 Yuan Tian1†
Yuan Tian1† Jing Zhou1†
Jing Zhou1† Juanli Qiao1Zhaojun Liu1Liankun Gu1
Juanli Qiao1Zhaojun Liu1Liankun Gu1 Baozhen Zhang1Youyong Lu2
Baozhen Zhang1Youyong Lu2 Rui Xing2*
Rui Xing2* Dajun Deng1*
Dajun Deng1*- 1Key Laboratory of Carcinogenesis and Translational Research (MOE/Beijing), Division of Etiology, Peking University Cancer Hospital and Institute, Beijing, China
- 2Key Laboratory of Carcinogenesis and Translational Research (MOE/Beijing), Division of Tumor Biology, Peking University Cancer Hospital and Institute, Beijing, China
Background: A feasible method to detect somatic copy number deletion (SCND) of genes is still absent to date.
Methods: Interstitial base-resolution deletion/fusion coordinates for CDKN2A were extracted from published articles and our whole genome sequencing (WGS) datasets. The copy number of the CDKN2A gene was measured with a quantitative multiplex PCR assay P16-Light and confirmed with whole genome sequencing (WGS).
Results: Estimated common deletion regions (CDRs) were observed in many tumor suppressor genes, such as ATM, CDKN2A, FAT1, miR31HG, PTEN, and RB1, in the SNP array-based COSMIC datasets. A 5.1 kb base-resolution CDR could be identified in >90% of cancer samples with CDKN2A deletion by sequencing. The CDKN2A CDR covers exon-2, which is essential for P16INK4A and P14ARF synthesis. Using the true CDKN2A CDR as a PCR target, a quantitative multiplex PCR assay P16-Light was programmed to detect CDKN2A gene copy number. P16-Light was further confirmed with WGS as the gold standard among cancer tissue samples from 139 patients.
Conclusion: The 5.1 kb CDKN2A CDR was found in >90% of cancers containing CDKN2A deletion. The CDKN2A CDR was used as a potential target for developing the P16-Light assay to detect CDKN2A SCND and amplification for routine clinical practices.
Background
Somatic copy number variations (SCNVs) of tumor-related genes are landmarks of human cancers (1, 2). Somatic copy number deletion (SCND) and amplification are two kinds of well-known SCNVs. However, current gene copy number detection methods, including microsatellite instability (MSI), loss/gain of heterozygosity (LOH/GOH), fluorescence-in situ hybridization (FISH), whole genome sequencing (WGS) or whole exome sequencing (WES), are not sensitive enough or too costly for routine clinical use. While the amplification of oncogenes (such as EGFR, c-ERBB2, c-MYC, and c-MET) is increasingly driving decision-making for precise cancer treatments, clinical applications of SCND of tumor suppressor genes, including CDKN2A, are still rare owing to the lack of a feasible detection assay.
The frequency of CDKN2A SCND detected by single nucleotide polymorphism (SNP) microarray, WGS or WES was found to range from 30% to 60% in bladder cancer, melanoma, head and neck cancer, pleural mesothelioma, glioblastoma, and esophageal squamous cell cancer (ESCC), with an average frequency of 13% in pan-cancer datasets in The Cancer Genome Atlas (TCGA) (Figure S1A) (2–6). CDKN2A deep deletion is associated with downregulation of CDKN2A gene expression, while CDKN2A amplification is associated with upregulation of CDKN2A gene expression in Pan-TCGA cancers (Figure S1B). It is well known that genetic CDKN2A inactivation contributes to malignant transformation, cancer metastasis, and therapeutic sensitivity of cancers to drugs, including CDK4/6 inhibitors and their combination with PD-1 blockade (7–11). Recently, it was reported that CDKN2A copy number deletion often accompanied with deletion of a type I interferon gene cluster. Codeletion of the interferon cluster promoted immune evasion, metastasis and immunotherapy resistance of pancreatic cancer in mice (12). Therefore, a convenient and sensitive assay to detect CDKN2A SCND is eagerly awaited.
In the present study, we characterized patterns of estimated genomic coordinates for SCNDs in a set of tumor suppressor genes using the public Catalogue Of Somatic Mutations In Cancer (COSMIC) SCNV datasets and found common deletion regions (CDRs) in many frequently deleted genes. Then, we further defined a 5.1 kb base-resolution CDR within the CDKN2A gene using sequencing data for the first time. A sensitive P16-Light assay targeting the CDKN2A CDR was established for clinical practice.
Materials and methods
COSMIC and TCGA SCNV datasets
SNP6 array-based estimated genomic coordinates of interstitial copy number deletion/fusion of the CDKN2A gene in cancer cell lines (n=273) with homozygous CDKN2A deletion and estimated genomic coordinates of deep-deleted fragments of CDKN2A, PTEN, RB1, and other frequently deleted genes in cancer tissues were downloaded from the Copy Number Analysis (CONA) datasets in the COSMIC project (Data Files 1-11) (13).
Patients, tissues, and DNA preparation
Frozen fresh gastric carcinoma (GC) and paired surgical margin (SM) tissue samples were collected from 156 patients in the WGS study (14). These samples were frozen in liquid nitrogen approximately 30 min after surgical dissection and then stored in a -80°C freezer for 2-5 yrs. Clinicopathological information was also obtained. The 2010 UICC tumor-node-metastasis (TNM) system was used to classify these GCs (15). Genomic DNA was extracted from these samples with a phenol/chloroform method coupled with RNase treatment. Concentrations of these DNA samples were determined with NanoVue Plus (Biochrom LTD, Cambridge, UK). DNA samples with OD260nm/OD280nm ratios ranging from 1.7 to 1.9 were used for the detection of gene copy number as described below.
Optimized quantitative multiplex PCR assay (P16-Light) to detect CDKN2A copy number
Multiplex primer and probe combinations were designed based on the best multiplex primer probe scores for conserved sequences within the CDR in the CDKN2A (HGNC: 1787) and GAPDH (HGNC: 4141) gene sequences by Bacon Designer 8 software. Multiplex PCR assays were established according to the Applied Biosystems (ABI) TaqMan universal PCR master mix manual. The performance of these assays for the detection of CDKN2A copy numbers was compared with each other. Finally, a multiplex primer and probe combination targeting CDKN2A intron-2 was selected (Table 1), and the concentrations of the components were optimized. Each multiplex PCR assay was carried out in a total volume of 20 μL that included 5-10 ng of input DNA, 10 μM of forward and reverse primers and probe for CDKN2A intron-2, 10 μM forward and reverse primers and probe for GAPDH, and 10 μL of 2x TaqMan Universal Master Mix II with uracil-N-glycosylase (Kit-4440038, ABI, Lithuania). The PCRs were performed in triplicate in a MicroAmp Fast Optical 96-Well Reaction Plate with a barcode (0.1 mL; ABI, China) with an ABI 7500 Fast Real-Time PCR System. The specific conditions of the PCR were as follows: initial incubation for 10 min at 95°C, followed by 40 cycles of 95°C for 20 sec and 58°C for 60 sec. When the Ct value for GAPDH input for a sample was 34 or fewer cycles, this sample was considered CDKN2A SCNV informative. The specificity of the PCR was monitored through running the gel. Distilled water was used as a no-template control for each experiment.

Table 1 Oligo sequences.
Definitions of CDKN2A CDR deletion positivity and amplification positivity
We used genomic DNA from A549 cells containing no CDKN2A allele to dilute genomic DNA from RKO cells containing 2 wild-type CDKN2A alleles, and then we set the standard curve according to the relative copy number of the CDKN2A gene at different dilutions. The ΔCt value and relative copy number for the CDKN2A gene were calculated using the GAPDH gene as the internal reference. When the CDKN2A copy number in the A549-diluted template was consistently lower than that in the RKO control template and the difference was statistically significant (t test, p<0.05), it was judged that the lowest dilution concentration was the detection limit of CDKN2A deletion (the difference in CDKN2A copy number between the 100% RKO template and 80% RKO template spiked with 20% A549 DNA). When the CDKN2A relative copy number in a tissue sample was significantly lower or higher than that of the paired SM sample, the sample was defined as somatic CDKN2A CDR deletion-positive or amplification-positive, respectively. For each experiment, the 100% A549, 100% RKO, and 20% A549 + 80% RKO DNA mix controls were analyzed.
Quantitative detection of CDKN2A/P14ARF exon-1β copy number by PCR assay (P14-qPCR)
The copy number of CDKN2A exon-1β was detected using the primer and probe set (Table 1) as previously reported (16). When the relative copy number of CDKN2A exon-1β in a tissue sample was significantly lower or higher than that of the paired SM sample, the sample was defined as somatic CDKN2A/P14ARF deletion-positive or amplification-positive, respectively.
Call for CDKN2A interstitial deletion/fusion and calculate the purity of cancer cells in the GC WGS datasets
We used Meerkat to predict somatic SVs and their breakpoints in WGS datasets (accession numbers, EGAD00001004811 with 36× of sequencing depth) for gastric adenocarcinoma samples from 168 patients using the suggested parameters (14). This method used soft-clipped and split reads to identify candidate breakpoints, and precise breakpoints were refined by local alignments. CDKN2A deletion information of 157 GC samples was obtained from WGS datasets. We also estimated copy number profiling over 10 kb windows with Patchwork 28 and calculated the ratio of standardized average depth between normal tissue and tumor tissue (log2R ratio). The purity and ploidy of each tumor were calculated using ABSOLUTE software (17).
Cell lines and cultures
The CDKN2A allele homozygously deleted cell line A549 (kindly provided by Dr. Zhiqian Zhang of Peking University Cancer Hospital and Institute) was grown in RPMI-1640 medium, and the RKO cell line containing two wild-type CDKN2A alleles was purchased from American Type Culture Collection and grown in DMEM. The medium was supplemented with 10% (v/v) fetal bovine serum (FBS). These cell lines were tested and authenticated by Beijing JianLian Genes Technology Co., Ltd. before they were used in this study. A Goldeneye 20A STR Identifier PCR Amplification kit was used to analyze the STR patterns.
Statistical analysis
Chi-square or Fisher’s exact tests were used to compare the positive rates of CDKN2A SCND or amplification between different groups of tissue samples. Student’s t test was used to compare the proportion of the CDKN2A gene copy number between genomic DNA samples. All statistical tests were two-sided, and a p value less than 0.05 was considered to be statistically significant.
Results
Prevalence of estimated CDRs within various tumor suppressor genes
It has been previously reported that homozygous deletion of approximately 170 kilobase pairs (kb), including the CDKN2A locus, can be detected in human cancers by MSI analyses (18). SCND inactivates the CDKN2A gene in 273 human cancer cell lines according to the COSMIC dataset (Data File 1). We found that an 8 kb estimated CDKN2A CDR could be detected among these cell lines by ordering “start” genomic coordinates of these breaking points (Figure S2). To investigate the prevalence of CDRs within tumor suppressor genes in human cancer tissues with a high deletion frequency (1, 2), we further downloaded the estimated genomic coordinates for deletion fragments that overlapped with these genes. We found that CDRs could be detected not only within the CDKN2A gene (Figure 1A; approximately 17 kb) but also within the ATM, FAT1, RB1, PTEN, and miR31HG genes (Figure 2; approximately 1232 kb, 50 kb, 12 kb, 33 kb, and 46 kb, respectively) (Data Files 2-7). No CDR could be observed within CCSER1, FHIT, LRP1B, and WWOX genes according to the SNP-array data (Data Files 8-11).
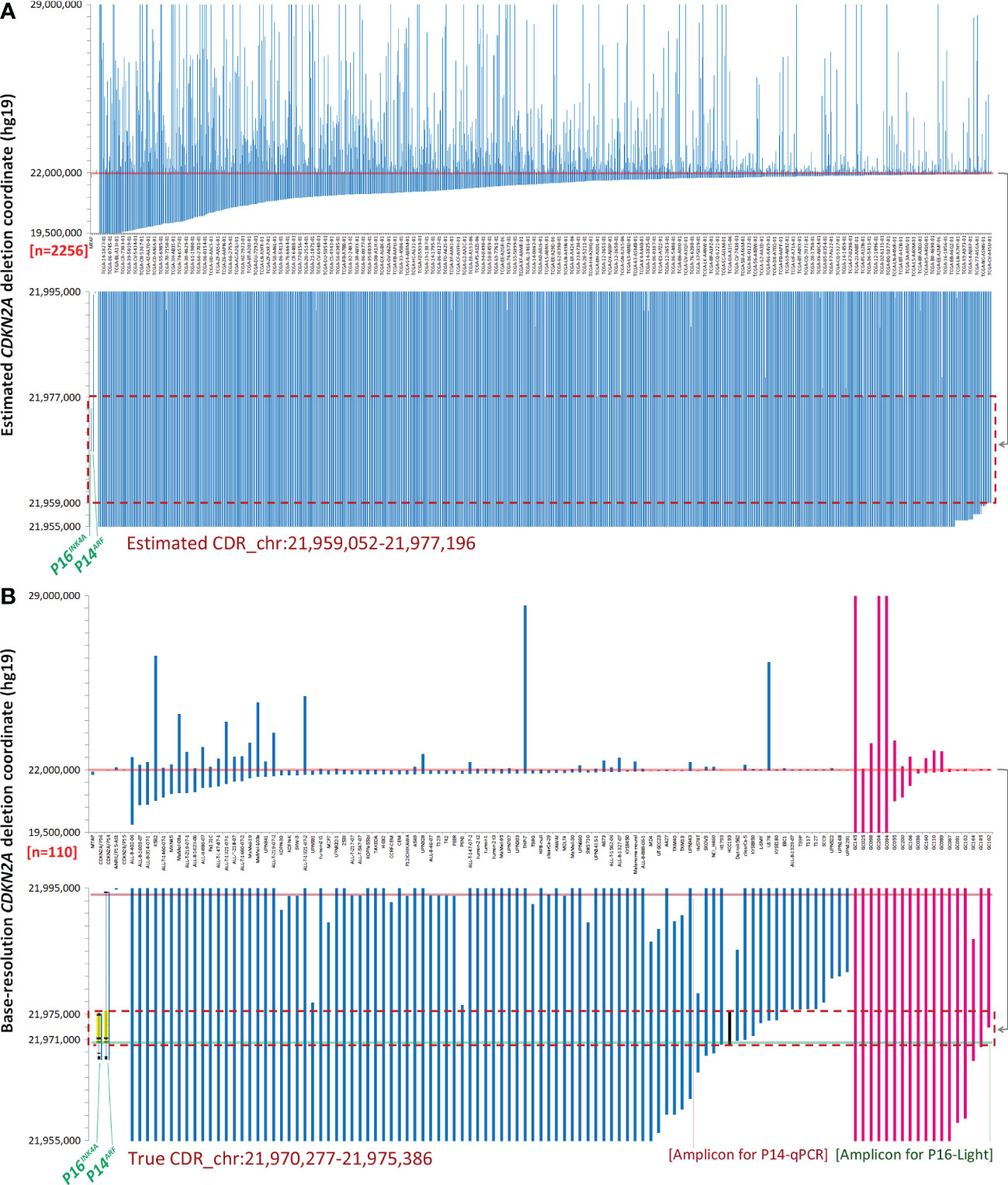
Figure 1 Genomic coordinates of interstitial CDKN2A deletion/fusion in human cancer genomes. (A) Estimated coordinates of CDKN2A deep deletion in cancer tissues according to the COSMIC data. (B) True coordinates at the base resolution of CDKN2A deletion in cancer cell lines (n=92, blue lines) and gastric cancer (n=18, purple lines) according to sequencing data. The two top charts display the coordinates of most deletion fragments. The sample ID is labeled under each column in charts (B) or some columns in charts (A). The two bottom charts display the amplified view of these deletion fragments, where the 17 kb and 5.1 kb common deletion regions (CDRs) are highlighted with a red dashed line rectangle in chart (A)and chart (B). The 5.1 kb true CDR from the P16INK4A promoter to intron-2 is exactly the same region as the deleted CDKN2A fragment in the HCC193 lung cancer cell line (highlighted with a black line). Each line represents a CDKN2A deletion fragment. The locations of P16INK4A and P14ARF (gray shadow) and exon-1α/1β/2/3 (black dots) are also labeled as landmarks. The positions of amplicons for P16-Light and P14-qPCR are illustrated with half-transparent green and red lines, respectively. The detailed deletion coordinates for each sample are listed in Data Files 3, 12.
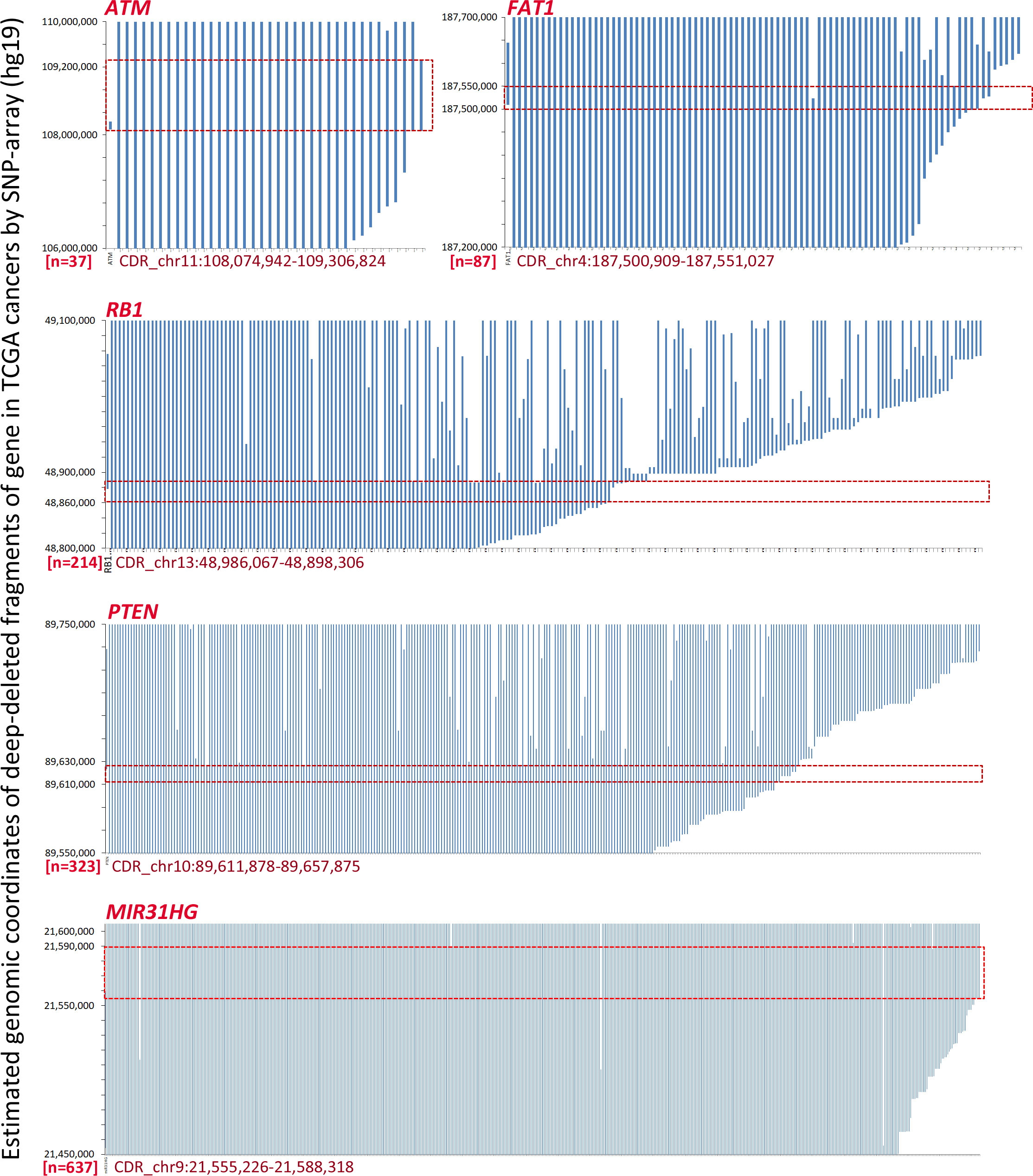
Figure 2 The estimated coordinates of loss of heterozygosity (LOH) and homozygous deletion (HD) fragments overlapped with tumor suppressor genes ATM, FAT1, RB1, PTEN, and miR31HG according to the COSMIC data. The common deletion region (CDR) for each gene is highlighted with a red line rectangle. The detailed deletion coordinates for each sample are listed in Data Files 2, 4–7.
Characterization of a true CDKN2A CDR at base resolution in human cancers
It was reported that the error in CDKN2A breakpoint estimation based on SNP-array data is approximately 10 kb (19). To characterize the true genomic coordinates of CDKN2A deletion fragments in cancers, we extracted base-resolution sequence information of interstitial CDKN2A deletions from available published articles and our sequencing data (Data File 12) (20–29). We found a 5.1 kb CDR (chr9: 21,970,277 - 21,975,386, hg19) that spanned from the P16INK4A promoter to intron-2 in 83 (90%) of 92 reported cancer cell lines or tissue samples containing interstitial CDKN2A deletions (Figure 1B, blue lines). This CDR sequence is the same as the CDKN2A deletion fragment in the HCC193 lung cancer cell line (26). The CDR coordinates were also confirmed in our WGS datasets (average sequencing depth, 36×) of 18 (100%) of 18 GCs (14), in which interstitial CDKN2A deletions/fusions were identified (Figure 1B, purple lines; Data File 12).
It is well known that germline CDKN2A inactivation can lead to a high predisposition for melanoma and pancreatic cancer (30–32). Interestingly, we found that 14 (93.3%) of 15 CDKN2A allelic variants in the Online Mendelian Inheritance in Man (OMIM) database are located within the CDR sequence, especially in CDKN2A exon-2 (Figure S3) (33, 34).
In addition, both P16INK4A and P14ARF mRNAs are transcribed from the human CDKN2A gene at chromosome 9p21 but with different transcription start sites; they share the same exon-2 but have different translation reading frames. Because CDKN2A exon-2 located within the true CDR is the essential exon for coding P16INK4A and P14ARF proteins, the above findings indicate that P16INK4A and P14ARF are coinactivated in 87% (96/110) of human cancer cell lines and tissues containing CDKN2A CDR deletion (Figure 1B).
Establishment of a convenient PCR assay (P16-Light) to detect somatic CDKN2A CDR deletion
The current clinical method FISH for detecting SCND is composed of a set of biotin-labeled probes that should cover at least 50 kb DNA sequences. Thus, FISH is not a suitable method for detecting the copy number deletion of the 5.1 kb CDKN2A CDR. To provide a convenient method for routine clinical use, we designed and experimentally evaluated a set of multiplex quantitative PCR assays and finally optimized the CDKN2A CDR-specific quantitative multiplex PCR assay called P16-Light for detecting the copy number of a 129-bp amplicon within the CDKN2A intron-2 (Figure 3A), which covers 86% (94/110) of known CDKN2A deletion fragments (Figure 1B, green line).
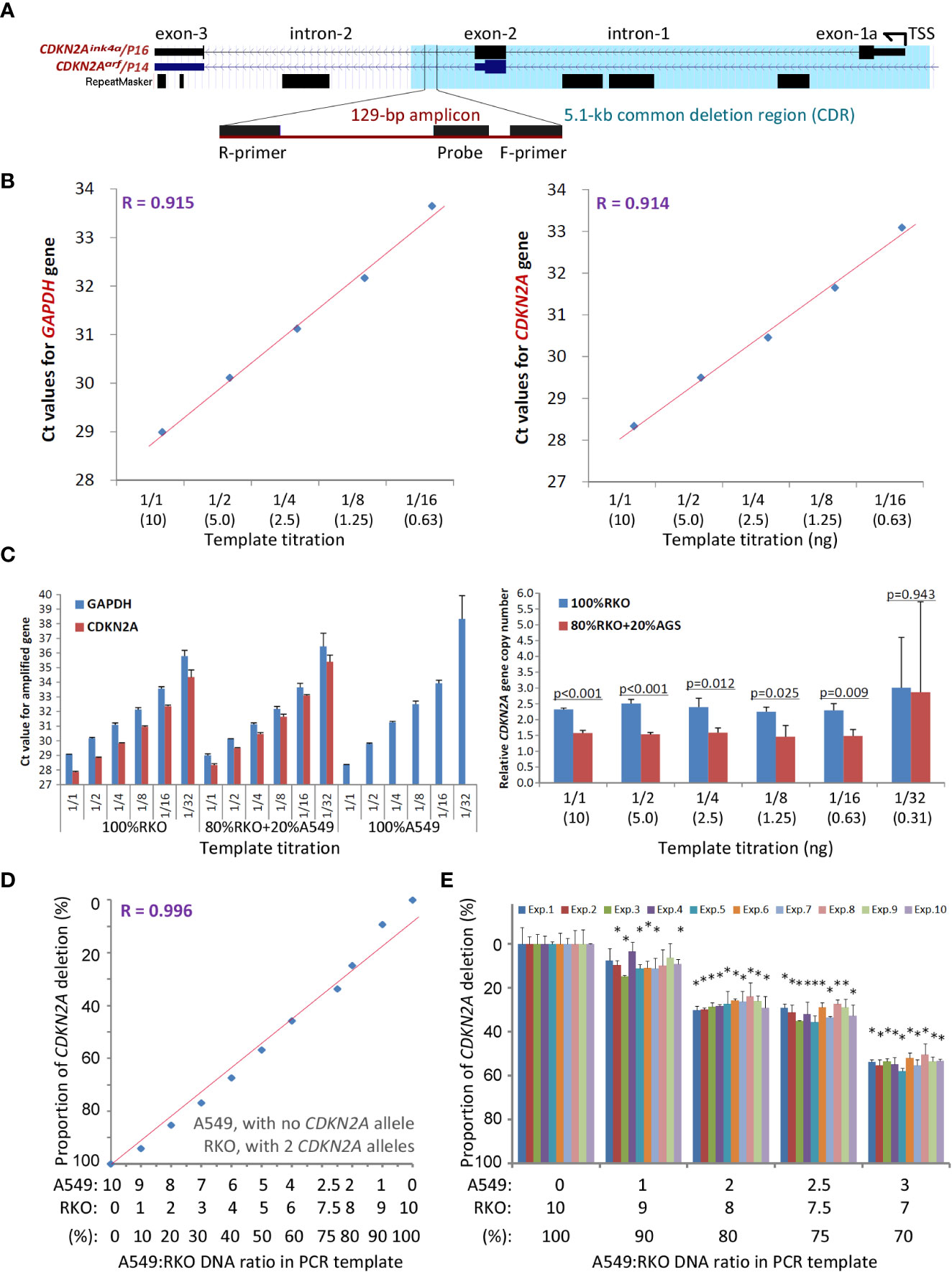
Figure 3 Detection of the copy number of CDKN2A intron-2 with quantitative gene-specific multiplex PCR (P16-Light). (A) The location of the 129-bp amplicon within the common deletion region (CDR) and its host genes. (B) The amplification efficiency of two amplicons for the GAPDH and CDKN2A genes in the template titration assays using standard DNA samples from RKO cells (with two wild-type CDKN2A alleles) and A549 cells (with a homozygous CDKN2A deletion). (C) Effects of the amount of template DNA on the efficiency of PCR amplification for amplicons in the CDKN2A and GAPDH genes (left chart) and detection of the relative CDKN2A gene copy number (right chart). The p-value in Student’s t-test is labelled for each template titration. (D) The linear relationship between the proportion of CDKN2A copy number deletion and ratios of RKO cells spiked with A549 cells. (E) Stability of the proportion of the CDKN2A copy number deletion by P16-Light during ten experiments over different days. The RKO cell DNA templates were spiked with 0, 10%, 20%, 25%, and 30% A549 cell DNA. Each column represents the average proportion of CDKN2A copy number deletions in triplicate. Exp. 1 - 10: the results of 10 repeated experiments performed on different days. When the difference in the proportion of CDKN2A deletion between 100% of RKO DNA and detected concentrations of A549 DNA in each experiment reaches to a statistically significant (p < 0.05) in Student’s t test, the sample containing A549 DNA was marked with a star “*”.
The copy number of the GAPDH gene was used as the internal reference. Genomic DNA from human A549 cells (with homozygous deletion of CDKN2A alleles) and RKO cells (with 2 wild-type CDKN2A alleles) were used as CDKN2A CDR deletion-positive and deletion-negative controls, respectively. The amplification efficiencies of the two amplicons in GAPDH and CDKN2A were very similar (Figure 3B). No template inhibition was observed when the amount of template DNA ranged from 10 to 0.63 ng (Figure 3C). The proportions of CDKN2A CDR copy number were linearly correlated with the ratios (0 - 100%) of RKO cell DNA and A549 cell DNA in the input mixtures (10 ng/reaction) when the A549 DNA was spiked in at different proportions for the P16-Light analyses (Figure 3D). Furthermore, there was a high reproducibility when DNA with homozygous deletion of CDKN2A was present in ≥20% of the cells verified in ten experimental repeats performed on different days (Figure 3E). Thus, when the proportion of CDKN2A copy number was significantly decreased (or increased) in a sample relative to the paired normal control (Student’s t test, p<0.05) in the P16-Light analyses, the sample was defined as CDKN2A SCND-positive (or amplification-positive).
Comparison of P16-Light with WGS datasets
As we described above, information on interstitial copy number deletion/fusion of the CDKN2A gene was extracted from WGS datasets for 156 of 168 GC patients enrolled in a GC genome study (14), and a total of 18 CDKN2A deletion/fusion coordinates at the base resolution were detected in 17 (10.8%) GCs (Data Files 12, 13). To compare the performance of P16-Light with WGS, we analyzed the status of SCNVs, including SCND and amplification, of the CDKN2A gene in 156 of these GCs with enough genomic DNA samples with P16-Light using the paired SM sample as the diploid reference (Data File 13). CDKN2A SCND and amplification were detected in 40 (25.6%) and 34 (21.8%) of these GCs, respectively. The P16-Light analysis was confirmed by the WGS results: the frequency of CDKN2A SCND (or amplification) by P16-Light was significantly higher (or lower) in 17 GCs containing interstitial CDKN2A deletion/fusion than in 139 GCs without interstitial CDKN2A deletion/fusion (chi-square test, p<0.05; Figure 4A). These results also indicate that there is a significantly higher sensitivity for detecting CDKN2A SCND by the quantitative P16-Light assay than the hemi-quantitative WGS.
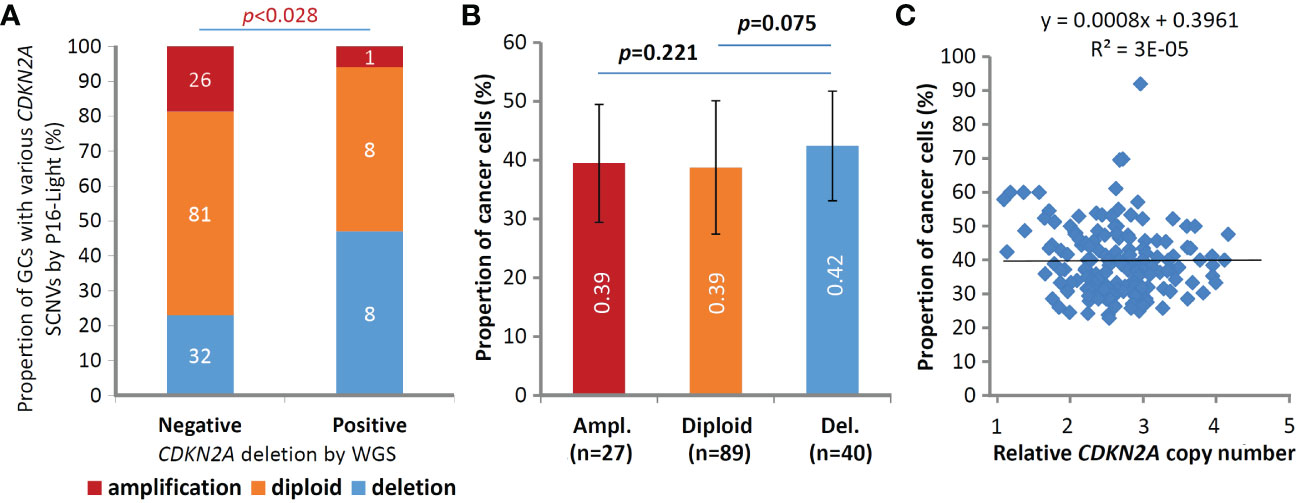
Figure 4 Comparisons of somatic copy number variations (SCNVs) of the CDKN2A gene in gastric carcinoma samples (GCs) from 156 patients in the P16-Light and WGS (30×) analyses. (A) The states of CDKN2A SCNVs by P16-Light (relative to paired surgical margin reference) in GC groups with and without CDKN2A deletion/fusion in the WGS analysis. (B) Comparison of the proportion of cancer cells (or sample purity; by WGS) in GC groups with various CDKN2A SCNVs, including amplification (Ampl.), diploid, and deletion (Del.), by P16-Light. The average proportion of cancer cells in each GC group is labeled. (C) Correlation analysis between the proportion and relative copy number of the CDKN2A gene in GCs.
Moreover, it is well known that the proportion of cancer cells in tissue samples (i.e., sample purity) may affect the detection values of various genome data. To study whether the cancer cell proportion disturbs the detection of CDKN2A SCNVs, we calculated the cancer cell proportion in the above GC samples using WGS data (Data File 13). We found that the difference in sample purity between GC subgroups with different CDKN2A SCNV statuses was not statistically significant (t test, p=0.075; Figure 4B), although the proportion was slightly higher in GCs with CDKN2A SCND than in those without CDKN2A SCND. No correlation was observed between the proportion of cancer cells and the relative copy number of the CDKN2A gene among these GCs (Figure 4C).
Comparison of P16-Light with P14-qPCR assay
The P14-qPCR assay was previously established for detecting the copy number of CDKN2A/P14ARF exon-1β (16). Two amplicons in the P16-Light and P14-qPCR assays cover 98% (108/110) of known CDKN2A deletion fragments (Figure 1B, red and green lines). Therefore, we further compared the performance of P16-Light, P14-qPCR, and their combination using GC and paired SM samples from patients who were recently included in the cross-sectional cohort in our association study (35). GC samples (n=139) with enough genomic DNA were used in P14-qPCR analysis (Data File 14). The SCND-positive rate for P14ARF was similar to that for the CDKN2A CDR (31.7% vs. 36.7%) (Table 2). CDKN2A SCND was found only in 19 GCs by both assays. While CDKN2A CDR SCND by P16-Light was significantly associated with distant metastasis of GC (odds ratio=4.09, p<0.001), no association was observed between GC metastasis and P14ARF SCND by P14-qPCR. Using merged CDKN2A SCND data (CDKN2A CDR SCND-positive and/or P14ARF SCND-positive), only a weaker association was observed. These results suggest that individual P16-Light alone may be good enough for detecting CDKN2A SCND in tissue samples.
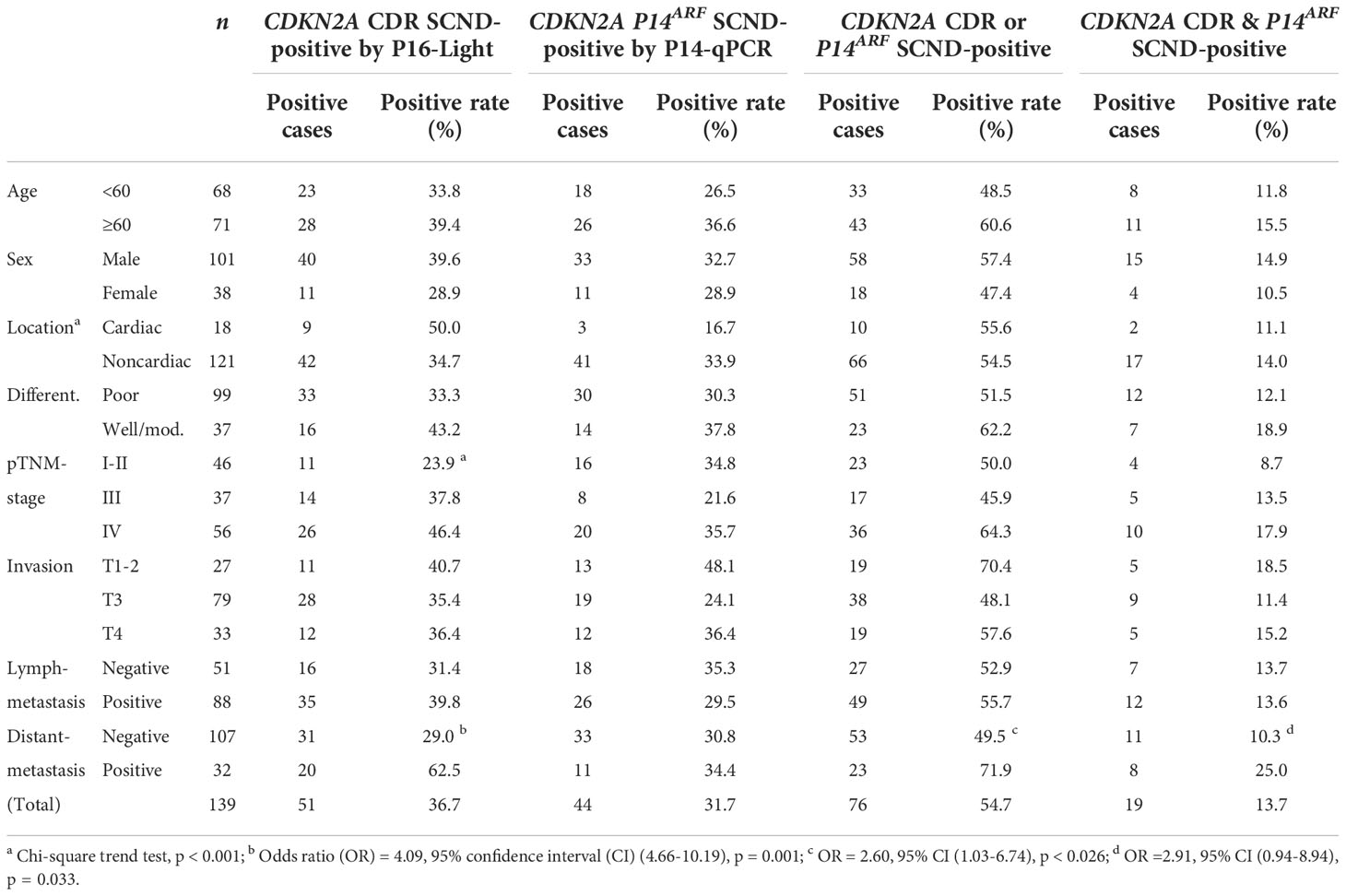
Table 2 Association between clinicopathological characteristics and CDKN2A SCND detected by P16-Light and P14-qPCR.
Discussion
Somatic copy number deletion and amplification are two main kinds of SCNVs. The detection of copy number amplification of oncogenes is routinely used for precise cancer treatments. However, the detection of SCND of tumor suppressor genes is absent, and its significance in clinical practice is not well studied. The reason should be the lack of feasible detection approaches. Here, we report that there are CDRs in many tumor suppressor genes, such as CDKN2A, miR31HG, PTEN, and RB1, which are commonly inactivated by SCND in various human cancers (1, 2). Notably, we characterized, for the first time, the 5.1 kb true CDR from the CDKN2A/P16INK4A promoter to intron-2 in >90% of cancers containing CDKN2A deletion. Using the CDKN2A CDR as a PCR target, we further established a feasible P16-Light assay to detect CDKN2A SCND and amplification. These findings indicate that CDRs are prevalent sequences in tumor suppressor genes, and characterization of the base-resolution genomic coordinates of CDRs could enable us to establish convenient methods for SCND detection of genes.
Interstitial deletion/fusion is the main type of CDKN2A SCND, and the breaking/fusing coordinates for CDKN2A SCNDs in cancer genomes are diverse, which blocks the establishment of a feasible detection assay for CDKN2A SCND, although many efforts have been made (21). In the present study, we initially found the 8~17 kd estimated CDKN2A CDR in both monoclonal cancer cell lines and cell-heterogeneous cancer tissues with CDKN2A copy number deletion according to the SNP-array datasets from COSMIC and TCGA projects (1, 13). Then, we further characterized the 5.1 kb true CDR at the base resolution within the CDKN2A gene in cancer genomes using DNA sequencing data (20–29) and confirmed the CDR using WGS datasets in all 18 GCs containing CDKN2A SCND (14). Because the true CDKN2A CDR was observed in more than 90% of CDKN2A-deleted cancer samples and the P16-Light assay is highly reproducible and convenient, the quantitative P16-Light assay should be considered a viable assay for detecting CDKN2A SCNVs in clinical practice. This is supported by the result that CDKN2A SCND detected by P16-Light was significantly associated with GC metastasis and further supported by the results of our prospective study, in which CDKN2A SCND was closely associated with hematogenous metastasis of GCs (35). In another long-term prospective study, we also found that CDKN2A SCND and amplification by P16-Light were significantly associated with malignant transformation and complete regression of mild or moderate esophageal squamous cell dysplasia, respectively [Fan et al. submitted for publication]. The results of these studies also suggest that the sensitivity of 20% for the P16-Light assay may be good enough for routine clinical use.
WGS is generally used as a kind of golden standard to study structural variations of genomic DNAs, especially for interstitial gene copy deletion/fusions. However, WGS is a cost assay, and its accuracy depends on sequencing depth. WGS at sequencing depth 36× would be considered a hemi-quantitative assay. In our calling of CDKN2A SCND coordinate processes, it was found that 18 CDKN2A SCND coordinates were identified in 17 (10.8%) of 157 GCs, which was consistent with the frequency (11.4% =50/438) of homozygous deletion of CDKN2A in GCs in WES or WGS sequencing datasets (Data File 14) (36). The positive rate (25.6%) of CDKN2A SCND in 156 GCs with enough genomic DNA samples in the P16-Light analysis was more than twice that of WGS. The results of P16-Light analyses were significantly correlated with those of WGS. These phenomena suggest that P16-Light is a much more sensitive, convenient, and less expensive assay than WGS.
P14-qPCR is a method used to detect the copy number of CDKN2A/P14ARF exon-1β (16). Although the combination of P16-Light with P14-qPCR may detect both SCNDs overlapping with the CDKN2A CDR and not overlapping with the CDKN2A CDR, the results of our comparison analysis among 139 GC patients showed that detecting CDKN2A SCND by individual P16-Light may be good enough for clinical practice because combination with P14-qPCR could not improve the performance of P16-Light. However, for other genes, such as RB1 and PTEN, whether a qPCR array needs to be employed for detecting SCNVs should be studied case by case.
Generally, IHC is a popular method used to detect expression changes in protein-coding genes. For example, P16INK4A overexpression in cervical mucosa samples is currently used for rapid HPV infection screening. We compared the status of P16INK4A expression by IHC between GCs with CDKN2A SCND (n=4) and GCs without CDKN2A SCND (n=12) and did not find any difference in the P16INK4A positive-staining rate between these GCs (3/4 vs. 9/12). The expression level of CDKN2A/P16INK4A is not only affected by SCNVs but also regulated by the methylation status of CpG islands, histone modifications, and high-risk HPV infection (37, 38). These factors may partially account for the inconsistency between IHC and P16-Light.
The driver function of the CDKN2A gene in cancer development is enigmatic. P16ink4a inactivation contributes less than P19arf (the murine counterpart of human P14ARF) inactivation to cancer development in mice, while P16INK4A inactivation contributes more than P14ARF inactivation to cancer development in humans (39, 40). The exact mechanisms leading to the difference among species are still unclear. Here, we reported that approximately 87% of genetic P16INK4A inactivation by CDKN2A SCND is accompanied by P14ARF inactivation in human cancer cell lines or tissues. This may account for the species-related functional difference in the CDKN2A gene. The report supports this explanation that knocking out both p16ink4a and p19arf leads to more cancer development than individual inactivation in mice (41). This also may account for the fact that P14ARF exon-1β deletion was not associated with GC metastasis, whereas CDKN2A CDR deletion was significantly associated with GC metastasis, as described above.
In conclusion, we have found estimated CDRs in many tumor suppressor genes in the cancer genome. There is a 5.1 kb CDR region within the CDKN2A gene, and most CDKN2A deletions lead to P16INK4A and P14ARF inactivation in human cancers. Using the CDKN2A CDR as a target sequence, we developed a convenient quantitative multiplex PCR assay, P16-Light, to detect CDKN2A SCNVs for clinical practice, suggesting that the strategy to detect CDKN2A SCNVs may be suitable for the establishment of SCNV detection methods for other tumor suppressor genes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
This study was approved by the Institution Review Board of Peking University Cancer Hospital & Institute and carried out in accordance with the principles outlined in the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Author contributions
YT: Methodology, writing- original draft preparation. JZ: Methodology, formal analysis, writing- original draft preparation. JQ: Investigation. ZL: Data curation. LG: Investigation. BZ: Investigation. YL: Supervision, resources. RX: Conceptualization, resources, data curation, validation, writing- original draft preparation. DD: Conceptualization, supervision, funding acquisition, methodology, data curation, visualization, writing- Original draft preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Beijing Natural Science Foundation (grant number 7181002 to DD), Beijing Capital’s Funds for Health Improvement and Research (grant number 2018-1-1021 to DD), and National Natural Science Foundation of China (grant number 91640108 to DD). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
We thank Dr. Sanford Dawsey at NCI, NIH, Bethesda, Maryland for critical comments on the manuscript. We also thank Miss Gina Mckeown in New York, USA for English language editing.
Conflict of interest
DD, YT, JZ, and ZL are the creators for the pending patent “A quantitative method for detection of human CDKN2A gene copy number using a primer set and their applications” PCT/CN2019/087172; WO2020228009.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1038380/full#supplementary-material
References
1. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi: 10.1038/nature08822
2. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
3. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol (2011) 12:R41. doi: 10.1186/gb-2011-12-4-r41
4. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:pl1. doi: 10.1126/scisignal.2004088
5. Network CGAR, University AWGA, Agency BC, Hospital BaWs, Institute B, University B, et al. Integrated genomic characterization of oesophageal carcinoma. Nature (2017) 541:169–75. doi: 10.1038/nature20805
6. Cui Y, Chen H, Xi R, Cui H, Zhao Y, Xu E, et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res (2020) 30:902–13. doi: 10.1038/s41422-020-0333-6
7. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho R. Role of the INK4a locus in tumor suppression and cell mortality. Cell (1996) 85:27–37. doi: 10.1016/S0092-8674(00)81079-X
8. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discovery (2018) 8:216–33. doi: 10.1158/2159-8290.CD-17-0915
9. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell (2018) 175:984–997.e24. doi: 10.1016/j.cell.2018.09.006
10. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin d-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature (2018) 553:91–5. doi: 10.1038/nature25015
11. Yu J, Yan J, Guo Q, Chi Z, Tang B, Zheng B, et al. Genetic aberrations in the CDK4 pathway are associated with innate resistance to PD-1 blockade in Chinese patients with non-cutaneous melanoma. Clin Cancer Res (2019) 25:6511–23. doi: 10.1158/1078-0432.CCR-19-0475
12. Barriga FM, Tsanov KM, Ho Y-J, Sohall N, Zhang A, Baslan T, et al. MACHETE identifies interferon-encompassing chromosome 9p21.3 deletions as mediators of immune evasion and metastasis. Nat Cancer (2022). doi: 10.1038/s43018-022-00443-5
13. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucl Acids Res (2019) 47:D941–7. doi: 10.1093/nar/gky1015
14. Xing R, Zhou Y, Yu J, Yu Y, Nie Y, Luo W, et al. Whole-genome sequencing reveals novel tandem-duplication hotspots and a prognostic mutational signature in gastric cancer. Nat Commun (2019) 10:2037. doi: 10.1038/s41467-019-09644-6
15. Sobin L, Cummins R, Leader M, Kay E, et al. TNM classification of malignant tumours. 7th edn. New York, USA: International Union Against Cancer (UICC). Wiley Press (2009).
16. Berggren P, Kumar R, Sakano S, Hemminki L, Wada T, Steineck G, et al. Detecting homozygous deletions in the CDKN2A(p16INK4a)/ARF(p14ARF) gene in urinary bladder cancer using real-time quantitative PCR. Clin Cancer Res (2003) 9:235–42.
17. Carter SL, Cibulskis K, Helman E, Mckenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol (2012) 30:413–21. doi: 10.1038/nbt.2203
18. Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet (1995) 11:210–2. doi: 10.1038/ng1095-210
19. Novara F, Beri S, Bernardo ME, Bellazzi R, Malovini A, Ciccone R, et al. Different molecular mechanisms causing 9p21 deletions in acute lymphoblastic leukemia of childhood. Hum Genet (2009) 126:511–20. doi: 10.1007/s00439-009-0689-7
20. Xie H, Rachakonda PS, Heidenreich B, Nagore E, Sucker A, Hemminki K, et al. Mapping of deletion breakpoints at the CDKN2A locus in melanoma: detection of MTAP-ANRIL fusion transcripts. Oncotarget (2016) 7:16490–504. doi: 10.18632/oncotarget.7503
21. Patel A, Schwab R, Liu YT, Bafna V. Amplification and thrifty single-molecule sequencing of recurrent somatic structural variations. Genome Res (2014) 24:318–28. doi: 10.1101/gr.161497.113
22. Norris AL, Kamiyama H, Makohon-Moore A, Pallavajjala A, Morsberger LA, Lee K, et al. Transflip mutations produce deletions in pancreatic cancer. Genes Chromosomes Cancer (2015) 54:472–81. doi: 10.1002/gcc.22258
23. Guney S, Bertrand P, Jardin F, Ruminy P, Kerckaert JP, Tilly H, et al. Molecular characterization of 9p21 deletions shows a minimal common deleted region removing CDKN2A exon 1 and CDKN2B exon 2 in diffuse large b-cell lymphomas. Genes Chromosomes Cancer (2011) 50:715–25. doi: 10.1002/gcc.20893
24. Florl AR, Schulz WA. Peculiar structure and location of 9p21 homozygous deletion breakpoints in human cancer cells. Genes Chromosomes Cancer (2003) 37:141–8. doi: 10.1002/gcc.10192
25. Kitagawa Y, Inoue K, Sasaki S, Hayashi Y, Matsuo Y, Lieber MR, et al. Prevalent involvement of illegitimate V(D)J recombination in chromosome 9p21 deletions in lymphoid leukemia. J Biol Chem (2002) 277:46289–97. doi: 10.1074/jbc.M208353200
26. Sasaki S, Kitagawa Y, Sekido Y, Minna JD, Kuwano H, Yokota J, et al. Molecular processes of chromosome 9p21 deletions in human cancers. Oncogene (2003) 22:3792–8. doi: 10.1038/sj.onc.1206589
27. Cayuela JM, Gardie B, Sigaux F. Disruption of the multiple tumor suppressor gene MTS1/p16(INK4a)/CDKN2 by illegitimate V(D)J recombinase activity in T-cell acute lymphoblastic leukemias. Blood (1997) 90:3720–6. doi: 10.1182/blood.V90.9.3720
28. Pasmant E, Laurendeau I, Héron D, Vidaud M, Vidaud D, Bièche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res (2007) 67:3963–9. doi: 10.1158/0008-5472.CAN-06-2004
29. Raschke S, Balz V, Efferth T, Schulz WA, Florl AR, et al. Homozygous deletions of CDKN2A caused by alternative mechanisms in various human cancer cell lines. Genes Chromosomes Cancer (2005) 42:58–67. doi: 10.1002/gcc.20119
30. Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 mutations in familial melanoma. Nat Genet (1994) 8:15–21. doi: 10.1038/ng0994-15
31. Freedberg DE, Rigas SH, Russak J, Gai W, Kaplow M, Osman I, et al. Frequent p16-independent inactivation of p14ARF in human melanoma. J Natl Cancer Inst (2008) 100:784–95. doi: 10.1093/jnci/djn157
32. Harinck F, Kluijt I, van der Stoep N, Oldenburg RA, Wagner A, Aalfs CM, et al. Indication for CDKN2A-mutation analysis in familial pancreatic cancer families without melanomas. J Med Genet (2012) 49:362–5. doi: 10.1136/jmedgenet-2011-100563
33. Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucl Acids Res (2005) 33:D514–7. doi: 10.1093/nar/gki033
34. Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's online mendelian inheritance in man (OMIM). Nucl Acids Res (2009) 37:D793–6. doi: 10.1093/nar/gkn665
35. Qiao JL, Tian Y, Cheng XJ, Liu ZJ, Zhou J, Gu LK, et al. CDKN2A deletion leading to hematogenous metastasis of human gastric carcinoma. Front Oncol (2021) 11:801219. doi: 10.3389/fonc.2021.801219
36. Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell (2018) 33:721–735.e8. doi: 10.1016/j.ccell.2018.03.010
37. Peters G. Tumor suppression for ARFicionados: the relative contributions of p16INK4a and p14ARF in melanoma. J Natl Cancer Inst (2008) 100:757–9. doi: 10.1093/jnci/djn156
38. Cui CH, Gan Y, Gu L, Wilson J, Liu Z, Zhang B, et al. P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol (2015) 16:252. doi: 10.1186/s13059-015-0819-6
39. Li Q, Wang XH, Lu ZM, Zhang BZ, Guan ZP, Liu ZJ, et al. Polycomb CBX7 directly controls trimethylation of histone H3 at lysine 9 at the p16 locus. PloS One (2010) 5:e13732. doi: 10.1371/journal.pone.0013732
40. Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature (2009) 460:1136–9. doi: 10.1038/nature08290
Keywords: CDKN2A, somatic copy number deletion, common deletion region, multiplex PCR, whole genome sequencing, gastric carcinoma
Citation: Tian Y, Zhou J, Qiao J, Liu Z, Gu L, Zhang B, Lu Y, Xing R and Deng D (2022) Detection of somatic copy number deletion of the CDKN2A gene by quantitative multiplex PCR for clinical practice. Front. Oncol. 12:1038380. doi: 10.3389/fonc.2022.1038380
Received: 07 September 2022; Accepted: 17 November 2022;
Published: 02 December 2022.
Edited by:
Shuai Cheng Li, City University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Fei Yuan, Baylor College of Medicine, United StatesPan Zhang, Sanford Burnham Prebys Medical Discovery Institute, United States
Copyright © 2022 Tian, Zhou, Qiao, Liu, Gu, Zhang, Lu, Xing and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Xing, eGluZ3J1aUBiam11LmVkdS5jbg==; Dajun Deng, ZGVuZ2RhanVuQGJqbXUuZWR1LmNu
†These authors have contributed equally to this work