
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 29 September 2022
Sec. Genitourinary Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1016894
This article is part of the Research TopicReviews in Genitourinary OncologyView all 19 articles
 Simona Camero1†
Simona Camero1† Matteo Cassandri2,3†
Matteo Cassandri2,3† Silvia Pomella3,4†Luisa Milazzo5Francesca Vulcano5
Silvia Pomella3,4†Luisa Milazzo5Francesca Vulcano5 Antonella Porrazzo2,6
Antonella Porrazzo2,6 Giovanni Barillari4
Giovanni Barillari4 Cinzia Marchese7Silvia Codenotti8Miriam Tomaciello2
Cinzia Marchese7Silvia Codenotti8Miriam Tomaciello2 Rossella Rota3
Rossella Rota3 Alessandro Fanzani8
Alessandro Fanzani8 Francesca Megiorni7*‡
Francesca Megiorni7*‡ Francesco Marampon2*‡
Francesco Marampon2*‡Management of rhabdomyosarcoma (RMS), the most common soft tissue sarcoma in children, frequently accounting the genitourinary tract is complex and requires a multimodal therapy. In particular, as a consequence of the advancement in dose conformity technology, radiation therapy (RT) has now become the standard therapeutic option for patients with RMS. In the clinical practice, dose and timing of RT are adjusted on the basis of patients’ risk stratification to reduce late toxicity and side effects on normal tissues. However, despite the substantial improvement in cure rates, local failure and recurrence frequently occur. In this review, we summarize the general principles of the treatment of RMS, focusing on RT, and the main molecular pathways and specific proteins involved into radioresistance in RMS tumors. Specifically, we focused on DNA damage/repair, reactive oxygen species, cancer stem cells, and epigenetic modifications that have been reported in the context of RMS neoplasia in both in vitro and in vivo studies. The precise elucidation of the radioresistance-related molecular mechanisms is of pivotal importance to set up new more effective and tolerable combined therapeutic approaches that can radiosensitize cancer cells to finally ameliorate the overall survival of patients with RMS, especially for the most aggressive subtypes.
Rhabdomyosarcoma (RMS) is a highly aggressive soft tissue sarcoma (STS) that primarily affects pediatric patients, accounting for 5% of all childhood cancers and representing 3% of STS in adult, for whom it has a worse prognosis. As shown in Figure 1, the most common RMS location is in the head and neck region (35%–40%), genitourinary tract (bladder/prostate, 11%), genitourinary tract non-bladder/prostate (male, 12%; female, 5%), and limbs (16%). Signs and symptoms at presentation will depend on the site of the primary tumor, whether there is extension into contiguous organs, and, in some cases, the presence of metastatic disease (1). Originally, two major subtypes of RMS were recognized: embryonal RMS (ERMS), preferring male children, and alveolar RMS (ARMS), which remains constant throughout childhood and adolescence, showing the worse prognosis. Others two rarer RMS subtypes are the pleomorphic RMS and the spindle cell/sclerosing RMS, which typically occur in adults and children, respectively (2, 3). ARMSs more frequently carry t (2;13)(q35;q14) or t(1;13)(p36;q14) chromosomal translocations that, juxtaposing Paired box gene 3 (PAX3) on chromosome 2 or PAX7 on chromosome 1 with Forkhead box protein O1 (FOXO1) on chromosome 13, generate PAX3–FOXO1 and PAX7–FOXO1 fusion genes, respectively, and finally transcribe/translate into pro-oncogenic fusion proteins with an aberrantly enhanced transcriptional activity (4, 5). Because fusion protein presence correlates with a poorer prognosis, nowadays, the preferred RMS classification is expressing, i.e., “fusion positive” (FP-RMS), or not expressing fusion protein, i.e., “fusion negative” (FN-RMS) (6). ERMSs (FN-RMS tumors), more frequently present various mutations largely converging on a limited number of pathways, also perturbed in FP-RMSs, indicating some commonality in the molecular driving forces in RMS (2). FN-RMSs often harbor a mutation affecting mitogen-activated protein kinases and/or PI3K–AKT–mTOR pathways (7, 8), aberrantly activated also in FP-RMS, to the ability of fusion proteins to activate several cell surface receptor tyrosine kinases upstream of these pathways (5). Notably, patients with FN-ARMS are clinically and molecularly indistinguishable from ERMS (9).
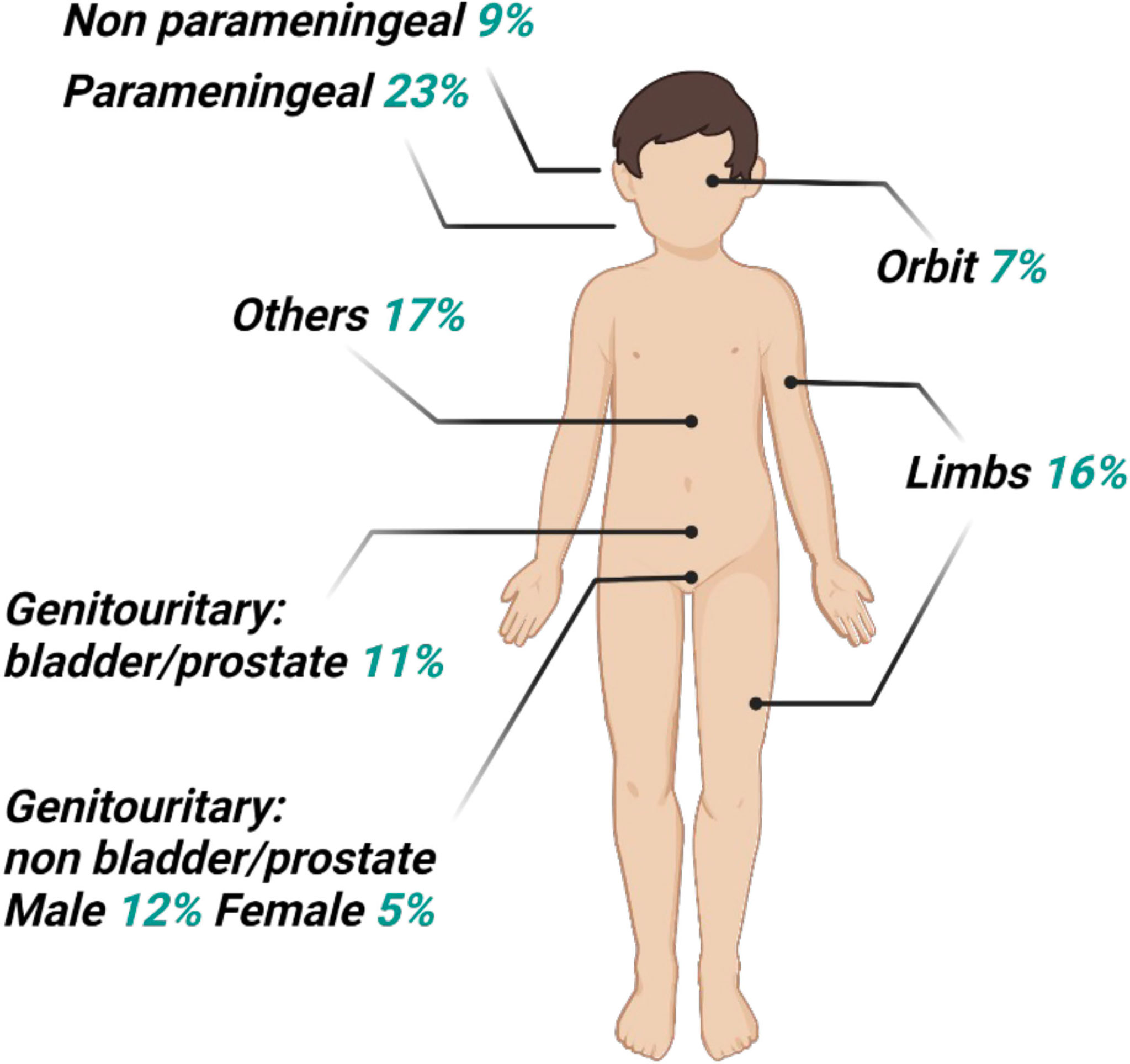
Figure 1 Distribution of primary sites for rhabdomyosarcoma. The head and neck site may be subdivided as 7% orbit, 8% other head, 23% parameningeal, and 9% non-parameningeal. The pelvic sites may be subdivided as 11% bladder and prostate, and 5% female genital or 12% male non-bladder/prostate.
This review presents a brief overview of the guidelines for the diagnosis and treatment of RMS, with particular emphasis on the role of radiotherapy (RT) and on the molecular mechanisms mainly responsible for radioresistance, focusing on possible candidate radiosensitizing strategies in RMS. In particular, after summarizing the key principles of the management of the patient with RMS, from diagnosis to treatment, focusing on the role of RT and on the novelties in terms of indications, therapy schemes, and treatment techniques, we will analyze the principles of radiobiology and of the RMS and, therefore, the molecular mechanisms of radioresistance.
After the head and neck site (35%–50%), the genitourinary tract represents the primary site for around 20%–25% of RMS pediatric patients, resulting as exceedingly rare in adults (10, 11). Vagina or uterus, favorable sites, are more frequently involved, followed by the kidney, bladder, or prostate, considered as unfavorable sites (12, 13). At the onset, hematuria and urinary obstruction represent the signs and symptoms more frequent, whereas 10%–20% of pediatric and 40% of adult patients present distant metastases. RMS diagnosis requires, in addition to standard laboratory (complete blood counts, electrolytes, renal function tests, liver function tests, and urinalysis), the direct evaluation of tumor tissue derived from either an incisional/excisional biopsy or a core needle biopsy (1) magnetic resonance imaging, for local staging, and computed tomography (CT) and [F-18]2-fluoro-2-deoxyglucose positron emission tomography (18F-FDG PET)/CT), for systemic staging and the risk stratification (14, 15). TNM staging is based on the anatomic location and invasiveness of the primary tumor, tumor size, nodal status, and extent of metastasis (Tables 1, 2). Intergroup Rhabdomyosarcoma Study Group (IRSG) establishes risk group stratification, identifying low-, standard-, high, or very high–risk patients. The clinical subgroup is primarily determined by IRSG group, lymph node involvement, fusion protein expression, site, and age (16, 17). Risk stratification is summarized in Table 3. The bioptic samples must be subjected to a series of histology and molecular pathology studies aimed at measuring myogenic markers like desmin, skeletal alpha-actin, myosin, and myoglobin and early myogenesis transcription factors like MyoD and myogenin (18). The analysis of these markers is achieved with immunohistochemical assays, and it is combined with cell morphological assessment in light microscopy, used to distinguish RMS from other childhood neoplasms also expressing myogenic proteins (18). More recently, molecular analysis has become an essential tool for differential diagnosis and classification of RMS (Figure 2). Specifically, Real-Time PCR (RT-PCR) and Fluorescence in situ hybridization (FISH) assays designed to measure the expression of fusion gene PAX3–FOXO1 or PAX7–FOXO1 are very useful to identify subsets of ARMS, and microarray genome-wide RNA expression techniques have been shown to generate, through various statistical algorithms, “diagnostic signatures” of the FP-RMS and FN-RMS categories (2).

Table 1 TNM classification for rhabdomyosarcoma.
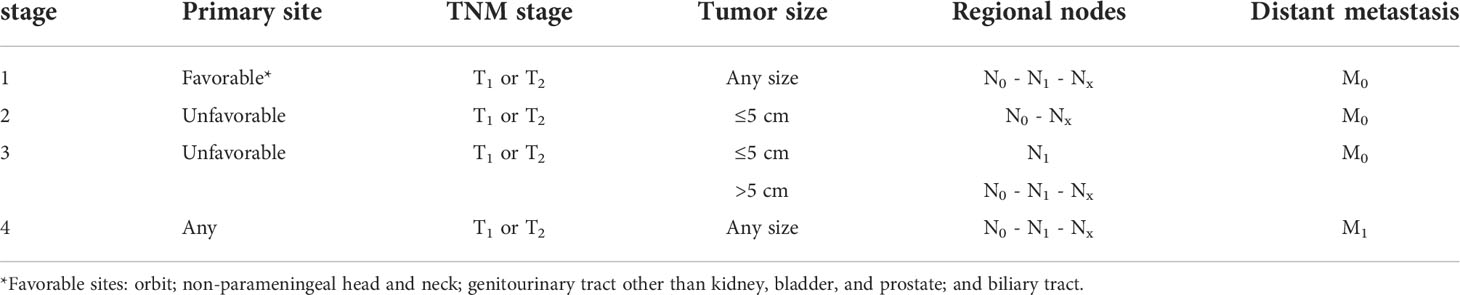
Table 2 TNM stage for rhabdomyosarcoma.
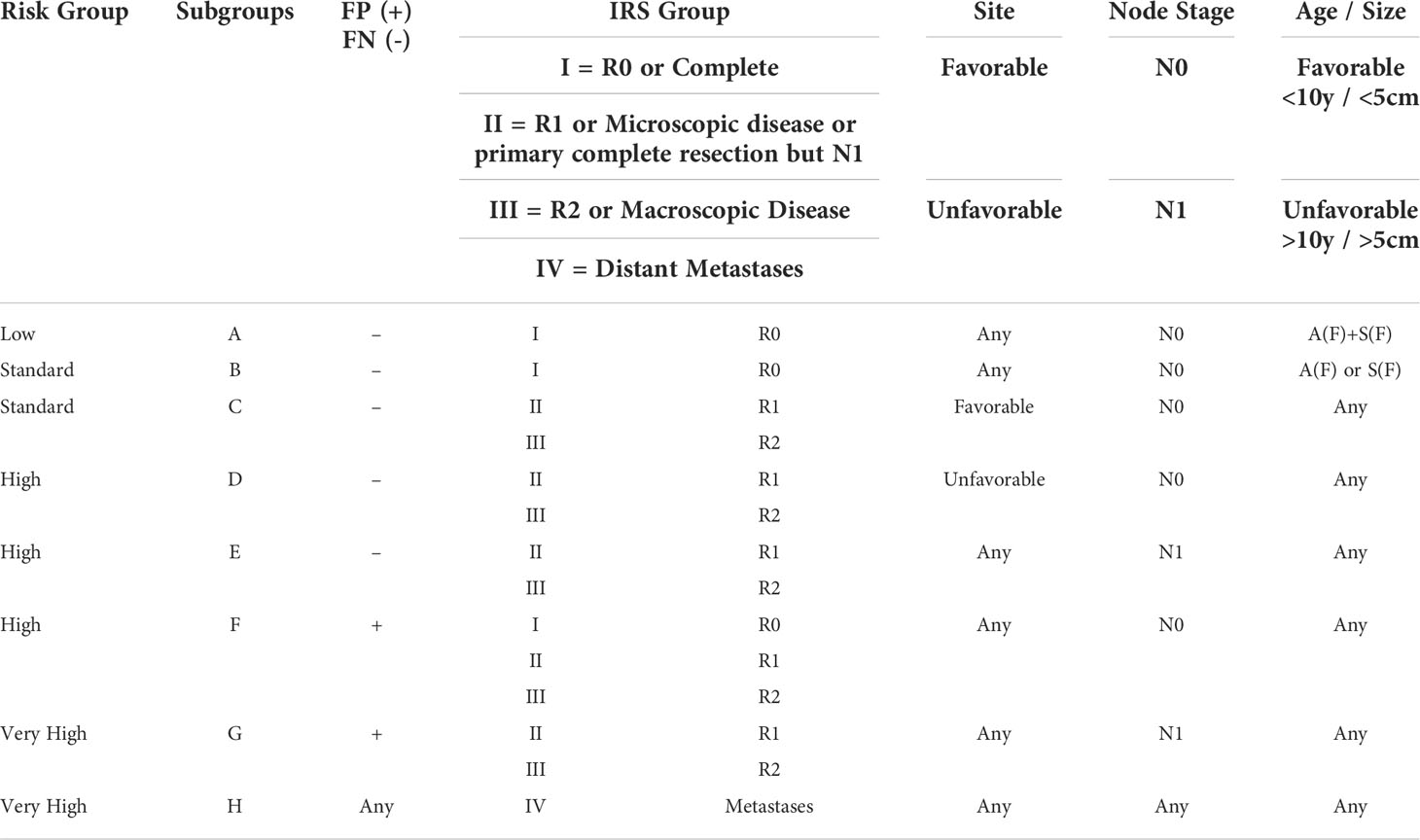
Table 3 European Paediatric Soft Tissue Sarcoma Study Group staging of rhabdomyosarcoma.
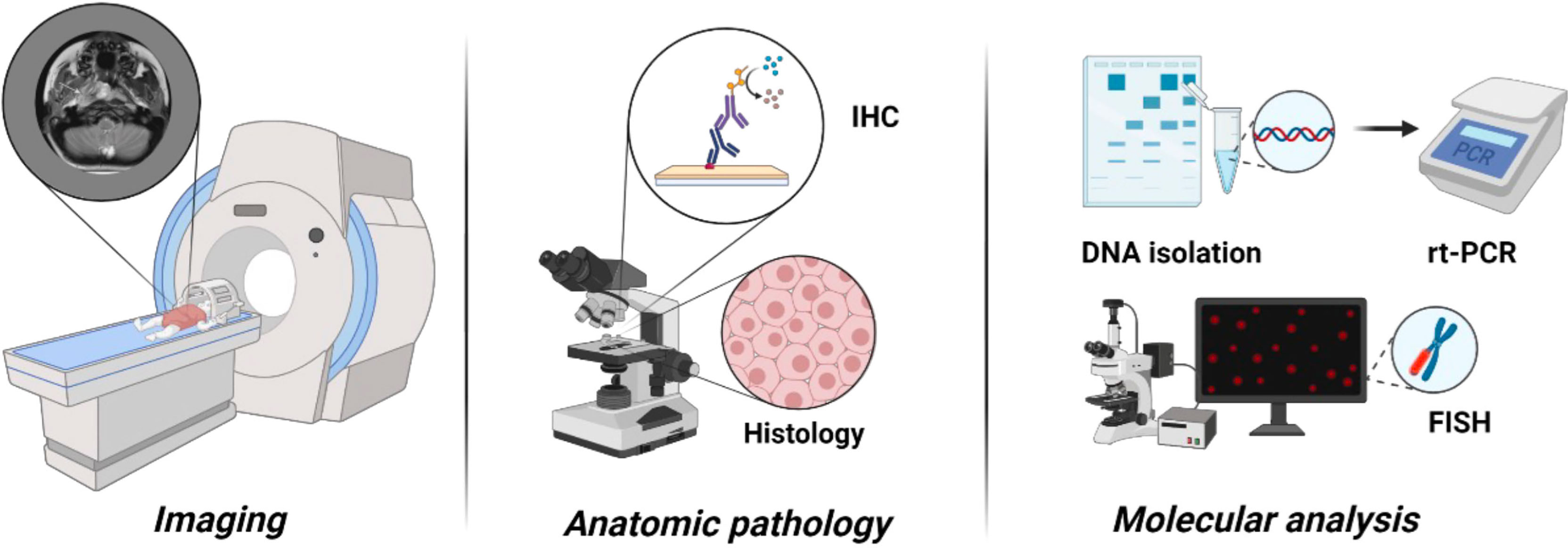
Figure 2 The role of molecular analysis for differential diagnosis and classification of RMS. Molecular analysis helps to identify the subtypes and classify RMS.
Treatment of locally and locally advanced RMS is mainly based on surgery (14, 15), although, aggressive surgery, often necessary to achieve tumor debulking and negative microscopic margins, is no longer recommended (19). This is particularly true for genitourinary RMS, to avoid significant long-term morbidities such as urinary diversion, infertility, and sexual dysfunction particularly. Therefore, except for paratesticular tumors (20), the standard care of RMS, genitourinary and non-genitourinary, usually provides neoadjuvant chemotherapy (CHT) followed by RT or concomitant CHT/RT followed or not by excision (14, 15). In 20% of patients with genitourinary RMS, a close follow-up with imaging is a reasonable alternative to aggressive surgery (14, 15). In support of a delayed surgery, it has been shown that, despite RMS can persist after a neoadjuvant approach (21, 22), the rhabdomyoblasts found in subsequent biopsies progressively decrease and their presence do not necessarily predict local recurrence (23, 24). Thus, aggressive surgical resection, followed or not by RT, is usually performed for recurrent and metastatic RMS (25, 26). CHT is based on IVA (ifosfamide, vincristine, and actinomycin D) or VAC (vincristine, actinomycin D, and cyclophosphamide), respectively used, with no difference, in Europe or in North America (27, 28). Combining doxorubicin improves IVA, inducing several treatment-related adverse events (29). Low-dose maintenance CHT has been shown to improve outcome (30). Trabectedin is commonly used as a second line (31), whereas several clinical trials conducted to test the effects of several molecular targeted drugs combined or not with other targeted therapies or CHT had not shown significant clinical improvement (32–40). However, because of CHT-induced toxicities, pharmacological treatments are often interrupted (41).
Achieving local tumor control with the first-line treatment is crucial for patients with RMS (42). RT plays an increasingly critical role in the management of RMS for local control both at primary and metastatic sites and continues to be a major treatment modality for genitourinary RMS (43–49). Because routine use has been encouraged by the EpSSG RMS 2005 study (44), RT has been shown to improve the event-free survival rate of patients, whereas local failures have been shown to be more frequent when the irradiation is omitted (50). The dose, duration, and timing of external beam radiation therapy, in which x-rays can penetrate deeper in the tissues while minimizing skin irradiation and side effects (51), depend on the patient’s age, RMS type and histology, the site of origin of the tumor, how much tumor remained after surgery, and the local lymph nodes involvement. Table 4 resumes treatment schedules (15, 52, 53). In case of adult patients with RMS, RT can provide a total dose from 50 up to 70 Gy, conventionally delivered (54–56). The use of hyperfractionated (hFRT) regimen, smaller doses per single fraction, performed by the large, randomized IRS-IV study, failed (57). This failure, however, has permitted to revise the radiobiology of RMS, as later discussed. On the other hand, the use of hyperfractionated (HFRT) regimen, larger doses per single fraction, delivered in combination or not with CHT, did not give significant advantages (58–70). Thus, the RMS response to RT appears to go far beyond the simple dose problem because, as for other highly radioresistant tumor types, RMS appears capable of activating a complex biological response supporting radioresistance.
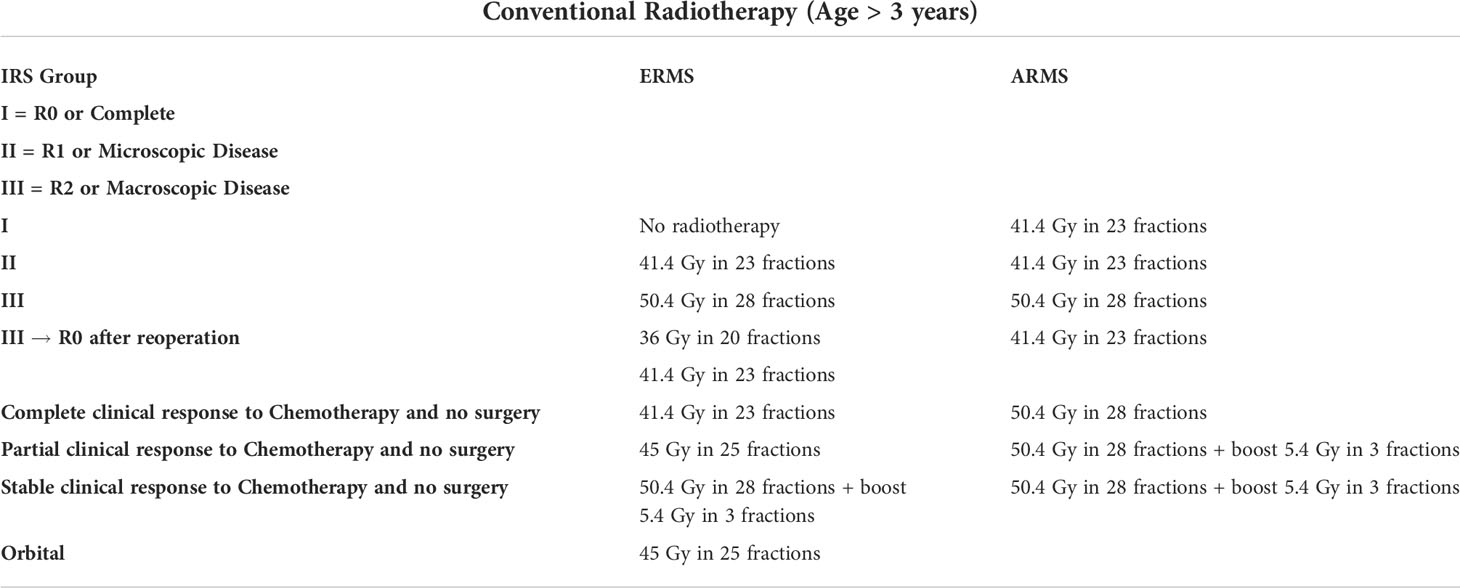
Table 4 Doses and fractions of radiotherapy for patients over 3 years of age.
Radiobiology has been classically focused on achieving the greatest possible difference between a high probability of local tumor control [tumor control probability (TCP)] and a low risk of normal tissue complications [normal tissue complication probability (NTCP)], namely, therapeutic window. In fact, whether increasing the dose improves TCP, because of the lack of technology able to spare normal tissues, it also increases NTCP. Thus, RT has been long delivered by using daily fractions of 1.8–2.2 Gy, the conventional fractionation, which is still largely used today. The reason why conventional fractionation guarantees the best therapeutic window depends on the concept that normal cells repair sublethal damages more efficiently than cancerous cells, as shown from the linear quadratic model (LQ) (71) and from of “4Rs” of radiobiology (72). Briefly, LQ is a mathematical model describing the relationship between cell survival and delivered dose, and it is represented by the equation S = e− (71). The probability to survive (S) of a cell/tissue type to a single dose of radiation depends on the ratio between two factors: i) the number of cells directly killed by double-strand breaks (DSBs), namely, a; and ii) the number of cells that, having saturated the repair mechanisms, die for the accumulation of sublethal unrepaired single-strand breaks (SSBs), namely, b. The a/b ratio indicates the fraction size sensitivity of a tissue, with b indicating the ability of cell to repair SSBs. Hence, cells with a low a/b ratio efficiently repair SSBs, contrary to cells with a high a/b ratio (71). Notably, doses of RT close to 1.8–2.2 Gy induce thousands of repairable SSBs and few DSBs (73–75), thus indicating that the proportion of cells surviving to conventional fractionation strictly depends on the ability to repair SSBs. Thus, because cancer cells less efficiently “R”epair SSBs than normal cells, cancer cells slower “R”edistribute cell cycle from RT-induced G2/M arrest, less efficiently “R”epopulate killed cancer cells, and result more affected by the “R”eoxygenation of the central portions of the tumor induced by the progressive reduction of the peripheral regions. Those are the “4Rs” of radiobiology, historically supporting the efficiency of the conventional fractionation (72). However, after a long time, it was shown that not all cells within cancer population and not all patients with the same tumor have the same sensitivity to RT, introducing the fifth “R”, the “R”adiosensitivity (76). Thus, considering the technological evolution of RT that nowadays permits the safety delivery of larger fractions (77), the use of HFRT or Stereotactic Body Radiation Therapy (SBRT), ablative dose of radiation, has been proposed as strategy to overcome the intrinsic radioresistance of cancer. Furthermore, this choice is also supported by the fact that increasing evidence shows the ability of HFRT and SBRT to “R”eactivate the anti-tumor immune response, the sixth “R” of radiobiology (78). As previously discussed, the use of higher dose per fraction has been also proposed for RMS as a consequence of the low a/b ratio (2.8 Gy) shown for this cancer type (79). However, the use of HFRT for the treatment of RMS did not lead to any improvement in efficacy (58–70), as already described for other cancer types (80–84). Despite being a milestone in the multimodal treatment of pediatric RMS, RT is still significantly associated with local failure in most cases of tumor relapse, and the RMS response to radiation appears to go far beyond the simple dose problem. Indeed, together with the development of more sophisticated and effective technologies, overcoming radioresistance seems to be not just a question of dose but rather of understanding the cellular mechanisms that support radioresistance to identify future radiosensitizing strategies.
As other highly radioresistant tumor types, RMS appears capable of activating a complex biological response that makes them capable of resisting even high radiation doses. Therefore, it is necessary to deeply elucidate the precise mechanisms that are responsible for the radioresistance in RMS to identify new radiosensitizing therapeutic strategies. Over the last years, different studies have identified several key cellular and molecular factors, including DNA damage and repair, oxidative stress, tumor microenvironment, cancer stem cells (CSCs), and tumor heterogeneity (Figure 3), which are implied in RMS radioresistance and are discussed in detail in the following subsections.
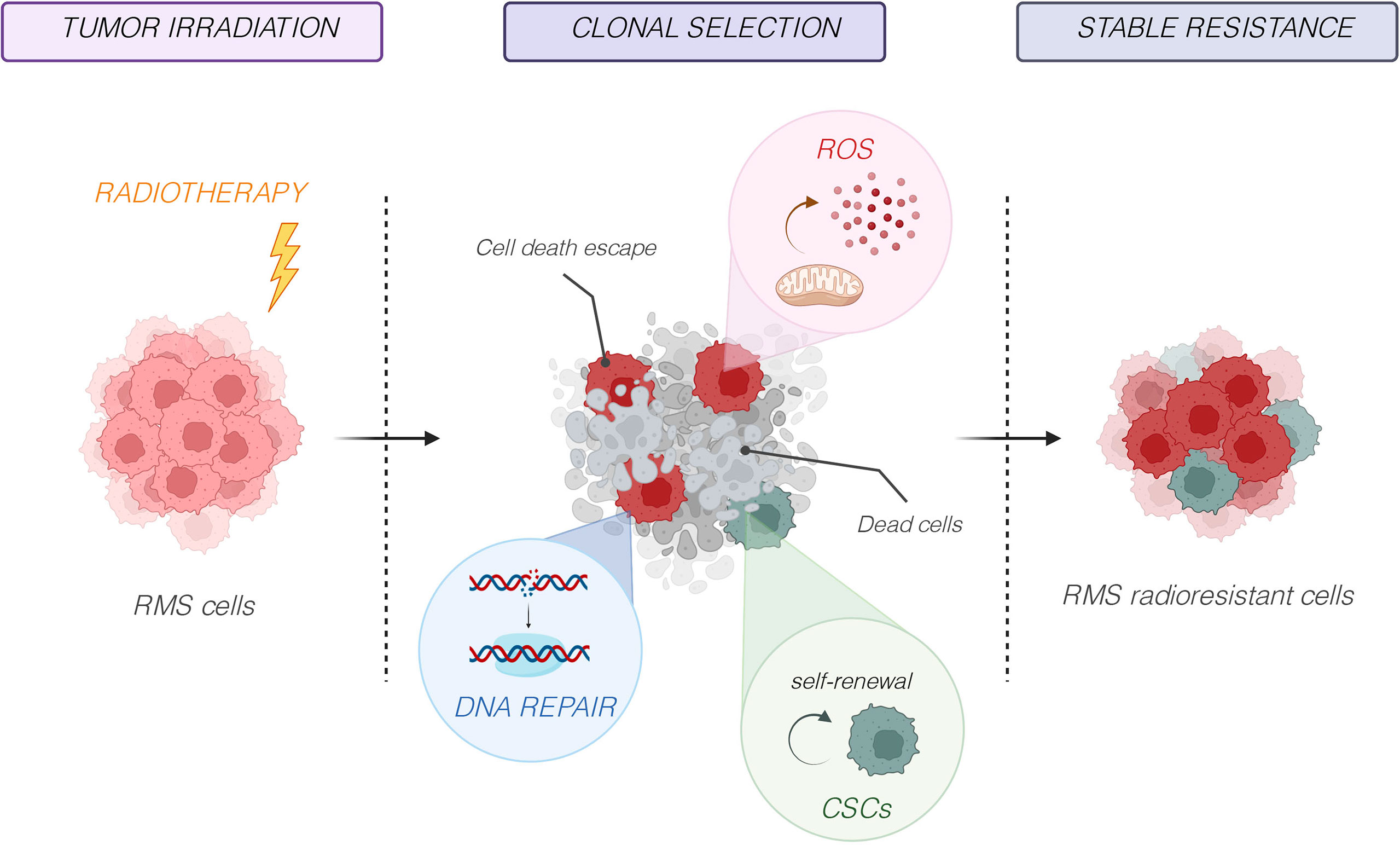
Figure 3 Molecular mechanisms responsible of radioresistance. Several key cellular and molecular factors, including DNA damage and repair, oxidative stress, tumor microenvironment, cancer stem cells (CSCs), and tumor heterogeneity, are implied in RMS radioresistance.
Ionizing radiations used in RT are electrically charged particles, which deposit energy in the tissues that they pass through, killing cancer cells or causing genetic changes that lead to cancer cell death (85). On a cellular level, the biological target of radiation is DNA, by inducing several types of DNA damages involving one strand (SSBs) or both strands of DNA (DSBs) (86, 87). Although radiations damage both cancer and normal cells, cancer cells are generally less efficient in repairing damages caused by RT, resulting in differential killing outcomes (88). DSBs, the most lethal form of DNA damage and a primary cause of cell death induced by RT, can be divided in simple and complex types. Simple DSBs are two-ended breaks of DNA, usually directly consequent to the action of radiation, whereas complex DSBs are clusters of different DNA damages including single-base mutations, insertions, and deletions and/or SSBs around DSBs, generally indirectly induced by radiation through the production of reactive oxygen species (ROS) (89–93). Thus, contrary to SSBs and simplex DSBs, complex DSBs are usually inefficiently repaired, determining genomic instability and cell death (94–96) although cancer cells can activate specific DNA damage repair mechanisms, thus surviving the following irradiation (92). The homologous recombination (HR) and the non-homologous end joining (NHEJ) mechanisms represent the most prominent pathways, orchestrating the DNA damage response (DDR) in eukaryotic cells. HR uses homologous sequences of undamaged sister chromatid as a template to repair DSBs, thus resulting in an error-free DDR mechanism (97). HR is mainly regulated by the MRN complex (Mre11–Rad50–Nbs1), which recognizes DSBs recruiting activated ataxia-telangiectasia mutated (ATM) that, in turn, orchestrates the activity of Breast Cancer gene 1 (BRCA1), Breast Cancer gene 2 (BRCA2), Checkpoint kinase 2 (CHK2), RAD51, and tumor protein 53 (p53)—key factors involved in HR. Parallelly, ATR (ataxia-telangiectasia mutated and Rad3-related) kinase attenuates DSB-induced ATM activation by switching DSB ends from an ATM-activating mode to an ATR-activating model (98) and activates CHK1 that slows or arrests cell cycle progression, thus allowing more time for DNA repair (99). Mutation inactivating BRCA1 and/or BRCA2 and/or other(s) gene(s) of the HR pathway, namely, the “BRCAness” status, permits to stratify HR-deficient (HRD) from HR-proficient (HRP) cancers (100) and to identify HRD as more sensitive to “synthetic lethality” mediated by PARP inhibitor (PARPi) (101, 102). The “BRCAness” phenotype has been shown in several types of sarcomas (103–105), including RMS (106, 107), although the ability of RMS to activate HR-mediated DDR is not excluded (108, 109). On the benchside, RMS biopsies overexpressing PARP1, PARP2, and PARP3 mRNAs compared with normal skeletal muscle and PARPi have been demonstrated to affect growth, survival, and radiation susceptibility of human ARMS and ERMS cell lines (110, 111). However, on the bedside, clinical trials testing PARPi on sarcomas, not including RMS, failed (112, 113), whereas a recent phase I trial (NCT02787642) combining the PARPi with RT in locally advanced/unresectable STS, including RMS, is going to give encouraging downstaging and survival rates (114). Therefore, using PARPi could radiosensitize RMS independently of HRD or HRP phenotype because conventional RT, causing thousands of SSBs, would saturate the HR mechanisms inducing, in the presence of PARPi, RMS death, as already shown for other cancer types (115). Another potential target to affect ERMS radiosensitivity is c-Myc, whose downregulation through the inhibition of the MEK/ERK pathway has been demonstrated to in vitro and in vivo cause cell death by promoting the radiation-induced DNA DSB damage and impairing the DNA DSB repair machinery (116). NHEJ is the major DDR pathway activated by RT (117). Unlike HR, NHEJ re-ligates two broken DNA strands mainly through DNA-dependent Protein Kinase catalitic subunit (DNA-PKcs) that, complexing with Ku70/Ku80 heterodimer and DNA polymerase μ (Pol μ) and Pol λ, and in collaboration with XRCC4, XLF, LIG4, and PAXX, orchestrates this prone-error repair process. Moreover, DNA-PKcs has been shown to interplay with HR pathway, suggesting its pleiotropic role in regulating DDR (118). Targeting DNA-PKcs has been supposed to be a critical radiosensitizing strategy (89, 119), and, nowadays, several inhibitors, with a high selectivity and a valid pharmacokinetics, are available (118), including peposertib that is investigated by several clinical trials, in combination with RT or CHT plus RT, across a variety of cancer types (NCT02516813, NCT02316197, NCT03770689, NCT04555577, NCT04533750, and NCT03907969). Preclinical evidence shows that inhibiting DNA-PKcs sensitizes sarcoma to RT (120, 121), although no RMS cells have been investigated. However, several studies suggest a role for DNA-PKcs in RMS radioresistance. Specifically, DNA-PKcs has been shown to promote sarcomagenesis (122) and to sustain the activity of c-Myc (123) and AKTs (124), which are known to foster radioresistance in ERMS (116, 125–127) and ARMS tumors (128). Thus, it seems unlikely that DNA-PKcs targeting will not lead to an RMS radiosensitization. Notably, several molecules have been identified as upstream regulators of DDR in RMS including ERKs (126, 129), DNA methyltransferases 3A (DNMT3A) and DNMT3B (130), BET proteins (131), ephrin-A2 (132), caveolin-1 (CAV-1) (128), nuclear factor erythroid 2–related factor 2 (NRF-2) (133), c-Myc (116), SNAI2 (134), FAK (135), androgen receptor (136), and HDAC (137–139). Thus, another strategy to target DDR could be inhibiting these upstream molecules.
RT mainly kills cancer cells by inducing the generation of ROS, which, in turn, represents the main induction mechanism of DSBs (140). Furthermore, the production of ROS can persist for several months after RT, thus enhancing the curative effects of treatment (141). However, cancer cells can activate an antioxidant stress response able to protect cells against ROS injury during RT exposure (142, 143). Kelch-like ECH-associated protein 1 and NRF2, respectively, inhibits and promotes the antioxidant response by upregulating the expression of downstream genes, such as peroxiredoxins (PRDXs), superoxide dismutases (SODs), catalase (CAT), and glutathione peroxidases 4 (GPx4) (144). The radiosensitizing effects of targeting antioxidant response in cancer cells shown on the benchside (145) have been recently confirmed on the bedside (146–148). ROS levels are critical for RMS homeostasis (149), and their modulation results are critical for the response to therapies (150). Irradiated RMS upregulates NRF2, SODs, CAT, and GPx4 expression, whereas NRF2 silencing counteracts RMS radioresistance by increasing DSBs and impairing DDR (133). Furthermore, we have recently shown that CAV-1, a tumor promoter sustaining rhabdomyosarcomagenesis (151–153), promotes radioresistance in RMS through increased oxidative stress protection (128) and that RMS surviving to RT more efficiently detoxifies from ROS (109). Increasing oxidative stress has been shown to efficiently kill RMS (154). RMS antioxidant response is finely regulated by molecular epigenetic mechanisms (137–139), known to be critical regulator of adaptive responses to stress (155, 156), including RT (157). Interestingly, the ability of RMS to detoxify from ROS increases in parallel with the acquisition of a more radioresistant phenotype (109), suggesting that, for these cells, ROS detoxification is critical to survive to radiation. However, clinical trials working by inducing ROS levels (158–160) have not included RMS. No preclinical and/or clinical data related to the use of directly targeting redox proteins drugs have been collected on RMS. However, several pieces of evidence suggest the use of drugs able to increase ROS beyond targeting redox proteins (137–139). On this regard, ROS generation has been identified as a mediator of histone deacetylase (HDAC) inhibitor (HDACi)–induced cell death (161), and the combination of HDACi with RT brings to RMS radiosensitization through increased ROS accumulation (137–139).
Radiobiology defines cell death as the loss of replicative capacity determined by clonogenic assays, thus including apoptosis, necrosis, mitotic catastrophe, and mitotic death, autophagy, and tumor dormancy (162, 163), although increasing evidence indicates that RT-induced tumor dormancy may not be reversible (164, 165). Apoptosis caused by RT can be mediated by the following: i) intrinsic apoptotic pathway, through the activation of the cytochrome c-caspase 9/8/3 cascade (166); ii) extrinsic apoptotic pathway, through TNF-α/TNF-R1– (167) or TRAIL/Apo2L/TRAIL–receptor–caspase 8/3 cascade (168); and iii) ceramide accumulation that, acting as second messenger, initiates a complex apoptotic program (169). Mitotic catastrophe and mitotic death, defined as the failure to undergo complete mitosis after DNA damage, coupled to defective checkpoints, are usually mediated by intrinsic apoptosis (170). In addition, cell death can be induced by inducing necroptosis, pyroptosis, and ferroptosis (162, 163). Necroptosis, mediated by TNF-α/TNF-R1/RIP1/RIP3/MLKL cascade, in the absence of caspase 8 activation (171) and pyroptosis, triggered by cytoplasmic damaged-associated molecular patterns (DAMPs) and mediated by NLRP1/NLRP3/NLRC4/caspase 1/gasdermin cascade, leads to pore formation at the cytoplasmatic membrane. Ferroptosis, induced by excessive lipid peroxidation that leads to Fe3+ accumulation-induced oxidative stress, is mediated by SLC11A2 and negatively regulated by GSH/GPx4 cascade (172). RMS is resistant to apoptosis (173) and necrosis (174), including from RT, as we have shown in preclinical in vitro and in vivo models (130, 137–139, 175). The tumor suppressor p53, a master promoter of apoptosis (176) and programmed necrosis (177), is frequently mutated in ERMS (178) and downregulated in ARMS (179). Recently, p53 mutations and/or pathway alterations have been associated with the increase of RMS radioresistance (180). Furthermore, RMS expresses high levels of anti-apoptotic Bcl-2 family members (181) and inactivation of caspase 8 expression by hypermethylation (182–184). On the other hand, RMS has been shown to differently modulate the expression of several factors, restraining the activation of programmed necrosis (185–188), whereas a programmed necrosis–related gene signature has been recently identified as novel prognostic biomarker for sarcoma (189). Thus, altogether, these alterations could explain the RMS resistance to RT-induced apoptosis and necrosis. Targeting cell death pathways regulating molecules has been supposed to be an opportunity for the development of innovative treatment strategies also in RMS (190). Targeting TRAIL (184, 191) and Bcl-2 (192) and reactivating caspase 8 expression (183) have been shown to promote apoptosis in RMS, alone or in combination with cytotoxic agents. The depletion of endogenous GSH by sorafenib has shown encouraging result in vitro and in vivo (193) but failed on the bedside (37), whereas others GSH inhibitors have shown anti-RMS therapeutic potential (194, 195). No data have been collected on combining pro-apoptotic or pro-necrotic agents with RT in treating RMS; however, our group has recently showed that pre-treatment with the BET inhibitor (BETi) OTX015 radiosensitizes RMS cells by inhibiting DDR and concomitantly inducing cell death as demonstrated by the strong activation of the apoptotic marker cleaved PARP (131). Death is not the only response from irradiated cells. Autophagy is a catabolic pathway for lysosomal-mediated cellular components degradation, basally inhibited by the mammalian target of rapamycin (TOR) complex 1 (mTORC1) pathway and tightly regulated by autophagy-related proteins (196). Physiologically considered as a cell survival mechanism that can also promote cell death (197), autophagy plays dual roles in cancer (198), including in RMS (199–204). Similarly, RT-induced autophagy has been shown to be cytoprotective or not (205, 206). However, increasing evidence suggests that autophagy cannot be restricted to a single cytoprotective or cytotoxic function, although it is more correct to speak about “autophagic switch”. Thus, autophagy can switch its function even within the context of a specific cancer type and/or with the respect to external stress type (207). Notably, RMS aberrantly expresses guanine nucleotide exchange factor T (208) recently shown to protects cells by inhibiting autophagy and apoptosis (204). Thus, the reduction in autophagy, which we recently shown on irradiated RMS (175), could be a mechanism of radioresistance. There is a lack of evidence on the effects of combining autophagy and RT promoters or inhibitors in the treatment of RMS. Senescence is classically defined as an irreversible form of growth arrest, mainly induced by p53, p21WAF1, p27KIP1, and p16INK4A and the inhibition of cyclin-dependent kinases and RB (209). The induction of senescence represents a therapeutic advantage, thus preventing further proliferation. However, senescent cells can escape from the irreversible growth arrest status and re-enter the cell cycle, boosting tumor growth (210). In particular, these “post-senescent” cells retain stem cell–related features, also known as senescence-associated stemness, suggesting a more aggressive behavior and favoring tumor relapse (210). Senescence represents the most common cellular response after RT (211–213). However, it has been recently shown that RT-induced accumulation of senescent cells can interfere with the therapy and encourage tumor regrowth (211). Thus, the use of senolytics, small molecules that can selectively induce apoptosis of senescent cells, has been supposed to be a valid radiosensitizing strategy (214). The role of RT-induced senescence in RMS is under investigation. Our group has recently shown that DNMT3A promotes radioresistance of RMS by restraining RT-induced senescence (130), probably for the ability of DNMTs to promote DNA repair activity (215), as summarized in Figure 4. Furthermore, we have also found that the expression of CAV-1 protects against RT-induced cell senescence (128), thus indicating the modulation of specific regulators of cellular senescence as a promising tool to set up new and effective therapeutic intervention against RMS, mainly for overcoming tumor radioresistance-related mechanisms.

Figure 4 Effects of the combined treatment with DNMT3A/3B silencing and radiotherapy. Visual representation of the different radiosensitizing mechanisms observed upon (A) DNMT3A and (B) DNMT3B knocking down and RT co-treatment.
Immunotherapies, largely used in several cancer types, resulting effective in a significant fraction of standard therapy refractory patients (216), have not shown objective response in patients with RMS (217, 218). Sarcoma and particularly RMS are considered “cold” to underline the immunologically inert nature of these tumors. Thus, identifying new strategies to turn “cold” in “hot” tumors could be the way to induce immunogenic cell death (ICD) and promote the use of immunotherapies for treating sarcoma (219). RT has been shown to convert malignant cells into endogenous anticancer vaccines, thus resulting in the main strategy able to trigger ICD and boost immunotherapies. RT promotes the release of DAMPs, triggering the chemotaxis of antigen-presenting cells (APCs), dendritic cells, macrophages, and B cells, finally determining cross priming of CD8+ effector T cells. Parallelly, RT-induced cell death modifies the tumor microenvironment through the cytokine release and the expression on endothelium of cell adhesion molecules. In response, cancer cells can rapidly trigger anti-immune response by different signals including PD1/PD-L1 and CTLA4/B7-1 or B7-2 (220, 221), resulting in an opportunity for combining immunotherapies (222). However, cancer cells surviving to RT express mutated genes, increasing the presentation to APCs of neoantigens potentially able to refresh the immune response (223). This process could lead to an “antigen recycling” potentially able to re-boost the ICD-induced anticancer immune response. In this context, repeated exposure to tumor antigens released by “pulsed-RT” has been recently shown to amplify the adaptive immune response by expanding the tumor-specific T-cell receptor repertoire, the production of high-affinity tumor antibodies, and the generation of memory lymphocytes and thereby improve immune control of systemic disease (224). Therefore, the dose and fractionation seem to be the variables mainly affecting pro-immunogenic effects of RT (225, 226). Actually, HFRT and SBRT seem to improve the ability of RT to promote immune responses to tumors (225, 226). A very small percentage of RMS or RMS-infiltrating immune cells express PD-L1, more frequently in low-stage RMS and related to an increased 5-year overall survival rate (227–229). The use of immunotherapies for the treatment of pediatric advanced solid tumors has been limited (227–229), and no patients with RMS have been included. We have recently shown that Transforming growth factor beta (TGF-β), Macrophage migration inhibitory factor (MIF), C-C Motif Chemokine Ligand 2 (CCL2), C-X-C motif chemokine 5 (CXCL5), CXCL8, and CXCL12, are key players of both intrinsic and acquired radioresistance in RMS (109). Thus, targeting these cytokines, known to be mediators of radioresistance (230), could be another strategy to radiosensitize RMS tumors.
An important evidence of cancer management is the impact of RT-mediated strategies on CSCs, which are characterized by a slowly dividing subpopulation of tumoral cells capable of self-renewal features that have a critical role in tumor maintenance and metastasis as well as in resistance phenomena to conventional treatments in many cancer types (231). Recent evidence suggests that CSCs of several malignancies, also comprehending RMS, can resist ionizing radiation because of their peculiar metabolic status, associated with high expression of genes and pathways related to stem-like features, activated DNA repair mechanisms (232), and altered levels of free radical scavenger levels (233). Specifically, several studies have demonstrated the specific molecular pathways contributing to the CSC intrinsic radioresistance, such as PI3K/Akt/mTOR and NOTCH ones (234–237), which upregulates ROS scavenging enzymes (238). Thus, inhibiting NOTCH could be the efficient strategy to radiosensitize CSCs, bypassing their ability to detoxify from ROS. To date, clinical trials testing NOTCH inhibitors have not included RMS tumors as well as the combination with RT, but their use as radiosensitizers has been recently encouraged (239). More recently, boron neutron capture therapy and carbon-ion particle therapy have been proposed in combination with PARPi as effective strategies for the treatment of radioresistant clear cell sarcoma and osteosarcoma, opening up the possibility of successfully treating patients with RMS by combined treatment with RT and PARPi.
Epigenetic alterations, mainly DNA methylation and histone modifications, characterize various cancers (240), including RMS (8, 241, 242). This evidence raises the question about whether the regulation of DNA methylation activity might represent a useful target for radiation sensitization. Targeting epigenetic molecules may therefore be significant in development of novel therapies, including the development of radiosensitizers (243, 244). Notably, deregulated epigenetic mechanisms have been shown to sustain different mechanisms of radioresistance including DNA repair (245, 246), antioxidant response (247, 248), cancer cell life and death decisions (249), as well as anti-cancer immune response (250). Therefore, identifying the molecules and epigenetic reprogramming pathways used by cancer cells could lead to the development of promising targeted therapies able of weakening the different mechanisms of radioresistance, also in the context of RMS. Indeed, targeting specific DNMTs or HDACs has been demonstrated to reverse RMS phenotype, counteracting stemness and inducing radiosensitization (137–139). Specifically, our group has recently demonstrated the overexpression of DNMT3A and DNMT3B in ERMS primary tumor biopsies (251) and highlighted the synergic impact of DNMT3A or DNMT3B silencing and irradiation on viability and aggressiveness of RMS cells, suggesting that DNMT inhibitors could have a clinical application in combination with standard RT in the clinical management of patients with RMS. Other strategies to reduce radioresistance-mediated mechanisms have been recently shown in ARMS tumor, the most aggressive type of RMS (137, 252). The BETi OTX015, an orally drug able to bind and block histones’ acetylated lysines, can downregulate GNL3 gene, encoding for the G nucleolar 3 protein, which is overexpressed in different malignancies, and it has been associated with uncontrolled proliferation, inhibition of programmed cell death, and resistance to therapies. Interestingly, our preclinical data also indicate that OTX015 exposure can enhance the radiosensitivity of ARMS cells by inducing a drastic G2 cell cycle arrest, which was correlated to a permanent DNA damage (upregulation of γ-H2AX) and to the inability of tumoral cells to repair it (alteration of RAD51, ATM, and DNA-PK protein expression). Moreover, OTX015 and irradiation (IR) synergistically downregulated the expression of GNL3 gene, thus suggesting a potential role of BETi in reducing cell cycle progression and maintenance of cell stemness with the potential to counteract the radioresistance phenomena. Similar remarkable radiosensitizing effects were exerted on FP-RMS cells by targeting class I and IV HDACs through MS-275 in vitro and in vivo treatment (137), confirming the crucial role of epigenetic deregulation in RMS onset and progression. Interestingly, the immunological effects of epigenetic modifiers could be used for stimulating therapeutically relevant anticancer immunity when used as stand-alone treatments or in combination with established immunotherapies, favoring the RT-induced presentation of new antigens. Thus, it is possible to assume that the antigenic recycling induced by pulsed radiotherapy, for example, could be further enhanced in presence of an epigenetic remodulation and that, therefore, the tumor can be, in this way, more easily “heated”. To this date, growing evidence suggests that radiation exposure is also related to substantial epigenetics changes of cancer cells (253). Different studies demonstrated that RT could affect DNA methylation patterns and promote a decrease in the expression level of DNMTs (254), with that genomic hypomethylation resulting in enhanced radiation sensitivity in colon carcinoma (255).
Years of oncology research have demonstrated the great ability of cancer cells to adapt to various therapies, including RT. Technological advances in delivering radiation have improved tumor targeting, limiting radiation exposure of healthy tissues. Thus, nowadays, the largest part of cancer patients receives RT, which results to be curative in the most cases. RT is integrated into the primary treatment of most patients with RMS. However, despite more than 90% of children with non-metastatic RMS achieves complete remission, up to one-third of them experiences a recurrence, whereas the outcome of adult patients treated with RT has not been improved. Thus, RMS remains a very deadly cancer. The use of HFRT, based on single larger dose fractions, has not led to the desired results, suggesting that the improvement of the therapeutic potential of RT goes far beyond the question of the dose, requiring knowledge and counteracting of the molecular mechanisms responsible for radioresistance. Thus, radiosensitizers remain a viable option for improving the outcome of therapy in RMS. More research is necessary to fully understand the mechanisms of RMS radioresistance and improve the outcomes of patients with this deadly disease.
SCa, MC, and SP: study design, data analysis, data collection, and writing; LM, FV, AP, GB, CM, SCo, and MT: data analysis and data collection; RR and AF: review and editing; FMa and FMe: review and editing, and supervision. All authors contributed to the article and approved the submitted version.
The research leading to these results has received funding from AIRC under IG 2020 - ID. 24696 project – P.I. FMa.
The authors are grateful to Dr. Giulia Vitali for supporting with the images’ creation performed by using BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McDowell HP. Update on childhood rhabdomyosarcoma. Arch Dis Child (2003) 88:354–7. doi: 10.1136/adc.88.4.354
2. Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers (2019) 5(1):1. doi: 10.1038/s41572-018-0051-2
3. Rudzinski ER, Anderson JR, Hawkins DS, Skapek SX, Parham DM, Teot LA. The world health organization classification of skeletal muscle tumors in pediatric rhabdomyosarcoma: A report from the children’s oncology group. Arch Pathol Lab Med (2015) 139:1281–7. doi: 10.5858/arpa.2014-0475-OA
4. Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, et al. Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis. Am J Pathol (2009) 174:550–64. doi: 10.2353/ajpath.2009.080631
5. Sorensen PHB, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol (2002) 20:2672–9. doi: 10.1200/JCO.2002.03.137
6. Rudzinski ER, Anderson JR, Chi Y-Y, Gastier-Foster JM, Astbury C, Barr FG, et al. Histology, fusion status, and outcome in metastatic rhabdomyosarcoma: A report from the children’s oncology group. Pediatr Blood Cancer (2017) 64:e26645. doi: 10.1002/pbc.26645
7. Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun (2015) 6:7557. doi: 10.1038/ncomms8557
8. Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell (2018) 34:411–426.e19. doi: 10.1016/j.ccell.2018.07.012
9. Williamson D, Missiaglia E, de Reyniès A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene–negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol (2010) 28:2151–8. doi: 10.1200/JCO.2009.26.3814
10. Patel SR, Hensel CP, He J, Alcalá NE, Kearns JT, Gaston KE, et al. Epidemiology and survival outcome of adult kidney, bladder, and prostate rhabdomyosarcoma: A SEER database analysis. Rare Tumors (2020) 12:203636132097740. doi: 10.1177/2036361320977401
11. Wu H-Y, Snyder HM, Womer RB. Genitourinary rhabdomyosarcoma: Which treatment, how much, and when? J Pediatr Urol (2009) 5:501–6. doi: 10.1016/j.jpurol.2009.06.011
12. Rodeberg DA, Anderson JR, Arndt CA, Ferrer FA, Raney RB, Jenney ME, et al. Comparison of outcomes based on treatment algorithms for rhabdomyosarcoma of the bladder/prostate: Combined results from the children’s oncology group, German cooperative soft tissue sarcoma study, Italian cooperative group, and international society of pediatric oncology malignant mesenchymal tumors committee. Int J Cancer (2011) 128:1232–9. doi: 10.1002/ijc.25444
13. Walterhouse DO, Meza JL, Breneman JC, Donaldson SS, Hayes-Jordan A, Pappo AS, et al. Local control and outcome in children with localized vaginal rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the children’s oncology group. Pediatr Blood Cancer (2011) 57:76–83. doi: 10.1002/pbc.22928
14. PDQ Pediatric Treatment Editorial Board. Childhood rhabdomyosarcoma treatment (PDQ®): Health professional version. (2002).
15. Gallego S, Bernabeu D, Garrido-Pontnou M, Guillen G, Hindi N, Juan-Ribelles A, et al. GEIS-SEHOP clinical practice guidelines for the treatment of rhabdomyosarcoma. Clin Trans Oncol (2021) 23:2460–73. doi: 10.1007/s12094-021-02654-1
16. Lawrence W, Anderson JR, Gehan EA, Maurer H. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma study group. children’s cancer study group. pediatric oncology group. Cancer (1997) 80:1165–70.
17. Weiss AR, Lyden ER, Anderson JR, Hawkins DS, Spunt SL, Walterhouse DO, et al. Histologic and clinical characteristics can guide staging evaluations for children and adolescents with rhabdomyosarcoma: A report from the children’s oncology group soft tissue sarcoma committee. J Clin Oncol (2013) 31:3226–32. doi: 10.1200/JCO.2012.44.6476
18. Ruymann FB, Grovas AC. Progress in the diagnosis and treatment of rhabdomyosarcoma and related soft tissue sarcomas. Cancer Invest (2000) 18:223–41. doi: 10.3109/07357900009031827
19. Cecchetto G, Bisogno G, de Corti F, Dall’Igna P, Inserra A, Ferrari A, et al. Biopsy or debulking surgery as initial surgery for locally advanced rhabdomyosarcomas in children? Cancer (2007) 110:2561–7. doi: 10.1002/cncr.23079
20. Rogers TN, Seitz G, Fuchs J, Martelli H, Dasgupta R, Routh JC, et al. Surgical management of paratesticular rhabdomyosarcoma: A consensus opinion from the children’s oncology group, European paediatric soft tissue sarcoma study group, and the cooperative weichteilsarkom studiengruppe. Pediatr Blood Cancer (2021) 68(4):e28938. doi: 10.1002/pbc.28938
21. Borinstein SC, Steppan D, Hayashi M, Loeb DM, Isakoff MS, Binitie O, et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr Blood Cancer (2018) 65:e26809. doi: 10.1002/pbc.26809
22. Rodeberg DA, Stoner JA, Hayes-Jordan A, Kao SC, Wolden SL, Qualman SJ, et al. Prognostic significance of tumor response at the end of therapy in group III rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol (2009) 27:3705–11. doi: 10.1200/JCO.2008.19.5933
23. Arndt CAS, Hammond S, Rodeberg D, Qualman S. Significance of persistent mature rhabdomyoblasts in Bladder/Prostate rhabdomyosarcoma. J Pediatr Hematol Oncol (2006) 28:563–7. doi: 10.1097/01.mph.0000212978.21372.97
24. Ortega JA, Rowland J, Monforte H, Malogolowkin M, Triche T. Presence of well-differentiated rhabdomyoblasts at the end of therapy for pelvic rhabdomyosarcoma: Implications for the outcome. J Pediatr Hematol Oncol (2000) 22:106–11. doi: 10.1097/00043426-200003000-00005
25. Hayes-Jordan A, Doherty DK, West SD, Raney RB, Blakely ML, Cox CS, et al. Outcome after surgical resection of recurrent rhabdomyosarcoma. J Pediatr Surg (2006) 41:633–8. doi: 10.1016/j.jpedsurg.2005.12.002
26. Heske CM, Mascarenhas L. Relapsed rhabdomyosarcoma. J Clin Med (2021) 10:804. doi: 10.3390/jcm10040804
27. Crist WM, Anderson JR, Meza JL, Fryer C, Raney RB, Ruymann FB, et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J Clin Oncol (2001) 19:3091–102. doi: 10.1200/JCO.2001.19.12.3091
28. Miwa S, Yamamoto N, Hayashi K, Takeuchi A, Igarashi K, Tsuchiya H. Recent advances and challenges in the treatment of rhabdomyosarcoma. Cancers (Basel) (2020) 12:1758. doi: 10.3390/cancers12071758
29. Bisogno G, Jenney M, Bergeron C, Gallego Melcón S, Ferrari A, Oberlin O, et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol (2018) 19:1061–71. doi: 10.1016/S1470-2045(18)30337-1
30. Bisogno G, de Salvo GL, Bergeron C, Gallego Melcón S, Merks JH, Kelsey A, et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2019) 20:1566–75. doi: 10.1016/S1470-2045(19)30617-5
31. Baruchel S, Pappo A, Krailo M, Baker KS, Wu B, Villaluna D, et al. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: A report from the children’s oncology group. Eur J Cancer (2012) 48:579–85. doi: 10.1016/j.ejca.2011.09.027
32. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the children’s oncology group. Pediatr Blood Cancer (2014) 61:452–6. doi: 10.1002/pbc.24605
33. Mascarenhas L, Chi Y-Y, Hingorani P, Anderson JR, Lyden ER, Rodeberg DA, et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol (2019) 37:2866–74. doi: 10.1200/JCO.19.00576
34. Geoerger B, Kieran MW, Grupp S, Perek D, Clancy J, Krygowski M, et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer (2012) 48:253–62. doi: 10.1016/j.ejca.2011.09.021
35. Schöffski P, Wozniak A, Leahy MG, Aamdal S, Rutkowski P, Bauer S, et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European organisation for research and treatment of cancer phase 2 trial 90101 ‘CREATE.’. Eur J Cancer (2018) 94:156–67. doi: 10.1016/j.ejca.2018.02.011
36. Santoro A, Comandone A, Basso U, Soto Parra H, de Sanctis R, Stroppa E, et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann Oncol (2013) 24:1093–8. doi: 10.1093/annonc/mds607
37. Kim A, Widemann BC, Krailo M, Jayaprakash N, Fox E, Weigel B, et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the children’s oncology group. Pediatr Blood Cancer (2015) 62:1562–6. doi: 10.1002/pbc.25548
38. Okada K, Yamasaki K, Tanaka C, Fujisaki H, Osugi Y, Hara J. Phase I study of bevacizumab plus irinotecan in pediatric patients with Recurrent/Refractory solid tumors. Jpn J Clin Oncol (2013) 43:1073–9. doi: 10.1093/jjco/hyt124
39. van der Graaf WT, Blay J-Y, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2012) 379:1879–86. doi: 10.1016/S0140-6736(12)60651-5
40. Sleijfer S, Ray-Coquard I, Papai Z, le Cesne A, Scurr M, Schöffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European organisation for research and treatment of cancer–soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol (2009) 27:3126–32. doi: 10.1200/JCO.2008.21.3223
41. Gupta AA, Anderson JR, Pappo AS, Spunt SL, Dasgupta R, Indelicato DJ, et al. Patterns of chemotherapy-induced toxicities in younger children and adolescents with rhabdomyosarcoma. Cancer (2012) 118:1130–7. doi: 10.1002/cncr.26358
42. Chisholm JC, Marandet J, Rey A, Scopinaro M, de Toledo JS, Merks JHM, et al. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: A nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol (2011) 29:1319–25. doi: 10.1200/JCO.2010.32.1984
43. Ludin A, Macklis RM. RADIOTHERAPY FOR PEDIATRIC GENITOURINARY TUMORS. Urologic Clinics North America (2000) 27:553–62. doi: 10.1016/S0094-0143(05)70102-6
44. Mandeville HC. Radiotherapy in the management of childhood rhabdomyosarcoma. Clin Oncol (2019) 31:462–70. doi: 10.1016/j.clon.2019.03.047
45. Curtis AE, Okcu MF, Chintagumpala M, Teh BS, Paulino AC. Local control after intensity-modulated radiotherapy for head-and-Neck rhabdomyosarcoma. Int J Radiat OncologyBiologyPhysics (2009) 73:173–7. doi: 10.1016/j.ijrobp.2008.03.029
46. Wen Y, Huang D, Zhang W, Zhang Y, Hu H, Li J. Radiation therapy is an important factor to improve survival in pediatric patients with head and neck rhabdomyosarcoma by enhancing local control: a historical cohort study from a single center. BMC Pediatr (2020) 20:265. doi: 10.1186/s12887-020-02165-y
47. Skamene S, Abish S, Mitchell D, Freeman C. Radiotherapy is important for local control at primary and metastatic sites in pediatric rhabdomyosarcoma. Cureus (2015) 7(11):e388. doi: 10.7759/cureus.388
48. Terezakis SA, Wharam MD. Radiotherapy for rhabdomyosarcoma: Indications and outcome. Clin Oncol (2013) 25:27–35. doi: 10.1016/j.clon.2012.07.009
49. Shen W, Sakamoto N, Yang L. Model to predict the survival benefit of radiation for patients with rhabdomyosarcoma after surgery: A population-based study. Int J Oncol (2014) 45:549–57. doi: 10.3892/ijo.2014.2466
50. Ferrari A, Casanova M, Bisogno G, Zanetti I, Cecchetto G, de Bernardi B, et al. Rhabdomyosarcoma in infants younger than one year old. Cancer (2003) 97:2597–604. doi: 10.1002/cncr.11357
51. Abshire D, Lang MK. The evolution of radiation therapy in treating cancer. Semin Oncol Nurs (2018) 34:151–7. doi: 10.1016/j.soncn.2018.03.006
52. Wolden SL, Lyden ER, Arndt CA, Hawkins DS, Anderson JR, Rodeberg DA, et al. Local control for intermediate-risk rhabdomyosarcoma: Results from D9803 according to histology, group, site, and size: A report from the children’s oncology group. Int J Radiat OncologyBiologyPhysics (2015) 93:1071–6. doi: 10.1016/j.ijrobp.2015.08.040
53. Raney RB, Anderson JR, Barr FG, Donaldson SS, Pappo AS, Qualman SJ, et al. Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades of life: A selective review of intergroup rhabdomyosarcoma study group experience and rationale for intergroup rhabdomyosarcoma study V. J Pediatr Hematol Oncol (2001) 23:215–20. doi: 10.1097/00043426-200105000-00008
54. Gerber NK, Wexler LH, Singer S, Alektiar KM, Keohan ML, Shi W, et al. Adult rhabdomyosarcoma survival improved with treatment on multimodality protocols. Int J Radiat OncologyBiologyPhysics (2013) 86:58–63. doi: 10.1016/j.ijrobp.2012.12.016
55. Bergamaschi L, Bertulli R, Casanova M, Provenzano S, Chiaravalli S, Gasparini P, et al. Rhabdomyosarcoma in adults: analysis of treatment modalities in a prospective single-center series. Med Oncol (2019) 36:59. doi: 10.1007/s12032-019-1282-0
56. Zhao R, Yu X, Feng Y, Wang J, Chen Y, Mao Y, et al. The survival benefit of radiotherapy in localized primary adult rhabdomyosarcoma. Asia Pac J Clin Oncol (2020) 16:266–72. doi: 10.1111/ajco.13331
57. Donaldson SS, Meza J, Breneman JC, Crist WM, Laurie F, Qualman SJ, et al. Results from the IRS-IV randomized trial of hyperfractionated radiotherapy in children with rhabdomyosarcoma–a report from the IRSG 1 1For a complete list of the members of the children’s oncology group soft tissue sarcoma committee (formerly intergroup rhabdomyosarcoma group) representing the children’s oncology group and the quality assurance review center, see the appendix. Int J Radiat OncologyBiologyPhysics (2001) 51:718–28. doi: 10.1016/S0360-3016(01)01709-6
58. Soyfer V, Corn BW, Kollender Y, Issakov J, Dadia S, Flusser G, et al. Hypofractionated adjuvant radiation therapy of soft-tissue sarcoma achieves excellent results in elderly patients. Br J Radiol (2013) 86:20130258. doi: 10.1259/bjr.20130258
59. Spencer RM, Aguiar Junior S, Ferreira FO, Stevanato Filho PR, Kupper BE, Silva ML, et al. Neoadjuvant hypofractionated radiotherapy and chemotherapy in high-grade extremity soft tissue sarcomas: Phase 2 clinical trial protocol. JMIR Res Protoc (2017) 6:e97. doi: 10.2196/resprot.6806
60. Kalbasi A, Kamrava M, Chu F-I, Telesca D, van Dams R, Yang Y, et al. A phase II trial of 5-day neoadjuvant radiotherapy for patients with high-risk primary soft tissue sarcoma. Clin Cancer Res (2020) 26:1829–36. doi: 10.1158/1078-0432.CCR-19-3524
61. Parsai S, Lawrenz J, Kilpatrick S, Rubin B, Hymes C, Gray M, et al. Early outcomes of preoperative 5-fraction radiation therapy for soft tissue sarcoma followed by immediate surgical resection. Adv Radiat Oncol (2020) 5:1274–9. doi: 10.1016/j.adro.2020.06.024
62. Spałek M, Koseła-Paterczyk H, Borkowska A, Wągrodzki M, Szumera-Ciećkiewicz A, Pałucki J, et al. Hypofractionated radiotherapy in locally advanced myxoid liposarcomas of extremities or trunk wall: Results of a single arm prospective clinical trial. Int J Radiat OncologyBiologyPhysics (2019) 105:S63. doi: 10.1016/j.ijrobp.2019.06.506
63. Pennington JD, Eilber FC, Eilber FR, Singh AS, Reed JP, Chmielowski B, et al. Long-term outcomes with ifosfamide-based hypofractionated preoperative chemoradiotherapy for extremity soft tissue sarcomas. Am J Clin Oncol (2018) 41:1154–61. doi: 10.1097/COC.0000000000000443
64. Koseła-Paterczyk H, Szacht M, Morysiński T, Ługowska I, Dziewirski W, Falkowski S, et al. Preoperative hypofractionated radiotherapy in the treatment of localized soft tissue sarcomas. Eur J Surg Oncol (EJSO) (2014) 40:1641–7. doi: 10.1016/j.ejso.2014.05.016
65. Meyer JM, Perlewitz KS, Hayden JB, Doung Y-C, Hung AY, Vetto JT, et al. Phase I trial of preoperative chemoradiation plus sorafenib for high-risk extremity soft tissue sarcomas with dynamic contrast-enhanced MRI correlates. Clin Cancer Res (2013) 19:6902–11. doi: 10.1158/1078-0432.CCR-13-1594
66. MacDermed DM, Miller LL, Peabody TD, Simon MA, Luu HH, Haydon RC, et al. Primary tumor necrosis predicts distant control in locally advanced soft-tissue sarcomas after preoperative concurrent chemoradiotherapy. Int J Radiat OncologyBiologyPhysics (2010) 76:1147–53. doi: 10.1016/j.ijrobp.2009.03.015
67. Ryan CW, Montag AG, Hosenpud JR, Samuels B, Hayden JB, Hung AY, et al. Histologic response of dose-intense chemotherapy with preoperative hypofractionated radiotherapy for patients with high-risk soft tissue sarcomas. Cancer (2008) 112:2432–9. doi: 10.1002/cncr.23478
68. Temple WJ, Temple CLF, Arthur K, Schachar NS, Paterson AHG, Crabtree TS. Prospective cohort study of neoadjuvant treatment in conservative surgery of soft tissue sarcomas. Ann Surg Oncol (1997) 4:586–90. doi: 10.1007/BF02305541
69. Kılıç L, Ekenel M, Karabulut S, Ağaoğlu F, Darendeliler E. Neoadjuvant sequential chemoradiotherapy versus radiotherapy alone for treatment of high-risk extremity soft tissue sarcoma: a single-institution experience. Współczesna Onkologia (2017) 1:60–5. doi: 10.5114/wo.2017.66658
70. Spalek M, Koseła-Paterczyk H, Borkowska A, Wągrodzki M, Szumera-Ciećkiewicz A, Cieszanowski A, et al. OC-0069 5x5 gy with chemotherapy in borderline resectable soft tissue sarcomas: early results of a trial. Radiother Oncol (2019) 133:S31–2. doi: 10.1016/S0167-8140(19)30489-X
71. Fowler JF. The linear-quadratic formula and progress in fractionated radiotherapy. Br J Radiol (1989) 62:679–94. doi: 10.1259/0007-1285-62-740-679
72. Withers HR. “The four r’s of radiotherapy.” Add Adv Radiat Biol (1975). 5:241–71. doi: 10.1016/B978-0-12-035405-4.50012-8.
73. Balagurumoorthy P, James Adelstein S, Kassis AI. Novel method for quantifying radiation-induced single-strand-break yields in plasmid DNA highlights 10-fold discrepancy. Anal Biochem (2011) 417:242–6. doi: 10.1016/j.ab.2011.06.023
74. Tounekti O, Kenani A, Foray N, Orlowski S, Mir LM. The ratio of single- to double-strand DNA breaks and their absolute values determine cell death pathway. Br J Cancer (2001) 84:1272–9. doi: 10.1054/bjoc.2001.1786
75. Goodhead DT. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int J Radiat Biol (1994) 65:7–17. doi: 10.1080/09553009414550021
76. Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. Int J Radiat Biol (1989) 56:1045–8. doi: 10.1080/09553008914552491
77. Pacelli R, Caroprese M, Palma G, Oliviero C, Clemente S, Cella L, et al. Technological evolution of radiation treatment: Implications for clinical applications. Semin Oncol (2019) 46:193–201. doi: 10.1053/j.seminoncol.2019.07.004
78. Boustani J, Grapin M, Laurent P-A, Apetoh L, Mirjolet C. The 6th r of radiobiology: Reactivation of anti-tumor immune response. Cancers (Basel) (2019) 11:860. doi: 10.3390/cancers11060860
79. Mendonca M, Timmerman RD. In regard to Donaldson et al: results from the IRS-IV randomized trial of hyperfractionated radiotherapy in children with rhabdomyosarcoma–a report from the IRSG. IJROBP (2001) 51:718–28. doi: 10.1016/S0360-3016(02)03015-8
80. Chen F, Hui TSK, Ma L, Nong Y, Han Y, Jing H, et al. Real-world practice of hypofractionated radiotherapy in patients with invasive breast cancer. Front Oncol (2022) 12:811794. doi: 10.3389/fonc.2022.811794
81. Trone J-C, Vallard A, Sotton S, ben Mrad M, Jmour O, Magné N, et al. Survival after hypofractionation in glioblastoma: a systematic review and meta-analysis. Radiat Oncol (2020) 15:145. doi: 10.1186/s13014-020-01584-6
82. Murgic J, Jaksic B, Prpic M, Kust D, Bahl A, Budanec M, et al. Comparison of hypofractionation and standard fractionation for post-prostatectomy salvage radiotherapy in patients with persistent PSA: single institution experience. Radiat Oncol (2021) 16:88. doi: 10.1186/s13014-021-01808-3
83. Chin S, Fatimilehin A, Walshaw R, Argarwal A, Mistry H, Elliott T, et al. Ten-year outcomes of moderately hypofractionated salvage postprostatectomy radiation therapy and external validation of a contemporary multivariable nomogram for biochemical failure. Int J Radiat OncologyBiologyPhysics (2020) 107:288–96. doi: 10.1016/j.ijrobp.2020.01.008
84. Arcangeli S, Strigari L, Gomellini S, Saracino B, Petrongari MG, Pinnarò P, et al. Updated results and patterns of failure in a randomized hypofractionation trial for high-risk prostate cancer. Int J Radiat OncologyBiologyPhysics (2012) 84:1172–8. doi: 10.1016/j.ijrobp.2012.02.049
85. Baskar R, Dai J, Wenlong N, Yeo R, Yeoh K-W. Biological response of cancer cells to radiation treatment. Front Mol Biosci (2014) 1:24. doi: 10.3389/fmolb.2014.00024
86. Hei T K, Zhou H, Chai Y, Ponnaiya B, N. Ivanov V. Radiation induced non-targeted response: Mechanism and potential clinical implications. Curr Mol Pharmacol (2011) 4:96–105. doi: 10.2174/1874467211104020096
87. Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin Oncol (2013) 25:578–85. doi: 10.1016/j.clon.2013.06.007
88. Barnett GC, West CML, Dunning AM, Elliott RM, Coles CE, Pharoah PDP, et al. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer (2009) 9:134–42. doi: 10.1038/nrc2587
89. Huang R-X, Zhou P-K. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther (2020) 5:60. doi: 10.1038/s41392-020-0150-x
90. Nikjoo H, Emfietzoglou D, Liamsuwan T, Taleei R, Liljequist D, Uehara S. Radiation track, DNA damage and response–a review. Rep Prog Phys (2016) 79:116601. doi: 10.1088/0034-4885/79/11/116601
91. Sage E, Shikazono N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic Biol Med (2017) 107:125–35. doi: 10.1016/j.freeradbiomed.2016.12.008
92. Mladenov E, Magin S, Soni A, Iliakis G. DNA Double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol (2013) 3:113. doi: 10.3389/fonc.2013.00113
93. Groth P, Orta ML, Elvers I, Majumder MM, Lagerqvist A, Helleday T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res (2012) 40:6585–94. doi: 10.1093/nar/gks315
94. Li Z, Pearlman AH, Hsieh P. DNA Mismatch repair and the DNA damage response. DNA Repair (Amst) (2016) 38:94–101. doi: 10.1016/j.dnarep.2015.11.019
95. Goldstein M, Kastan MB. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu Rev Med (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
96. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
97. Li X, Heyer W-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res (2008) 18:99–113. doi: 10.1038/cr.2008.1
98. Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell (2009) 33:547–58. doi: 10.1016/j.molcel.2009.01.024
99. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by atr and required for the G(2)/M DNA damage checkpoint. Genes Dev (2000) 14:1448–59. doi: 10.1101/gad.14.12.1448
100. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer (2016) 16:110–20. doi: 10.1038/nrc.2015.21
101. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434:917–21. doi: 10.1038/nature03445
102. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434:913–7. doi: 10.1038/nature03443
103. Kovac M, Blattmann C, Ribi S, Smida J, Mueller NS, Engert F, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun (2015) 6:8940. doi: 10.1038/ncomms9940
104. Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, et al. EWS–FLI1 increases transcription to cause r-loops and block BRCA1 repair in Ewing sarcoma. Nature (2018) 555:387–91. doi: 10.1038/nature25748
105. Stewart E, Goshorn R, Bradley C, Griffiths LM, Benavente C, Twarog NR, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep (2014) 9:829–40. doi: 10.1016/j.celrep.2014.09.028
106. Walsh MF, Kennedy J, Harlan M, Kentsis A, Shukla N, Musinsky J, et al. Germline BRCA2 mutations detected in pediatric sequencing studies impact parents’ evaluation and care. Mol Case Stud (2017) 3:a001925. doi: 10.1101/mcs.a001925
107. Zhang P, Bhakta KS, Puri PL, Newbury R, Feramisco J, Wang J. Association of ataxia telangiectasia mutated (ATM) gene Mutation/Deletion with rhabdomyosarcoma. Cancer Biol Ther (2003) 2:88–92. doi: 10.4161/cbt.231
108. Sun X, Guo W, Shen JK, Mankin HJ, Hornicek FJ, Duan Z. Rhabdomyosarcoma: Advances in molecular and cellular biology. Sarcoma (2015) 2015:1–14. doi: 10.1155/2015/232010
109. Petragnano F, Pietrantoni I, Camero S, Codenotti S, Milazzo L, Vulcano F, et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: functional, molecular and immune-related characterization. J BioMed Sci (2020) 27:90. doi: 10.1186/s12929-020-00683-6
110. Mangoni M, Sottili M, Salvatore G, Meattini I, Desideri I, Greto D, et al. Enhancement of soft tissue sarcoma cell radiosensitivity by Poly(ADP-ribose) polymerase-1 inhibitors. Radiat Res (2018) 190:464. doi: 10.1667/RR15035.1
111. Camero S, Ceccarelli S, de Felice F, Marampon F, Mannarino O, Camicia L, et al. PARP inhibitors affect growth, survival and radiation susceptibility of human alveolar and embryonal rhabdomyosarcoma cell lines. J Cancer Res Clin Oncol (2019) 145:137–52. doi: 10.1007/s00432-018-2774-6
112. Choy E, Butrynski JE, Harmon DC, Morgan JA, George S, Wagner AJ, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer (2014) 14:813. doi: 10.1186/1471-2407-14-813
113. Schafer ES, Rau RE, Berg SL, Liu X, Minard CG, Bishop AJR, et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A children’s oncology group phase 1 consortium study (ADVL1411). Pediatr Blood Cancer (2020) 67(2):e28073. doi: 10.1002/pbc.28073
114. Vatner R, James CD, Sathiaseelan V, Bondra KM, Kalapurakal JA, Houghton PJ. Radiation therapy and molecular-targeted agents in preclinical testing for immunotherapy, brain tumors, and sarcomas: Opportunities and challenges. Pediatr Blood Cancer (2021) 68(Suppl 2):e28439. doi: 10.1002/pbc.28439
115. Jannetti SA, Zeglis BM, Zalutsky MR, Reiner T. Poly(ADP-Ribose)Polymerase (PARP) inhibitors and radiation therapy. Front Pharmacol (2020) 11:170. doi: 10.3389/fphar.2020.00170
116. Gravina GL, Festuccia C, Popov VM, di Rocco A, Colapietro A, Sanità P, et al. C-myc sustains transformed phenotype and promotes radioresistance of embryonal rhabdomyosarcoma cell lines. Radiat Res (2016) 185:411–22. doi: 10.1667/RR14237.1
117. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
118. Mohiuddin IS, Kang MH. DNA-PK as an emerging therapeutic target in cancer. Front Oncol (2019) 9:635. doi: 10.3389/fonc.2019.00635
119. Feng W, Smith CM, Simpson DA, Gupta GP. Targeting non-homologous and alternative end joining repair to enhance cancer radiosensitivity. Semin Radiat Oncol (2022) 32:29–41. doi: 10.1016/j.semradonc.2021.09.007
120. Vormoor B, Schlosser YT, Blair H, Sharma A, Wilkinson S, Newell DR, et al. Sensitizing Ewing sarcoma to chemo- and radiotherapy by inhibition of the DNA-repair enzymes DNA protein kinase (DNA-PK) and poly-ADP-ribose polymerase (PARP) 1/2. Oncotarget (2017) 8:113418–30. doi: 10.18632/oncotarget.21300
121. Mamo T, Mladek AC, Shogren KL, Gustafson C, Gupta SK, Riester SM, et al. Inhibiting DNA-PKCS radiosensitizes human osteosarcoma cells. Biochem Biophys Res Commun (2017) 486:307–13. doi: 10.1016/j.bbrc.2017.03.033
122. Sharpless NE, Ferguson DO, O’Hagan RC, Castrillon DH, Lee C, Farazi PA, et al. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol Cell (2001) 8:1187–96. doi: 10.1016/S1097-2765(01)00425-7
123. An J, Yang D-Y, Xu Q-Z, Zhang S-M, Huo Y-Y, Shang Z-F, et al. DNA-Dependent protein kinase catalytic subunit modulates the stability of c-myc oncoprotein. Mol Cancer (2008) 7:32. doi: 10.1186/1476-4598-7-32
124. Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H, et al. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia (2011) 13:1069–IN35. doi: 10.1593/neo.111032
125. Marampon F, Gravina GL, di Rocco A, Bonfili P, di Staso M, Fardella C, et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells In vitro and In vivo by downregulating growth and DNA repair signals. Mol Cancer Ther (2011) 10:159–68. doi: 10.1158/1535-7163.MCT-10-0631
126. Marampon F, Bossi G, Ciccarelli C, di Rocco A, Sacchi A, Pestell RG, et al. MEK/ERK inhibitor U0126 affects in vitro and in vivo growth of embryonal rhabdomyosarcoma. Mol Cancer Ther (2009) 8:543–51. doi: 10.1158/1535-7163.MCT-08-0570
127. Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer (2006) 5:31. doi: 10.1186/1476-4598-5-31
128. Codenotti S, Marampon F, Triggiani L, Bonù ML, Magrini SM, Ceccaroli P, et al. Caveolin-1 promotes radioresistance in rhabdomyosarcoma through increased oxidative stress protection and DNA repair. Cancer Lett (2021) 505:1–12. doi: 10.1016/j.canlet.2021.02.005
129. Ciccarelli C, Vulcano F, Milazzo L, Gravina GL, Marampon F, Macioce G, et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol Cancer (2016) 15:16. doi: 10.1186/s12943-016-0501-y
130. Camero S, Vitali G, Pontecorvi P, Ceccarelli S, Anastasiadou E, Cicchetti F, et al. DNMT3A and DNMT3B targeting as an effective radiosensitizing strategy in embryonal rhabdomyosarcoma. Cells (2021) 10:2956. doi: 10.3390/cells10112956
131. Camero S, Camicia L, Marampon F, Ceccarelli S, Shukla R, Mannarino O, et al. BET inhibition therapy counteracts cancer cell survival, clonogenic potential and radioresistance mechanisms in rhabdomyosarcoma cells. Cancer Lett (2020) 479:71–88. doi: 10.1016/j.canlet.2020.03.011
132. Megiorni F, Gravina GL, Camero S, Ceccarelli S, del Fattore A, Desiderio V, et al. Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells. J Hematol Oncol (2017) 10(1):161. doi: 10.1186/s13045-017-0530-z
133. Marampon F, Codenotti S, Megiorni F, del Fattore A, Camero S, Gravina GL, et al. NRF2 orchestrates the redox regulation induced by radiation therapy, sustaining embryonal and alveolar rhabdomyosarcoma cells radioresistance. J Cancer Res Clin Oncol (2019) 145(4):881–93. doi: 10.1007/s00432-019-02851-0
134. Wang L, Hensch NR, Bondra K, Sreenivas P, Zhao XR, Chen J, et al. SNAI2-mediated repression of BIM protects rhabdomyosarcoma from ionizing radiation. Cancer Res (2021) 81:5451–63. doi: 10.1158/0008-5472.CAN-20-4191
135. Pomella S, Cassandri M, Braghini MR, Marampon F, Alisi A, Rota R. New insights on the nuclear functions and targeting of FAK in cancer. Int J Mol Sci (2022) 23:1998. doi: 10.3390/ijms23041998
136. Giannattasio S, Megiorni F, di Nisio V, del Fattore A, Fontanella R, Camero S, et al. Testosterone-mediated activation of androgenic signalling sustains in vitro the transformed and radioresistant phenotype of rhabdomyosarcoma cell lines. J Endocrinol Invest (2019) 42:183–97. doi: 10.1007/s40618-018-0900-6
137. Cassandri M, Pomella S, Rossetti A, Petragnano F, Milazzo L, Vulcano F, et al. MS-275 (Entinostat) promotes radio-sensitivity in PAX3-FOXO1 rhabdomyosarcoma cells. Int J Mol Sci (2021) 22(19):10671. doi: 10.3390/ijms221910671
138. Rossetti A, Petragnano F, Milazzo L, Vulcano F, Macioce G, Codenotti S, et al. Romidepsin (FK228) fails in counteracting the transformed phenotype of rhabdomyosarcoma cells but efficiently radiosensitizes, in vitro and in vivo, the alveolar phenotype subtype. Int J Radiat Biol (2021) 97(7):943–57. doi: 10.1080/09553002.2021.1928786
139. Marampon F, di Nisio V, Pietrantoni I, Petragnano F, Fasciani I, Scicchitano BM, et al. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer Lett (2019) 461:90–101. doi: 10.1016/j.canlet.2019.07.009
140. Bielski BHJ, Cabelli DE. Highlights of current research involving superoxide and perhydroxyl radicals in aqueous solutions. Int J Radiat Biol (1991) 59:291–319. doi: 10.1080/09553009114550301
141. Tamminga J, Kovalchuk O. Role of DNA damage and epigenetic DNA methylation changes in radiation-induced genomic instability and bystander effects in germline. In Vivo Curr Mol Pharmacol (2011) 4:115–25. doi: 10.2174/1874467211104020115
142. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discovery (2013) 12:931–47. doi: 10.1038/nrd4002
143. Sun J, Chen Y, Li M, Ge Z. Role of antioxidant enzymes on ionizing radiation resistance. Free Radic Biol Med (1998) 24:586–93. doi: 10.1016/S0891-5849(97)00291-8
144. Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev (2018) 98:1169–203. doi: 10.1152/physrev.00023.2017
145. Jiang H, Wang H, de Ridder M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett (2018) 438:154–64. doi: 10.1016/j.canlet.2018.09.004
146. Hidayat Y, Wagey F, Suardi D, Susanto H, Laihad BJ, Tobing M. Analysis of curcumin as a radiosensitizer in cancer therapy with serum survivin examination: Randomised control trial. Asian Pac J Cancer Prev (2021) 22:139–43. doi: 10.31557/APJCP.2021.22.1.139
147. Mehta MP, Shapiro WR, Phan SC, Gervais R, Carrie C, Chabot P, et al. Motexafin gadolinium combined with prompt whole brain radiotherapy prolongs time to neurologic progression in non–Small-Cell lung cancer patients with brain metastases: Results of a phase III trial. Int J Radiat OncologyBiologyPhysics (2009) 73:1069–76. doi: 10.1016/j.ijrobp.2008.05.068
148. Carde P, Timmerman R, Mehta MP, Koprowski CD, Ford J, Tishler RB, et al. Multicenter phase Ib/II trial of the radiation enhancer motexafin gadolinium in patients with brain metastases. J Clin Oncol (2001) 19:2074–83. doi: 10.1200/JCO.2001.19.7.2074
149. Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell (2013) 24:710–24. doi: 10.1016/j.ccr.2013.11.002
150. Zhang M, Linardic CM, Kirsch DG. RAS and ROS in rhabdomyosarcoma. Cancer Cell (2013) 24:689–91. doi: 10.1016/j.ccr.2013.11.015
151. Faggi F, Mitola S, Sorci G, Riuzzi F, Donato R, Codenotti S, et al. Phosphocaveolin-1 enforces tumor growth and chemoresistance in rhabdomyosarcoma. PloS One (2014) 9:e84618. doi: 10.1371/journal.pone.0084618
152. Codenotti S, Faggi F, Ronca R, Chiodelli P, Grillo E, Guescini M, et al. Caveolin-1 enhances metastasis formation in a human model of embryonal rhabdomyosarcoma through erk signaling cooperation. Cancer Lett (2019) 449:135–44. doi: 10.1016/j.canlet.2019.02.013
153. Faggi F, Chiarelli N, Colombi M, Mitola S, Ronca R, Madaro L, et al. Cavin-1 and caveolin-1 are both required to support cell proliferation, migration and anchorage-independent cell growth in rhabdomyosarcoma. Lab Invest (2015) 95:585–602. doi: 10.1038/labinvest.2015.45
154. Pal A, Chiu HY, Taneja R. Genetics, epigenetics and redox homeostasis in rhabdomyosarcoma: Emerging targets and therapeutics. Redox Biol (2019) 25:101124. doi: 10.1016/j.redox.2019.101124
155. Camiña N, Penning TM. Genetic and epigenetic regulation of the NRF2-KEAP1 pathway in human lung cancer. Br J Cancer (2022) 126:1244–52. doi: 10.1038/s41416-021-01642-0
156. Rossnerova A, Izzotti A, Pulliero A, Bast A, Rattan SIS, Rossner P. The molecular mechanisms of adaptive response related to environmental stress. Int J Mol Sci (2020) 21:7053. doi: 10.3390/ijms21197053
157. Peng Q, Weng K, Li S, Xu R, Wang Y, Wu Y. A perspective of epigenetic regulation in radiotherapy. Front Cell Dev Biol (2021) 9:624312. doi: 10.3389/fcell.2021.624312
158. Steiner R, Manasanch EE. Carfilzomib boosted combination therapy for relapsed multiple myeloma. Onco Targets Ther (2017) 10:895–907. doi: 10.2147/OTT.S102756
159. Rajkumar SV, Richardson PG, Lacy MQ, Dispenzieri A, Greipp PR, Witzig TE, et al. Novel therapy with 2-methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin Cancer Res (2007) 13:6162–7. doi: 10.1158/1078-0432.CCR-07-0807
160. Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, et al. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol Ther (2006) 5:22–7. doi: 10.4161/cbt.5.1.2349
161. Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets (2005) 9:809–24. doi: 10.1517/14728222.9.4.809
162. Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, vanden Berghe T, et al. Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death. Cell Death Dis (2020) 11:1003. doi: 10.1038/s41419-020-03209-y
163. Sia J, Szmyd R, Hau E, Gee HE. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front Cell Dev Biol (2020) 8:41. doi: 10.3389/fcell.2020.00041
164. Damen MPF, Rheenen J, Scheele CLGJ. Targeting dormant tumor cells to prevent cancer recurrence. FEBS J (2021) 288:6286–303. doi: 10.1111/febs.15626
165. Wang S, Lin S-Y. Tumor dormancy: potential therapeutic target in tumor recurrence and metastasis prevention. Exp Hematol Oncol (2013) 2:29. doi: 10.1186/2162-3619-2-29
166. Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol (2007) 35:495–516. doi: 10.1080/01926230701320337
167. Palata O, Hradilova Podzimkova N, Nedvedova E, Umprecht A, Sadilkova L, Palova Jelinkova L, et al. Radiotherapy in combination with cytokine treatment. Front Oncol (2019) 9:367. doi: 10.3389/fonc.2019.00367
168. Niemoeller OM, Belka C. Radiotherapy and TRAIL for cancer therapy. Cancer Lett (2013) 332:184–93. doi: 10.1016/j.canlet.2011.07.003
169. Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2002) 1585:114–25. doi: 10.1016/S1388-1981(02)00331-1
170. Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene (2004) 23:2825–37. doi: 10.1038/sj.onc.1207528
171. Dondelinger Y, Darding M, Bertrand MJM, Walczak H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci (2016) 73:2165–76. doi: 10.1007/s00018-016-2191-4
172. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther (2022) 7:196. doi: 10.1038/s41392-022-01046-3
173. Fulda S. Targeting apoptosis resistance in rhabdomyosarcoma. Curr Cancer Drug Targets (2008) 8:536–44. doi: 10.2174/156800908785699333
174. Meager A. A cytotoxicity assay for tumour necrosis using a human rhabdomyosarcoma cell line. J Immunol Methods (1991) 144:141–3. doi: 10.1016/0022-1759(91)90239-C
175. Petragnano F, Pietrantoni I, di Nisio V, Fasciani I, del Fattore A, Capalbo C, et al. Modulating the dose-rate differently affects the responsiveness of human epithelial prostate- and mesenchymal rhabdomyosarcoma-cancer cell line to radiation. Int J Radiat Biol (2020) 96:823–35. doi: 10.1080/09553002.2020.1739774
176. Shen Y, White E. “p53-dependent apoptosis pathways.” Add Adv Cancer Res (2001) 82:55–84. doi: 10.1016/S0065-230X(01)82002-9.
177. Napoletano F, Gibert B, Yacobi-Sharon K, Vincent S, Favrot C, Mehlen P, et al. p53-dependent programmed necrosis controls germ cell homeostasis during spermatogenesis. PloS Genet (2017) 13:e1007024. doi: 10.1371/journal.pgen.1007024
178. Felix CA, Kappel CC, Mitsudomi T, Nau MM, Tsokos M, Crouch GD, et al. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res (1992) 52:2243–7.
179. Stuart ET, Haffner R, Oren M, Gruss P. Loss of p53 function through PAX-mediated transcriptional repression. EMBO J (1995) 14:5638–45. doi: 10.1002/j.1460-2075.1995.tb00251.x
180. Casey DL, Pitter KL, Wexler LH, Slotkin EK, Gupta GP, Wolden SL. TP53 mutations increase radioresistance in rhabdomyosarcoma and Ewing sarcoma. Br J Cancer (2021) 125:576–81. doi: 10.1038/s41416-021-01438-2
181. Boman F, Brel D, Antunes L, Alhamany Z, Floquet J, Boccon-Gibod L. Gene alterations and apoptosis in rhabdomyosarcoma. Pediatr Pathol Lab Med (1997) 17:233–47.
182. Takita J, Yang HW, Bessho F, Hanada R, Yamamoto K, Kidd V, et al. Absent or reduced expression of thecaspase 8 gene occurs frequently in neuroblastoma, but not commonly in Ewing sarcoma or rhabdomyosarcoma. Med Pediatr Oncol (2000) 35:541–3. doi: 10.1002/1096-911X(20001201)35:6<541::AID-MPO9>3.0.CO;2-T
183. Fulda S, Küfer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin K-M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene (2001) 20:5865–77. doi: 10.1038/sj.onc.1204750
184. Petak I, Vernes R, Szucs KS, Anozie M, Izeradjene K, Douglas L, et al. A caspase-8-independent component in TRAIL/Apo-2L-induced cell death in human rhabdomyosarcoma cells. Cell Death Differ (2003) 10:729–39. doi: 10.1038/sj.cdd.4401232
185. Codenotti S, Poli M, Asperti M, Zizioli D, Marampon F, Fanzani A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J Cancer Res Clin Oncol (2018) 144:1717–30. doi: 10.1007/s00432-018-2699-0
186. Fanzani A, Poli M. Iron, oxidative damage and ferroptosis in rhabdomyosarcoma. Int J Mol Sci (2017) 18:1718. doi: 10.3390/ijms18081718
187. Dächert J, Ehrenfeld V, Habermann K, Dolgikh N, Fulda S. Targeting ferroptosis in rhabdomyosarcoma cells. Int J Cancer (2020) 146:510–20. doi: 10.1002/ijc.32496
188. Yang L, Kong D, He M, Gong J, Nie Y, Tai S, et al. MiR-7 mediates mitochondrial impairment to trigger apoptosis and necroptosis in rhabdomyosarcoma. Biochim Biophys Acta (BBA) - Mol Cell Res (2020) 1867:118826. doi: 10.1016/j.bbamcr.2020.118826
189. Wei D, Lan X, Huang Z, Tang Q, Wang Z, Ma Y, et al. Pyroptosis-related gene signature is a novel prognostic biomarker for sarcoma patients. Dis Markers (2021) 2021:1–13. doi: 10.1155/2021/9919842
190. Fulda S. Cell death pathways as therapeutic targets in rhabdomyosarcoma. Sarcoma (2012) 2012:1–5. doi: 10.1155/2012/326210
191. Petak I, Douglas L, Tillman DM, Vernes R, Houghton JA. Pediatric rhabdomyosarcoma cell lines are resistant to fas-induced apoptosis and highly Sensitive to TRAIL-induced apoptosis. Clin Cancer Res (2000) 6:4119–27.
192. Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer (2008) 50:1181–9. doi: 10.1002/pbc.21433
193. Maruwge W. Sorafenib inhibits tumor growth and vascularization of rhabdomyosarcoma cells by blocking IGF-1R-mediated signaling. Onco Targets Ther (2008) 1:67–8. doi: 10.2147/OTT.S3833
194. Habermann KJ, Grünewald L, van Wijk S, Fulda S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell Death Dis (2017) 8:e3067–7. doi: 10.1038/cddis.2017.412
195. Castro B, Alonso-Varona A, del Olmo M, Bilbao P, Palomares T. Role of γ-glutamyltranspeptidase on the response of poorly and moderately differentiated rhabdomyosarcoma cell lines to buthionine sulfoximine-induced inhibition of glutathione synthesis. Anticancer Drugs (2002) 13:281–91. doi: 10.1097/00001813-200203000-00010
196. Mizushima N. Autophagy: process and function. Genes Dev (2007) 21:2861–73. doi: 10.1101/gad.1599207
197. Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol (2012) 4:a008813–a008813. doi: 10.1101/cshperspect.a008813
198. Yun C, Lee S. The roles of autophagy in cancer. Int J Mol Sci (2018) 19:3466. doi: 10.3390/ijms19113466
199. Araki M, Motojima K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem Biophys Res Commun (2008) 367:462–7. doi: 10.1016/j.bbrc.2007.12.166
200. Wang C, Qu J, Yan S, Gao Q, Hao S, Zhou D. PFK15, a PFKFB3 antagonist, inhibits autophagy and proliferation in rhabdomyosarcoma cells. Int J Mol Med (2018) 42(1):359–367. doi: 10.3892/ijmm.2018.3599
201. Miyagaki S, Kikuchi K, Mori J, Lopaschuk GD, Iehara T, Hosoi H. Inhibition of lipid metabolism exerts antitumor effects on rhabdomyosarcoma. Cancer Med (2021) 10:6442–55. doi: 10.1002/cam4.4185
202. Moghadam AR, da Silva Rosa SC, Samiei E, Alizadeh J, Field J, Kawalec P, et al. Autophagy modulates temozolomide-induced cell death in alveolar rhabdomyosarcoma cells. Cell Death Discovery (2018) 4:52. doi: 10.1038/s41420-018-0115-9
203. Almasi S, Crawford Parks TE, Ravel-Chapuis A, MacKenzie A, Côté J, Cowan KN, et al. Differential regulation of autophagy by STAU1 in alveolar rhabdomyosarcoma and non-transformed skeletal muscle cells. Cell Oncol (2021) 44:851–70. doi: 10.1007/s13402-021-00607-y
204. Li C, Li Z, Song L, Meng L, Xu G, Zhang H, et al. GEFT inhibits autophagy and apoptosis in rhabdomyosarcoma via activation of the Rac1/Cdc42-mTOR signaling pathway. Front Oncol (2021) 11:656608. doi: 10.3389/fonc.2021.656608
205. Tam SY, Wu VWC, Law HKW. Influence of autophagy on the efficacy of radiotherapy. Radiat Oncol (2017) 12:57. doi: 10.1186/s13014-017-0795-y
206. Li L, Liu W-L, Su L, Lu Z-C, He X-S. The role of autophagy in cancer radiotherapy. Curr Mol Pharmacol (2020) 13:31–40. doi: 10.2174/1874467212666190809154518
207. Gewirtz DA. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem Pharmacol (2014) 90:208–11. doi: 10.1016/j.bcp.2014.05.016
208. Liu Y, Qi S, Meng L, Zhang L, Pang Y, Cui W, et al. GEFT aberrant expression in soft tissue sarcomas. Transl Cancer Res (2019) 8:141–9. doi: 10.21037/tcr.2019.01.16
209. Kumari R, Jat P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol (2021) 9:645593. doi: 10.3389/fcell.2021.645593
210. Medema JP. Escape from senescence boosts tumour growth. Nature (2018) 553:37–8. doi: 10.1038/d41586-017-08652-0
211. Tabasso AFS, Jones DJL, Jones GDD, Macip S. Radiotherapy-induced senescence and its effects on responses to treatment. Clin Oncol (2019) 31:283–9. doi: 10.1016/j.clon.2019.02.003
212. Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, et al. Senescence and cancer: A review of clinical implications of senescence and senotherapies. Cancers (Basel) (2020) 12:2134. doi: 10.3390/cancers12082134
213. Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The roles of autophagy and senescence in the tumor cell response to radiation. Radiat Res (2020) 194:103. doi: 10.1667/RADE-20-00009
214. di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
215. Kim H, Kim J, Chie E, DaYoung P, Kim I, Kim I. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat Oncol (2012) 7:39. doi: 10.1186/1748-717X-7-39
216. Sun Q, Melino G, Amelio I, Jiang J, Wang Y, Shi Y. Recent advances in cancer immunotherapy. Discover Oncol (2021) 12:27. doi: 10.1007/s12672-021-00422-9
217. Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res (2016) 22:1364–70. doi: 10.1158/1078-0432.CCR-15-0491
218. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol (2020) 21:541–50. doi: 10.1016/S1470-2045(20)30023-1
219. Rytlewski J, Milhem MM, Monga V. Turning ‘Cold’ tumors ‘Hot’: Immunotherapies in sarcoma. Ann Transl Med (2021) 9:1039–9. doi: 10.21037/atm-20-6041
220. Zhu M, Yang M, Zhang J, Yin Y, Fan X, Zhang Y, et al. Immunogenic cell death induction by ionizing radiation. Front Immunol (2021) 12:705361. doi: 10.3389/fimmu.2021.705361
221. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
222. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: Current challenges and future directions. Front Pharmacol (2018) 9:185. doi: 10.3389/fphar.2018.00185
223. Lhuillier C, Rudqvist N-P, Elemento O, Formenti SC, Demaria S. Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med (2019) 11:40. doi: 10.1186/s13073-019-0653-7
224. He K, Barsoumian HB, Sezen D, Puebla-Osorio N, Hsu EY, Verma V, et al. Pulsed radiation therapy to improve systemic control of metastatic cancer. Front Oncol (2021) 11:737425. doi: 10.3389/fonc.2021.737425
225. Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: The importance of dose and fractionation. Front Oncol (2014) 4:1. doi: 10.3389/fonc.2014.00001
226. Demaria S, Guha C, Schoenfeld J, Morris Z, Monjazeb A, Sikora A, et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J Immunother Cancer (2021) 9:e002038. doi: 10.1136/jitc-2020-002038
227. Gabrych A, Pęksa R, Kunc M, Krawczyk M, Izycka-Swieszewska E, Biernat W, et al. The PD-L1/PD-1 axis expression on tumor-infiltrating immune cells and tumor cells in pediatric rhabdomyosarcoma. Pathol Res Pract (2019) 215:152700. doi: 10.1016/j.prp.2019.152700
228. Kim C, Kim EK, Jung H, Chon HJ, Han JW, Shin K-H, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer (2016) 16:434. doi: 10.1186/s12885-016-2451-6
229. Bertolini G, Bergamaschi L, Ferrari A, Renne SL, Collini P, Gardelli C, et al. PD-L1 assessment in pediatric rhabdomyosarcoma: a pilot study. BMC Cancer (2018) 18:652. doi: 10.1186/s12885-018-4554-8
230. di Maggio FM, Minafra L, Forte GI, Cammarata FP, Lio D, Messa C, et al. Portrait of inflammatory response to ionizing radiation treatment. J Inflammation (2015) 12:14. doi: 10.1186/s12950-015-0058-3
231. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J BioMed Sci (2018) 25:20. doi: 10.1186/s12929-018-0426-4
232. Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: The 4 r’s of radiobiology revisited. Stem Cells (2010) 28:639–48. doi: 10.1002/stem.318
233. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature (2009) 458:780–3. doi: 10.1038/nature07733
234. Ignatius MS, Hayes MN, Lobbardi R, Chen EY, McCarthy KM, Sreenivas P, et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep (2017) 19:2304–18. doi: 10.1016/j.celrep.2017.05.061
235. Rota R, Ciarapica R, Miele L, Locatelli F. Notch signaling in pediatric soft tissue sarcomas. BMC Med (2012) 10:141. doi: 10.1186/1741-7015-10-141
236. Slemmons KK, Crose LES, Riedel S, Sushnitha M, Belyea B, Linardic CM. A novel notch–YAP circuit drives stemness and tumorigenesis in embryonal rhabdomyosarcoma. Mol Cancer Res (2017) 15:1777–91. doi: 10.1158/1541-7786.MCR-17-0004
237. Perrone C, Pomella S, Cassandri M, Pezzella M, Milano GM, Colletti M, et al. MET inhibition sensitizes rhabdomyosarcoma cells to NOTCH signaling suppression. Front Oncol (2022) 12:835642. doi: 10.3389/fonc.2022.835642
238. Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood (2010) 115:4030–8. doi: 10.1182/blood-2009-09-241000
239. Thippu Jayaprakash K, Michael A. Notch inhibition: a promising strategy to improve radiosensitivity and curability of radiotherapy. Clin Oncol (2021) 33:e44–9. doi: 10.1016/j.clon.2020.06.015
240. Lu Y, Chan Y-T, Tan H-Y, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol Cancer (2020) 19:79. doi: 10.1186/s12943-020-01197-3
241. Megiorni F. Epigenetics in rhabdomyosarcoma: cues to new biomarkers and targeted therapies. EBioMedicine (2020) 52:102673. doi: 10.1016/j.ebiom.2020.102673
242. Cieśla M, Dulak J, Józkowicz A. MicroRNAs and epigenetic mechanisms of rhabdomyosarcoma development. Int J Biochem Cell Biol (2014) 53:482–92. doi: 10.1016/j.biocel.2014.05.003
243. Pucci G, Forte GI, Cavalieri V. Evaluation of epigenetic and radiomodifying effects during radiotherapy treatments in zebrafish. Int J Mol Sci (2021) 22:9053. doi: 10.3390/ijms22169053
244. de Schutter H, Nuyts S. Radiosensitizing potential of epigenetic anticancer drugs. Anticancer Agents Med Chem (2009) 9:99–108. doi: 10.2174/187152009787047707
245. Christmann M, Kaina B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat Research/Reviews Mutat Res (2019) 780:15–28. doi: 10.1016/j.mrrev.2017.10.001
246. Garner IM, Brown R. Is there a role for epigenetic therapies in modulating DNA damage repair pathways to enhance chemotherapy and overcome drug resistance? Cancers (Basel) (2022) 14:1533. doi: 10.3390/cancers14061533
247. Hayes JD, Dinkova-Kostova AT. Epigenetic control of NRF2-directed cellular antioxidant status in dictating life-death decisions. Mol Cell (2017) 68:5–7. doi: 10.1016/j.molcel.2017.09.023
248. García-Guede Á, Vera O, Ibáñez-de-Caceres I. When oxidative stress meets epigenetics: Implications in cancer development. Antioxidants (2020) 9:468. doi: 10.3390/antiox9060468
249. Hajji N, Joseph B. Epigenetic regulation of cell life and death decisions and deregulation in cancer. Essays Biochem (2010) 48:121–46. doi: 10.1042/bse0480121
250. Saleh R, Toor SM, Sasidharan Nair V, Elkord E. Role of epigenetic modifications in inhibitory immune checkpoints in cancer development and progression. Front Immunol (2020) 11:1469. doi: 10.3389/fimmu.2020.01469
251. Megiorni F, Camero S, Ceccarelli S, McDowell HP, Mannarino O, Marampon F, et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget (2016) 7:79342–56. doi: 10.18632/oncotarget.12688
252. Megiorni F, Camero S, Pontecorvi P, Camicia L, Marampon F, Ceccarelli S, et al. OTX015 epi-drug exerts antitumor effects in ovarian cancer cells by blocking GNL3-mediated radioresistance mechanisms: Cellular, molecular and computational evidence. Cancers (Basel) (2021) 13:1519. doi: 10.3390/cancers13071519
253. Kim J-G, Park M-T, Heo K, Yang K-M, Yi J. Epigenetics meets radiation biology as a new approach in cancer treatment. Int J Mol Sci (2013) 14:15059–73. doi: 10.3390/ijms140715059
254. Pogribny I, Raiche J, Slovack M, Kovalchuk O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem Biophys Res Commun (2004) 320:1253–61. doi: 10.1016/j.bbrc.2004.06.081
Keywords: rhabdomyosarcoma, radiotherapy, radiation therapy, radioresistance, radiosensitizers
Citation: Camero S, Cassandri M, Pomella S, Milazzo L, Vulcano F, Porrazzo A, Barillari G, Marchese C, Codenotti S, Tomaciello M, Rota R, Fanzani A, Megiorni F and Marampon F (2022) Radioresistance in rhabdomyosarcomas: Much more than a question of dose. Front. Oncol. 12:1016894. doi: 10.3389/fonc.2022.1016894
Received: 11 August 2022; Accepted: 12 September 2022;
Published: 29 September 2022.
Edited by:
Andrea Lancia, San Matteo Hospital Foundation (IRCCS), ItalyReviewed by:
Venturina Stagni, Department of Biomedical Sciences (CNR), ItalyCopyright © 2022 Camero, Cassandri, Pomella, Milazzo, Vulcano, Porrazzo, Barillari, Marchese, Codenotti, Tomaciello, Rota, Fanzani, Megiorni and Marampon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Marampon, ZnJhbmNlc2NvLm1hcmFtcG9uQHVuaXJvbWExLml0; Francesca Megiorni, ZnJhbmNlc2NhLm1lZ2lvcm5pQHVuaXJvbWExLml0
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.