
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 October 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1020011
This article is part of the Research TopicApplied Precision Medicine in Plasma Cell Dyscrasias: From Prevention to TreatmentView all 7 articles
 Mateo Mejia Saldarriaga
Mateo Mejia Saldarriaga Walaa Darwiche
Walaa Darwiche David JayabalanJorge MongeCara RosenbaumRoger N. PearseRuben Niesvizky
David JayabalanJorge MongeCara RosenbaumRoger N. PearseRuben Niesvizky Mark Bustoros*
Mark Bustoros*Recent insight in the genomic landscape of newly diagnosed multiple myeloma (NDMM) and its precursor conditions, monoclonal gammopathy of uncertain significance (MGUS), and smoldering myeloma have allowed the identification of patients with precursor conditions with a high risk of progression. These cases with “progressor” MGUS/SMM have a higher average mutation burden, have higher rates of mutations in specific genes such as MAPK, DNA repair, MYC, DIS3, and are enriched for specific mutational signatures when compared to non-progressors and are comparable to those found in NDMM. The highly preserved clonal heterogeneity seen upon progression of SMM, combined with the importance of these early variables, suggests that the identification of progressors based on these findings could complement and enhance the currently available clinical models based on tumor burden. Mechanisms leading to relapse/refractory multiple myeloma (RRMM) are of clinical interest given worse overall survival in this population. An Increased mutational burden is seen in patients with RRMM when compared to NDMM, however, there is evidence of branching evolution with many of these mutations being present at the subclonal level. Likewise, alterations in proteins associated with proteosome inhibitor and immunomodulatory drugs activity could partially explain clinical resistance to these agents. Evidence of chromosomal events leading to copy number changes is seen, with the presence of TP53 deletion, mutation, or a combination of both being present in many cases. Additional chromosomal events such as 1q gain and amplification may also interact and lead to resistance.
Multiple Myeloma (MM) represents 1.8% of all new cancers in the United States (US) and is the second most common hematologic malignancy in the US with roughly 34,000 new cases/year (1), with a higher incidence and earlier age of diagnosis in African American (2 fold increase), while Asian population have a lower incidence (2–4). Advances in understanding of MM biology have led to therapeutic improvements over the last two decades, with over 10 new molecules targeting novel pathways and mechanism of action, and with a significant improvement in overall survival and quality of life since the introduction of these agents into clinical practice (5, 6).
MM is preceded by monoclonal gammopathy of undetermined significance (MGUS) and smoldering myeloma (SMM), collectively known as precursor conditions. More recently, it became clear MGUS/SMM precede the appearance of MM by several years (7, 8), representing a precursor state mirroring other hematological malignancies such as monoclonal B cell lymphocytosis and clonal hematopoiesis of indeterminate potential. Similarly, to MM, MGUS incidence is higher and at an earlier age in African American when compared to white population, suggesting the increase rate of MM seen in this population is the product of increased incidence of MGUS (7).
Understanding of the biology of plasma cell dyscrasias has been fundamental for the creation of risk scores used to identify patients at risk for progression or poor clinical outcomes, development of new drugs targeting plasma cell specific pathways, and ultimately improve the outcomes of the patients living with MM and precursor conditions. As the genomic landscape continues to be discovered, new strategies, improvements on the diagnosis, prognostication, and treatment of these patients are developed. We will review the current state and areas of interest in patients with MM and MGUS/SMM.
In contrast to patients with NDMM, who exhibit significant end organ damage and poor outcomes, a (9) subgroup of patients with a monoclonal protein, but no evidence of end-organ damage and an indolent course was identified first by Dr. Jan Waldeström. Later, Dr. Robert Kyle at Mayo Clinic demonstrated that although these patients usually are indolent, their risk of developing MM is significantly higher than the rest of the population, leading to the recognition of monoclonal gammopathy of undetermined significance. Likewise, patients with increased or marked monoclonal hypergammaglobulinemia with bone marrow plasmacytosis but with no end-organ damage, with a higher rate of progression to MM when compared to MGUS, led to the identification of smoldering multiple myeloma (SMM) as a clinical entity (10).
Given the increased risk of developing MM in these patients, clinical variables were developed to identify patients at higher risk. Two of the early and widely used clinical models in SMM were the Mayo Clinic model, which relied on the presence of ≥ 10% bone marrow plasma cells (BMPC), an M protein ≥3gr/dL, and kappa/lambda free light chain ratio ≥8 or <0.125, and the PETHEMA (Spanish) model, which included ≥95% of abnormal plasma cells on BMPC identified through immunophenotype and the presence of immunoparesis (defined as of ≥1 non-involved immunoglobulin with a >25% below lower limit of normal). High risk patients (those with 3/3 and 2/2 in the Mayo Clinic and PETHEMA models, respectively) had a median time to progression of about 24 months (11, 12). More recently, the 2/20/20 model, which relies on >2 gr/dL monoclonal protein, >20% BMPC, and an involved/uninvolved FLC ratio >20 was developed and validated in a cohort of 1996, with high-risk (2-3 factors) patients having a 2-year progression rate of 44.2% vs 6.2% in low-risk (0 factors) patients (13). Similarly, models based on immunoglobulin isotype, the presence of M protein >1.5gr/dL, and FLC ratio >8 have been used to stratify the risk of progression to MM in non-IgM MGUS (14).
Although these models are helpful to determine the risk of progression, they have several inherent limitations. Since they rely on surrogates of tumor burden, they do not indicate the actual molecular underpinnings of disease, which translate into the clinical findings in patients exhibiting indolent disease without progression despite high-risk features or patients with rapid progression despite low-risk criteria at diagnosis. Changes in hemoglobin or M-protein concentration are prognostic (15, 16), however, they are not currently adapted in any model. Recent advances have increased our understanding of the genetic events leading to myelomagenesis and have arisen as potential markers to further establish the true progressors from those with indolent disease.
Historically, the use of conventional cytogenetics and fluorescence in-situ hybridization (FISH) allowed the identification of two distinct groups of MM. One with recurrent translocations involving the IGH locus with another partner gene (such as CCND1 in t(11;14) and MMSET in t(4;14)), and another group of patients with trisomies of whole chromosomes. One of these two alterations is almost universally present in MM, with 40-50% having IGH translocations and 50-60% of cases classified as hyperdiploid.
The time of acquisition of these alterations can be inferred based on the principle that earlier mutations will be present in a larger population of cancer cells (also known as the cancer cell fraction). Translocations and hyperdiploidy MM are clonal (CCF of >95%) and mostly mutually exclusive, suggesting they occur early during myelomagenesis and represent important driver events; this is also supported by a similar proportion of these alterations in precursor states (SMM/MGUS). However, other recurrent cytogenetic abnormalities seen in MM are subclonal and suggest a later acquisition, with each cytogenetic alteration exhibiting a variable degree of sub-clonality. For example, deletion 17p, deletion 18q, and deletion 1p were noted to be highly subclonal, whereas deletion 13q and 1q gain, are mostly clonal events suggesting they occur earlier during the malignant transformation of plasma cells (17). These cytogenetics alterations not only served as an important clue to the early events leading to MM, but also serve as the basis for the identification of patient subgroups with more aggressive phenotype and worse clinical outcomes, as it is the case of deletion 17p, IGH-MMSET (t(4;14), and 1q amplification (18, 19).
The introduction of next-generation sequencing and the increasing availability of these platforms has led to a new understanding of the characteristics of MGUS/SMM and the evolution towards overt MM. Whole-exome sequencing (WES) allowed the identification of several mutations in MM, with the most common pathways involved the MAPK pathway (with mutations in the KRAS, NRAS, and BRAF genes present in roughly 40% of cases), DNA damage response (including TP53 and ATM), and the NF-kB response pathway (20, 21) Figure 1. Unlike acute myeloid leukemia in which point mutations are main driver events and define a clinical phenotype, mutations in MM are often subclonal (later events) (25) and are largely variable from patient to patient, with only a small fraction of patients sharing common mutations, However, there is evidence some of the most common mutations described above are pathogenic and have clinical impact, as they are associated with certain cytogenetic subtypes as is the case for DIS3 and t(4;14), and have been associated with worse clinical outcomes as is the case for TP53 and ATM mutations (21), whereas many of the less frequent mutations encountered in few patients do no have particular associattion with disease subtype or cytogenetic group, suggesting they are rather “passenger” events that occur as a result of subclonal evolution.
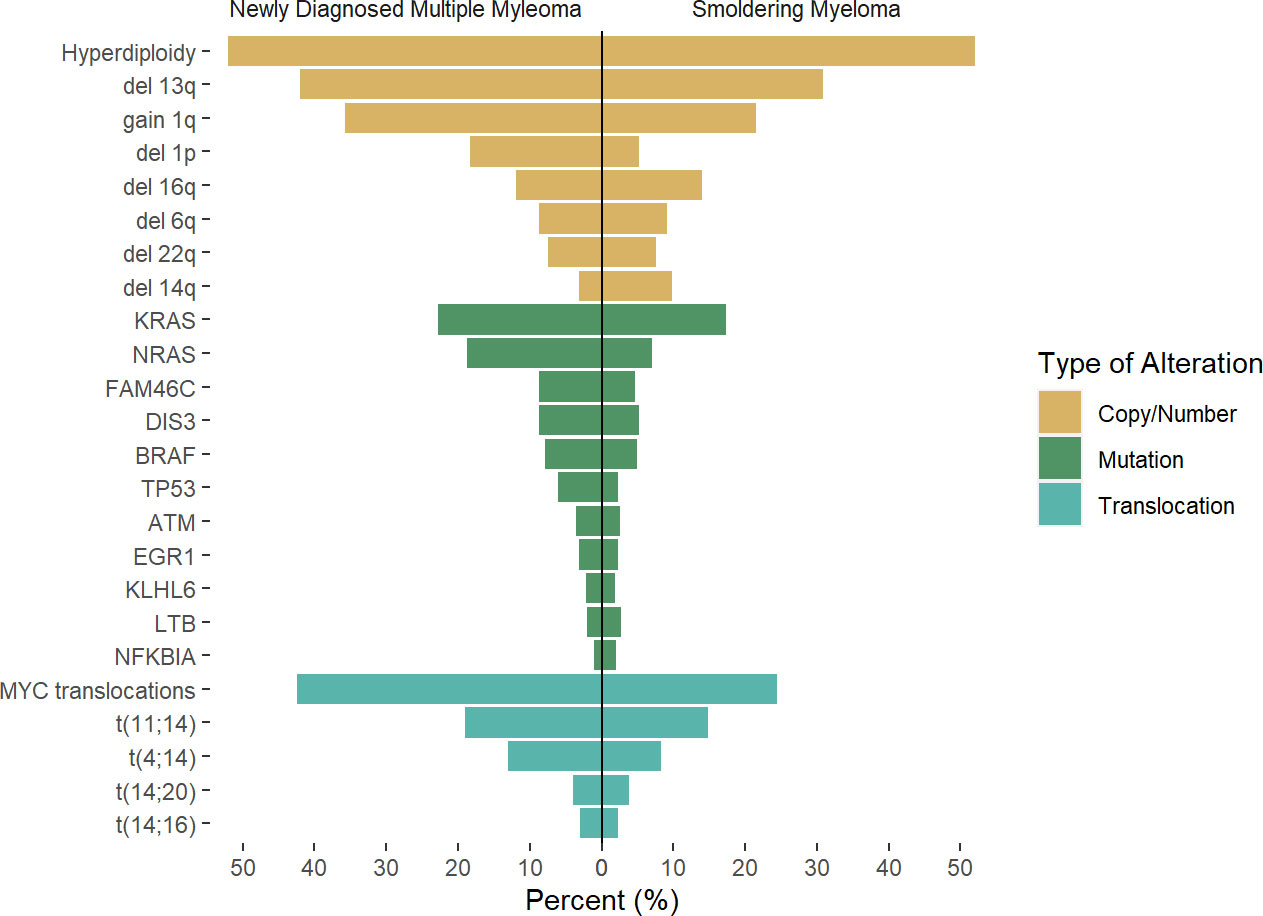
Figure 1 Rate of genetic alterations across newly diagnosed multiple myeloma and smoldering myeloma. Results of previous reports by Boyle et al (22), Bustoros et al (17), Misund et al (23), Walker et al (24) were pooled to the described average rate of the described alteration, as there is variability in likely secondary to differences in methodology used. Differences across studies and methodology used may account for differences.
Although WES was widely used initially in MM, one of the limitations is missing structural variants (SV’s) that may also play an important role. Large SV’s events have been widely described in both hematology and solid malignancies and are associated with chromosomal events leading to deletion, translocation, and tandem duplication causing known pathologic copy number/number or enhancer hijacking in oncogenes and tumor suppressor genes (26, 27). In the case of MM, these events were initially considered to be rare, however, the use of long-read whole-genome sequencing (WGS) platforms have allowed the identification of 1 or more complex SV’s in 68% of cases (28). Chromotrypsis, chromoplexy, and templated insertions were present, in 38%, 11%, and 19% of newly diagnosed patients, and have been associated with worse clinical outcomes in addition to close association with certain disease subgroups, as is the case for t(11;14). Furthermore, novel hotspots in previously considered non-coding regions have been identified, and have been linked to complex SV’s associated with other clinically relevant alterations such as 17p deletion and 1q gain (28, 29). Importantly, these complex SV’s have been associated with simultaneous acquisition of >1 driver event, often in unrelated regions that would otherwise be considered as independent events.
After the initial description of the genetic landscape in MM was described and expanded in multiple studies (21, 24), a similar approach was taken for MGUS/SMM. As mentioned before, patients with SMM have a similar prevalence of recurrent translocations and hyperdiploidy as those seen in MM, highlighting the clonal nature of these events.
Recent efforts to identify the mutational landscape in SMM have shown very similar results to those seen in NDMM. A cohort of 214 patients with SMM who underwent WES mutations in the MAPK pathway (KRAS, NRAS, and BRAF) was the most common finding, seen in 48% of patients, followed by mutations involving DNA repair pathways (TP53, ATM, and deletion 17p, 21% of cases), and NF-kB pathway (10% of cases), while other mutations such as DIS3, FAMC46C, ZNF292, RB1, CDKN2C were present in less than 10% of cases. Again, some of these mutations are highly subclonal, suggesting the acquisition of these events occurred at a later stage than foundational events, but often present to provide fitness to the tumor cells. Based on these findings, the mutations in the MAPK pathway, DNA repair, and MYC alterations were identified as risk factors for progression, and when added to the 2/20/20 model, they increased the performance when compared to the model based on clinical variables (17). The importance of these alterations in the progression of SMM was confirmed as well in additional studies (22, 23), also highlighting the higher prevalence of MYC alterations in progressor SMM and NDMM (Figure 1).
The role of SVs in the progression of precursor conditions was evaluated in a cohort of 18 MGUS and 14 SMM and compared to 80 MM. Cases with “stable” MGUS/SMM had a lower SV’s burden overall. This difference was even more striking for complex SV’s, with only 1 stable case having evidence of chromothripsis vs 47% and 41% of progressor precursors having evidence of chromothripsis and templated insertions, respectively, and with a similar burden of SV’s when compared to established MM cases (30). Similar findings were seen for copy number alterations, with a higher rate of MM recurrent chromosomal events such as gain 1q, deletion 8p, deletion 6q in cases of progressor precursors, despite no significant difference in aneuploidies or recurrent translocation between these two groups.
Mutational signatures analysis can inform on the mechanism leading to mutations and SV’s. In the case of MM, several mutational signatures have been identified and seem to be relatively constant across cases. These include evidence of activity of the enzyme activation-induced cytidine deaminase (AID) across immunoglobulin locus (known as canonical AID, c-AID) and in non-immunoglobulin areas (known as non-canonical AID, nc-AID), APOBEC, “clock-like” signatures (related to constant mutation over time (31)). Patients with progressor precursors have been associated with higher evidence on nc-AID and APOBEC when compared to stable precursors, suggesting differences in underlying mutational events in both subgroups. In addition, the overall mutational rate for progressor conditions mirrors the one seen in NDMM (17, 30). In addition, in matched samples of a patient with progressive SMM, most cases (>70%) had preserved subclonal structure with no new subclones and no significant subclonal expansion, suggesting the necessary events for malignant transformation are already present at the SMM/MGUS stage, and that subclonal evolution may be a marker of higher risk but not necessarily causal of progression, following a model of static evolution, with a minority of cases exhibiting acquisition of new lesions and/or changes in subclone structure (17, 22, 30).
Improvement in the understanding of the genetic events present in NDMM has led to changes in the established model of myelomagenesis. Initially, MM development was considered to follow a step-wise model, such as colon cancer, where specific mutations occurred over time leading to precursor conditions and eventual progression to malignancy. However, this model seems to not be representative of MGUS/SMM and MM, especially given the relatively preserved subclonal composition of most patients with progressive precursors when compared to matched NDMM samples. Early events leading to hyperdiploidy and translocations involving the IGH locus seem to be related to exposure to AID in the germinal center, while APOBEC and “clock-like” signatures contribute to later events, presumably once migration to the bone marrow has been completed (Figure 2). This conceptual framework is not only relevant to understanding the events leading to myelomagenesis but also provides the basis for the identification of potential progressor vs stable precursors clinically, potentially improving our stratification of these patients. Additionally, other cytogenetic alterations such as deletion 17p, 1q gain, t(4;14), t(14;16), and 13q deletion/13 monosomy have also been associated with an increased risk of progression to MM, leading to the addition of these findings to the previously described 2/20/20 model (13). The frequent co-occurrence of copy number abnormalities, recurrent translocations, and mutations led to the identification of well-defined molecular subgroups of NDMM and RRMM (32, 33), which not only differ by expression of certain markers (such as CD20 enrichment in CD-1, associated with t(11;14), but also exhibit differences in clinical phenotype and prognosis. Recently, using a set of 42 alterations (single nucleotide variants, translocations, and copy number alterations), 6 well-defined molecular subtypes of SMM were identified, which not only had significant overlap in terms of co-existing abnormalities with previously defined MM genomic subgroups but were also prognostic in terms of TTP to active MM. Furthermore, the use of these genomic classifications coupled with the 20/2/20 model improved the prediction performance of the model. In addition, the presence of high-risk subgroups identified patients at higher risk of progression within the clinically defined high and intermediate risk (34).
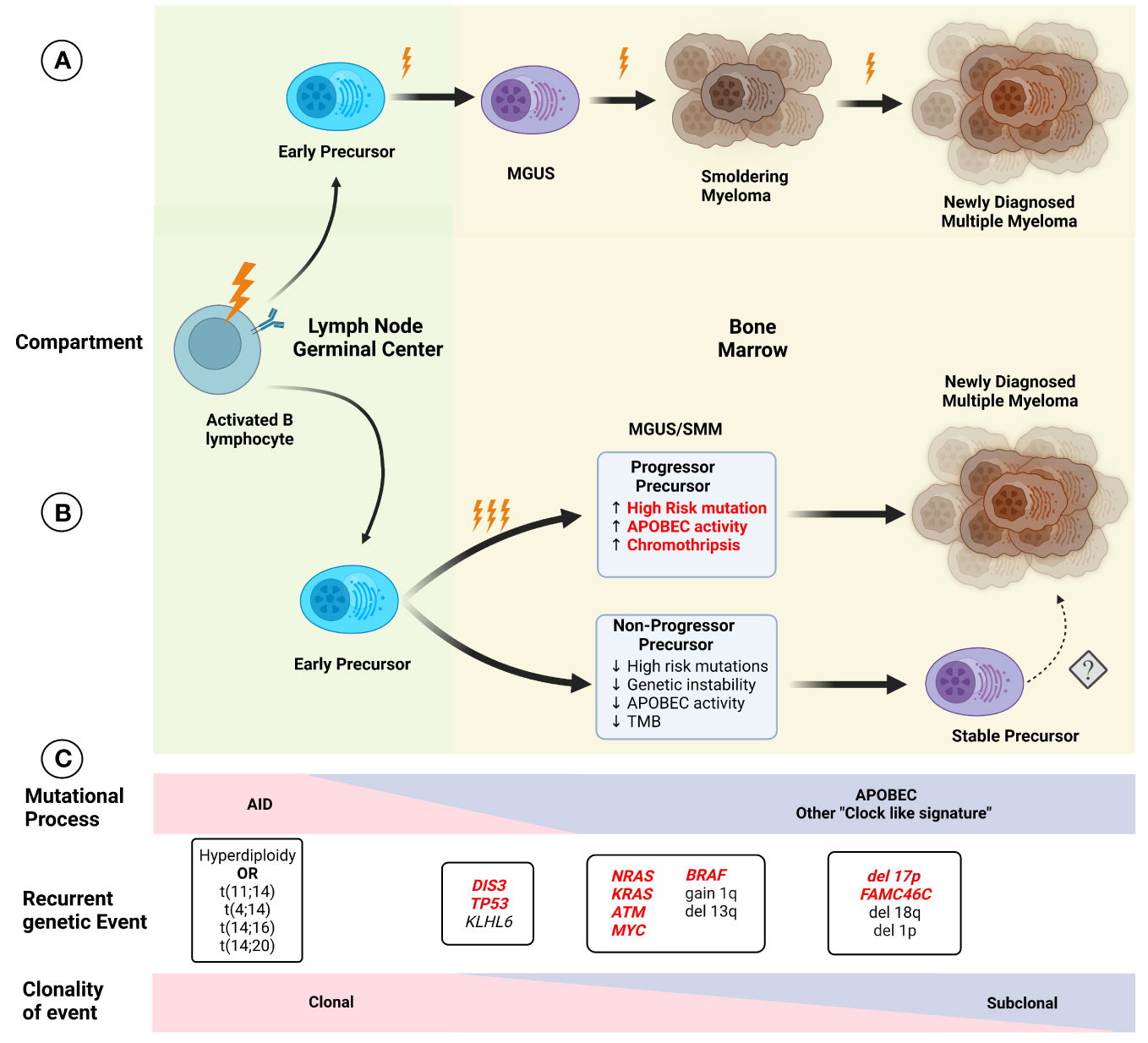
Figure 2 Initial models of (A) myelomagenesis postulated the appearance of a precursor clone which required acquisition of mutations and/or chromosomal abnormalities in a stepwise fashion leading to the observe progression from monoclonal gammopathy of uncertain significance (MGUS), to smoldering myeloma (SMM) and finally multiple myeloma (MM) after enough pathogenic event have occurred. (B) Based on recent findings, the paradigm of myelomagenesis has shifted, with highly clonal events, such as recurrent translocations involving IGH gene and hyperdiploidy representing fundational events. The role of AID initially, and later APOBEC as important mutational process likely contribute to acquisition of additional events (and possibly several during one event, described as “catastrophic evolution”). At the moment of clinically evident precursors (MGUS/SMM), two groups of are present, one characterized by higher genomic instability, with higher tumor mutation burden (TMB), increased chromoplexis and other complex structural events, and increased in pathogenic events, whereas the “non-progressor” precursor have low risk of progression over time and lack some of the features described before, although the potential risk of conversion to a more aggressive disease with progression to MM is not clear (denoted by dashed arrow). Several characteristics in the precursor clone (highlighted in red) indicate a higher risk of progression and potentially could be used clinically for risk stratification. (C) The effect of different mutational process potentially varies over time, with an increased in AID, while APOBEC and other process such as “clock-like” signatures are present at a later time. Except for early foundational events described before, other alterations display different degrees of sub clonality, with most cases of MM exhibiting relatively preserved clonal structure upon progression.
Additionally, recent insights on additional high-risk alterations in NDMM, suggest that not all chromosomal cytogenetic alterations confer the same degree of risk. Biallelic TP53 loss (either through deletion or mutation) was associated poor overall survival when compared to patients with no or only 1 TP53 event, suggesting biallelic events are relevant during clonal evolution and potentially more informative than isolated, mono-allelic alteration currently being captured (35). The importance of 1q copy number gains as a prognostic factor in NDMM has also been highlighted recently as these patients have poor outcomes and mirror those considered to have high risk cytogenetic features such as del 17p and t(4;14). In addition, the “burden” of 1q gain seems to be relevant, as patient with 1q amplification (>3 copies) have worse outcomes when compared to patients with 1q gain (3 copies) (18, 36). Proteosome inhibitors (PI’s) are thought to abrogate the effect of high risk cytogenetic features (del 17p and t(4;14)) on outcomes, however, 1q amplification may indicate therapeutic resistance, as patients with 1q amplification enrolled in the FORTE trial had poor outcomes despite upfront Carfilzomib use, whereas those with del 17p and t(4;14) had similar outcomes to those with standard risk cytogenetic (37, 38).
Limited panels detecting known clinically relevant mutations and fusions have been already developed and are widely used in other disease settings, such as acute myeloid leukemia (39). These panels could identify prognostic mutations in MAPK pathway, DNA repair, MYC, and other potential genes. The identification of specific genomic signatures, tumor mutational burden, and complex SV’s, although associated with higher rates of progression, has the limitation of requiring much broader coverage of the genome, potentially limiting the current clinical application of these markers. However, high-throughput next-generation platforms are evolving quickly with increasing efficiency and decreasing cost per patient, potentially opening the doors for a future wide-scale implementation of this technology in clinical practice.
The use of circulating tumor DNA (ctDNA) has the potential to identify clinically relevant mutations, and indicate response or residual disease without the need for an invasive procedure, hence the term “liquid biopsy”. It has been applied for several solid and hematologic malignancies and is widely used in clinical practice (40). In the case of MM, ctDNA burden correlates with higher 70-GEP and prognosis, with patients with higher burden having worse PFS and OS (41). Furthermore, it has been proposed as a tool to detect treatment failure and subsequent relapse in the RRMM setting (42). As mentioned before, the integration of SNV’s, translocation, and copy number changes into defined molecular subgroups of SMM improve the performance of 2/20/20 model (34), but will lack of widespread access to this technology remains a limitation.
The availability of a minimally invasive test that could inform on disease stratification and potentially provide information on progression would be of great use in SMM. However, some limitations exist for ctDNA in the case of MGUS/SMM. First, patients with precursor conditions tend to have much lower ctDNA, possibly related to a lower tumor burden. Second, there is a lack of correlation between the findings in the bone marrow and ctDNA, with the latter missing recurrent translocation and other point mutations, as described by Deshpande et al. in a cohort of 25 SMM patients with 14 patients having translocations in the bone marrow and none found in ctDNA. Similarly, there was only matching of mutations in 4/13 patients (41), suggesting ctDNA may be limited either by DNA quantity or lack of correlation. Upon follow up, 2/25 patients had an increasing burden of ctDNA and expansion or appearance of new mutations, however, only 1 of these patients progressed to MM.
More promising is the use of circulating tumor cells (CTC) as a marker for high risk in precursor conditions. CTC’s are frequently found in NDMM, with some cohorts describing up to 95% of NDMM cases having detectable CTC using highly sensitive methods such as next generation flow cytometry (43). Additionally, higher burden of CTC’s correlates with higher ISS stage in NDMM (43), and worse outcomes in NDMM and RRMM, representing an atractive biomarker (44, 45). Furthermore, differences in RNA expression profile of CTC’s when compared to bone marrow or extramedullary (EMD) matched samples demostrate the complex and dynamic process of malignant plasma cell egress and potentially serve as the basis for markers to identify patients at high risk of developing EMD disease (46). CTC’s can be detected in SMM, with up to 78% of patients having detectable CTC’s using next generation flow cytometry (43), and higher burden of CTC’s are associated with an increased risk of of progression (43, 47), however, the treshold to define this high risk population has not been well defined and likely will vary depending on the type of assay used, however, in a recent cohort of 230 SMM with detectable CTC’s, patients with ≥ 0.02% CTC’s had a median TTP of 11 months (43), which is similar to what was known as “ultra-high risk” SMM, and were later classified as NDMM after the 2014 updated IMWG diagnostic criteria (48). Similarly, higher rates of CTC’s correlated with MGUS progression, again highlighting the potential role of this marker in evaluating precursor conditions (49).
The need for refinement of existing ctDNA assay or development of new technologies is needed in this population, as the use of ctDNA in precursor conditions seems to be limited at this point. Some of the limitations of CTC’s include the lack of widespread access to next generation flow cytometry, and lack of standarization of optimal tresholds to indentify high and low risk subgroups have varied, as prior results have varied likely secondary to differences in methdology and patient selection. However, both CTC’s and ctDNA represents attractive areas for research as they represent minimally invasive methodologies which could allow sequential sampling allowing for longitudinal follow up of patients. Additionally, they do not have the sampling bias that is seen with bone marrow samples, potentially allowing to capture the heterogeneity of MM.
Advances in MM therapy have come about due to therapies that target vulnerabilities of the plasma cell such as high protein load (proteasome inhibitor (PI); bortezomib, ixazomib and carfilzomib), dependence on specific transcription factors such as IKZF1 and IKZF3 which are degraded by immunomodulatory drugs (IMiDs; thalidomide, lenalidomide, and pomalidomide), the susceptibility of B cells to glucocorticoids (Dexamethasone) and the presence of specific B cell markers that can serve as targets for monoclonal antibodies and CAR-T cells (BCMA). Survival of patients with MM has significantly improved over the past decade with the introduction of these therapeutic strategies (5). However, these therapies are not curative, and nearly most of the patients with MM eventually relapse and require further therapy. In this part, we will discuss the genetic aberrations related to resistance to various therapies in MM (Table 1).
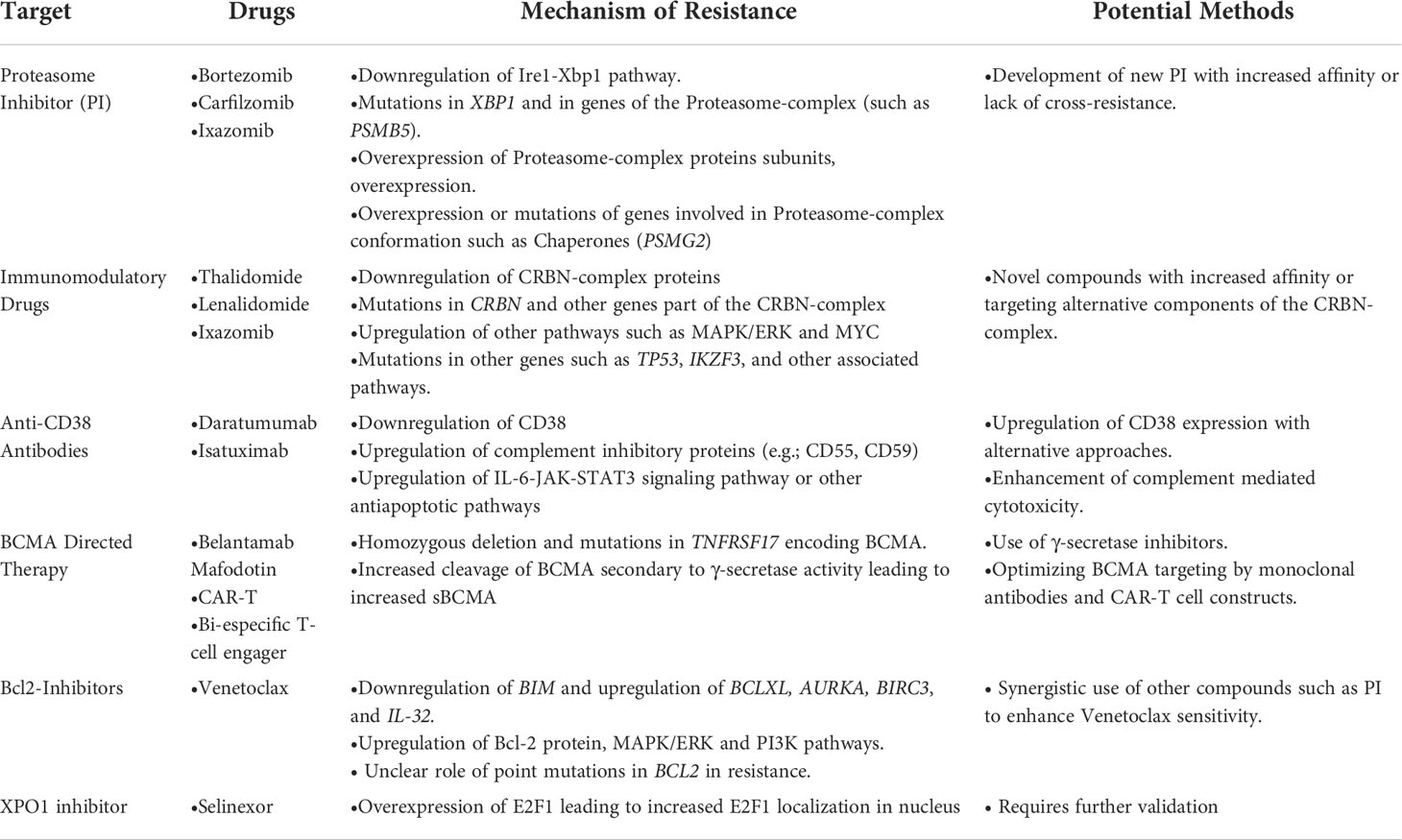
Table 1 Proposed mechanism of resistance and possible.
Proteasome inhibitors (PIs) were developed for the treatment of MM, and it has dramatically improved survival and treatment responses (50). These molecules driven apoptosis are related to the role of the ubiquitin proteasome pathway (UPP) in protein turnover. The UPP degrades intracellular aberrant or unnecessary proteins (misfolded and potentially toxic proteins) in eukaryotic cells. This process is essential to maintain cellular homeostasis and UPP is involved in apoptosis, cell survival and cell cycle control (51). It includes different steps: polyubiquitylation, deubiquitylation, and degradation of the target protein. The polyubiquitylation is mediated by ubiquitin activating enzyme 1 (E1), multiple ubiquitin-conjugating enzymes (E2) and ubiquitin-protein ligases (E3). The deubiquitylation is mediated by the proteasome regulatory subunits, which include both the 19S particle in the constitutive proteasome and the 11S particle in the immunoproteasome. The proteasome degrades the proteins via the function of the 20S core particle catalytic sites, and release oligopeptides. The catalytic sites of the 20S core particle have chymotrypsin-like (β5), trypsin-like (β2), and post-glutamyl peptide hydrolyzing, or caspase-like, (β1) activities (50).
Three proteasome inhibitors (Bortezomib, Carfilzomib and Ixazomib) have been approved by the US Food and Drug Administration (FDA) for treatment of MM. These molecules are administered subcutaneously, intravenously and orally, respectively and inhibit the 20S proteolytic core of the proteasome with different degree of inhibition of β5/chymotrypsin-like, the β2/trypsin-like or β1/post-glutamyl peptide hydrolyzing activity.
Proteasome inhibitors dramatically improved survival in myeloma patients; however, relapses are frequent and acquired resistance to treatment eventually emerges. The exacerbation of ER stress-related cytotoxicity induced by PIs could be reversed by the downregulation of Ire1-Xbp1 pathway. When stimulated by ER stress, Ire1 (one of three ER transmembrane proteins) transduces stress signals to the nucleus and activates the UPR via the activation of a transcription factor, Xbp1. Two XBP1 mutations have been detected in PI refractory patients. It was also reported that Xbp1s-negative tumor cells are resistant to PI; The mechanism of this resistance was explained by the decommitment to plasma cell maturation, and the lower IgG secretion in Xbp1-negative cells in comparison to PI-sensitive cells, as plasma cells depend on Xbp1s for Ig synthesis (52). Furthermore, bortezomib resistance was associated with mutations in PSMB5, gene encoding proteasome subunit β5, in myeloma cell lines (53). These mutations affect the PI-binding pocket S1 leading to conformational and steric changes to the proteasome drug- binding. However, somatic mutations of PSMB5 were rarely detected in patients and Carfilzomib response was less affected by PSMB5 mutations, due to its unique structure and binding (54). Due to their different targets and mode of action, MM patients who relapse on bortezomib are responders to treatment with Carfilzomib. Carfilzomib targets the 20S subunit and chaperones and stress-response regulators (55). Another mutation in proteasome assembly chaperone 2, PSMG2 gene has been also detected in one patient refractory to bortezomib (56). This protein is involved in mammalian 20S proteasome maturation, and exonic deletion of PSMG2 has also been reported in MM (57). Mutations in XBP1 (p.L314Ffs) was found in one patient resistant to PI at the time of tumor sampling (58). However, these potential findings need to be verified in larger studies to determine the scale and effect of such mutations in response to PIs.
Other mechanisms of PI resistance include overexpression of the proteasome subunit β5, overexpression of other subunits such as β2 and β1, downregulation of 19S proteasomal subunits in MM cell lines and MM patients (55), or overexpression of microenvironmental proteins (IL-6 and IGF-1) and chaperones (59, 60). Very recently, Li et al. showed that a deubiquitylase USP12 (Ubiquitin specific protease-12) is involved in bortezomib resistance in myeloma cells (61). In addition, proteins involved in proteasome function, oxidative stress, defense response and regulation of apoptotic process could be potential biomarkers of resistance to PIs (62).
Immunomodulatory agents are cornerstone in MM therapy. The first clinical trial assessing the effects of thalidomide, an immunomodulatory drug (IMiD), in 1999 showed that it is active against advanced myeloma in patients who had failed stem cell transplantation (63). Similar clinical trial was then conducted with 16 patients with relapsed myeloma and confirmed the previous results (64). An analogue or derivative of thalidomide (now known as lenalidomide) was tested in 2002 and showed good response in relapsed MM patients without the neurotoxicity and other adverse effects described after thalidomide treatment (somnolence, constipation, and neuropathy) (65). This drug increased TNF-alpha production by both CD4+ and CD8+ T cells (66). Another study showed that IMiD overcomes CTLA-4-Ig inhibitory effects of T cell proliferation (67). lenalidomide has been used in combination with dexamethasone for the treatment of adult patients with MM. It has also been used in combination with proteasome inhibitors. IMiDs not only target myeloma cells but also exert an indirect effect by activating the proliferation of the cytotoxic T and NK cells (65).The mechanism of IMiDs action was clearly described in studies, elucidating that cereblon (CRBN) plays an important role in mediating IMiDs anti-tumor effects (68). CRBN is a ubiquitously expressed E3 ligase protein, and it is the primary target of IMiDs (69). CRBN is part of a functional E3 ubiquitin ligase complex together with the DNA damage-binding protein-1 (DDB1), Cullin 4A and Roc1 (CUL4-RBX1-DDB1-CRBN) and acts as a substrate receptor for client proteins to be ubiquitinated and degraded by proteasome. IMiDs binding to cereblon E3 ligase complex induces the ubiquitination and degradation of transcription factors Ikaros and Aiolos, and subsequently downregulates IRF4 and c-MYC (70–72). These factors are essential for myeloma cells proliferation and survival. Furthermore, Ikaros and Aiolos are repressors of IL-2 (73), leading to T cell expansion in the tumor microenvironment.
In human MM cells, CRBN down-regulation resulted in the development of IMiD resistance (72, 74, 75). In a xenograft model, resistance to pomalidomide plus dexamethasone, but not lenalidomide plus dexamethasone was related to decreased CRBN expression but the resistance to both combinations is accompanied by upregulation of MEK/ERK pathway; this was confirmed by the inhibition of ERK which sensitizes resistant cells (76). In patient samples treated and became refractory to lenalidomide, a decrease in CRBN and increase in MYC expression were observed (77, 78).. In another study, higher CRBN expression level was associated with better PFS in patients treated with pomalidomide + low-dose dexamethasone (79). CRBN downregulation could be related to epigenetic modifications as epigenetic therapy re-sensitized IMiDs-resistant cell lines and changed the global chromatin accessibility associated with IMiDs resistance (75).
Recently, using a MM gene panel, Kortüm et al. showed an increased prevalence of mutations in CRBN and CRBN pathway genes (CRBN, CULB4, IRF4, IKZF1) in IMiDs (lenalidomide) refractory patients compared with newly diagnosed MM patients (58). These mutations in CRBN were either located within the IMiD-binding domain or occurred at sites truncating the protein. These mutations were not detected at the earlier time point when the patients were sensitive to IMiDs. In vitro, these mutations introduced in OCI-MY5 cell line caused a lack of response to lenalidomide. Another study showed other mutations in CRBN in MM patient who was initially responsive to thalidomide and lenalidomide treatment, who acquired resistance to lenalidomide over the disease course, and who was finally unresponsive to pomalidomide treatment at the time of tumor sampling. In this patient, a point mutation (p.Arg283Lys) and a truncating mutation (p.Glu99X) were detected within CRBN gene (56). Suggesting an association to IMiD resistance, another CRBN mutation has been identified in a patient unresponsive to initial lenalidomide and later pomalidomide treatment, this mutation is located in close proximity to the IMiD-binding site of the gene (58). A specific CRBN mutation (p.Cys326Gly) has also been detected at relapse in a patient treated with lenalidomide showing that this mutation contributed to lenalidomide resistance and clinical relapse (80). The cysteine amino acid caused by this mutation is involved in the Zinc finger motif, leading to protein misfolding and aggregation which destabilizes the IMiD binding domain of the protein (81). In a larger cohort of 198 newly diagnosed (ND) patients, 203 lenalidomide-refractory and 54 pomalidomide-refractory patients, WGS revealed that CRBN mutations occurred in 2.2% of lenalidomide-refractory and 9% of pomalidomide-refractory patients and 0.5% in ND-patients (82). Furthermore, resistance to IMiDs is also due to other genetic aberrations in CRBN such as copy number loss and structural variations (translocation or inversion) (82). Additionally, exon 10 splicing or the deletion of pomalidomide/lenalidomide binding domain is also associated with IMiDs resistance. The ratio of exon 10 spliced/full-length CRBN transcripts was increased in pomalidomide-refractory compared to ND or lenalidomide-refractory patients affects lenalidomide response in myeloma cell lines (82, 83). The presence of these CRBN aberrations were associated with significantly reduced PFS and OS in lenalidomide-refractory MM (82). Other mutations were detected in genes coding for the core CRL4CRBN E3-ligase complex and COP9 signalosome (80). There are 42 genes termed “CRBN/IMiD genes”, and 12/42 genes showed mutations in a dataset of 56 patients from the UK National Cancer Research Institute Myeloma XI trial at presentation, relapse or at both time points (80). These mutations are associated with relapse after lenalidomide treatment since 9/17 mutations arose in patients who had received lenalidomide and 6/9 had a higher cancer clonal fraction (CCF) in the relapse sample, suggesting that they may have been selected for by exposure to treatment. Furthermore, mutations in other related genes (TP53 and IKZF3) have been also detected in patients’ samples after lenalidomide treatment (77). An overview of the mechanisms of resistance to IMiD’s are described in Figure 3.
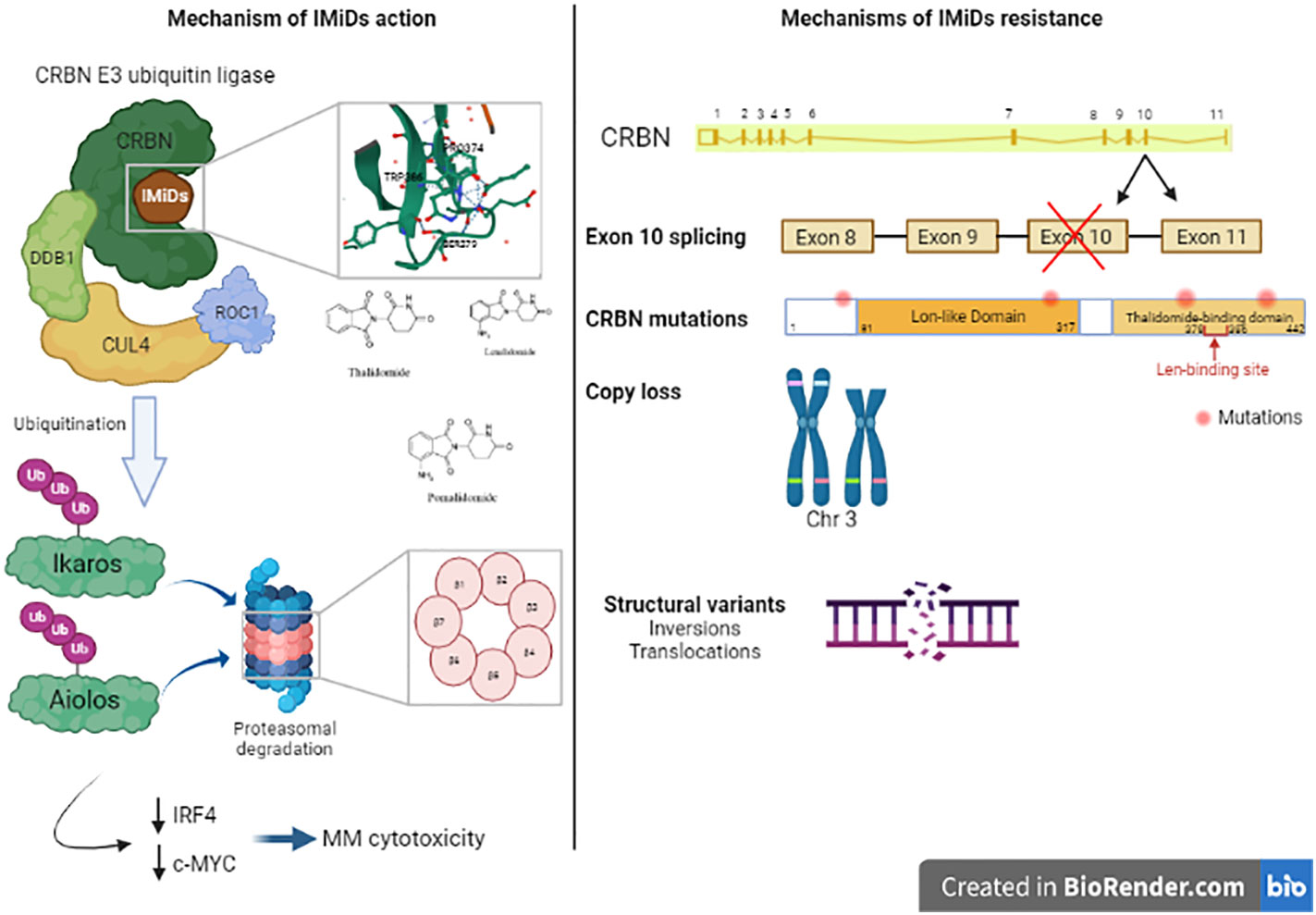
Figure 3 Mechanism of action of Immunomodulatory drugs (IMiDs): Immunomodulatroy drugs interacts with Cereblon (CRBN), which is a part of CRBN E3 ubiquitin ligase and induces the ubiquitination of Ikaros and Aiolos and their degradation which decreases the expression of IRF4 and c-MYC, two important factors in MM cell’s survival. This leads to MM cytotoxicity. Mechanisms of IMiDs resistance: Acquired resistance to IMiDs is associated with different genetic alterations in CRBN gene including exon 10 splicing, CRBN gene mutations, copy loss or structural variants like inversions or translocations in CRBN locus. These alterations impair CRBN function and consequently, MM cell’s response to IMiDs.
The development of immunotherapies in the treatment of MM has emerged with the use of monoclonal antibodies anti-CD38 (daratumumab (DARA) and Isatuximab) and the development of Anti-BCMA CAR-T cells and bispecific antibodies.
DARA, a human CD38-specific IgG1 antibody, was first approved in 2015 as a single agent therapy in RRMM patients (84). CD38 is a transmembrane glycoprotein expressed on MM cells and at low levels on normal lymphoid and myeloid cells (85). CD38 has ectoenzymatic activity in the catabolism of extracellular nucleotides and adhesion function related to its interaction with CD31 or hyaluronic acid which regulates the cell’s migration. CD38-targeting antibodies kill MM tumor cells via Fc-dependent immune effector mechanisms including complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and apoptosis upon secondary cross-linking (86). These mechanisms are dependent on the interaction of the Fc region of the antibody with Fcγ receptors (FcγRs) expressed on immune effector cells.
DARA combination with IMiDs (lenalidomide) and PIs (bortezomib) improved anti-MM activity and led to significant improvements in clinical outcomes in RRMM patients (87, 88) or untreated myeloma (89, 90), and DARA combinations were approved by FDA for the treatment of MM patients with at least one prior line of therapy in 2016. Moreover, further analysis of these trials showed an improvement of outcome of high-risk patients (del17p, t(4:14) or t(14:16)) in comparison to Rd and Vd only (91, 92).
DARA efficacy is correlated to the CD38 expression on MM cells (93). Several case reports showed CD38 downregulation in MM patient relapsing after DARA treatment as an escape mechanism (94, 95). In a subset of 102 patients treated with DARA monotherapy, CD38 pretreatment levels on MM cells were significantly higher in responders to DARA vs. non-responders (96). This study suggested the implication of CD38 downregulation and complement-inhibitory proteins CD55 and CD59 upregulation in the resistance to daratumumab. Another study from the same group suggested the implication of trogocytosis, cell membrane transfer from MM cells to monocytes and granulocytes, in the CD38 downregulation on the MM cell surface (97). CD38 downregulation was observed during DARA treatment and at the time of progression but CD38 expression increase after stopping DARA (96). Preclinical studies showed that all-trans retinoic acid (ATRA) increased CD38 expression and reduces CD55 and CD59 expression on MM cells and reverts DARA-resistance ex vivo (96, 98). However, ATRA-combined treatment did not enhance daratumumab response in MM patients, this may be explained by the transient upregulation of CD38 expression (99). In addition, HDAC inhibitor, Panobinostat also induces CD38 upregulation on MM cells ex vivo, leading to increased antimyeloma efficacy of daratumumab through an increase in ADCC (100). CD38 downregulation and reduced DARA-mediated antibody-dependent cellular cytotoxicity are observed in presence of bone marrow stromal cells (BMSCs) supernatant and IL-6 via JAK-STAT3 signaling, suggesting that bone marrow microenvironment could have an important role in protecting myeloma cells from DARA-induced cytotoxicity (101). In addition, BMSCs confer protection of MM cells against Dara-induced ADCC; this resistance is possibly related to the upregulation of anti-apoptotic proteins survivin and Mcl-1 (102).
BCMA (B-cell maturation antigen) is another ideal target for MM immunotherapy, it is expressed on MM cell lines and malignant plasma cells with high prevalence and its expression increases during disease progression from MGUS to SMM to MM (103–105). BCMA is involved in tumor proliferation via the delivery of pro-survival signals in MM cells and is ubiquitously expressed on the surface of MM cells. Anti-BCMA Chimeric Antigen Receptor (CAR) T cell therapy has been developed in MM and improved outcome in poor prognosis population like triple-refracted patients. The KarMMa study assessed the efficacy of idecabtagene vicleucel (ide-cel), a BCMA-directed CAR T cell therapy, in 128 refractory MM patients (≥3 prior regimens). This study showed that 76% of patients achieved CR and 29% MRD-negative status maintained for up to 12 months (in 60% of MRD-negative patients) (106). However, some patients relapsed following this CAR-T cell therapy. Da Vià et al. identified a homozygous deletion of BCMA-encoding gene (TNFRSF17) as a mechanism of escape from CAR-T cell therapy in relapsed patient (107). They observed a heterozygous deletion of this gene in MM patients even before treatment which can be a predicting marker for the BCMA-targeting therapies. Furthermore, another group showed that deletion of 16p, including the BCMA locus, and a truncating mutation in BCMA gene occur after anti-BCMA CAR-T cell therapy causing lack of BCMA expression at the time of relapse and explaining the lack of response to second CAR T cell infusion (108). The efficacy of BCMA-targeting therapies could be also dependent on the soluble BCMA (sBCMA) levels. Soluble BCMA release in the serum is due to the γ-secretase-induced shedding, a ubiquitous intramembranous protease that sheds membrane bound BCMA (109). sBCMA can bind and interfere with anti-BCMA antibodies, thus, drugs inhibiting gamma-secretase could enhance the efficacy of BCMA antibodies by reducing shedding of BCMA form the cell surface and subsequent interference of BCMA-specific antibodies by sBCMA (110). Soluble BCMA is also a biomarker for myeloma prognosis (111). The level of serum sBCMA is correlated with bone marrow plasma cell levels (112). sBCMA levels were higher in MGUS, and SMM patients who progressed to active MM showing that sBCMA may be a useful prognostic biomarker in MM (113).
Venetoclax (ABT-199) is a selective antiapoptotic protein B-cell lymphoma 2 inhibitor that induces apoptosis by displacing proapoptotic proteins from the antiapoptotic protein Bcl-2 (114). Venetoclax efficacy in inducing cell death ex vivo of MM cells form 76 patients showed higher sensitivity in samples from patients with t(11:14) than samples from t(11:14)-negative patients (115). MM patients with t(11;14) translocations, who represents 15-20% of MM patients be more sensitive to venetoclax as they have high ratios of BCL-2/MCL-1 in their tumor cells (116, 117). In human myeloma cell lines (HMCLs), a synergism between of compounds individually inhibiting Bcl-2 and Mcl-1 has been confirmed, and the combination of these drugs overcomes Mcl-1 resistance in MM (118, 119). Moreover, mTOR inhibition (everolimus) increases Bcl-2/Mcl-1 ratio in HMCLs by enhancing the binding of transcription factors IKZF3 and Blimp-1 to the BCL2 promoter and therefore enhances venetoclax anti-MM effects (120). Transcriptomic analysis in a panel of 31 myeloma cell lines and 25 patient samples showed an enrichment of specific B-cell genes and specific B cell surface markers, including CD20 and CD79A in venetoclax- sensitive myeloma cells (121). These results could help identifying patients who will respond to treatment with venetoclax. Recently, Todoerti et al (122) showed difference in expression pattern in BCL2 gene family members between MM cells with t(11:14) compared with MM cells without translocation. There is a need to determine potential predictive biomarkers for venetoclax -sensitive samples. Response to venetoclax correlated with higher BCL2:MCL1 and BCL2:BCL2L1 mRNA expression ratios as confirmed by biomarker analysis (BCL2 overexpression, BCL2L1 (coding for Bcl-xL) downregulation); these ratios are essential response predictors for venetoclax and were significantly higher in t(11:14) MM cells compared to MM cells without t(11:14) translocation (116, 123). In addition to Mcl-1, Bcl-xL may be a potential resistance factor to venetoclax (124) as single agent or in combination with bortezomib. The 1q21 gain is also a high-risk biomarker of resistance to venetoclax through the upregulation of MCL-1 (125). Dual targeting of Mcl-1 and Bcl-2 in vitro and ex vivo increased cell death in resistant cells and showed a synergy between these drugs (126, 127). Functional profiling of BCL2 dependence can also predict clinical response in MM, as an alternative approach to precision medicine that utilizes preclinical BH3 profiling and ex vivo testing with venetoclax to determine what level of ex vivo drug sensitivity is associated with clinical response (115). Furthermore, disease relapse after treatment resulted in increased NFkB activity and increased BCL2A1 (coding for BFL-1) as well as Bcl-2 and Bcl-xL (128).
A single-center retrospective study assessing the OS from time of venetoclax refractoriness in MM with t(11;14) or high BCL2 profile showed that the median PFS from the initiation of venetoclax was significantly longer when venetoclax was used as earlier line of treatment (fewer than three prior lines) at 23.2 months vs. 10.4 months in those with three or more prior lines, supporting the use of venetoclax as earlier lines of therapy in t(11:14) MM patients (129). Another study reported that venetoclax sensitivity is correlated to low Electron Transport Chain (ETC) complex I and II activities which propose the use of succinate ubiquinone reductase (SQR) activity as functional biomarker in MM patients for the prediction of venetoclax (130). SQR could be a metabolic target to sensitize resistant MM cells to venetoclax. Venetoclax in patients with t(11;14)(q13;q32) is considered the first example of personalized/precision therapy in the field of MM; suggesting that with more clinical studies this agent will have its place in MM patients management.
Venetoclax has found more success in combination with dexamethasone and bortezomib in comparison to its use as single agent in MM patients (131, 132). Bortezomib inhibits Mcl-1 by stabilizing the Mcl-1-neutralizing protein NOXA (133), dexamethasone up-regulates BCL2 in addition to the proapoptotic Bim, leaving Bcl-2–inhibited cells with an abundance of free Bim (134), therefore the combination of venetoclax, bortezomib, and dexamethasone showed promising effects in R/RMM patients especially those harboring t(11:14) with 78% of partial response (131). In a single-center retrospective study, low dose of venetoclax (≤250mg/day) in combination with DARA, Bortezomib and dexamethasone, 22 RR patients who had received a least 2 lines of prior therapy including at least one PI, showed an overall response rate of 80% in patients harboring t(11:14) vs. 31% in t(11:14)-negative patients, and most importantly, this response was without frequent infection-related serious adverse effects (135). In another phase I multicentric study, the combination of venetoclax, Dara, Dexamethasoe (Ven-Dd in R/RMM t(11:14) patients was compared to the combination of ven, DARA, dexamethasone and bortezomib (Ven-DVd) (cytogenetically unselected patients). The CR was 58% in Ven-Dd group vs. 46% in Ven-DVd group with 33% of patients achieved MRD- vs 21% in Ven-DVd group showing that the addition of DARA with or without bortezomib resulted in deep and durable responses and a higher MRD negativity rate and with no treatment related deaths has been observed in this study (136).
The export of several cargo molecules (proteins, mRNA) is heavily regulated by the protein XPO1, which serves as a mediator of nuclear export by the nuclear pore complex of several potential tumor suppressor proteins and other potential tumor driver proteins such as p53, RB1, and p27 (137). Selinexor is the first XPO1 inhibitor approved by the FDA for used in RRMM in combination with bortezomib after the phase 3 BOSTON trial showed an increased PFS in favor of patients treated with Selinexor bortezomib dexamethasone when compared to bortezomib and dexamethasone (138). Although some preliminary data MM cell lines and patient samples suggest overexpression of E2F1 as a potential mechanism of resistance (139), there is paucity of data on mechanism of resistance in MM to XPO1 inhibitors.
Our understanding of the genetic landscape of MGUS, SMM, and MM have increased significantly in recent years, resulting in fundamental changes in our model for myeloma development. Furthermore, understanding of the molecular and genetic events associated with a high risk of progression, such as mutations involving the MAPK pathway, DIS3, DNA damage repair, and MYC, are promising markers that could be incorporated in clinical practice and identify a high-risk subgroup that could benefit from early treatment. Additionally, the presence of biallelic inactivation of TP53 and 1q amplification identify a subgroup of patients with newly diagnosed MM with poor outcomes, and routine use of this alterations, along with previously defined cytogenetic and clinical models could help refine our stratification models and introduce intense regimens to this patient group. Additionally, the use of CTC’s represents an attractive prognostic biomarker that could be implemented to indicate risk of progression (in the case of MGUS/SMM) and prognosis (for MM), however, global definitions, standardization of the technology used, and cutoffs used are required before this is used routinely in clinical practice.
To this date, targeting t(11;14) or high Bcl2 expression using venetoclax is the most clear example of the target therapy principle in MM, and the worse outcomes in unselected population treated with venetoclax further highlight the selective activity of these targeted agents. As our understanding of the driver events in each subgroup of MM improve and new targeted therapies are developed, the routine identification of these alterations will become more relevant in clinical practice. Similarly, recent molecular studies on the mechanism of resistance to IMiD’s, PI’s, CD38 and BCMA directed therapies have shed light on potential mechanisms of resistance; however, they also show that other mechanisms of resistance are yet to be discovered.
To conclude, the advances in our understanding of the characteristics of MM and precursor conditions have paved the way for the therapeutic and diagnostic advances achieved over the last 20 years. The use of genetic and molecular alterations as prognostic and predictive tools for disease progression and therapeutic response could improve patient care and management. With the increasing affordability of these technologies, we expect they will become an important factor in decision-making in patient care.
MM, WD, and MB participated in manuscript design and conception. MM and WD elaborated the manuscript and figures. All authors participated in manuscript review and correction.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Myeloma — cancer stat facts. published march 16, 2022 . Available at: https://seer.cancer.gov/statfacts/html/mulmy.html (Accessed March 15, 2022).
2. Waxman A, Mink P, Devesa S, Anderson W, Weiss B, Kristinsson S, et al. Racial disparities in incidence and outcome in multiple myeloma: a population-based study. Blood (2010) 116(25):5501–6. doi: 10.1182/BLOOD-2010-07-298760
3. Kaur G, Mejia Saldarriaga M, Shah N, Catamero D, Yue L, Ashai N. Multiple myeloma in hispanics: Incidence, characteristics, survival, results of discovery, and validation using real-world and connect MM registry data. Clin Lymphoma Myeloma Leuk. (2021) 21(4):e384–97. doi: 10.1016/j.clml.2020.11.013
4. Benjamin M, Reddy S, Brawley OW. Myeloma and race: a review of the literature. Cancer Metastasis Rev (2003) 22(1):87–93. doi: 10.1023/A:1022268103136
5. Fonseca R, Abouzaid S, Bonafede M, Cai Q, Parikh K, Cosler L, et al. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia (2017) 31:9. doi: 10.1038/leu.2016.380
6. Kumar SK, Dispenzieri A, Lacy MQ, Gertz M, Buadi F, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia (2014) 28:5. doi: 10.1038/leu.2013.313
7. Landgren O, Gridley G, Turesson I, Caporaso N, Goldin L, Baris D, et al. Risk of monoclonal gammopathy of undetermined significance (MGUS) and subsequent multiple myeloma among African American and white veterans in the united states. Blood (2006) 107(3):904–6. doi: 10.1182/BLOOD-2005-08-3449
8. Landgren O, Kyle RA, Pfeiffer RM, Katzmann J, Caporaso N, Hayes R, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood (2009) 113(22):5412–7. doi: 10.1182/BLOOD-2008-12-194241
9. Kyle RA, Rajkumar SV. Multiple myeloma. Blood (2008) 111(6):2962–72. doi: 10.1182/BLOOD-2007-10-078022
10. Kyle RA, Greipp PR. Smoldering multiple myeloma. N Engl J Med (1980) 302(24):1347–9. doi: 10.1056/NEJM198006123022405
11. Dispenzieri A, Kyle RA, Katzmann JA, Therneau T, Larson D, Benson J, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood (2008) 111(2):785–9. doi: 10.1182/blood-2007-08-108357
12. Pérez-Persona E, Vidriales MB, Mateo G, García-Sanz R, Mateos M, De Coca A, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood (2007) 110(7):2586–92. doi: 10.1182/blood-2007-05-088443
13. Mateos MV, Kumar S, Dimopoulos MA, González-Calle V, Kastritis E, Hajek R, et al. International myeloma working group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J (2020) 10(10):102. doi: 10.1038/s41408-020-00366-3
14. Rajkumar SV, Kyle RA, Therneau TM, Melton L, Bradwell A, Clark R, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood (2005) 106(3):812–7. doi: 10.1182/BLOOD-2005-03-1038
15. Ravi P, Kumar S, Larsen JT, Gonsalves W, Buadi F, Lacy M, et al. Evolving changes in disease biomarkers and risk of early progression in smoldering multiple myeloma. Blood Cancer J (2016) 6(7):e454. doi: 10.1038/bcj.2016.65
16. de Larrea CF, Isola I, Pereira A, Cibeira M, Magnano L, Tovar N, et al. Evolving m-protein pattern in patients with smoldering multiple myeloma: Impact on early progression. Leukemia (2018) 32(6):1427–34. doi: 10.1038/s41375-018-0013-4
17. Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford A, et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J Clin Oncol (2020) 38:2380–9. doi: 10.1200/JCO.20.00437
18. Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J (2021) 11:4. doi: 10.1038/s41408-021-00474-8
19. Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia (2014) 28(2):269–77. doi: 10.1038/leu.2013.247
20. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel A, et al. Initial genome sequencing and analysis of multiple myeloma. Nature (2011) 471(7339):467–72. doi: 10.1038/nature09837
21. Walker BA, Boyle EM, Wardell CP, Murison A, Begum D, Dahir N, et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol (2015) 33(33):3911–20. doi: 10.1200/JCO.2014.59.1503
22. Boyle EM, Deshpande S, Tytarenko R, Ashby C, Wang Y, Bauer M, et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat Commun (2021) 12(1):1–13. doi: 10.1038/s41467-020-20524-2
23. Misund K, Keane N, Stein CK, Asmann Y, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia (2020) 34(1):322–6. doi: 10.1038/s41375-019-0543-4
24. Walker BA, Mavrommatis K, Wardell CP, Cody Ashby T, Bauer M, Davies F, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood (2018) 132(6):587–97. doi: 10.1182/blood-2018-03-840132
25. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence M, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell (2014) 25(1):91–101. doi: 10.1016/J.CCR.2013.12.015/ATTACHMENT/F8CA6BBE-57E7-46FB-840C-20C0C12E93BA/MMC10.XLSX
26. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell G, Mudie L, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell (2011) 144(1):27–40. doi: 10.1016/J.CELL.2010.11.055/ATTACHMENT/F05F620E-45EC-41D3-81B9-1AFB409EE44F/MMC6.PDF
27. Li Y, Roberts ND, Wala JA, Shapira O, Schumacher S, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature (2020) 578:7793. doi: 10.1038/s41586-019-1913-9
28. Rustad EH, Yellapantula VD, Glodzik D, Maclachlan K, Diamond B, Boyle E, et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov (2020) 1(3):258–73. doi: 10.1158/2643-3230.BCD-20-0132
29. Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood (2011) 118(3):675–8. doi: 10.1182/blood-2011-03-344069
30. Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Abascal F, Zheng-Lin B, et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun (2021) 12:1. doi: 10.1038/s41467-021-22140-0
31. Alexandrov LB, Jones PH, Wedge DC, Sale J, Campbell P, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet (2015) 47:12. doi: 10.1038/ng.3441
32. Zhan F, Huang Y, Colla S, Stewart J, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood (2006) 108(6):2020–8. doi: 10.1182/BLOOD-2005-11-013458
33. Broyl A, Hose D, Lokhorst H, De Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood (2010) 116(14):2543–53. doi: 10.1182/BLOOD-2009-12-261032
34. Bustoros M, Anand S, Sklavenitis-Pistofidis R, Redd R, Boyle E, Zhitomirsky B, et al. Genetic subtypes of smoldering multiple myeloma are associated with distinct pathogenic phenotypes and clinical outcomes. Nat Commun (2022) 13:1. doi: 10.1038/s41467-022-30694-w
35. Weinhold N, Ashby C, Rasche L, Chavan S, Stein C, Stephens O, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood (2016) 128(13):1735–44. doi: 10.1182/BLOOD-2016-06-723007
36. Weinhold N, Salwender HJ, Cairns DA, Raab M, Waldron G, Blau I, et al. Chromosome 1q21 abnormalities refine outcome prediction in patients with multiple myeloma - a meta-analysis of 2,596 trial patients. Haematologica. (2021) 106(10):2754–8. doi: 10.3324/HAEMATOL.2021.278888
37. D’Agostino M, Ruggeri M, Aquino S, Giuliani N, Arigoni M, Gentile M, et al. Impact of gain and amplification of 1q in newly diagnosed multiple myeloma patients receiving carfilzomib-based treatment in the forte trial. Blood (2020) 136(Supplement 1):38–40. doi: 10.1182/BLOOD-2020-137060
38. Gay F, Musto P, Rota-Scalabrini D, Bertamini L, Belotti A, Galli M, et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. Lancet Oncol (2021) 22(12):1705–20. doi: 10.1016/S1470-2045(21)00535-0/ATTACHMENT/3D490F77-13D8-475C-AEC6-5D03249A3F62/MMC2.PDF
39. Heuser M, Ofran Y, Boissel N, Brunet Mauri S, Craddock C, Janssen J, et al. Acute myeloid leukaemia in adult patients: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol (2020) 31(6):697–712. doi: 10.1016/J.ANNONC.2020.02.018/ATTACHMENT/8FD9C8AD-B48C-472C-8302-DCF911D402B8/MMC1.DOCX
40. Cescon DW, Bratman S v., SM C, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer (2020) 1:3. doi: 10.1038/s43018-020-0043-5
41. Deshpande S, Tytarenko RG, Wang Y, Boyle E, Ashby C, Schinke C, et al. Monitoring treatment response and disease progression in myeloma with circulating cell-free DNA. Eur J Haematol (2021) 106(2):230–40. doi: 10.1111/EJH.13541
42. Waldschmidt JM, Yee AJ, Vijaykumar T, Pinto R, Frede J, Anand P, et al. Cell-free DNA for the detection of emerging treatment failure in relapsed/ refractory multiple myeloma. Leukemia (2021) 2022:1–10. doi: 10.1038/s41375-021-01492-y
43. Garcés JJ, Puig N, Termini R, Cedena M, Moreno C, Pérez J, et al. Circulating tumor cells (CTCs) in smoldering and active multiple myeloma (MM): Mechanism of egression, clinical significance and therapeutic endpoints. Blood (2021) 138(Supplement 1):76–6. doi: 10.1182/BLOOD-2021-146535
44. Gonsalves W, Rajkumar S, Dispenzieri A, Dingli D, Timm M, Morice W, et al. Quantification of clonal circulating plasma cells in relapsed multiple myeloma. Br J Haematol (2014) 167(4):500–5. doi: 10.1111/BJH.13067
45. Nowakowski GS, Witzig TE, Dingli D, Tracz M, Gertz M, Lacy M, et al. Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood (2005) 106(7):2276–9. doi: 10.1182/BLOOD-2005-05-1858
46. Garcés JJ, Bretones G, Burgos L, Valdes-Mas R, Puig N, Cedena M, et al. Circulating tumor cells for comprehensive and multiregional non-invasive genetic characterization of multiple myeloma. Leukemia (2020) 34(11):3007–18. doi: 10.1038/S41375-020-0883-0
47. Gonsalves WI, Rajkumar S v, Dispenzieri A, Dingli D, Timm M, Morice W, et al. Quantification of circulating clonal plasma cells via multiparametric flow cytometry identifies patients with smoldering multiple myeloma at high risk of progression. Leukemia (2017) 31(1):130–5. doi: 10.1038/LEU.2016.205
48. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M, et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol (2014) 15(12):e538–48. doi: 10.1016/S1470-2045(14)70442-5
49. Sanoja-Flores L, Flores-Montero J, Garcés JJ, Paiva B, Puig N, García-Mateo A, et al. Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J (2018) 8:12. doi: 10.1038/s41408-018-0153-9
50. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol (2017) 14(7):417–33. doi: 10.1038/NRCLINONC.2016.206
51. Manasanch E, Korde N, Zingone A, Tageja N, Fernandez De Larrea C, Bhutani M, et al. The proteasome: mechanisms of biology and markers of activity and response to treatment in multiple myeloma. Leuk Lymphoma. (2014) 55(8):1707–14. doi: 10.3109/10428194.2013.828351
52. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats J, Stewart A, Reece D, Chung K, et al. Xbp1s-negative tumor b cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell (2013) 24(3):289–304. doi: 10.1016/J.CCR.2013.08.009
53. Balsas P, Galán-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMβ5 overexpression and polyploidy. Leuk Res (2012) 36(2):212–8. doi: 10.1016/J.LEUKRES.2011.09.011
54. Barrio S, Stühmer T, Da-Viá M, Barrio-Garcia C, Lehners N, Besse A, et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia. (2019) 33(2):447–56. doi: 10.1038/S41375-018-0216-8
55. Acosta-Alvear D, Cho MY, Wild T, Buchholz T, Lerner A, Simakova O, et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife (2015) 4. doi: 10.7554/ELIFE.08153
56. Egan J, Kortuem K, Kurdoglu AIzatt T, Aldrich J, Reiman R. Extramedullary myeloma whole genome sequencing reveals novel mutations in cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br J Haematol (2013) 161(5):748–51. doi: 10.1111/BJH.12291
57. Walker BA, Wardell CP, Melchor L, Hulkki S, Potter N, Johnson D, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood (2012) 120(5):1077–86. doi: 10.1182/BLOOD-2012-03-412981
58. Kortüm KM, Mai EK, Hanafiah NH, Shi C, Zhu Y, Bruins L, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and ras pathway genes. Blood (2016) 128(9):1226–33. doi: 10.1182/BLOOD-2016-02-698092
59. Chauhan D, Li G, Shringarpure R, Podar K, Ohtake Y, Hideshima T, et al. Blockade of Hsp27 overcomes bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res (2003) 63(19):6174–7.
60. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund C, Ma W, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood (2012) 120(16):3260–70. doi: 10.1182/BLOOD-2011-10-386789
61. Li H, Roy M, Liang L, Cao W, Hu B, Li Y, et al. Deubiquitylase USP12 induces pro-survival autophagy and bortezomib resistance in multiple myeloma by stabilizing HMGB1. Oncogene (2022) 41(9): 1298–308. doi: 10.1038/s41388-021-02167-9
62. Dytfeld D, Luczak M, Wrobel T, Usnarska-Zubkiewicz L, Brzezniakiewicz K, Jamroziak K, et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget (2016) 7(35):56726–36. doi: 10.18632/oncotarget.11059
63. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med (1999) 341(21):1565–71. doi: 10.1056/NEJM199911183412102
64. Vincent Rajkumar S, Fonseca R, Dispenzieri A, Lacy M, Lust J, Witzig T, et al. Thalidomide in the treatment of relapsed multiple myeloma. Mayo Clin Proc (2000) 75(9):897–901. doi: 10.4065/75.9.897
65. Richardson PG, Schlossman RL, Weller E, Hideshima T, Mitsiades C, Davies F, et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood (2002) 100(9):3063–7. doi: 10.1182/BLOOD-2002-03-0996
66. Marriott JB, Clarke IA, Dredge K, Muller G, Stirling D, Dalgleish AG. Thalidomide and its analogues have distinct and opposing effects on TNF-alpha and TNFR2 during co-stimulation of both CD4(+) and CD8(+) T cells. Clin Exp Immunol (2002) 130(1):75–84. doi: 10.1046/J.1365-2249.2002.01954.X
67. LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood (2004) 103(5):1787–90. doi: 10.1182/BLOOD-2003-02-0361
68. Stanková M, Bešše L, Sedlarikova L, Vrábel D, Hájek R, Sevcikove S. Cereblon - a new target of therapy in the treatment of multiple myeloma. Klin Onkol (2014) 27(5):326–30. doi: 10.14735/AMKO2014326
69. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science (2010) 327(5971):1345–50. doi: 10.1126/SCIENCE.1177319
70. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst S, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science (2014) 343(6168):301–5. doi: 10.1126/SCIENCE.1244851
71. Gandhi A, Kang J, Havens C, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors ikaros and aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol (2014) 164(6):811–21. doi: 10.1111/BJH.12708
72. Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi A, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia (2012) 26(11):2326–35. doi: 10.1038/LEU.2012.119
73. Thomas RM, Chunder N, Chen C, Umetsu SE, Winandy S, Wells AD. Ikaros enforces the costimulatory requirement for IL2 gene expression and is required for anergy induction in CD4+ T lymphocytes. J Immunol (2007) 179(11):7305–15. doi: 10.4049/JIMMUNOL.179.11.7305
74. Zhu YX, Braggio E, Shi CX, Bruins L, Schmidt J, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood (2011) 118(18):4771–9. doi: 10.1182/BLOOD-2011-05-356063
75. Dimopoulos K, Søgaard Helbo A, Fibiger Munch-Petersen H, Sjö L, Christensen J, Sommer Kristensen L, et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol (2018) 12(2):180–95. doi: 10.1002/1878-0261.12157
76. Ocio EM, Fernández-Lázaro D, San-Segundo L, López-Corral L, Corchete L, Gutiérrez N, et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia. (2015) 29(3):705–14. doi: 10.1038/LEU.2014.238
77. Tachita T, Kinoshita S, Ri M, Aoki S, Asano A, Kanamori T, et al. Expression, mutation, and methylation of cereblon-pathway genes at pre- and post-lenalidomide treatment in multiple myeloma. Cancer Sci (2020) 111(4):1333–43. doi: 10.1111/CAS.14352
78. Rychak E, Mendy D, Shi T, Ning Y, Leisten J, Lu L, et al. Pomalidomide in combination with dexamethasone results in synergistic anti-tumour responses in pre-clinical models of lenalidomide-resistant multiple myeloma. Br J Haematol (2016) 172(6):889–901. doi: 10.1111/BJH.13905
79. Qian X, Dimopoulos MA, Amatangelo M, Bjorklund C, Towfic F, Flynt E, et al. Cereblon gene expression and correlation with clinical outcomes in patients with relapsed/refractory multiple myeloma treated with pomalidomide: an analysis of STRATUS. Leuk Lymphoma (2019) 60(2):462–70. doi: 10.1080/10428194.2018.1485915
80. Jones J, Barber A, Le Bihan Y v, Weinhold N, Ashby C, Walker B, et al. Mutations in CRBN and other cereblon pathway genes are infrequently associated with acquired resistance to immunomodulatory drugs. Leukemia (2021) 35(10):3017–20. doi: 10.1038/S41375-021-01373-4
81. Akuffo AA, Alontaga AY, Metcalf R, Beatty M, Becker A, McDaniel J, et al. Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J Biol Chem (2018) 293(16):6187–200. doi: 10.1074/JBC.M117.816868
82. Gooding S, Ansari-Pour N, Towfic F, Estévez M, Chamberlain P, Tsai K, et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood (2021) 137(2):232–7. doi: 10.1182/BLOOD.2020007081
83. Neri P, Maity R, Keats JJ, Tagoug I, Simms J, Auclair D, et al. Cereblon splicing of exon 10 mediates IMiDs resistance in multiple myeloma: Clinical validation in the CoMMpass trial. Blood (2016) 128(22):120. doi: 10.1182/BLOOD.V128.22.120.120
84. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis N, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet (2016) 387(10027):1551–60. doi: 10.1016/S0140-6736(15)01120-4
85. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein A, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev (2008) 88(3):841–86. doi: 10.1152/PHYSREV.00035.2007
86. van de Donk NWCJ, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood (2018) 131(1):13–29. doi: 10.1182/BLOOD-2017-06-740944
87. Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis N, Usmani S, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375(14):1319–31. doi: 10.1056/NEJMOA1607751
88. Palumbo A, Chanan-Khan A, Weisel K, Nooka A, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375(8):754–66. doi: 10.1056/NEJMOA1606038
89. Facon T, Kumar S, Plesner T, Orlowski R, Moreau P, Bahlis N, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med (2019) 380(22):2104–15. doi: 10.1056/NEJMOA1817249
90. Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med (2018) 378(6):518–28. doi: 10.1056/NEJMOA1714678
91. Dimopoulos MA, San-Miguel J, Belch A, White D, Benboubker L, Cook G, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica (2018) 103(12):2088–96. doi: 10.3324/HAEMATOL.2018.194282
92. Spencer A, Lentzsch S, Weisel K, Avet-Loiseau H, Mark T, Spicka I, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica (2018) 103(12):2079–87. doi: 10.3324/HAEMATOL.2018.194118
93. van de Donk NWCJ, Usmani SZ. CD38 antibodies in multiple myeloma: Mechanisms of action and modes of resistance. Front Immunol (2018) 9:2134. doi: 10.3389/FIMMU.2018.02134
94. Minarik J, Novak M, Flodr P, Balcarkova J, Mlynarcikova M, Krhovska P, et al. CD38-negative relapse in multiple myeloma after daratumumab-based chemotherapy. Eur J Haematol (2017) 99(2):186–9. doi: 10.1111/EJH.12902
95. Ise M, Matsubayashi K, Tsujimura H, Kumagai K. Loss of CD38 expression in relapsed refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. (2016) 16(5):e59–64. doi: 10.1016/J.CLML.2016.02.037
96. Nijhof IS, Casneuf T, van Velzen J, Van Kessel B, Axel A, Syed K, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood (2016) 128(7):959–70. doi: 10.1182/BLOOD-2016-03-703439
97. Krejcik J, Frerichs KA, Nijhof IS, Van Kessel B, Van Velzen J, Bloem A, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res (2017) 23(24):7498–511. doi: 10.1158/1078-0432.CCR-17-2027
98. Nijhof IS, Groen RWJ, Lokhorst HM, Van Kessel B, Bloem A, Van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia (2015) 29(10):2039–49. doi: 10.1038/LEU.2015.123
99. Frerichs K, Minnema M, Levin M, Broijl A, Bos G, Kersten M, et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma. Blood Adv (2021) 5(23):5128–39. doi: 10.1182/BLOODADVANCES.2021005220
100. García-Guerrero E, Gogishvili T, Danhof S, Schreder M, Pallaud C, Pérez-Simón J, et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood (2017) 129(25):3386–8. doi: 10.1182/BLOOD-2017-03-770776
101. Ogiya D, Liu J, Ohguchi H, Kurata K, Samur M, Tai Y, et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic implications. Blood (2020) 136(20):2334–45. doi: 10.1182/BLOOD.2019004332
102. de Haart S, Holthof L, Noort W, Minnema M, Emmelot M, Aarts-Riemens T, et al. Sepantronium bromide (YM155) improves daratumumab-mediated cellular lysis of multiple myeloma cells by abrogation of bone marrow stromal cell-induced resistance. Haematologica (2016) 101(8):e339–43. doi: 10.3324/HAEMATOL.2015.139667
103. Yong K, Germaschewski F, Rodriguez-Justo M, Bounds D, Lee L, Mayes P, et al. Evaluation of bcma as a therapeutic target in multiple myeloma using an antibody-drug conjugate. Blood (2013) 122(21):4447–7. doi: 10.1182/BLOOD.V122.21.4447.4447
104. Carpenter R, Evbuomwan M, Pittaluga S, Rose J, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res (2013) 19(8):2048–60. doi: 10.1158/1078-0432.CCR-12-2422
105. Novak A, Darce J, Arendt B, Harder B, Henderson K, Kindsvogel W, et al. Expression of BCMA, TACI, and BAFF-r in multiple myeloma: a mechanism for growth and survival. Blood (2004) 103(2):689–94. doi: 10.1182/BLOOD-2003-06-2043
106. Munshi N, Anderson L, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMOA2024850
107. Da Vià M, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med (2021) 27(4):616–9. doi: 10.1038/S41591-021-01245-5
108. Samur M, Fulciniti M, Aktas Samur A, Bazarbachi A, Tai Y, Prabhala R, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun (2021) 12(1). doi: 10.1038/S41467-021-21177-5
109. Laurent S, Hoffmann F, Kuhn P, Cheng Q, Chu Y, Schmidt-Supprian M, et al. γ-secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun (2015) 6:7333. doi: 10.1038/NCOMMS8333
110. Chen H, Li M, Xu N, Ng N, Sanchez E, Soof C, et al. Serum b-cell maturation antigen (BCMA) reduces binding of anti-BCMA antibody to multiple myeloma cells. Leuk Res (2019) 81:62–6. doi: 10.1016/J.LEUKRES.2019.04.008
111. Jew S, Chang T, Bujarski S, Soof C, Chen H, Safaie T, et al. Normalization of serum b-cell maturation antigen levels predicts overall survival among multiple myeloma patients starting treatment. Br J Haematol (2021) 192(2):272–80. doi: 10.1111/BJH.16752
112. Ghermezi M, Li M, Vardanyan S, Harutyunyan N, Gottlieb J, Berenson A, et al. Serum b-cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica (2017) 102(4):785–95. doi: 10.3324/HAEMATOL.2016.150896
113. Visram A, Soof C, Rajkumar S, Kumar S, Bujarski S, Spektor T, et al. Serum BCMA levels predict outcomes in MGUS and smoldering myeloma patients. Blood Cancer J (2021) 11(6). doi: 10.1038/S41408-021-00505-4
114. Souers A, Leverson J, Boghaert E, Ackler S, Catron N, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19(2):202–8. doi: 10.1038/NM.3048
115. Matulis S, Gupta V, Neri P, Bahlis N, Maciag P, Leverson J, et al. Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia (2019) 33(5):1291–6. doi: 10.1038/S41375-018-0374-8
116. Kumar S, Kaufman J, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood (2017) 130(22):2401–9. doi: 10.1182/BLOOD-2017-06-788786
117. Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson J, Souers A, et al. The bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia (2014) 28(1):210–2. doi: 10.1038/LEU.2013.216
118. Wong KY, Chim CS. Venetoclax, bortezomib and S63845, an MCL1 inhibitor, in multiple myeloma. J Pharm Pharmacol (2020) 72(5):728–37. doi: 10.1111/JPHP.13240
119. Siu K, Huang C, Panaroni C, Mukaihara K, Fulzele K, Soucy R, et al. BCL2 blockade overcomes MCL1 resistance in multiple myeloma. Leukemia (2019) 33(8):2098–102. doi: 10.1038/S41375-019-0421-0
120. Osada N, Kikuchi J, Koyama D, Kuroda Y, Yasui H, Leverson J, et al. mTOR inhibitors sensitize multiple myeloma cells to venetoclax via IKZF3- and blimp-1-mediated BCL-2 upregulation. Haematologica (2021) 106(11):3008–13. doi: 10.3324/HAEMATOL.2021.278506
121. Gupta V, Barwick B, Matulis S, Shirasaki R, Jaye D, Keats J, et al. Venetoclax sensitivity in multiple myeloma is associated with b-cell gene expression. Blood (2021) 137(26):3604–15. doi: 10.1182/BLOOD.2020007899
122. Todoerti K, Taiana E, Puccio N, Favasuli V, Lionetti M, Silvestris I, et al. Transcriptomic analysis in multiple myeloma and primary plasma cell leukemia with t(11;14) reveals different expression patterns with biological implications in venetoclax sensitivity. Cancers (Basel) (2021) 13(19):4898. doi: 10.3390/CANCERS13194898
123. Punnoose E, Leverson J, Peale F, Boghaert E, Belmont L, Tan N, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther (2016) 15(5):1132–44. doi: 10.1158/1535-7163.MCT-15-0730
124. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez M, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood (2018) 132(25):2656–69. doi: 10.1182/BLOOD-2018-03-836718
125. Slomp A, Moesbergen L, Gong J, Cuenca M, von dem Borne P, Sonneveld P, et al. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv (2019) 3(24):4202–14. doi: 10.1182/BLOODADVANCES.2019000702
126. Algarín E, Díaz-Tejedor A, Mogollón P, Hernández-García S, Corchete L, San-Segundo L, et al. Preclinical evaluation of the simultaneous inhibition of MCL-1 and BCL-2 with the combination of S63845 and venetoclax in multiple myeloma. Haematologica (2020) 105(3):E116–20. doi: 10.3324/HAEMATOL.2018.212308
127. Seiller C, Maiga S, Touzeau C, Bellanger C, Kervoëlen C, Descamps G, et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/BAK hetero-complexes. Cell Death Dis (2020) 11(5). doi: 10.1038/S41419-020-2505-1
128. Spaan I, van de Stolpe A, Raymakers RA, Peperzak V. Multiple myeloma relapse is associated with increased NFκB pathway activity and upregulation of the pro-survival BCL-2 protein BFL-1. Cancers (Basel) (2021) 13(18):4668. doi: 10.3390/CANCERS13184668
129. Maples K, Nooka A, Gupta V, Joseph N, Heffner L, Hofmeister C, et al. Natural history of multiple myeloma patients refractory to venetoclax: A single center experience. Am J Hematol (2021) 96(3):E68–71. doi: 10.1002/AJH.26064
130. Bajpai R, Sharma A, Achreja A, Edgar C, Wei C, Siddiqa A, et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat Commun (2020) 11(1). doi: 10.1038/S41467-020-15051-Z
131. Moreau P, Chanan-Khan A, Roberts A, Agarwal A, Facon T, Kumar S, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood (2017) 130(22):2392–400. doi: 10.1182/BLOOD-2017-06-788323
132. Kumar S, Harrison S, Cavo M, de la Rubia J, Popat R, Gasparetto C, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol (2020) 21(12):1630–42. doi: 10.1016/S1470-2045(20)30525-8
133. Qin J, Ziffra J, Stennett L, Bodner B, Bonish B, Chaturvedi V, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res (2005) 65(14):6282–93. doi: 10.1158/0008-5472.CAN-05-0676
134. Matulis S, Gupta V, Nooka A, Hollen H, Kaufman J, Lonial S, et al. Dexamethasone treatment promotes bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia (2016) 30(5):1086–93. doi: 10.1038/LEU.2015.350
135. Regidor B, Goldwater M, Wang J, Bujarski S, Swift R, Eades B, et al. Low dose venetoclax in combination with bortezomib, daratumumab, and dexamethasone for the treatment of relapsed/refractory multiple myeloma patients-a single-center retrospective study. Ann Hematol (2021) 100(8):2061–70. doi: 10.1007/S00277-021-04555-3
136. Bahlis N, Baz R, Harrison S, Quach H, Ho S, Vangsted A, et al. Phase I study of venetoclax plus daratumumab and dexamethasone, with or without bortezomib, in patients with relapsed or refractory multiple myeloma with and without t(11;14). J Clin Oncol (2021) 39(32):3602–12. doi: 10.1200/JCO.21.00443
137. Azmi AS, Uddin MH, Mohammad RM. The nuclear export protein XPO1 — from biology to targeted therapy. Nat Rev Clin Oncol (2020) 18:3. doi: 10.1038/s41571-020-00442-4
138. Grosicki S, Simonova M, Spicka I, Pour L, Kriachok I, Gavriatopoulou M, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet (2020) 396(10262):1563–73. doi: 10.1016/S0140-6736(20)32292-3
Keywords: myeloma and other plasma cell dyscrasias, resistance, molecular subclassification, smoldering myeloma (SMM), MGUS, risk stratification
Citation: Mejia Saldarriaga M, Darwiche W, Jayabalan D, Monge J, Rosenbaum C, Pearse RN, Niesvizky R and Bustoros M (2022) Advances in the molecular characterization of multiple myeloma and mechanism of therapeutic resistance. Front. Oncol. 12:1020011. doi: 10.3389/fonc.2022.1020011
Received: 15 August 2022; Accepted: 07 October 2022;
Published: 27 October 2022.
Edited by:
Evdoxia Hatjiharissi, University General Hospital of Thessaloniki AHEPA, GreeceReviewed by:
Song Xu, Tianjin Medical University General Hospital, ChinaCopyright © 2022 Mejia Saldarriaga, Darwiche, Jayabalan, Monge, Rosenbaum, Pearse, Niesvizky and Bustoros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Bustoros, bWFiNDAzM0BtZWQuY29ybmVsbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.