
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 20 December 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1038678
This article is part of the Research Topic Identification, Risk Stratification, and Optimized Management for Lynch Syndrome View all 14 articles
 Ayushi Jain1
Ayushi Jain1 Maryam Alimirah2Heather Hampel3Rachel Pearlman4Jianing Ma5
Maryam Alimirah2Heather Hampel3Rachel Pearlman4Jianing Ma5 Jing Peng5Matthew F. Kalady4,6Peter P. Stanich2*
Jing Peng5Matthew F. Kalady4,6Peter P. Stanich2*Background: Lynch syndrome has not traditionally been considered to have a high colorectal adenoma burden. However, with increasing adenoma detection rates in the general population, the incidence of adenoma detection in Lynch syndrome may also be increasing and leading to higher cumulative adenoma counts.
Aim: To clarify the prevalence and clinical impact of multiple colorectal adenomas (MCRA) in Lynch syndrome.
Methods: A retrospective review of patients with Lynch syndrome at our institution was performed to assess for MCRA (defined as ≥10 cumulative adenomas).
Results: There were 222 patients with Lynch syndrome among whom 14 (6.3%) met MCRA criteria. These patients had increased incidence of advanced neoplasia (OR 10, 95% CI: 2.7-66.7).
Conclusions: MCRA is not unusual in Lynch syndrome and is associated with a significantly increased likelihood of advanced colon neoplasia. Consideration should be given to differentiating colonoscopy intervals based on the presence of polyposis in Lynch syndrome.
Inherited colorectal cancer syndromes are often categorized into “polyposis” and “nonpolyposis” syndromes (1). Lynch syndrome is a hereditary cancer syndrome characterized by heterozygous pathogenic variants in MLH1, MSH2, MSH6, or PMS2 or an EPCAM deletion. Existing literature in Lynch syndrome initially suggested the majority of patients carry MLH1 or MSH2 pathogenic variants, although analysis of multigene panel testing results suggest that the frequency is likely similar across the genes and analysis of international registries predicts that MSH6 and PMS2 pathogenic variants may be more common (2, 3). Lynch syndrome has classically been associated with a lower colorectal adenoma burden and considered a nonpolyposis syndrome (4, 5). There is limited data on the adenoma burden in patients with Lynch syndrome, though one previous estimate suggested a mean of 7 adenomas by age 80 (6). Another study reported 4% of patients with Lynch syndrome had ≥ 10 cumulative lifetime adenomas and qualified as having a clinical oligopolyposis syndrome, contrary to the traditional assumption that patients with Lynch syndrome do not meet this criteria (7). In addition, with recent reports of nationwide increases in adenoma detection rates, the cumulative lifetime adenoma counts in the Lynch syndrome population are likely increasing along with the general population (8). As such, Lynch syndrome should also be considered in the differential of patients meeting multiple colorectal adenomas criteria, defined as 10 or more adenomas (9).
Lynch syndrome patients have long been recommended to have a colonoscopy every 1-2 years in the United States (1, 10). The latest guidelines from the Mallorca group are now recommending colonoscopy surveillance intervals of 2 to 3 years for most genotypes and up to 5 years for those with PMS2 mutations (11). However, recent studies have shown that patients with ≥ 10 cumulative lifetime adenomas are at higher risk of advanced neoplasia and colorectal cancer (12). It is unclear if Lynch syndrome patients that meet this criterion would also have additional increased risks.
Our aim was to assess the prevalence of multiple colorectal adenomas in Lynch syndrome and assess for an association with advanced colorectal neoplasia and colorectal cancer.
This was a retrospective study assessing patients with Lynch syndrome followed in the Hereditary and High-Risk Gastroenterology Clinic. Institutional Review Board approval was obtained prior to study initiation.
Inclusion criteria for the study were age 18 years or greater, a documented pathogenic or likely pathogenic variant in a mismatch repair gene (MLH1, MSH2, MSH6, PMS2) or in EPCAM on germline genetic testing, the completion of at least one colonoscopy at our institution and a clinic visit from August 2014 through December 2020. Exclusion criteria included a known pathogenic or likely pathogenic variant in additional hereditary cancer genes and a history of total colectomy prior to identification of first adenomatous polyp. All available colonoscopy and pathology records were reviewed to identify polyp characteristics. Colonoscopies were performed with the available endoscopic technology at the time of completion, which included both standard definition and high-definition white light endoscopy and virtual chromoendoscopy. Dye chromoendoscopy was not utilized.
The primary study outcome was the prevalence of multiple colorectal adenomas, defined as ≥ 10 lifetime tubular adenomas (9). The secondary outcomes of interest included prevalence of previous advanced colorectal neoplasia and colorectal cancer in those with and without MCRA. Advanced colorectal neoplasia was defined as a lifetime history of colorectal cancer, advanced adenoma, or advanced sessile serrated lesion. Advanced adenomas were defined as adenomas ≥ 10 mm in size, villous or tubulovillous adenomas or adenomas with high-grade dysplasia. Advanced sessile serrated lesions were defined as sessile serrated polyps ≥ 10 mm in size or with features of high-grade or low-grade dysplasia.
Statistical analysis included univariable analysis to compare patients with 0-9 tubular adenomas and ≥ 10 tubular adenomas. Odds ratio and 95% confidence interval was reported using univariable logistic regression for all variables. Multivariable analysis was utilized to adjust for age.
Two hundred and twenty-two patients met study criteria and were included in the analysis with demographic and clinical details available in Table 1. There were 142 patients (64%) with adenomas in the cohort and 14 patients (6.3%) met criteria for MCRA with 10 or more cumulative adenomatous polyps. The patients with MCRA had a mean of 7 colonoscopies available for review but the majority of patients with MCRA required 3 or less procedures to reach this criterion (13/14, 92.9%). The highest MCRA count was 28 adenomas. The MCRA patients had a mean age of 62 and the two most common mutated genes were MSH6 (8/14) and MSH2 (4/14). Of note, 12 (86%) had a history of advanced neoplasia including 5 (36%) with colorectal cancer.
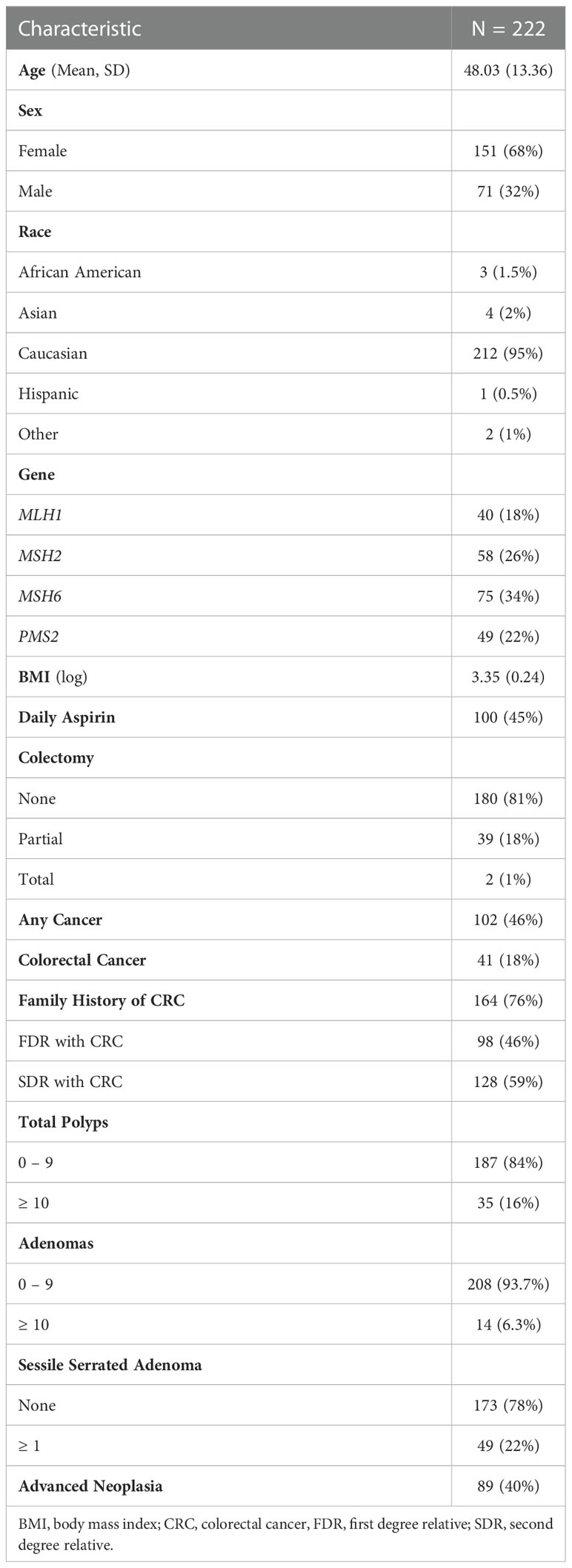
Table 1 Characteristics of patients with Lynch syndrome.
Univariable analysis was performed to compare the cohorts with 0-9 MCRA and ≥ 10 MCRA. Patients with ≥ 10 MCRA were older with a mean age of 62 as compared to 47 in patients with 0-9 MCRA (OR: 1.09, 95% CI: 1.04, 1.15), otherwise the cohorts had similar demographics including similar rates when compared within mismatch repair gene cohorts (Table 2). Patients with ≥ 10 MCRA were significantly more likely to have a history of advanced colorectal neoplasia as compared to patients with only 0-9 MCRA (OR: 10.2, CI: 2.69, 66.7). Notably, this remained true on multivariable analysis adjusted for age (OR: 5.35, CI: 1.34, 35.88). Patients with ≥ 10 MCRA were also more likely to have a personal history of malignancy (OR: 3.12, CI: 1.01, 11.7), although there was not a detected significant difference in a history of colorectal cancer.
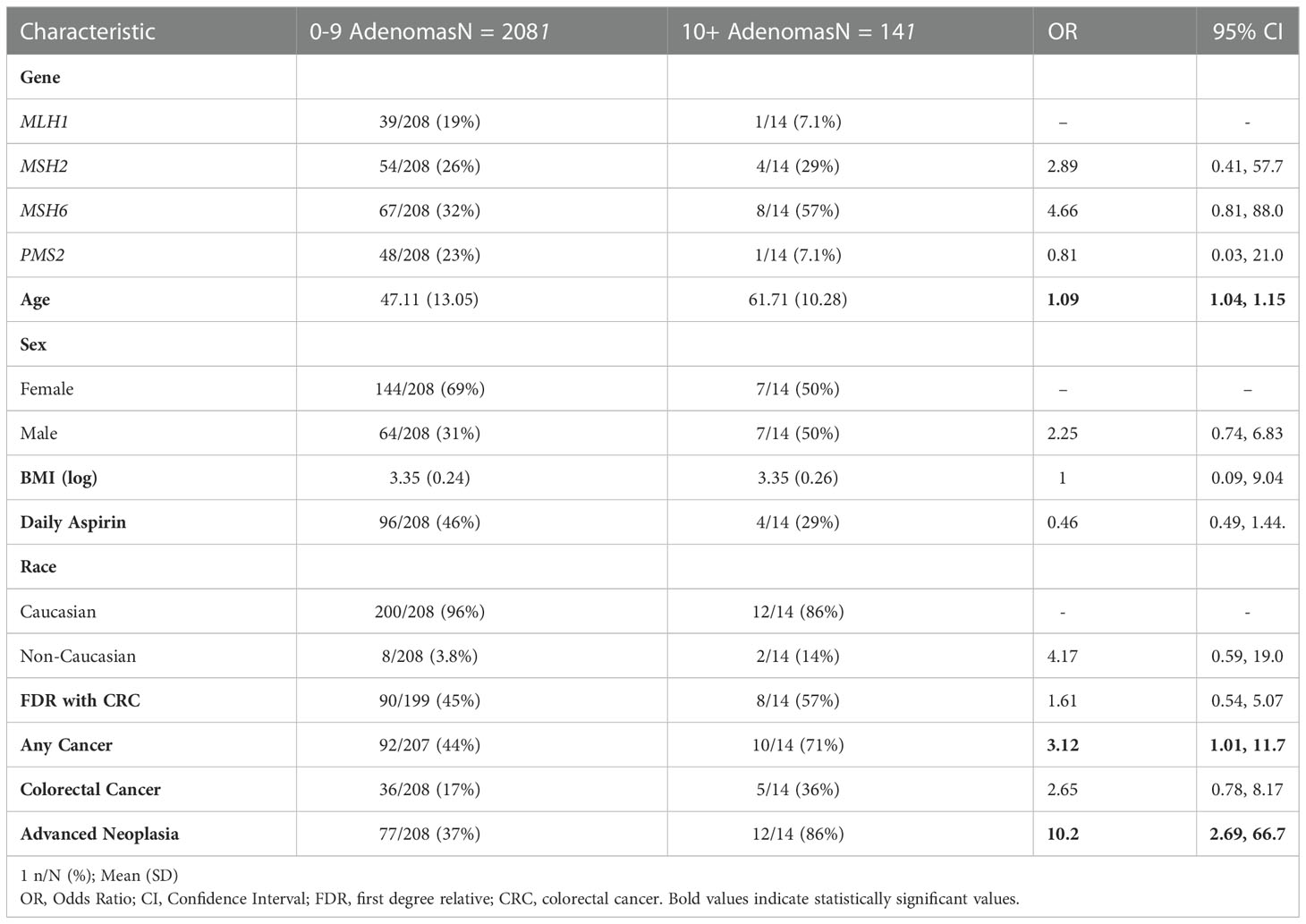
Table 2 Univariable analysis of patients with adenomatous oligopolyposis.
Lynch syndrome has traditionally been defined as a non-polyposis syndrome and differentiated from polyposis syndromes through a lower adenoma burden (4). In this study, we found that 6% of patients with Lynch syndrome had 10 or more cumulative adenomatous polyps and met MCRA criteria. This is similar to rates of MCRA seen in the general population in a recent study by Sullivan et al. (12). Although this is still a minority of patients with Lynch syndrome, these findings are notable as it is likely that the prevalence of colorectal adenomas in Lynch syndrome will continue to rise in conjunction with increases in adenoma detection rates in the general population. Given this, the mismatch repair genes should be included in multigene panel testing conducted for patients with MCRA as reflected in recent guidelines (13).
Our analysis also revealed that 86% of patients with Lynch syndrome and MCRA had a history of advanced neoplasia and that this was significantly higher than Lynch syndrome patients without polyposis even with adjustment for age. Similarly, Sullivan et al. found that patients without known inherited colorectal syndromes with ≥ 10 MCRA were 17 times more likely to have a history of advanced neoplasia (12). Together, these results indicate that MCRA is a high-risk phenotype for advanced neoplasia independent of genotype and should be taken into consideration to guide individualized recommendations for colorectal cancer surveillance.
A previous international cohort study presented evidence of the existence of unknown familial risk factors that result in wide variations in the risk of colorectal cancer across patients with Lynch syndrome (14). We propose that an MCRA phenotype may be a risk factor for advanced neoplasia and malignancy in patients with Lynch syndrome and should be given consideration when determining surveillance colonoscopy intervals. As guidelines are shifting toward longer colonoscopy intervals, we should consider that patients with a polyposis phenotype independent of Lynch syndrome genotype are likely high risk for advanced neoplasia and malignancy and may benefit from continued close monitoring.
Limitations of our study include the risk of ascertainment bias due to its use of genetic testing results as an inclusion criterion. Additionally, the majority of patients were Caucasian which may limit generalizability across diverse populations. It should also be noted that the majority of our patients carried MSH6 and PMS2 variants and thus may not be representative of all Lynch syndrome cohorts. In addition, the use of aspirin may have impacted advanced neoplasia in our patient population and could have a potential confounding effect. Large multi-center and prospective studies are needed to confirm the risks of polyposis in Lynch syndrome and to assess optimal colonoscopy intervals for these patients.
In summary, MCRA phenotype is not unusual in Lynch syndrome and was associated with a significant increase in history of advanced colon neoplasia. Given this, consideration should be given to individualizing colonoscopy intervals based on the presence of polyposis in Lynch syndrome.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ohio State Institutional Review Board. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
AJ and PS: conception and design, data collection, drafting of the article, critical revision of the article for important intellectual content, analysis of data, and final approval of the article. MA: data collection, analysis of the data, critical revision of the article for important intellectual content and final approval. JM and JP: Data analysis and final approval. HH, RP, and MK: critical revision of the article for important intellectual content and final approval. All authors contributed to the article and approved the submitted version.
Guarantor of the article: PS.
PS receives research support from Emtora Biosciences, Janssen Pharmaceuticals Inc., Pfizer Inc. and the PTEN Research Foundation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol (2015) 110(2):223–62. doi: 10.1038/ajg.2014.435
2. Espenschied CR, LaDuca H, Li S, McFarland R, Gau C-L, Hampel H, et al. Multigene panel testing provides a new perspective on lynch syndrome. J Clin Oncol (2017) 35(22):2568–75. doi: 10.1200/JCO.2016.71.9260
3. Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev (2017) 26(3):404–12. doi: 10.1158/1055-9965.EPI-16-0693
4. Lindgren G, Liljegren A, Jaramillo E, Rubio C, Lindblom A. Adenoma prevalence and cancer risk in familial non-polyposis colorectal cancer. Gut (2002) 50(2):228–34. doi: 10.1136/gut.50.2.228
5. Idos G, Valle L. Lynch syndrome. In: Adam MP, et al, editors. GeneReviews(®). Seattle (WA (1993). University of Washington, Seattle Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle, Seattle (WA). All rights reserved.
6. Edelstein DL, Axilbund J, Baxter M, Hylind LM, Romans K, Griffin CA, et al. Rapid development of colorectal neoplasia in patients with lynch syndrome. Clin Gastroenterol Hepatol (2011) 9(4):340–3. doi: 10.1016/j.cgh.2010.10.033
7. Kalady MF, Kravochuck SE, Heald B, Burke CA, Church JM. Defining the adenoma burden in lynch syndrome. Dis Colon Rectum (2015) 58(4):388–92. doi: 10.1097/DCR.0000000000000333
8. Shaukat A, Holub J, Pike IM, Pochapin M, Greenwald D, Schmitt C, et al. Benchmarking adenoma detection rates for colonoscopy: Results from a US-based registry. Am J Gastroenterol (2021) 116(9):1946–9. doi: 10.14309/ajg.0000000000001358
9. Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, et al. Guidelines for the management of hereditary colorectal cancer from the British society of gastroenterology (BSG)/Association of coloproctology of great Britain and Ireland (ACPGBI)/United kingdom cancer genetics group (UKCGG). Gut (2020) 69(3):411–44. doi: 10.1136/gutjnl-2019-319915
10. Weiss JM, Gupta S, Burke CA, Axell L, Chen L-M, Chung DC, et al. NCCN guidelines® insights: Genetic/Familial high-risk assessment: Colorectal, version 1.2021. J Natl Compr Canc Netw (2021) 19(10):1122–32. doi: 10.1164/jnccn.2021.0048
11. Seppälä TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sánchez-Guillén L, et al. European Guidelines from the EHTG and ESCP for lynch syndrome: an updated third edition of the mallorca guidelines based on gene and gender. Br J Surg (2021) 108(5):484–98. doi: 10.1002/bjs.11902
12. Sullivan BA, Redding TS 4th, Qin X, Gellad ZF, Hauser ER, O'Leary C M, et al. Ten or more cumulative lifetime adenomas are associated with increased risk for advanced neoplasia and colorectal cancer. Dig Dis Sci (2021) 6:2526–34. doi: 10.1007/s10620-021-07069-0
13. Heald B, Hampel H, Church J, Dudley B, Hall MJ, Mork ME, et al. Collaborative group of the americas on inherited gastrointestinal cancer position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam Cancer (2020) 19(3):223–39. doi: 10.1007/s10689-020-00170-9
Keywords: lynch syndrome, adenomas, colon cancer, multiple adenomas, Lynch
Citation: Jain A, Alimirah M, Hampel H, Pearlman R, Ma J, Peng J, Kalady MF and Stanich PP (2022) Multiple colorectal adenomas in Lynch syndrome. Front. Oncol. 12:1038678. doi: 10.3389/fonc.2022.1038678
Received: 07 September 2022; Accepted: 30 November 2022;
Published: 20 December 2022.
Edited by:
Toni T. Seppälä, Helsinki University Central Hospital, FinlandReviewed by:
Dimitri Krizzuk, Aurelia Hospital, ItalyCopyright © 2022 Jain, Alimirah, Hampel, Pearlman, Ma, Peng, Kalady and Stanich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter P. Stanich, cGV0ZXIuc3RhbmljaEBvc3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.