
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 04 January 2023
Sec. Breast Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1037531
This article is part of the Research TopicWomen in Breast Cancer: 2022, volume IIView all 18 articles
 Zsuzsanna Nagy1,2,3*
Zsuzsanna Nagy1,2,3* Rinath Jeselsohn1,2,3,4*
Rinath Jeselsohn1,2,3,4*Breast cancer is the most frequent female malignant tumor, and the leading cause of cancer death in women worldwide. The most common subtype of breast cancer is hormone receptor positive that expresses the estrogen receptor (ER). Targeting ER with endocrine therapy (ET) is the current standard of care for ER positive (ER+) breast cancer, reducing mortality by up to 40% in early- stage disease. However, resistance to ET represents a major clinical challenge for ER+ breast cancer patients leading to disease recurrence or progression of metastatic disease. Salient drivers of ET resistance are missense mutations in the ER gene (ESR1) leading to constitutive transcriptional activity and reduced ET sensitivity. These mutations are particularly prominent and deleterious in metastatic breast cancer (MBC). In addition to activating ESR1 point mutations, emerging evidence imposes that chromosomal translocation involving the ESR1 gene can also drive ET resistance through the formation of chimeric transcription factors with constitutive transcriptional activity. Although these ESR1 gene fusions are relatively rare, they are enriched in ET resistant metastatic disease. This review discusses the characteristics of ER fusion proteins and their association with clinical outcomes in more aggressive and metastatic breast cancer. The structure and classification of ER fusion proteins based on function and clinical significance are also addressed. Finally, this review summarizes the metastatic phenotypes exhibited by the ER fusion proteins and their role in intrinsic ET resistance.
Despite significant advances in breast cancer screening and treatment, mortality rates remain high with nearly 2.3 million new cases diagnosed and more than 650 000 patients dying each year worldwide according to the World Health Organization (1). The most common breast cancer subtype is hormone receptor positive, expressing the ER and/or progesterone receptor, accounting for approximately 75% of breast cancers (2). ER is a nuclear transcription factor that drives breast cancer development and growth. ER is comprised of four domains (3), an N-terminal activation function-1 (AF-1), a central DNA binding domain followed by a hinge region and the C-terminal ligand binding domain (LBD) that contains the ligand- dependent activation function (AF-2). Following estrogen binding, ER dimerizes and translocate to the nucleus where it binds to DNA at estrogen response elements (ERE) to regulate the transcription of multiple genes involved in tumor progression (3). ER functions as part of a transcriptional complex including (1) other transcription factors, such as Activator Protein 1 (AP1), Transcription Factor SP1 (SP1), Nuclear Factor-κB (NF-κB) and E2F Transcription Factor 1 (E2F1) (4–8); (2) co-factors that regulate chromatin structure, such as Nuclear Receptor Coactivator 1 (SRC-1), Nuclear Receptor Coactivator 2 (TIF2), Glutamate Receptor Interacting Protein 1 (GRIP-1), Amplified in Breast Cancer 1 (AIB1), CREB binding protein (CBP), p300 and the p300/CBP-associated factor (pCAF) (9–16); (3) pioneer factors that modulate ER binding to chromatin, such as Forkhead Box A1 (FOXA1), GATA Binding Protein 3 (GATA3), Pre-B-cell Leukemia Transcription Factor 1 (PBX1), and transducin-like enhancer protein 1 (TLE1) (17–23). ET is the mainstay treatment in ER+ breast cancer (24), and these treatment options include selective estrogen receptor modulators (SERMs), aromatase inhibitors (AI), selective estrogen receptor degraders (SERDs), and selective estrogen receptor covalent antagonists (SERCAs). SERMs, such as tamoxifen antagonize ER by reducing co-factor binding (25). AIs block the conversion of testosterone to estrogen and SERDs, such as fulvestrant, competitively bind ER and lead to proteasomal degradation (25). SERCA H3B-5942 inactivates ER by targeting Cys530 to enforce a distinct antagonist conformation (26, 27). Combination of ET with inhibitors for cyclin dependent kinase 4/6 (CDK4/6), mammalian target of rapamycin (mTOR), or phosphatidylinositol-4,5- biphosphate 3-kinase catalytic subunit alpha (PIK3CA) is integral part of the treatment in metastatic ER+ breast cancer and represent major improvements in progression free survival (28).
While ET reduces mortality by up to 40% in early-stage disease and highly effective in controlling metastatic disease, therapeutic resistance remains a momentous clinical issue (29, 30). At most, 20% of resistant cases lose ER expression (31) and in many patients the ER transcriptional axis remains active, however, in an altered fashion. To date, multiple mechanisms of acquired resistance to ET have been investigated and identified. These include (1) altered expression of transcription factors and co-regulatory proteins (e.g. SP1, AP1, NF-κB, SRC-1, AIB1, FOXA1) (18, 32–37), (2) modification of ER by miRNAs (e.g. miR-148, miR-152 and miR-221/222) (38–41), (3) increased crosstalk between ER/HER2/SRC3 (42), (4) amplification of tyrosine kinase receptors (e.g. fibroblast growth factor receptor 1 and 2 or insulin-like growth factor receptor 1) (43–46), (5) aberrant expression of cell cycle proteins (e.g. c-Myc, p21 and p27) (47–52) and (6) immune system-dependent resistance regulated via the NF-κB pathway (53) or chemokines activated PI3K/Akt/mTOR signaling (54, 55). Additional mechanism of ET resistance is the acquisition of somatic mutations in ESR1 that are present in up to 50% of MBC patients (56–61). Widely studied examples are point mutations in the LBD (Y537S and D538G) that confer ER constitutive activity and exhibit decreased ET sensitivity (57, 59).
Studies have shown that chimeric proteins are powerful drivers of cancer with tremendous clinical impact (62). Larotrectinib, the first pan-cancer drug against the NTRK gene fusions demonstrated rapid responses in both adult and pediatric cancer patients (63–68). Driven by deep transcriptomic sequencing studies, several pathological gene fusions have been identified in aggressive (luminal B, basal like, or endocrine resistant breast cancer) breast cancers (69–72). These include fusion proteins associated with ER such as ESR1-CCDC170 (73–75), and ESR1-YAP1 (76) and non ER related fusions such as CTNNBL1-RAF1, ACTL6A-PIK3CA, S6KCI-AKT3 (71), SEC16A-NOTCH1 (77), SEC22B-NOTCH2 (72), and ETV6-NTRK3 (78). A number of these fusions promote tumor growth, and patients expressing these fusion proteins have more rapid disease progression and shorter survival than fusion-negative patients (70, 71, 75, 79). Identifying the full spectrum of the ESR1 gene fusions and characterizing their role in intrinsic ET resistance is critical for developing novel and effective targeted therapies.
RNA-seq analysis conducted by Veeraraghavan and colleagues on 990 primary TCGA breast samples identified the first ESR1 gene fusion, ESR1-e2>CCDC170 (Table 1), in a subset (2.1%) of Luminal B breast tumor samples (75, 82). This fusion is formed by tandem-duplication, it retains the first two non-coding exons of ESR1 (ESR1-e2) connected to various sequences from the coiled-coil domain containing 170 (CCDC170) gene (Figure 1). The promoter trap drives aberrant expression of CCDC170 and produces N-terminal truncated forms of the CCDC170 protein (ΔCCDC170) (75). The authors also provided functional evidence that this fusion promotes more aggressive oncogenic phenotypes in ER+ breast cancer cells, such as increased cell migration, invasion, and reduced tamoxifen sensitivity. Utilizing RNA sequencing, Li and colleagues (76) described the first inter-chromosomal ESR1 fusion in a patient-derived xenograft from ET resistant MBC (Luminal A subtype, skin metastasis). This fusion is formed by a translocation event that brought ESR1 exons 1 to 6 (ESR1-e6) on chromosome (chr) 6q into the yes associated protein 1 gene YAP1 locus on chr11q (ESR1- e6>YAP1; Table 1), replacing the LBD of ESR1 with the transactivation domain (TAD) sequence from YAP1 (Figure 1). Although Li and colleagues conducted limited functional studies, overexpression of ESR1-e6>YAP1 in ER+ breast cancer cells conferred estradiol-independent growth in their study (Table 1). Lei and colleagues (80) and Gou and colleagues (81) provided additional mechanistic data for the ESR1-e6>YAP1 fusion and described its functional properties in driving estrogen-independent growth, constitutive expression of ER target genes, and anti-estrogen resistance. Several years later, two additional ESR1 fusions, ESR1-e2 fusion with the acidic residue methyltransferase 1 gene, C6orf211/ARMT1 (ESR1-e2>C6orf211/ARMT1) and ESR1-e6 fusion with a-kinase anchoring protein 12 gene, AKAP12 (ESR1-e6>AKAP12) were identified in AI resistant breast cancer by Giltnane and colleagues (Table 1) (82) with no functional data available (Table 1). Using whole genome sequencing, Robinson and colleagues (83) identified three additional ESR1 fusions (Table 1) including fusionsaryl hydrocarbon receptor nuclear translator 2 gene, ARNT2 (ESR1-e6>ARNT2-e18); protein-L-isoaspartate O-methyltransferase gene, PCMT1 (ESR1-e6>PCMT1); AT-rich interaction domain 1B gene, ARID1B (ESR1-e6>ARID1B), but the functional properties of these fusions were investigated only later on by Gou and colleagues (81). Hartmaier and colleagues also described the expression of ESR1-e6>AKAP12 in ER+ MBC and identified several novel ESR1 fusions (84). The authors also established that ESR1 fusion proteins are enriched in ER+ MBC (Table 1) and contribute to ET resistance. Hartmaier and colleagues described eight novel ESR1 fusion proteins (Table 1), all with junctions between ESR1 exon 6 and 7. ESR1-e6>fusions included DAB adaptor protein 2 gene, DAB2 (ESR1-e6>DAB2); glycogenin-1 gene, GYG (ESR1-e6>GYG1); SRY-box transcription factor 9 gene, SOX9 (ESR1-e6>SOX9); pleckstrin homology and RhoGEF domain containing G1, PLEKHG1 (ESR1-e6>PLEKHG1); trafficking from ER to Golgi regulator, TFG (ESR1-e6>TFG); and sodium/potassium transporting ATPase interacting 2, NKAIN2 (ESR1-e6>NKAIN2). ESR1-e7>fusions included mitochondrial isozyme of C1-tetrahydrofolate (THF) synthase, MTHFD1L (ESR1-e7>MTHFD1L) and cyclin dependent kinase 13, CDK13 (ESR1-e7>CDK13). The authors emphasized that many genetic rearrangement events are not expressed or translated into functional protein products, therefore they utilized an array of techniques (DNA and/or RNA sequencing, PCR and immunoblot) to investigate fusion protein expression (Table 1). As example, ESR1-e6>DAB2 and ESR1-e6>GYG1 were detected by DNA and/or RNA sequencing, and immunoblot, and in vitro studies established that these fusions were stable and active. The authors were able to detect ESR1-e6>SOX9, ESR1-e7>MTHFD1L, ESR1-e6>PLEKHG1, ESR1-e6>NKAIN2, ESR1-e6>AKAP12, and ESR1-e7>CDK13 only by DNA sequencing with low confidence in producing fusion transcripts. Utilizing anchored multiplex PCR, Matissek and colleagues (71) identified an additional ESR1 fusion protein (Table 1) with junction between ESR1 exon 4 and 3’ fusion partner cytochrome C oxidase assembly factor 5, COA5 (ESR1-e4>COA5). The role of this fusion in MBC and ET resistance is currently unknown. Lei and colleagues (80) conducted a comprehensive study and identified several novel ESR1 fusions (Table 1). The protocadherin 11 X-linked fusion (ESR1-e6>PCDH11X) was identified from a male patient with ER+ MBC. Inter-chromosomal ESR1 translocations included the nucleolar protein 2 homolog gene, NOP2 (ESR1-e6>NOP2), and aldo-keto reductase family 1 member D1 (ESR1-e6>AKR1D1). Fusion with DNA polymerase eta gene, POLH (ESR1-e7>POLH) was formed by intra-chromosomal translocation. In this study, the authors also identified the ESR1-e4>CCDC170 and ESR1-e5>CCDC170 fusions. Lei and colleagues conducted functional studies and provided further evidence that ESR1-e6>YAP1 and ESR1-e6>PCDH11X, identified from ER+ MBC, encoded stable and functional fusion proteins and promoted estrogen-independent growth, induced cellular motility, constitutive expression of ER target genes, and anti-estrogen resistance. Seven additional ESR1-e6>fusions (Table 1) were identified by Priestley and colleagues in ER+ MBC, including aqryl hydrocarbon receptor nuclear translocator 2, ARNT2 (ESR1-e6>ARNT2); LIM domain containing preferred translocation partner in lipoma, LPP (ESR1-e6>LPP); nuclear receptor coactivator 1, NCOA1 (ESR1-e6>NCOA1); transcription factor 12, TFC12 (ESR1-e6>TCF12); clathrin interactor 1, CLINT1 (ESR1-e6>CLINT1); glutamate receptor interacting protein 1, GRIP1 (ESR1-e6>GRIP1) and trinucleotide repeat containing adaptor 6B, TNRC6B (ESR1-e6>TNRC6B). Functional characterization of these fusions were investigated by Gou and colleagues (81, 85) (Table 1). Except ESR1-e6>TCF12, all ESR1-e6>fusions promoted estrogen-independent growth. It is noteworthy that up to date, only few ESR1 fusions (ESR1-e2>CCDC170, ESR1-e4>CCDC170, ESR1-e5>CCDC170, ESR1-e6>NOP2, ESR1-e6>AKR1D1, ESR1-e6>POLH) were detected in primary breast cancer samples (Table 1).
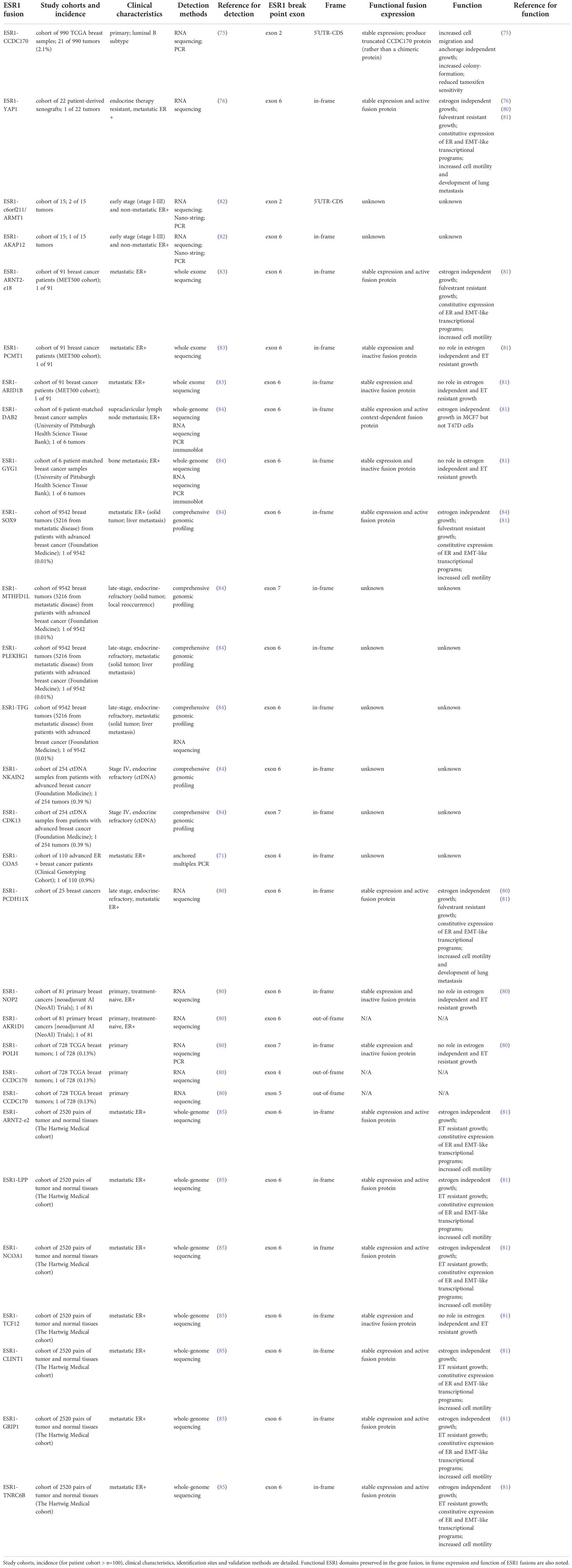
Table 1 Summary of ESR1 gene fusions identified in breast cancers.
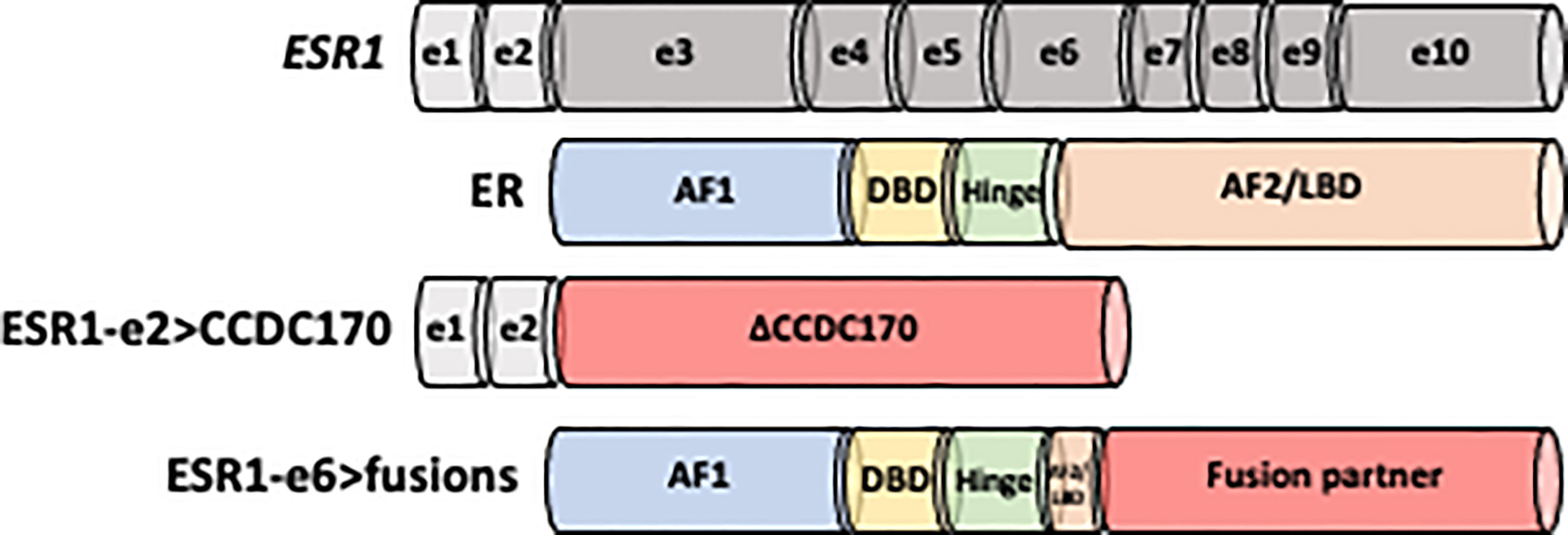
Figure 1 Schematic structure of ESR1-e2>CCDC170 and ESR1-e6>fusion proteins. Non-coding exons (e) 1 and 2 are shown as white boxes, while encoding domains in the ESR1 codon structure are presented in gray. ER is comprised of four domains: N-terminal activation function-1 (AF-1), DNA binding domain (DBD), hinge region and C-terminal ligand binding domain (AF-2/LBD). ESR1-e2>CCDC170 fusion proteins retain the first two non-coding exons of ESR1 (ESR1-e2) and link to the coiled-coil domain containing 170 (CCDC170) gene generating truncated CCDC170 proteins (ΔCCDC170). ESR1-e6>fusions preserve the first 6 exons of ESR1 (ESR1-e6), while replace the LBD by a 3’ fusion partner.
The ESR1-e2>CCDC170 fusion protein consists of the 5′ untranslated region of ESR1 to the coding region of CCDC170, generating N-terminally truncated CCDC170 proteins (ΔCCDC170) expressed under the ESR1 promoter (Figure 1) (75, 82). Structural studies have revealed that this structure is distinct from the ESR1-e6>fusions identified from ET resistant MBC. Despite the diversity among the ESR1-e6>fusions, they share a common structure whereby the first 6 exons of ESR1 (ESR1-e6) are preserved, retaining the hormone-independent transactivation domain (TAD) as well as the DNA-binding domain of ER whereas the LBD is lost and replaced with a functional domain of the 3’ fusion partner (Figure 1) (76, 81, 84). This structure is strongly associated with estrogen independent growth and ET resistant metastatic ER+ breast tumors. The loss of a functional LBD suggests a clear pathological impact, leading to complete resistance to the activity of current ER antagonists, which all bind to the LBD. As expected, several ESR1-e6>fusions (ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18) remained stably expressed in the presence of fulvestrant and promoted ET-resistant growth of T-47D and MCF7 cells (80, 81). In contrast, the expression of ER mutant constructs that lack the LBD had decreased transcriptional activity, suggesting that the presence of the 3’ partner is essential for the ER fusion activity (80, 81, 84). The fact that multiple different 3’ partners have the same effect and drive ET resistance and malignant phenotypes, indicates that the enhanced activity of the ER fusions is not dependent on a specific 3’ partners. These findings suggest that the 3’ partner may be important for the stability of ER and possibly the dimerization of ER, however, an intact LBD is not required for ER activity in the context of the ER fusions.
It is important to note that not all ESR1-e6>fusions produce stable proteins with clear transcription factor (TF) or co-activator (CoA) functions, and only a subset of the ESR1-e6>fusions are activating fusions. The number of studies investigating the activity of ESR1-e6>fusions is limited, the function of some fusions are still unknown. Further studies are required to investigate and fully validate the stability and activity of ESR1-e6>fusions. Some ESR1-e6>fusions such as ESR1-e6>YAP1, ESR1-e6>SOX9, ESR1- e6>ARNT2, ESR1-e6>LPP, ESR1-e6>NCOA1, ESR1-e6->PCDH11X, ESR1-e6>CLINT1, ESR1-e6>GRIP1 and ESR1-e6>TNRC6B produce stable and active fusion proteins that are positive regulators of transcription (80, 81). ESR1-e6->DAB2 has cell type specific transcriptional activity- active in MCF7 but not T47D cells. In contrast to transcriptionally active ESR1-e6>fusions, multiple ESR1- e6>fusions (e.g. ESR1-e6>TCF12, ESR1-e6>ARID1B, ESR1-e6>NOP2) were identified as transcriptionally inactive despite producing stable fusion protein, adding to the complex landscape of ESR1- e6>fusion proteins. None of the 3’ partner genes of these latter ESR1-e6>fusions are known to be a TF or CoA and the wild-type protein is not nuclear-localized (80, 81). As hypothesized by Gou and colleagues (81), these transcriptionally inactive ESR1-e6>fusions could (1) encode tumor suppressors, (2) be active exclusively in the presence of a specific set of coactivators, or (3) act as dominant negative regulators interrupting the function of the remaining intact wild-type protein activity.
When first described, ESR1-e2>CCDC170 in ER+ breast cancer cells led to enhanced growth and reduced sensitivity to tamoxifen (75) suggesting a role for ESR1-e2>CCDC170 in ET resistance. Additional pre-clinical studies (74, 75, 79) showed that the expression of ESR1-e2>CCDC170 fusions in ER-positive breast cancer cells resulted in increased cell migration, increased colony formation, and increased cell proliferation as evidenced by the increase in the number of cells in S-G2/M phase. Li and colleagues (74) provided detailed evidence supporting the function of ESR1-e2>CCDC170 in promoting breast cancer cell survival and endocrine resistance both in vitro and in xenograft models. Their mechanistic study suggests that ESR1-e2>CCDC170 fusions bind and stabilize the HER2/HER3/SRC complex and enhance the activation of SRC/PI3K/AKT signaling during ET in vitro and in vivo. This study also suggested a potential strategy to manage ESR1-e2>CCDC170 positive patients by combining the HER2 inhibitor lapatinib and/or SRC inhibitor dasatinib with ET.
A series of publications clearly demonstrated that ESR1-e6>fusions were identified from ER+ MBC patients and most ESR1-e6>fusion proteins are drivers of ET resistance (69, 76, 80, 81, 84). Functional properties of these ESR1-e6>fusions include estradiol-independent growth and constitutive expression of ER target genes leading to ET-resistant proliferation and epithelial-mesenchymal transition (EMT) genes facilitating metastasis. The latest and most comprehensive study by Gou and colleagues (81) functionally screened multiple ESR1-e6>fusions and 4 were found to promote estradiol-independent cell growth, migration, EMT and resistance to fulvestrant. The ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9, and ESR1-e6>ARNT2-e18 fusions promoted cell proliferation and migration in a hormone-independent and fulvestrant-resistant manner in multiple ER+ cell models. Although the four other ESR1-e6>fusions included in this study (ESR1-e6>DAB2, ESR1-e6>GYG1, ESR1-e6>PCMT1, and ESR1-e6>ARID1B) produced stable proteins, they did not promote ET-resistant growth. Moreover, RNA-seq showed that ER-positive breast cancer cells expressing ESR1-e6>YAP1, ESR1-e6>PCDH11X, ESR1-e6>SOX9 and ESR1-e6>ARNT2-e18 fusions upregulated the same cluster of ER target genes that were observed in the control cells stimulated by estradiol and drove constitutive expression of these ER target genes in the absence of estrogen. Pathway analysis also revealed that these transcriptionally active ESR1-e6>fusion proteins upregulated two EMT-related genes, SNAI1 (Snail) and VCAN (versican). The other ESR1-e6>fusions (ESR1-e6>GYG1, ESR1-e6>PCMT1, and ESR1-e6>ARID1B) did not induce estradiol-independent activation of ER and EMT target genes, despite the fact that they translocated to the nucleus. Additional functional studies showed the transcriptionally active ESR1-e6>YAP1 and ESR1-e6>PCDH11X fusions induced cell motility in vitro and promoted metastasis to the lung in cell-line xenograft models as well as in a PDX model harboring the ESR1-YAP1 fusion.
Next generation sequencing (NGS) methods such as DNA-based comprehensive whole genome (WGS) or RNA-based transcriptome (WTS) sequencing have been extensively used to describe gene fusions in multiple cancer types (86–90). WGS detects gene fusions based on hybrid-capture methods and still considered the most unbiased approach to identify fusion events, especially in large gene panels. WGS is highly sensitive and can be used on fresh, snap frozen and formalin fixed specimens. Still and all, WGS does not indicate the expression of the gene fusions, and the detection of fusion variants involving large DNA intronic regions is poor (86, 91). WTS have the overall advantage of detecting transcriptionally expressed gene fusions and the sequencing is not affected by intronic regions. Moreover, WTS does not require a priori knowledge of gene fusion partners, can distinguish splicing isoforms, quantify fusion transcripts, and it requires low input material. WTS approaches can be based on hybrid-capture or amplicon-based methods using classical or anchor multiplex PCR (86, 92–94). Matissek and colleagues reported anchored multiplex PCR (AMP) as an effective approach to identify gene fusions in cancer, including ER+ metastatic breast cancer (71). AMP was also validated in their study and applied to cohorts of (1) 110 of early-stage and advanced ER+ breast cancer patients (Clinical Genotyping Cohort) and (2) 63 of advanced ER+ breast cancer patients with matched primary and metastatic samples (Matched Primary/Metastasis Cohort). 14 patients in the Clinical Genotyping Cohort harbored intergenic exon–exon fusions, including the in-frame fusion of ESR1 to CCDC170. The authors emphasized that the identified fusion junction sequences involved at least one precise exon boundary. 10 of 63 patients in the Matched Primary/Metastasis Cohort harbored gene fusions in either the primary or metastatic samples. Collectively, AMP detected fusions in 24 of 173 breast cancer patients (14%) in this study, including 11 primary tumors. Combined with complementary “break-apart” fluorescence in situ hybridization (FISH) analysis, they further validated AMP as detection technique for clinically relevant fusions. As example, break-apart of ESR1 was present in primary tumors and metastases from a patient whose tumors demonstrated the ESR1-e4>COA5 fusion upon AMP analysis. Additionally, the ESR1-e2>CCDC170 fusion detected by AMP was also confirmed upon FISH analysis for ESR1. A disadvantage of WTS is that it only identifies expressed fusion genes and not adequate for gene fusion analysis at DNA level. Moreover, biological material is often short, resulting in poor quality RNA and false positive sequencing results. For multiplex PCR approach, the primer design and PCR bias like allele dropout can also impact analysis result (86, 92–94).
ESR1-e6>fusion genes have been detected by NGS methods (Table 1), techniques that are not yet employed routinely in the clinic. Analysis of plasma circulating tumor DNA to detect ESR1 point mutations by droplet digital PCR (ddPCR) is now done in the clinic and clinical trials are investigating the use of these assays in real-time (95–97). Obtaining circulating DNA from liquid biopsies conserves the genomic landscape of the tumor suggesting that this less invasive detection methods may efficiently identify ESR1 fusions, particularly in metastatic ET resistant ER+ breast cancer. Indeed, Hartmaier and colleagues (84) used this approach to provide additional evidence of ESR1 fusion recurrence following extensive ER-targeted endocrine therapies. They obtained target capture sequencing data and examined a cohort of 9542 solid breast tumors and a cohort of 254 ctDNA samples from patients with advanced breast cancer. They successfully identified the ESR1-e6>SOX9, ESR1-e7>MTHFD1L, ESR1-e6>PLEKHG1, and ESR1-e6>TFG fusions (Table 1) in four solid tumors and the ESR1-e6>NKAIN2, ESR1-e6>AKAP12, and ESR1-e7>CDK13 (Table 1) fusions in 3 ctDNA samples. While there are several commercial platforms available and the cost of ctDNA assay is acceptable in the clinic, the concentration of ctDNA in plasma correlates with tumor size and stage, thus this assay is likely to be useful for late-stage breast cancer patients only (98).
As aforementioned, several ESR1-e6>fusion proteins are inactive and therefore not clinically actionable. As a potential efficient approach for screening samples for the presence of ESR1 fusions that drive ET failure in MBC, Gou and colleagues (81) developed a 24-gene expression signature that is specific for the presence of transcriptionally active ESR1 fusion proteins. Specifically, they identified 24 Hallmark genes, including 19 genes in the estrogen response gene set (CHST8, MAPT, OLFM1, PDZK1, RASGRP1, MPPED2, GREB1, MYB, GFRA1, PGR, ELOVL2, ADCY1, NPY1R, TFF1, ACOX2, SGK1, STC2, CALCR and KRT13), two genes in the EMT gene set (VCAN and COL3A1), and three genes shared in both gene sets (CXCL12, GJA1 and TGM2). To compare the transcriptional profile of ESR1-e6>fusions with known activating ESR1 LBD point mutations (Y537S and D538G), Gou and colleagues performed RNA-sequencing on T47D cells that overexpressed either several ESR1 fusion proteins (ESR1-e6>ARNT2-e2, ESR1-e6>LPP, ESR1-e6>NCOA1, ESR1-e6>CLINT1, ESR1-e6>TNRC6B and ESR1-e6>GRIP1), or the Y537S and D538G point mutations. ESR1-e6>ARNT2-e2, ESR1-e6>LPP, ESR1-e6>NCOA1, ESR1-e6>CLINT1 and ESR1-e6>TNRC6B demonstrated elevated expression of this gene signature with expression levels comparable to the Y537S and D538G point mutants. Since the LBD point mutants and translocated ESR1 fusions activate a similar pathogenic transcriptional pattern, the gene signature was named “MOTERA” for Mutant or Translocated Estrogen Receptor Alpha. This signature was examined in 20 ER+ patient-derived xenografts and in 55 ER+ MBC samples and successfully identified cases harboring ESR1 fusions.
Gou and colleagues further confirmed the overlap in the transcriptional properties of ESR1-e6>fusions and ESR1 LBD point mutants in several PDX models and MBC cases. ET-resistant PDXs harboring LBD point mutations (e.g. BCM15100, WHIM20, WHIM40, and HCI013 for ESR1-Y537S; WHIM37 and WHIM43 for ESR1-D538G) highly expressed the MOTERA signature, similar to the PDX naturally expressing the ESR1-e6>YAP1 (WHIM18) fusion. The expression levels of the MOTERA genes were not affected by E2 supplementation in the ESR1-e6>YAP1 expressing PDX or PDXs harboring the LBD point mutations. Furthermore, the MOTERA scores of PDXs expressing WT ESR1 was significantly lower than those of expressing the LBD mutations or the ESR1-e6>YAP1 fusion. Similar to the PDX models, MOTERA gene expression was significantly elevated in MBC tumors harboring the Y537S and D538G point mutations or the ESR1-e6>ARNT2-e18 fusion, and the signature score distinguished the LBD point mutations and the ESR1-e6>ARNT2-e18 fusion from WT ESR1.
In addition to the mechanistic studies, evaluating the potential of targeting these fusion proteins for the development of new targeted therapies is critical. Due to the formation and unique structure of ESR1-e6>fusions, all known ET options that target the LBD are ineffective. Lei and colleagues (80) targeted ER signaling regulated by ESR1 fusions by using Palbociclib, a CDK4/6 inhibitor for MBC. ESR1-e6>YAP1 and ESR1-e6>PCDH11X induced cell proliferation was sensitive to a CDK4/6 inhibition, and a PDX naturally expressing the ESR1-e6>YAP1 fusion was also responsive to Palbociclib treatment. Since ESR1 fusion driven growth of ER-positive breast cancers remained sensitive to CDK4/6 inhibition, the presence of an ESR1-e6>fusion could be a putative biomarker to stratify patients for CDK4/6 inhibitor therapy after resistance to endocrine treatment or continued CDK4/6 inhibitor therapy with a second targeted therapy after resistance to first line treatment for metastatic disease with endocrine therapy in combination with a CDK4/6 inhibitor. It is also hypothesized that CDK4/6 inhibition could be beneficial for patients with ESR1 fusions. Further mechanistic and preclinical studies are expected to introduce additional ESR1 fusions sensitive to CDK4/6 inhibitor therapy and expand on the data from patients harboring ESR1 fusions to examine their CDK4/6 inhibitor responses.
To further explore therapeutic strategies that target ESR1 fusions, Gates and colleagues (99) showed that pharmacological inhibition of ESR1-e6>YAP1 fusion with the proteosome inhibitor MG132, blocked ESR1-e6>YAP1 mediated activation of ER target genes. In the same study, bortezomib, a specific 26S proteasome inhibitor, also suppressed growth driven by the ESR1-e6>YAP1 fusion. Bortezomib was tested in a phase II clinical trial in postmenopausal women with ER+ MBC who had progressive disease after prior aromatase inhibitor therapy. The patients were randomized to fulvestrant and bortezomib versus fulvestrant alone groups. In this study, there was no significant difference in progression free survival, which was the primary end point. However, the combination was overall well tolerated and may have enhanced activity in patients who have an ESR1 fusion (100).
ET resistance in ER+ breast cancer patients remain a significant clinical problem. The ESR1 fusion proteins are emerging as a mechanism of ET resistance and the studies discussed in this review, deepened our understanding of the prevalence of the ESR1 fusion proteins and the mechanisms by which they drive resistance. The most prevalent and clinically significant ESR1 fusions can be divided into the ESR1-e2>CCDC170 and ESR1-e6>fusion genes. ΔCCDC170, identified in Luminal B breast cancer and generated by ESR1-e2>CCDC170 led to enhanced growth and reduced sensitivity to ET in MBC. ESR1- e6>fusions were identified in ET-resistant MBC and are formed by inter-chromosomal translocation fusing ESR1 exons 1 to 6 into a 3’ fusion partner, replacing the LBD of ESR1. ESR1-e6>fusions drive estradiol-independent growth and constitutive expression of ER target genes leading to ET-resistance.
ESR1 fusions were identified in more aggressive forms of breast cancer (ET resistant MBC and Luminal B breast cancer) and can guide the diagnosis and the development of therapeutic strategies to treat a subset of patients with tumors that harbor these ESR1 alterations. As for the ESR1-e6>fusion genes, only a handful of functionally active ESR1 fusion proteins have been studied to date and therefore ESR1 fusion events remain an understudied form of somatic mutation in breast cancer. The incidence of these ESR1 fusions is still not well understood, but the studies discussed here collectively suggest that the frequency of ESR1 fusions may be higher in heavily pre-treated metastatic samples and when using more sensitive detection techniques.
The discovery of the ESR1 LBD point mutations has sparked enthusiasm for the development of a new generation of compounds that not only combat existing ER mutants but also inhibit secondary mutations in ER. Indeed, novel oral SERDs and SERCAs are being developed, and likely to be approved in the clinic. Similar to the emergence of ESR1 LBD mutations that render ligand independent activity, it is likely that the adoption of more potent SERDs and SERCAs will lead to adaptive mechanisms of resistance that are either ER independent or ER dependent but independent of the LBD. It is currently unclear which mechanisms of resistance may emerge following novel SERD and SERCA treatments.
Although, ESR1 gene fusions are rare, the frequency of these fusions may increase under the selective pressure of more effective SERDs and SERCAs. Therefore, better understanding of the mechanism of action of these fusions that lack the LBD, yet drive tumor progression in ER+ MBC, will be critical for the identification of vulnerabilities to target these fusions.
All authors listed have made a substantial contribution to the work and approved it for publication.
RJ receives NIH/NCI support (5RO1CA237414-02) and support from the Adams Barr Award (DFCI).
RJ received research funding from Pfizer and Lilly and is on an advisory board for GE Healthcare.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Organization WH. Cancer. Available at: https://www.who.int/news-room/fact-sheets/detail/cancer.
2. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol (1999) 17(5):1474–81. doi: 10.1200/JCO.1999.17.5.1474
3. Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, Michalides R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci (2010) 123(Pt 8):1253–61. doi: 10.1242/jcs.061135
4. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, et al. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol (2000) 74(5):311–7. doi: 10.1016/s0960-0760(00)00108-4
5. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science (1997) 277(5331):1508–10. doi: 10.1126/science.277.5331.1508
6. Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm (2001) 62:231–52. doi: 10.1016/s0083-6729(01)62006-5
7. Safe S, Kim K. Nuclear receptor-mediated transactivation through interaction with sp proteins. Prog Nucleic Acid Res Mol Biol (2004) 77:1–36. doi: 10.1016/S0079-6603(04)77001-4
8. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol (1999) 13(10):1672–85. doi: 10.1210/mend.13.10.0357
9. Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science (1997) 277(5328):965–8. doi: 10.1126/science.277.5328.965
10. Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, et al. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev (1998) 12(11):1638–51. doi: 10.1101/gad.12.11.1638
11. Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, et al. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell (1997) 90(3):569–80. doi: 10.1016/S0092-8674(00)80516-4
12. Hanstein B, Eckner R, DiRenzo J, Halachmi S, Liu H, Searcy B, et al. p300 is a component of an estrogen receptor coactivator complex. Proc Natl Acad Sci U.S.A. (1996) 93(21):11540–5. doi: 10.1073/pnas.93.21.11540
13. Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin d receptors. Mol Cell Biol (1997) 17(5):2735–44. doi: 10.1128/MCB.17.5.2735
14. Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci U.S.A. (1997) 94(16):8479–84. doi: 10.1073/pnas.94.16.8479
15. Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science (1995) 270(5240):1354–7. doi: 10.1126/science.270.5240.1354
16. Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, et al. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature (1997) 387(6634):677–84. doi: 10.1038/42652
17. Holmes KA, Hurtado A, Brown GD, Launchbury R, Ross-Innes CS, Hadfield J, et al. Transducin-like enhancer protein 1 mediates estrogen receptor binding and transcriptional activity in breast cancer cells. Proc Natl Acad Sci U.S.A. (2012) 109(8):2748–53. doi: 10.1073/pnas.1018863108
18. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet (2011) 43(1):27–33. doi: 10.1038/ng.730
19. Kong SL, Li G, Loh SL, Sung WK, Liu ET. Cellular reprogramming by the conjoint action of ERalpha, FOXA1, and GATA3 to a ligand-inducible growth state. Mol Syst Biol (2011) 7:526. doi: 10.1038/msb.2011.59
20. Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol (2008) 20(2):164–70. doi: 10.1016/j.ceb.2008.02.003
21. Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell (2006) 127(5):1041–55. doi: 10.1016/j.cell.2006.09.048
22. Lee JH, Bae SB, Oh MH, Cho HD, Jang SH, Hong SA, et al. Clinicopathologic and prognostic significance of transducin-like enhancer of split 1 protein expression in invasive breast cancer. J Breast Cancer (2017) 20(1):45–53. doi: 10.4048/jbc.2017.20.1.45
23. Magnani L, Ballantyne EB, Zhang X, Lupien M. PBX1 genomic pioneer function drives ERalpha signaling underlying progression in breast cancer. PloS Genet (2011) 7(11):e1002368. doi: 10.1371/journal.pgen.1002368
24. Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther (2018) 186:1–24. doi: 10.1016/j.pharmthera.2017.12.012
25. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
26. Furman C, Hao MH, Prajapati S, Reynolds D, Rimkunas V, Zheng GZ, et al. Estrogen receptor covalent antagonists: The best is yet to come. Cancer Res (2019) 79(8):1740–5. doi: 10.1158/0008-5472.CAN-18-3634
27. Puyang X, Furman C, Zheng GZ, Wu ZJ, Banka D, Aithal K, et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERalpha(WT) and ERalpha(MUT) breast cancer. Cancer Discovery (2018) 8(9):1176–93. doi: 10.1158/2159-8290.CD-17-1229
28. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol (2016) 13(7):417–30. doi: 10.1038/nrclinonc.2016.26
29. Early Breast Cancer Trialists' Collaborative, G. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet (2015) 386(10001):1341–52. doi: 10.1016/S0140-6736(15)61074-1
30. Early Breast Cancer Trialists' Collaborative, G. Aromatase inhibitors versus tamoxifen in premenopausal women with oestrogen receptor-positive early-stage breast cancer treated with ovarian suppression: a patient-level meta-analysis of 7030 women from four randomised trials. Lancet Oncol (2022) 23(3):382–92. doi: 10.1016/S1470-2045(21)00758-0
31. Aurilio G, Disalvatore D, Pruneri G, Bagnardi V, Viale G, Curigliano G, et al. A meta-analysis of oestrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 discordance between primary breast cancer and metastases. Eur J Cancer (2014) 50(2):277–89. doi: 10.1016/j.ejca.2013.10.004
32. Browne AL, Charmsaz S, Vareslija D, Fagan A, Cosgrove N, Cocchiglia S, et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene (2018) 37(15):2008–21. doi: 10.1038/s41388-017-0042-x
33. Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U.S.A. (2016) 113(43):E6600–9. doi: 10.1073/pnas.1612835113
34. O'Hara J, Vareslija D, McBryan J, Bane F, Tibbitts P, Byrne C, et al. AIB1:ERalpha transcriptional activity is selectively enhanced in aromatase inhibitor-resistant breast cancer cells. Clin Cancer Res (2012) 18(12):3305–15. doi: 10.1158/1078-0432.CCR-11-3300
35. Walsh CA, Qin L, Tien JC, Young LS, Xu J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int J Biol Sci (2012) 8(4):470–85. doi: 10.7150/ijbs.4125
36. Zhou J, Xu M, Le K, Ming J, Guo H, Ruan S, et al. SRC promotes tamoxifen resistance in breast cancer via up-regulating SIRT1. Onco Targets Ther (2020) 13:4635–47. doi: 10.2147/OTT.S245749
37. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa b and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer (2007) 7:59. doi: 10.1186/1471-2407-7-59
38. Alamolhodaei NS, Behravan J, Mosaffa F, Karimi G. MiR 221/222 as new players in tamoxifen resistance. Curr Pharm Des (2016) 22(46):6946–55. doi: 10.2174/1381612822666161102100211
39. Chen MJ, Cheng YM, Chen CC, Chen YC, Shen CJ. MiR-148a and miR-152 reduce tamoxifen resistance in ER+ breast cancer via downregulating ALCAM. Biochem Biophys Res Commun (2017) 483(2):840–6. doi: 10.1016/j.bbrc.2017.01.012
40. Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem (2008) 283(44):29897–903. doi: 10.1074/jbc.M804612200
41. Zhang W, Xu J, Shi Y, Sun Q, Zhang Q, Guan X. The novel role of miRNAs for tamoxifen resistance in human breast cancer. Cell Mol Life Sci (2015) 72(13):2575–84. doi: 10.1007/s00018-015-1887-1
42. Chang M. Tamoxifen resistance in breast cancer. Biomol Ther (Seoul) (2012) 20(3):256–67. doi: 10.4062/biomolther.2012.20.3.256
43. Christopoulos PF, Msaouel P, Koutsilieris M. The role of the insulin-like growth factor-1 system in breast cancer. Mol Cancer (2015) 14:43. doi: 10.1186/s12943-015-0291-7
44. Fan P, Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist (2019) 2:198–209. doi: 10.20517/cdr.2019.13
45. Turczyk L, Kitowska K, Mieszkowska M, Mieczkowski K, Czaplinska D, Piasecka D, et al. FGFR2-driven signaling counteracts tamoxifen effect on ERalpha-positive breast cancer cells. Neoplasia (2017) 19(10):791–804. doi: 10.1016/j.neo.2017.07.006
46. Zhou Y, Wu C, Lu G, Hu Z, Chen Q, Du X. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer (2020) 11(8):2000–7. doi: 10.7150/jca.40531
47. Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med (2008) 10:e19. doi: 10.1017/S1462399408000744
48. Abukhdeir AM, Vitolo MI, Argani P, De Marzo AM, Karakas B, Konishi H, et al. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proc Natl Acad Sci U.S.A. (2008) 105(1):288–93. doi: 10.1073/pnas.0710887105
49. Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, et al. p27 phosphorylation by src regulates inhibition of cyclin e-Cdk2. Cell (2007) 128(2):281–94. doi: 10.1016/j.cell.2006.11.049
50. Jankevicius F, Goebell P, Kushima M, Schulz WA, Ackermann R, Schmitz-Drager BJ. p21 and p53 immunostaining and survival following systemic chemotherapy for urothelial cancer. Urol Int (2002) 69(3):174–80. doi: 10.1159/000063949
51. Venditti M, Iwasiow B, Orr FW, Shiu RP. C-myc gene expression alone is sufficient to confer resistance to antiestrogen in human breast cancer cells. Int J Cancer (2002) 99(1):35–42. doi: 10.1002/ijc.10269
52. Yu L, Wang L, Mao C, Duraki D, Kim JE, Huang R, et al. Estrogen-independent myc overexpression confers endocrine therapy resistance on breast cancer cells expressing ERalphaY537S and ERalphaD538G mutations. Cancer Lett (2019) 442:373–82. doi: 10.1016/j.canlet.2018.10.041
53. Kastrati I, Joosten SEP, Semina SE, Alejo LH, Brovkovych SD, Stender JD, et al. The NF-kappaB pathway promotes tamoxifen tolerance and disease recurrence in estrogen receptor-positive breast cancers. Mol Cancer Res (2020) 18(7):1018–27. doi: 10.1158/1541-7786.MCR-19-1082
54. Joffroy CM, Buck MB, Stope MB, Popp SL, Pfizenmaier K, Knabbe C. Antiestrogens induce transforming growth factor beta-mediated immunosuppression in breast cancer. Cancer Res (2010) 70(4):1314–22. doi: 10.1158/0008-5472.CAN-09-3292
55. Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, et al. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci (2020) 111(1):47–58. doi: 10.1111/cas.14230
56. Hermida-Prado F, Jeselsohn R. The ESR1 mutations: From bedside to bench to bedside. Cancer Res (2021) 81(3):537–8. doi: 10.1158/0008-5472.CAN-20-4037
57. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell (2018) 33(2):173–186 e5. doi: 10.1016/j.ccell.2018.01.004
58. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol (2015) 12(10):573–83. doi: 10.1038/nrclinonc.2015.117
59. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res (2013) 73(23):6856–64. doi: 10.1158/0008-5472.CAN-13-1197
60. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet (2013) 45(12):1446–51. doi: 10.1038/ng.2823
61. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet (2013) 45(12):1439–45. doi: 10.1038/ng.2822
62. Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep (2018) 23(1):227–238 e3. doi: 10.1016/j.celrep.2018.03.050
63. Dunn DB. Larotrectinib and entrectinib: TRK inhibitors for the treatment of pediatric and adult patients with NTRK gene fusion. J Adv Pract Oncol (2020) 11(4):418–23. doi: 10.6004/jadpro.2020.11.4.9
64. Filippi R, Depetris I, Satolli MA. Evaluating larotrectinib for the treatment of advanced solid tumors harboring an NTRK gene fusion. Expert Opin Pharmacother (2021) 22(6):677–84. doi: 10.1080/14656566.2021.1876664
65. Groussin L, Theodon H, Bessiene L, Bricaire L, Bonnet-Serrano F, Cochand-Priollet B, et al. Redifferentiating effect of larotrectinib in NTRK-rearranged advanced radioactive-iodine refractory thyroid cancer. Thyroid (2022) 32(5):594–8. doi: 10.1089/thy.2021.0524
66. Hempel D, Wieland T, Solfrank B, Grossmann V, Steinhard J, Frick A, et al. Antitumor activity of larotrectinib in esophageal carcinoma with NTRK gene amplification. Oncologist (2020) 25(6):e881–6. doi: 10.1634/theoncologist.2019-0641
67. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol (2018) 19(5):705–14. doi: 10.1016/S1470-2045(18)30119-0
68. Ricciuti B, Genova C, Crino L, Libra M, Leonardi GC. Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: a short review on the current evidence. Onco Targets Ther (2019) 12:3171–9. doi: 10.2147/OTT.S177051
69. Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat (2019) 5. doi: 10.20517/2394-4722.2019.12
70. Loo SK, Yates ME, Yang S, Oesterreich S, Lee AV, Wang XS. Fusion-associated carcinomas of the breast: Diagnostic, prognostic, and therapeutic significance. Genes Chromosomes Cancer (2022) 61(5):261–73. doi: 10.1002/gcc.23029
71. Matissek KJ, Onozato ML, Sun S, Zheng Z, Schultz A, Lee J, et al. Expressed gene fusions as frequent drivers of poor outcomes in hormone receptor-positive breast cancer. Cancer Discovery (2018) 8(3):336–53. doi: 10.1158/2159-8290.CD-17-0535
72. Veeraraghavan J, Ma J, Hu Y, Wang XS. Recurrent and pathological gene fusions in breast cancer: current advances in genomic discovery and clinical implications. Breast Cancer Res Treat (2016) 158(2):219–32. doi: 10.1007/s10549-016-3876-y
73. Jeong JH, Yun JW, Kim HY, Heo CY, Lee S. Elucidation of novel therapeutic targets for breast cancer with ESR1-CCDC170 fusion. J Clin Med (2021) 10(4). doi: 10.3390/jcm10040582
74. Li L, Lin L, Veeraraghavan J, Hu Y, Wang X, Lee S, et al. Therapeutic role of recurrent ESR1-CCDC170 gene fusions in breast cancer endocrine resistance. Breast Cancer Res (2020) 22(1):84. doi: 10.1186/s13058-020-01325-3
75. Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, Chamness GC, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun (2014) 5:4577. doi: 10.1038/ncomms5577
76. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep (2013) 4(6):1116–30. doi: 10.1016/j.celrep.2013.08.022
77. Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, et al. Functionally recurrent rearrangements of the MAST kinase and notch gene families in breast cancer. Nat Med (2011) 17(12):1646–51. doi: 10.1038/nm.2580
78. Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell (2002) 2(5):367–76. doi: 10.1016/S1535-6108(02)00180-0
79. Vitale SR, Ruigrok-Ritstier K, Timmermans AM, Foekens R, Trapman-Jansen A, Beaufort CM, et al. The prognostic and predictive value of ESR1 fusion gene transcripts in primary breast cancer. BMC Cancer (2022) 22(1):165. doi: 10.1186/s12885-022-09265-1
80. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep (2018) 24(6):1434–1444 e7. doi: 10.1016/j.celrep.2018.07.009
81. Gou X, Anurag M, Lei JT, Kim BJ, Singh P, Seker S, et al. Transcriptional reprogramming differentiates active from inactive ESR1 fusions in endocrine therapy-refractory metastatic breast cancer. Cancer Res (2021) 81(24):6259–72. doi: 10.1158/0008-5472.CAN-21-1256
82. Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med (2017) 9(402):eaai7993. doi: 10.1126/scitranslmed.aai7993
83. Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature (2017) 548(7667):297–303. doi: 10.1038/nature23306
84. Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Vanden Borre P, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann Oncol (2018) 29(4):872–80. doi: 10.1093/annonc/mdy025
85. Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature (2019) 575(7781):210–6. doi: 10.1038/s41586-019-1689-y
86. Bruno R, Fontanini G. Next generation sequencing for gene fusion analysis in lung cancer: A literature review. Diagnostics (Basel) (2020) 10(8):521. doi: 10.3390/diagnostics10080521
87. Heyer EE, Blackburn J. Sequencing strategies for fusion gene detection. Bioessays (2020) 42(7):e2000016. doi: 10.1002/bies.202000016
88. Schroder J, Kumar A, Wong SQ. Overview of fusion detection strategies using next-generation sequencing. Methods Mol Biol (2019) 1908:125–38. doi: 10.1007/978-1-4939-9004-7_9
89. Singh RR. Next-generation sequencing in high-sensitive detection of mutations in tumors: Challenges, advances, and applications. J Mol Diagn (2020) 22(8):994–1007. doi: 10.1016/j.jmoldx.2020.04.213
90. Supplee JG, Milan MSD, Lim LP, Potts KT, Sholl LM, Oxnard GR, et al. Sensitivity of next-generation sequencing assays detecting oncogenic fusions in plasma cell-free DNA. Lung Cancer (2019) 134:96–9. doi: 10.1016/j.lungcan.2019.06.004
91. Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci (2018) 14(8):843–8. doi: 10.7150/ijbs.24626
92. Prodduturi N, Bhagwate A, Kocher JA, Sun Z. Indel sensitive and comprehensive variant/mutation detection from RNA sequencing data for precision medicine. BMC Med Genomics (2018) 11(Suppl 3):67. doi: 10.1186/s12920-018-0391-5
93. Seager M, Aisner DL, Davies KD. Oncogenic gene fusion detection using anchored multiplex polymerase chain reaction followed by next generation sequencing. J Vis Exp (2019) 2019(149):10.3791/59895. doi: 10.3791/59895
94. Teixido C, Gimenez-Capitan A, Molina-Vila MA, Peg V, Karachaliou N, Rodriguez-Capote A, et al. RNA Analysis as a tool to determine clinically relevant gene fusions and splice variants. Arch Pathol Lab Med (2018) 142(4):474–9. doi: 10.5858/arpa.2017-0134-RA
95. Ahn SG, Bae SJ, Kim Y, Ji JH, Chu C, Kim D, et al. Primary endocrine resistance of ER+ breast cancer with ESR1 mutations interrogated by droplet digital PCR. NPJ Breast Cancer (2022) 8(1):58. doi: 10.1038/s41523-022-00424-y
96. Callens C, Bidard FC, Curto-Taribo A, Trabelsi-Grati O, Melaabi S, Delaloge S, et al. Real-time detection of ESR1 mutation in blood by droplet digital PCR in the PADA-1 trial: Feasibility and cross-validation with NGS. Anal Chem (2022) 94(16):6297–303. doi: 10.1021/acs.analchem.2c00446
97. Jeannot E, Darrigues L, Michel M, Stern MH, Pierga JY, Rampanou A, et al. A single droplet digital PCR for ESR1 activating mutations detection in plasma. Oncogene (2020) 39(14):2987–95. doi: 10.1038/s41388-020-1174-y
98. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6(224):224ra24. doi: 10.1038/s41388-020-1174-y
99. Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA, et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene (2018) 37(33):4581–98. doi: 10.1038/s41388-018-0284-2
100. Adelson K, Ramaswamy B, Sparano JA, Christos PJ, Wright JJ, Raptis G, et al. Randomized phase II trial of fulvestrant alone or in combination with bortezomib in hormone receptor-positive metastatic breast cancer resistant to aromatase inhibitors: a new York cancer consortium trial. NPJ Breast Cancer (2016) 2:16037. doi: 10.1038/npjbcancer.2016.37
Keywords: breast cancer, estrogen receptor, ESR1 fusion, endocrine therapy resistance, SERD
Citation: Nagy Z and Jeselsohn R (2023) ESR1 fusions and therapeutic resistance in metastatic breast cancer. Front. Oncol. 12:1037531. doi: 10.3389/fonc.2022.1037531
Received: 06 September 2022; Accepted: 22 November 2022;
Published: 04 January 2023.
Edited by:
Ariella Hanker, University of Texas Southwestern Medical Center, United StatesReviewed by:
Angel Luis Guerrero-Zotano, Instituto Valenciano de Oncologia, SpainCopyright © 2023 Nagy and Jeselsohn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rinath Jeselsohn, UmluYXRoX0plc2Vsc29obkBkZmNpLmhhcnZhcmQuZWR1; Zsuzsanna Nagy, WnN1enNhbm5hX05hZ3lAZGZjaS5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.