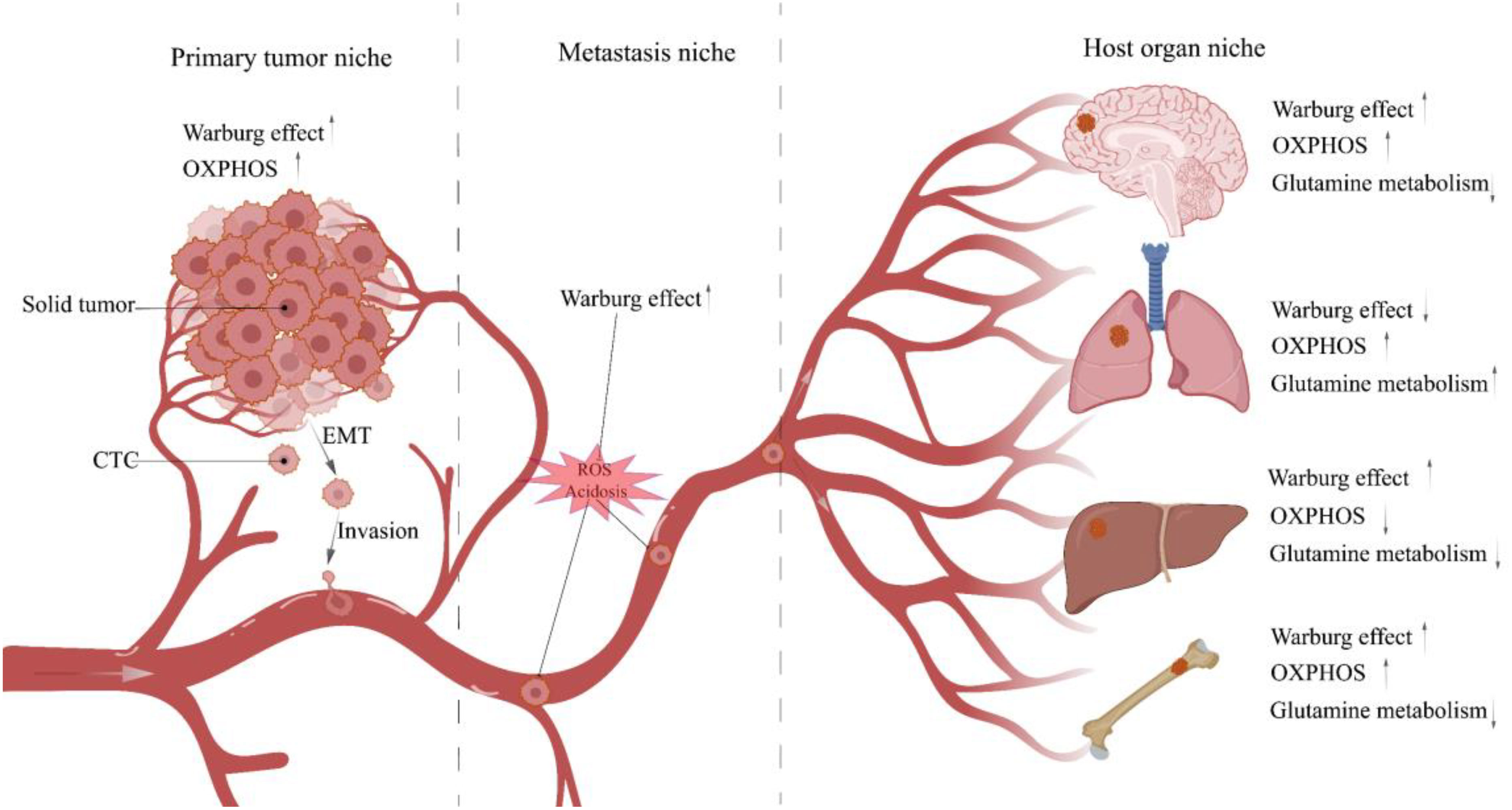
Figure 3 Metabolic characteristics of TNBC cells in different niches. When tumor cells leave the tumor bed and begin the metastasis process, they will traverse multiple niches with diverse microenvironments. Different microenvironments impose distinct survival restrictions, and tumor cells must change their metabolic pathways dynamically to resist these pressures. CTC, Circulating tumor cell; EMT, Epithelial-mesenchymal transition.
Next is an overlap of host organ colonization and metastatic dormancy, in which the majority of tumors metastasize to specific organs, called “organotropism” (96). Studies have shown that the lung, bones, and brain are more likely to serve as TNBC target organs (97, 98). After reaching the targeted organ, CTCs occupy the favorable niche and become disseminated tumor cells (DTCs). Due to the balance between tumor cell proliferation and apoptosis, DTCs enter the reversible growth arrest of single cells located in the G0/G1 phase of the cell cycle, called cell dormancy. This persists for years or even decades, and DTCs reactivate when the balance swings toward cell proliferation, either because of proangiogenic substances or because tumor cells defy immune suppression (77, 79, 99). For instance, CD39 PD-1 CD8 T cells were demonstrated to mediate metastatic dormancy in TNBC lung-oriented DTCs, and depletion of CD8 T cells may induce DTCs reactivation (100). Activated DTCs destroy host organs and form metastases, such as up-regulation of the Warburg effect by MDA-MB-231 cells found in bone TME to produce excessive lactate, leading to MCT4/MCT1-mediated osteolytic lesions (101).
Minimal residual disease (MRD) is considered the basis of TNBC recurrence (102), and postoperative circulating tumor DNA (ctDNA) sequencing and CTCs counting have been used to detect MRD and evaluate the likelihood of patients suffering from recurrence (103). Cell dormancy is critical for MRD survival and reactivation (104, 105). Genetic heterogeneity, TME regulation, ecological niche, and immunosuppression drive MRD into a dormancy (106) characterized by cell cycle arrest, ecological niche dependence, drug resistance, immune escape, and reversibility (107). When conditions such as surgery, drugs, and metabolic remodeling intervene, MRD exits dormancy and causes recurrence (108). Glycolysis is possibly involved in the reactivation of dormant MRD. Studies have shown that dormant cells exhibit an increase in mitochondrial mass and mitochondrial ROS. Therefore, dormant cells inhibit oxidative stress-induced apoptosis via autophagy-related 7 (ATG7) mediated autophagy (109), in which up-regulated AGT7 suppresses the glycolytic phenotype (110). During the transitional period of dormant cells toward reactivation, autophagic inhibition and increased abundance of PFKFB3, a key mediator of glycolysis, facilitate dormant escape (111). The argument that a negative correlation between autophagy and the Warburg phenotype differs from Radic Shechter et al., who found that MRD retains the metabolic memory of original tumor cells and maintains high aerobic glycolytic flux to ensure survival (112). A possible explanation is that MRDs dynamically reshape the balance between autophagy and glycolysis in response to varied ecological niches (113). In addition, consistent with the primary tumor, the metabolism of the cells that undergo macroscopical metastasis after escaping the dormant state continues to be biased toward glycolysis to sustain growth (114). Remarkably, the tricarboxylic acid cycle (TCA cycle) metabolites succinate, fumarate, and 2-hydroxyglutarate (2-HG) were associated with post-treatment tumor reconstruction (115), and glycolytic product lactate functions as a substrate to support TCA cycle running (116).
As evident from recent reviews (106, 108, 117), despite the emerging roles of the Warburg phenotype in metastatic dormancy of tumor cells, there is a considerable blankness in the relevant research, including how the Warburg phenotype controls DTCs to enter a dormancy state? How does the Warburg phenotype help dormant DTCs survive? Furthermore, how the Warburg phenotype regulates dormant DTCs reactivation? Metastatic dormancy is essential for DTCs survival and reactivation as a protective mechanism (118), so filling these gaps may provide new clinical opportunities for TNBC.
From the above discussion, it can be clarified that while TNBCs adaptively modulate their metabolic phenotype following different conditions, TNBCs up-regulate the Warburg phenotype to maintain their invasion activity is still a high probability event. Accordingly, high glycolytic flux means an adequate supply of extra-membrane glucose is necessary. Increasing evidence that most immune cells present in TME (Figure 4), including innate immune cells such as NK cells, macrophages, and dendritic cells; adaptive immune cells, such as T cells in the activated state, mimic the metabolic patterns of tumor cells (119, 120). Therefore, the competition between tumor cells and immune cells for metabolic substrates is inevitable (121), and the consequence is always that immune cell function is curbed due to the lack of fuel (122). Because tumor cells will induce immune cells’ glycolytic suppression, for example, IFN-γ secreted by T cells or the glycolytic phenotype of the tumor itself triggers PD-L1 expression in tumor cells. In turn, PD-L1 mediates up-regulation of the glycolytic pathway in tumor cells via the Akt/mTOR pathway, imposing a nutrient limitation on T cells. Prolonged exposure to glucose limitation will lead to irreversible functional impairment that cannot be corrected by simple nutrient supplementation (122, 123). In addition, the expression of CTL-4 associated with immune suppression and its principal ligand CD80/CD86 were detected in TNBCs (124), and a study of CTL-4 blockade by ipilimumab linked Treg instability to its glycolysis, suggesting the involvement of CTL-4 in metabolic competition between TNBCs and immune cells (125).

Figure 4 The Warburg effect enhances immune escape of TNBC. Glycolysis facilitates tumor immune evasion via multiple mechanisms. MDSC, Myeloid-derived suppressor cell.
Lactate mediates multiple pro-tumor behaviors, including immune escape (126, 127). For instance, lactate works directly with mitochondrial antiviral signaling proteins (MAVS), attenuates retinoic acid-inducible gene I-like receptors (RLRs) signal transduction and downstream type I interferons (IFNs) production, and induces failure of immune surveillance (128). Unfortunately, this mechanism has not been characterized in TNBC cell lines, but it is a potential research direction. Furthermore, the acidic microenvironment due to lactate accumulation significantly down-regulates macrophage-mediated Programmed Cell Removal (PrCR) damage to MDA-MB-231 cells (129).
Indeed, the Warburg phenotype derived from TNBC can also evade immune surveillance by activating immunosuppressive cells. For example, tumor glycolysis regulates the molecular network of AMPK-ULK1, autophagy, and CEBPB pathways. This way, myeloid-derived suppressor cells (MDSC) are activated, and immune suppression is maintained (130). Moreover, Microenvironmental lactic acidosis induces tumor-associated macrophages (TAM) to skew toward an M2-like phenotype, resulting in the obstacle of immuno-detection of tumor cells by macrophages (129, 131). A recent study found that TNBC CTCs recruit neutrophils to form CTC-neutrophil clusters, leading to an accelerated metastatic process (132). However, further studies are needed to determine whether the Warburg phenotype is involved in neutrophil recruitment and whether CTC-neutrophil clusters contribute to immune evasion (133).
Currently, chemotherapy based on anthracyclines and taxanes is the standard treatment for TNBC. The effectiveness of platinum compounds in neoadjuvant chemotherapy has been confirmed by evidence. Research on drugs related to targeted therapy, immunotherapy, and endocrine therapy has also collected initial results (134, 135). Nonetheless, multi-drug resistance deepens the conflict between TNBC therapy options and adverse outcomes (136). Studies have indicated that primary resistance genes pre-exist and are adaptively selected by treatment regimens. Acquired resistance gene transcriptional profiles are obtained by reprogramming in response to treatment in TNBC patients (137). Various mechanisms mediate drug resistance in TNBC (138), including the Warburg phenotype (25, 139, 140). First, the Warburg effect confers enhanced DNA repair capacity to tumor cells. Rac1 enhances nucleotide production to defend MDA-MB-231 cells from chemotherapeutic drug-induced DNA damage by triggering the aldolase A and ERK signaling cascades and upregulating aerobic glycolysis, especially its branching Pentose phosphate pathway (PPP) fluxes (141). A recent study demonstrating that overactivation of the ERK pathway promotes apoptosis in TNBC cells shows the dual role of ERK in TNBC biology, which should therefore be considered when selecting ERK as a therapeutic target (142). Second, the Warburg effect gives tumor cells a more robust antioxidant capacity. ROS up-regulation induced by chemotherapy drugs exposed tumor cells to oxidative stress, oxidative damage provoked the overexpression of the PPP rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PD), accelerated PPP enhanced the production of nicotinamide adenine dinucleotide phosphate (NADPH), thereby up-regulating the synthesis rate of the antioxidant glutathione (GSH). Ultimately, the oxidative damage caused by ROS can be inhibited (143). In addition, autophagy’s clearance of injured organelles and macromolecules is essential for inhibiting drug-induced apoptosis in TNBCs (144, 145). A recent study identified Proviral Insertion in Murine Lymphomas 2 (PIM2) directly binding to hexokinase-II (HK-2) and phosphorylating HK2 on Thr473. Phosphorylated HK2 up-regulates glycolytic flux and induces autophagy to exert chemoresistance by promoting protein stability (146). The Warburg effect also manifests resistance by inhibiting drug influx and promoting drug efflux. Glycolysis generates a large amount of H+ accumulation in the cytoplasm, and if it is not removed in time, the consequent acidosis threatens cell survival. The existence of a net acid extrusion system ignores such events. It leads to the rise of intracellular pH (pHi) and the reduction of extracellular pH (pHe) simultaneously. This pH gradient has been confirmed to be conducive to the formation of drug resistance (147). Elevated pHi not only inhibits the influx of alkaline drugs such as doxorubicin but also induces active drug efflux by coordinating the transporter ABCB1 (148). In addition, ABC transporters are ATP-dependent (149), and suppressing glycolysis leads to transporter inactivation (150), so it is reasonable to speculate that glycolysis provides energy support for the actions of ABC transporters. Based on the above theory, Omran Z et al. recommend that the intervention of proton pump and transporter inhibitors is vital to manage resistance (151).
Notably, with the intervention of compounds other than chemotherapy, the resistance spectrum of TNBC expands, such as lactate modulating anti-PD-1/PD-L1 drug activity (152) via activating TAMs (153). In summary, targeting the Warburg effect seems perfect for curbing resistance; however, this is not the case; silencing of GLUT1 does not ablate TNBC resistance as expected but further induces drug resistance via Akt/GSK-3β/β-catenin/survivin signaling (154). This opposite phenomenon may involve dynamic remodeling of metabolism (155) since switching metabolism to mitochondrial or lipid metabolism equally mediates resistance, depending on the molecular phenotype of the interventional drug (156, 157). Hence, careful considerations should be taken into account in sequential drug resistance control programs.
In the early stages of TNBC, hypoxia gives birth to the Warburg phenotype, and mutations or epigenetic modifications result in constitutive expression of the Warburg effect. However, the pressure on TNBC’s survival is ever-changing, necessitating that it maintain a varied metabolic pattern (15). Therefore, glycolysis inhibitors are only effective against a subset of TNBCs (34). Hence, it is necessary to identify Warburg-dependent TNBC and administer tailored treatment for it. With the advent of omics approaches, several teams have stratified TNBC based on various omics analyses and obtained experimental findings (34, 158–161) (Table 1). However, the road is always tortuous; stratification schemes relying on genomics and transcriptomics failed to break through the barrier of LAR and BLIS tumors in later FUTURE experiments (162). Of note, Xiao Y et al. integrated metabolomics and genomics to classify TNBC into three subtypes: C1, with ceramide and fatty acid enrichment; C2, with overexpression of oxidative processes and glycosyl transfer-related metabolites; and C3, with low-level metabolic dysregulation, and proposed that genomics-based LAR subtypes overlap with C1 subtypes and BLIS subtypes overlap with C2 and C3 subtypes, finding a new solution for refractory TNBC (158, 163). Moreover, by investigating metabolic dysregulation in TNBC, Gong Y et al. identified the lipogenic subtype MPS1 with up-regulated lipid metabolism, the glycolytic subtype MPS2 with up-regulated carbohydrate and nucleotide metabolism, and the mixed subtype MPS3 with partially dysregulated pathways, each of MPS showed consistent sensitivity to inhibitors targeting the same metabolic pathway (34). Accordingly, stratified management of TNBC patients is crucial for minimizing inefficient medical treatment and reducing the cancer burden among patients (7).
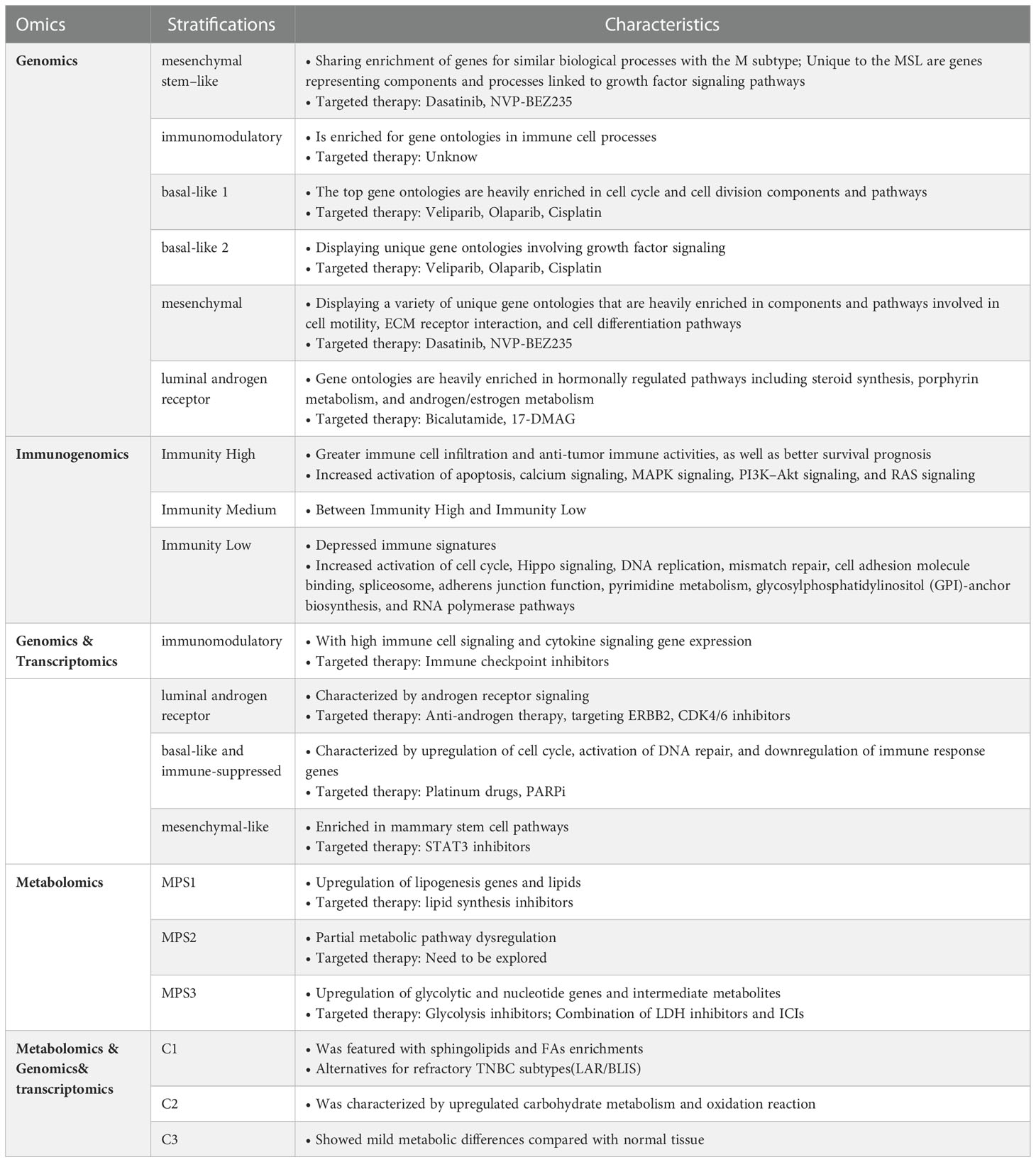
Table 1 TNBC stratification schemes based on different omics.
Over the past few decades, significant progress has been made in studying compounds targeting glycolysis (Table 2). Certain compounds target upstream regulators of glycolysis. For example, Cardamonin and Honokiol restricted glycolysis in MDA-MB-231 cells through the HIF-1α pathway (174, 175). Betulinic Acid inhibits glycolytic flux via GRP78/β-catenin/c-Myc or CAV-1/NF-κB/c-Myc signaling pathways (176, 177). Furthermore, some compounds target the critical regulators of the Warburg phenotype. Examples include GNE-140 and FX-11, which down-regulate glycolysis by blocking lactate dehydrogenase (LDH) (34). Elemene (β-elemene) suppresses breast cancer metastasis by obstructing pyruvate kinase M2 (PKM2) dimerization and nuclear translocation.
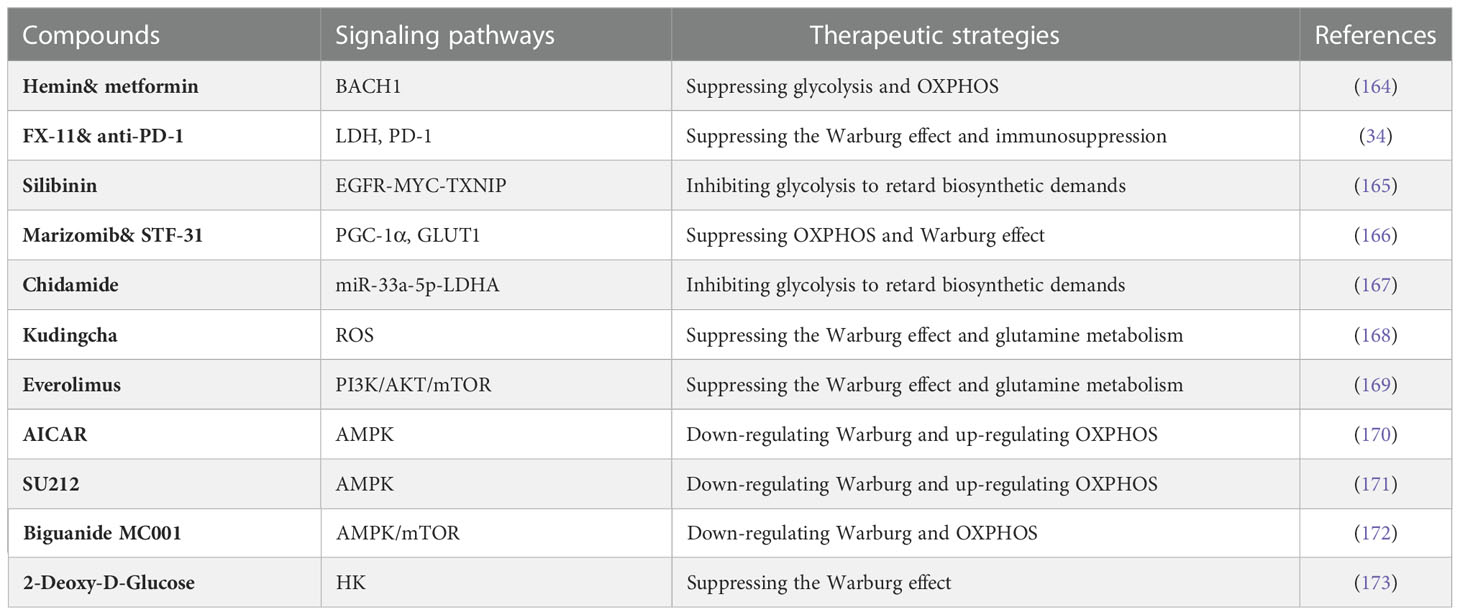
Table 2 Compounds targeting the Warburg effect in TNBC.
In addition, based on understanding the effect of ECM remodeling on TNBC invasive behavior, targeting ECM is becoming an emerging fulcrum for TNBC treatment protocols. For example, the TME-tunable BAGM therapeutic nanoplatform activates a cyclic cascade that degrades the dense ECM and converts matrix collagen into a loose state to exert antitumor effects (178). LOX inhibitor chemically linked lipid-based nanoparticles loaded with chemotherapeutic epirubicin have been demonstrated to prolong survival in patients with TNBC (179). This is probably because LOX inhibitors ablate the dense ECM and enhance chemotherapy drug permeability (62). Moreover, it has been demonstrated that inhibiting tumor development by targeting ECM to impact tumor cell metabolism is achievable (69, 180); the exact application of this technique to TNBC requires additional investigation.
The Warburg phenotype also functions as a strategy for eliminating transformed cells. RasV12-, Src-, or Erbb2-transformed cells are in a relatively rigid microenvironment formed by compacted epithelial cells, which are frequently apically extruded from epithelial tissues in search of more survival space, after which the epithelial defense against cancer (EDAC) induces epithelial protein lost in neoplasm (EPLIN)-mediated metabolic transformation (OXPHOS to glycolysis) to remove dissociative transformed cells (41, 181). This mechanism must be tested further to determine if it can be used to prevent the development of TNBC.
Worthy of note is that glycolysis suppression possibly triggers the up-regulation of compensatory metabolic pathways, such as OXPHOS (21), and drugs that simultaneously target both glycolysis and OXPHOS pathways, marizomib, and gracillin, have been successful in the mouse model (166, 182). It is necessary to clarify that while the inhibitory effect of targeted aerobic glycolysis on TNBC is undeniable, metabolic plasticity poses a new and pressing challenge for this therapeutic option (183). Perhaps targeting the metabolic switch is the key to tackling metabolic plasticity in the future (184).
Hypoxia is the source of the Warburg phenotype, whereas mutations and epigenetic modifications are the primary causes of its constitutive expression. The Warburg effect regulates TNBC invasion, including proliferation, metastasis, recurrence, treatment resistance, and immune evasion. Consequently, targeting the Warburg effect seems to be a promising method for treating refractory TNBC. Numerous in vitro investigations have also demonstrated the efficacy of this treatment protocol. Nevertheless, because of metabolic plasticity, the outcome of using a single glycolytic inhibitor is frequently disappointing (21, 183). Consequently, future research may focus less on a single glycolytic inhibitor delivery strategy and more on the following topics: a. A stratified approach to the management of TNBC patients based on metabolomics. b. Continuous innovation for auxiliary diagnostic technology of glycolytic-dependent TNBC. The simultaneous characterization of the Warburg effect using PET, MRI, and hyperpolarized 13C-MRSI is a notable advancement in this field (185). c. Targeting hypoxia. A hypoxic environment induces HIF to catalyze the Warburg phenotype, hence inhibiting HIF holds the promise of blocking glycolytic dysregulation at the root. d. Targeting ECM. The ECM regulates the expression of the Warburg effect, and the compacted ECM impacts chemotherapeutic drugs’ penetration; thus, the ECM-targeting protocol is significant for TNBC. e. Targeting multiple metabolic pathways. Since a single application of glycolysis inhibitors activates compensatory energy metabolic pathways, simultaneous inhibition of multiple metabolic pathways hopefully promotes apoptosis in cancer cells; Marizomib and gracillin, compounds that target both glycolysis and OXPHOS, have been successfully tested in mice. f. To develop new strategies to overcome the chemoresistance of TNBC by inhibiting the Warburg effect. g. Combined therapies. Chemoresistance of TNBC is the greatest barrier to chemotherapy treatments, and the Warburg effect certainly exacerbates this resistance. Theoretically, combining chemotherapeutic medicines and glycolytic inhibitors could increase the effectiveness. In addition, compacted ECM impedes drug delivery; hence ECM remodeling inhibitors paired with tumor cytotoxic medicines have the potential to boost TNBC patient survival.
ML provided direction and guidance throughout the preparation of this manuscript. QS provided guidance throughout the revision of this manuscript. SJL collected and prepared the related literature and drafted the manuscript. YXL designed the imagines and tables. MY corrected the language ML and QS reviewed and made significant revisions to the manuscript. All authors contributed to the article and approved the submitted version article and approved the submitted version.
This work was financially supported by the National Natural Science Foundation of China (82274423).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
TNBC, triple-negative breast cancer; ER, estrogen receptor; PR, progesterone receptor; (Her-2), human epidermal growth factor receptor-2; OXPHOS, oxidative phosphorylation; ECM, extracellular matrix; COL17A1, collagen Type XVII Alpha 1 Chain; HIF, hypoxia-induced factor; GLUT, glucose transporter; HK, hexokinase; PFK, phosphofructokinase; ENO, enolase; MCT, monocarboxylate transporter; AMPK, adenosine 5’-monophosphate (AMP)-activated protein kinase; LOX, lysyl oxidase; CAF, carcinoma-associated fibroblasts; EDAC, epithelial defense against cancer; EPLIN, epithelial protein lost in neoplasm; ROCK, Rho-associated coiled-coil kinase; CTGF, connective tissue growth factor; PM, plasma membrane; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PKM2, pyruvate kinase isozyme typeM2; MIC, metastasis-initiating cell; CSC, cancer stem cell; EMT, epithelial-mesenchymal transition; YBX1, Y-box binding protein 1;CTC, circulating tumor cell; LDHA, lactate dehydrogenase; MRD, Minimal residual disease; TAM, tumor-associated macrophages; PPP, pentose phosphate pathway; ROS, reactive oxygen species.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Biancolella M, Testa B, Baghernajad Salehi L, D'Apice MR, Novelli G. Genetics and genomics of breast cancer: Update and translational perspectives. Semin Cancer Biol (2021) 72:27–35. doi: 10.1016/j.semcancer.2020.03.013
3. Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet (London England) (2021) 397(10286):1750–69. doi: 10.1016/s0140-6736(20)32381-3
4. Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: Disease entity or title of convenience? Nat Rev Clin Oncol (2010) 7(12):683–92. doi: 10.1038/nrclinonc.2010.154
5. Borri F, Granaglia A. Pathology of triple negative breast cancer. Semin Cancer Biol (2021) 72:136–45. doi: 10.1016/j.semcancer.2020.06.005
6. Yau C, Osdoit M, van der Noordaa M, Shad S, Wei J, de Croze D, et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: A multicentre pooled analysis of 5161 patients. Lancet Oncol (2022) 23(1):149–60. doi: 10.1016/s1470-2045(21)00589-1
7. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discovery (2019) 9(2):176–98. doi: 10.1158/2159-8290.Cd-18-1177
8. Li X, Yang J, Peng L, Sahin AA, Huo L, Ward KC, et al. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-Triple-Negative breast cancer. Breast Cancer Res Treat (2017) 161(2):279–87. doi: 10.1007/s10549-016-4059-6
9. Smolarz B, Nowak AZ, Romanowicz H. Breast cancer-epidemiology, classification, pathogenesis and treatment (Review of literature). Cancers (2022) 14(10):2569. doi: 10.3390/cancers14102569
10. Li Y, Zhan Z, Yin X, Fu S, Deng X. Targeted therapeutic strategies for triple-negative breast cancer. Front Oncol (2021) 11:731535. doi: 10.3389/fonc.2021.731535
11. Hwang SY, Park S, Kwon Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol Ther (2019) 199:30–57. doi: 10.1016/j.pharmthera.2019.02.006
12. Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat Rev Clin Oncol (2022) 19(2):91–113. doi: 10.1038/s41571-021-00565-2
13. Kanwal B. Untangling triple-negative breast cancer molecular peculiarity and chemo-resistance: Trailing towards marker-based targeted therapies. Cureus (2021) 13(7):e16636. doi: 10.7759/cureus.16636
14. Gupta GK, Collier AL, Lee D, Hoefer RA, Zheleva V, Siewertsz van Reesema LL, et al. Perspectives on triple-negative breast cancer: Current treatment strategies, unmet needs, and potential targets for future therapies. Cancers (2020) 12(9):2392. doi: 10.3390/cancers12092392
15. Martínez-Reyes I, Chandel NS. Cancer metabolism: Looking forward. Nat Rev Cancer (2021) 21(10):669–80. doi: 10.1038/s41568-021-00378-6
16. Sun L, Suo C, Li ST, Zhang H, Gao P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the warburg effect. Biochim Biophys Acta Rev Cancer (2018) 1870(1):51–66. doi: 10.1016/j.bbcan.2018.06.005
17. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
18. Weinhouse S. On respiratory impairment in cancer cells. Sci (New York NY) (1956) 124(3215):267–9. doi: 10.1126/science.124.3215.267
19. Guha M, Srinivasan S, Raman P, Jiang Y, Kaufman BA, Taylor D, et al. Aggressive triple negative breast cancers have unique molecular signature on the basis of mitochondrial genetic and functional defects. Biochim Biophys Acta Mol basis Dis (2018) 1864(4 Pt A):1060–71. doi: 10.1016/j.bbadis.2018.01.002
20. Reda A, Refaat A, Abd-Rabou AA, Mahmoud AM, Adel M, Sabet S, et al. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hormone-responsive breast cancer cells. Sci Rep (2019) 9(1):13748. doi: 10.1038/s41598-019-50141-z
21. Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci United States America (2019) 116(9):3909–18. doi: 10.1073/pnas.1816391116
22. Damaghi M, West J, Robertson-Tessi M, Xu L, Ferrall-Fairbanks MC, Stewart PA, et al. The harsh microenvironment in early breast cancer selects for a warburg phenotype. Proc Natl Acad Sci United States America (2021) 118(3). doi: 10.1073/pnas.2011342118
23. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4(11):891–9. doi: 10.1038/nrc1478
24. Romani P, Valcarcel-Jimenez L, Frezza C, Dupont S. Crosstalk between mechanotransduction and metabolism. Nat Rev Mol Cell Biol (2021) 22(1):22–38. doi: 10.1038/s41580-020-00306-w
25. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the warburg effect supports aggressiveness and drug resistance of cancer cells? Drug resistance updates Rev commentaries antimicrobial Anticancer chemother (2018) 38:1–11. doi: 10.1016/j.drup.2018.03.001
26. Liu C, Jin Y, Fan Z. The mechanism of warburg effect-induced chemoresistance in cancer. Front Oncol (2021) 11:698023. doi: 10.3389/fonc.2021.698023
27. Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, et al. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther (2019) 195:162–71. doi: 10.1016/j.pharmthera.2018.11.004
28. Lu J. The warburg metabolism fuels tumor metastasis. Cancer metastasis Rev (2019) 38(1-2):157–64. doi: 10.1007/s10555-019-09794-5
29. Cho ES, Kim NH, Yun JS, Cho SB, Kim HS, Yook JI. Breast cancer subtypes underlying emt-mediated catabolic metabolism. Cells (2020) 9(9):2064. doi: 10.3390/cells9092064
30. Yu TJ, Ma D, Liu YY, Xiao Y, Gong Y, Jiang YZ, et al. Bulk and single-cell transcriptome profiling reveal the metabolic heterogeneity in human breast cancers. Mol Ther J Am Soc Gene Ther (2021) 29(7):2350–65. doi: 10.1016/j.ymthe.2021.03.003
31. Abad E, Samino S, Yanes O, Potesil D, Zdrahal Z, Lyakhovich A. Activation of glycogenolysis and glycolysis in breast cancer stem cell models. Biochim Biophys Acta Mol basis Dis (2020) 1866(10):165886. doi: 10.1016/j.bbadis.2020.165886
32. Wang Z, Jiang Q, Dong C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol Med (2020) 17(1):44–59. doi: 10.20892/j.issn.2095-3941.2019.0210
33. Hesketh RL, Wang J, Wright AJ, Lewis DY, Denton AE, Grenfell R, et al. Magnetic resonance imaging is more sensitive than pet for detecting treatment-induced cell death-dependent changes in glycolysis. Cancer Res (2019) 79(14):3557–69. doi: 10.1158/0008-5472.Can-19-0182
34. Gong Y, Ji P, Yang YS, Xie S, Yu TJ, Xiao Y, et al. Metabolic-Pathway-Based subtyping of triple-negative breast cancer reveals potential therapeutic targets. Cell Metab (2021) 33(1):51–64.e9. doi: 10.1016/j.cmet.2020.10.012
35. Fujita M, Imadome K, Somasundaram V, Kawanishi M, Karasawa K, Wink DA. Metabolic characterization of aggressive breast cancer cells exhibiting invasive phenotype: Impact of non-cytotoxic doses of 2-dg on diminishing invasiveness. BMC Cancer (2020) 20(1):929. doi: 10.1186/s12885-020-07414-y
36. Cui L, Gouw AM, LaGory EL, Guo S, Attarwala N, Tang Y, et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat Biotechnol (2021) 39(3):357–67. doi: 10.1038/s41587-020-0707-9
37. Ramchandani D, Berisa M, Tavarez DA, Li Z, Miele M, Bai Y, et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat Commun (2021) 12(1):7311. doi: 10.1038/s41467-021-27559-z
38. Sgroi DC. Preinvasive breast cancer. Annu Rev Pathol (2010) 5:193–221. doi: 10.1146/annurev.pathol.4.110807.092306
39. Pavel M, Renna M, Park SJ, Menzies FM, Ricketts T, Füllgrabe J, et al. Contact inhibition controls cell survival and proliferation Via Yap/Taz-autophagy axis. Nat Commun (2018) 9(1):2961. doi: 10.1038/s41467-018-05388-x
40. Dann SG, Golas J, Miranda M, Shi C, Wu J, Jin G, et al. P120 catenin is a key effector of a ras-pkcε oncogenic signaling axis. Oncogene (2014) 33(11):1385–94. doi: 10.1038/onc.2013.91
41. Kozawa K, Sekai M, Ohba K, Ito S, Sako H, Maruyama T, et al. The Cd44/Col17a1 pathway promotes the formation of multilayered, transformed epithelia. Curr Biol CB (2021) 31(14):3086–97.e7. doi: 10.1016/j.cub.2021.04.078
42. Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell (2022) 185(2):299–310.e18. doi: 10.1016/j.cell.2021.12.023
43. Briggs KJ, Koivunen P, Cao S, Backus KM, Olenchock BA, Patel H, et al. Paracrine induction of hif by glutamate in breast cancer: Egln1 senses cysteine. Cell (2016) 166(1):126–39. doi: 10.1016/j.cell.2016.05.042
44. Tirpe AA, Gulei D, Ciortea SM, Crivii C, Berindan-Neagoe I. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of hif genes. Int J Mol Sci (2019) 20(24):6140. doi: 10.3390/ijms20246140
45. Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
46. Liu C, Li M, Dong ZX, Jiang D, Li X, Lin S, et al. Heterogeneous microenvironmental stiffness regulates pro-metastatic functions of breast cancer cells. Acta biomaterialia (2021) 131:326–40. doi: 10.1016/j.actbio.2021.07.009
47. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol (2020) 82:103–26. doi: 10.1146/annurev-physiol-021119-034627
48. Corbet C, Feron O. Tumour acidosis: From the passenger to the driver's seat. Nat Rev Cancer (2017) 17(10):577–93. doi: 10.1038/nrc.2017.77
49. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol (2017) 27(11):863–75. doi: 10.1016/j.tcb.2017.06.003
50. Madan E, Peixoto ML, Dimitrion P, Eubank TD, Yekelchyk M, Talukdar S, et al. Cell competition boosts clonal evolution and hypoxic selection in cancer. Trends Cell Biol (2020) 30(12):967–78. doi: 10.1016/j.tcb.2020.10.002
51. Zolota V, Tzelepi V, Piperigkou Z, Kourea H, Papakonstantinou E, Argentou MI, et al. Epigenetic alterations in triple-negative breast cancer-the critical role of extracellular matrix. Cancers (2021) 13(4):713. doi: 10.3390/cancers13040713
52. Cox TR. The matrix in cancer. Nat Rev Cancer (2021) 21(4):217–38. doi: 10.1038/s41568-020-00329-7
53. Feinberg TY, Zheng H, Liu R, Wicha MS, Yu SM, Weiss SJ. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev Cell (2018) 47(2):145–60.e6. doi: 10.1016/j.devcel.2018.08.025
54. Stowers RS, Shcherbina A, Israeli J, Gruber JJ, Chang J, Nam S, et al. Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat Biomed Eng (2019) 3(12):1009–19. doi: 10.1038/s41551-019-0420-5
55. Kim J, Cao Y, Eddy C, Deng Y, Levine H, Rappel WJ, et al. The mechanics and dynamics of cancer cells sensing noisy 3d contact guidance. Proc Natl Acad Sci United States America (2021) 118(10). doi: 10.1073/pnas.2024780118
56. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat Rev Cancer (2014) 14(6):430–9. doi: 10.1038/nrc3726
57. Wicks EE, Semenza GL. Hypoxia-inducible factors: Cancer progression and clinical translation. J Clin Invest (2022) 132(11). doi: 10.1172/jci159839
58. Mohammadi H, Sahai E. Mechanisms and impact of altered tumour mechanics. Nat Cell Biol (2018) 20(7):766–74. doi: 10.1038/s41556-018-0131-2
59. Lv Y, Wang H, Li G, Zhao B. Three-dimensional decellularized tumor extracellular matrices with different stiffness as bioengineered tumor scaffolds. Bioactive materials (2021) 6(9):2767–82. doi: 10.1016/j.bioactmat.2021.02.004
60. Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for rhogtpases in leading and following cells. Nat Cell Biol (2007) 9(12):1392–400. doi: 10.1038/ncb1658
61. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and yap-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol (2013) 15(6):637–46. doi: 10.1038/ncb2756
62. Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, et al. Targeting lysyl oxidase (Lox) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun (2020) 11(1):2416. doi: 10.1038/s41467-020-16199-4
63. Becker LM, O'Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (2020) 31(9):107701. doi: 10.1016/j.celrep.2020.107701
64. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab (2019) 29(1):124–40.e10. doi: 10.1016/j.cmet.2018.09.012
65. Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, et al. Extracellular matrix remodeling regulates glucose metabolism through txnip destabilization. Cell (2018) 175(1):117–32.e21. doi: 10.1016/j.cell.2018.08.017
66. Kim H, Son S, Ko Y, Shin I. Ctgf regulates cell proliferation, migration, and glucose metabolism through activation of fak signaling in triple-negative breast cancer. Oncogene (2021) 40(15):2667–81. doi: 10.1038/s41388-021-01731-7
67. Peng Y, Chen Z, Chen Y, Li S, Jiang Y, Yang H, et al. Rock isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta biomaterialia (2019) 88:86–101. doi: 10.1016/j.actbio.2019.02.015
68. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant P53 drives the warburg effect. Nat Commun (2013) 4:2935. doi: 10.1038/ncomms3935
69. Buchheit CL, Rayavarapu RR, Schafer ZT. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol (2012) 23(4):402–11. doi: 10.1016/j.semcdb.2012.04.007
70. Ayub A, Yip WK, Seow HF. Dual treatments targeting igf-1r, Pi3k, mtorc or mek synergize to inhibit cell growth, induce apoptosis, and arrest cell cycle at G1 phase in mda-Mb-231 cell line. Biomed pharmacother = Biomed pharmacother (2015) 75:40–50. doi: 10.1016/j.biopha.2015.08.031
71. Icard P, Fournel L, Wu Z, Alifano M, Lincet H. Interconnection between metabolism and cell cycle in cancer. Trends Biochem Sci (2019) 44(6):490–501. doi: 10.1016/j.tibs.2018.12.007
72. Roy D, Sheng GY, Herve S, Carvalho E, Mahanty A, Yuan S, et al. Interplay between cancer cell cycle and metabolism: Challenges, targets and therapeutic opportunities. Biomed pharmacother = Biomed pharmacother (2017) 89:288–96. doi: 10.1016/j.biopha.2017.01.019
73. Icard P, Simula L. Metabolic oscillations during cell-cycle progression. Trends Endocrinol metabolism: TEM (2022) 33(7):447–50. doi: 10.1016/j.tem.2022.04.006
74. Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O'Neal J, et al. 6-Phosphofructo-2-Kinase (Pfkfb3) promotes cell cycle progression and suppresses apoptosis Via Cdk1-mediated phosphorylation of P27. Cell Death Dis (2014) 5(7):e1337. doi: 10.1038/cddis.2014.292
75. Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-Phosphofructo-2-Kinase (Pfkfb3) increases proliferation Via cyclin-dependent kinases. J Biol Chem (2009) 284(36):24223–32. doi: 10.1074/jbc.M109.016816
76. Apostolidi M, Vathiotis IA, Muthusamy V, Gaule P, Gassaway BM, Rimm DL, et al. Targeting pyruvate kinase M2 phosphorylation reverses aggressive cancer phenotypes. Cancer Res (2021) 81(16):4346–59. doi: 10.1158/0008-5472.Can-20-4190
77. Massagué J, Ganesh K. Metastasis-initiating cells and ecosystems. Cancer Discovery (2021) 11(4):971–94. doi: 10.1158/2159-8290.Cd-21-0010
78. Suhail Y, Cain MP, Vanaja K, Kurywchak PA, Levchenko A, Kalluri R, et al. Systems biology of cancer metastasis. Cell Syst (2019) 9(2):109–27. doi: 10.1016/j.cels.2019.07.003
79. Hen O, Barkan D. Dormant disseminated tumor cells and cancer Stem/Progenitor-like cells: Similarities and opportunities. Semin Cancer Biol (2020) 60:157–65. doi: 10.1016/j.semcancer.2019.09.002
80. Pastushenko I, Blanpain C. Emt transition states during tumor progression and metastasis. Trends Cell Biol (2019) 29(3):212–26. doi: 10.1016/j.tcb.2018.12.001
81. Sousa B, Pereira J, Paredes J. The crosstalk between cell adhesion and cancer metabolism. Int J Mol Sci (2019) 20(8):1933. doi: 10.3390/ijms20081933
82. Lai YW, Hsu WJ, Lee WY, Chen CH, Tsai YH, Dai JZ, et al. Prognostic value of a glycolytic signature and its regulation by y-Box-Binding protein 1 in triple-negative breast cancer. Cells (2021) 10(8):1890. doi: 10.3390/cells10081890
83. Schuster E, Taftaf R, Reduzzi C, Albert MK, Romero-Calvo I, Liu H. Better together: Circulating tumor cell clustering in metastatic cancer. Trends Cancer (2021) 7(11):1020–32. doi: 10.1016/j.trecan.2021.07.001
84. Chang PH, Chen MC, Tsai YP, Tan GYT, Hsu PH, Jeng YM, et al. Interplay between Desmoglein2 and hypoxia controls metastasis in breast cancer. Proc Natl Acad Sci United States America (2021) 118(3). doi: 10.1073/pnas.2014408118
85. Thews O, Riemann A. Tumor ph and metastasis: A malignant process beyond hypoxia. Cancer metastasis Rev (2019) 38(1-2):113–29. doi: 10.1007/s10555-018-09777-y
86. Deepak KGK, Vempati R, Nagaraju GP, Dasari VR, S N, Rao DN, et al. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. . Pharmacol Res (2020) 153:104683. doi: 10.1016/j.phrs.2020.104683
87. Tsai TH, Yang CC, Kou TC, Yang CE, Dai JZ, Chen CL, et al. Overexpression of Glut3 promotes metastasis of triple-negative breast cancer by modulating the inflammatory tumor microenvironment. J Cell Physiol (2021) 236(6):4669–80. doi: 10.1002/jcp.30189
88. Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG, et al. Itgb4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene (2020) 39(3):664–76. doi: 10.1038/s41388-019-1014-0
89. Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized atm-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine (2019) 41:370–83. doi: 10.1016/j.ebiom.2019.02.025
90. Wang L, Zhang S, Wang X. The metabolic mechanisms of breast cancer metastasis. Front Oncol (2020) 10:602416. doi: 10.3389/fonc.2020.602416
91. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer (2021) 21(3):162–80. doi: 10.1038/s41568-020-00320-2
92. Qin Q, Yang B, Liu J, Song E, Song Y. Polychlorinated biphenyl quinone exposure promotes breast cancer aerobic glycolysis: An in vitro and in vivo examination. J hazardous materials (2022) 424(Pt C):127512. doi: 10.1016/j.jhazmat.2021.127512
93. Labuschagne CF, Cheung EC, Blagih J, Domart MC, Vousden KH. Cell clustering promotes a metabolic switch that supports metastatic colonization. Cell Metab (2019) 30(4):720–34.e5. doi: 10.1016/j.cmet.2019.07.014
94. Park HA, Brown SR, Kim Y. Cellular mechanisms of circulating tumor cells during breast cancer metastasis. Int J Mol Sci (2020) 21(14):5040. doi: 10.3390/ijms21145040
95. Jin L, Chun J, Pan C, Alesi GN, Li D, Magliocca KR, et al. Phosphorylation-mediated activation of ldha promotes cancer cell invasion and tumour metastasis. Oncogene (2017) 36(27):3797–806. doi: 10.1038/onc.2017.6
96. Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH. Metastasis organotropism: Redefining the congenial soil. Dev Cell (2019) 49(3):375–91. doi: 10.1016/j.devcel.2019.04.012
97. Ignatov A, Eggemann H, Burger E, Ignatov T. Patterns of breast cancer relapse in accordance to biological subtype. J Cancer Res Clin Oncol (2018) 144(7):1347–55. doi: 10.1007/s00432-018-2644-2
98. Press DJ, Miller ME, Liederbach E, Yao K, Huo D. De novo Metastasis in breast cancer: Occurrence and overall survival stratified by molecular subtype. Clin Exp metastasis (2017) 34(8):457–65. doi: 10.1007/s10585-017-9871-9
99. Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci (2019) 40(2):128–41. doi: 10.1016/j.tips.2018.12.004
100. Tallón de Lara P, Castañón H, Vermeer M, Núñez N, Silina K, Sobottka B, et al. Cd39(+)Pd-1(+)Cd8(+) T cells mediate metastatic dormancy in breast cancer. Nat Commun (2021) 12(1):769. doi: 10.1038/s41467-021-21045-2
101. Lemma S, Di Pompo G, Porporato PE, Sboarina M, Russell S, Gillies RJ, et al. Mda-Mb-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim Biophys Acta Mol basis Dis (2017) 1863(12):3254–64. doi: 10.1016/j.bbadis.2017.08.030
102. Foldi J, Rozenblit M, Park TS, Knowlton CA, Golshan M, Moran M, et al. Optimal management for residual disease following neoadjuvant systemic therapy. Curr Treat options Oncol (2021) 22(9):79. doi: 10.1007/s11864-021-00879-4
103. Radovich M, Jiang G, Hancock BA, Chitambar C, Nanda R, Falkson C, et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: Preplanned secondary analysis of the Bre12-158 randomized clinical trial. JAMA Oncol (2020) 6(9):1410–5. doi: 10.1001/jamaoncol.2020.2295
104. Ruth JR, Pant DK, Pan TC, Seidel HE, Baksh SC, Keister BA, et al. Cellular dormancy in minimal residual disease following targeted therapy. Breast Cancer Res BCR (2021) 23(1):63. doi: 10.1186/s13058-021-01416-9
105. Manjili MH, Khazaie K. Pattern recognition of tumor dormancy and relapse beyond cell-intrinsic and cell-extrinsic pathways. Semin Cancer Biol (2022) 78:1–4. doi: 10.1016/j.semcancer.2021.12.008
106. Ramamoorthi G, Kodumudi K, Gallen C, Zachariah NN, Basu A, Albert G, et al. Disseminated cancer cells in breast cancer: Mechanism of dissemination and dormancy and emerging insights on therapeutic opportunities. Semin Cancer Biol (2022) 78:78–89. doi: 10.1016/j.semcancer.2021.02.004
107. Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer (2020) 20(7):398–411. doi: 10.1038/s41568-020-0263-0
108. Werner S, Heidrich I, Pantel K. Clinical management and biology of tumor dormancy in breast cancer. Semin Cancer Biol (2022) 78:49–62. doi: 10.1016/j.semcancer.2021.02.001
109. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun (2018) 9(1):1944. doi: 10.1038/s41467-018-04070-6
110. Li M, Liu J, Li S, Feng Y, Yi F, Wang L, et al. Autophagy-related 7 modulates tumor progression in triple-negative breast cancer. Lab investigation; J Tech Methods Pathol (2019) 99(9):1266–74. doi: 10.1038/s41374-019-0249-2
111. La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun (2019) 10(1):3668. doi: 10.1038/s41467-019-11640-9
112. Radic Shechter K, Kafkia E, Zirngibl K, Gawrzak S, Alladin A, Machado D, et al. Metabolic memory underlying minimal residual disease in breast cancer. Mol Syst Biol (2021) 17(10):e10141. doi: 10.15252/msb.202010141
113. Lim AR, Ghajar CM. Thorny ground, rocky soil: Tissue-specific mechanisms of tumor dormancy and relapse. Semin Cancer Biol (2022) 78:104–23. doi: 10.1016/j.semcancer.2021.05.007
114. Shinde A, Wilmanski T, Chen H, Teegarden D, Wendt MK. Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res BCR (2018) 20(1):76. doi: 10.1186/s13058-018-1008-9
115. Dando I, Pozza ED, Ambrosini G, Torrens-Mas M, Butera G, Mullappilly N, et al. Oncometabolites in cancer aggressiveness and tumour repopulation. Biol Rev Cambridge Philos Soc (2019) 94(4):1530–46. doi: 10.1111/brv.12513
116. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the tca cycle Via circulating lactate. Nature (2017) 551(7678):115–8. doi: 10.1038/nature24057
117. Dittmer J. Mechanisms governing metastatic dormancy in breast cancer. Semin Cancer Biol (2017) 44:72–82. doi: 10.1016/j.semcancer.2017.03.006
118. Risson E, Nobre AR, Maguer-Satta V, Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer (2020) 1(7):672–80. doi: 10.1038/s43018-020-0088-5
119. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi: 10.1016/j.immuni.2013.04.005
120. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer (2020) 20(9):516–31. doi: 10.1038/s41568-020-0273-y
121. Dias AS, Almeida CR, Helguero LA, Duarte IF. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer (Oxford Engl 1990) (2019) 121:154–71. doi: 10.1016/j.ejca.2019.09.002
122. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
123. Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine (2019) 42:431–42. doi: 10.1016/j.ebiom.2019.03.068
124. Navarrete-Bernal MGC, Cervantes-Badillo MG, Martínez-Herrera JF, Lara-Torres CO, Gerson-Cwilich R, Zentella-Dehesa A, et al. Biological landscape of triple negative breast cancers expressing ctla-4. Front Oncol (2020) 10:1206. doi: 10.3389/fonc.2020.01206
125. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. Ctla-4 blockade drives loss of T(Reg) stability in glycolysis-low tumours. Nature (2021) 591(7851):652–8. doi: 10.1038/s41586-021-03326-4
126. Brooks GA. The tortuous path of lactate shuttle discovery: From cinders and boards to the Lab and icu. J sport Health Sci (2020) 9(5):446–60. doi: 10.1016/j.jshs.2020.02.006
127. Brown TP, Ganapathy V. Lactate/Gpr81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and warburg phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
128. Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. Lactate is a natural suppressor of rlr signaling by targeting mavs. Cell (2019) 178(1):176–89.e15. doi: 10.1016/j.cell.2019.05.003
129. Chen J, Cao X, Li B, Zhao Z, Chen S, Lai SWT, et al. Warburg effect is a cancer immune evasion mechanism against macrophage immunosurveillance. Front Immunol (2020) 11:621757. doi: 10.3389/fimmu.2020.621757
130. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity Via a specific cebpb isoform in triple-negative breast cancer. Cell Metab (2018) 28(1):87–103.e6. doi: 10.1016/j.cmet.2018.04.022
131. Jiang H, Wei H, Wang H, Wang Z, Li J, Ou Y, et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis (2022) 13(3):206. doi: 10.1038/s41419-022-04632-z
132. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature (2019) 566(7745):553–7. doi: 10.1038/s41586-019-0915-y
133. Guo B. Oliver TG. partners in crime: Neutrophil-ctc collusion in metastasis. Trends Immunol (2019) 40(7):556–9. doi: 10.1016/j.it.2019.04.009
134. Wu SY, Wang H, Shao ZM, Jiang YZ. Triple-negative breast cancer: New treatment strategies in the era of precision medicine. Sci China Life Sci (2021) 64(3):372–88. doi: 10.1007/s11427-020-1714-8
135. Zhu Y, Hu Y, Tang C, Guan X, Zhang W. Platinum-based systematic therapy in triple-negative breast cancer. Biochim Biophys Acta Rev Cancer (2022) 1877(1):188678. doi: 10.1016/j.bbcan.2022.188678
136. Saha T, Lukong KE. Breast cancer stem-like cells in drug resistance: A review of mechanisms and novel therapeutic strategies to overcome drug resistance. Front Oncol (2022) 12:856974. doi: 10.3389/fonc.2022.856974
137. Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell (2018) 173(4):879–93.e13. doi: 10.1016/j.cell.2018.03.041
138. Bai X, Ni J, Beretov J, Graham P, Li Y. Triple-negative breast cancer therapeutic resistance: Where is the achilles' heel? Cancer Lett (2021) 497:100–11. doi: 10.1016/j.canlet.2020.10.016
139. Gonçalves AC, Richiardone E, Jorge J, Polónia B, Xavier CPR, Salaroglio IC, et al. Impact of cancer metabolism on therapy resistance - clinical implications. Drug resistance updates Rev commentaries antimicrobial Anticancer chemother (2021) 59:100797. doi: 10.1016/j.drup.2021.100797
140. Marcucci F, Rumio C. Glycolysis-induced drug resistance in tumors-a response to danger signals? Neoplasia (New York NY) (2021) 23(2):234–45. doi: 10.1016/j.neo.2020.12.009
141. Li Q, Qin T, Bi Z, Hong H, Ding L, Chen J, et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat Commun (2020) 11(1):1456. doi: 10.1038/s41467-020-15308-7
142. Mehraj U, Mir IA, Hussain MU, Alkhanani M, Wani NA, Mir MA. Adapalene and doxorubicin synergistically promote apoptosis of tnbc cells by hyperactivation of the Erk1/2 pathway through ros induction. Front Oncol (2022) 12:938052. doi: 10.3389/fonc.2022.938052
143. Luo M, Fu A, Wu R, Wei N, Song K, Lim S, et al. High expression of G6pd increases doxorubicin resistance in triple negative breast cancer cells by maintaining gsh level. Int J Biol Sci (2022) 18(3):1120–33. doi: 10.7150/ijbs.65555
144. Wen J, Yeo S, Wang C, Chen S, Sun S, Haas MA, et al. Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res Treat (2015) 149(3):619–29. doi: 10.1007/s10549-015-3283-9
145. Chittaranjan S, Bortnik S, Dragowska WH, Xu J, Abeysundara N, Leung A, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2014) 20(12):3159–73. doi: 10.1158/1078-0432.Ccr-13-2060
146. Yang T, Ren C, Qiao P, Han X, Wang L, Lv S, et al. Pim2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene (2018) 37(45):5997–6009. doi: 10.1038/s41388-018-0386-x
147. Amith SR, Fliegel L. Na(+)/H(+) exchanger-mediated hydrogen ion extrusion as a carcinogenic signal in triple-negative breast cancer etiopathogenesis and prospects for its inhibition in therapeutics. Semin Cancer Biol (2017) 43:35–41. doi: 10.1016/j.semcancer.2017.01.004
148. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated ph: A perfect storm for cancer progression. Nat Rev Cancer (2011) 11(9):671–7. doi: 10.1038/nrc3110
149. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: Role of atp-dependent transporters. Nat Rev Cancer (2002) 2(1):48–58. doi: 10.1038/nrc706
150. Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, et al. Glycolysis inhibition inactivates abc transporters to restore drug sensitivity in malignant cells. PloS One (2011) 6(11):e27222. doi: 10.1371/journal.pone.0027222
151. Omran Z, Scaife P, Stewart S, Rauch C. Physical and biological characteristics of multi drug resistance (Mdr): An integral approach considering ph and drug resistance in cancer. Semin Cancer Biol (2017) 43:42–8. doi: 10.1016/j.semcancer.2017.01.002
152. Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M, et al. Triple negative breast cancer: Key role of tumor-associated macrophages in regulating the activity of anti-Pd-1/Pd-L1 agents. Biochim Biophys Acta Rev Cancer (2018) 1869(1):78–84. doi: 10.1016/j.bbcan.2017.10.007
153. Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta Rev Cancer (2020) 1874(2):188427. doi: 10.1016/j.bbcan.2020.188427
154. Oh S, Kim H, Nam K, Shin I. Silencing of Glut1 induces chemoresistance Via modulation of Akt/Gsk-3β/B-Catenin/Survivin signaling pathway in breast cancer cells. Arch Biochem biophysics (2017) 636:110–22. doi: 10.1016/j.abb.2017.08.009
155. Wu Q, Ba-Alawi W, Deblois G, Cruickshank J, Duan S, Lima-Fernandes E, et al. Glut1 inhibition blocks growth of Rb1-positive triple negative breast cancer. Nat Commun (2020) 11(1):4205. doi: 10.1038/s41467-020-18020-8
156. Abad E, García-Mayea Y, Mir C, Sebastian D, Zorzano A, Potesil D, et al. Common metabolic pathways implicated in resistance to chemotherapy point to a key mitochondrial role in breast cancer. Mol Cell Proteomics MCP (2019) 18(2):231–44. doi: 10.1074/mcp.RA118.001102
157. Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, et al. Targeting immunosuppressive macrophages overcomes parp inhibitor resistance in Brca1-associated triple-negative breast cancer. Nat Cancer (2021) 2(1):66–82. doi: 10.1038/s43018-020-00148-7
158. Xiao Y, Ma D, Yang YS, Yang F, Ding JH, Gong Y, et al. Comprehensive metabolomics expands precision medicine for triple-negative breast cancer. Cell Res (2022) 32(5):477–90. doi: 10.1038/s41422-022-00614-0
159. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest (2011) 121(7):2750–67. doi: 10.1172/jci45014
160. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: Subtypes and treatment strategies. Cancer Cell (2019) 35(3):428–40.e5. doi: 10.1016/j.ccell.2019.02.001
161. He Y, Jiang Z, Chen C, Wang X. Classification of triple-negative breast cancers based on immunogenomic profiling. J Exp Clin Cancer Res CR (2018) 37(1):327. doi: 10.1186/s13046-018-1002-1
162. Jiang YZ, Liu Y, Xiao Y, Hu X, Jiang L, Zuo WJ, et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: The future trial. Cell Res (2021) 31(2):178–86. doi: 10.1038/s41422-020-0375-9
163. Tripp A, Poulogiannis G. Banking on metabolomics for novel therapies in tnbc. Cell Res (2022) 32(5):423–4. doi: 10.1038/s41422-022-00637-7
164. Lee J, Yesilkanal AE, Wynne JP, Frankenberger C, Liu J, Yan J, et al. Effective breast cancer combination therapy targeting Bach1 and mitochondrial metabolism. Nature (2019) 568(7751):254–8. doi: 10.1038/s41586-019-1005-x
165. Iqbal MA, Chattopadhyay S, Siddiqui FA, Ur Rehman A, Siddiqui S, Prakasam G, et al. Silibinin induces metabolic crisis in triple-negative breast cancer cells by modulating egfr-Myc-Txnip axis: Potential therapeutic implications. FEBS J (2021) 288(2):471–85. doi: 10.1111/febs.15353
166. Raninga PV, Lee A, Sinha D, Dong LF, Datta KK, Lu X, et al. Marizomib suppresses triple-negative breast cancer Via proteasome and oxidative phosphorylation inhibition. Theranostics (2020) 10(12):5259–75. doi: 10.7150/thno.42705
167. Bai X, Jiang H, Han G, He Q. Chidamide suppresses the glycolysis of triple negative breast cancer cells partially by targeting the Mir−33a−5p−Ldha axis. Mol Med Rep (2019) 20(2):1857–65. doi: 10.3892/mmr.2019.10425
168. Zhu S, Wei L, Lin G, Tong Y, Zhang J, Jiang X, et al. Metabolic alterations induced by kudingcha lead to cancer cell apoptosis and metastasis inhibition. Nutr Cancer (2020) 72(4):696–707. doi: 10.1080/01635581.2019.1645865
169. Euceda LR, Hill DK, Stokke E, Hatem R, El Botty R, Bièche I, et al. Metabolic response to everolimus in patient-derived triple-negative breast cancer xenografts. J Proteome Res (2017) 16(5):1868–79. doi: 10.1021/acs.jproteome.6b00918
170. Tripathi V, Jaiswal P, Assaiya A, Kumar J, Parmar HS. Anti-cancer effects of 5-Aminoimidazole-4-Carboxamide-1-B-D-Ribofuranoside (Aicar) on triple-negative breast cancer (Tnbc) cells: Mitochondrial modulation as an underlying mechanism. Curr Cancer Drug Targets (2022) 22(3):245–56. doi: 10.2174/1568009622666220207101212
171. Tailor D, Going CC, Resendez A, Kumar V, Nambiar DK, Li Y, et al. Novel aza-podophyllotoxin derivative induces oxidative phosphorylation and cell death Via ampk activation in triple-negative breast cancer. Br J Cancer (2021) 124(3):604–15. doi: 10.1038/s41416-020-01137-4
172. Fu J, Liu S, Hu M, Liao X, Wang X, Xu Z, et al. Biguanide Mc001, a dual inhibitor of oxphos and glycolysis, shows enhanced antitumor activity without increasing lactate production. ChemMedChem (2022) 17(6):e202100674. doi: 10.1002/cmdc.202100674
173. O'Neill S, Porter RK, McNamee N, Martinez VG, O'Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep (2019) 9(1):3788. doi: 10.1038/s41598-019-39789-9
174. Jin J, Qiu S, Wang P, Liang X, Huang F, Wu H, et al. Cardamonin inhibits breast cancer growth by repressing hif-1α-Dependent metabolic reprogramming. J Exp Clin Cancer Res CR (2019) 38(1):377. doi: 10.1186/s13046-019-1351-4
175. Yi X, Qi M, Huang M, Zhou S, Xiong J. Honokiol inhibits hif-1α-Mediated glycolysis to halt breast cancer growth. Front Pharmacol (2022) 13:796763. doi: 10.3389/fphar.2022.796763
176. Zheng Y, Liu P, Wang N, Wang S, Yang B, Li M, et al. Betulinic acid suppresses breast cancer metastasis by targeting Grp78-mediated glycolysis and er stress apoptotic pathway. Oxid Med Cell Longevity (2019) 2019:8781690. doi: 10.1155/2019/8781690
177. Jiao L, Wang S, Zheng Y, Wang N, Yang B, Wang D, et al. Betulinic acid suppresses breast cancer aerobic glycolysis Via caveolin-1/Nf-Kb/C-Myc pathway. Biochem Pharmacol (2019) 161:149–62. doi: 10.1016/j.bcp.2019.01.016
178. Su Y, Zhang X, Lei L, Liu B, Wu S, Shen J. Tumor microenvironment-activatable cyclic cascade reaction to reinforce multimodal combination therapy by destroying the extracellular matrix. ACS Appl materials interfaces (2021) 13(11):12960–71. doi: 10.1021/acsami.1c02011
179. De Vita A, Liverani C, Molinaro R, Martinez JO, Hartman KA, Spadazzi C, et al. Lysyl oxidase engineered lipid nanovesicles for the treatment of triple negative breast cancer. Sci Rep (2021) 11(1):5107. doi: 10.1038/s41598-021-84492-3
180. Mason JA, Hagel KR, Hawk MA, Schafer ZT. Metabolism during ecm detachment: Achilles heel of cancer cells? Trends Cancer (2017) 3(7):475–81. doi: 10.1016/j.trecan.2017.04.009
181. Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol (2017) 19(5):530–41. doi: 10.1038/ncb3509
182. Min HY, Pei H, Hyun SY, Boo HJ, Jang HJ, Cho J, et al. Potent anticancer effect of the natural steroidal saponin gracillin is produced by inhibiting glycolysis and oxidative phosphorylation-mediated bioenergetics. Cancers (2020) 12(4):913. doi: 10.3390/cancers12040913
183. Gandhi N, Das GM. Metabolic reprogramming in breast cancer and its therapeutic implications. Cells (2019) 8(2):89. doi: 10.3390/cells8020089
184. Tseng CW, Kuo WH, Chan SH, Chan HL, Chang KJ, Wang LH. Transketolase regulates the metabolic switch to control breast cancer cell metastasis Via the A-ketoglutarate signaling pathway. Cancer Res (2018) 78(11):2799–812. doi: 10.1158/0008-5472.Can-17-2906
Keywords: Warburg effect, glycolysis, metabolic plasticity, triple-negative breast cancer, basal-like breast cancer
Citation: Liu S, Li Y, Yuan M, Song Q and Liu M (2023) Correlation between the Warburg effect and progression of triple-negative breast cancer. Front. Oncol. 12:1060495. doi: 10.3389/fonc.2022.1060495
Received: 03 October 2022; Accepted: 06 December 2022;
Published: 27 January 2023.
Edited by:
Wimal Pathmasiri, University of North Carolina at Chapel Hill, United StatesReviewed by:
Umar Mehraj, University of Kashmir, IndiaCopyright © 2023 Liu, Li, Yuan, Song and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Liu, bGl1bWluNjk1OEAxNjMuY29t; Qing Song, c29uZ3FpbmcxMjAxNjA4NEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.