
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 15 November 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1014859
This article is part of the Research Topic Identification, Risk Stratification, and Optimized Management for Lynch Syndrome View all 14 articles
 Jiaying Liu1†Xiaona Chang1†Guixiang Xiao1Jingmin Zhong1
Jiaying Liu1†Xiaona Chang1†Guixiang Xiao1Jingmin Zhong1 Bo Huang1
Bo Huang1 Jiwei Zhang1Beibei Gao1
Jiwei Zhang1Beibei Gao1 Gang Peng2*
Gang Peng2* Xiu Nie1*
Xiu Nie1*Background: Patients with Lynch syndrome are at an increased risk of developing simultaneous or metachronous tumors, while sarcomas have been occasionally reported. Sarcomas are generally not considered part of the common Lynch syndrome tumor spectrum. However, more and more studies and case reports suggested that sarcoma could be a rare clinical manifestation of Lynch syndrome, leading to new treatment strategies for sarcoma.
Case summary: We report the case of a 74-year-old male patient with Lynch syndrome who had rectal mucinous adenocarcinoma and prostate adenocarcinoma and then developed undifferentiated sarcoma of the left neck two years later. Mismatch repair deficiency (dMMR) was confirmed by immunohistochemical staining for the mismatch repair proteins MSH2, MSH6, MLH1 and PMS2. The result of polymerase chain reaction (PCR) microsatellite instability (MSI) testing of sarcoma showed high-level microsatellite instability (MSI-H). Additionally, a pathogenic germline mutation in MSH2 (c.2459-12A>G) was detected by next-generation sequencing (NGS). Taking into account HE morphology, immunohistochemical phenotype, MSI status, NGS result, medical history and germline MSH2 gene mutation, the pathological diagnosis of left neck biopsy tissue was Lynch syndrome related undifferentiated sarcoma with epithelioid morphology. The patient has been receiving immunotherapy (sintilimab) combined with chemotherapy (tegafur, gimeracil and oteracil potassium capsules) and currently has stable disease. We also reviewed the literature to understand the association between sarcoma and Lynch syndrome.
Conclusion: Sarcoma may now be considered a rare clinical manifestation of Lynch syndrome. Attention and awareness about the association between Lynch syndrome and sarcoma need to be increased. Therefore, timely detection of MMR proteins and validation at the gene level for suspicious patients are the keys to avoiding missed or delayed diagnosis and to identifying patients suited for immunotherapy, which may also help to provide appropriate genetic counseling and follow-up management for patients.
Lynch syndrome is a hereditary cancer predisposition syndrome caused by a germline mutation in one of several DNA mismatch repair (MMR) genes (including MLH1, MSH2, MSH6, PMS2) or loss of expression of MSH2 due to deletion in the EPCAM gene (1, 2). Individuals with Lynch syndrome are at an increased risk of developing simultaneous or metachronous tumors, predominantly colorectal cancer and endometrial cancer (3, 4), and are also at increased risk of cancer of the ovary, prostate, stomach, genitourinary system, and hepatobiliary system (2). Moreover, sarcomas are generally not considered part of the common Lynch syndrome tumor spectrum. However, patients with Lynch syndrome have been occasionally reported to develop sarcomas (5–11). As more and more studies and case reports published, the opinion that sarcoma could be a rare clinical manifestation of Lynch syndrome is getting more and more attention, leading to new treatment strategies for sarcoma.
We reported a case in which undifferentiated sarcoma of the neck was identified two years later in a patient with Lynch syndrome who had rectal mucinous adenocarcinoma and prostate adenocarcinoma. The patient has been receiving immunotherapy (sintilimab) combined with chemotherapy (tegafur, gimeracil and oteracil potassium capsules) and currently has stable disease. Furthermore, we also reviewed the literature to understand the association between sarcoma and Lynch syndrome. The report aims to raise awareness of Lynch syndrome-related sarcomas and to identify patients suited for immunotherapy.
We present the case according to the CARE reporting checklist (Supplementary Figure S1; available at https://www.care-statement.org/checklist).
A 74-year-old male patient with left neck swelling for one month, without tenderness, and without fever or other symptoms was admitted to Union Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology in July 2021. A CT-scan of the neck showed a 66 mm×54 mm round soft tissue mass shadow in the left neck (Figure 1A), supraclavicular area and superior mediastinum, with multiple enlarged lymph nodes around, and the trachea, thyroid and esophagus were pushed to the right, with local tracheal narrowing. Further contrast-enhanced CT scans showed ring enhancement (Figure 1B), and a lack of clear demarcation between the mass and the esophageal wall. In addition, tumor markers were normal.
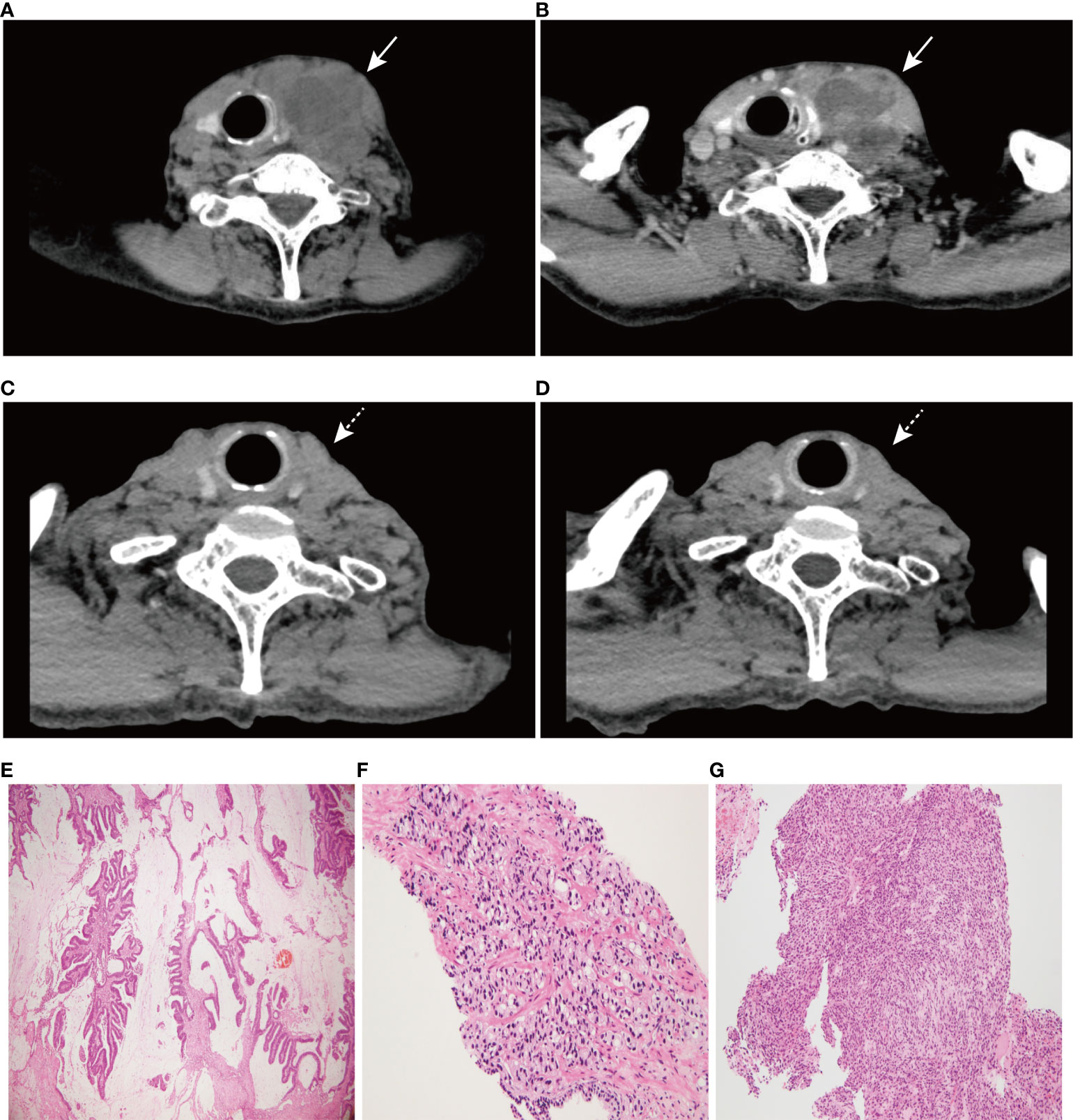
Figure 1 CT scan images of neck and hematoxylin-eosin (HE) staining of three tumors (×100). (A) A 66 mm×54 mm round soft tissue mass shadow in the left neck. (B) Further contrast-enhanced CT scan showed ring enhancement. (C) CT scan image of neck at 6 months post-treatment. (D) CT scan image of neck at 10 months post-treatment. (E) HE staining of rectal mucinous adenocarcinoma, (F) prostate adenocarcinoma, and (G) undifferentiated sarcoma of the left neck. Solid arrows indicate tumor masses; dashed arrows indicate significant tumor regression.
It is noteworthy that the patient was diagnosed with rectal mucinous adenocarcinoma 30 months ago and subsequently underwent surgery. And there was no special treatment after surgery. Meanwhile, the patient was diagnosed with prostate adenocarcinoma on biopsy (Gleason 3 + 4 = 7) and then received castration therapy. The patient reported no family history of tumors.
More specifically, the surgical pathology confirmed a diagnosis of rectal mucinous adenocarcinoma (Figure 1E), while the tumor invaded through the muscularis propria and into the adipose tissue outside the intestinal wall (pT3), without tumor vascular thrombus or perineural invasion around the tumor, with negative surgical margins. There was no evidence of lymph node involvement (14 lymph nodes were resected), while there was one peri-intestinal cancer nodule. Mismatch repair deficiency (dMMR) was confirmed by immunohistochemical staining for the mismatch repair proteins MSH2, MSH6, MLH1 and PMS2. Immunohistochemistry showed complete loss of MSH2 and MSH6 expression but normal MLH1 and PMS2 expression (Figure 2). Germline MSH2 gene mutation (c.2459-12A>G) was detected by next generation sequencing (NGS). NGS result of MSH2 also showed that tumor tissues had higher mutation abundance than the control (84% vs 48%, respectively) (Supplementary Figure S2). This genetic variant was located within an intron and did not generally affect the function of protein. It was not represented in the large population databases (1000 Genomes, gnomAD, and ExAC), indicating that this mutation was a rare variant. In addition, the ClinVar database contained six records for the variant, where the pathogenicity was recorded as likely pathogenic of three records and uncertain significance of three remaining records (12–16). In summary, MSH2 gene mutation (c.2459-12A>G) was classified as likely pathogenic according to American College of Medical Genetics and Genomics guidelines (ACMG, 2015). Moreover, gene mutations strongly associated with treatment and prognosis in rectal cancer were also detected by NGS, including KRAS (p. G12D), PIK3CA (p. E545G) and TP53 (p. R248W). Moreover, NGS test of rectal mucinous adenocarcinoma indicated high TMB (68.79 mutations/Mb) and mutations in other mismatch repair related genes (ATM, CDK12, FANCA and MRE11).
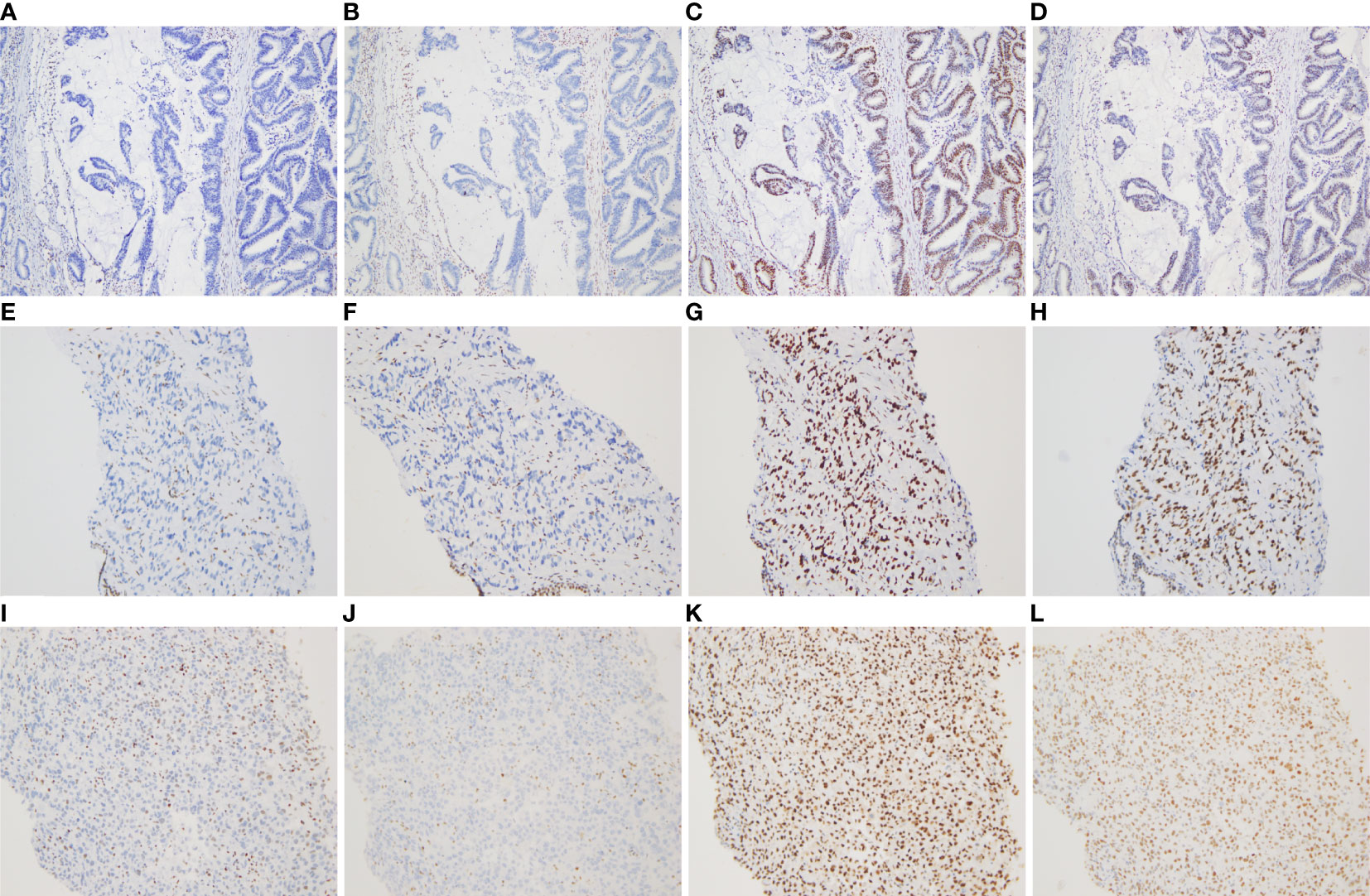
Figure 2 Immunohistochemical staining for DNA mismatch repair proteins (MMR proteins MSH6, MSH2, MLH1, and PMS2) of three tumors (×100). (A–D) Absence of MSH6 and MSH2 staining, and positive staining for MLH1 and PMS2 in the rectal mucinous adenocarcinoma; (E–H) Absence of MSH6 and MSH2 staining, and positive staining for MLH1 and PMS2 in the prostate adenocarcinoma; (I–L) Absence of MSH6 and MSH2 staining, and positive staining for MLH1 and PMS2 in the undifferentiated sarcoma of left neck.
The pathological diagnosis of prostate biopsy revealed prostate adenocarcinoma with Gleason 3 + 4 (Figure 1F). Similarly, dMMR was also confirmed by immunohistochemical staining. Immunohistochemistry showed complete loss of MSH2 and MSH6 expression but normal MLH1 and PMS2 expression (Figure 2). The deficiency of mismatch repair function has several important consequences, such as gain of growth advantage, increase in the point mutation rate, MSI-H and abnormal MMR protein expression by IHC. In summary, we reached consensus that the prostate adenocarcinoma and rectal mucinous adenocarcinoma were associated with Lynch syndrome.
After comprehensive consideration of the patient’s history and status, the patient underwent biopsy of the left neck mass. Microscopically, tumor cells displayed striking atypia and epithelioid morphology, infiltrating into skeletal muscle, without lymph node structure detected (Figure 1G). In addition, the lack of differentiation of the immunohistochemical phenotype led to difficulty in understanding the tumor cell of origin. For more details, see Supplementary Table 1. Furthermore, NGS, including genes and mutations associated with soft tissue sarcoma typing (57 genes, 236 types of gene fusions and 14 gene mutations), was performed. TP53 mutation (p. Arg175His) was detected, but we were still unable to determine the tumor cell of origin. As expected, dMMR was also confirmed by immunohistochemical staining, consistent with the phenotypes of rectal mucinous adenocarcinoma and prostate adenocarcinoma (Figure 2). In addition, the result of polymerase chain reaction (PCR) microsatellite instability (MSI) testing of sarcoma showed high-level microsatellite instability (MSI-H). For more details, see Supplementary Figure S3.
In summary, taking into account HE morphology, immunohistochemical phenotype, NGS result, MSI result, medical history and germline MSH2 gene mutation (c.2459-12A>G), the pathological diagnosis of left neck biopsy tissue was Lynch syndrome-related undifferentiated sarcoma with epithelioid morphology.
Among the differential diagnoses, the diagnosis of metastatic cancer of the left neck was ruled out due to a lack of expression of epithelial markers (PCK, CK8/18, CK7, CK20, Villin, CDX2, PSAP, etc.). Lack of expression of malignant melanoma markers (S100, SOX10, HMB45, MelanA, etc.) made it impossible to make the diagnosis of malignant melanoma. Similarly, the absence of detection of lymphatic and hematopoietic system markers (LCA, CD3, CD20, CD38, CD138, MUM1, Kappa, Lambda, MPO, CD43, CD117, etc.) was unable to support the diagnosis of lymphatic and hematopoietic cancer. In addition, the diagnosis of sarcoma with certain differentiation was hard to make due to the absence of lineage-specific markers (Desmin, ERG, CD34, or corresponding fusion genes and mutant genes). In addition, it was unreasonable to make a diagnosis of sporadic undifferentiated sarcoma, which barely demonstrated immunohistochemical absence of MMR proteins and had no pathogenic or likely pathogenic germline gene mutations.
The patient has been receiving 15 cycles of immunotherapy (sintilimab, 200 mg i.v. every three weeks) combined with oral chemotherapy (tegafur, gimeracil and oteracil potassium capsules) and well tolerated. Reassuringly, significant regression of the left neck tumor was observed after two cycles of treatment, and the curative effect was evaluated as partial response (PR) according to the RECIST criteria and then maintained the state of PR during a follow-up of 14 months, which further supported our diagnosis (Figures 1C, D). The timeline scheme of the major clinical event of the patient is represented in Figure 3.
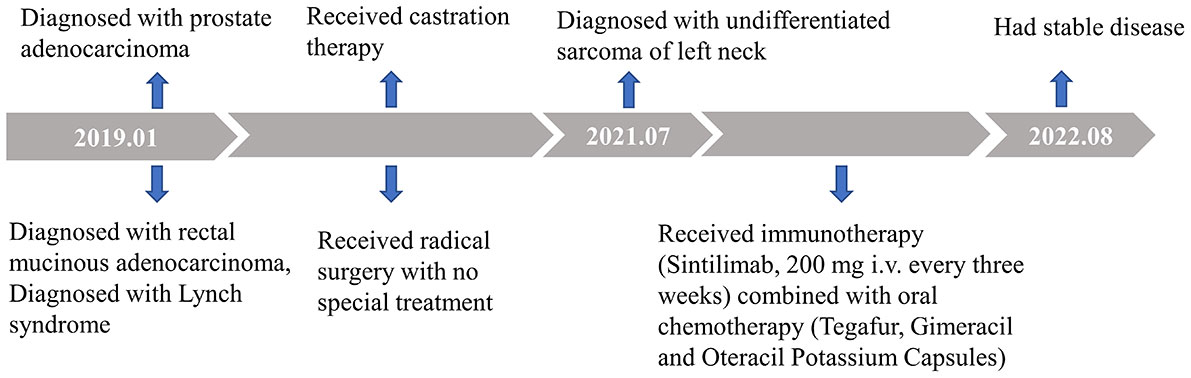
Figure 3 Timeline scheme of the major clinical event of the patient.
We present a case report of a male patient who was diagnosed with rectal mucinous adenocarcinoma and prostate adenocarcinoma at age 71 and left neck undifferentiated sarcoma at age 74. Immunohistochemical staining for MMR proteins of three tumors yielded consistent results, MSH6 (–), MSH2 (-), MLH1 (+), and PMS2 (+), indicating the presence of dMMR. In addition, the result of PCR MSI testing of sarcoma showed MSI-H. Moreover, the patient carries a germline likely pathogenic MSH2 gene mutation (c.2459-12A>G). All things considered, the final pathological diagnosis of the left neck tumor was Lynch syndrome-related undifferentiated sarcoma with epithelioid morphology.
Sarcoma is a rare clinical manifestation of Lynch syndrome (8, 17, 18). We summarized sarcomas reported in conjunction with Lynch syndrome (except for occasional cases reported in non-English literature) in Table 1. A previous study in the Prospective Lynch Syndrome Database showed an increase in the incidence and lifetime risk of sarcoma, although details of specific illness risk and mutated genes were not reported (17). The study enrolled 6,350 patients with Lynch syndrome, and 16 of them developed sarcomas (12 osteosarcomas and 4 soft tissue sarcomas) after 51,646 follow-up years (17), which meant that patients with Lynch syndrome had more than 50-fold and 1.2-fold higher incidence of osteosarcomas and soft tissue sarcomas compared with the expected rates in the general population (osteosarcomas 0.34 per 100,000, soft tissue sarcomas 5.03 per 100,000), respectively (37–39). An Asian study demonstrated tumor development in 55 Japanese Lynch syndrome patients and reported a patient developing sarcoma with germline MLH1 mutation (8). Recently, a cohort study by de Angelis et al. evaluated the occurrence of sarcomas in a cohort of patients with tumors on the Lynch syndrome spectrum and finally identified five eligible cases, three of which carried MSH2 pathogenic variants (18).
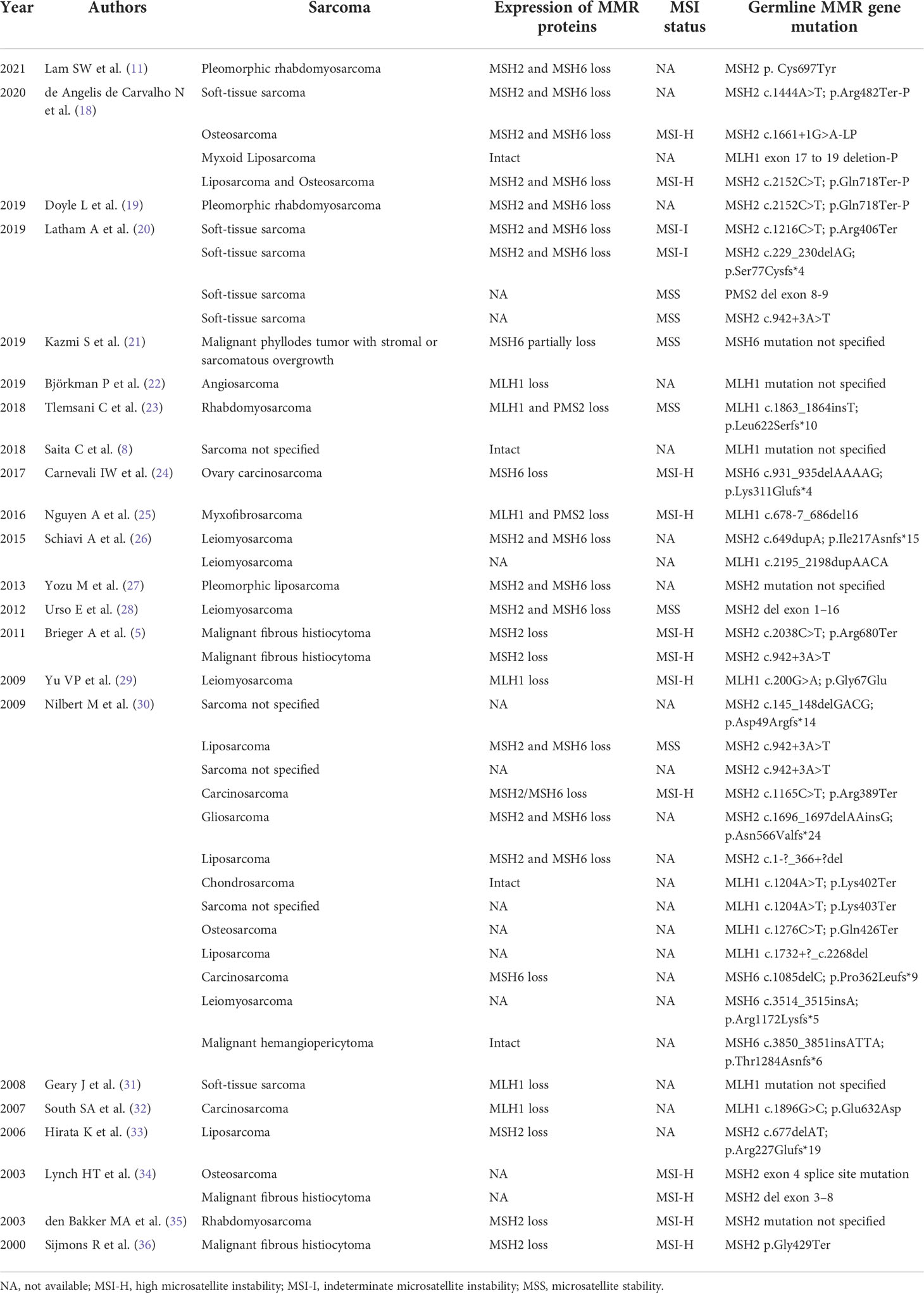
Table 1 Overview of sarcomas linked to Lynch syndrome published in the literature.
Some previous studies indicated that the development of sarcoma in patients with Lynch syndrome was associated with the expression of MMR proteins, thereby connecting sarcoma with MMR genes (5, 7, 9–11, 28, 33). Furthermore, previous studies have shown that MMR genes may be associated with sarcoma risk (18, 40–42). A study of cancer susceptibility variants based on The Cancer Genome Atlas (TCGA) data described that two MSH2 mutation carriers were detected in an unselected sarcoma population (225 patients) and classified MSH2 as potentially associated with sarcoma risk according to variant burden analysis (odds ratio, 9.9; p = 0.02; false discovery rate, 0.09) (40). In addition, Mirabello et al. analyzed pathogenic germline variants in cancer-susceptibility genes in 1244 patients with osteosarcoma and found more germline MSH2 pathogenic variants in patients with osteosarcoma than in the control group (p < 0.05) (41). Moreover, a previous study showed that sarcoma tended to be more associated with pathogenic variants of MSH2 than other MMR genes, as 25 of 43 (58.1%) tested cases had MSH2 germline mutations (18). It was a significantly higher frequency in patients with sarcoma than in unselected patients with Lynch syndrome, where MSH2 was usually the second most frequently mutated gene (seen in approximately 40% of patients) (17, 43).
With the advent of the era of tumor immunity, immune checkpoint inhibitor therapy has become an effective treatment for microsatellite instability-high (MSI-H) or dMMR tumors (44, 45). Latham A et al. assessed the MSI status of 15,045 patients (more than 50 cancer types) based on NGS data, and the incidence of MSI-H and MSI-indeterminate (MSI-I) in soft tissue sarcomas was found to be 5.7% (45/785), while two of them were diagnosed with Lynch syndrome with pathogenic MSH2 variants (20). Similarly, another recent study based on NGS data reported that the incidence of dMMR in an unselected cohort of adult soft tissue and bone sarcomas was 2.3% (7/304) (19). Somatic mutation analysis showed that all seven patients had MMR gene mutations (4 of MSH2 or EPCAM, 2 of PMS2, 1 of MSH6), and further germline sequencing of three patients (2 of MSH2, 1 of MSH6) suggested that one patient had pathogenic MSH2 germline mutation and was also diagnosed with Lynch syndrome (19). Tlemsani C et al. highlighted the importance of identifying Lynch syndrome in patients with sarcoma (23). The article described a 19-year-old male patient who presented with metastatic chemoresistant pleomorphic rhabdomyosarcoma. Then, the patient received anti- programmed death (PD)-1 antibody therapy (nivolumab) due to detection of the MLH1 germline pathogenic variant and achieved a rapid complete response of the lung metastases, which appeared sustained after a 1-year follow-up (23). Furthermore, data from the phase II KEYNOTE-158 study of pembrolizumab (an anti-PD-1 monoclonal antibody) in patients with previously treated, advanced noncolorectal MSI-H/dMMR cancer (including 14 sarcomas) demonstrated the clinical benefit of anti-PD-1 therapy among patients with sarcoma (46).
In the present case, undifferentiated sarcoma of the left neck was identified two years later in a 74-year-old male patient with Lynch syndrome who had rectal mucinous adenocarcinoma and prostate adenocarcinoma. The conventional chemotherapy drugs for undifferentiated sarcoma were adriamycin, ifosfamide, gemcitabine, paclitaxel, etc (47–49). The patient refused all intravenous chemotherapy due the older age. Despite the lack of reliable evidence, there were several studies showed that the fluorouracil was effective against undifferentiated sarcoma (50, 51). Therefore, the patient has been receiving immunotherapy (sintilimab) combined with chemotherapy (tegafur, gimeracil and oteracil potassium capsules). Reassuringly, significant regression of the left neck tumor was observed, and the patient was in good condition after a follow-up of 14 months.
In conclusion, sarcoma may now be considered a rare clinical manifestation of Lynch syndrome. Although the risk of sarcoma was significantly lower than that of other common Lynch syndrome-associated tumors, attention to and awareness of the association between Lynch syndrome and sarcoma need to be increased. Therefore, timely detection of MMR proteins by IHC and validation at the gene level for suspicious patients are the keys to avoiding missed or delayed diagnosis and to identifying patients suited for immunotherapy, which may also help to provide appropriate genetic counseling and follow-up management for patients.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Conception/Design: XN and XC; Provision of study material or patients: GP, GX and JinZ; Collection and/or assembly of data: XC and JL; Data analysis and interpretation: JL; Manuscript writing: JL and XC; Final approval of manuscript: GP and XN. All authors have read and approved the submitted version of the manuscript.
The research was supported by Union Hospital, Tongji Medical College, Huazhong University of Science and Technology Research Fund. (2021xhyn080).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1014859/full#supplementary-material
1. Ligtenberg MJ, Kuiper RP, Geurts van Kessel A, Hoogerbrugge N. Epcam deletion carriers constitute a unique subgroup of lynch syndrome patients. Fam Cancer (2013) 12(2):169–74. doi: 10.1007/s10689-012-9591-x
2. Liccardo R, De Rosa M, Izzo P, Duraturo F. Novel implications in molecular diagnosis of lynch syndrome. Gastroenterol Res Pract (2017) 2017:2595098. doi: 10.1155/2017/2595098
3. Lee HS, Kim WH, Kwak Y, Koh J, Bae JM, Kim KM, et al. Molecular testing for gastrointestinal cancer. J Pathol Transl Med (2017) 51(2):103–21. doi: 10.4132/jptm.2017.01.24
4. Zhao S, Chen L, Zang Y, Liu W, Liu S, Teng F, et al. Endometrial cancer in lynch syndrome. Int J Cancer (2022) 150(1):7–17. doi: 10.1002/ijc.33763
5. Brieger A, Engels K, Schaefer D, Plotz G, Zeuzem S, Raedle J, et al. Malignant fibrous histiocytoma is a rare lynch syndrome-associated tumor in two German families. Familial Cancer (2011) 10(3):591–5. doi: 10.1007/s10689-011-9455-9
6. Yozu M, Symmans P, Dray M, Griffin J, Han C, Ng D, et al. Muir–Torre syndrome-associated pleomorphic liposarcoma arising in a previous radiation field. Virchows Archiv (2013) 462(3):355–60. doi: 10.1007/s00428-012-1369-x
7. Lee N, Luthra R, Lopez-Terrada D, Wang WL, Lazar AJ. Retroperitoneal undifferentiated pleomorphic sarcoma having microsatellite instability associated with Muir-torre syndrome: Case report and review of literature. J Cutan Pathol (2013) 40(8):730–3. doi: 10.1111/cup.12172
8. Saita C, Yamaguchi T, Horiguchi SI, Yamada R, Takao M, Iijima T, et al. Tumor development in Japanese patients with lynch syndrome. PloS One (2018) 13(4):e0195572. doi: 10.1371/journal.pone.0195572
9. Shigematsu Y, Yamashita K, Takamatsu M, Tanizawa T, Togashi Y, Nakajima T, et al. Primary intramucosal synovial sarcoma of the sigmoid colon in a patient with a germline mutation in the Msh2 gene: A case report. Pathol Int (2020) 70(12):1015–9. doi: 10.1111/pin.13020
10. Liu Y, GuLiBaHa M, Yue YB, Li MW, Cao SB, Yan M. An isolated childhood myeloid sarcoma with germline Msh6 mutation-a case report. Transl Pediatr (2021) 10(8):2136–43. doi: 10.21037/tp-21-326
11. Lam SW, Kostine M, de Miranda N, Schoffski P, Lee CJ, Morreau H, et al. Mismatch repair deficiency is rare in bone and soft tissue tumors. Histopathology (2021) 79(4):509–20. doi: 10.1111/his.14377
12. Mangold E, Pagenstecher C, Friedl W, Fischer HP, Merkelbach-Bruse S, Ohlendorf M, et al. Tumours from Msh2 mutation carriers show loss of Msh2 expression but many tumours from Mlh1 mutation carriers exhibit weak positive Mlh1 staining. J Pathol (2005) 207(4):385–95. doi: 10.1002/path.1858
13. Shirts BH, Konnick EQ, Upham S, Walsh T, Ranola JMO, Jacobson AL, et al. Using somatic mutations from tumors to classify variants in mismatch repair genes. Am J Hum Genet (2018) 103(1):19–29. doi: 10.1016/j.ajhg.2018.05.001
14. Mangold E, Pagenstecher C, Friedl W, Mathiak M, Buettner R, Engel C, et al. Spectrum and frequencies of mutations in Msh2 and Mlh1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer (2005) 116(5):692–702. doi: 10.1002/ijc.20863
15. Barrow E, Jagger E, Brierley J, Wallace A, Evans G, Hill J, et al. Semiquantitative assessment of immunohistochemistry for mismatch repair proteins in lynch syndrome. Histopathology (2010) 56(3):331–44. doi: 10.1111/j.1365-2559.2010.03485.x
16. Pope BJ, Clendenning M, Rosty C, Mahmood K, Georgeson P, Joo JE, et al. Germline and tumor sequencing as a diagnostic tool to resolve suspected lynch syndrome. J Mol Diagn (2021) 23(3):358–71. doi: 10.1016/j.jmoldx.2020.12.003
17. Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the prospective lynch syndrome database. Genet Med (2020) 22(1):15–25. doi: 10.1038/s41436-019-0596-9
18. de Angelis de Carvalho N, Niitsuma BN, Kozak VN, Costa FD, de Macedo MP, Kupper BEC, et al. Clinical and molecular assessment of patients with lynch syndrome and sarcomas underpinning the association with Msh2 germline pathogenic variants. Cancers (Basel) (2020) 12(7):1848. doi: 10.3390/cancers12071848
19. Doyle LA, Nowak JA, Nathenson MJ, Thornton K, Wagner AJ, Johnson JM, et al. Characteristics of mismatch repair deficiency in sarcomas. Modern Pathol (2019) 32(7):977–87. doi: 10.1038/s41379-019-0202-3
20. Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol Off J Am Soc Clin Oncol (2019) 37(4):286–95. doi: 10.1200/jco.18.00283
21. Kazmi S, Wagner S, Heintzelman R, Corbman M. Malignant phyllodes tumor in lynch syndrome: A case report. J Med Case Rep (2019) 13(1):216. doi: 10.1186/s13256-019-2138-0
22. Björkman P, Kantonen I, Blomqvist C, Venermo M, Albäck A. En bloc resection of visceral aorta and right kidney due to aortic sarcoma using temporary extracorporeal bypass grafting. J Vasc Surg cases innovative techniques (2019) 5(4):589–92. doi: 10.1016/j.jvscit.2019.08.002
23. Tlemsani C, Leroy K, Gimenez-Roqueplo A-P, Mansuet-Lupo A, Pasmant E, Larousserie F, et al. Chemoresistant pleomorphic rhabdomyosarcoma: Whole exome sequencing reveals underlying cancer predisposition and therapeutic options. J Med Genet (2020) 57(2):104–8. doi: 10.1136/jmedgenet-2018-105594
24. Carnevali IW, Cimetti L, Sahnane N, Libera L, Cavallero A, Formenti G, et al. Two cases of carcinosarcomas of the ovary involved in hereditary cancer syndromes. Int J gynecol Pathol Off J Int Soc Gynecol Pathol (2017) 36(1):64–70. doi: 10.1097/pgp.0000000000000290
25. Nguyen A, Bougeard G, Koob M, Chenard MP, Schneider A, Maugard C, et al. Msi detection and its pitfalls in cmmrd syndrome in a family with a bi-allelic Mlh1 mutation. Fam Cancer (2016) 15(4):571–7. doi: 10.1007/s10689-016-9894-4
26. Schiavi A, Lavigne J, Turcotte R, Kasprzak L, Dumas N, Chong G, et al. Using a family history questionnaire to identify adult patients with increased genetic risk for sarcoma. Curr Oncol (Toronto Ont) (2015) 22(5):317–25. doi: 10.3747/co.22.2588
27. Yozu M, Symmans P, Dray M, Griffin J, Han C, Ng D, et al. Muir-Torre syndrome-associated pleomorphic liposarcoma arising in a previous radiation field. Virchows Archiv an Int J Pathol (2013) 462(3):355–60. doi: 10.1007/s00428-012-1369-x
28. Urso E, Agostini M, Pucciarelli S, Bedin C, D'Angelo E, Mescoli C, et al. Soft tissue sarcoma and the hereditary non-polyposis colorectal cancer (Hnpcc) syndrome: Formulation of an hypothesis. Mol Biol Rep (2012) 39(10):9307–10. doi: 10.1007/s11033-012-1729-2
29. Yu VP, Novelli M, Payne SJ, Fisher S, Barnetson RA, Frayling IM, et al. Unusual presentation of lynch syndrome. Hereditary Cancer Clin Pract (2009) 7(1):12. doi: 10.1186/1897-4287-7-12
30. Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcomas associated with hereditary nonpolyposis colorectal cancer: Broad anatomical and morphological spectrum. Fam Cancer (2009) 8(3):209–13. doi: 10.1007/s10689-008-9230-8
31. Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne SJ, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (Hnpcc). Fam Cancer (2008) 7(2):163–72. doi: 10.1007/s10689-007-9164-6
32. South SA, Hutton M, Farrell C, Mhawech-Fauceglia P, Rodabaugh KJ. Uterine carcinosarcoma associated with hereditary nonpolyposis colorectal cancer. Obstet gynecol (2007) 110(2 Pt 2):543–5. doi: 10.1097/01.Aog.0000275262.60526.01
33. Hirata K, Kanemitsu S, Nakayama Y, Nagata N, Itoh H, Ohnishi H, et al. A novel germline mutation of Msh2 in a hereditary nonpolyposis colorectal cancer patient with liposarcoma. Am J Gastroenterol (2006) 101(1):193–6. doi: 10.1111/j.1572-0241.2005.00308.x
34. Lynch HT, Deters CA, Hogg D, Lynch JF, Kinarsky Y, Gatalica Z. Familial sarcoma: Challenging pedigrees. Cancer (2003) 98(9):1947–57. doi: 10.1002/cncr.11743
35. den Bakker MA, Seynaeve C, Kliffen M, Dinjens WN. Microsatellite instability in a pleomorphic rhabdomyosarcoma in a patient with hereditary non-polyposis colorectal cancer. Histopathology (2003) 43(3):297–9. doi: 10.1046/j.1365-2559.2003.01681.x
36. Sijmons R, Hofstra R, Hollema H, Mensink R, van der Hout A, Hoekstra H, et al. Inclusion of malignant fibrous histiocytoma in the tumour spectrum associated with hereditary non-polyposis colorectal cancer. Genes Chromosomes Cancer (2000) 29(4):353–5. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1042>3.0.co;2-t
37. Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer (2009) 125(1):229–34. doi: 10.1002/ijc.24320
38. Misaghi A, Goldin A, Awad M, Kulidjian AA. Osteosarcoma: A comprehensive review. SICOT J (2018) 4:12. doi: 10.1051/sicotj/2017028
39. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer (2006) 119(12):2922–30. doi: 10.1002/ijc.22239
40. Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell (2018) 173(2):355–70 e14. doi: 10.1016/j.cell.2018.03.039
41. Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol (2020) 6(5):724–34. doi: 10.1001/jamaoncol.2020.0197
42. Chan SH, Lim WK, Ishak NDB, Li ST, Goh WL, Tan GS, et al. Germline mutations in cancer predisposition genes are frequent in sporadic sarcomas. Sci Rep (2017) 7(1):10660. doi: 10.1038/s41598-017-10333-x
43. Vaccaro CA, López-Kostner F, Adriana DV, Palmero EI, Rossi BM, Antelo M, et al. From colorectal cancer pattern to the characterization of individuals at risk: Picture for genetic research in Latin America. Int J Cancer (2019) 145(2):318–26. doi: 10.1002/ijc.31920
44. Lee V, Murphy A, Le DT, Diaz LA Jr. Mismatch repair deficiency and response to immune checkpoint blockade. oncol (2016) 21(10):1200–11. doi: 10.1634/theoncologist.2016-0046
45. Fanale D, Corsini LR, Scalia R, Brando C, Cucinella A, Madonia G, et al. Can the tumor-agnostic evaluation of Msi/Mmr status be the common denominator for the immunotherapy treatment of patients with several solid tumors? Crit Rev oncology/hematol (2022) 170:103597. doi: 10.1016/j.critrevonc.2022.103597
46. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite Instability/Mismatch repair-deficient cancer: Results from the phase ii keynote-158 study. J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(1):1–10. doi: 10.1200/jco.19.02105
47. Lange SS, Novetsky AP, Powell MA. Recent advances in the treatment of sarcomas in gynecology. Discovery Med (2014) 18(98):133–40.
48. Goy BW, Syed S, Padmanabhan A, Burchette RJ, Helmstedter CS. The role of ifosfamide-doxorubicin chemotherapy in histology-specific, high grade, locally advanced soft tissue sarcoma, a 14-year experience. Radiother Oncol J Eur Soc Ther Radiol Oncol (2021) 165:174–8. doi: 10.1016/j.radonc.2021.10.019
49. Novetsky AP, Powell MA. Management of sarcomas of the uterus. Curr Opin Oncol (2013) 25(5):546–52. doi: 10.1097/CCO.0b013e328363e0ef
50. Lobo-Sanahuja F, Garcia I, Carranza A, Camacho A. Treatment and outcome of undifferentiated carcinoma of the nasopharynx in childhood: A 13-year experience. Med Pediatr Oncol (1986) 14(1):6–11. doi: 10.1002/mpo.2950140103
Keywords: undifferentiated sarcoma, immune checkpoint inhibitor, sintilimab, lynch syndrome, MSH2, mismatch repair deficiency
Citation: Liu J, Chang X, Xiao G, Zhong J, Huang B, Zhang J, Gao B, Peng G and Nie X (2022) Case report: Undifferentiated sarcoma with multiple tumors involved in Lynch syndrome: Unexpected favorable outcome to sintilimab combined with chemotherapy. Front. Oncol. 12:1014859. doi: 10.3389/fonc.2022.1014859
Received: 09 August 2022; Accepted: 26 October 2022;
Published: 15 November 2022.
Edited by:
Christina Therkildsen, Copenhagen University Hospital, DenmarkReviewed by:
Haiping Jiang, Zhejiang University, ChinaCopyright © 2022 Liu, Chang, Xiao, Zhong, Huang, Zhang, Gao, Peng and Nie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiu Nie, bmlleGl1eWlzaGlAMTI2LmNvbQ==; Gang Peng, cGVuZ2dhbmcxOTc3QGFsaXl1bi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.