 Guanghua Liu
Guanghua Liu Jiangwen Long1,2
Jiangwen Long1,2 Yuyu Chen
Yuyu Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 26 January 2023
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1049473
This article is part of the Research Topic Acute Promyelocytic Leukemia - Towards a Chemotherapy-Free Approach to Cure in All Patients, Volume II View all 9 articles
Background: Acute promyelocytic leukemia (APL) is typically characterized by the presence of coagulopathy and the PML::RARA fusion gene. The FIP1L1::RARA has been reported as a novel fusion gene, but studies on its pathogenesis are limited.
Objectives: A FIP1L1::RARA fusion in a child finally diagnosed as APL was reported. RNA sequencing (RNA-seq) of six patients (three cases of acute lymphoblastic leukemia (ALL), one case of myelodysplastic syndrome (MDS), one case of acute megakaryoblastic leukemia (M7), and one case of APL with FIP1L1::RARA) were performed.
Methods: Transcriptome analysis of six patients was performed by RNA-seq. The heat map was used for showing the RNA expression profile, the volcano plot for identifying differential expression genes (DEGs), and the KEGG Orthology-Based Annotation System (KOBAS) online biological information database for KEGG pathway enrichment analysis.
Results: Obvious differences between APL with FIP1L1::RARA and hematologic malignancies were identified. 1060 common differentially expressed genes (co-DEGs) were detected between APL with FIP1L1::RARA vs ALL and APL with FIP1L1::RARA vs myeloid neoplasms (MDS, M7), the up-regulated genes were mainly mapped into platelet activation, cancer, AMPK signaling pathway, PI3K-Akt signaling pathway, and MAPK signaling pathway. The down-regulated genes were significantly associated with TNF signaling pathway, Rap1 signaling pathway, Age-RAGE signaling pathway, and apoptosis.
Conclusion: A FIP1L1::RARA fusion in a child finally diagnosed as APL was reported. RNA-seq may provide a new diagnostic method when RARA rearrangements fail to be identified by conventional methods. In the analysis of co-DEGs between case vs ALL and case vs myeloid neoplasms, the up-regulated and down-regulated genes were enriched in different signaling pathways. Further experimental studies are needed to identify pathogenesis and treatment for APL with FIP1L1::RARA.
Acute promyelocytic leukemia (APL) is characterized by abnormal promyelocytic proliferation and the presence of coagulopathy (1, 2). About 95% of APL is typically characterized by the fusion between promyelocytic leukemia (PML) and retinoic acid receptor alpha (RARA) and sensitive to differentiation induction therapy containing all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) with a good clinical outcome. Rarely, about 2% of APL is characterized by atypical rearrangements with PLZF, NPM1, NUMA, STAT5B, FIP1L1, PRKAR1A, BCOR, OBFC2A, TBLR1, GTF2I, IRF2BP2, FNDC3B, TFGA, NUP98, TNRC18, STAT3 and TFG (3, 4). The RARA fusion partner plays an important role in the morphology and clinical features (5), alternative fusion types demonstrate varying sensitivity to ATRA (6).
FIP1L1::RARA is a rare alternative fusion type associated with myeloid neoplasm composed of 426 amino acids (AA) from the N terminus of FIP1L1 and 403 AA from the C terminus of RARA (7). 5 cases with FIP1L1::RARA have been reported in the literature at present. The FIP1L1::RARA fusion gene was first reported as a novel RARA-associated fusion gene in a 20-month-old child with juvenile myelomonocytic leukemia (JMML), no other mutations in N-Ras, K-Ras, or PTPN11 were detected (8). In addition, a FIP1L1::RARA fusion gene was detected in a 90-year-old woman, who was finally diagnosed with APL and achieved complete remission after ATRA therapy alone (9). Another 77-year-old woman was also reported as APL with FIP1L1::RARA and the FLT3-ITD, FLT3-D835Y, CEBPA and NPM1 mutations were not detected, PML::RARA, AML1::ETO, and CBFβ::MYH11 fusion genes were negative. Unfortunately, she died after ten days of treatment according to Programa para el Tratamiento de Hemopatias Malignas (PETHEMA) APL 2005 protocol (10). Besides, a FIP1L1::RARA fusion was reported in a 28-month-old girl with APL who presented with an extramedullary tumor in the skull without the classic karyotype. An initial complete remission was achieved by a treatment combining ATRA with DA regimen (daunorubicin and cytarabine) and kept leukemia-free after a 5-month follow-up by continual therapy (11).
Despite reports of five patients with the FIP1L1::RARA fusion gene, among which three were diagnosed as APL, studies on its pathogenesis and treatment are limited. Table 1 provides patient characteristics of all reported cases, including the current case. In this study, three cases of ALL and two cases of myeloid neoplasm were collected to explore the differences between APL with FIP1L1::RARA and other hematologic malignancies at the level of transcriptome and lay a certain molecular basis for further research on the treatment and prognosis.
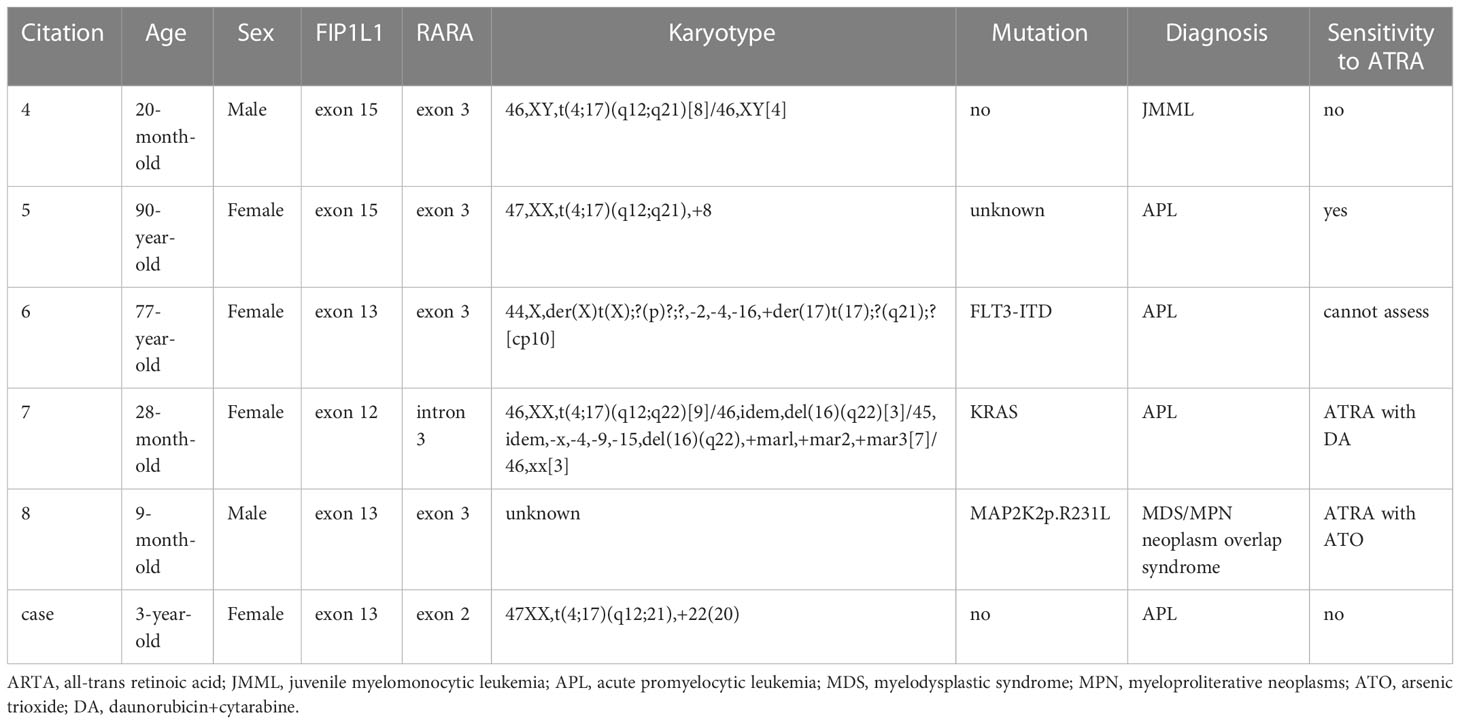
Table 1 Characteristics of reported myeloid neoplasms with FIP1L1::RARA.
In total, six patients admitted to the pediatric department of Hunan Provincial People’s Hospital were included in this study, of which three were B-cell acute lymphoblastic leukemia (B-ALL, female, 4 years old).T he other three were myelodysplastic syndrome (MDS, male, 17 years old), acute megakaryoblastic leukemia (M7, female, 11 months old), and APL with FIP1L1::RARA (female, 4 years old). None of the six patients received neoadjuvant chemotherapy before transcriptome sequencing (RNA-Seq). This study was approved by Hunan Provincial People’s Hospital Medical Ethics Committee.
PAXgene Blood RNA Tube (BD, cat.762165) was used for bone marrow sample collection. RNA was isolated by MagMAX for Stabilized Blood Tubes RNA Isolation Kit, compatible with PAXgene Blood RNA Tubes (Invitrogen, cat.4451894). NanodropOne (Thermofisher) was used for RNA concentration and purity. For RNA quality control, Qseq400 (BIOPTIC) with R1 cartridge (BIOPTIC), RQN≥5 is acceptable for following library preparation. Libraries were prepared with the KAPA RNA HyperPrep Kit (Kapabiosystems, cat. KK8540) after rRNA depletion with the KAPA RiboErase (HMR) Kit(Kapabiosystems, cat. KK8482), library preparation was started with 1ug total RNA per sample as described in kit handbook manually.
For Library quality control, Qubit 3.0 with qubit dsDNA HS Assay Kit (Thermofisher) was used for concentration, Qseq400 (BIOPTIC) with S2 cartridge (BIOPTIC) for library size, KAPA Library Quantification Kit (KAPA biosystems) for library quantification and novaseq6000, PE150, 15G raw data for sequencing.
Cytogenetic analysis of BM samples after short-term culture (24 h) was carried out according to a standard procedure. G-banded chromosome was determined according to International System for Human Cytogenetic Nomenclature. Commercially available PML::RARa dual color dual fusion DNA probes were used for FISH analysis.
The primer sequences of the FIP1L1::RARA fusion gene are GTCCTTTCTGAAAGATCTGCTACTGAAGTAGACAACAATTTTAGCAAACCACCTCCGTTTTTCCCTCCAGGAGCTCCTCCCACTCACCTTCCACCTCCTCCATTTCTTCCACCTCCTCCGACTGTCAGCACTGCTCCACCTCTGATTCCACCACCGGCCATTGAGACCCAGAGCAGCAGTTCTGAAGAGATAGTGCCCAGCCCTCCCTCGCCACCCCCTCTACCCCGCATCTACAAGCCTTGCTTTGTCTGTCAGGACAAGTCCTCAGGCTACCACTATGGGGTCAGCGCCTGTGAGGGCTGCAAG. The primer sequences of the WT1 gene are as follows: WT1 forword: ACAGGGTACGAGAGCGATAACCA; WT1 reverse: CACACGTCGCACATCCTGAAT; WT1 probe: CAACGCCCATCCTCTGCGGAGCCCA. The primer sequences of the PML::RARA fusion gene are as follows: bcr1-ENF903: TCTTCCTGCCCAACAGCAA; bcr2-ENF906: ACCTGGATGGACCGCCTAG; bcr3-ENF905: CCGATGGCTTCGACGAGTT; RARa-1099R ACATGCCCACTTCAAAGCAC. The primer sequences of the CBFβ::MYH11 fusion gene are as follows: CBFB-C: GGGCTGTCTGGAGTTTGATG; MYH11-D1:
TCCCTGTGACGCTCTCAACT; MYH11-D2: CTTGAGCGCCTGCATGTT. Illumina Hiseq X sequencing platform was used to detect RNA-level gene fusions and SNVs/Indels variants at the whole transcriptome level. The bioinformatics analysis methods of gene mutation and gene fusion are as follows: the sequencing fragment was compared with the EnsemblGRCh37 reference genome by STAR software. Mutation detection was performed by VarScan software, including mutation threshold screening and SNVs mutation detection and typing. The fusion gene was analyzed by Arriba software. Mutation results were annotated by AnnovAR for downstream data, and the annotation databases mainly included Clinvar, dbSNP, 1000 Genome, genomeAD, ExAC, COSMIC, etc. The fusion gene databases include but are not limited to COSMIC, Fusion can cer, Atlas of Genetics and Cytogenetics in Oncology and Haematology, My Cancer Genome, etc.
Differential gene expression and heat maps were performed by the DESeq2 and pheatmap packages in R (1.16.1). Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology-Based Annotation System (KOBAS) (version 3.0) online biological information database was used for KEGG pathway enrichment analysis of differentially expressed genes (DEGs). At least log FC > 1.5 (up-regulated) or log FC < -1.5 (down-regulated) and adjusted P-value <0.05 were considered statistically significant.
Case report: a 3-year-old girl presented with intermittent pain in her lower limb for half a month, and fever for 3 days. Physical examination was significant for scattered hemorrhagic spots on the face, mild hepatomegaly, and enlarged cervical lymph node. Brain magnetic resonance imaging showed inflammation or effusion in the bilateral middle ear mastoid process, and no obvious abnormalities in the brain. Initial laboratory evaluation revealed a high white blood cell (WBC) count of 246 × 109/L, hemoglobin of 6.6g/dl, and a platelet count of 54 × 109/L. Coagulation function showed a prothrombin time of 15.6s, international normalized ratio of 1.38, activated partial thromboplastin time of 22.6s, thrombin time of 19.2s, fibrinogen at 2.61g/L, fibrinogen degradation products at 100.4 ug/ml, d-dimer at 37.76 mg/L, while lactate dehydrogenase was 4179 U/L. The peripheral blood cell differential count revealed a predominance of abnormal promyelocyte cells (60%) (Figure 1A). Bone marrow examination revealed 39.5% promyelocytes that had numerous hypergranularity without evidence of Auer rods (Figure 1B), the rate of positive peroxidase staining was 100% (Figure 1C), the rate of naphythol AS-D chloroacetate esterase (AS-D-CE) was 86% (Figure 1D), α-naphthol acetate esterace (α-NAE) was negative (Figure 1E). The immunophenotype was positive for MPO, CD33, CD13, CD15, CD123, CD38, and CD64 and negative for CD34, CD117, CD19, CD7, CD56, CD11b, and HLA-DR. Fluorescence in situ hybridization (FISH) analysis with the PML::RARA dual-fusion translocation probe identified no dual fusion signal (Figure 1F), and the BCR::ABL, AML1::ETO, and CBFβ::MYH11fusion genes were negative. Quantitative detection of the WT1 gene showed it was highly expressed, and the positive rate was 98.2771%. Reverse transcriptase polymerase chain reaction (RT-PCR) revealed PML::RARA fusion gene (Bcr1, Bcr2, Bcr3) was negative. Besides, conventional karyotype analysis revealed a karyotype in 20 metaphase cells with the following formula: 47XX,t(4;17)(q12;21),+22(20) according to ISCN2016 (Figure 1G). To further characterize, RNA-seq was arranged for hematopoietic malignancies, sequencing of this sample confirmed the FIP1L1::RARA and RARA::FIP1L1 fusion transcripts, the FIP1L1::RARA fusion gene was generated between exon 13 of FIP1L1 and exon2 of RARA (Figure 2). Whole exon sequencing (WES) and copy number variation-sequencing (CNV-Seq) revealed dup(22)(q11.1q13.33), which can occur in hematologic malignancies such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). This patient was finally diagnosed with APL according to bone marrow morphology, immunophenotype, cytogenetics and RNA-seq. Besides, single-drug chemotherapies/chemotherapy regimens based on an ATP-based tumor chemosensitivity assay (ATP-TCA) were used to evaluate the in vitro chemosensitivity (Table 2). Tumor cells are moderately sensitive to single-drug chemotherapies including epirubicin, mitoxantrone, homoharringtonine, arsenic trioxide, selinexor, and chemotherapy regimens including FLAG (fludarabine and cytarabine), CLAG (clavibine and cytarabine) and ME (mitoxantrone and etoposide) and no sensitivity to ATRA. The body weight of the child was 16.3 kg, the height was 105 cm, and the body surface area was 0.69 m2. After a treatment combining ATRA (16.7 mg/d, divided twice a day) with hydroxycarbamide (1.5 g/d, divided three times a day) for 3 days, WBC count decreased from 246×109 to 241×109, indicating no response to ATRA. The protocol was changed to a FALG chemotherapy regimen (fludarabine 20 mg/d, Day1-Day5; cytarabine 1.3g/d, Day1-Day5; granulocyte colony-stimulating factor 80ug/d, Day0-Day7, idarbicin 5mg/d, Day4-Day6). After chemotherapy, the WBC of the patient dropped to 0.15 × 109, suggested that the patient benefited from this chemotherapy. She received the same chemotherapy regimen after 40 days, and achieved a complete remission with minimal residue detection (MRD) negative. FIP1L1::RARA was negative by digital-PCR analysis. Conventional karyotype analysis revealed a normal karyotype in 20 metaphase cells. Quantitative detection of Wilms tumor 1 (WT1) gene showed the expression was high with a positive rate of 0.7548%. Then, she received an HAE chemotherapy regimen (homoharringatonine, 2 mg/d, Day1-Day5; cytarabine, 134 mg/d, Day1-Day5; etoposide, 67 mg/d, Day1-Day5) 30 days after the last chemotherapy, MRD detection showed no immunophenotypic abnormalities. FIP1L1::RARA was negative by digital-PCR analysis. Quantitative detection of WT1 gene showed the expression was low, and the positive rate was 0.1636%. Last chemotherapy treatment, she received a MidAC chemotherapy regimen (mitoxantrone, 20 mg/d, Day1-Day5; cytarabine, 0.7g/d, Day1-Day3) 40 days after the last chemotherapy. MRD and FIP1L1::RARA fusion gene were negative. Quantitative detection of WT1 gene showed the expression was high, and the positive rate was 0.7563%.
Figure 1 The clinical information of the APL patient. (A) Peripheral blood cell morphology at initial diagnosis. ×400; (B) Bone marrow morphology at initial diagnosis. ×400; (C) Peroxidase staining.×400; (D) Naphythol AS-D chloroacetate esterase staining.×400; (E) α-naphthol acetate esterace staining.×400; (F) FISH using the PML::RARA dual-color, dual fusion translocation probe. (G) The chromosome karyotype of the APL patient, the red arrows indicate the chromosome translocation. The PML probes are labeled with orange fluorescein and RARA probes with green fluorescein, the green and orange signal form a yellow fusion signal when PML::RARA fusion gene is present.
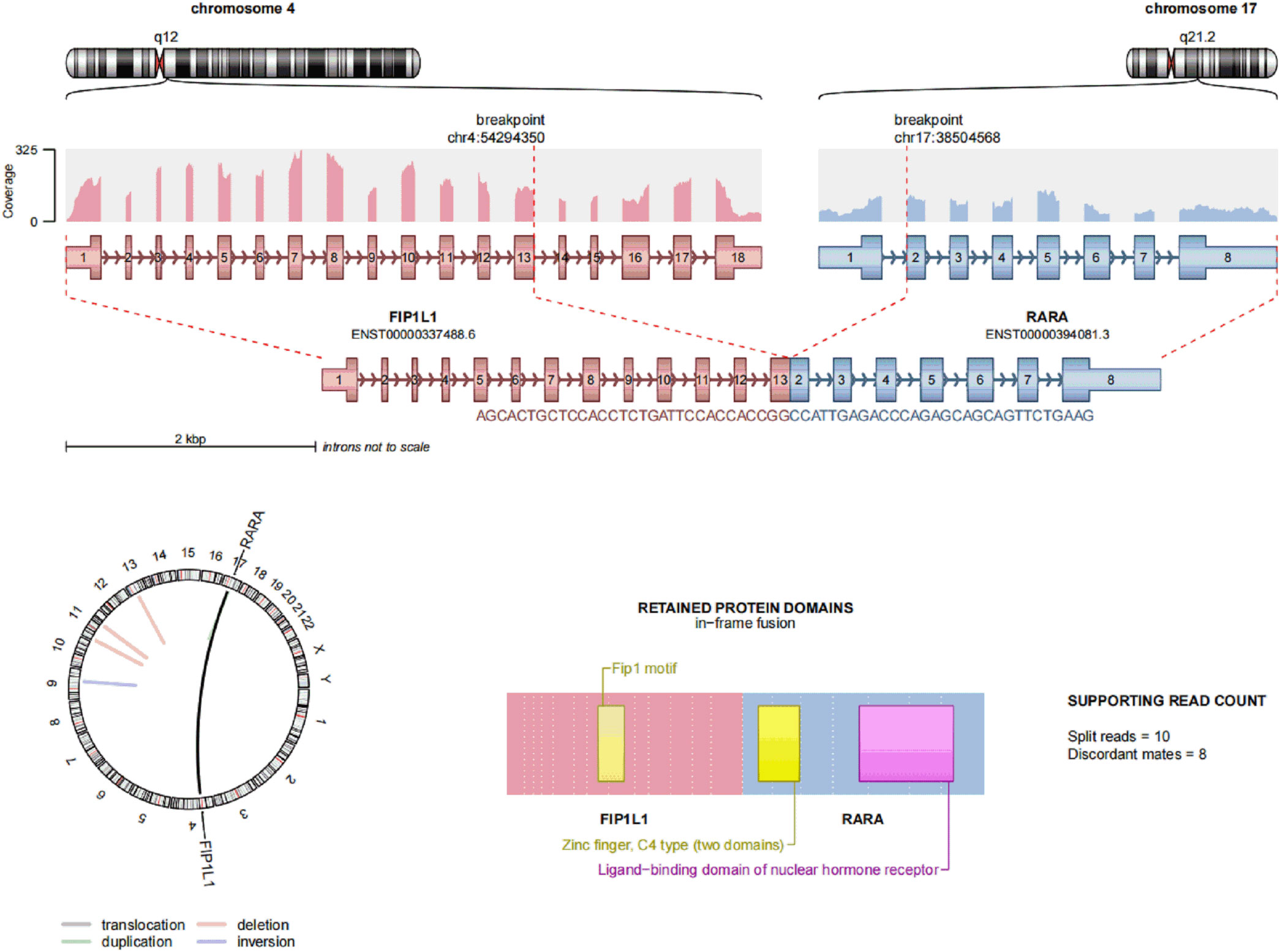
Figure 2 Transcriptome sequencing of FIP1L1::RARA fusion gene at onset, a fusion between exon 13 of FIP1L1 and exon 3 of RARA was identified.
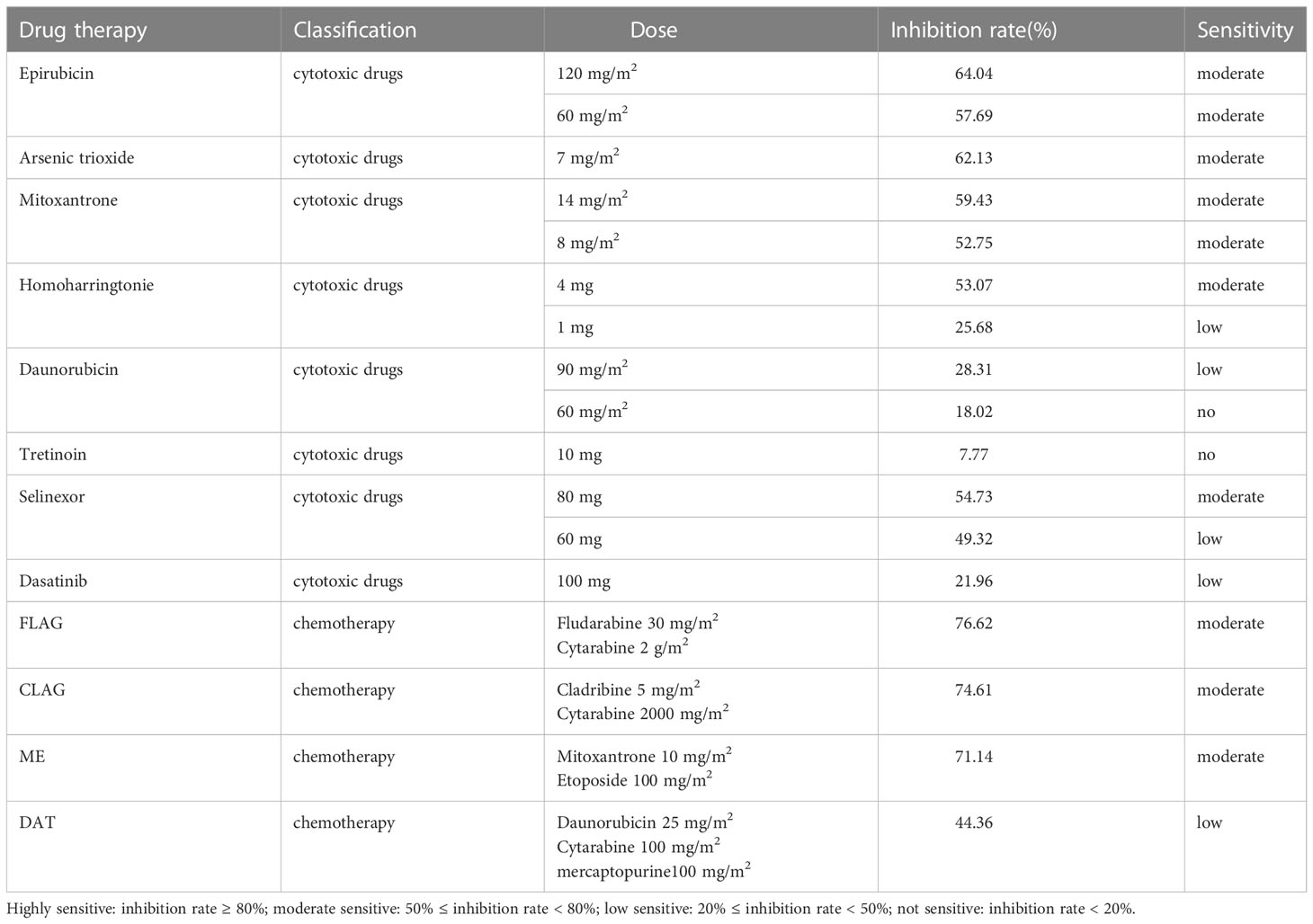
Table 2 Evaluating the in vitro chemosensitivity of single drug chemotherapies/chemotherapy regimens to APL with FIP1L1::RARA.
In order to understand the different RNA expression profile of each case, transcriptome analysis of six bone marrow specimens was performed by RNA-seq. The heat map was used to show the total RNA expression profile of each patient. Obvious differences between APL with FIP1L1::RARA and other hematologic malignancies were identified (Figure 3).
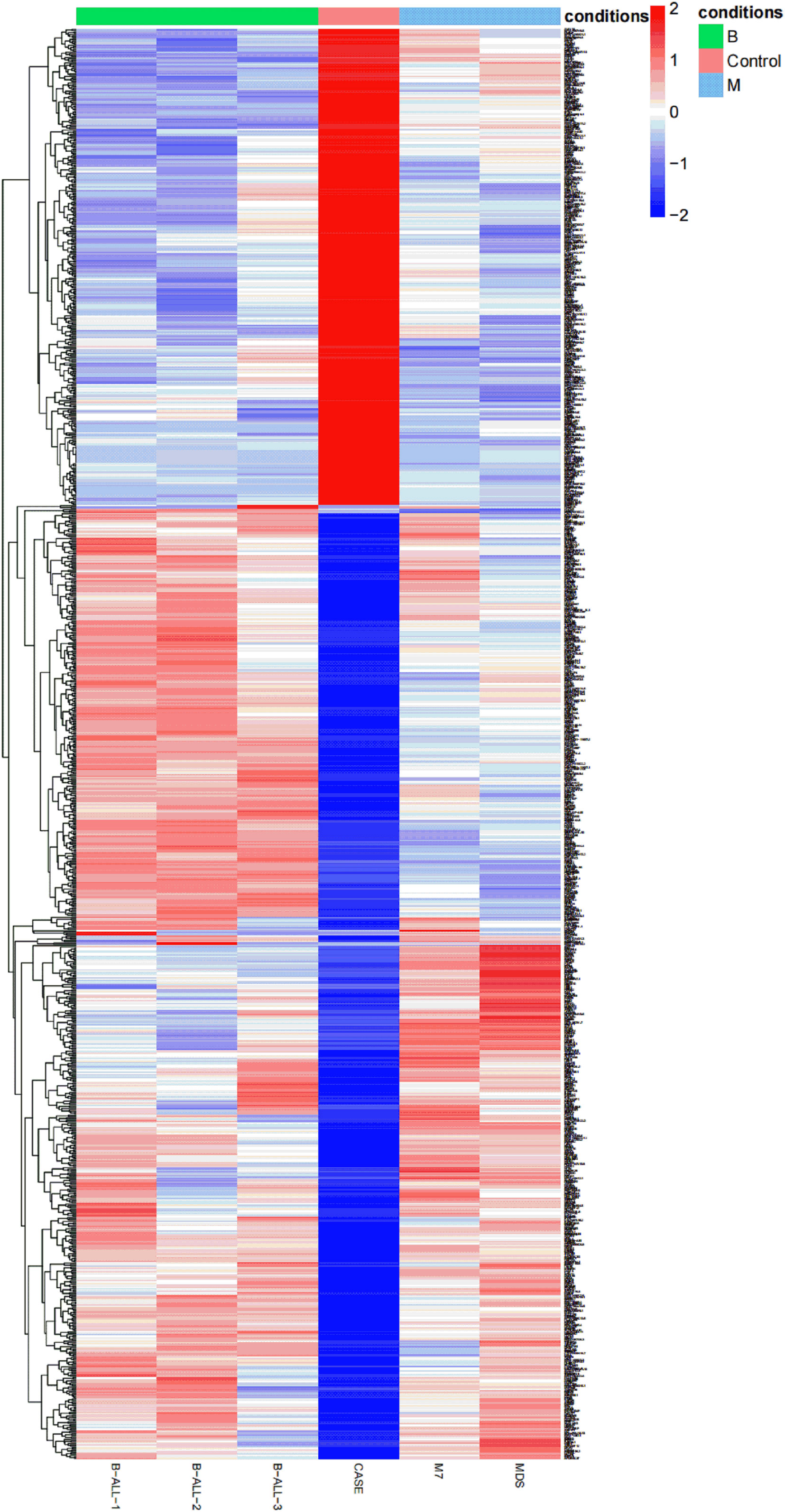
Figure 3 A heat map is created based on the total RNA expression of six patients with hematologic malignancies: the relative expression abundance of each gene in B-ALL, APL with FIP1L1::RARA, M7, MDS. The coloured scale varies from deep blue to deep red, which indicates low or high expression.
To identify the DEGs between different hematological malignancies, the patients were separated into three groups: case group (APL with FIP1L1::RARA), ALL group (three cases of B-ALL), and myeloid neoplasms (M) group (M7 and MDS). After analyzing the transcriptomic changes of the case group and M group, a total of 4181 DEGs (|fold change>2| and p<0.05) were identified, including 2110 up-regulated and 2071 down-regulated genes. Between the case group vs ALL group, 2268 DEGs were observed, among which 1639 were up-regulated genes and 629 were down-regulated genes. A total of 1060 common DEGs (co-DEGs) were identified between case vs ALL and case vs M, including 706 up-regulated and 354 down-regulated genes (Figure 4A). To make it more intuitive, the volcano plot was used for identifying DEGs (Figure 4B).
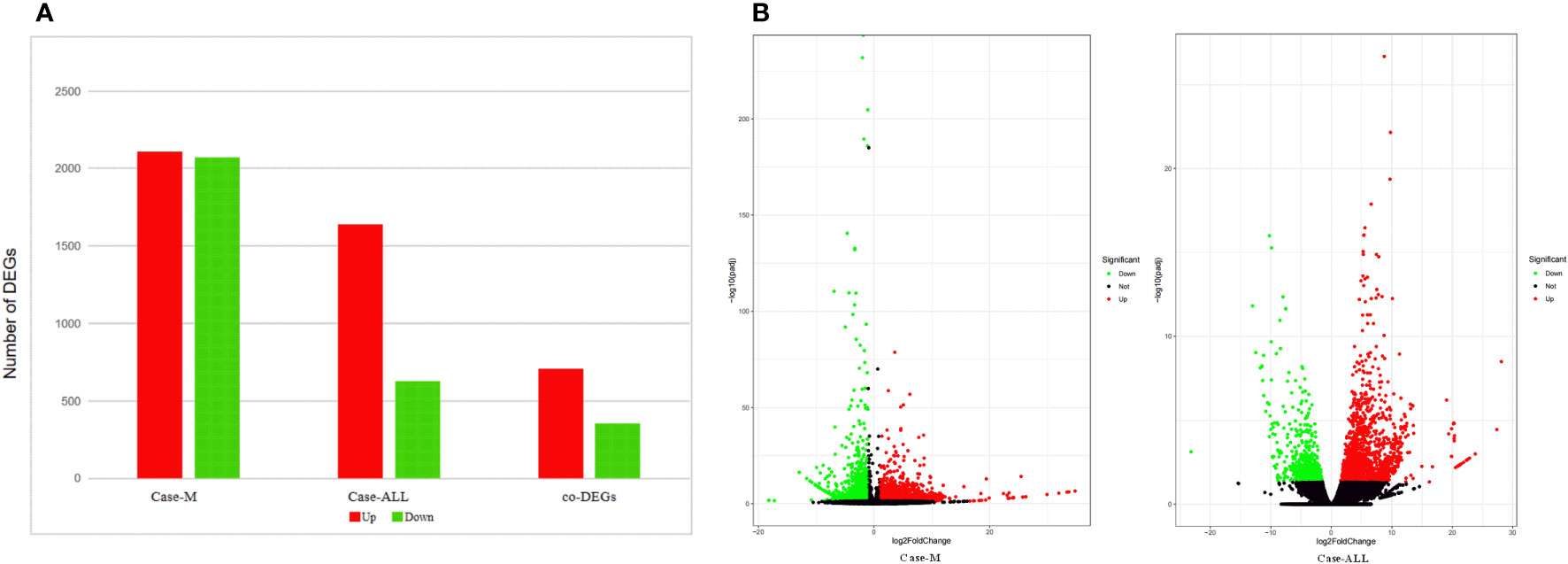
Figure 4 (A) DEGs between case group and M group, case group vs ALL group, co-DEGs between case vs ALL and case vs M; (B) Volcano map of DEGs. The x-axis is the log2 scale of the fold change of gene expression in case, ALL and M (log2(fold change)). Negative values indicate down regulation; positive values indicate up regulation. The y-axis is the minus log10 scale of the adjusted p values (elog10 (padj)), which indicates the significant level of expression difference. The red dots represent significantly up-regulated genes with at least two fold change, while the green dots represent significantly down-regulated genes with at least two fold change.
A total of 1060 co-DEGs (|fold change|>2 and p < 0.05) were detected between case vs ALL and case vs M, a KEGG pathway enrichment analysis was conducted to explore the most significantly enriched pathways for co-DEGs. The up-regulated genes were mainly mapped into platelet activation, cancer, AMPK signaling pathway, PI3K-Akt signaling pathway, and MAPK signaling pathway. The down-regulated genes were significantly associated with TNF signaling pathway, Rap1 signaling pathway, Age-RAGE signaling pathway, and apoptosis (Figure 5).
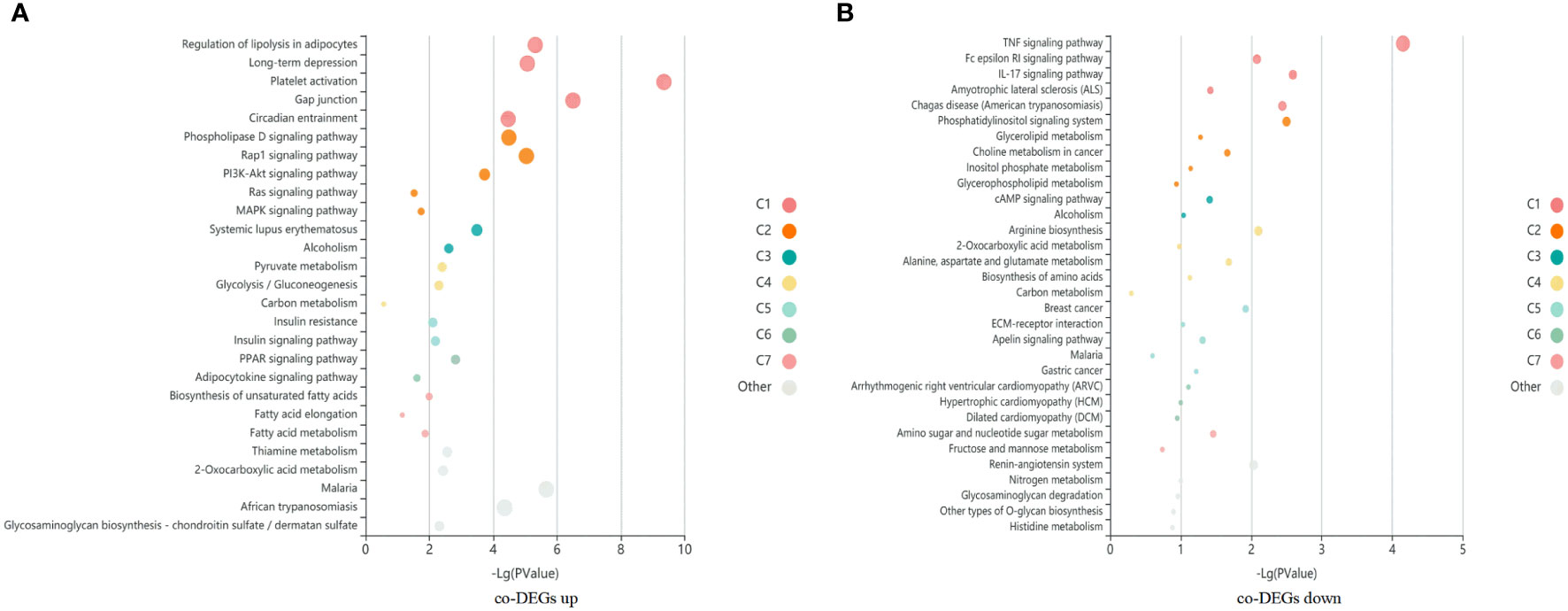
Figure 5 A KEGG pathway enrichment analysis was conducted to explore the most significantly enriched pathways for co-DEGs. Each bubble represents an enriched function, and the size of the bubble from small to large: [0.05,1], [0.01,0.05), [0.001,0.01), [0.0001,0.001), [1e-10,0.0001), [0,1e-10). The color of the bar is the same as the color in the circular network, which represents different clusters. For each cluster, if there are more than 5 terms, top 5 with the highest enrich ratio will be displayed. (A) KEGG enrichment bubble map of co-upregulated differential genes in case compared with M and ALL groups. (B) KEGG enrichment bubble map of co-downregulated differential genes in case compared with M and ALL groups.
In this study, a patient with the FIP1L1::RARA fusion gene was reported and eventually diagnosed as APL. Only the FIP1L1::RARA fusion gene was identified, with no evidence of an established oncogenic RAS pathway mutation. Although APL with atypical rearrangements usually has a clinical presentation and morphological and immunophenotypic picture similar to classical APL, several different features exist compared with classical APL including very young age, and absence of laboratory of coagulopathy.
Due to there being few reports on the FIP1L1::RARA fusion gene, studies on the pathogenesis, treatment, and prognosis of APL with FIP1L1::RARA are limited. Since it is an aggressive disease (12), there exists a high possibility of early mortality if clinicians are unable to identify an abnormal karyotype that appeared singularly or as part of a complex mutation (13). Identifying variant RARA rearrangements is critical for the diagnosis and treatment of patients with APL (14). In order to lay a molecular basis for further research on the occurrence and treatment of this disease, RNA-Seq was conducted to explore the differences between APL with FIP1L1::RARA and other hematologic malignancies from the level of transcriptome. Obvious transcriptomic differences between APL with FIP1L1::RARA and myeloid neoplasms were identified. In an analysis of co-DEGs, the up-regulated and down-regulated genes were enriched in different signaling pathways.
Many genes involved in APL have similar cellular functions, but lack recurrence and consistency, except for FLT3, WT1, and KRAS. The physical and functional interactions of the mutated genes have been useful to discover driver mutations in cancers, the damage of these genes in APL is weaker than the interaction between mutant genes and different functionally related classes (15). In this case, the molecular basis behind APL has been focused on the role of the FIP1L1::RARA fusion gene. The study indicated the retinoic acid receptor alpha (RARα) forms a heterodimer with another form of a nuclear hormone receptor protein called retinoid X receptors (RXR), the RARα- RXR heterodimer binds to regions of DNA referred to as retinoic acid response elements (RAREs) to mediate the transcription of hundreds of genes. Many of the RAREs are involved in self-renewal and differentiation (16). RARA has been implicated as a key regulator of normal as well as transformed blood cells, and APL is driven by fusion genes involving retinoic acid receptors, primarily RARA. Besides, RARA breakpoints are highly fixed in variant APL, mainly responsible for regulating bone marrow microenvironment and hematopoietic function. The constant involvement of RARA or other retinoic acid receptor super family members stresses the importance of deregulated retinoic acid signaling in driving APL pathogenesis (17). FIP1L1 is an integral subunit for cleavage and polyadenylation specificity factor for stimulation of poly(A) polymerase. It was found that the FIP1L1 is necessary for activating STAT5 and PKB/Akt in human hematopoietic progenitor cells (18), indicating it is a contributing factor to the higher proliferating activity. As for the roles and mechanisms of these genes in the different signaling pathways, further studies are needed to elucidate.
In therapy, patients with FIP1L1::RARA present different drug sensitivities to ATRA. A 90-year-old woman was reported as APL with FIP1L1::RARA fusion achieving a complete remission by oral administration of ATRA alone in a dose of 50 mg daily without further follow-up information, indicating a high sensitivity to ATRA (9). Another 77-year-old woman reported as APL with FP1L1::RARA fusion gene was treated according to the PETHEMA APL 2005 protocol. Unfortunately, she died after ten days of treatment, probably due to retinoic acid syndrome, and response to ATRA treatment could not be assessed in this case due to early death (10). A 28-month-old girl with FIP1L1::RARA and complex karyotype was diagnosed with APL complicated by the de novo myeloid sarcoma. After a treatment combining ATRA with DA regimen as induction chemotherapy, the patient had achieved an initial complete remission by day 30. However, FIP1L1::RARA was still positive after the second course. Then she received three cycles of ATRA and idarubicin in the following consolidation treatment and continual therapy with ATRA with chemotherapy as previously described and kept leukemia-free after a 5-month follow-up (11). During this case, the efficacy of the combination ATRA-chemotherapy therapy is positive, but the response to ATRA could not be assessed. These three patients with FIP1L1::RARA fusion were eventually diagnosed as APL. Several patients with FIP1L1::RARA were diagnosed as other myeloid neoplasms. A 20-month-old child with the FIP1L1::RARA fusion gene was first reported with a final diagnosis of JMML (8). Despite two allogeneic stem cell transplantations, the patient still died from relapse of JMML 17 months after presentation. No ATRA sensitivity was tested and there was no material to perform in vitro ATRA-mediated differentiation assays in this case. A 9-month-old boy with a FIP1L1::RARA fusion-associated myelodysplastic/myeloproliferative neoplasm-like overlap syndrome was also reported. After combined chemotherapy (ATRA, ATO, and idarubicin) and differentiating agents (ATRA, ATO), he achieved cytogenetic remission. After a consolidation cycle (mitoxantrone and cytarabine) and maintenance therapy (ATRA, ATO), he replased with new-onset facial swelling and palpable skull-based masses before allogeneic hematopoietic stem cell transplant (allo-HCT). Then he received achieved a cytogenetic remission after the Capizzi II regimen (high-dose cytarabine) reinduction therapy and topotecan, vinorelbine, thiotepa, and clofarabine chemotherapy combined with the synthetic retinoid agent tamibarotthis. After that, he proceeded to a peripheral blood stem cell transplant and achieved remission 20 months after allo-HCT (19). In this study, a 3-year-old girl with the FIP1L1::RARA fusion gene was eventually diagnosed as APL. At the beginning, she received ATRA treatment with no reducing of WBC, indicating no sensitivity to ATRA. Then she underwent a chemotherapy regimen for AML (two courses of FALG, one course of HAE, one course of MidAC). She achieved cytogenetic remission during every course, indicating high sensitivity to this chemotherapy regimen.
Reasons for patients with the FIP1L1::RARA fusion gene showing different sensitivities to ATRA are poorly understood. Study shows FIP1L1::RARA functions as a transcriptional repressor in retinoic acid-dependent transcription and can form a homodimer and suppress retinoic acid-dependent transcriptional activity, resulting in low sensitivity to ATRA (9). FIP1L1::RARA functions as a transcriptional repressor in retinoic acid-dependent transcription, FIP1 motif is a protein interaction domain and plays a pivotal role in homodimerization, which is crucial for retinoic acid-dependent transcriptional activity (20). Further studies are needed to elucidate the reasons for patients with the FIP1L1::RARA fusion gene exhibiting different drug sensitivities to ATRA. Since APL with FIP1L1::RARA has demonstrated different sensitivities to ATRA, combined chemotherapy rather than differentiation induction therapy may be the appropriate treatment for APL with FIP1L1::RARA with no sensitivity to ATRA. Reasons for different phenotypes of leukemia caused by FIP1L1::RARA remain unknown. Study indicates FIP1L1::RARA may interfere with FIP1L1 function, resulting in leukemia with monocytic features. FIP1L1 promoter regulated expression may influence FIP1L1::RARA-mediated leukemogenesis. Low-level expression of the fusion protein may have contributed to the non-APL phenotype (8). Moreover, the homodimerization ability of RARA fusion proteins is also critical for leukemic transformation (21). The different signal pathways enriched in this case may be the mechanism of FIP1L1-RARA leading to different types of leukemia in different ways, further studies are needed to elucidate the mechanism of different phenotypes of leukemia caused by FIP1L1::RARA. Since not all patients with FIP1L1::RARA are APL, it is important to combine morphology with flow cytometry, karyotype analysis, FISH, and RT-PCR for accurate diagnosis. In practice, when the APL phenotype was presented with a negative PML::RARA fusion gene and the translocation cannot be identified by conventional methods, APL with variant rearrangements of RARA needed to be considered and diagnosed as early as possible by fusion gene screening and RNA-Seq. RNA-seq may be a new diagnostic method when RARA rearrangements are failed to be identified by conventional methods, because of the obvious transcriptomic difference between APL with FIP1L1::RARA and myeloid neoplasms.
In this study, there are several limitations that need to be clarified. Firstly, there is no clinical data regarding ATO sensitivity, so whether the patient with FIP1L1::RARA will respond to ATO remains unknown. Secondly, the number of APL patients with FIP1L1::RARA and other myeloid neoplasms is extremely limited, and the credibility of the results is limited. In the follow-up work, the collection of cases and various subtypes will be expanded, further experimental verification will be carried out, and a diagnostic model based on transcriptome is expected to be constructed. Thirdly, the in vitro study data to confirm the association of FIP1L1::RARA with APL was insufficient. We plan to construct the expression plasmid and transfect to establish a stable cell line, further studying the association of FIP1L1::RARA with APL and the mechanism of APL induced by the FIP1L1::RARA fusion gene. Besides, we will explore the differences in different fusion sites and construct the cell models of a single agent/therapeutic regimen intervention in vitro studying the mechanism of drug resistance of APL to ATRA.
In conclusion, a child with FIP1L1::RARA fusion diagnosed as APL was reported. RNA-seq may be a new diagnostic method for patients with other RARA rearrangements. The up-regulated and down-regulated co-DEGs between case vs ALL and case vs myeloid neoplasms were enriched in different signaling pathways. Further experimental studies are needed to identify pathogenesis and treatment for APL with FIP1L1::RARA
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject, accession number PRJNA911738.
The studies involving human participants were reviewed and approved by Hunan Provincial People's Hospital Medical Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
GL and PL developed the study design; GL wrote the original draft. PL reviewed the manuscript; YC was responsible for data acquisition and revising; JL, LL, and XH were responsible for data analysis and methodology. All authors approved the final version to be published.
This work was supported by Hunan Provincial Natural Science Foundation of China (Grant No. 2021JJ70098) and Changsha Science and Technology Project (Grant No. kq2004136).
The authors thank all the patients participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hambley BC, Tomuleasa C, Ghiaur G. Coagulopathy in acute promyelocytic leukemia: Can we go beyond supportive care? Front Med (2021) 8:722614. doi: 10.3389/fmed.2021.722614
2. Xiao MY, Zhou P, Liu YH, Wei SJ, Li D, Li WY, et al. Predictive factors for differentiating thrombohemorrhagic disorders in high-risk acute promyelocytic leukemia. Thromb Res (2022) 210:33–41. doi: 10.1016/j.thromres.2021.12.020
3. Zhang X, Sun JW, Yu WJ, Jin J. Current views on the genetic landscape and management of variant acute promyelocytic leukemia. biomark Res (2021) 9(1):33. doi: 10.1186/s40364-021-00284-x
4. Guarnera L, Ottone T, Fabiani E, Divona M, Savi A, Travaglini S, et al. Atypical rearrangements in APL-like acute myeloid leukemias: Molecular characterization and prognosis. Front Oncol (2022) 12:871590. doi: 10.3389/fonc.2022.871590
5. Nakanishi T, Nakaya A, Nishio Y, Fujita S, Satake A, Azuma Y, et al. A variant of acute promyelocytic leukemia with t(4;17)(q12;q21) showed two different clinical symptoms. Hematol Rep (2019) 11(3):7971. doi: 10.4081/hr.2019.7971
6. Baba SM, Pandith AA, Shah ZA, Baba RA. Pathogenetic implication of fusion genes in acute promyelocytic leukemia and their diagnostic utility. Clin Genet (2019) 95(1):41–52. doi: 10.1111/cge.13372
7. Leguit RJ, Orazi A, Kucine N, Kvasnicka HM, Gianelli U, Arber DA, et al. EAHP 2020 workshop proceedings, pediatric myeloid neoplasms. Virchows Archiv (2022) 481(4):621–46. doi: 10.1007/s00428-022-03375-8
8. Buijs A, Bruin M. Fusion of FIP1L1 and RARA as a result of a novel t(4;17)(q12;q21) in a case of juvenile myelomonocytic leukemia. Leukemia (2007) 21(5):1104–8. doi: 10.1038/sj.leu.2404596
9. Kondo T, Mori A, Darmanin S, Hashino S, Tanaka J, Asaka M. The seventh pathogenic fusion gene FIP1L1::RARA was isolated from a t(4;17)-positive acute promyelocytic leukemia. Haematologica (2008) 93(9):1414–6. doi: 10.3324/haematol.12854
10. Menezes J, Acquadro F, Perez-Pons de la Villa C, García-Sánchez F, Álvarez S, Cigudosa JC. FIP1L1::RARA with breakpoint at FIP1L1 intron 13: a variant translocation in acute promyelocytic leukemia. Haematologica (2011) 96(10):1565–6. doi: 10.3324/haematol.2011.047134
11. Wang YR, Rui YY, Shen Y, Li J, Liu PN, Lu Q, et al. Myeloid sarcoma type of acute promyelocytic leukemia with a cryptic insertion of RARA into FIP1L1: The clinical utility of NGS and bioinformatic analyses. Front Oncol (2021) 11:688203. doi: 10.3389/fonc.2021.688203
12. Jiang XW, Chen SZ, Zhu XY, Xu XX, Liu Y. Development and validation of a droplet digital PCR assay for the evaluation of PML-RARα fusion transcripts in acute promyelocytic leukemia. Mol Cell Probes (2020) 53:101617. doi: 10.1016/j.mcp.2020.101617
13. Chen C, Huang X, Wang K, Chen K, Gao D, Qian S. Early mortality in acute promyelocytic leukemia: Potential predictors. Oncol Lett (2018) 15(4):4061–9. doi: 10.3892/ol.2018.7854
14. Peterson JF, He RR, Nayer H, Cuevo RS, Smadbeck JB, Vasmatzis G, et al. Characterization of a rarely reported STAT5B/RARA gene fusion in a young adult with newly diagnosed acute promyelocytic leukemia with resistance to ATRA therapy. Cancer Genet (2019) 237:51–4. doi: 10.1016/j.cancergen.2019.06.007
15. Ibáñez M, Carbonell-Caballero J, García-Alonso L, Such E, Jiménez-Almazán J, Vidal E, et al. The mutational landscape of acute promyelocytic leukemia reveals an interacting network of Co-occurrences and recurrent mutations. PLoS One (2016) 11(2):e0148346. doi: 10.1371/journal.pone.0148346
16. Jimenez JJ, Chale RS, Abad AC, Schally AV. Acute promyelocytic leukemia (APL): a review of the literature. Oncotarget (2020) 11(11):992–1003. doi: 10.18632/oncotarget
17. Geoffroy MC, de Thé H. Classic and variants APLs, as viewed from a therapy response. Cancers (Basel) (2020) 12(4):967. doi: 10.3390/cancers12040967
18. Buitenhuis M, Verhagen LP, Cools J, Coffer PJ. Molecular mechanisms underlying FIP1L1-PDGFRA-mediated myeloproliferation. Cancer Res (2007) 67(8):3759–66. doi: 10.1158/0008-5472
19. Miltiadous O, Petrova-Drus K, Kaicker S, Mathew S, Kluk M, Geyer JT, et al. Successful treatment and integrated genomic analysis of an infant with FIP1L1::RARA fusion-associated myeloid neoplasm. Blood Adv (2022) 6(4):1137–42. doi: 10.1182/bloodadvances.2021004966
20. Iwasaki J, Kondo T, Darmanin S, Ibata M, Onozawa M, Hashimoto D, et al. FIP1L1 presence in FIP1L1::RARA or FIP1L1-PDGFRA differentially contributes to the pathogenesis of distinct types of leukemia. Ann Hematol (2014) 93(9):1473–81. doi: 10.1007/s00277-014-2085-1
Keywords: acute promyelocytic leukemia, FIP1L1::RARA, transcriptome sequencing, bioinformatic analyses, differential expression genes
Citation: Liu G, Long J, Chen Y, Li L, Huan X and Long P (2023) Acute promyelocytic leukemia with FIP1L1::RARA fusion gene: The clinical utility of transcriptome sequencing and bioinformatic analyses. Front. Oncol. 12:1049473. doi: 10.3389/fonc.2022.1049473
Received: 20 September 2022; Accepted: 29 December 2022;
Published: 26 January 2023.
Edited by:
Harinder Gill, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Susanna Akiki, Hamad Medical Corporation, QatarCopyright © 2023 Liu, Long, Chen, Li, Huan and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panpan Long, Mjg2Mzk3NzE1QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.