 Pattanaik Smita
Pattanaik Smita Patil Amol Narayan
Patil Amol Narayan Kumaravel J
Kumaravel J Prakash Gaurav
Prakash Gaurav
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 December 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1015200
This article is part of the Research Topic Therapeutic Drug Monitoring and Clinical Toxicology of Anti-Cancer Drugs, volume II View all 13 articles
Cytotoxic drugs are highly efficacious and also have low therapeutic index. A great degree of caution needs to be exercised in their usage. To optimize the efficacy these drugs need to be given at maximum tolerated dose which leads to significant amount of toxicity to the patient. The fine balance between efficacy and safety is the key to the success of cytotoxic chemotherapeutics. However, it is possibly more rewarding to obtain that balance for this class drugs as the frequency of drug related toxicities are higher compared to the other therapeutic class and are potentially life threatening and may cause prolonged morbidity. Significant efforts have been invested in last three to four decades in therapeutic drug monitoring (TDM) research to understand the relationship between the drug concentration and the response achieved for therapeutic efficacy as well as drug toxicity for cytotoxic drugs. TDM evolved over this period and the evidence gathered favored its routine use for certain drugs. Since, TDM is an expensive endeavor both from economic and logistic point of view, to justify its use it is necessary to demonstrate that the implementation leads to perceivable improvement in the patient outcomes. It is indeed challenging to prove the utility of TDM in randomized controlled trials and at times may be nearly impossible to generate such data in view of the obvious findings and concern of compromising patient safety. Therefore, good quality data from well-designed observational study do add immense value to the scientific knowledge base, when they are examined in totality, despite the heterogeneity amongst them. This article compiles the summary of the evidence and the best practices for TDM for the three cytotoxic drug, busulfan, 5-FU and methotrexate. Traditional use of TDM or drug concentration data for dose modification has been witnessing a sea change and model informed precision dosing is the future of cytotoxic drug therapeutic management.
Cytotoxic drugs are the oldest class of anticancer drugs. They are highly efficacious and also have low therapeutic index; therefore, a great degree of caution needs to be exercised in their usage. To optimize the efficacy, these drugs need to be given at the maximum tolerated dose. This dose leads to a significant amount of toxicity to the patient. The fine balance between efficacy and safety is the key to the success of cytotoxic chemotherapeutics. However, it is possibly more rewarding to obtain that balance for this class drugs as the frequency of drug-related toxicities is higher compared to the other therapeutic class and is potentially life-threatening and may cause prolonged morbidity. Significant efforts have been invested in the last three to four decades in therapeutic drug monitoring (1) research to understand the relationship between the drug concentration and the response achieved for therapeutic efficacy as well as drug toxicity for cytotoxic drugs. TDM evolved over this period and the evidence gathered favored its routine use for certain drugs. Since TDM is an expensive endeavor, both from an economic and a logistic point of view, to justify its use, it is necessary to demonstrate that the implementation leads to perceivable improvement in patient outcomes. It is indeed challenging to prove the utility of TDM in randomized controlled trials and at times may be nearly impossible to generate such data in view of the obvious findings and concern of compromising patient safety. Therefore, good quality data from a well-designed observational study add immense value to the scientific knowledge base, when they are examined in totality, despite the heterogeneity among them. This article compiles the summary of the evidence and the best practices for TDM for the three cytotoxic drug, busulfan, 5-FU, and methotrexate (Mtx) (Figure 1).
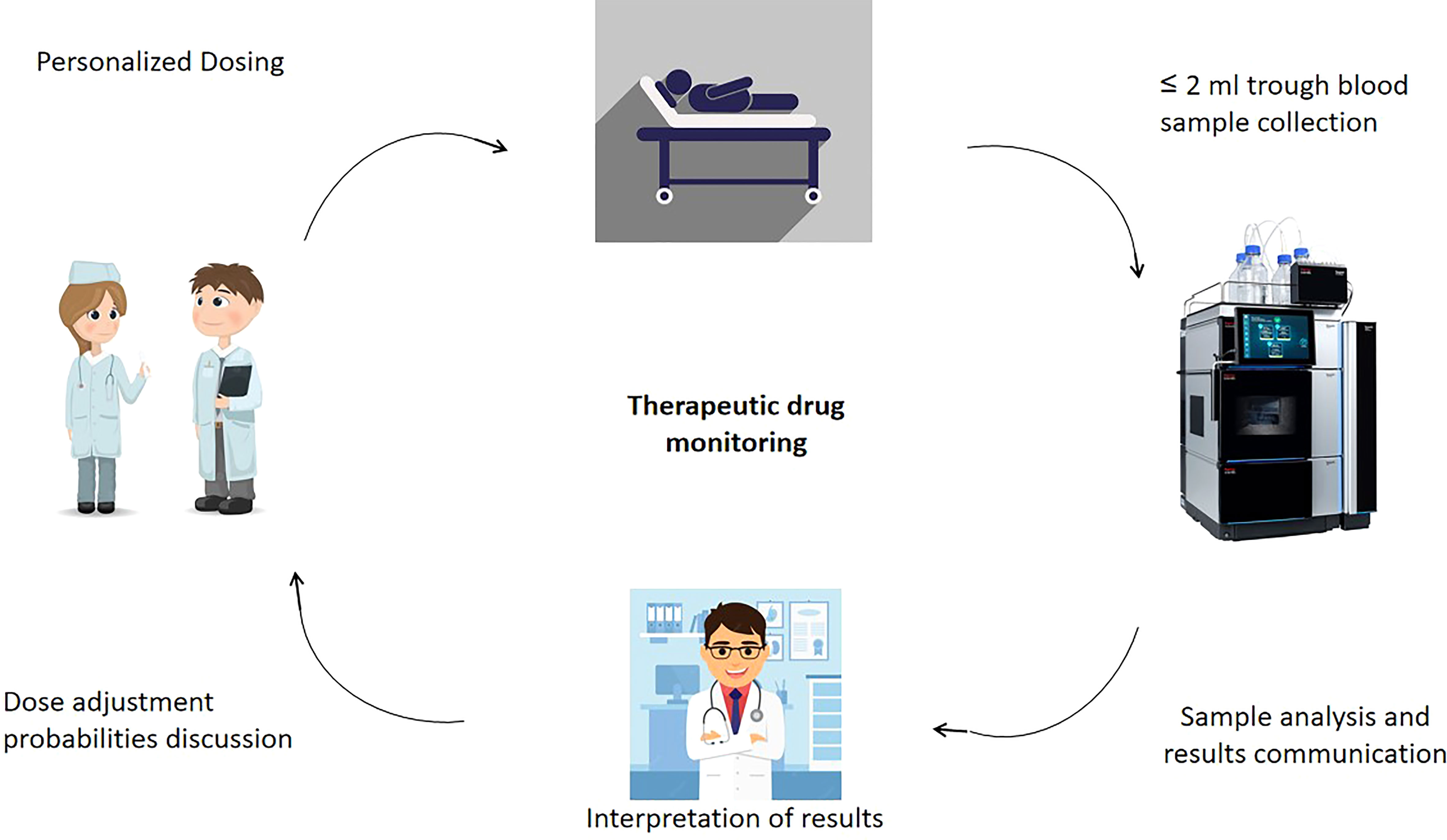
Figure 1 Overview of TDM applications in cytotoxic anticancer drug therapy.
This article compiles the summary of the evidence for three cytotoxic drugs, busulfan, 5-FU, and Mtx, where the case for TDM is much established. Though there is evolving evidence for TDM in favor of the platinum group of drugs (paclitaxel, docetaxel, and carboplatin), tyrosine kinase inhibitors (1), and others like abiraterone and tamoxifen, they are beyond the scope of this review as we primarily intend to bring out the TDM best practices for the established cytotoxic drugs only (1–4).
Busulfan is a bifunctional DNA alkylating anticancer agent and acts in a cell cycle nonspecific manner. After systemic absorption, the carbonium ions are rapidly formed, which react with guanine molecules of the DNA through a nucleophilic substitution reaction (SN2) (5) forming intra- and interstrand crosslinks. This leads to breaks in the DNA molecule as well as crosslinking of the twin strands, resulting in interference of DNA replication and transcription of RNA and hence cell proliferation (6, 7).
Oral busulfan had been used for the treatment of chronic myeloid leukemia (CML) (8, 9) and other myeloproliferative disorders since the 1950s due to the inhibitory activity of the sulfonic derivatives of the drug on hematopoiesis (10, 11). In the last few decades, this use has become less popular due to the unsatisfactory curative potential despite initial good cytoreduction, thus reducing its use as a palliative therapy only. It was also found to have dismal efficacy in certain subsets of patients like those who have Philadelphia negative CML (12). In the present time, the use of oral busulfan is limited to palliative treatment of CML (myeloid, myelocytic, and granulocytic) and myeloproliferative neoplasms (13). In the late 1980s, oral busulfan was started to be used as a pretransplant myeloablative agent along with cyclophosphamide (8, 14). Oral administration of busulfan results in significant gastrointestinal irritability leading to nausea and vomiting that typically led to unpredictable systemic bioavailability (15, 16). Erratic absorption from the intestine coupled with GI complications substantially increased the systemic exposure variability to more than 10-fold (reported bioavailability ranged from 20% to 99%) (9). This unpredictability was responsible for poor efficacy and higher failure rate of the oral regimen and therefore paved the way for a major refinement in busulfan delivery. The i.v. formulations of busulfan were developed by Anderson and colleagues at the MD Anderson Cancer Center in the USA and was approved by the US FDA in 1999 (17, 18). Later on, intravenous busulfan replaced the oral route, as it provided better control over drug administration, bringing down the intradose exposure variation to 2- to 2.5-fold and maximizing the antitumor efficacy (19).
Though the use of oral busulfan was short-lived and it has now been replaced by intravenous use, it provided great insight to understand the pharmacokinetics (PK) of the drug and helped in the evolution of the TDM strategy. The PK profile of the drug is best described as a single compartment model, with rapid absorption and maximum plasma concentration achieved within 1 h. Majority of the drug is metabolized in the liver by the glutathione-S-transferase (20) enzyme and the metabolites are excreted via urine. A minimal amount of the drug (<2%) is excreted unchanged in the urine. The drug is eliminated by a log-linear fashion. We will discuss the PK of i.v. administered drug especially in the context of high-dose busulfan therapy as a part of myeloablative regimen before hematopoietic stem cell transplantation (HSCT). It is generally administered at a dose of 0.8 mg/kg in normal saline or 5% dextrose 6 hourly infusion over 2 h (or 3.2 mg/kg daily). Sixteen such doses (or four doses for once daily doses) are administered over 4 days (−7, −6, −5, and −4 days when day 0 is the day for infusion of the hematopoietic stem cell transfusion). The PK profile of the i.v. drug is similar to that of oral administration, which skips the fast-pass metabolism, providing 100% bioavailability.
High-dose intravenous busulfan has now been the preferred choice for myeloablation at most of the HSCT centers compared to total body irradiation (TBI) (12). It is currently the standard of care in the pretransplant myeloablative conditioning regimens along with other lymphotoxic chemotherapeutic agents (cyclophosphamide and fludarabine), for hematopoietic stem cell transplantation (HSCT). HSCT is a widely used potentially curative treatment strategy for several diseases, including hematological malignancies (e.g., leukemia and lymphoma), solid tumors, and nonmalignant disorders (e.g., thalassemia and sickle cell anemia).
As described above, though busulfan is a highly efficacious cytotoxic drug, it poses several challenges in optimum dosing. Many factors qualify busulfan as an ideal candidate for TDM. The need for optimization of the dose was realized right at the beginning of the therapeutic failure of the oral regimen due to inadequate exposure. On the other hand, the serious adverse drug effects associated with higher exposure of busulfan lead to both hematological and nonhematological events. The onset of some of the ADRs are acute and some lead to chronic effects due to exposure over longer periods (21). The most common ADR include acute graft versus host disease (GVHD), mucositis, myelosuppression, seizures, hepatic veno-occulsive disease (HVOD, also known as sinusoidal occlusion syndrome), bronchopulmonary dysplasia, pulmonary fibrosis, and embryo–fetal toxicity. Robust evidence from several clinical studies support direct relationship between exposure–efficacy–toxicity. The introduction of the i.v. formulation led to better control over the concentration of the drug achieved in the systemic circulation circumventing the unpredictable intestinal absorption and hepatic fast-pass metabolism. However, the need for TDM was realized because of the bimodal distribution in the posttransplant mortality. Those who achieved inadequate concentration or those who achieved undesirably high concentration succumbed. With 6 hourly regimen, the threshold for therapeutic response is an exposure over one dosing interval, which is found to be 900–950 μM·min, whereas the threshold for toxic adverse effects is 1,200–1,500 μM·min. Hence, it can be concluded that busulfan has a narrow therapeutic window (900–1,200 mM·min) and consequently has a low therapeutic index (1,500/900 is <2).
With the use of i.v. formulation, the systemic concentration of busulfan had attained better predictability. Previously, interindividual variability of busulfan bioavailability with oral administration varied more than 10 fold, whereas it reduced to 2- to 2.5-fold with the use of i.v. formulation (19, 21). The reported coefficient of variation in busulfan maximal plasma concentration, clearance, and AUC was 18%, 25%, and 25% (13, 18) (Otsuka America Pharmaceutical Inc., Rockville,). Several subsequent clinical studies also confirmed the same (19, 22). The globally reported interindividual variability with i.v. use of busulfan is about 30%. Moreover, the intraindividual variability in exposure for the same dose may be as high as fivefold. Pharmacogenetic alterations in the metabolizing enzymes, drug transporters, and chronopharmacology have been studied in this context. Single-nucleotide polymorphisms (SNPs) of the GST isoenzymes (GSTA1, GSTM1, and GSTP1) have been demonstrated to significantly affect the PK (clearance) of busulfan, thereby contributing to interindividual variability in small-sized studies and mostly in a pediatric population (23–25). However, their effect on the pharmacodynamics and incidence of therapeutic success vis-à-vis adverse events is not clear and hence their clinical relevance has not yet been demonstrated (26, 27). Similarly, polymorphic variants of CYP 2C9, 2B6, and membrane transporters like ABCB1 and SLC have also been studied for their association with busulfan disposition. Nevertheless, their role has not been found to be consequential (28). Chronopharmacologic variability in GST and CYP enzyme activity have also been studied in the context of variability in PK of busulfan. It has been postulated that this may contribute to intraindividual variability of AUC for the same dose of busulfan. However, these studies were conducted in the earlier period with oral busulfan and had several operational confounding factors, most importantly changes in intestinal motility. Moreover, the results from other studies have also yielded conflicting results (29, 30). To summarize, it was observed that the interindividual variability is significant enough with high-dose intravenous busulfan therapy to merit practice of TDM.
In addition to the above factors, potentially important drug–drug interactions may be kept in mind to optimize busulfan exposure and TDM could help in dose and exposure optimization. Several classes of drugs have been studied (Table 1). The most important class that needs a cautious approach is antifungal drugs, and the strategy of spacing the prescription is adopted at many centers to avoid the interaction. Another drug that merits mention is phenytoin, which had been the most common antiseizure prophylaxis prescribed during high-dose busulfan therapy, which potentially induces busulfan clearance. The newer antiepileptics like levetiracetam is used these days to avoid the interaction. Nevertheless, TDM can be a very useful tool in case of inadvertent drug interaction.
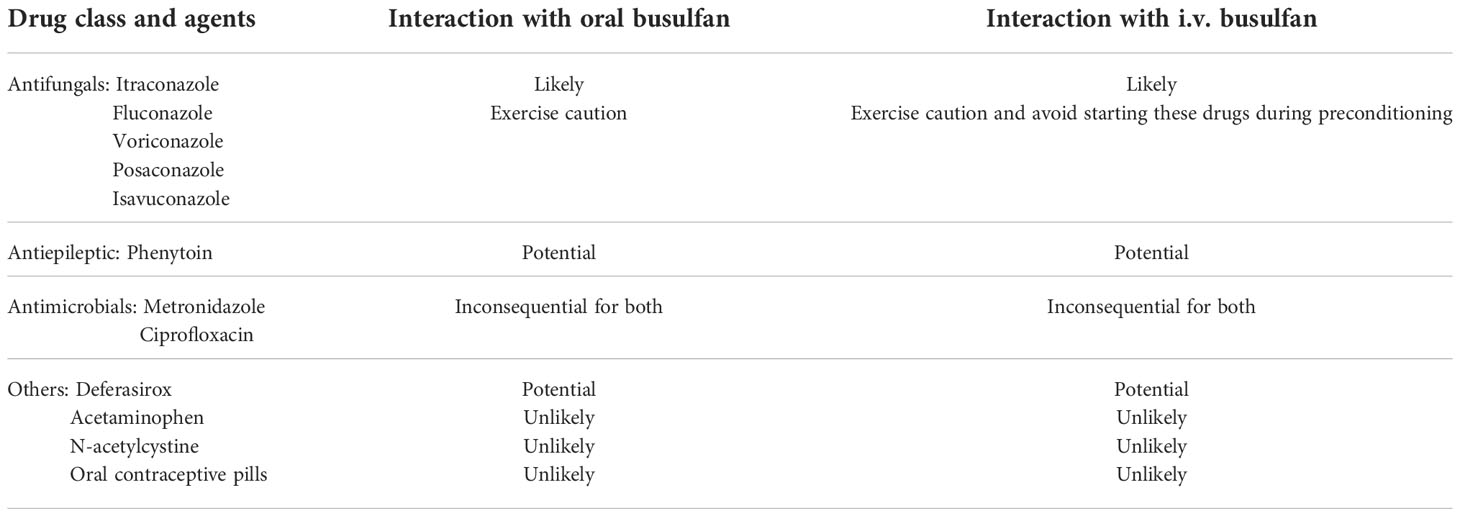
Table 1 Potential drug interactions with busulfan.
Other factors contributing to variability in busulfan exposure are age and body composition. Clearance of busulfan is 30% higher in children compared to adults, which has been postulated to be due to the higher physiological activity of the GST in young age groups. Patients with higher fat composition have a prolonged elimination half-life for busulfan as the fat tissue behaves as a temporary storage compartment for busulfan due to its high lipid solubility (31).
At the advent of the oral conditioning regimen by Santos et al., a classical veno-occlusive disease of the liver was reported in the early 1980s, and by the end of the 1980s, it was suggested that TDM could have a potential role in reducing such adverse events (32–36). The oral dose regimen underwent several modifications; however, it was increasingly realized that several adverse events other than HVOD were associated with higher exposure to busulfan like acute GVHD (a cytokine storm that damages the normal organs in the body), seizures (typically of generalized type), and total incidence of treatment-related mortality. Though a multivariate analysis confirmed that oral administration is the single most important factor for HVOD, several other concomitant drugs like cyclophosphamide used in the preconditioning regimen, individual patient susceptibility, and disease type were also found to contribute to the event (12). Nevertheless, HVOD and hepato-renal failures are considered to be a classical trademark toxicity of high-dose busulfan therapy (12). Grochow and Dix demonstrated that the hepatic events are related to the drug concentration (34, 37). Both the studies indicated that an AUC of >1,500 μM·min and a steady-state plasma concentration (Css) above 1,025 ng/ml were significantly associated with HVOD after 6 hourly oral administration of busulfan. The evidence from these studies confirmed that the threshold to HVOD is about 1,500 μM·min for a 6 hourly dosing (22, 38–40). Other studies, which used single daily dose administration, showed that the incidence of death due to HVOD was significantly increased above the exposure level of 6,000 μM·min (41, 42), hence confirming a full concordance between the findings. Similarly, the incidence seizure occurrence during treatment were significantly more during the higher exposures (30, 43). This is clearly explained by the PK profile of the drug, busulfan being lipophilic, and small molecules crossing the blood–brain barrier, and a higher concentration is achieved in the brain (1.3:1.0), hence causing acute neurotoxic effects. Apart from reducing toxicity, a pooled analysis of data of 674 patients who underwent stem cell transplant has shown that maintaining busulfan concentration within a therapeutic range led to better posttransplant survival. In the study, event-free survival at 2 years was 77.0% in patients with an optimum intravenous busulfan AUC of 78–101 mg·h/L compared with 66.1% in patients at the low historical target of 58–86 mg·h/L. Moreover, acute toxicity (p = 0.011) and transplant-related mortality (p < 0.0001) were significantly higher in high (>101 mg·h/L) busulfan AUC (44).
A substantial number of studies in the early period during the oral busulfan use documented therapeutic failures. The patients with nonmalignant disease were especially prone to failed engraftment due to the intact immune system and faster clearance of busulfan. On the other hand, the patients with malignant disease failed engraftment, which led to the relapse and recurrence (45, 46) of the disease (47, 48). Slattery et al. suggested a concentration cutoff of 1,250 μM·min and a Css of 925 ng/ml for optimal therapeutic effect in bone marrow transplantation (39). Subsequently, they performed the study in patients with CML and showed that targeting an AUC of >1,350 μM·min led to treatment success (38). Radich et al. also showed similar findings; with AUC >1,350 μM·min, excellent outcome was observed (49). More recently, Anderson et al. showed that an AUC value <900 μM·min was associated with higher failure rates in the 6 hourly i.v. busulfan regimen (22). There was another interesting observation that confirmed the exposure–response relationship for busulfan. Children who clinically tolerated oral busulfan better than adults perhaps experienced more relapse after HSCT due to graft failure. As the understanding in busulfan PK was consolidated for its effect related to age, it was found that these children had faster clearance of busulfan and hence had reduced exposure to the drug, leading to better tolerance as well as therapeutic failure (48, 50, 51). Hence, the TDM-based dose adjustment to keep the AUC above 900 μM·min has been the consensus agreement for achieving desirable therapeutic effect. Several large retrospective studies established a therapeutic window for clinical response between 900 and 1,500 μM·min for AUC over 6 h in a 16-dose administration schedule. The corresponding cumulative AUC over 16 doses was set at 144,000 to 240,000 mM·min (32, 38, 39, 52, 53).
In the 1980s, it was realized that single time point concentration would not be enough due to erratic absorption. Hence, exposure needs to be determined. With intravenous dosing, the concept of monitoring steady-state concentration was floated. Researchers came up with several formulas to calculate the Css and extrapolate a Css curve with first-dose AUC assuming that an ideal true steady state is reached. However, this was not a universal phenomenon, and a variation of 500% is noted between days 1 and 4 of infusion, as the volume of distribution (Vd) appeared to change with time. The hepatic enzyme activity decreases with progressive administration; hence, higher concentrations are observed in pretreated patients. Therefore, targeting the Css was found to be difficult and only 55% could achieve it even with best possible dose titration. Subsequent studies demonstrated that the exposure over a single dose is a better parameter to target. Hence, a limited sampling approach is adopted and the exposure is assessed by the calculation of area under the curve over the dosing interval (AUC0-τ), which is the parameter of interest for TDM (54). The current best practices for sample collection is described in the subsequent section.
The published literature that constitutes the evidence base for deployment of TDM in routine patient care is very heterogeneous. Adequately powered controlled clinical studies are sparse. Despite this, the expert opinion fully favors practice of TDM for personalized busulfan therapy so as to optimize the exposure, maximize the therapeutic effect, and minimize the adverse events. This is applicable only in the setting of high-dose busulfan for preconditioning before HSCT but not reduced intensity preconditioning (55). The landmark randomized controlled study by Grochow et al. demonstrated a reduction of HVOD to reduce from 75% to 15% when the busulfan dose titration was done based on the TDM data to keep the AUC below 1,500 μM·min and only 5% of the patients were maintained within therapeutic range. Thereafter, it had been really difficult to refute this finding by conducting randomized controlled studies, as it involved concerns of patient safety. Several studies conducted in different settings (different age groups like adult and children; several disease conditions like CML, AML, and multiple myeloma; and different co-conditioning agents like cyclophosphamide and melphalan) encountered similar findings (5, 38, 41, 49, 56–59). Traditionally, oral busulfan was administered in 6 hourly doses to minimize the GI adverse events. The same schedule continued for i.v. drug also due to the short half-life of the drug. The subsequent phase I/II PK studies did not demonstrate that there is a significant difference in the busulfan volume of distribution, half-life, and clearance between 6 hourly and 24 hourly dosing (60). As expected, the Cmax values achieved with once daily doses were higher compared to the 6 hourly doses, but it was found that the clinical outcomes like rates of disease relapse (61), overall survival (61, 62), and HVOD (61–63) were not significantly different. Though definitive conclusions were impossible to arrive at, due to significant heterogeneity between the studies with regard to the disease population, concomitant medications, and inconstant use of TDM, QID dosing and OD dosing are in general considered to be equivalent.
A number of analytical techniques have been used for the quantification of busulfan (e.g., HPLC, LC/MS, and GC/MS) to measure circulating plasma concentrations. Liquid chromatography methods coupled with mass spectrometry have been the most commonly used technique for clinical care because of their sensitivity, specificity, and accuracy of results (64, 65). However, it is technically demanding and the sample processing and analysis may be time-consuming. Busulfan degrades quickly at room temperature; hence, the samples have to be analyzed quickly. It is also desirable to have a short turnaround time as the dose modification decisions have to be provided within a window of as less as 6 h. For delivering an efficient TDM service, these factors must be taken into consideration. Recently, several automated assays have been developed. Nanoparticle- and microparticle-based immunoassays are the two significant additions. These assays can be quickly performed without any sample processing steps. It has also been shown that they are in good conformity with the LC-MS/MS methods (20, 66) and are gaining acceptance.
The steady-state plasma concentration for busulfan is not achieved over 6–24 h even if the half-life is 2–3 h. Therefore, several samples are needed to determine the exposure to the drug over a dosing interval. A limited sampling approach is adopted, and the best schedule for sample collection as per the FDA monograph for the first dose is 2 h (end of infusion), 4 h, and 6 h (just before the start of next infusion) after the start of the infusion. For subsequent doses another sampling time point (baseline, before the start of the infusion) is added. The blood sample should be collected in heparinized vials and placed on wet ice till centrifugation. The plasma should be separated within 1 h and transferred to cryo vials. All plasma samples should be frozen at −20°C until analysis. The samples are stable for 3–4 days at 2–8°C and up to 6 months at −20°C. The AUC over the dosing interval is calculated by the simple trapezoidal rule. For once daily dosing, the terminal part of the elimination curve is calculated from a preinfusion “0” concentration for the first dose or the preinfusion concentration for the next dose. AUC0–24 h is calculated by adding AUC0-6 h and AUC6–24 h. The FDA-recommended AUC and Css values are similar to the consensus statement by the American Society for Bone Marrow Transplantation Practice Guideline Committee. The AUC and Css targets for high-dose busulfan therapy is enumerated in Table 2. The attempt to optimize the exposure is started as early as the first day dose modifications.

Table 2 Summary of dose and exposure–concentration targets for high-dose busulfan therapy.
Model informed precision dosing (MPID) has been gaining popularity over the last decade. These methods are attractive as they use less number of samples to predict the AUC and suggest dose modification using Bayesian forecasting. The model-informed dosing also takes into account the body size, age, organ function, etc. for dose prediction unlike the traditional dosing, which is weight based. Examples of some of the available software are insightRX and NextDose. The web-based services are www.insight-rx.com and https://doseme-rx.com. It has been shown to have relatively less bias in AUC estimations of busulfan compared to the conventional trapezoidal method (78, 79) and may be less prone for error that may creep in due to inadequate documentation of sampling time (80). The MIPD strategy may be software or web-based platforms.
5-fluorouracil (5-FU) is a pyrimidine analogue used in the treatment of several solid tumors (breast, colorectal, stomach, head, and neck). It is typically administered intravenously as prolonged infusion due to its very short half-life (20 min) (81).
After administration, 5-FU penetrates cells by a facilitated transport route, where it is transformed into fluorodeoxyuridine monophosphate (FdUMP). FdUMP inhibits the synthesis of the deoxythymidine monophosphate (dTMP) by forming complexes with the enzyme thymidylate synthase (TS). dTMP is integral to DNA replication and repair, and its depletion causes an imbalance of intracellular nucleotides, which allows the enzyme endonuclease to cause double-stranded breaks in the DNA (82).
When used orally, 5-FU shows poor absorption in the gastrointestinal tract. To maximize systemic absorption, parenteral administration of 5-FU is used when treating visceral cancers. 5-FU can be given intravenously as a bolus infusion over a period of days or as a “protracted” infusion using an ambulatory pump for 1 to 2 weeks. Fluorouracil is distributed throughout the body, including the liver, brain, bone marrow, CSF, and intestinal mucosa. About 80% of 5-FU is metabolized in the liver by dihydropyrimidine dehydrogenase (DPD) to the inactive metabolite dihydrofluorouracil (5-FUH2) (83). Due to the fast catabolism in the liver, the terminal half-life of 5-FU when administered intravenously is about 8 to 20 min. This finding can be explained by the saturation of 5-FU metabolism by DPD as plasma concentrations approach the Km of DPD, which is known to be approximately 4.6 mg/L, which subsequently causes a more than proportionate increase in 5-FU plasma concentrations with dosage (73). 5-FU undergoes dose-dependent elimination and is excreted via urine as unchanged drug within 6 h of 5% to 20% and metabolites over 3 to 4 h.
Diarrhea was the most often reported side effect in patients undergoing systemic 5-FU treatment. Dehydration, nausea, and vomiting are typical side effects. Neutropenia, pyrexia, pulmonary embolism, thrombocytopenia, and leukopenia are more serious side effects that need to be monitored in individuals receiving systemic 5-FU chemotherapy (82).
5-FU therapy meets the most important criteria for TDM, i.e., an established exposure–toxicity and exposure–clinical activity relationship. Currently, body surface area (BSA)-based calculation is used for dosing the fluoropyrimidine 5-FU (84). At typical doses, 5-FU’s therapeutic effectiveness is moderate, as the dosing is often limited by the safety profile, with myelosuppression and gastrointestinal toxicity being the most common side effects. The conservative dose adjustment steps without TDM support result in delayed nonattainment of requisite exposure and, therefore, lead to suboptimal therapeutic success. The exposure–toxicity and exposure–clinical activity relationship has been reported in several clinical studies both prospective and retrospective (Table 3).
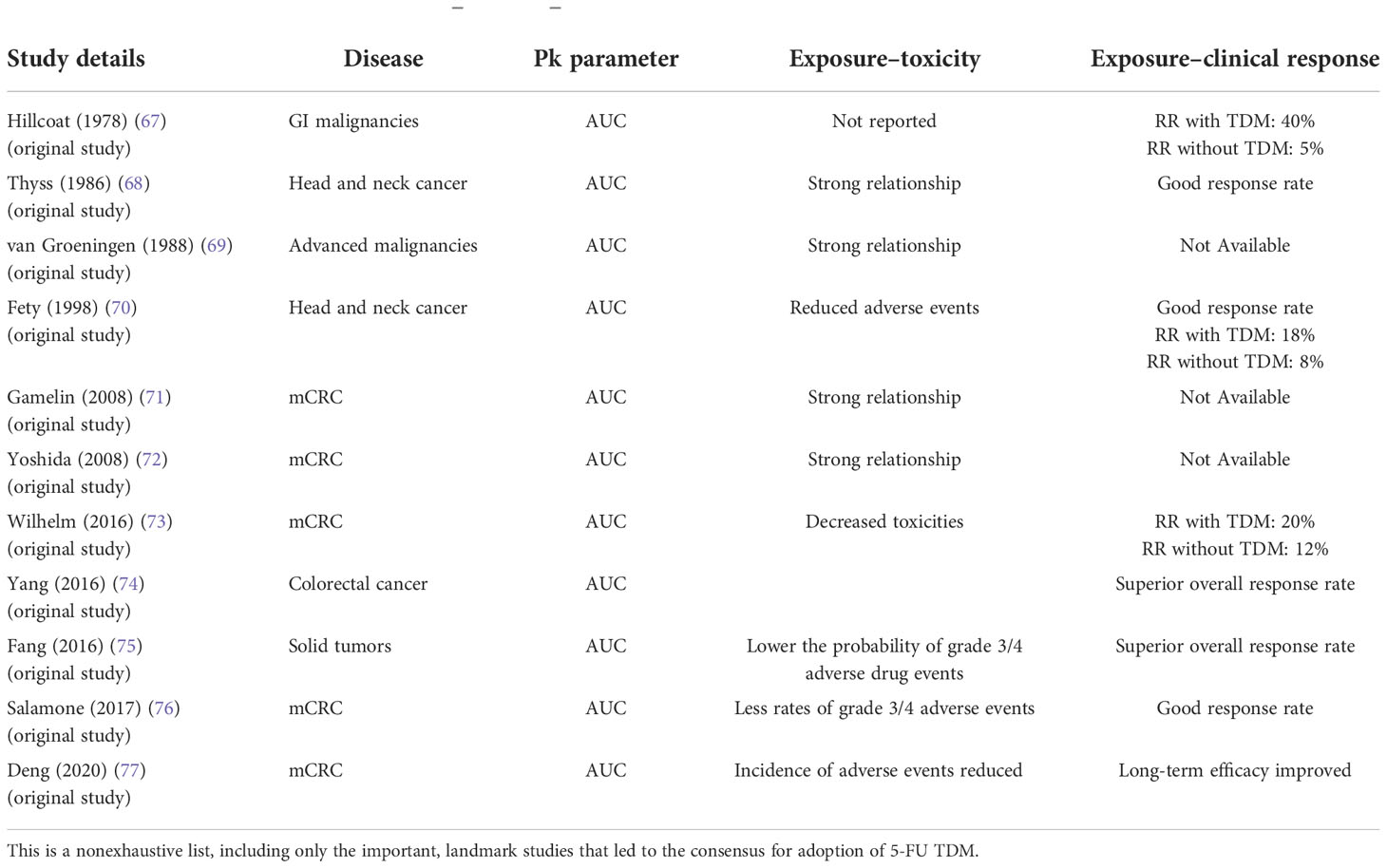
Table 3 Evidence base in support of exposure–response–toxicity for 5-FU.
The second factor that adds to the justification for TDM is extremely high interindividual variability and intraindividual variability. While it was brainstormed in several professional meetings of physicians and laboratory scientists that there might be methodological variability in studies reporting 5-FU blood concentration, it would be important to evaluate more closely the technical and pharmacological issues while interpreting the results. The technical and pharmacological issues include the use of elastomeric pump balloons, which are sensitive to pressure, temperature, season, and patient activity, causing variability in infusion pump speed and resulting in variability in steady-state plasma concentrations of 5-FU (85, 86). Variability in infusion pump speed will also occur when using portable pumps, which essentially deliver a series of boluses. Due to the fact that even trace levels of DPD are present in blood, particularly the buffy coat, which makes 5-FU unstable after sample collection (87–90), it is crucial to properly separate the plasma and/or add DPD inhibitors. A sampling time of five half-lives following the start of the infusion does not yet equate to a sample at genuine steady state since, biologically, the elimination of 5-FU seems to fluctuate upon dosing. In fact, reaching steady-state 5-FU levels could take several hours (91–93). Furthermore, as DPD activity and maybe that of other 5-FU metabolizing enzymes exhibit some circadian rhythm (94, 95), variations in the timing of samples may further contribute to variability 84–88.
Nevertheless, it was unanimously agreed that by using the existing BSA-based dosing techniques and administering 5-FU by prolonged infusion, the interindividual variability in 5-FU plasma concentrations reached 40% and intraindividual variability reached 20%. Causes of interindividual variability include significant variation in DPD-mediated catabolism due to pharmacogenetic factors like SNPs in DPYD gene encoding DPD activity and environmental factors like nutritional status (68, 76).
Discounting technical and pharmacological issues, the intraindividual variability is considered to be modest and less than interindividual variability but significant enough for disproportionate systemic exposure (69) [22].
There are some important metrics of exposure that can correlate the clinical outcome, including AUC, time above threshold concentration, and Cmax (73). The exposure to the drug (AUC) over the dosing interval is the most important PK correlate from a TDM perspective. The cumulative dosage and the exposure AUC are two metrics that have been shown to be substantially linked to clinical outcomes. Determining AUC for bolus 5-FU dosages is logistically challenging given the amount of samples that must be obtained in a short period of time. AUC measurements with 5-FU infusion schedules are relatively easier as only one sample is necessary with advanced modeling techniques, which is usually collected at steady state to predict the exposure (AUC) (84). The current consensus is a single sample at 18 h after the commencement of the infusion is the best parameter of evaluation, and the target therapeutic window is 20–30 mg/h/L (96).
In 1989, a study done by Santini et al. is the first study of TDM for 5-FU (71). In that PK study, AUC was measured and considered as an important PK parameter for predicting toxicity. AUC was used as a significant parameter for dosing in the second half of the cycle. The monitoring of these individuals’ PK has proven to be a reliable way to objectively improve the therapeutic index. AUC is reported as a measure of exposure in the majority of studies while Css is only rarely reported in earlier studies.
Using the existing BSA-based dosing techniques, there is about a 40% interindividual variability in 5-FU plasma concentrations via infusion. There is about 20% intraindividual variability in 5-FU plasma concentrations. Every drug has an exposure–response relationship that can be observed when comparing proximal biochemical effects brought on by target modification to drug concentration at the target site. Therefore, before attempting to change dose based on measures of such exposure, it is crucial to assess the exposure–response relationship.
Hillcoat et al. found a strong relationship between 5-FU plasma concentrations and tumor response in patients with gastrointestinal malignancies. In this study, patients received a 5-day continuous infusion of 5-FU given every 6 weeks at a dose of 1,200 mg/m2/day on days 1–5. The patients’ plasma 5-FU concentrations were estimated and found to differ greatly. Furthermore, plasma 5-FU area under the plasma concentrations × time curve (AUC) was shown to be considerably larger in patients with either a partial response (PR) or stable disease (SD) compared to individuals who did not have a tumor response. This was the first evidence of clinical data correlating 5-FU plasma exposure to clinical activity (72).
Martin et al. demonstrated the efficacy of therapeutic drug management (TDM) in individualized 5-FU dose in patients with metastatic colorectal cancer in routine clinical practice. Study results suggested that there was a decreased incidence of 5-FU-related toxicities and a much improved 5-FU exposure if TDM is adopted in clinical practice (90).
Yang et al. (2016) performed a meta-analysis to assess the efficacy and possible side effects of 5-FU using PK-guided vs. BSA-based dose modification in advanced malignancies using data from two randomized control trials (RCTs) and three observational studies involving 654 patients. A significant improvement in overall response rate (odds ratio = 2.04) compared with the conventional BSA technique was found to be an outcome of PK-monitored 5-FU therapy. The study revealed that PK-monitored 5-FU dosage has the potential to be used in colorectal cancer personalized therapy. The researchers concluded that PK-based 5-FU dosage showed superior overall response rate when compared to BSA-based dosing and improved toxicities irrespective of significant difference (77).
Fang et al. (2016) conducted a meta-analysis to evaluate the PKG-based algorithm for 5-FU compared with BSA-based methodology (5-FU). There were four studies (n = 504) included. The objective response rate to 5-FU treatment was “substantially” higher with the PKG algorithm than with the BSA-based method. Additionally, PKG was shown to “significantly” lower the probability of grade 3/4 potential adverse drug events (70). Salamone et al. (2017) conducted a study to validate the use of TDM to adjust 5-FU dose in mCRC patients under regular clinical settings. A total of 75 patients with metastatic colorectal cancer (mCRC) from eight German medical facilities participated in this trial. They were given up to six administrations of 5-FU infusions using the AIO (n = 16), FOLFOX6 (n = 26), or FUFOX (n = 33) regimens based on BSA. The subsequent 5-FU infusion dosages were modified in accordance with the 5-FU AUC of the preceding cycle in order to reach the desired AUC of 20 to 30 mg·h/L. The main goal was to demonstrate that TDM of 5-FU enhanced the proportion of patients in the desired AUC range at the fourth treatment compared to the first dose. Average 5-FU AUC at the time of the first administration was 18 + 6 mg·h/L, with 64%, 33%, and 3% of the patients having AUCs that were below, within, or beyond the target range, respectively. By the fourth dosage, 54% of patients were within the desired 5-FU AUC range (p = 0.0294), and the average 5-FU AUC was 25 + 7 mg·h/L (p < 0.001). Even though 55% of patients had their doses raised, the rates of grade 3–4 diarrhea (4.6%), nausea (3.4%), fatigue (0.0%), and mucositis (0.2%) associated with 5-FU were less compared to the historical data (97).
A PK/PD study by Thyss determined the plasma concentration of 5-FU in 29 patients with head and neck cancer who were receiving a combination chemotherapy regimen that included cisplatin at a dose of 100 mg/m2 on day 1 and a continuous 5-day infusion of 5-FU at a dose of 1,000 mg/m2/24 h on days 2–6. In this study, a strong relationship was found between the incidence of toxicities such myelosuppression, mucositis, and diarrhea and greater systemic exposure to 5-FU (5-FU AUC > 30 mg·h/L) (98).
Twenty-one patients with advanced malignancies underwent 5-FU PK analysis by van Groeningen et al. from the Free University in Amsterdam, the Netherlands (99). 5-FU was given as an i.v. bolus once a week at a starting dose of 500 mg/m2, increasing the dose by 20% every 4 weeks until dose-limiting toxicity was noticed. The prevalence of toxicities was found to be strongly linked to 5-FU versus AUC using the logistic regression approach (96).
The first significant, prospective, multicenter, phase III randomized trial was carried out by Gamelin and colleagues in patients with mCRC receiving a weekly, 8-h infusion of 5-FU at a dose of 1,500 mg/m2. These patients would have been more likely to experience toxicity if the 5-FU dose had not been altered (100). This work is significant because it established for the first time that PK-guided 5-FU dose adjustment for the treatment of mCRC patients is practicable in clinical practice on an individual basis. The best way to ensure the right dose intensity for better results while limiting toxicity appears to be to evaluate plasma 5-FU AUC values. This method is more efficient and safe than dose adjustment based solely on clinical evaluation (100).
Yoshida and colleagues evaluated if there might be a relationship between the amount of 5-FU administered and the level of toxicity in 19 patients with mCRC. Patients in this study received a continuous i.v. infusion of 5-FU for 7 days straight at a dosage of 190–600 mg/m2/day. Nine patients (toxic group) out of the total study participants experienced toxic dermatitis, anorexia, nausea/vomiting, and >grade 2 stomatitis. AUC from 0 to 72 h after the start of 5-FU infusion (AUC0–72 h) and steady-state concentrations (CSS) of 5-FU were calculated. Between the individuals who experienced toxicity (n = 9) and those who did not (n = 10), there was a roughly twofold difference in the serum 5-FU CSS, AUC72 h, and total body clearance (101).
Deng et al. (2020) studied the efficacy of PK-based 5-FU dosing. A total of 153 patients with advanced colorectal cancer were randomly assigned to undergo either 5-FU chemotherapy with BSA-guided dose or a double-week chemotherapy regimen with 5-FU using PK dosing. When using AUC-based dosage, oral mucositis incidence dropped and the frequency of diarrhea was dramatically reduced. The researchers found that for patients with advanced CRC, PK-based dose control of 5-FU lowers the toxicity of chemotherapy and increases its long-term efficacy when compared to BSA-based dosing (102).
A randomized multicentric trial was conducted by Fety et al. (1998) to assess the clinical value of 5-FU dose adaptation guided by PK. A total of 122 patients with head and neck cancer were randomized to receive either the normal dose of cisplatin (100 mg/m2, day 1) and 5-FU (96-h continuous infusion) or a dose adjusted for the 5-FU level (AUC0–48 h; PKarm). A total of 106 patients were evaluated for toxicity and response. In comparison to the St-arm (n = 57), the 5-FU dosages and AUC were considerably lower in the PK-arm (n = 49) during cycles 2 and 3. Grade 3–4 neutropenia and thrombopenia were substantially more common in the St-arm compared to the PK-arm. In both treatment arms, the objective response rate was similar: 77.2% in the St-arm and 81.7% in the PK-arm. The authors concluded that therapeutic index can be improved by PK-based individual dose adaptation for 5-FU (103).
Pharmacoeconomic considerations of cancer therapy become crucial while choosing a chemotherapeutic regimen. When patients frequently cannot receive therapy because of considerable financial burden labeled as “financial toxicity,” cost-effectiveness has now become an even more crucial factor to take into account. There are two studies [conducted by Goldstein (2014) and Soh (2015)] that evaluated the cost-effectiveness of TDM, and it has been observed that 5-FU TDM is economical in the management of both mCRC and SCCHN (104, 105). Associated toxicities with 5-FU, such as febrile neutropenia, nausea/vomiting, and diarrhea, can have serious negative clinical and cost effects on patients and the healthcare system. It is possible to modify the dosage of 5-FU depending on the quantity of 5-FU in the plasma, and it is obvious that PK-based dosing can greatly enhance clinical outcomes by lowering toxicities and boosting efficacy (104). In the UK, patients with mCRC treated with various 5-FU combination regimens had the potential cost benefit of PK-based versus BSA dosing of 5-FU examined (106). The cost-effectiveness of administering 5-FU by PK versus BSA in various standard chemotherapy regimens in the UK population was counterfactually simulated using a decision tree model, and all patients were presumptively treated with first-line therapy for 6 or 12 cycles or until disease progression. From the viewpoint of the national health system, the model’s costs were calculated, and it was found to be effective (106).
The use of 5-FU TDM in patients with mCRC resulted in significant cost savings and a gain in quality-adjusted life-years (QALYs), according to emerging data. 5-FU TDM should be regarded as a clinically significant and essential component of individualized therapy in the routine care of cancer patients in this era of precision medicine (73).
Compared to the majority of other anticancer medicines, 5-FU TDM faces obstacles that start much earlier in the preanalytical stage. Due to the widespread presence of the catabolic enzyme (DPD), which quickly converts 5-FU to its metabolite dihydro-5-FU, 5-FU is incredibly unstable in whole blood and plasma at ambient temperature. In order to separate plasma from cells, blood samples should typically be promptly put on ice and plasma extracted (73).
The last 40 years have seen huge breakthroughs in 5-FU TDM, and today, there is a validated algorithm of 5-FU dose adjustment based on plasma 5-FU levels to decrease toxicity and improve 5-FU efficacy. The levels of 5-FU in peripheral blood can be measured directly using a number of techniques, such as HPLC, GC-MS, and LC-MS/MS. Recently, a sensitive and accurate immunoassay for measuring 5-FU has become available. Comparing this test to conventional HPLC and LC-MS/MS procedures reveals significant logistical benefits. The immunoassay has a number of potential advantages over conventional HPLC and/or LCMS assays, including (1) a quicker turnaround time for clinical samples, (2) a smaller sample size requirement, and (3) automated quantitation using a reputable, validated clinical chemistry analyzer that can test a large number of samples at once. Conventional chromatographic techniques need sophisticated equipment and a higher level of staff training to operate the equipment. Given the necessity of more sample preparation stages and the lengthier time required to analyze samples and all calibrators before the sample result can be completed than immunoassay, the turnaround time for HPLC and/or LC-MS/MS procedures is often longer than immunoassay. Cross-reactivity of the antibody to analytes structurally related to the target analyte has historically been one of the possible drawbacks of immunoassays (107).
There are a number of crucial factors that must be taken into account while performing TDM 5 FU. Since 5-FU has a short half-life of only 10–15 min, steady-state conditions are expected 1 h after 5-FU infusion begins. Clinical studies actually suggest getting TDM samples at least 18 h following the onset of a 5-FU infusion (73, 91–93, 108). In view of this, the majority of the current protocols for 5-FU TDM suggest sampling be done on day 2 of a 48-h 5-FU infusion. Regarding the latter, blood sample is not advised if the infusion pump is empty or if it is thought to be within 30 min of emptying. Patients should be called back to the facility around 4 h before the expected completion of the drug infusion if blood collection for TDM is planned in order to prevent a significant fraction of TDM failures due to empty drug pumps. For patients undergoing 5-FU TDM, electric pumps are preferred over elastomeric pumps because they provide better timing accuracy, as the balloons of elastomeric pumps are sensitive to pressure, temperature, season, and patient activity (73, 85, 86). The blood sampled should be collected from a peripheral vein at a distance from the central port being used for 5-FU infusion. 5-FU is unstable in whole blood at room temperature due to ex vivo catabolism by DPD in the RBC; therefore, the collected blood sample should be placed on ice and plasma should be separated as soon as possible. Alternatively, a DPD inhibitor (gimeracil) may be added to the sample that allows centrifugation up to 24 h. However, a minimal turnaround time is important to continue the dose modification by modifying the infusion rate.
The conventional 5-FU dose based on BSA produces a wide range of 5-FU systemic exposure that correlates with a wide range of effectiveness measurements. BSA-based dosing allows for more customized 5-FU administration, although in adults, BSA does not correspond well with any PK measures (67). Many US centers use PK-based dosing to achieve a target concentration, and there are solid comparative data about the mortality benefit of TDM-guided 5-FU dose [32]. By lowering toxicities and boosting efficacy, dose modification of 5-FU is doable and can greatly enhance therapeutic outcomes (90). Two prominent dose modification algorithms have been proposed. Gamerlin et al. recommended dose adjustment over a 5-FU concentration range on <4 to >31 mg·h/L (91) while the Kaldate algorithm is proposed for a wider range of concentration; 8–10 to >40 mg·h/L (108). The proposed dose modification algorithm by Kaldate has been prospectively validated in a single cohort of patients in a clinical trial setting (90) and hence also the algorithm of choice as recommended by the IATDMCT consensus statement for TDM of 5-FU in oncological practices (73). Bayesian forecasting based on PB-PK modeling and simulation is an upcoming strategy proposed for MPID of 5-FU. The greatest advantage it may offer is to do away with the DPD genotyping or phenotyping for dose optimization of 5-FU. The MPID has been proposed to overcome genotype or phenotypic misclassification, by predicting the clearance of the drug accurately (109).
Mtx is an antifolate drug used in low doses (7.5–25 mg/week) as a disease-modifying antirheumatic drug (DMRD) whereas it is used at large doses (i.e., >500 mg/m2 intravenously) to treat a variety of malignancies [(acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma (NHL), osteosarcoma, and medulloblastoma]. The majority (90%) of Mtx is cleared unchanged via the kidneys, with the hepatic metabolism producing only a minor amount of metabolite 7-hydroxy methotrexate. The systemic clearance is roughly 50–135 ml/min/m2, and the terminal half-life ranges from 8 to 15 h. The PK, pharmacodynamic, and toxicity profiles depend on the dose (110). Upon entering the cells, Mtx is converted into a series of Mtx-polyglutamates (Mtx-PGs). High-dose Mtx (HDMtx) acts through inhibition of the dihydrofolate reductase (DHFR) enzyme with very high potency and hence blocks the purine synthesis in the s-phase of the cell cycle, thereby preventing cell proliferation in both tumor and normal cells. At low doses, Mtx-PGs prevent the inflammatory processes in the white blood cells and hence prevent the immune-mediated damage. Both low-dose and high-dose Mtx may cause life-threatening events such as hepatotoxicity, myelosuppression, and pulmonary toxicity, but the high-dose therapy may cause potentially life-threatening nephrotoxicity due to crystallization of the drug in the nephrons, which is rarely seen with low-dose therapy (111). Therefore, dose reduction is recommended when the creatinine clearance is decreased (112). The guiding principle is maintaining hydration, urinary alkalinization, monitoring serum creatinine, and a pharmacokinetically guided leucovorin rescue with TDM.
HDMtx therapy meets the most important criteria for TDM, a large inter- and intraindividual variability in the systemic concentration achieved (113–115). The extent of interpatient variability is 52% and intrapatient variability is as high as 48% (116). This is reflected in the wide range of incidence of nephrotoxicity from 1.8% to 10.7% (110, 117). Pharmacogenetic factors like SNPs of MRP2 and OATP1B1 affecting the expression of these transport proteins partly contribute to the interindividual variability of disposition of Mtx. Several drug–drug interactions (NSAIDs, ciprofloxacin, probenecid, sulfamethoxazole, trimethoprim, amiodarone, tyrosine kinase inhibitors, and proton pump inhibitors) and known disease conditions like Down’s syndrome. Coupled with all these known factors that affect Mtx elimination, there is an unexplained and variable delay in the renal clearance of Mtx despite all preventive measures taken. Maintaining the concentration in a nontoxic and efficient target range is made possible by using TDM and optimizing the doses of MTX and leucovorin. HDMtx is one of the prototype oncologic scenarios for which TDM had been employed (118).
Classically, the purpose of Mtx TDM has been to monitor the plasma concentration of the drug to avoid toxicity. Most lines of evidence come from observational studies and not from the randomized controlled clinical trials for obvious reasons. The plasma levels that define toxic exposure evolved over time. In one of the earliest studies by Stoller et al., it was suggested that a concentration >0.9 μM/ml at 48-h increases the risk of myelotoxicity (119, 120). These levels are important to guide the dose of folinic rescue therapy. Pediatric ALL consensus statement recommends a standard 50 mg/m2 infusion of leucovorin every 6 h, if the Mtx concentration is >20 μM/ml at 36 h, >10 μM/ml at 42 h, and >5 μM/ml at 48 h. Supplemental leucovorin must be administered until the Mtx level falls to 0.1 to 0.05 μM/ml. A delay in measurement of plasma Mtx concentration by 24–36 h equates to having an uncompromised therapeutic action of the drug. It would be interesting to note that while Mtx nephrotoxicity has been most consistently associated with plasma concentration across studies, it has been found to be a poor predictor of other toxicities like myelotoxicity and hepatotoxicity with variable results reported from studies (118). If acute kidney injury occurs with high-dose Mtx therapy, or an extremely high concentration of Mtx is found in the blood, glucapidase (carboxypeptidase G2) is the drug of choice, and not leucovorin, as it is extremely efficient and has removed 98% of the Mtx in first 30 min.
Mtx has long been used as a disease-modifying agent in low doses (<50 mg/m2) in many rheumatic and autoimmune diseases. Due to its low cost, it is still the drug of choice in such chronic diseases (121, 122). However, 40% of the patients do not show clinical response to Mtx. Some of them have early nonresponse and some cease to respond after an initial period of nonresponse. Apart from the reasons like noncompliance and inadequate dosing, the variation in Mtx uptake and metabolism has been postulated to play a role. The cellular uptake of Mtx and their polyglutamation rate may be important contributors for such variable response. The activity of the folypolyglutamase synthase enzyme, which is responsible for polyglutamation, and the levels of Mtx-PGs have been proposed to be the biomarkers for detecting patients who are at the risk of nonresponse (123). Sensitive LC-MS/MS-based methods have been developed to detect them with precision. Studies have shown that there is a great variation in the levels of polyglutamates in the blood cells for patients receiving the same dose of Mtx (123). De Rotte et al. have proposed a multivariate model including age, gender, folate status, and genotype to correlate the disease activity with the polyglutamate levels (124). Recently, a randomized controlled study showed that daily therapy is as effective as weekly therapy when the polyglutamate-3 levels are similar. This study also showed that obese patients achieve a lower concentration of Mtx-PG3 (125). Clinical response in several other diseases like juvenile rheumatoid arthritis, juvenile dermatomyositis, and inflammatory bowel disease was also reported to be affected by the Mtx-PG concentrations. However, this is still investigated and not yet ready for clinical use; more concrete evidence is needed.
For the 24-h infusion schedule, it is advised to use TDM at least 24 h (the end of the infusion), 48 h, and 72 h following the start of high-dose MTX infusion, until the concentration is below 0.1–0.2 mol/L. The TDM protocols can be divided into two modes: The steady-state concentration for the 24-h infusion regimen is thought to be C24h (the end of infusion), which is correlated to efficacy and safety, whereas C48h and C72h are mostly related to safety. C3-6h (the end of the infusion), for the rapid infusion (less than 6 h) regimen, is thought to represent the peak concentration in terms of efficacy and safety, while C24h, C48h, and C72h are primarily connected to safety. In most circumstances, the safe MTX concentration range is below 0.1–0.2 mol/L, and TDM can be discontinued once it is attained. It is important to note that MTX levels less than 0.05 mol/L can be regarded as a stricter safe range for patients with delayed MTX elimination and/or indications of acute renal dysfunction.
Both MTX and its metabolites can be distinguished and detected using the HPLC and HPLC-MS/MS procedures, although they are expensive and require sophisticated sample processing. Automated immunoassay platforms are also available for Mtx concentration measurement. Both liquid chromatography and immunoassay could be used in a TDM laboratory. It must be kept in mind that the immunoassays have the disadvantage of slightly overestimating the concentrations due to nonspecific interactions with the Mtx metabolites. Another instance where immunoassay can falter is during treatment with glucarpidase, as Mtx degradation products interfere with the analysis and true estimates may not be found. Therefore, chromatographic methods must be employed during this situation.
Apart from the pharmacokinetically dosing leucovorin, Mtx TDM data have also been utilized to build dose prediction models taking into account covariates like creatinine clearance and alanine transaminase levels to forecast the dose. This is to facilitate a personalized dose for the patients, which attempts to attain the desired fall in the concentration of Mtx in a desired time frame and minimizing hospital stay (126, 127). These studies reported the lower incidence of nephrotoxicity, the therapeutic concentration of Mtx was within the therapeutic range, and the clearance of Mtx was as anticipated (94, 95).
The main purpose of using HDMtx administration in patients with ALL and NHL is to address the CNS disease and to reduce risk of CNS relapse. Most studies with Mtx TDM have been performed on serum samples. A study on 138 CSF samples from children with ALL and NHL was conducted to evaluate the Mtx concentration in the cerebrospinal fluid (CSF). Serum Mtx concentrations at the end of infusion were assessed by routine TDM. Cytotoxic Mtx concentrations of 1 μM or greater were detected in 81.2% of CSF samples before administering intrathecal Mtx. One micromolar concentration is proposed to provide the desired antileukemic effect to prevent CNS relapse. This important study made a case for reducing the need for intrathecal injection of Mtx and, hence, reducing the risk of multiple lumbar puncture in children suffering from ALL and NHL (128).
Bayesian modeling and simulation-based MIPD are some of the noteworthy precision medicine tools likely to be implemented in routine patient care in the near future. Busulfan has made a significant progress in terms of clinical testing for MIPD, though widespread application of Bayesian adaptive dosing is yet to be achieved. For 5-FU, it is still in the evolution phase and a through work in this direction is needed. For Mtx, two extremes of doses are practiced; thus, this calls for an Mtx TDM separately for autoimmune diseases apart from the well-established practice for HDMtx. Utilizing the chronic toxicity data of Mtx TDM could be tuned in for understanding the Mtx nonresponder profile in patients with rheumatic diseases. Though a few model informed dosing are in practice, it is time to refine these strategies and the development of user-friendly clinical decision support tools and their wider application. The reduction in the turnaround time in TDM reporting along with the implementation of MIPD should be aimed for in the future to make quicker as well as smarter therapeutic decisions in patient care. In addition, pharmacometabolomic approaches are gaining momentum and their association with different clinical end-points have also been demonstrated (129, 130).
PS accepted the invitation and built the team. She conceived the idea, compiled the first draft with PA and JK. PG provided the clinical inputs and practice oriented points. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Groenland SL, Mathijssen RHJ, Beijnen JH, Huitema ADR, Steeghs N. Individualized dosing of oral targeted therapies in oncology is crucial in the era of precision medicine. Eur J Clin Pharmacol (2019) 75(9):1309–18. doi: 10.1007/s00228-019-02704-2
2. Knezevic CE, Clarke W. Cancer chemotherapy: The case for therapeutic drug monitoring. Ther Drug Monit (2020) 42(1):6–19. doi: 10.1097/FTD.0000000000000701
3. Mueller-Schoell A, Groenland SL, Scherf-Clavel O, van Dyk M, Huisinga W, Michelet R, et al. Therapeutic drug monitoring of oral targeted antineoplastic drugs. Eur J Clin Pharmacol (2021) 77(4):441–64. doi: 10.1007/s00228-020-03014-8
4. Westerdijk K, Desar IME, Steeghs N, van der Graaf WTA, van Erp NP. Imatinib, sunitinib and pazopanib: From flat-fixed dosing towards a pharmacokinetically guided personalized dose. Br J Clin Pharmacol (2020) 86(2):258–73. doi: 10.1111/bcp.14185
5. Bleyzac N, Souillet G, Magron P, Janoly A, Martin P, Bertrand Y, et al. Improved clinical outcome of paediatric bone marrow recipients using a test dose and Bayesian pharmacokinetic individualization of busulfan dosage regimens. Bone Marrow Transplant (2001) 28(8):743–51. doi: 10.1038/sj.bmt.1703207
6. Aspen Pharmacare Canada Inc. MYLERAN® product monograph. Toronto OJ. 8-1155 North Service Road West Oakville, Ontario, L6M 3E3.
7. Otsuka Pharmaceutical Inc. BUSULFEX® product monograph. Saint Laurent QM. Otsuka Pharmaceutical Co., Ltd. Tokyo, 101-8535 Japan.
8. Tutschka PJ, Copelan EA, Klein JP. Bone marrow transplantation for leukemia following a new busulfan and cyclophosphamide regimen. Blood (1987) 70(5):1382–8. doi: 10.1182/blood.V70.5.1382.1382
10. Haddow A, Timmis GM. Myleran in chronic myeloid leukaemia; chemical constitution and biological action. Lancet (1953) 264(6753):207–8. doi: 10.1016/S0140-6736(53)90884-8
11. Galton DA. Myleran in chronic myeloid leukaemia; results of treatment. Lancet (1953) 264(6753):208–13. doi: 10.1016/S0140-6736(53)90885-X
12. Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2009) 15(5):523–36. doi: 10.1016/j.bbmt.2008.12.489
13. MYLERAN®. (Busulfan, 2-mg Scored tablets) US-FDA prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/09386slr023_myleran_lb%20l.pdf.
14. Santos GW, Tutschka PJ, Brookmeyer R, Saral R, Beschorner WE, Bias WB, et al. Marrow transplantation for acute nonlymphocytic leukemia after treatment with busulfan and cyclophosphamide. N Engl J Med (1983) 309(22):1347–53. doi: 10.1056/NEJM198312013092202
15. Krivoy N, Hoffer E, Lurie Y, Bentur Y, Rowe JM. Busulfan use in hematopoietic stem cell transplantation: Pharmacology, dose adjustment, safety and efficacy in adults and children. Curr Drug Saf (2008) 3(1):60–6. doi: 10.2174/157488608783333899
16. Buggia I, Locatelli F, Regazzi MB, Zecca M. Busulfan. Ann Pharmacother (1994) 28(9):1055–62. doi: 10.1177/106002809402800911
17. Bhagwatwar HP, Phadungpojna S, Chow DS, Andersson BS. Formulation and stability of busulfan for intravenous administration in high-dose chemotherapy. Cancer Chemother Pharmacol (1996) 37(5):401–8. doi: 10.1007/s002800050404
19. Madden T, de Lima M, Thapar N, Nguyen J, Roberson S, Couriel D, et al. Pharmacokinetics of once-daily IV busulfan as part of pretransplantation preparative regimens: a comparison with an every 6-hour dosing schedule. Biol Blood Marrow Transplant (2007) 13(1):56–64. doi: 10.1016/j.bbmt.2006.08.037
20. Verougstraete N, Stove V, Verstraete AG, Oyaert M. Automation in busulfan therapeutic drug monitoring: Evaluation of an immunoassay on two routine chemistry analyzers. Ther Drug Monit (2022) 44(2):335–9. doi: 10.1097/FTD.0000000000000933
21. Geddes M, Kangarloo SB, Naveed F, Quinlan D, Chaudhry MA, Stewart D, et al. High busulfan exposure is associated with worse outcomes in a daily i.v. busulfan and fludarabine allogeneic transplant regimen. Biol Blood Marrow Transplant (2008) 14(2):220–8. doi: 10.1016/j.bbmt.2007.10.028
22. Andersson BS, Thall PF, Madden T, Couriel D, Wang X, Tran HT. , 477–85. toxicity eaBsertr-r, window aag-v-hddat, Blood fivBicmlBMT.
23. Ansari M, Lauzon-Joset JF, Vachon MF, Duval M, Théoret Y, Champagne MA, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant (2010) 45(2):261–7. doi: 10.1038/bmt.2009.143
24. Ansari M, Rezgui MA, Théoret Y, Uppugunduri CR, Mezziani S, Vachon MF, et al. Glutathione s-transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplant (2013) 48(7):939–46. doi: 10.1038/bmt.2012.265
25. Andersson BS, Thall PF, Madden T, Couriel D, Wang X, Tran HT, et al. Busulfan systemic exposure relative to regimen-related toxicity and acute graft-versus-host disease: defining a therapeutic window for i.v. BuCy2 in chronic myelogenous leukemia. Biol Blood Marrow Transplant (2002) 8(9):477–85. doi: 10.1053/bbmt.2002.v8.pm12374452
26. Kim MG, Kwak A, Choi B, Ji E, Oh JM, Kim K. Effect of glutathione s-transferase genetic polymorphisms on busulfan pharmacokinetics and veno-occlusive disease in hematopoietic stem cell transplantation: A meta-analysis. Basic Clin Pharmacol Toxicol (2019) 124(6):691–703. doi: 10.1111/bcpt.13185
27. Nishikawa T, Yamaguchi H, Ikawa K, Nakayama K, Higashi E, Miyahara E, et al. Influence of GST polymorphisms on busulfan pharmacokinetics in Japanese children. Pediatr Int (2019) 61(6):558–65. doi: 10.1111/ped.13859
28. Myers AL, Kawedia JD, Champlin RE, Kramer MA, Nieto Y, Ghose R, et al. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: A comprehensive review. Expert Opin Drug Metab Toxicol (2017) 13(9):901–23. doi: 10.1080/17425255.2017.1360277
29. Hassan M, Oberg G, Bekassy AN, Aschan J, Ehrsson H, Ljungman P, et al. Pharmacokinetics of high-dose busulphan in relation to age and chronopharmacology. Cancer Chemother Pharmacol (1991) 28(2):130–4. doi: 10.1007/BF00689702
30. Vassal G, Deroussent A, Hartmann O, Challine D, Benhamou E, Valteau-Couanet D. , 6203–7. eaD-dnoh, study. bicacapCR.
31. Matar KM, Alshemmari SH, Refaat S, Anwar A. UPLC-tandem mass spectrometry for quantification of busulfan in human plasma: Application to therapeutic drug monitoring. Sci Rep (2020) 10(1):8913. doi: 10.1038/s41598-020-65919-9
32. Grochow LB, Jones RJ, Brundrett RB, Braine HG, Chen TL, Saral R, et al. Pharmacokinetics of busulfan: correlation with veno-occlusive disease in patients undergoing bone marrow transplantation. Cancer Chemother Pharmacol (1989) 25(1):55–61. doi: 10.1007/BF00694339
33. Shulman HM, Hinterberger W. Hepatic veno-occlusive disease–liver toxicity syndrome after bone marrow transplantation. Bone Marrow Transplant (1992) 10(3):197–214.
34. Dix SP, Wingard JR, Mullins RE, Jerkunica I, Davidson TG, Gilmore CE, et al. Association of busulfan area under the curve with veno-occlusive disease following BMT. Bone Marrow Transplant (1996) 17(2):225–30.
35. Jones RJ, Lee KS, Beschorner WE, Vogel VG, Grochow LB, Braine HG, et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation (1987) 44(6):778–83. doi: 10.1097/00007890-198712000-00011
36. McDonald GB, Sharma P, Matthews DE, Shulman HM, Thomas ED. Venocclusive disease of the liver after bone marrow transplantation: diagnosis, incidence, and predisposing factors. Hepatology (1984) 4(1):116–22. doi: 10.1002/hep.1840040121
37. Grochow LB. Busulfan disposition: the role of therapeutic monitoring in bone marrow transplantation induction regimens. Semin Oncol (1993) 20(4 Suppl 4):18–25.
38. Slattery JT, Clift RA, Buckner CD, Radich J, Storer B, Bensinger WI. , 3055–60. eaMtfcml, transplantation. tiopblotooB.
39. Slattery JT, Sanders JE, Buckner CD, Schaffer RL, Lambert KW, Langer FP. , 31–42. eaG-ratfbm, Bone tirtbpMT.
40. Slattery JT, Sanders JE, Buckner CD, Schaffer RL, Lambert KW, Langer FP, et al. Graft-rejection and toxicity following bone marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Transplant (1995) 16(1):31–42.
41. Williams CB, Day SD, Reed MD, Copelan EA, Bechtel T, Leather HL. (2004), 614–23. eaDmpuib, allogeneic Bacfbao, with pbsctiphmBBMT.
42. Geddes M, Kangarloo SD, Naveed F, Quinlan D, Chaudhry MA, Stewart D. , 220–8. eaHbeiaww, transplant oiadivbafarBBMT.
44. Bartelink IH, Lalmohamed A, van Reij EML, Dvorak CC, Savic RM, Zwaveling J, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: A multicentre, retrospective cohort analysis. Lancet Haematol (2016) 3(11):e526–36. doi: 10.1016/S2352-3026(16)30114-4
45. Tran HT, Madden T, Petropoulos D, Worth LL, Felix EA, Sprigg-Saenz HA. 463–70. eaIh-dobp, allogeneic daiappu, malignancies. sctfahBMT.
46. McCune JS, Gibbs JP, Slattery JT. , 155–65. Plasma concentration monitoring, Pharmacokinet obdiicoC.
47. Hobbs JR, Hugh-Jones K, Shaw PJ, Downie CJ, Williamson S. , 201–8. dosages Errtbac, children. fdbmtifBMT.
48. Pawlowska AB, Blazar BR, Angelucci E, Baronciani D, Shu XO. (1997), 915–20. high-dose BBRoppo, transplantation obttooabmicwtBMT.
49. Radich JP, Gooley T, Bensinger W, Chauncey T, Clift R, Flowers M, et al. HLAmatched related hematopoietic cell transplantation for chronic-phase CML using a targeted busulfan and cyclophosphamide preparative regimen. Blood (2003) 102(1):31–5. doi: 10.1182/blood-2002-08-2619
50. Poonkuzhali B, Srivastava A, Quernin MH, Dennison D, Aigrain EJ, Kanagasabapathy AS, et al. (1999), 5–11. Pharmacokinetics of oral busulphan, allogeneic icwbtmubmtBMT.
51. Bolinger AM, Zangwill AB, Slattery JT, Glidden D, DeSantes K, Heyn L. 25:925–30. eaAeoe, toxicity and busulfan, for cicrbmtlogdBMT.
52. Grochow LB. Busulfan disposition: The role of therapeutic monitoring in bone marrow transplantation induction regimens. Semin Oncol (1993) 20(4 Suppl 4):18–25.
53. Ljungman P, Hassan M, Békássy AN, Ringdén O, Oberg G. High busulfan concentrations are associated with increased transplant-related mortality in allogeneic bone marrow transplant patients. Bone Marrow Transplant (1997) 20(11):909–13. doi: 10.1038/sj.bmt.1700994
54. Russell JA, Kangarloo SB. Therapeutic drug monitoring of busulfan in transplantation. Curr Pharm Des (2008) 14(20):1936–49. doi: 10.2174/138161208785061382
55. Palmer J, McCune JS, Perales M-A, Marks D, Bubalo J, Mohty M, et al. Personalizing busulfan-based conditioning: Considerations from the American society for blood and marrow transplantation practice guidelines committee. Biol Blood Marrow Transplant (2016) 22(11):1915–25. doi: 10.1016/j.bbmt.2016.07.013
56. Slattery JT, Risler LJ. Therapeutic monitoring of busulfan in hematopoietic stem cell transplantation. Ther Drug Monit (1998) 20(5):543–9. doi: 10.1097/00007691-199810000-00017
57. Tran HT, Madden T, Petropoulos D, Worth LL, Felix EA, Sprigg-Saenz HA, et al. Individualizing high-dose oral busulfan: prospective dose adjustment in a pediatric population undergoing allogeneic stem cell transplantation for advanced hematologic malignancies. Bone Marrow Transplant (2000) 26(5):463–70. doi: 10.1038/sj.bmt.1702561
58. Deeg HJ, Storer B, Slattery JT, Anasetti C, Doney KC, Hansen JA, et al. Conditioning with targeted busulfan and cyclophosphamide for hemopoietic stem cell transplantation from related and unrelated donors in patients with myelodysplastic syndrome. Blood (2002) 100(4):1201–7. doi: 10.1182/blood-2002-02-0527
59. Clopés A, Sureda A, Sierra J, Queraltó JM, Broto A, Farré R, et al. Absence of venoocclussive disease in a cohort of multiple myeloma patients undergoing autologous stem cell transplantation with targeted busulfan dosage. Eur J Haematol (2006) 77(1):1–6. doi: 10.1111/j.0902-4441.2006.t01-1-EJH2478.x
60. Parmar S, Rondon G, de Lima M, Thall P, Bassett R, Anderlini P, et al. Dose intensification of busulfan in the preparative regimen is associated with improved survival: a phase I/II controlled, randomized study. Biol Blood Marrow Transplant (2013) 19(3):474–80. doi: 10.1016/j.bbmt.2012.12.001
61. Bartelink IH, Bredius RGM, Belitser SV, Suttorp MM, Bierings M, Knibbe CAJ, et al. Association between busulfan exposure and outcome in children receiving intravenous busulfan before hematologic stem cell transplantation. Biol Blood Marrow Transplant (2009) 15(2):231–41. doi: 10.1016/j.bbmt.2008.11.022
62. Mellgren K, Nilsson C, Fasth A, Abrahamsson J, Winiarski J, Ringdén O, et al. Safe administration of oral BU twice daily during conditioning for stem cell transplantation in a paediatric population: a comparative study between the standard 4-dose and a 2-dose regimen. Bone Marrow Transplant (2008) 41(7):621–5. doi: 10.1038/sj.bmt.1705947
63. Ryu S-G, Lee J-H, Choi S-J, Lee J-H, Lee Y-S, Seol M, et al. Randomized comparison of four-Times-Daily versus once-daily intravenous busulfan in conditioning therapy for hematopoietic cell transplantation. Biol Blood Marrow Transplant (2007) 13(9):1095–105. doi: 10.1016/j.bbmt.2007.06.005
64. Andersen AM, Bergan S, Gedde-Dahl T, Buechner J, Vethe NT. Fast and reliable quantification of busulfan in blood plasma using two-channel liquid chromatography tandem mass spectrometry: Validation of assay performance in the presence of drug formulation excipients. J Pharm Biomed Anal (2021) 203:114216. doi: 10.1016/j.jpba.2021.114216
65. De Gregori S, Tinelli C, Manzoni F, Bartoli A. Comparison of two analytical methods for busulfan therapeutic drug monitoring. Eur J Drug Metab Pharmacokinet (2021) 46(1):155–9. doi: 10.1007/s13318-020-00660-2
66. Hilaire MR, Gill RV, Courtney JB, Baburina I, Gardiner J, Milone MC, et al. Evaluation of a nanoparticle-based busulfan immunoassay for rapid analysis on routine clinical analyzers. Ther Drug Monit (2021) 43(6):766–71. doi: 10.1097/FTD.0000000000000883
67. Jiang H, Lu J, Ji J. Circadian rhythm of dihydrouracil/uracil ratios in biological fluids: a potential biomarker for dihydropyrimidine dehydrogenase levels. Br J Pharmacol (2004) 141(4):616–23. doi: 10.1038/sj.bjp.0705651
68. Davis LE, Lenkinski RE, Shinkwin MA, Kressel HY, Daly JM. The effect of dietary protein depletion on hepatic 5-fluorouracil metabolism. Cancer (1993) 72(12):3715–22. doi: 10.1002/1097-0142(19931215)72:12<3715::AID-CNCR2820721225>3.0.CO;2-W
69. Kline CL, Sheikh HS, Scicchitano A, Gingrich R, Beachler C, Finnberg NK, et al. Preliminary observations indicate variable patterns of plasma 5-fluorouracil (5-FU) levels during dose optimization of infusional 5-FU in colorectal cancer patients. Cancer Biol Ther (2011) 12(7):557–68. doi: 10.4161/cbt.12.7.18059
70. Fang L, Xin W, Ding H, Zhang Y, Zhong L, Luo H, et al. Pharmacokinetically guided algorithm of 5-fluorouracil dosing, a reliable strategy of precision chemotherapy for solid tumors: a meta-analysis. Sci Rep (2016) 6:25913. doi: 10.1038/srep25913
71. Santini J, Milano G, Thyss A, Renee N, Viens P, Ayela P, et al. 5-FU therapeutic monitoring with dose adjustment leads to an improved therapeutic index in head and neck cancer. Br J Cancer (1989) 59(2):287–90. doi: 10.1038/bjc.1989.59
72. Hillcoat BL, McCulloch PB, Figueredo AT, Ehsan MH, Rosenfeld JM. Clinical response and plasma levels of 5-fluorouracil in patients with colonic cancer treated by drug infusion. Br J Cancer (1978) 38(6):719–24. doi: 10.1038/bjc.1978.278
73. Beumer JH, Chu E, Allegra C, Tanigawara Y, Milano G, Diasio R, et al. Therapeutic drug monitoring in oncology: International association of therapeutic drug monitoring and clinical toxicology recommendations for 5-fluorouracil therapy. Clin Pharmacol Ther (2019) 105(3):598–613. doi: 10.1002/cpt.1124
74. Milano G, Etienne MC. Individualizing therapy with 5-fluorouracil related to dihydropyrimidine dehydrogenase: theory and limits. Ther Drug Monit (1996) 18(4):335–40. doi: 10.1097/00007691-199608000-00004
75. Milano G, Chamorey AL. Clinical pharmacokinetics of 5-fluorouracil with consideration of chronopharmacokinetics. Chronobiol Int (2002) 19(1):177–89. doi: 10.1081/CBI-120002597
76. Fleming RA, Milano G, Thyss A, Etienne MC, Renée N, Schneider M, et al. Correlation between dihydropyrimidine dehydrogenase activity in peripheral mononuclear cells and systemic clearance of fluorouracil in cancer patients. Cancer Res (1992) 52(10):2899–902.
77. Yang R, Zhang Y, Zhou H, Zhang P, Yang P, Tong Q, et al. Individual 5-fluorouracil dose adjustment via pharmacokinetic monitoring versus conventional body-Area-Surface method: A meta-analysis. Ther Drug Monit (2016) 38(1):79–86. doi: 10.1097/FTD.0000000000000238
78. Shukla P, Goswami S, Keizer RJ, Winger BA, Kharbanda S, Dvorak CC, et al. Assessment of a model-informed precision dosing platform use in routine clinical care for personalized busulfan therapy in the pediatric hematopoietic cell transplantation (HCT) population. Front Pharmacol (2020) 11:888. doi: 10.3389/fphar.2020.00888
79. Lawson R, Paterson L, Fraser CJ, Hennig S. Evaluation of two software using Bayesian methods for monitoring exposure and dosing once-daily intravenous busulfan in paediatric patients receiving haematopoietic stem cell transplantation. Cancer Chemother Pharmacol (2021) 88(3):379–91. doi: 10.1007/s00280-021-04288-0
80. Dadkhah A, Alihodzic D, Broeker A, Kröger N, Langebrake C, Wicha SG. Evaluation of the robustness of therapeutic drug monitoring coupled with Bayesian forecasting of busulfan with regard to inaccurate documentation. Pharm Res (2021) 38(10):1721–9. doi: 10.1007/s11095-021-03115-8
81. Paci A, Veal G, Bardin C, Levêque D, Widmer N, Beijnen J, et al. Review of therapeutic drug monitoring of anticancer drugs part 1–cytotoxics. Eur J Cancer (2014) 50(12):2010–9. doi: 10.1016/j.ejca.2014.04.014
82. Zhang N, Yin Y, Xu SJ, Chen WS. 5-fluorouracil: mechanisms of resistance and reversal strategies. Molecules (2008) 13(8):1551–69. doi: 10.3390/molecules13081551
83. Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet (1989) 16(4):215–37. doi: 10.2165/00003088-198916040-00002
84. Salman B, Al-Khabori M. Applications and challenges in therapeutic drug monitoring of cancer treatment: A review. J Oncol Pharm Pract (2021) 27(3):693–701. doi: 10.1177/1078155220979048
85. Blaschke M, Cameron S, Emami K, Blumberg J, Wegner U, Nischwitz M, et al. Measurement of 5-FU plasma levels in patients with advanced cancer: correct approach to practical procedures is essential. Int J Clin Pharmacol Ther (2011) 49(1):83–5.
86. Salman D, Barton S, Nabhani-Gebara S. Effect of environmental conditions on performance of elastomeric pumps. Am J Health Syst Pharm (2013) 70(13):1100. doi: 10.2146/ajhp130150
87. Lu Z, Zhang R, Diasio RB. Dihydropyrimidine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient patients, and clinical implication in 5-fluorouracil chemotherapy. Cancer Res (1993) 53(22):5433–8.
88. Wattanatorn W, McLeod HL, Macklon F, Reid M, Kendle KE, Cassidy J. Comparison of 5-fluorouracil pharmacokinetics in whole blood, plasma, and red blood cells in patients with colorectal cancer. Pharmacotherapy (1997) 17(5):881–6.
89. Highlights from: 5-fluorouracil drug management pharmacokinetics and pharmacogenomics workshop; Orlando, florida; January 2007. Clin Colorectal Cancer (2007) 6(6):407–22. doi: 10.1016/s1533-0028(11)70480-7
90. Wilhelm M, Mueller L, Miller MC, Link K, Holdenrieder S, Bertsch T, et al. Prospective, multicenter study of 5-fluorouracil therapeutic drug monitoring in metastatic colorectal cancer treated in routine clinical practice. Clin Colorectal Cancer (2016) 15(4):381–8. doi: 10.1016/j.clcc.2016.04.001
91. Gamelin E, Boisdron-Celle M, Delva R, Regimbeau C, Cailleux PE, Alleaume C, et al. Long-term weekly treatment of colorectal metastatic cancer with fluorouracil and leucovorin: results of a multicentric prospective trial of fluorouracil dosage optimization by pharmacokinetic monitoring in 152 patients. J Clin Oncol (1998) 16(4):1470–8. doi: 10.1200/JCO.1998.16.4.1470
92. Mueller F, Büchel B, Köberle D, Schürch S, Pfister B, Krähenbühl S, et al. Genderspecific elimination of continuous-infusional 5-fluorouracil in patients with gastrointestinal malignancies: results from a prospective population pharmacokinetic study. Cancer Chemother Pharmacol (2013) 71(2):361–70. doi: 10.1007/s00280-012-2018-4
93. Terret C, Erdociain E, Guimbaud R, Boisdron-Celle M, McLeod HL, Féty-Deporte R, et al. Dose and time dependencies of 5-fluorouracil pharmacokinetics. Clin Pharmacol Ther (2000) 68(3):270–9. doi: 10.1067/mcp.2000.109352
94. Harris BE, Song R, Soong SJ, Diasio RB. Relationship between dihydropyrimidine dehydrogenase activity and plasma 5-fluorouracil levels with evidence for circadian variation of enzyme activity and plasma drug levels in cancer patients receiving 5-fluorouracil by protracted continuous infusion. Cancer Res (1990) 50(1):197–201.
95. Kuwahara A, Yamamori M, Nishiguchi K, Okuno T, Chayahara N, Miki I, et al. Effect of dose-escalation of 5-fluorouracil on circadian variability of its pharmacokinetics in Japanese patients with stage III/IVa esophageal squamous cell carcinoma. Int J Med Sci (2010) 7(1):48–54. doi: 10.7150/ijms.7.48
96. Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol (2016) 78(3):447–64. doi: 10.1007/s00280-016-3054-2
97. Salamone SJ, Jaehde U, Mueller L, Link K, Holdenrieder S, Bertsch T, et al. Prospective, multi-center study of 5-fluorouracil (5-FU) therapeutic drug management (TDM) in metastatic colorectal cancer (mCRC) patients treated in routine clinical practice. J Clin Oncol (2017) 35(4_suppl):650–. doi: 10.1200/JCO.2017.35.4_suppl.650
98. Thyss A, Milano G, Renée N, Vallicioni J, Schneider M, Demard F. Clinical pharmacokinetic study of 5-FU in continuous 5-day infusions for head and neck cancer. Cancer Chemother Pharmacol (1986) 16(1):64–6. doi: 10.1007/BF00255288
99. van Groeningen CJ, Pinedo HM, Heddes J, Kok RM, de Jong AP, Wattel E, et al. Pharmacokinetics of 5-fluorouracil assessed with a sensitive mass spectrometric method in patients on a dose escalation schedule. Cancer Res (1988) 48(23):6956–61.
100. Gamelin E, Delva R, Jacob J, Merrouche Y, Raoul JL, Pezet D, et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol (2008) 26(13):2099–105. doi: 10.1200/JCO.2007.13.3934
101. Yoshida T, Araki E, Iigo M, Fujii T, Yoshino M, Shimada Y, et al. Clinical significance of monitoring serum levels of 5-fluorouracil by continuous infusion in patients with advanced colonic cancer. Cancer Chemother Pharmacol (1990) 26(5):352–4. doi: 10.1007/BF02897292
102. Deng R, Shi L, Zhu W, Wang M, Guan X, Yang D, et al. Pharmacokinetics-based dose management of 5-fluorouracil clinical research in advanced colorectal cancer treatment. Mini Rev Med Chem (2020) 20(2):161–7. doi: 10.2174/1389557519666191011154923
103. Fety R, Rolland F, Barberi-Heyob M, Hardouin A, Campion L, Conroy T, et al. Clinical impact of pharmacokinetically-guided dose adaptation of 5-fluorouracil: results from a multicentric randomized trial in patients with locally advanced head and neck carcinomas. Clin Cancer Res (1998) 4(9):2039–45.
104. Goldstein DA, Chen Q, Ayer T, Howard DH, Lipscomb J, Harvey RD, et al. Cost effectiveness analysis of pharmacokinetically-guided 5-fluorouracil in FOLFOX chemotherapy for metastatic colorectal cancer. Clin Colorectal Cancer (2014) 13(4):219–25. doi: 10.1016/j.clcc.2014.09.007
105. Soh IPT, Mogro MJ, Soo RA, Pang A, Tan CS, Chuah B, et al. The optimization of 5fluorouracil (5FU) dose by pharmacokinetic (PK) monitoring in Asian patients with advanced-stage gastrointestinal (GI) cancer. J Clin Oncol (2015) 33(3_suppl):770. doi: 10.1200/jco.2015.33.3_suppl.770
106. Becker R, Hollenbeak CS, Choma A, Kenny P, Salamone SJ. Cost-effectiveness of pharmacokinetic dosing of 5Fluorouracil in metastatic colorectal cancer in the united kingdom. Vale Health (2013) 16:A139. doi: 10.1016/j.jval.2013.03.680
107. Beumer JH, Boisdron-Celle M, Clarke W, Courtney JB, Egorin MJ, Gamelin E, et al. Multicenter evaluation of a novel nanoparticle immunoassay for 5-fluorouracil on the Olympus AU400 analyzer. Ther Drug Monit (2009) 31(6):688–94. doi: 10.1097/FTD.0b013e3181b9b8c0
108. Kaldate RR, Haregewoin A, Grier CE, Hamilton SA, McLeod HL. Modeling the 5fluorouracil area under the curve versus dose relationship to develop a pharmacokinetic dosing algorithm for colorectal cancer patients receiving FOLFOX6. Oncologist (2012) 17(3):296–302. doi: 10.1634/theoncologist.2011-0357
109. Darwich AS, Ogungbenro K, Hatley OJ, Rostami-Hodjegan A. Role of pharmacokinetic modeling and simulation in precision dosing of anticancer drugs. Trans Cancer Res (2017) 6(Suppl 10):S1512–S29. doi: 10.21037/tcr.2017.09.14
110. Widemann BC, Adamson PC. Understanding and managing methotrexate nephrotoxicity. Oncologist (2006) 11(6):694–703. doi: 10.1634/theoncologist.11-6-694
111. Comandone A, Passera R, Boglione A, Tagini V, Ferrari S, Cattel L. High dose methotrexate in adult patients with osteosarcoma: Clinical and pharmacokinetic results. Acta Oncol (2005) 44(4):406–11. doi: 10.1080/02841860510029770
112. Kintzel PE, Dorr RT. Anticancer drug renal toxicity and elimination: dosing guidelines for altered renal function. Cancer Treat Rev (1995) 21(1):33–64. doi: 10.1016/0305-7372(95)90010-1
113. Fukuhara K, Ikawa K, Morikawa N, Kumagai K. Population pharmacokinetics of high-dose methotrexate in Japanese adult patients with malignancies: a concurrent analysis of the serum and urine concentration data. J Clin Pharm Ther (2008) 33(6):677–84. doi: 10.1111/j.1365-2710.2008.00966.x
114. Graf N, Winkler K, Betlemovic M, Fuchs N, Bode U. Methotrexate pharmacokinetics and prognosis in osteosarcoma. J Clin Oncol (1994) 12(7):1443–51. doi: 10.1200/JCO.1994.12.7.1443
115. Joerger M, Ferreri AJ, Krähenbühl S, Schellens JH, Cerny T, Zucca E, et al. Dosing algorithm to target a predefined AUC in patients with primary central nervous system lymphoma receiving high dose methotrexate. Br J Clin Pharmacol (2012) 73(2):240–7. doi: 10.1111/j.1365-2125.2011.04084.x
116. Levêque D, Becker G, Toussaint E, Fornecker L-M, Paillard C. Clinical pharmacokinetics of methotrexate in oncology. Int J Pharmacokinet (2017) 2(2):137–47. doi: 10.4155/ipk-2016-0022
117. Wiczer T, Dotson E, Tuten A, Phillips G, Maddocks K. Evaluation of incidence and risk factors for high-dose methotrexate-induced nephrotoxicity. J Oncol Pharm Pract (2016) 22(3):430–6. doi: 10.1177/1078155215594417
118. Howard SC, McCormick J, Pui CH, Buddington RK, Harvey RD. Preventing and managing toxicities of high-dose methotrexate. Oncologist (2016) 21(12):1471–82. doi: 10.1634/theoncologist.2015-0164
119. Stoller RG, Hande KR, Jacobs SA, Rosenberg SA, Chabner BA. Use of plasma pharmacokinetics to predict and prevent methotrexate toxicity. N Engl J Med (1977) 297(12):630–4. doi: 10.1056/NEJM197709222971203
120. Schmiegelow K, Attarbaschi A, Barzilai S, Escherich G, Frandsen TL, Halsey C, et al. Consensus definitions of 14 severe acute toxic effects for childhood lymphoblastic leukaemia treatment: a Delphi consensus. Lancet Oncol (2016) 17(6):e231–e9. doi: 10.1016/S1470-2045(16)30035-3
121. Saag KG, Teng GG, Patkar NM, Anuntiyo J, Finney C, Curtis JR, et al. American College of rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Care Res (2008) 59(6):762–84. doi: 10.1002/art.23721
122. Ramanan A, Whitworth P, Baildam E. Use of methotrexate in juvenile idiopathic arthritis. Arch Dis Child (2003) 88(3):197–200. doi: 10.1136/adc.88.3.197
123. Muller IB, Hebing RF, Jansen G, Nurmohamed MT, Lems WF, Peters GJ, et al. Personalized medicine in rheumatoid arthritis: methotrexate polyglutamylation revisited. Expert Rev Precis Med Drug Dev (2018) 3(6):331–4. doi: 10.1080/23808993.2018.1517025
124. de Rotte MCFJ, Pluijm SMF, de Jong PHP, Bulatovic Calasan M, Wulffraat NM, Weel AEAM, et al. Development and validation of a prognostic multivariable model to predict insufficient clinical response to methotrexate in rheumatoid arthritis. PloS One (2018) 13(12):e0208534. doi: 10.1371/journal.pone.0208534
125. Rodríguez-Báez AS, Huerta-García AP, Medellín-Garibay SE, Rodríguez-Pinal CJ, Martínez-Martínez MU, Herrera-Van Oostdam D, et al. Disease activity and therapeutic drug monitoring of polyglutamates of methotrexate after daily or weekly administration of lowdose methotrexate in patients recently diagnosed with rheumatoid arthritis. Basic Clin Pharmacol Toxicol (2022) 130(6):644–54. doi: 10.1111/bcpt.13728
126. Dupuis C, Mercier C, Yang C, Monjanel-Mouterde S, Ciccolini J, Fanciullino R, et al. High-dose methotrexate in adults with osteosarcoma: a population pharmacokinetics study and validation of a new limited sampling strategy. Anticancer Drugs (2008) 19(3):267–73. doi: 10.1097/CAD.0b013e3282f21376
127. Foster JH, Thompson PA, Bernhardt MB, Margolin JF, Hilsenbeck SG, Jo E, et al. A prospective study of a simple algorithm to individually dose high-dose methotrexate for children with leukemia at risk for methotrexate toxicities. Cancer Chemother Pharmacol (2019) 83(2):349–60. doi: 10.1007/s00280-018-3733-2
128. Niemann A, Mühlisch J, Frühwald MC, Gerss J, Hempel G, Boos J. Therapeutic drug monitoring of methotrexate in cerebrospinal fluid after systemic high-dose infusion in children: can the burden of intrathecal methotrexate be reduced? Ther Drug Monit (2010) 32(4):467–75. doi: 10.1097/FTD.0b013e3181e5c6b3
129. Shanker Kasudhan K, Patial A, Mehra N, Verma Attri S, Malhotra P, Pattanaik S, et al. Cyclophosphamide, hydroxycyclophosphamide and carboxyethyl phosphoramide mustard quantification with liquid chromatography mass spectrometry in a single run human plasma samples: A rapid and sensitive method development. Journal of Chromatography B (2022) 1198:123228.
Keywords: therapeutic drug monitoring, cytotoxic drugs reconstitution, busulfan, methotrexate, 5-FU, 5 fluorouracil, precision medicine
Citation: Pattanaik S, Patil AN, Kumaravel J and Prakash G (2022) Therapeutic drug monitoring for cytotoxic anticancer drugs: Principles and evidence-based practices. Front. Oncol. 12:1015200. doi: 10.3389/fonc.2022.1015200
Received: 09 August 2022; Accepted: 24 October 2022;
Published: 08 December 2022.
Edited by:
Robert Clarke, University of Minnesota Twin Cities, United StatesReviewed by:
Dominique Leveque, Hôpital d’Hautepierre, FranceCopyright © 2022 Pattanaik, Patil, Kumaravel and Prakash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pattanaik Smita, cGF0dGFuYWlrLnNtaXRhQHBnaW1lci5lZHUuaW4=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.