
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 27 December 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1014592
This article is part of the Research TopicImplementation of Genomic and Epigenomic Innovation in Clinical Cancer DiagnosticsView all 8 articles
 Ariane Hallermayr1,2,3
Ariane Hallermayr1,2,3 Teresa M. Neuhann1
Teresa M. Neuhann1 Verena Steinke-Lange1,4
Verena Steinke-Lange1,4 Florentine Scharf1
Florentine Scharf1 Andreas Laner1Roland Ewald5Ben Liesfeld5Elke Holinski-Feder1,4
Andreas Laner1Roland Ewald5Ben Liesfeld5Elke Holinski-Feder1,4 Julia M. A. Pickl1,4*
Julia M. A. Pickl1,4*Background: Liquid biopsy (LB) is a promising complement to tissue biopsy for detection of clinically relevant genetic variants in cancer and mosaic diseases. A combined workflow to enable parallel tissue and LB analysis is required to maximize diagnostic yield for patients.
Methods: We developed and validated a cost-efficient combined next-generation sequencing (NGS) workflow for both tissue and LB samples, and applied Duplex sequencing technology for highly accurate detection of low frequency variants in plasma. Clinically relevant cutoffs for variant reporting and quantification were established.
Results: We investigated assay performance characteristics for very low amounts of clinically relevant variants. In plasma, the assay achieved 100% sensitivity and 92.3% positive predictive value (PPV) for single nucleotide variants (SNVs) and 91.7% sensitivity and 100% PPV for insertions and deletions (InDel) in clinically relevant hotspots with 0.5-5% variant allele frequencies (VAFs). We further established a cutoff for reporting variants (i.e. Limit of Blank, LOB) at 0.25% VAF and a cutoff for quantification (i.e. Limit of Quantification, LOQ) at 5% VAF in plasma for accurate clinical interpretation of analysis results. With our LB approach, we were able to identify the molecular cause of a clinically confirmed asymmetric overgrowth syndrome in a 10-year old child that would have remained undetected with tissue analysis as well as other molecular diagnostic approaches.
Conclusion: Our flexible and cost-efficient workflow allows analysis of both tissue and LB samples and provides clinically relevant cutoffs for variant reporting and precise quantification. Complementing tissue analysis by LB is likely to increase diagnostic yield for patients with molecular diseases.
LB enables identification of genetic sequence variants in circulating free DNA (cfDNA) from plasma and allows stratification of patients that will benefit from targeted therapies. One example is the detection of oncogenic driver variants in plasma of non-small cell lung cancer (NSCLC) patients that is associated with response to tyrosine kinase inhibitor therapy (1).
LB is an alternative to standard tissue biopsy, and is increasingly applied when tissue accessibility is limited, in case tissue biopsy leads to insufficient quality or quantity of material, or the result of a tissue biopsy analysis is expected to take longer than a LB assay (2). Accordingly, LB has been acknowledged as possible alternative to tissue biopsy by U.S. and European guidelines and position papers (3–6). With ~16,000 advanced NSCLC patients per year in Europe who are eligible for biomarker analysis but do not receive biomarker testing due to unsuccessful tissue biopsy, there is a huge potential of LB to increase the number of patients that benefit from personalized treatment (2). These include not only NSCLC or other solid tumor patients, but also patients with mosaic diseases.
A major advantage of LB compared to tissue biopsy is that LB is able to cover the genetic heterogeneity of disease (7–9). Cells carrying the disease-causing variant release their cfDNA into circulation, which can then be analyzed using LB. In contrast, these cells may be missed by tissue analysis, as it is commonly performed at one site at a singular time point. Accordingly, addition of LB to tissue biopsy has shown to add significant diagnostic value (1). Nevertheless, LB has not widely been implemented into routine clinical practice to date.
Aiming at offering LB to as many patients as possible, clinical laboratories must 1) cover the important therapy relevant variants, 2) use validated highly sensitive and accurate methods for detection of very low variant frequencies in plasma with well-defined cutoffs for variant reporting and quantification, and 3) offer both complementary and combined analysis of plasma and tissue biopsies for full flexibility and to maximize diagnostic yield.
The aim of this study is to develop aNGS-based assay that combines liquid and tissue biopsy (including tumor tissue and fibroblasts) to maximize flexibility for both patients and clinicians and to increase diagnostic yield at acceptable cost. We selected the most important therapy relevant variants and considered latest technological improvements for detection of very low frequency variants commonly present in plasma (3, 5, 10–14). We performed analytical validation of our workflows including definition of cutoffs for variant reporting and quantification, and describe a clinical case where LB was able to provide molecular diagnosis of overgrowth syndrome that could not be identified by tissue biopsy and other molecular analyses approaches.
The study was approved by the ethics commission of the Bavarian Medical Association (No. 17059) and is registered with the German registry for clinical trials (trial registration ID: DRKS00012890). All participants or their legal guardian provided informed written consent prior to blood and tissue specimen collection. For the case report, the legal guardian provided written consent for publication of pictures. The study was performed in accordance with the Declaration of Helsinki.
Skin fibroblast and plasma samples were obtained in parallel from a ten-year old girl with clinically diagnosed asymmetric overgrowth syndrome. Another plasma sample was obtained from one tumor patient and FFPE tissue samples were collected from a total of 12 tumor patients (Supplementary Table 1).
Information on reference materials, DNA extraction, kit design, library preparation and sequencing, karyotyping, microarray analysis, and whole-exome sequencing (WES) are provided in the Supplementary Methods.
Raw data (FASTQ.GZ format) was uploaded to the VARVIS® platform and aligned against the hg38 reference genome followed by variant calling using the bioinformatics pipeline VARFEED worker 1.5.1 with in silico validated standard settings. Within the LB workflow the Duplex consensus was built by extraction and processing of Duplex sequencing barcodes according to Schmitt et al. (14) and the manufacturer's "analysis guidelines, version 1" (15). A minimum of two reads were used to construct a strand-specific consensus read. Strand-specific consensus reads were then combined to create a final consensus read. No consensus was built within the tissue analysis workflow.
To allow 95% confidence for detection of variants present with low VAFs, we aimed to analyze at least 60 low frequency variants with both LB and tissue analysis (16–18). The LB analysis was validated using the Seraseq® ctDNA Complete™ Reference Materials (LGC seracare) with 0%, 0.05%, 0.1%, 0.5%, 1% and 5% VAF. Performance was evaluated based on the single well-characterized genomic background (GM24385) (19) including 59 SNVs and 1 InDel, and spike-in variants present in the respective VAF including 8 SNVs and 7 InDels. The analysis of the wild type (WT) once and each of the spike-in reference materials in duplicates covered a total of 660 germline variants (649 SNVs, 11 InDels) and 90 spike-in variants present in VAFs above the LOB (48 SNVs, 42 InDels).
Validation of the tissue analysis was performed using the Quantitative Multiplex Reference Standard FFPE (Horizon), ten well-characterized clinical samples and the Ashkenazim Son FFPE Reference Standard NA24385 (SensID) as WT control. Further, in silico dilutions of three of the well-characterized clinical samples in the Ashkenazim Son FFPE Reference Standard NA24385 (SensID) were generated with 8% to 16% VAF of spike-in variants. Performance was evaluated based on a total of 573 somatic variants (546 SNVs, 27 InDels).
To limit the number of false positive (FP) variants, we established the LOB at 0.25% VAF based on the background noise at genomic positions expected to be WT in all samples as cutoff for variant calling. With a target region of 102 kb the number of positions in each of the validation samples for both LB and tissue analysis exceeds the required 60 WT positions for determination of the LOB (16). Further, we established that variants are expected to be true positives (TP), when they are present in at least eight consensus reads.
For determination of the LOD we investigated the detection rate of 90 spike-in variants from 0.5% to 5% VAF for LB analysis and of 573 variants with 8% to 16% VAF for tissue analysis. The number of analyzed spike-in variants exceeds the required 60 variants for determination of the LOD with 95% confidence (16, 17). Since we detected 27/30 variants with 0.5% VAF the LOD could be established at 0.5% VAF with 90% confidence. All missed variants were InDels, therefore the performance for InDel detection is expected to be significantly lower than for SNVs.
Variants were detected above the LOB of the respective analysis (LB: 0.25%, tissue: 5%) as cutoff for true-positive variant detection. The variants detected in processed samples were compared to the intersected trusted regions of the GIAB version 3.3.2 (19) reference data set using vcfeval (Real Time Genomics) (20). Subsequently, sensitivity and PPV were separately assessed for SNVs and InDels. The number of InDels analyzed with both LB and tissue analysis is very low and thus does not yield accurate information. However, as described above, the performance for InDel detection is expected to be significantly lower than for SNVs.
In addition to sensitivity and PPV, we also assessed trueness, precision and the total error of 30 spike-in variants with 0.5% VAF, 1% VAF and 5% VAF, respectively, to establish the LOQ (Supplementary Methods). Guidelines recommend analyzing 40 variants of the target VAF to establish the LOQ (16), however with 30 variants at the respective VAF we were able to estimate the total error, required for determination of the LOQ.
Our aim was to develop a cost-efficient workflow for both LB and tissue analysis which covers the most important actionable genes and variants in solid tumors and mosaic diseases based on a custom hybrid-capture panel for targeted sequencing on the basis of European Society for Medical Oncology (ESMO) guidelines (3, 5, 11–13) and current clinical trials (21–27) (i.e. 30 genes and hotspots of three additional genes, Supplementary Methods, Supplementary Table 2). Our final panel targets in total 102 kb, and includes 500 exons of canonical and 33 exons of non-canonical transcripts. According to a comprehensive catalogue of hotspot variants identified in ~25.000 tumor samples (28) our assay interrogates 370 known variants in the target region (Supplementary Table 4).
To allow maximal flexibility we aimed at enabling plasma and tissue sample processing either in parallel or independently (Figure 1). After genomic DNA (gDNA) extraction from tissue followed by fragmentation and cfDNA extraction from plasma, both sample types are processed together beginning with library preparation, including the ligation of strand-aware barcodes (Duplex tags) for highly sensitive variant detection (29). While the combined workflow ensures a similar turnaround time, the additional fragmentation required for tissue analysis results in a slightly shorter turnaround time for LB analysis. To achieve ~90 Mio and ~4 Mio raw reads of plasma and tissue samples, respectively, target capture is performed in separate pre-pools for plasma and tissue samples followed by pooling in the appropriate ratio for sequencing. Depending on the number of plasma and tissue samples, various sequencing options are possible (Figure 1).
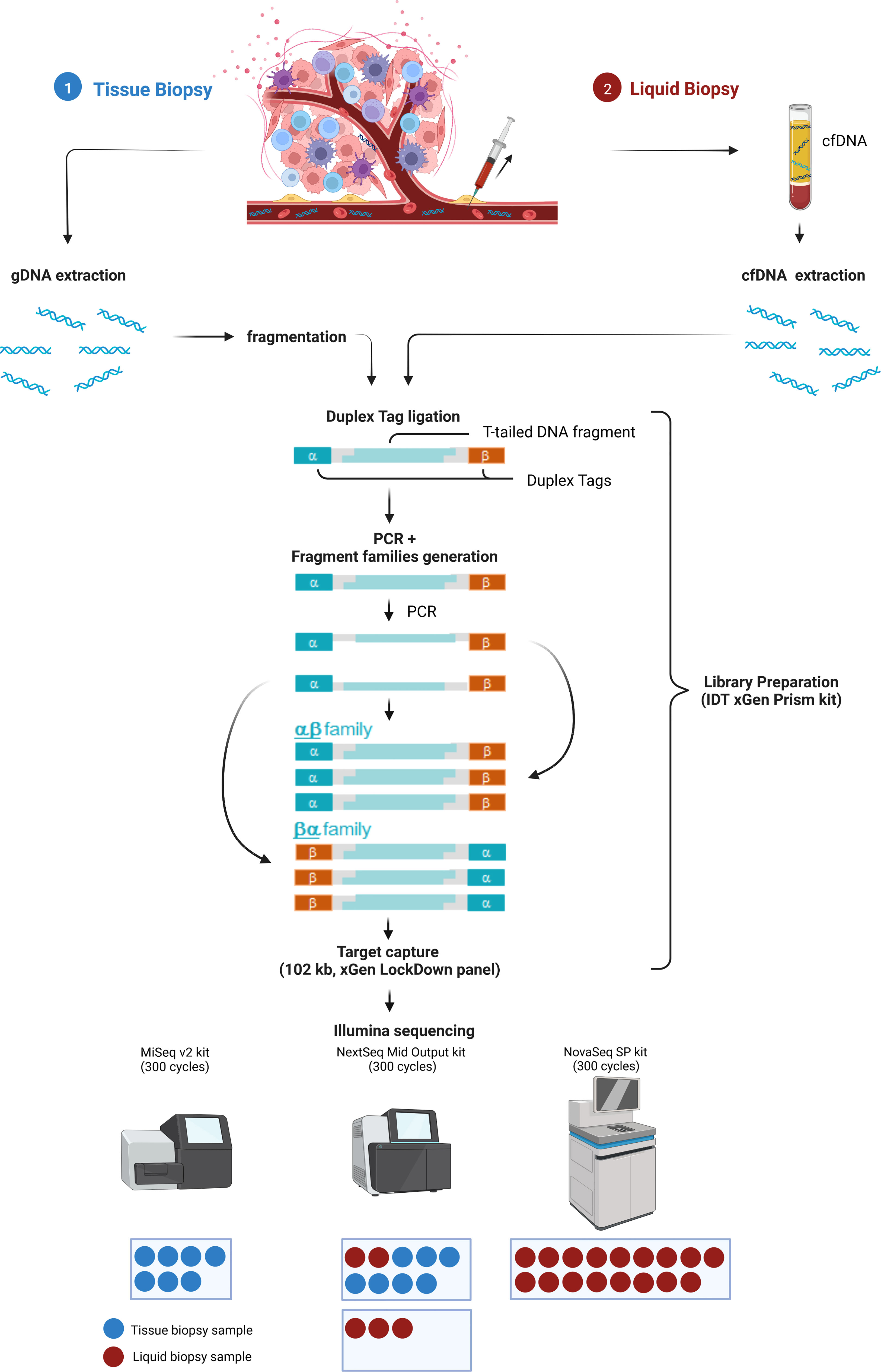
Figure 1 Combined analysis workflow for plasma and tissue samples. Created with BioRender.com.
The Duplex tags added during library preparation allow the bioinformatics identification of all amplified DNA fragments originating from a single strand of the original DNA molecule (29). PCR amplified fragments originating from the forward strand are assigned to the αβ fragment family, whereas PCR amplicons from the complementary strand are assigned to the βα fragment family (Figure 1). Each fragment family builds a single strand consensus to filter out false positives including sequencing errors and late PCR artifacts (Supplementary Figure 1). For LB analysis both strands are combined to build the Duplex consensus (29), which enables the detection of variants frequently present in very low VAFs in plasma by reducing the background noise (30). In our validation samples with a median ~89.8 million total reads were collapsed to a median of ~3.6 million Duplex consensus reads, leading to ~95.9% consensus reduction (Figure 2A; Supplementary Table 5). In contrast, for tissue samples a median of ~5.4 million total reads were achieved, for which no consensus was built (Figure 2B; Supplementary Table 5).
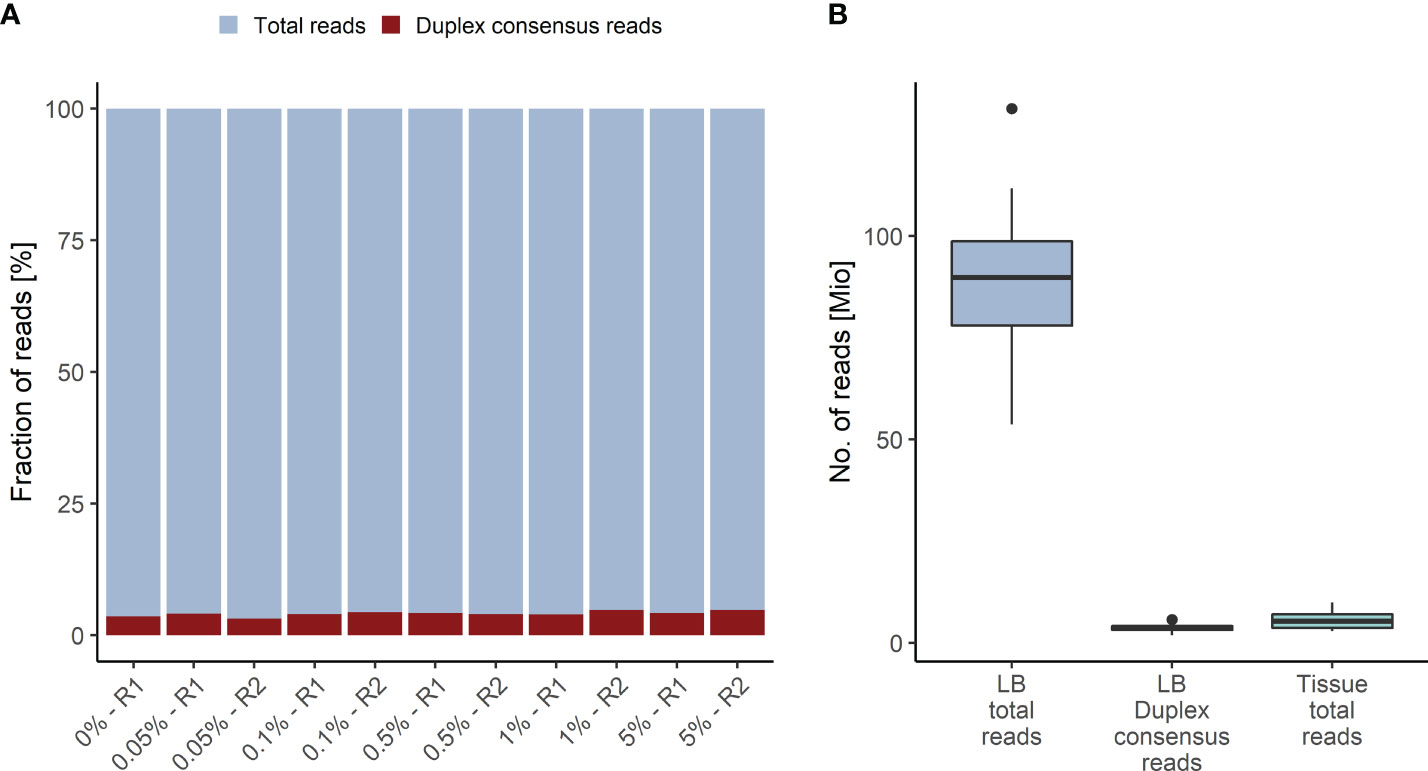
Figure 2 Duplex Consensus building. (A) Duplex consensus reads (red) as fraction of total reads (blue). (B) Total reads and Duplex consensus of plasma samples vs. total reads of tissue samples. R1, replicate 1; R2, replicate 2.
With our approach, we achieved a mean effective coverage of 1,735x and 1,496x for plasma and tissue samples, respectively. For plasma samples in median ~98.6% of regions were covered with >250x, and for tissue samples ~100% of regions were covered with >100x (Supplementary Table 6).
We assessed the detection rate in plasma samples using cfDNA isolated from Seraseq® ctDNA Complete™ Reference Materials (VAF 0%-5%) (Seracare). The Seraseq® ctDNA Complete™ Reference Material includes 40 clinically relevant variants across 28 genes at 0.5%, 1% and 5% and a WT control sample. Of these variants 15 (8 SNVs and 7 InDels) are located in the kit target region. Only variants with a VAF above the LOB of 0.25% for the LB Duplex workflow that are present in at least eight consensus reads were called. Using these parameters, we observed a SNV detection rate of 100% at 0.5%, 1.0% and 5.0% VAF and an InDel detection rate of 79% at 0.5% VAF (average of duplicate measurement), and of 100% at 1% and 5% VAF, with highly similar detection in forward and reverse reads (Figure 3; Supplementary Table 7). Notably, neither SNVs nor InDels contained in the reference material were detected in the WT sample.
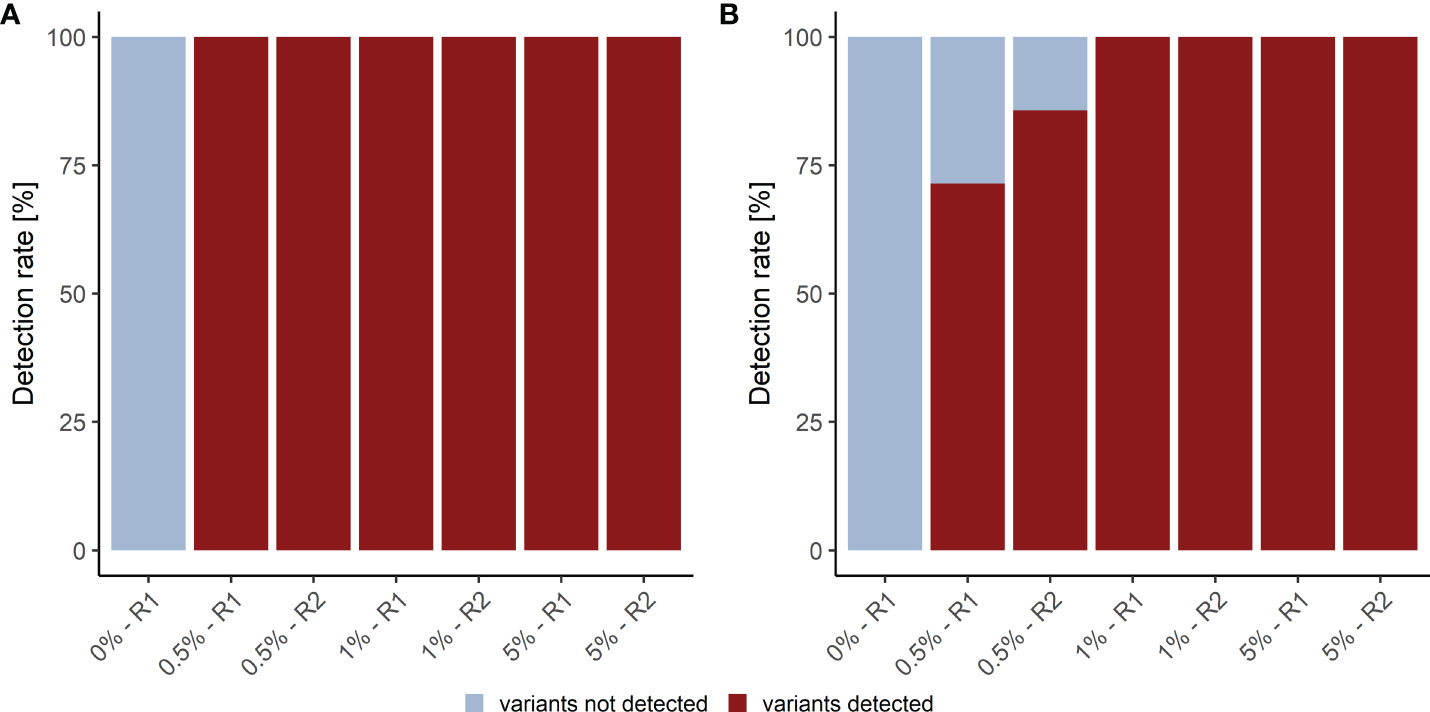
Figure 3 Detection rate of plasma analysis. (A) SNV and (B) InDel detection rate in plasma reference samples. R1, replicate 1; R2, replicate 2.
We further analyzed the detection rate in tissue samples using gDNA isolated from Quantitative Multiplex Reference Standard FFPE (Horizon), ten well-characterized clinical samples and the Ashkenazim Son FFPE Reference Standard NA24385 (SensID) as WT control. 7/11 clinically relevant variants of the Quantitative Multiplex Reference Standard FFPE (Horizon) and 11/11 clinically relevant variants in ten clinical samples were covered in the kit target region and above the LOB of 5% for the tissue analysis workflow. For tissue samples, a detection rate of 100% was observed since all 18 variants present in different reference materials with VAFs above the LOB of 5% were detected. As expected, no clinically relevant variants were detected in the Ashkenazim Son FFPE Reference Standard NA24385 (SensID) WT control (Supplementary Table 7).
To determine the sensitivity of LB analysis, we considered TP in all reference materials with 0.5%, 1.0% and 5.0% VAF. In total, 48/48 SNVs and 39/42 InDels were detected, resulting in 100% and 92.9% sensitivity for SNVs and InDels, respectively (Supplementary Table 7). In addition to sensitivity, we also determined the PPV considering TPs and FPs. Thereby, the PPV of the LB analysis was established to be 63.2% (48/76) for SNVs and 84.8% (39/46) for InDels (Table 1; Supplementary Table 7). One possible reason for the high number of FPs is the poor characterization of the germline background of the reference materials (19). Consequently, it is difficult to assess whether all FPs are indeed FPs or possibly TPs. To limit this issue, we further focused on the 2.5 kb of our target region overlapping with hotspot regions identified in 24.592 tumors reported by Chang et al. (28) (Supplementary Table 9). A total of 48/48 SNVs and 33/36 InDels with VAFs from 0.5%-5% were detected as TP variants, resulting in 100% and 91.7% sensitivity, respectively. Further, a PPV of 92.3% (48/52) for SNVs and of 100% (33/33) for InDels could be established in these hotspot regions (Table 1).

Table 1 Sensitivity and PPV of LB and tissue analyses.
To evaluate sensitivity and PPV of the tissue analysis, in silico dilutions of 10% and 20% of three internal reference materials (T-CRC-04, T-EC-01, and T-NF-01), previously characterized by WES, were generated in the Ashkenazim Son FFPE Reference Standard NA24385 (SensID) as well characterized background (19). Depending on the previously determined VAF of individual variants, present in the three internal reference materials, the generated in silico dilutions were expected to contain spike-in variants in the range from 8% to 16% VAF. For SNVs 99.4% (512/518) sensitivity and 100% (515/515) PPV was achieved. For InDels, 100% (22/22) sensitivity and 88% (22/25) PPV was obtained (Table 1).
To enable disease monitoring in cancer patients using LB, we established the LOQ for the LB Duplex sequencing workflow. The LOQ represents the cutoff above which VAFs can be accurately quantified based on acceptable trueness (>90%) and precision (>80%) (Supplementary Methods).
To estimate trueness, representing the closeness of agreement between measured and reference VAF, we calculated the bias between actual VAFs (confirmed by digital droplet PCR) and measured VAFs of variants present in each plasma reference material. Variants with ~0.5% VAF were determined with 85.0% trueness, variants with ~1% VAF with trueness of 92.5%, and variants with ~5% VAF with 99.9% trueness (Supplementary Figure 2A; Supplementary Table 10). Therefore, the goal for trueness was achieved with VAFs from 1%.
We further established precision in terms of repeatability based on each variant present in reference materials. Therefore, we calculated the pooled standard deviation over all variants between the two replicates of each reference material. For variants with ~0.5% VAF, ~1% and ~5% VAF repeatability was determined to be 34.4%, 65.2%, and 91.9%, respectively (Supplementary Figure 2B, Supplementary Table 10). Therefore, the goal for precision was achieved with VAFs from 5%.
Based on these results we were able to establish the LOQ as cutoff for VAF quantification at a VAF of 5% with a total error of 16.2%. These results indicate that variants determined with VAFs ≥5% can be reliably quantified with our LB Duplex sequencing workflow and therefore are informative for disease monitoring. Since tissue analysis cannot be used for disease monitoring, we only validated the tissue analysis with qualitative rather than quantitative intent. However, measured VAFs in two replicates for most of the variants deviated only 0.2% to 7.9%. Only for the PIK3CA (NM_006218.4) p.E545K, a VAF of ~34.2% higher than the true VAF of 8.3% was observed.
To test the diagnostic value of our approach, we applied plasma and tissue analysis with our combined workflow in samples of a ten-year old girl with clinically diagnosed asymmetric overgrowth syndrome including arteriovenous malformations in the right arm and right leg, but without molecular diagnosis that would support personalized treatment (Figure 4). For diagnosis of this phenotype standard procedure is a skin biopsy to identify mosaicism of pathogenic variants in associated genes (31). Since skin fibroblasts do not necessarily harbor the disease-causing variant in patients with vascular phenotypes, but the affected vascular tissue releases DNA into circulation, LB can be informative as well for the molecular characterization of disease.
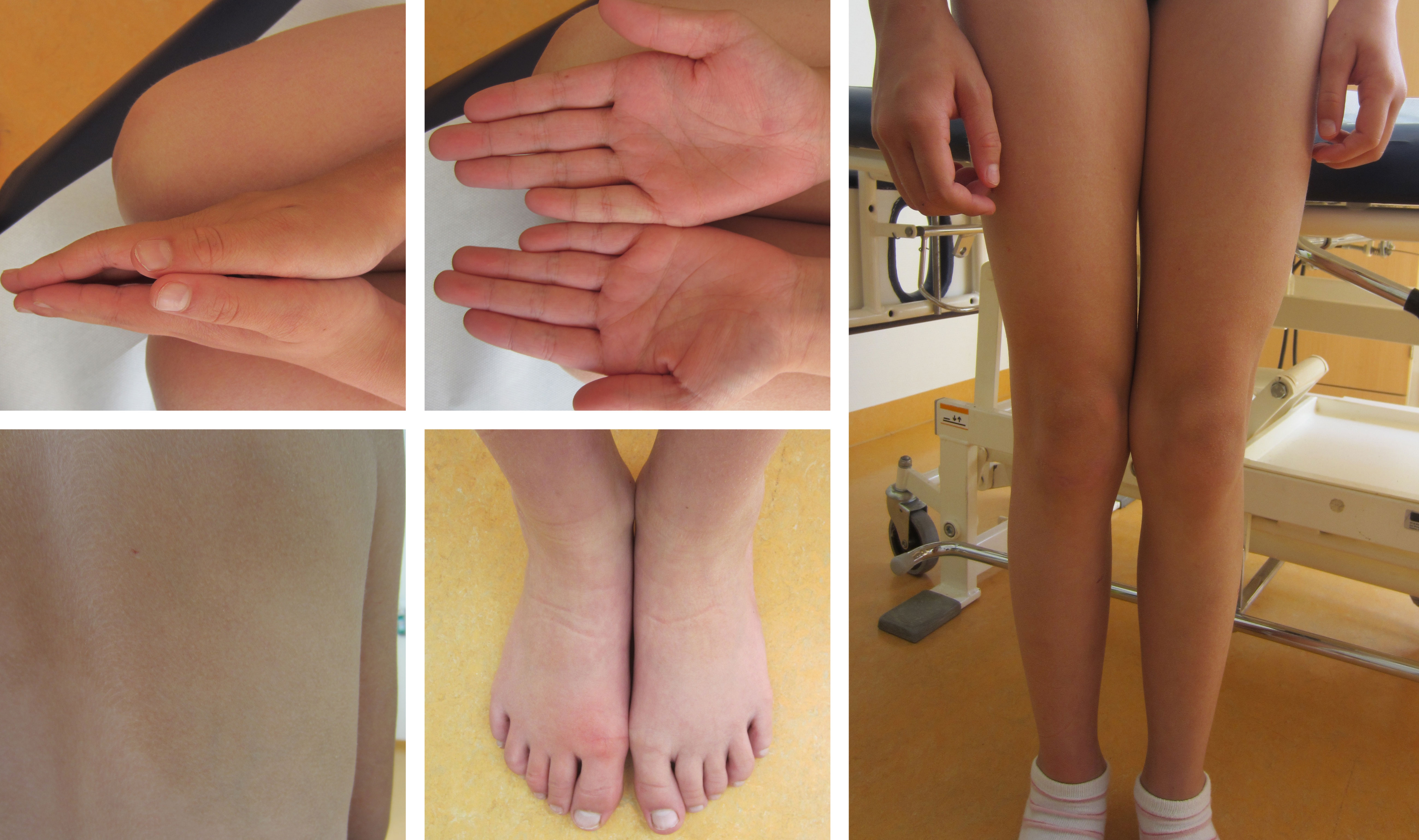
Figure 4 Clinically confirmed overgrowth syndrome in a 10-year-old girl to be analyzed for molecular clarification. Marked asymmetry of hands and legs. The feet have a difference in circumference. On the back, there is a large asymmetric reddish-brown area of skin, with temperature-depended hyperpigmentation, suggestive of capillary malformations in the area.
Initially, standard molecular diagnostics were performed including karyotyping from heparin blood, which provided inconspicuous results and microarray analysis from EDTA blood that identified a duplication of 15q13.3 not related to the phenotype. The following WES from skin fibroblasts did not identify any disease-causing variant. Using our tissue analysis workflow including 17 genes associated with overgrowth syndrome, also no pathogenic variant could be identified, whereas LB analysis identified KRAS (NM_004985.5) c.35G>A p.(Gly12Asp) in plasma with a measured VAF of 1.01% (Table 2). The KRAS c.35G>A, p.(Gly12Asp) variant leads to constitutive overactivation and increased signal transduction into downstream pathways and is associated with overgrowth including various types of congenital nevi and vascular malformations (so-called mosaic RASopathies) (32, 33). Our result is in line with a previous study identifying this variant as the molecular cause of asymmetric overgrowth syndrome and vascular malformation (33). Because all available cfDNA from the patient was used as input for LB analysis, the detected variant could not be verified using an orthogonal method such as droplet digital PCR (ddPCR). However, the detection of this variant in accordance with the previously validated performance criteria and the perfect genotype-phenotype correlation in combination with previous findings describing the respective variant as the cause of asymmetric overgrowth syndrome and vascular malformation suggest that this variant is a true positive. Taken together, detection of KRAS c.35G>A, p.(Gly12Asp) variant using LB that could not be identified with the tissue workflow, molecularly explained the clinically diagnosed overgrowth syndrome in our patient, hereby providing personalized treatment options including MEK inhibitors.

Table 2 Molecular analysis of clinically confirmed overgrowth syndrome.
LB is a promising tool in precision medicine. It is fast, non-invasive and represents disease heterogeneity at any desired time point (34–36). Several institutions including the National Comprehensive Cancer Network (NCCN), the College of American Pathologists (CAP), the International Association for the Study of Lung Cancer (IASLC), the Association for Molecular Pathology (AMP), the ESMO and the ESMO Precision Medicine Working Group (PMWG) have acknowledged the advantage of LB for patient management (3–6, 37). On national level, LB testing has already been included in clinical guidelines, and health insurances have started to reimburse LB analysis. Besides its high costs, a major challenge of LB for its usage in clinical practice, however, is the reliable detection with defined cutoffs for variant reporting and quantification of low variant allele frequencies in plasma that are especially common in patients with non-metastatic cancer and mosaic diseases (30). Here we developed a flexible workflow for parallel analysis of plasma and tissue samples to maximize diagnostic yield and provide LOB and LOQ as clinically relevant VAF reporting and quantification cutoffs.
A major challenge for implementation of highly sensitive LB analysis into clinical practice are the generally high costs due to the high sequencing coverage required. In our workflow, one to two LB samples can be pooled with seven tissue samples on an Illumina NextSeq flowcell (Mid output), at reasonable cost. Notably, combining 17 plasma samples on a NovaSeq SP flowcell (i.e. the maximal number of LB samples that can be pooled on this flowcell) reduces costs per sample significantly, which is in the range of targeted LB hotspot analysis using digital droplet PCR and tissue sample analysis. Notably, adding the option of LB analysis to clinical labs not only results in higher flexibility but also reduces costs for tissue sample analyses. Taken together, to the best of our knowledge this is the most cost-effective approach for a mid-size and highly sensitive NGS LB panel.
Using our workflow, LB samples can be processed in parallel to tissue samples similar to the MSI-ACCESS assay developed by the Memorial Sloan Kettering Cancer Center (MSK), processing LB samples from cancer patients in parallel to white blood cells (WBC) (38). This assay is also based on LB Duplex sequencing and achieved a slightly lower sensitivity for both hotspot and de novo variants, but with a higher PPV. Comparing performance characteristics of our workflow to five other commonly used LB SNV detection assays tested with the same reference material in Weber et al. (39), our assay was the only one with all SNVs detected at 0.5% VAF and none in the WT control, indicating a low number of false positives. Further, our 102 kb target region encompassed mainly the whole coding sequence of targeted genes, whereas the five investigated assays primarily focused on hotspot regions. The only assay targeting the whole coding sequence was the Oncomine Lung cfDNA assay (Thermo Fisher), encompassing 12 genes associated with lung cancer, but thereby restricting its application to a smaller patient population. This assay was also the best performing one, showing similar sensitivity and PPV values to our approach. However, this assay relies on the Ion Torrent platform, whereas our assay uses the Illumina platform, which enables broader application as it is more widespread. Further, our approach is the only one combining the analysis of LB and tissue samples for the detection of both tumor and mosaic diseases, making it an easily implementable workflow in clinical practice.
Rather than only including actionable variants for mosaic disease, we show with our case report a clinical proof-of-concept that LB can be extended from cancers to all heterogeneous diseases such as mosaic diseases, including asymmetric overgrowth syndromes (e.g. Proteus syndrome, Klippel-Trennaunay syndrome and PIK3CA-related overgrowth spectrum, PROS, as well as many others). Detection of somatic variants in cancer and PROS can guide personalized treatment including consideration of inhibitors of the PI3K/AKT/mTOR signaling pathway. Notably, simple blood draws required for LB are much more convenient for patients than tissue biopsies, especially for children, which are frequently affected by mosaic diseases.
However, there are limitations that need to be considered. Accurate detection of variants <0.5% VAF is challenging. This finding is in line with a recent study testing five leading commercial ctDNA assays, which show generally high performance for VAFs ≥0.5% (40). Accordingly, the main application is molecular stratification and profiling of tumor evolution in patients with advanced cancers, where variants are commonly detected with VAFs ranging from 1% to 10% (40). Another limitation represents the small sample size for the analytical validation of the LB analysis. However, there are only a limited number of reference materials with well-defined low-frequency variants available. Using the Seraseq® ctDNA Complete™ Reference Materials with different VAFs of spike-in enabled accurate validation of our approach based on a total of 90 spike-in variants and 660 germline variants. We further showed a clinical proof-of-concept for application of LB analysis for genotyping of mosaic disease in only one case. To show clinical validity in addition to analytical validity of LB analysis in patients with mosaic disease LB analysis in parallel to clinical evaluation in more patients would be required. Furthermore, the LB analysis was initially analytically validated by establishing the LOB as cutoff for variant detection and the LOQ as cutoff for variant quantification. However, it was not clinically validated for residual disease detection or treatment monitoring in cancer patients. To apply LB analysis also for residual disease detection based on the LOB or treatment monitoring by tracking changes in VAF above the LOQ, its prognostic value needs to be evaluated in an independent study using follow-up samples from patients with known somatic variants. Nevertheless, as described in previous studies, it is likely that the presence of post-surgery ctDNA is likely to indicate residual disease, and changes in VAFs throughout treatment are likely to indicate response or resistance (41–43). To determine clinical utility of LB analysis for treatment monitoring prospective studies would be required guiding treatment decisions based on presence of post-surgery ctDNA or changes in VAFs.
In conclusion, LB is capable to detect the complete mutational profile of both the primary tumor and metastatic lesions. LB Duplex sequencing pushes the boundaries for detection of low frequency variants in plasma with NGS based analysis. Our broad Duplex sequencing panel enables highly sensitive detection of therapy relevant variants in tumor and mosaic diseases. We were able to identify the KRAS c.35G>A, p.(Gly12Asp) variant in plasma as the molecular cause of the clinically confirmed overgrowth syndrome in a ten-year old girl, which could not be detected in the analysis of skin fibroblasts using our tissue workflow, which may be due to heterogeneity not depicted in the resected specimen. The identification of the KRAS variant may lead to novel therapy options, highlighting the diagnostic value of LB analysis for heterogeneous diseases in clinical practice. In summary, our workflow that easily combines tissue and LB analysis has the potential to increase the diagnostic yield, which is in line with previous results, identifying an increase of diagnostic yield by ~15% due to introduction of LB as tissue analysis alternative (1).
The sequence data have been deposited at the European Genome-phenome Archive (EGA) under accession number EGAS00001006805. The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by ethics commission of the Bavarian Medical Association. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
TN, VS-L, and JP researched the literature to identify cancer and PROS associated genes. FS established the target region of the sequencing panel. AH developed the experimental procedures, and performed all experiments. AH, AL, RE, BL, and JP analyzed and interpreted the data. AH, RE, and BL performed statistical analysis. AH and JP designed the study. JP supervised the work. EH-F provided financial and technical resources to enable conduction of the study. AH and JP wrote the manuscript. All authors contributed to the article and approved the submitted version.
We would like to thank the individuals providing their informed consent to use their DNA samples for research purposes.
Authors RE and BL were employed by Limbus Medical Technologies GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1014592/full#supplementary-material
1. Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol (2019) 5:173–80. doi: 10.1001/jamaoncol.2018.4305
2. Malapelle U, Tiseo M, Vivancos A, Kapp J, Serrano MJ, Tiemann M. Liquid biopsy for biomarker testing in non-small cell lung cancer: A European perspective. JMP (2021) 2:255–73. doi: 10.3390/jmp2030022
3. Cardoso F, Paluch-Shimon S, Senkus E, Curigliano G, Aapro MS, André F, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol (2020) 31:1623–49. doi: 10.1016/j.annonc.2020.09.010
4. Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO precision medicine working group. Ann Oncol (2020) 31:1491–505. doi: 10.1016/j.annonc.2020.07.014
5. Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2020) 21(Suppl 5):v116–9. doi: 10.1093/annonc/mdq189
6. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman JR, Bharat A, et al. Non-small cell lung cancer, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2022) 20(5):497–530. doi: 10.6004/jnccn.2022.0025
7. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature (2013) 501:355–64. doi: 10.1038/nature12627
8. Williams Parsons D, Jones S, Zhang X, Cheng-Ho Lin J, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi: 10.1126/science.1164382
9. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
10. Postmus PE, Kerr KM, Oudkerk MM, Senan S, Waller DA, Vansteenkiste J, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2017) 28(suppl_4):1–21. doi: 10.1093/annonc/mdx222
11. van Cutsem E, Cervantes A, Adam R, Sobrero A, van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
12. Michielin O, van Akkooi AC, Ascierto PA, Dummer R, Keilholz U. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol (2019) 30:1884–901. doi: 10.1093/annonc/mdz411
13. Colombo N, Sessa C, Du Bois A, Ledermann J, McCluggage WG, McNeish I, et al. ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease†. Ann Oncol (2019) 30:672–705. doi: 10.1093/annonc/mdz062
14. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U.S.A. (2012) 109:14508–13. doi: 10.1073/pnas.1208715109
15. Integrated DNA Technologies. Analysis guidelines: xGen prism DNA library prep kit - processing sequence data with unique molecular identifiers (UMIs). Version 2 (2020). Doc ID:RUO22-0723_001 04/22.
16. NCCLS. Protocols for determination of limits of detection and limits of QuantitationGuideline: Approved guideline. (Pennsylvania, USA: NCCLS document EP17-A) (2004). p. 52.
17. Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the association for molecular pathology and college of American pathologists. J Mol Diagn (2017) 19:341–65. doi: 10.1016/j.jmoldx.2017.01.011
18. Godsey JH, Silvestro A, Barrett JC, Bramlett K, Chudova D, Deras I, et al. Generic protocols for the analytical validation of next-generation sequencing-based ctDNA assays: A joint consensus recommendation of the BloodPAC's analytical variables working group. Clin Chem (2020) 66:1156–66. doi: 10.1093/clinchem/hvaa164
19. Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data (2016) 3:160025. doi: 10.1038/sdata.2016.25
20. Cleary JG, Braithwaite R, Gaastra K, Hilbush BS, Inglis S, Irvine SA, et al. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv (2015), 23754. doi: 10.1101/023754
21. Zhang X, Chang A. Somatic mutations of the epidermal growth factor receptor and non-small-cell lung cancer. J Med Genet (2007) 44:166–72. doi: 10.1136/jmg.2006.046102
22. Lang SM, Rachow T. Molekulare therapeutische stratifizierung des nichtkleinzelligen lungenkarzinoms. Wien klin Mag (2020) 23:288–96. doi: 10.1007/s00740-020-00371-z
23. Ma CX, Bose R, Gao F, Freedman RA, Pegram MD, Blackwell K, et al. Phase II trial of neratinib for HER2 mutated, non-amplified metastatic breast cancer (HER2 mut MBC). JCO (2016) 34:516. doi: 10.1200/JCO.2016.34.15_suppl.516
24. Kalinsky K, Hong F, McCourt CK, Sachdev JC, Mitchell EP, Zwiebel JA, et al. Effect of capivasertib in patients with an AKT1 E17K-mutated tumor: NCI-MATCH subprotocol EAY131-y nonrandomized trial. JAMA Oncol (2021) 7:271–8. doi: 10.1001/jamaoncol.2020.6741
25. Smyth LM, Tamura K, Oliveira M, Ciruelos EM, Mayer IA, Sablin M-P, et al. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1E17K-mutant, ER-positive metastatic breast cancer. Clin Cancer Res (2020) 26:3947–57. doi: 10.1158/1078-0432.CCR-19-3953
26. Dustin D, Gu G, Fuqua SA. ESR1 mutations in breast cancer. Cancer (2019) 125:3714–28. doi: 10.1002/cncr.32345
27. Breast cancer now (2022). Available at: https://breastcancernow.org/information-support/facing-breast-cancer/going-through-breast-cancer-treatment/hormone-therapy/aromatase-inhibitors-anastrozole-exemestane-letrozole.
28. Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MT, Jonsson P, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discovery (2018) 8:174–83. doi: 10.1158/2159-8290.CD-17-0321
29. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet (2018) 19:269–85. doi: 10.1038/nrg.2017.117
30. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6:224ra24. doi: 10.1126/scitranslmed.3007094
31. Mirzaa G, John M Graham JR, Keppler-Noreuil K. PIK3CA-related overgrowth spectrum. Univ Washington Seattle (2021).
32. Goss JA, Huang AY, Smith E, Konczyk DJ, Smits PJ, Sudduth CL, et al. Somatic mutations in intracranial arteriovenous malformations. PloS One (2019) 14:e0226852. doi: 10.1371/journal.pone.0226852
33. Schmidt VF, Wieland I, Wohlgemuth WA, Ricke J, Wildgruber M, Zenker M. Mosaic RASopathy due to KRAS variant G12D with segmental overgrowth and associated peripheral vascular malformations. Am J Med Genet A (2021) 185:3122–8. doi: 10.1002/ajmg.a.62386
34. Heitzer E, Haque IS, Roberts CE, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet (2019) 20:71–88. doi: 10.1038/s41576-018-0071-5
35. de Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol (2014) 25:1729–35. doi: 10.1093/annonc/mdu239
36. Murtaza M, Dawson S-J, Pogrebniak K, Rueda OM, Provenzano E, Grant J, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun (2015) 6:8760. doi: 10.1038/ncomms9760
37. Pascual J, Attard G, Bidard F-C, Curigliano G, de Mattos-Arruda L, Diehn M, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO precision medicine working group. Ann Oncol (2022) 0:750–68. doi: 10.1016/j.annonc.2022.05.520
38. Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun (2021) 12:3770. doi: 10.1038/s41467-021-24109-5
39. Weber S, Spiegl B, Perakis SO, Ulz CM, Abuja PM, Kashofer K, et al. Technical evaluation of commercial mutation analysis platforms and reference materials for liquid biopsy profiling. Cancers (Basel) (2020) 12:1–16. doi: 10.3390/cancers12061588
40. Deveson IW, Gong B, Lai K, LoCoco JS, Richmond TA, Schageman J, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol (2021) 39:1115–28. doi: 10.1038/s41587-021-00857-z
41. Zhou J, Wang C, Lin G, Xiao Y, Jia W, Xiao G, et al. Serial circulating tumor DNA in predicting and monitoring the effect of neoadjuvant chemoradiotherapy in patients with rectal cancer: A prospective multicenter study. Clin Cancer Res (2021) 27:301–10. doi: 10.1158/1078-0432.CCR-20-2299
42. Tie J, Cohen JD, Lo SN, Wang Y, Li L, Christie M, et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. Int J Cancer (2020) 148:1014–26. doi: 10.1002/ijc.33312
Keywords: liquid biopsy, tissue biopsy, circulating tumor DNA, analytical validation, Duplex sequencing, cancer, asymmetric overgrowth syndrome
Citation: Hallermayr A, Neuhann TM, Steinke-Lange V, Scharf F, Laner A, Ewald R, Liesfeld B, Holinski-Feder E and Pickl JMA (2022) Highly sensitive liquid biopsy Duplex sequencing complements tissue biopsy to enhance detection of clinically relevant genetic variants. Front. Oncol. 12:1014592. doi: 10.3389/fonc.2022.1014592
Received: 08 August 2022; Accepted: 14 November 2022;
Published: 27 December 2022.
Edited by:
Parvin Mehdipour, Tehran University of Medical Sciences, IranReviewed by:
Pankaj Kumar Ahluwalia, Augusta University, United StatesCopyright © 2022 Hallermayr, Neuhann, Steinke-Lange, Scharf, Laner, Ewald, Liesfeld, Holinski-Feder and Pickl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia M. A. Pickl, anVsaWEucm9taWMtcGlja2xAbWd6LW11ZW5jaGVuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.