 Edmond M. Kwan
Edmond M. Kwan Alexander W. Wyatt1,3
Alexander W. Wyatt1,3 Kim N. Chi
Kim N. Chi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 10 November 2022
Sec. Genitourinary Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1054497
This article is part of the Research Topic Prostate Cancer Genomics: Application at Different Stages of a Patient’s Journey View all 5 articles
Plasma circulating tumor DNA (ctDNA) represents short fragments of tumor-derived DNA released into the bloodstream primarily from cancer cells undergoing apoptosis. In metastatic castration-resistant prostate cancer (mCRPC), characterizing genomic alterations in ctDNA identifies mutations, copy number alterations, and structural rearrangements with predictive and prognostic biomarker utility. These associations with clinical outcomes have resulted in ctDNA increasingly incorporated into routine clinical care. In this review, we summarize current and emerging applications for ctDNA analysis in metastatic prostate cancer, including outcome prediction, treatment selection, and characterization of treatment resistance. We also discuss potential pitfalls with interpreting ctDNA findings, namely false negatives arising from low tumor content and optimal assay design, including correction for clonal hematopoiesis of indeterminate potential and germline variants. Understanding the influence of these limitations on interpretation of ctDNA results is necessary to overcome barriers to clinical implementation. Nevertheless, as assay availability and technology continue to improve, recognizing both opportunities and shortcomings of ctDNA analysis will retain relevance with informing the implementation of precision-oncology initiatives for metastatic prostate cancer.
Blood-based liquid biopsies are a minimally invasive tool to acquire molecular insights in metastatic prostate cancer. Diverse tumor-derived products can be detected in metastatic castration-resistant prostate cancer (mCRPC), including circulating tumor cells, circulating tumor DNA/RNA, proteins, extracellular vesicles, and tumor-educated platelets (1). Of these, plasma circulating tumor DNA (ctDNA) has attracted the greatest attention. Ease of sampling (including serial collection) and well-established isolation protocols have fueled considerable efforts to understand its value as a prognostic, predictive and response biomarker in the advanced disease setting. With the recent US Food and Drug Administration (FDA) approval of multiple commercial assays with companion diagnostic status, ctDNA is increasingly being used for the genomic profiling of patients as part of standard-of-care practice.
Plasma cell-free DNA (cfDNA) is highly fragmented, double-stranded extracellular DNA released into the bloodstream primarily from cells undergoing apoptosis (2). In healthy individuals, most cfDNA comes from normal turnover of white blood cells (3), with concentrations ranging from 1-10ng per milliliter of blood (4). In contrast, cfDNA concentration in patients with mCRPC is greater by two-to-three-fold (5). The abundance of ctDNA (i.e., tumor-derived cfDNA) as a proportion of total cell-free DNA can be expressed as the ctDNA fraction (ctDNA%) of a blood sample. The short half-life of ctDNA (minutes to hours) further lends itself as a real-time assessment of the current disease state.
With a growing number of therapy options now available to patients with metastatic prostate cancer, tools that guide treatment selection and sequencing remain an unmet clinical need. In this review, we summarize current and emerging ctDNA applications for outcome prediction, treatment selection and disease monitoring in metastatic prostate cancer. We focus on correlative studies that provide a framework for integrating ctDNA profiling into routine patient care. Importantly, we highlight potential pitfalls with interpreting ctDNA findings in challenging scenarios. As assay availability and technology improve, recognizing both opportunities and shortcomings of ctDNA analysis will become increasingly relevant to supporting precision-based care of patients with metastatic prostate cancer.
Genomic biomarker testing in metastatic prostate cancer normally requires analysis of tumor tissue from prostate primary specimens (6). However, disease-specific factors complicate using tissue as source material. In a recent analysis of over 4000 prostate cancer tissue specimens, sequencing failure rates approached 30-40% (7). Low pathological tumor content and poor DNA yield were frequently responsible for unsuccessful sequencing, an issue that pervasively impacts prostate core needle biopsies obtained at initial diagnosis. Furthermore, marked genomic spatial heterogeneity within the prostate can undermine the ability of a single prostate biopsy specimen to correctly identify the dominant genotype responsible for metastatic disease development (8, 9). In patients with metachronous metastatic disease, substantial time can separate initial diagnosis and subsequent development of clinical metastases (10). During this period, tumor evolution shaped by systemic therapy exposure to androgen deprivation therapy (ADT), docetaxel chemotherapy and/or potent androgen receptor pathway inhibitors (11) (ARPI) mean that archival tissue may not reflect the contemporaneous tumor genotype. One striking example involves genetic alterations in the androgen receptor (AR). AR alterations are rare in systemic therapy-naive prostate cancer, but highly enriched in the castration-resistant disease setting (5, 12–16). Furthermore, AR can exhibit heterogeneity between metastases within the same patient (17, 18).
Overcoming these limitations of primary tissue analysis by obtaining fresh biopsies from metastatic disease sites is similarly problematic. Not only are metastatic biopsies technically challenging, but serial sampling is impractical for patients and health services alike. Critically, single lesion sampling fails to represent the diversity of competing tumor clones present between different disease sites. In a recent study employing deep whole-genome sequencing of serial plasma samples and tissue biopsies of synchronous metastases in poor-risk mCRPC patients, the contribution of any singular metastasis to the total ctDNA pool was low, providing evidence to support the long held notion that ctDNA captures tumor material from multiple metastatic foci (19). This ability to capture spatial and temporal heterogeneity between metastases have led to a number of clinical applications for ctDNA in metastatic prostate cancer.
Considerable infrastructural, technological, and intellectual overhead generally accompanies most ctDNA analysis platforms. These resourcing requirements mean that a majority of clinicians around the world continue to have limited access to ctDNA profiling to inform patient care. Instead, clinicians typically encounter ctDNA testing in the clinical trial setting, where it is increasingly used alongside tumor tissue testing to screen for genomic alterations that may confer sensitivity to established and novel targeted therapies (20–25). Outside of clinical trials, US FDA-approved commercial platforms are available (e.g., FoundationOne Liquid CDx, Guardant360 CDx) (26, 27), but can be cost prohibitive for patients unable to acquire partial or full financial reimbursement for testing. While some academic research centers offer in-house ctDNA assays (28), these services are beyond reach for most community oncologists. Finally, where testing is available, there remains no consensus surrounding best practices for the post-analytic phase of ctDNA profiling. In contrast to considerable efforts to harmonize pre-analytical variables associated with cfDNA/ctDNA collection and isolation (29), the optimal approach to evaluation, interpretation, reporting and integration of patient results into clinical care is not established (30). Collectively, these factors must be addressed before widespread adoption of ctDNA testing to inform routine patient care. Despite these challenges, several clinical applications are likely to emerge as forerunners for ctDNA implementation in management of advanced prostate cancer.
Accurate prognostic information informs systemic therapy selection, dictates treatment urgency, stratifies participants in clinical trials, and shapes discussions with patients around expected long-term outcomes (31, 32). Current survival prediction models incorporate pathologic, biochemical and radiographic markers of disease burden, including performance status, Gleason score, prostate specific antigen (PSA) levels, lactate dehydrogenase, albumin, alkaline phosphatase, number of bone metastases, and visceral organ involvement (33–36). While these models are informative, performance is variable, and most were validated in an era with few life-prolonging treatment options. New methods of prognostication are therefore needed to account for a rapidly shifting therapeutic landscape.
In mCRPC, both total cfDNA concentration and ctDNA% correlate with many conventional clinical markers of disease burden, including visceral disease, lactate dehydrogenase, hemoglobin and alkaline phosphatase levels (5, 16, 37–39). However, even when accounting for these poor-risk clinical features, higher pretreatment cfDNA concentration and ctDNA% independently predicts shorter progression-free and overall survival in patients commencing contemporaneous systemic therapies. These associations are evident in men receiving next-generation ARPIs such as abiraterone and enzalutamide (5, 40–45), as well as taxane cytotoxic therapies docetaxel and cabazitaxel (16, 39, 46, 47). Less well understood is the precise relationship between these metrics and life expectancy. Despite knowledge of their negative prognostic potential, no clinically relevant thresholds for cfDNA concentration and ctDNA% currently exist. In addition, the influence of prior systemic treatment on the interpretation of ctDNA% has not been systematically investigated. This is particularly relevant in metastatic castration-sensitive prostate cancer (mCSPC), where ADT administration results in rapid ctDNA% decline (48). Understanding this, along with the additive contribution of pretreatment cfDNA concentration and ctDNA% to existing clinical prognostic models is needed before integration in the clinical setting.
A challenge with incorporating ctDNA% as a routine biomarker is that various methods exist for quantifying plasma ctDNA% (Figure 1). At its simplest form, ctDNA% can be estimated from the variant allele frequency (VAF) of somatic mutations detected within a patient sample. Typically, the highest VAF mutation(s) are chosen for ctDNA% approximation, on the assumption that they likely represent truncal mutation(s) present in most ctDNA-releasing cancer cells. Advances in molecular barcoding of individual cfDNA fragments, and custom-designed patient-specific custom panels (using mutations found in primary tissue) coupled with ultra-deep targeted sequencing have enabled mutations to be detected well below 1% VAF, allowing more refined estimates of ctDNA% (49). Mutation-based ctDNA% estimation methods can be complemented by systematic assessments of genomic regions affected by copy number variants (CNV). Current computational algorithms identify regions of copy gain or loss by evaluating sequencing read depth ratios (available from both targeted and shallow whole-genome sequencing approaches) and/or shifts in B-allele frequency at germline heterozygous SNP loci (requiring targeted sequencing across specific SNPs). The degree of signal from segment-level copy number alterations can then be used with overall tumor ploidy estimates to calculate ctDNA%, similar to how magnitude of somatic mutation VAF corresponds to ctDNA% (50, 51).
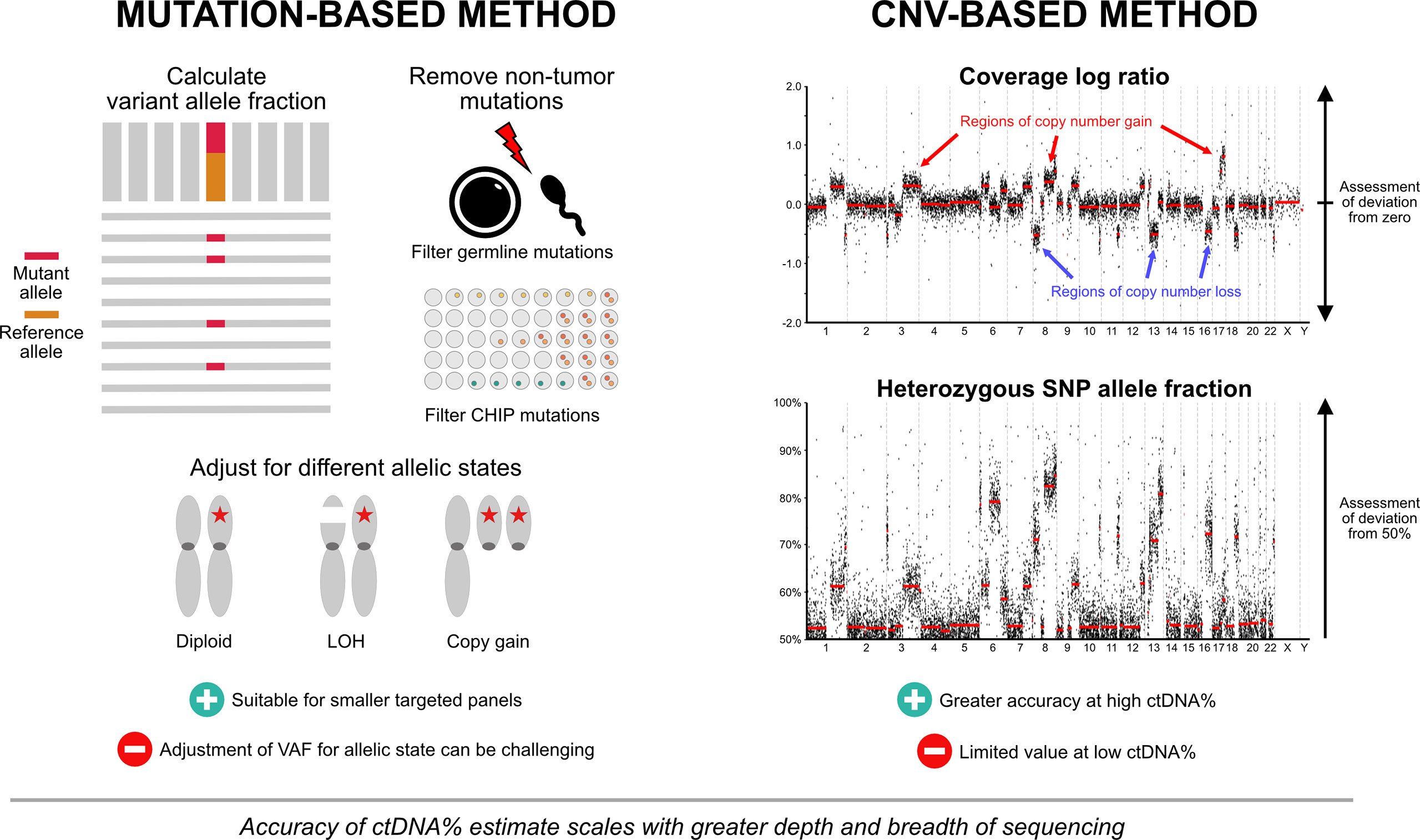
Figure 1 Approaches to ctDNA fraction estimation. Both mutation-based and copy number-based approaches are used for ctDNA% estimation. Mutation-based approaches typically utilize the variant allele fraction of truncal mutation(s), with or without adjustment for different allelic configurations. Germline mutations and mutations arising from clonal hematopoiesis of indeterminate potential are removed to prevent contribution to ctDNA% estimates. Mutation-based methods can be complemented by systematic assessments of genomic regions affected by copy number changes. Regions of copy gain or loss are identified by evaluating sequencing read depth ratios and/or shifts in B-allele frequency at germline heterozygous SNP loci. The degree of signal from segment-level copy number alterations can then be used with overall tumor ploidy estimates to calculate ctDNA%. Panels that provide both mutation and copy number data encompassing broad genomic territory are likely to yield the most accurate estimates across a spectrum of ctDNA%. Abbreviations: CHIP, clonal hematopoiesis of indeterminate potential; ctDNA%, circulating tumor DNA fraction; LOH, loss-of-heterozygosity; SNP, single nucleotide polymorphism; VAF, variant allele fraction.
No single method of ctDNA quantification is appropriate for all samples. Ideally, panels that provide both mutation and CNV data will yield the most accurate estimates across a spectrum of ctDNA%. Mutation-based methods are preferable in lower tumor content samples, as CNVs are difficult to differentiate from background signal at <10-15% ctDNA. Conversely, CNV-based (or adjusted) methods may be preferred in instances where use of mutation VAF alone will likely lead to ctDNA% overestimates, such as tumor aneuploidy (e.g., whole genome duplication), loss-of-heterozygosity across the wild-type allele, and copy gain of the allele carrying the mutation (known as mutant allele specific imbalance) (52). False positive mutations affecting ctDNA% estimates can also arise in patients with clonal hematopoiesis of indeterminate potential (CHIP). CHIP is defined by the presence of clonal expansion of somatic mutations in hematopoietic stem cells in the absence of any concurrent hematologic neoplasia. The risk of misattributing a CHIP-related somatic mutation as a legitimate prostate cancer mutation may be as high as 15-20% (53), and can lead to an overestimate of ctDNA%. Concurrent sequencing of matched white blood cells is a simple method to ensure that CHIP-related mutations are correctly identified, but surprisingly is not performed by current commercially available clinical grade ctDNA tests. Importantly, confidence in ctDNA% estimates scale with sequencing depth and breath. Sequencing approaches that encompass broad genomic territory (e.g., larger pan-cancer targeted panels or whole exome/genome assays) produce a greater amount of data that can be used for estimates. In turn, this must be balanced with the financial toxicity and substantial human capital required for successful implementation across a wide population group. While the real-world cost of shallow whole genome sequencing (0.1-5x depth) continues to fall with time, higher sequencing depth (>20-40x depth) is likely required to provide sufficient biologic insights that inform treatment decision-making (19, 54). Factoring in the expenditure associated with data analysis, interpretation and patient counseling, the sum cost of informative whole genome analysis is likely to exceed $10,000-$20,000 per patient (55), arguably not cost-effective for routine implementation in all advanced prostate cancer patients.
Serial PSA measurement is a mainstay of disease monitoring in metastatic prostate cancer. In most patients with mCSPC, PSA falls rapidly after starting ADT, with nadir levels prognostic for overall survival (56). In mCRPC, early PSA decline at four weeks following ARPI commencement predicts durability of treatment response and overall survival, irrespective of prior taxane chemotherapy (57). Nevertheless, PSA is an imperfect response marker. Up to 25% of patients receiving ADT +/- docetaxel for mCSPC experience clinical progression without PSA elevation (58). Similar findings have also been described in enzalutamide-treated mCRPC (59). Importantly, heavily pretreated mCRPC enriches AR-independent disease phenotypes well-recognized to secrete low levels of PSA (60).
Radiographic disease monitoring also has limitations. The sensitivity and specificity of CT and bone scan imaging is suboptimal for accurate identification of lymph node and skeletal metastases alike (61). The phenomenon of bone flare whereby effective treatments lead to the apparent appearance of new or more prominent bone lesions can be misinterpreted as true progression. While new radionuclide imaging techniques such as PSMA PET/CT may address the limitations of conventional imaging, high level evidence supporting prospective use as a treatment evaluation tool is lacking (62).
Tracking ctDNA% has the potential to address shortcomings of PSA and radiographic disease assessment. Notably, while on-treatment PSA and ctDNA% generally trend in the same direction, there is weak correlation for the magnitude of this change (38, 63). This suggests that PSA and ctDNA% do not reflect the same tumor biology, and ctDNA% could provide additional information to support current clinical assessment markers.
In patients receiving systemic therapy for mCRPC, early reductions in cfDNA concentration or ctDNA% (within 4-8 weeks of treatment commencement) are independently associated with longer progression-free survival and overall survival (47, 63–65). Conversely, a rise in ctDNA% at 12 weeks significantly increases the risk of early biochemical and radiographic progression on ARPI or cabazitaxel chemotherapy (16, 66). Examples where ctDNA% correctly differentiates between radiographic flare phenomenon and true progression have also been reported in patients receiving ARPI or immune checkpoint inhibitor therapy (66, 67).
The depth of ctDNA% decline may also be relevant. In an analysis of patients receiving abiraterone +/- ipatasertib, deeper reductions in ctDNA% were associated with higher rates of partial or complete radiographic soft tissue responses to therapy (64). Relatedly, two recent reports suggest that clearance of ctDNA from circulation may be the most informative predictor of long-term clinical outcomes. Tracking alterations in prostate cancer driver genes PTEN, TP53 and RB1, Jayaram et al. showed that persistent detection after four weeks of ARPI was independently associated with worse survival (68). Similarly, Tolmeijer et al. found that mCRPC patients with detectable ctDNA at both baseline and four-week timepoints following ARPI commencement selected individuals more likely to experience rapid acquired resistance within six months of starting treatment (65). Both studies found that patients that convert from ctDNA detectable to undetectable had outcomes similar to patients that were ctDNA undetectable at both timepoints. Note that ‘detectable’ versus ‘undetectable’ is a blood volume-dependent and assay-dependent variable. Greater volume of blood collected, and higher sequencing depth increases the probability of detecting rare tumor-derived cfDNA fragments. In future, more sensitive methodological approaches including personalized tumor-informed mutation panels and methylation-based cfDNA profiling are likely further reduce the absolute lower limit of detection for ctDNA (49), although tests that classify all patients as ctDNA-positive are unlikely to be useful in guiding patient management. Further work is required to understand exact clinically meaningful thresholds for ctDNA% detection or declines. In addition, we need to better ascertain the ideal on-treatment timepoint(s) to monitor ctDNA%. Finally, understanding how interpretation of ctDNA changes differs according to disease state (e.g., mCSPC vs mCRPC) and class of systemic therapy will require further evaluation as exploratory intermediate endpoints in large prospective cohorts.
The genomic landscape of metastatic prostate cancer has been established over a series of coordinated tissue sequencing efforts (12–15, 69, 70). These studies identified potentially actionable alterations in key growth pathways targetable using precision medicines. To date, two classes of targeted therapies have gained regulatory approval in the biomarker-selected mCRPC population: poly(ADP-ribose) polymerase (PARP) inhibitors olaparib and rucaparib in tumors with defective homologous recombination repair (HRR), and the anti-PD1 immune checkpoint inhibitor pembrolizumab in tumors with deficient mismatch repair (MMR) and/or high mutational burden.
The convenience of collecting ctDNA has spurred hopes that it could eventually replace tissue as a means for screening some patients for targeted therapies. The full spectrum of genomic alterations can be detected in ctDNA, including mutations, copy number alterations and structural rearrangements. However, like tumour tissue, liquid biopsy similarly suffers from sample insufficiency due to low levels of tumor material in the blood. Depending on prior treatment exposure and burden of disease, it is estimated that ctDNA fraction is below 1% in between 30-40% of patients with mCRPC (5, 41, 42); these patients are not amenable to tumor genotyping using ctDNA. In general, providing sufficient ctDNA% is present, ctDNA alterations from patients with mCRPC show strong concordance with matched metastatic tissue (19, 71), and reproduce the expected mutational landscape (5, 16, 42, 43, 71–73). Apparent differences are explained by low ctDNA% samples, variability in assay design and presence of subclonal alterations. The impact of these factors on identifying metastatic prostate cancer patients suitable for PARP inhibitors and immunotherapy is discussed below.
PARP inhibitors are standard-of-care in prostate cancer harboring pathogenic alterations in HRR-related genes (74–76). Platinum chemotherapy has also been shown to be effective in this genomic subgroup (77). 15-20% of mCRPC harbor deleterious HRR-related gene mutations, most commonly in BRCA2, ATM and CDK12. Concordance between tissue and ctDNA for HRR gene mutations is excellent, exceeding 90% in most studies (23, 64, 71, 78, 79). However, an HRR-related gene mutation alone may be insufficient to confer therapeutic vulnerability. Biallelic inactivation is increasingly recognized as a critical prerequisite of functional HRR deficiency, and by virtue, promoting greatest susceptibility to PARP inhibitors (76, 80). The genomic insult that establishes biallelic inactivation is gene-dependent. For example, the mechanism of biallelic BRCA2 inactivation in prostate cancer is primarily loss-of-heterozygosity (i.e., copy deletion of the wildtype allele), but with homozygous deletion a clear secondary mechanism (81). Conversely, biallelic CDK12 inactivation manifests almost exclusively as multiple separate somatic mutational events (78). As sensitivity for all major genomic alteration types in ctDNA is strongly influenced by tumor purity, confidence in ctDNA test results reporting HRR gene alterations must account for ctDNA%. Many commercial and academic plasma cfDNA assays are capable of detecting somatic mutations below 1% ctDNA, but heterozygous and homozygous deletions typically require far greater ctDNA to be reliably detected (78, 82, 83). Therefore, low ctDNA% may be a limiting factor in detecting HRR alterations in cfDNA, especially large structural rearrangements and homozygous deletion events (23).
Separate from somatic alterations, up to half of HRR-related gene alterations are germline in origin (84). Concurrent matched white blood cell DNA sequencing at the time of cfDNA profiling can readily discriminate between patients with germline versus somatic HRR gene mutations, with implications not just for PARP inhibitor candidacy, but also hereditary cancer screening. Unfortunately, many commercial ctDNA tests do not routinely perform concurrent germline screening. This failure also means that some CHIP-related mutations falling in HRR genes are at risk of being attributed to cancer and could influence PARP inhibitor eligibility. In a recent study of 69 men with advanced prostate cancer undergoing paired plasma cfDNA whole-blood germline control sample analysis, 10% had CHIP variants in genes used for US FDA-approved PARP inhibitors, most frequently in ATM (53). Without concurrent germline screening, these patients could inappropriately receive therapies including olaparib, rucaparib and niraparib, but would otherwise not be expected to respond to treatment.
MMR genes (MSH2, MSH6, MLH1 and PMS2) are responsible for encoding proteins that reverse base substitution errors arising from DNA replication. MMR-deficient prostate tumors are associated with unstable genomes, manifesting as somatic hypermutation and microsatellite instability (85). This increase in somatic mutations generates tumor neoantigens, which may improve immune system recognition and provides rationale for use of immune checkpoint inhibitors. Evidence to this, durable responses to PD-1 inhibitors have been reported in MMR-deficient mCRPC (86).
From tissue studies, deleterious mutations and inactivating structural rearrangements are present in 3-5% of metastatic prostate cancer (12, 87). Importantly, structural rearrangements in MSH2 and MSH6 are often large and complex, with breakpoints frequently in intronic and gene flanking regions (88). These characteristics present challenges for detection in ctDNA and tumor tissue alike. Assays that prioritize sequencing coverage of non-coding regions surrounding MMR genes are therefore helpful to identify these clinically relevant alterations that may sensitize to immunotherapy (89). Somatic hypermutation in metastatic prostate cancer is often associated with presence of these MMR gene defects (87, 89). In a recent non-randomized study assessing tumor mutational burden (TMB) via tissue biopsy, patients with high TMB mCRPC (defined as the FDA-approved cutoff ≥10 mutations per Mb) were shown to benefit greater from treatment with immune checkpoint inhibitors versus taxane chemotherapy (90). While TMB can be estimated in plasma cfDNA specimens (91, 92), thresholds for high TMB established in tissue studies may not be relevant for predicting checkpoint inhibitor sensitivity. Care should also be taken when interpreting any individual potentially clinically actionable mutation in patients with somatic hypermutation. Many represent subclonal passenger alterations (including in HRR-related genes), which are unlikely to result in overall disease sensitivity to targeted therapies.
Most contemporary basket and umbrella trials screen primary or metastatic tissue, typically formalin-fixed paraffin embedded (FFPE) specimens. However, plasma ctDNA screening is increasingly used to complement or replace existing screening procedures. Recent PARP inhibitor studies in mCRPC have endorsed ctDNA analysis alongside tumor tissue testing to cross-validate presence of HRR alterations (20–23). PC-BETS (IND.234) and ProBio are two ongoing biomarker-directed prospective mCRPC clinical trials utilizing cfDNA testing for treatment allocation (25, 93). Moving forward, efforts to improve reliability of alterations detected in ctDNA and the clinical contexts where ctDNA testing is appropriate will be essential for testing a broader range of hypotheses. Future biomarker-driven trials should also be aware of the bias cfDNA screening may introduce to patient selection. Trial eligibility predicated on minimum ctDNA% thresholds, or presence/absence of ctDNA can alter the risk profile of the study cohort, thereby impacting interpretation of targeted therapy efficacy.
Most patients with metastatic castration-naive prostate cancer exhibit exquisite sensitivity to ADT, with marked reductions in PSA and palliation of cancer-related symptoms. Despite this, continuous ADT results in inevitable treatment resistance and progression to mCRPC. In 75-80% of patients, sustained AR signaling drives ongoing tumor growth in mCRPC. Greater understanding of the genomic and non-genomic mechanisms of this AR-dependent phenotype have led to the development and successful implementation of next-generation ARPIs (94). In the remaining patients, AR-independent resistance mechanisms predominate. This subtype is characterized by aggressive disease features, low/absent AR protein expression or indifference to AR signaling. Importantly, prostate cancer exists on a spectrum between AR-dependent and AR-independent disease, and both can simultaneously exist within the same patient (60).
Clinically, this heterogeneity in intrinsic disease subtypes is evident in serial on-treatment imaging studies. Rarely are patterns of radiographic response uniform at all disease sites. On a single scan, certain lesions may grow despite treatment, while others shrink. This variation in response is indicative of distinct tumor populations that have developed acquired resistance to treatment. The ability of ctDNA to sample multiple cancer populations from spatially distinct metastatic foci presents opportunities to detect evolving genomic and epigenomic determinants of acquired treatment resistance (19). Specifically, the possibility of differentiating between AR-dependent and AR-independent metastatic prostate cancer is likely to have profound implications on optimal patient management.
The AR gene is the most frequently altered locus in mCRPC (12–15). Genomic alterations include copy number amplification, ligand binding domain missense mutations and intronic structural rearrangements. These alterations are readily identifiable in ctDNA, and their presence is linked to clinical outcomes (95). AR gene body amplification in ctDNA is associated with shorter response to ARPI in patients with mCRPC, independent of prior treatment exposure (5, 41, 96–101). The magnitude of AR amplification also appears to be relevant, with high-level amplification associated with worse outcomes (5, 99). Notably, with repeated exposure to intensive AR-directed therapies, resistance mechanisms continue to converge upon the AR gene body and upstream enhancer, selecting for tumor populations with a greater number of AR copies (19, 102). Mutations in the AR are concentrated in hotspot regions within the ligand binding domain. The resultant conformational change induced by each specific mutation disrupts ligand specificity, resulting in inappropriate activation by older first-generation antiandrogens and endogenous steroids alike. including estrogen, progesterone, and glucocorticoids (103, 104). Data on whether these mutations confer resistance to newer ARPIs is conflicting (5, 37, 43, 96, 102). AR gene structural rearrangements can encode abnormal AR isoforms that lack the ligand binding domain, but still retain constitutive activity via intact N-terminal and DNA-binding domains. Patients exhibiting gene structural rearrangements predicted to truncate the ligand binding domain have been shown to portend shorter progression-free survival in mCRPC patients treated with ARPIs (5, 72, 102). Collectively, AR genomic alterations frequently co-occur, and cumulative burden in ctDNA is likely to have important clinical ramifications (43, 72). It is anticipated that with greater use of ARPI in the earlier mCSPC disease state, AR alterations will be more strongly selected for at mCRPC development. Convenient detection of these alterations in later disease states will be critical to interpreting the efficacy of emerging therapies targeting the AR-signaling axis, including AR degraders, N-terminal/DNA-binding domain inhibitors and spliceosome-targeted therapeutic agents (105).
AR-independent prostate cancer is characterized by highly aggressive clinical features, including liver metastases, bulky lymphadenopathy, and lytic bone metastases. In most instances, it arises after long-standing intensive androgen suppression via ADT and/or ARPIs. Typically, these tumors begin as AR-dependent adenocarcinoma, before acquiring molecular events that promote tissue de-differentiation into small cell/neuroendocrine histologic features (e.g., chromogranin and/or synaptophysin expression). The process of transitioning from one developmental pathway to another is known as lineage plasticity. While the circumstances that drive lineage plasticity in prostate cancer are incompletely understood, the central facet involves loss of tumor suppressors RB1 and TP53, with accompanying transcriptional and epigenetic reprogramming (60).
Diagnosis of small cell/neuroendocrine prostate cancer (NEPC) usually requires tissue confirmation. More recently however, the role of ctDNA in detecting the emergence of this aggressive phenotype is being realized. Since biallelic loss of tumor suppressor genes in ctDNA is not specific to AR-independent prostate cancer (106), recent efforts have focused on using epigenomic techniques to improve NEPC detection. Adopting whole-genome bisulfite sequencing analysis from time-matched plasma cfDNA and metastatic tumor biopsies in a cohort of prostate adenocarcinoma and NEPC, Beltran et al. showed high concordance of ctDNA and tissue methylation patterns in patients with NEPC (107). The process of whole genome bisulfite sequencing is limited however by the requirement for high DNA input amounts. Newer approaches such as methylated DNA immunoprecipitation followed by high-throughput sequencing represent a highly sensitive method of obtaining genome-wide cfDNA methylation profiles, and have been successfully applied to identify NEPC with high specificity (108). Finally, cfDNA fragmentation patterns offer yet another alternate mechanism for NEPC diagnosis. In this method, nucleosome density at specific chromatin sites (e.g., transcriptional start sites, transcription factor binding sites) can be inferred by analyzing the genomic location of cfDNA fragment end points. How consistently nucleosomes are positioned in these regulatory sites appears to be a surrogate measure of relative RNA abundance. This approach has been applied to quantify relative RNA abundance in key NEPC regulatory genes (109) and AR binding sites to determine AR activity in prostate cancer cells (19). Overall, these results hold great prospects for the use of cfDNA epigenomic patterns to track emergence of distinct prostate cancer subtypes associated with poor outcomes, permitting earlier implementation of proven platinum-based chemotherapy treatments and novel agents currently in clinical trials.
Plasma ctDNA analysis has emerged as a key application of liquid biopsy for metastatic prostate cancer, with a wide range of potential and proven clinical utility to inform on optimal patient management (Figure 2). ctDNA% is highly prognostic and outperforms standard clinical prognostic factors. Furthermore, the ability to track changes in ctDNA% over time may represent a reliable early readout of treatment efficacy. With commercial assays increasingly reporting ctDNA%, understanding the implications of both static and dynamic readouts of ctDNA% will enable future production of point-of-care assessment tools in the clinic. More broadly, ctDNA genotyping to guide use of targeted precision oncology therapies is now a reality. As the cost of sequencing depreciates and the overhead of informatic analysis improves, it will become increasingly important to use serial sampling methods to understand evolving mechanisms of acquired treatment resistance.
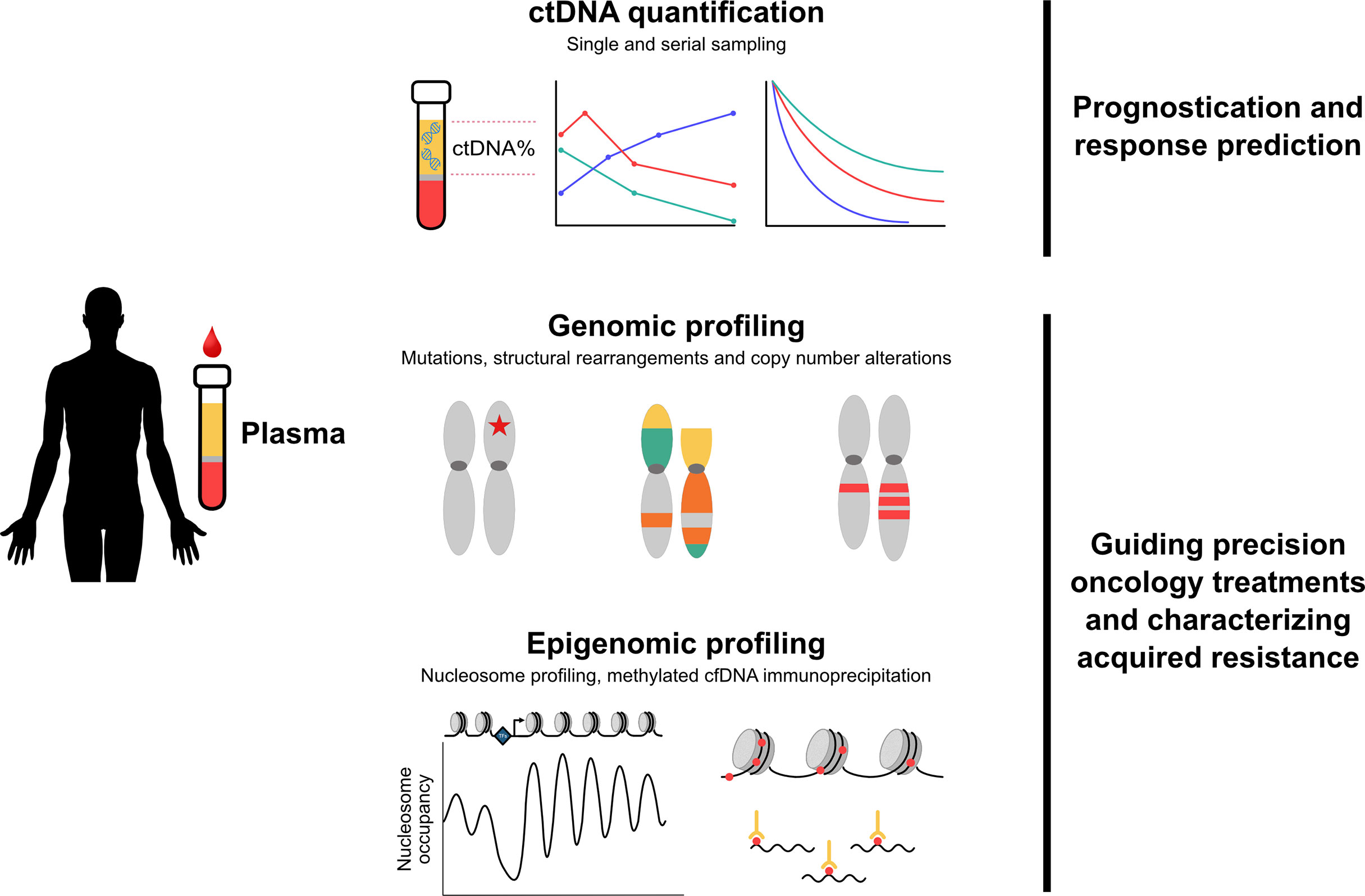
Figure 2 Overview of clinical applications of plasma ctDNA in metastatic prostate cancer. Quantification and genomic/epigenomic profiling of plasma ctDNA has identified multiple clinical applications relevant to metastatic prostate cancer. Top - ctDNA% is highly prognostic and outperforms standard clinical prognostic factors. Tracking changes in ctDNA% over time may represent a reliable early readout of treatment efficacy. Middle - characterization of mutations, structural rearrangements and copy number alterations guides use of targeted therapies and enables detection of evolving treatment resistance. Bottom - novel epigenomics approaches are capable of identifying aggressive subtypes of prostate cancer, including emergence of small cell/neuroendocrine disease. Abbreviations: cfDNA, cell-free DNA; ctDNA, circulating tumor DNA.
This manuscript was drafted and critically reviewed by EMK, AWW, and KNC. All authors contributed to the article and approved the submitted version.
This work was supported by a Prostate Cancer Foundation (PCF) Young Investigator Award and Killam Postdoctoral Fellowship to EMK.
EMK reports consulting roles with Astellas Pharma, Janssen and Ipsen; honoraria from Astellas Pharma, Janssen, Ipsen and Research Review, and research funding from AstraZeneca (institutional), Merck (institutional) and Astellas Pharma (institutional). AWW reports commercial research grants from Janssen and ESSA Pharma, and honoraria from AstraZeneca, Astellas, Janssen, and Merck. KNC reports grants and personal fees from Astellas, AstraZeneca, Janssen, Merck, Novartis, Pfizer, Point Biopharma, Roche, and Sanofi.
The handling editor AA declared a past co-authorship with the author KNC.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Casanova-Salas I, Athie A, Boutros PC, Del Re M, Miyamoto DT, Pienta KJ, et al. Quantitative and qualitative analysis of blood-based liquid biopsies to inform clinical decision-making in prostate cancer. Eur Urol (2021) 79:762–71. doi: 10.1016/j.eururo.2020.12.037
2. Vandekerkhove G, Wyatt A. Circulating tumour DNA as a biomarker source in metastatic prostate cancer. Société Internationale d’Urologie J (2020) 1:39–48. doi: 10.48083/VSOO5322
3. Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci USA (2016) 113:E1826–34. doi: 10.1073/pnas.1519286113
4. Breitbach S, Tug S, Helmig S, Zahn D, Kubiak T, Michal M, et al. Direct quantification of cell-free, circulating DNA from unpurified plasma. PloS One (2014) 9:e87838. doi: 10.1371/journal.pone.0087838
5. Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discovery (2018) 8:444–57. doi: 10.1158/2159-8290.CD-17-0937
6. Friedlander TW, Pritchard CC, Beltran H. Personalizing therapy for metastatic prostate cancer: The role of solid and liquid tumor biopsies. Am Soc Clin Oncol Educ Book (2017) 37:358–69. doi: 10.1200/EDBK_175510
7. Hussain M, Corcoran C, Sibilla C, Fizazi K, Saad F, Shore N, et al. Tumor genomic testing for >4000 men with metastatic castration-resistant prostate cancer in the phase III trial PROfound (Olaparib). Clin Cancer Res (2022) 28:1518–30. doi: 10.1158/1078-0432.CCR-21-3940
8. Cyrta J, Prandi D, Arora A, Hovelson DH, Sboner A, Rodriguez A, et al. Comparative genomics of primary prostate cancer and paired metastases: insights from 12 molecular case studies. J Pathol (2022) 257:274–84. doi: 10.1002/path.5887
9. Warner E, van der Eecken K, Murtha AJ, Kwan EM, Ng SW, Chen XE, et al. Abstract 41: Multi-focal genomic dissection of synchronous primary and metastatic tissue from de novo metastatic prostate cancer. Cancer Res (2022) 82:41–1. doi: 10.1158/1538-7445.AM2022-41
10. Johansson JE, Andren O, Andersson SO, Dickman PW, Holmberg L, Magnuson A, et al. Natural history of early, localized prostate cancer. JAMA (2004) 291:2713–9. doi: 10.1001/jama.291.22.2713
11. Kwan EM, Thangasamy IA, Teh J, Alghazo O, Sathianathen NJ, Lawrentschuk N, et al. Navigating systemic therapy for metastatic castration-naive prostate cancer. World J Urol (2021) 39:339–48. doi: 10.1007/s00345-019-03060-7
12. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
13. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol (2017) 2017:1–3. doi: 10.1200/PO.17.00029
14. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell (2018) 174:758–769.e9. doi: 10.1016/j.cell.2018.06.039
15. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
16. Annala M, Fu S, Bacon JVW, Sipola J, Iqbal N, Ferrario C, et al. Cabazitaxel versus abiraterone or enzalutamide in poor prognosis metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2 trial. Ann Oncol (2021) 32:896–905. doi: 10.1016/j.annonc.2021.03.205
17. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature (2015) 520:353–7. doi: 10.1038/nature14347
18. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med (2016) 22:369–78. doi: 10.1038/nm.4053
19. Herberts C, Annala M, Sipola J, Ng SWS, Chen XE, Nurminen A, et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature (2022) 608:199–208. doi: 10.1038/s41586-022-04975-9
20. Loehr A, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Response to rucaparib in BRCA-mutant metastatic castration-resistant prostate cancer identified by genomic testing in the TRITON2 study. Clin Cancer Res (2021) 27:6677–86. doi: 10.1158/1078-0432.CCR-21-2199
21. Clarke NW, Armstrong AJ, Thiery-Vuillemin A, Oya M, Shore N, Loredo E, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evidence (2022) 1. doi: 10.1056/evidoa2200043
22. Chi KN, Rathkopf DE, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol (2022) 40:12–2. doi: 10.1200/JCO.2022.40.6_suppl.012
23. Chi KN, Barnicle A, Sibilla C, Lai Z, Corcoran C, Barrett JC, et al. Detection of BRCA1, BRCA2, and ATM alterations in matched tumor tissue and circulating tumor DNA in patients with prostate cancer screened in PROfound. Clin Cancer Res (2022), OF1–OF11. doi: 10.1158/1078-0432.CCR-22-0931
24. Sumiyoshi T, Chi KN, Wyatt AW. Clinical implications of genomic alterations in metastatic prostate cancer. Prostate Cancer Prostatic Dis (2021) 24:310–22. doi: 10.1038/s41391-020-00308-x
25. De Laere B, Crippa A, Discacciati A, Larsson B, Oldenburg J, Mortezavi A, et al. Clinical trial protocol for ProBio: An outcome-adaptive and randomised multiarm biomarker-driven study in patients with metastatic prostate cancer. Eur Urol Focus (2022). doi: 10.1016/j.euf.2022.03.005
26. U.S. Food and Drug Administration. FoundationOne® liquid CDx 2020 . Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010S001C.pdfhttps://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032C.pdf.
27. U.S. Food and Drug Administration. Guardant360 CDx technical information . Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010S001C.pdf.
28. Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun (2021) 12:3770. doi: 10.1038/s41467-021-24109-5
29. Greytak SR, Engel KB, Parpart-Li S, Murtaza M, Bronkhorst AJ, Pertile MD, et al. Harmonizing cell-free DNA collection and processing practices through evidence-based guidance. Clin Cancer Res (2020) 26:3104–9. doi: 10.1158/1078-0432.CCR-19-3015
30. Sumanasuriya S, Omlin A, Armstrong A, Attard G, Chi KN, Bevan CL, et al. Consensus statement on circulating biomarkers for advanced prostate cancer. Eur Urol Oncol (2018) 1:151–9. doi: 10.1016/j.euo.2018.02.009
31. Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial design and objectives for castration-resistant prostate cancer: Updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol (2016) 34:1402–18. doi: 10.1200/JCO.2015.64.2702
32. Nuhn P, De Bono JS, Fizazi K, Freedland SJ, Grilli M, Kantoff PW, et al. Update on systemic prostate cancer therapies: Management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur Urol (2019) 75:88–99. doi: 10.1016/j.eururo.2018.03.028
33. Armstrong AJ, Garrett-Mayer E, de Wit R, Tannock I, Eisenberger M. Prediction of survival following first-line chemotherapy in men with castration-resistant metastatic prostate cancer. Clin Cancer Res (2010) 16:203–11. doi: 10.1158/1078-0432.CCR-09-2514
34. Halabi S, Lin C-Y, Small EJ, Armstrong AJ, Kaplan EB, Petrylak D, et al. Prognostic model predicting metastatic castration-resistant prostate cancer survival in men treated with second-line chemotherapy. J Natl Cancer Inst (2013) 105:1729–37. doi: 10.1093/jnci/djt280
35. Chi KN, Kheoh T, Ryan CJ, Molina A, Bellmunt J, Vogelzang NJ, et al. A prognostic index model for predicting overall survival in patients with metastatic castration-resistant prostate cancer treated with abiraterone acetate after docetaxel. Ann Oncol (2016) 27:454–60. doi: 10.1093/annonc/mdv594
36. Armstrong AJ, Lin P, Higano CS, Sternberg CN, Sonpavde G, Tombal B, et al. Development and validation of a prognostic model for overall survival in chemotherapy-naïve men with metastatic castration-resistant prostate cancer. Ann Oncol (2018) 29:2200–7. doi: 10.1093/annonc/mdy406
37. Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med (2015) 7:312re10. doi: 10.1126/scitranslmed.aac9511
38. Choudhury AD, Werner L, Francini E, Wei XX, Ha G, Freeman SS, et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI Insight (2018) 3:e122109. doi: 10.1172/jci.insight.122109
39. Mehra N, Dolling D, Sumanasuriya S, Christova R, Pope L, Carreira S, et al. Plasma cell-free DNA concentration and outcomes from taxane therapy in metastatic castration-resistant prostate cancer from two phase III trials (FIRSTANA and PROSELICA). Eur Urol (2018) 74:283–91. doi: 10.1016/j.eururo.2018.02.013
40. Torquato S, Pallavajjala A, Goldstein A, Valda Toro P, Silberstein JL, Lee J, et al. Genetic alterations detected in cell-free DNA are associated with enzalutamide and abiraterone resistance in castration-resistant prostate cancer. JCO Precis Oncol (2019) 3:1–14. doi: 10.1200/PO.18.00227
41. De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam PJ, Van Oyen P, et al. TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration-resistant prostate cancer. Clin Cancer Res (2019) 25:1766–73. doi: 10.1158/1078-0432.CCR-18-1943
42. Kohli M, Tan W, Zheng T, Wang A, Montesinos C, Wong C, et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine (2020) 54:102728. doi: 10.1016/j.ebiom.2020.102728
43. Fettke H, Kwan EM, Docanto MM, Bukczynska P, Ng N, Graham LK, et al. Combined cell-free DNA and RNA profiling of the androgen receptor: Clinical utility of a novel multianalyte liquid biopsy assay for metastatic prostate cancer. Eur Urol (2020) 78:173–80. doi: 10.1016/j.eururo.2020.03.044
44. Liu H, Gao Y, Vafaei S, Gu X, Zhong X. The prognostic value of plasma cell-free DNA concentration in the prostate cancer: A systematic review and meta-analysis. Front Oncol (2021) 11:599602. doi: 10.3389/fonc.2021.599602
45. Conteduca V, Casadei C, Scarpi E, Brighi N, Schepisi G, Lolli C, et al. Baseline plasma tumor DNA (ctDNA) correlates with PSA kinetics in metastatic castration-resistant prostate cancer (mCRPC) treated with abiraterone or enzalutamide. Cancers (2022) 14:2219. doi: 10.3390/cancers14092219
46. Kienel A, Porres D, Heidenreich A, Pfister D. cfDNA as a prognostic marker of response to taxane based chemotherapy in patients with prostate cancer. J Urol (2015) 194:966–71. doi: 10.1016/j.juro.2015.04.055
47. Sumanasuriya S, Seed G, Parr H, Christova R, Pope L, Bertan C, et al. Elucidating prostate cancer behaviour during treatment via low-pass whole-genome sequencing of circulating tumour DNA. Eur Urol (2021) 80:243–53. doi: 10.1016/j.eururo.2021.05.030
48. Vandekerkhove G, Struss WJ, Annala M, Kallio HML, Khalaf D, Warner EW, et al. Circulating tumor DNA abundance and potential utility in De novo metastatic prostate cancer. Eur Urol (2019) 75:667–75. doi: 10.1016/j.eururo.2018.12.042
49. Pantel K, Alix-Panabieres C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol (2019) 16:409–24. doi: 10.1038/s41571-019-0187-3
50. Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med (2013) 5:30. doi: 10.1186/gm434
51. Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun (2017) 8:1324. doi: 10.1038/s41467-017-00965-y
52. Loh JW, Guccione C, Di Clemente F, Riedlinger G, Ganesan S, Khiabanian H. All-FIT: allele-frequency-based imputation of tumor purity from high-depth sequencing data. Bioinformatics (2020) 36:2173–80. doi: 10.1093/bioinformatics/btz865
53. Jensen K, Konnick EQ, Schweizer MT, Sokolova AO, Grivas P, Cheng HH, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol (2021) 7:107–10. doi: 10.1001/jamaoncol.2020.5161
54. Weymann D, Laskin J, Roscoe R, Schrader KA, Chia S, Yip S, et al. The cost and cost trajectory of whole-genome analysis guiding treatment of patients with advanced cancers. Mol Genet Genomic Med (2017) 5:251–60. doi: 10.1002/mgg3.281
55. Pleasance E, Bohm A, Williamson LM, Nelson JMT, Shen Y, Bonakdar M, et al. Whole-genome and transcriptome analysis enhances precision cancer treatment options. Ann Oncol (2022) 33:939–49. doi: 10.1016/j.annonc.2022.05.522
56. Harshman LC, Chen YH, Liu G, Carducci MA, Jarrard D, Dreicer R, et al. Seven-month prostate-specific antigen is prognostic in metastatic hormone-sensitive prostate cancer treated with androgen deprivation with or without docetaxel. J Clin Oncol (2018) 36:376–82. doi: 10.1200/JCO.2017.75.3921
57. Rescigno P, Dolling D, Conteduca V, Rediti M, Bianchini D, Lolli C, et al. Early post-treatment prostate-specific antigen at 4 weeks and abiraterone and enzalutamide treatment for advanced prostate cancer: An international collaborative analysis. Eur Urol Oncol (2019) 3:176–82. doi: 10.1016/j.euo.2019.06.008
58. Bryce AH, Chen YH, Liu G, Carducci MA, Jarrard DM, Garcia JA, et al. Patterns of cancer progression of metastatic hormone-sensitive prostate cancer in the ECOG3805 CHAARTED trial. Eur Urol Oncol (2020) 3:717–24. doi: 10.1016/j.euo.2020.07.001
59. Bryce AH, Alumkal JJ, Armstrong A, Higano CS, Iversen P, Sternberg CN, et al. Radiographic progression with nonrising PSA in metastatic castration-resistant prostate cancer: post hoc analysis of PREVAIL. Prostate Cancer Prostatic Dis (2017) 20:221–7. doi: 10.1038/pcan.2016.71
60. Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res (2019) 25:6916–24. doi: 10.1158/1078-0432.CCR-19-1423
61. Trabulsi EJ, Bryan Rumble R, Alberto Vargas H. Optimum imaging strategies for advanced prostate cancer: ASCO guideline summary. JCO Oncol Pract (2020) 16:170–6. doi: 10.1200/jop.19.00699
62. Jadvar H, Calais J, Fanti S, Feng F, Greene KL, Gulley JL, et al. Appropriate use criteria for prostate-specific membrane antigen PET imaging. J Nucl Med (2022) 63:59–68. doi: 10.2967/jnumed.121.263262
63. Goodall J, Assaf ZJ, Shi Z, Seed G, Zhang L, Lauffer B, et al. Circulating tumor DNA (ctDNA) dynamics associate with treatment response and radiological progression-free survival (rPFS): Analyses from a randomized phase II trial in metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol (2020) 38:5508–8. doi: 10.1200/JCO.2020.38.15_suppl.5508
64. Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discovery (2017) 7:1006–17. doi: 10.1158/2159-8290.CD-17-0261
65. Tolmeijer SH, Boerrigter E, Sumiyoshi T, Ng S, Kwan EM, Annala M, et al. On-treatment plasma ctDNA fraction and treatment outcomes in metastatic castration-resistant prostate cancer. J Clin Oncol (2022) 40:5051–1. doi: 10.1200/JCO.2022.40.16_suppl.5051
66. Conteduca V, Wetterskog D, Scarpi E, Romanel A, Gurioli G, Jayaram A, et al. Plasma tumour DNA as an early indicator of treatment response in metastatic castration-resistant prostate cancer. Br J Cancer (2020) 123:982–7. doi: 10.1038/s41416-020-0969-5
67. Kwan EM, Spain L, Anton A, Gan CL, Garrett L, Chang D, et al. Avelumab combined with stereotactic ablative body radiotherapy in metastatic castration-resistant prostate cancer: The phase 2 ICE-PAC clinical trial. Eur Urol (2021) 81:253–62. doi: 10.1016/j.eururo.2021.08.011
68. Jayaram A, Wingate A, Wetterskog D, Wheeler G, Sternberg CN, Jones R, et al. Plasma tumor gene conversions after one cycle abiraterone acetate for metastatic castration-resistant prostate cancer: a biomarker analysis of a multicenter international trial. Ann Oncol (2021) 32:726–35. doi: 10.1016/j.annonc.2021.03.196
69. Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, et al. Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin Cancer Res (2020) 26:3230–8. doi: 10.1158/1078-0432.CCR-20-0168
70. Deek MP, van der Eecken K, Phillips R, Parikh NR, Isaacsson Velho P, Lotan TL, et al. The mutational landscape of metastatic castration-sensitive prostate cancer: The spectrum theory revisited. Eur Urol (2021) 80:632–40. doi: 10.1016/j.eururo.2020.12.040
71. Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst (2017) 109:78–86. doi: 10.1093/jnci/djx118
72. De Laere B, van Dam PJ, Whitington T, Mayrhofer M, Diaz EH, Van den Eynden G, et al. Comprehensive profiling of the androgen receptor in liquid biopsies from castration-resistant prostate cancer reveals novel intra-AR structural variation and splice variant expression patterns. Eur Urol (2017) 72:192–200. doi: 10.1016/j.eururo.2017.01.011
73. Mayrhofer M, De Laere B, Whitington T, Van Oyen P, Ghysel C, Ampe J, et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med (2018) 10:85. doi: 10.1186/s13073-018-0595-5
74. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med (2020) 382:2091–102. doi: 10.1056/NEJMoa1911440
75. Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol (2020) 38:3763–72. doi: 10.1200/JCO.20.01035
76. Smith MR, Scher HI, Sandhu S, Efstathiou E, Lara PN Jr, Yu EY, et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol (2022) 23:362–73. doi: 10.1016/S1470-2045(21)00757-9
77. Schmid S, Omlin A, Higano C, Sweeney C, Martinez Chanza N, Mehra N, et al. Activity of platinum-based chemotherapy in patients with advanced prostate cancer with and without DNA repair gene aberrations. JAMA Netw Open (2020) 3:e2021692. doi: 10.1001/jamanetworkopen.2020.21692
78. Warner E, Herberts C, Fu S, Yip S, Wong A, Wang G, et al. BRCA2, ATM, and CDK12 defects differentially shape prostate tumor driver genomics and clinical aggression. Clin Cancer Res (2021) 27:1650–62. doi: 10.1158/1078-0432.CCR-20-3708
79. Schweizer MT, Sivakumar S, Tukachinsky H, Coleman I, De Sarkar N, Yu EY, et al. Concordance of DNA repair gene mutations in paired primary prostate cancer samples and metastatic tissue or cell-free DNA. JAMA Oncol (2021) 7:1–5. doi: 10.1001/jamaoncol.2021.2350
80. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
81. Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature (2019) 571:576–9. doi: 10.1038/s41586-019-1382-1
82. Taavitsainen S, Annala M, Ledet E, Beja K, Miller PJ, Moses M, et al. Evaluation of commercial circulating tumor DNA test in metastatic prostate cancer. JCO Precis Oncol (2019) 12:1–9. doi: 10.1200/PO.19.00014
83. Herberts C, Wyatt AW. Technical and biological constraints on ctDNA-based genotyping. Trends Cancer Res (2021) 7:995–1009. doi: 10.1016/j.trecan.2021.06.001
84. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med (2016) 375:443–53. doi: 10.1056/NEJMoa1603144
85. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
86. Antonarakis ES, Shaukat F, Isaacsson Velho P, Kaur H, Shenderov E, Pardoll DM, et al. Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur Urol (2019) 75:378–82. doi: 10.1016/j.eururo.2018.10.009
87. Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol (2019) 5:471–8. doi: 10.1001/jamaoncol.2018.5801
88. Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun (2014) 5:4988. doi: 10.1038/ncomms5988
89. Ritch E, Fu SYF, Herberts C, Wang G, Warner EW, Schonlau E, et al. Identification of hypermutation and defective mismatch repair in ctDNA from metastatic prostate cancer. Clin Cancer Res (2020) 26:1114–25. doi: 10.1158/1078-0432.CCR-19-1623
90. Graf RP, Fisher V, Weberpals J, Gjoerup O, Tierno MB, Huang RSP, et al. Comparative effectiveness of immune checkpoint inhibitors vs chemotherapy by tumor mutational burden in metastatic castration-resistant prostate cancer. JAMA Netw Open (2022) 5:e225394. doi: 10.1001/jamanetworkopen.2022.5394
91. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med (2018) 24:1441–8. doi: 10.1038/s41591-018-0134-3
92. Peters S, Dziadziuszko R, Morabito A, Felip E, Gadgeel SM, Cheema P, et al. Atezolizumab versus chemotherapy in advanced or metastatic NSCLC with high blood-based tumor mutational burden: primary analysis of BFAST cohort c randomized phase 3 trial. Nat Med (2022) 28:1831–9. doi: 10.1038/s41591-022-01933-w
93. Chi KN, Mukherjee S, Saad F, Winquist E, Ong M, Kolinsky MP, et al. Prostate cancer biomarker enrichment and treatment selection (PC-BETS) study: A Canadian cancer trials group phase II umbrella trial for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol (2020) 38:5551–1. doi: 10.1200/JCO.2020.38.15_suppl.5551
94. Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol (2014) 11:365–76. doi: 10.1038/nrclinonc.2014.72
95. Kwan EM, Wyatt AW. Androgen receptor genomic alterations and treatment resistance in metastatic prostate cancer. Prostate (2022) 82(Suppl 1):S25–36. doi: 10.1002/pros.24356
96. Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res (2015) 21:2315–24. doi: 10.1158/1078-0432.CCR-14-2666
97. Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol (2016) 2:1598–606. doi: 10.1001/jamaoncol.2016.0494
98. Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol (2017) 28:1508–16. doi: 10.1093/annonc/mdx155
99. Jayaram A, Wingate A, Wetterskog D, Conteduca V, Khalaf D, Sharabiani MTA, et al. Plasma androgen receptor copy number status at emergence of metastatic castration-resistant prostate cancer: A pooled multicohort analysis. JCO Precis Oncol (2019) 3:1–13. doi: 10.1200/PO.19.00123
100. Tolmeijer SH, Boerrigter E, Schalken JA, Geerlings MJ, van Oort IM, van Erp NP, et al. A systematic review and meta-analysis on the predictive value of cell-free DNA–based androgen receptor copy number gain in patients with castration-resistant prostate cancer. JCO Precis Oncol (2020) 4:714–29. doi: 10.1200/PO.20.00084
101. Kwan EM, Dai C, Fettke H, Hauser C, Docanto MM, Bukczynska P, et al. Plasma cell–free DNA profiling of PTEN-PI3K-AKT pathway aberrations in metastatic castration-resistant prostate cancer. JCO Precis Oncol (2021) 5:622–37. doi: 10.1200/PO.20.00424
102. Annala M, Taavitsainen S, Khalaf DJ, Vandekerkhove G, Beja K, Sipola J, et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin Cancer Res (2021) 27:4610–23. doi: 10.1158/1078-0432.CCR-21-1625
103. Veldscholte J, Ris-Stalpers C, Kuiper GGJM, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun (1990) 173:534–40. doi: 10.1016/S0006-291X(05)80067-1
104. Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res (1997) 3:1383–8.
105. Westaby D, Maza M de LDF de L, Paschalis A, Jimenez-Vacas JM, Welti J, de Bono J, et al. A new old target: Androgen receptor signaling and advanced prostate cancer. Annu Rev Pharmacol Toxicol (2021) 62:131–53. doi: 10.1146/annurev-pharmtox-052220-015912
106. Maurice-Dror C, Fonseca N, Herberts C, Kwan EM, Kollmannsberger C, Tu W, et al. Biallelic loss of TP53, PTEN, and RB1 in association to aggressive clinical features and poor outcomes in metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol (2022) 40:5055–5. doi: 10.1200/JCO.2022.40.16_suppl.5055
107. Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest (2020) 130:1653–68. doi: 10.1172/JCI131041
108. Berchuck JE, Baca SC, McClure HM, Korthauer K, Tsai HK, Nuzzo PV, et al. Detecting neuroendocrine prostate cancer through tissue-informed cell-free DNA methylation analysis. Clin Cancer Res (2021) 28:928–38. doi: 10.1158/1078-0432.CCR-21-3762
Keywords: cell-free DNA, CtDNA, biomarker, resistance, targeted therapy
Citation: Kwan EM, Wyatt AW and Chi KN (2022) Towards clinical implementation of circulating tumor DNA in metastatic prostate cancer: Opportunities for integration and pitfalls to interpretation. Front. Oncol. 12:1054497. doi: 10.3389/fonc.2022.1054497
Received: 26 September 2022; Accepted: 25 October 2022;
Published: 10 November 2022.
Edited by:
Arun Azad, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Umang Swami, The University of Utah, United StatesCopyright © 2022 Kwan, Wyatt and Chi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim N. Chi, a2NoaUBiY2NhbmNlci5iYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.