 Sahar F. Bannoura
Sahar F. Bannoura Husain Yar Khan
Husain Yar Khan Asfar S. Azmi
Asfar S. Azmi- 1Cancer Biology Graduate Program, Wayne State University School of Medicine, Karmanos Cancer Institute, Detroit, MI, United States
- 2Department of Oncology, Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, MI, United States
KRAS mutations are among the most commonly occurring mutations in cancer. After being deemed undruggable for decades, KRAS G12C specific inhibitors showed that small molecule inhibitors can be developed against this notorious target. At the same time, there is still no agent that could target KRAS G12D which is the most common KRAS mutation and is found in the majority of KRAS-mutated pancreatic tumors. Nevertheless, significant progress is now being made in the G12D space with the development of several compounds that can bind to and inhibit KRAS G12D, most notably MRTX1133. Exciting advances in this field also include an immunotherapeutic approach that uses adoptive T-cell transfer to specifically target G12D in pancreatic cancer. In this mini-review, we discuss recent advances in KRAS G12D targeting and the potential for further clinical development of the various approaches.
Introduction
RAS genes (KRAS, HRAS, NRAS) are the most commonly mutated oncogenes in cancers, resulting in increased downstream signaling that drives incessant proliferation and tumorigenesis. Certain tumors are more dependent on KRAS mutation, especially pancreatic ductal adenocarcinoma (PDAC) which remains one of the most lethal cancers, with a 5-year survival rate of 11% in the United States (1). Advances in our understanding of PDAC molecular pathology and subtypes have not translated into significant improvements in patient outcomes (2). Genetic alterations in the oncogene KRAS, and the tumor suppressors CDKN2A, SMAD4, and TP53 are the most common mutational drivers in PDAC, in addition to various genes identified at low mutation frequency. This complex genetic landscape has created a tremendous challenge for therapeutic targeting.
KRAS mutations in PDAC
Mutations in KRAS, CDKN2A, TP53, and SMAD4 are the major genetic mutations that underly PDAC development (Figure 1A). The KRAS oncogene is the major oncogenic driver of PDAC (86 – 91%) (3), and is considered a master oncogenic regulator that drives cancer hallmarks including sustained proliferative signaling and evading growth suppression. KRAS mutations are found at the earliest stage of PDAC development in patient samples, and are required for the initiation and maintenance of PDAC in genetically engineered mouse models, suggesting that KRAS mutations are important for both PDAC initiation and progression (4–6). The most predominant KRAS mutation site in PDAC occurs at codon 12; most commonly G12D (45%), followed by G12V (35%), and G12R at 17% (Figure 1B) (3). Other mutations such as G12C and G12F occur at a lower frequency.
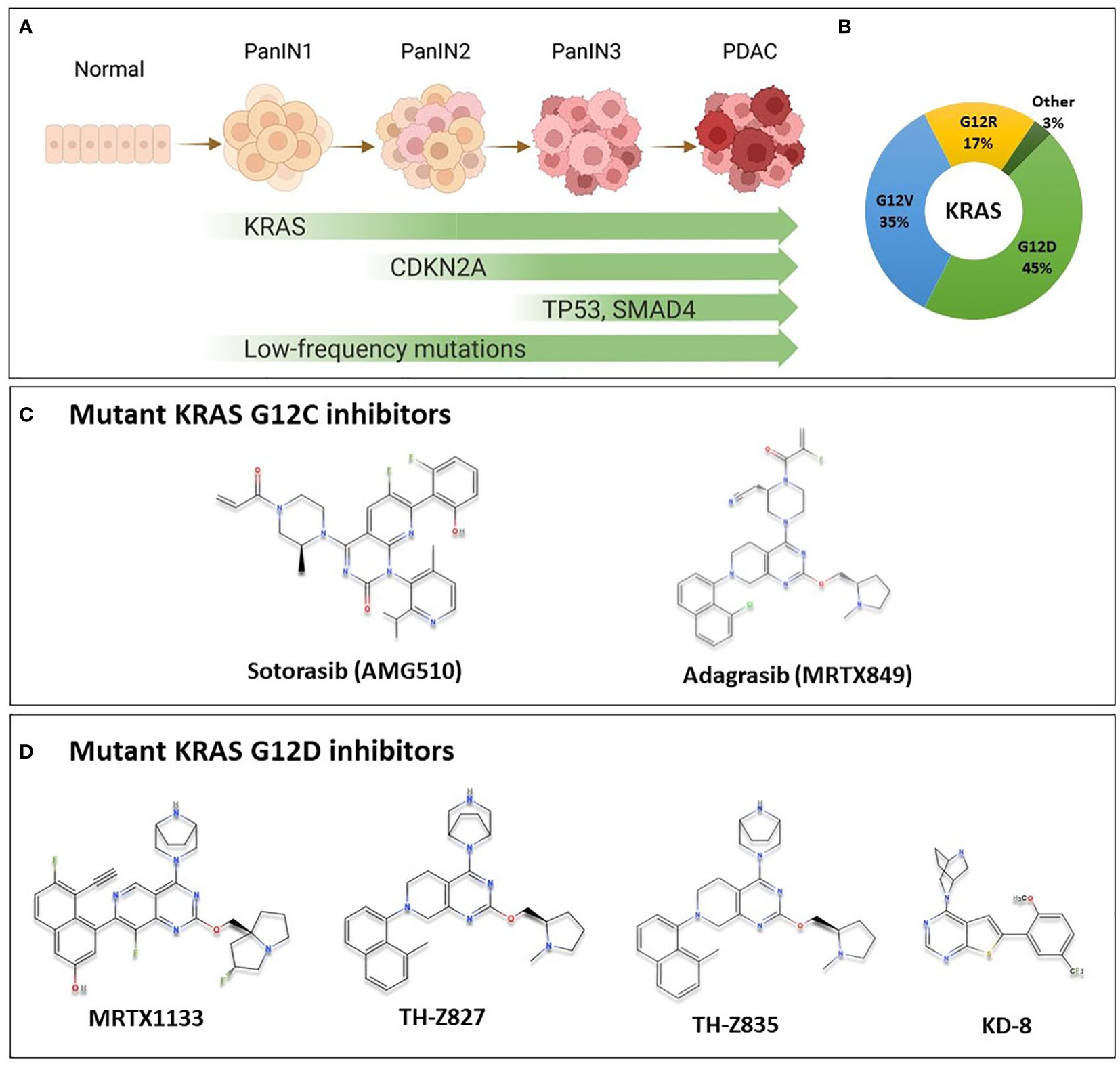
Figure 1 KRAS Oncogene in PDAC and the chemical structure of some of mutant KRAS small molecule inhibitors. (A) Multi-stage development model of KRAS-mutated PDAC. As cells acquire mutations in KRAS, CDKN2A, SMAD4 and TP53 in addition to less commonly mutated genes, the lesion progresses from low grade pancreatic intraepithelial neoplasia (PanIN1), through PanIN2, and high grade PanIN3 to become invasive carcinoma. (B) Most common KRAS mutations in PDAC are G12D (45%), G12V (35%), G12R (17%) representing important targets for PDAC patients. Other mutations such as G12C, G12A and others are less common (3%). (C) Sotorasib and adagrasib are mutant KRAS G12C inhibitors. They bind specifically to the mutant form by making covalent interactions with Cys12 in the switch-II pocket of mutant KRAS, locking it in the inactive state, and preventing downstream signaling. These agents have been investigated in clinical trials, and sotorasib is currently FDA approved for the treatment of KRAS G12C-mutated NSCLC. (D) Novel KRAS G12D inhibitors with available chemical structures, MRTX1133, TH-Z827, TH-Z835, and KD-8. These agents bind specifically and non-covalently to mutant KRAS G12D, thereby inhibiting proliferation in mutated cells. These agents are currently in preclinical development, and details on further clinical development are not currently known.
Mutant KRAS is notoriously difficult to target. It had been deemed undruggable until the recent success of inhibitors that target KRAS with a G12C mutation, AMG510 (sotorasib) and MRTX849 (adagrasib) (Figure 1C) (7). Sotorasib has been granted approval by the US Food and Drug Administration (FDA) for the treatment of certain patients with KRAS-mutated non-small cell lung cancer (NSCLC); and adagrasib has been given a breakthrough therapy designation by the FDA also for NSCLC (8). For PDAC patients, however, a mutation that replaces Glycine at codon 12 with Aspartic Acid (G12D) is the most common mutational event in KRAS, representing approximately 45% of KRAS mutations, as previously mentioned (3), and only a very modest proportion of patients would benefit from G12C targeted therapy. Nonetheless, since KRAS was found to be potentially druggable, multiple groups have aptly been working to develop inhibitors targeting the most common KRAS G12D mutation.
Overview of KRAS oncogene and targeting efforts
KRAS is a small GTPase protein that is encoded by the KRAS proto-oncogene (9). It is activated by extracellular growth signaling that is transmitted by growth receptors such as EGFR family (10). Growth signaling allows KRAS to exchange its GDP for GTP, thereby affecting conformational change and further transmission of downstream signals. KRAS is upstream of critical signaling pathways, such as RAF/MEK/ERK MAPK, and PI3K signaling pathways.
KRAS cycles between GDP and GTP with the activity of guanine exchange factors (GEFs) that catalyze the exchange of GDP for GTP, and GTPase activating proteins (GAPs) that enhance the intrinsic KRAS GTPase activity. Son of Sevenless 1 (SOS1) is the main GEF to act on KRAS, and together with GAPs, it is responsible for the regulation of the guanine exchange cycle, and maintenance of KRAS in an inactive state in the absence of proper growth signaling (11). Mutations in KRAS change the dynamics of the guanine exchange cycle, resulting in hyperactive KRAS and increased pools of GTP-bound KRAS. This leads to the constant activation of downstream signaling cascades such as MAPK and PI3K, and consequently incessant cellular proliferation.
KRAS is frequently mutated in pancreatic cancer, colorectal cancer (CRC), and lung cancer (3). Therefore, looking for ways to target KRAS, whether directly or indirectly, has garnered great attention from researchers (12). Directly targeting KRAS had been elusive for decades. KRAS lacks a suitable deep hydrophobic binding pocket to design small molecule inhibitors, other than the GTP/GDP binding pocket. However, GTP is highly abundant in the cell, thus precluding efficacious nucleotide competitive inhibitors. Additionally, indirect targeting of KRAS through upstream or downstream regulators or effectors was either ineffective or caused major toxicities (13). The major breakthrough in the field came when undruggable KRAS was finally drugged with the discovery of the first KRAS G12C inhibitors.
The Discovery of KRAS G12C inhibitors and its targetable pocket
Pioneering work by Shokat and colleagues paved the way for the clinical development of mutant KRAS G12C inhibitors (14). They were able to identify a druggable pocket termed the switch II pocket, which contains a reactive cysteine as a result of the G12C mutation in mutant KRAS (14). The co-crystal structure of hit compound and KRAS showed the compound binding in a pocket that lies beneath the switch II region of KRAS protein (14). Within the switch II allosteric pocket Cys12 is present, which contains a nucleophilic thiol group, making it a prime target for covalent drugs (15). It also affords the inhibitor high specificity over the other RAS isoforms (HRAS and NRAS) as well as wild-type KRAS, thereby reducing off-target effects and toxicities.
The early G12C inhibitor, compound 12, is a covalent inhibitor that binds in the switch II pocket of KRAS G12C in its GDP-bound form. Several other compounds were subsequently discovered and reported to have activity against cancer cells with KRAS G12C mutations (16, 17). An early compound, ARS-1620, was shown to inhibit tumor growth in cell-derived and patient-derived xenograft models (18). These efforts provided proof of concept for targeting KRAS G12C, which culminated in the development of the clinical agents sotorasib and adagrasib (19–23). Based on the safety, tolerability, and efficacy results of a phase 1/2 study of sotorasib (CodeBreaK 100, NCT03600883), it has received FDA approval for advanced NSCLC with KRAS G12C mutations. Results from the trial demonstrated an 80.6% disease control rate, with four patients achieving a complete response, and a median progression free survival of 6.3 months (22). A phase III trial (CodeBreaK 200, NCT04303780) is ongoing, which compares sotorasib with docetaxel chemotherapy in previously treated NSCLC patients. Sotorasib is also being investigated clinically as first-line therapy for stage 4 NSCLC patients (NCT04933695).
Adagrasib is being investigated in a phase I/II clinical trial (KRYSTAL-1, NCT03785249). In patients with measurable disease at baseline, the objective response rate was 42.9%, median progression free survival of 6.5 months, and overall survival of 12.6 months after 15.6 months of follow-up (23).
The limitation of any single agent treatment is the inevitable emergence of resistance. Studies have shown that resistance to G12C inhibitors could be inherent or acquired (24). Mechanisms of resistance include activation of receptor tyrosine kinase (RTKs), resulting in downstream activation of KRAS via SHP2 (25). Several mechanisms of resistance have been reported in patient samples treated with adagrasib including secondary activating mutations in KRAS, amplification of KRAS(G12C), and mutations in compensatory pathways that bypass KRAS such as MET amplification, and NRAS, BRAF, MAP2K1, and RET mutations (26). To combat resistance to G12C inhibitors, several trials are looking to combine mutation specific inhibitors with other agents. In CodeBreaK 101 (NCT04185883) several drugs are being investigated in combination with sotorasib, such as trametinib, TNO155, everolimus, palbociclib, pembrolizumab or several other agents. Adagrasib is also being evaluated in combination with other agents, such as pembrolizumab (NCT04613596), and TNO155 (NCT04330664). Although these trials would primarily benefit lung cancer patients, they provide valuable strategies for future targeting of non-G12C mutations.
As discussed, sotorasib has been given FDA approval for the treatment of certain patients with NSCLC (27). Despite encouraging results from the trials, and FDA approval, these drugs have provided little hope for the majority of PDAC patients. While KRAS G12C mutations are more prevalent in lung cancer, KRAS G12D mutation is the most common in PDAC as well as CRC. Available G12C inhibitors rely on the reactivity of the thiol group in Cys12 of mutant KRAS. To benefit PDAC patients and target the G12D mutation, an approach that does not rely on a reactive cysteine is required.
Small molecule inhibitors targeting KRAS G12D
Discovery of KRAS G12D inhibitor MRTX1133
As previously mentioned, G12D mutations are present in a large proportion of PDAC patients. The covalent G12C inhibitors relied on the presence of a strongly nucleophilic cysteine in the switch II pocket, as well as the unique biochemical properties of the KRAS G12C mutant, which has a higher rate of GTP hydrolysis compared to other mutants (28). The switch II pocket in KRAS G12D, however, lacks a reactive cysteine to target with a covalent inhibitor, and aspartic acid is not considered a good target for covalent attack.
A structure-based medicinal chemistry approach has been employed to identify compounds that can react through a salt bridge with Asp12 of the switch II pocket in KRAS G12D (29). Using adagrasib as a starting point, chemical modifications to the reactive warhead and various other groups in the compound were introduced to increase the binding affinity within the pocket. Optimization of hit compounds resulted in the discovery of MRTX1133 (Figure 1D), which selectively and reversibly binds KRAS G12D with low nanomolar affinity in cellular assays (30).
Binding of MRTX1133 to KRAS G12D prevents downstream signaling through inhibition of nucleotide exchange and binding of downstream effector RAF1 (30). This compound was shown to inhibit oncogenic KRAS signaling selectively in tumor cells (30). In a xenograft mouse model, MRTX1133 was able to significantly reduce tumor growth and decrease the phosphorylation of downstream signaling molecule ERK in a dose-dependent manner (30). So far, this agent remains an in vitro tool compound, and its clinical progress is not known.
Other KRAS G12D inhibitors
Besides MRTX1133 described above, other groups have been pursuing the development of KRAS G12D inhibitors as well. For example, using a medicinal chemistry approach to find inhibitors that bind to Asp12 in KRAS G12D, two inhibitors TH-Z827 and TH-Z835 were discovered (Figure 1D) (31). These inhibitors form a salt bridge with Asp12 within the switch-II pocket resulting in the inhibition of KRAS signaling in G12D mutant PDAC cell lines (31). These compounds were able to bind the G12D mutant specifically, and not KRAS G12C or WT. The compounds also showed in vivo inhibition of tumor growth in xenograft tumor mouse models (31). Another set of inhibitors was discovered using virtual combinatorial chemistry and compound screening approach (32). In this approach, they used the backbone of G12C inhibitors in combination with piperazine-based compounds as building blocks for the compound library, followed by molecular docking to discover compounds with predicted selective binding. Compound ‘KD-8’ was discovered, which resulted in the inhibition of cellular and tumor growth of KRAS G12D mutated cells (32). However, further development of these compounds is required to increase their potency and minimize off-target effects. While these agents are not yet ready for clinical development, they provide further proof of principle that several classes of G12D inhibitors could be potentially discovered as clinical anti-cancer agents.
A tricomplex inhibitor, RMC-9805, is a novel covalent KRAS G12D inhibitor that binds KRAS in the GTP-bound state, thus termed a KRAS-G12D(ON) inhibitor (33). Tricomplex inhibitors bind a chaperone protein, Cyclophilin A, which is ubiquitously found inside the cell (34), which then binds the target protein, creating a target-inhibitor-Cyclophilin-A complex. RMC-9805 reacts covalently with Asp12, thereby attenuating KRAS G12D downstream signaling specifically over KRAS WT and other KRAS mutants, and it restricts tumor growth in xenograft PDAC and CRC mouse models (33). Within this class of inhibitors, there are several other compounds being developed to target various KRAS mutants, as well.
An emerging strategy to target oncogenic RAS is using monobodies to target the α4-α5 interface of KRAS, thus preventing its dimerization and downstream signaling (35–37). A monobody termed NS-1 was able to inhibit growth of G12D mutated PDAC in mice (35). However, the limitation of this strategy is the low cellular permeability of these large molecules. Therefore, further optimization is required for this approach prior to clinical testing.
Various efforts are now focused on research and discovery of novel KRAS G12D inhibitors. The development of agents, such as MRTX1133, is exciting for the field of PDAC research and provides evidence that KRAS G12D can be effectively targeted with a small molecule inhibitor. As with the G12C inhibitors, perhaps this discovery could lead to more small molecules entering the arsenal against G12D. As we await to see further clinical development of MRTX1133, the optimization of hits and potential discovery of more targeted compounds is anticipated. These efforts are critical for patients with KRAS G12D mutations, which make up a large subset of Ras-mutated cancers, especially for PDAC where the therapeutics currently available are providing meager benefits in the majority of cases.
Immunotherapy targeting KRAS G12D in PDAC
Immunotherapeutic approaches have not had great success for PDAC patients (38). In other Ras-mutated cancers, immune checkpoint inhibitors (ICIs) have improved patient outcomes in clinical trials and thus have been approved by the FDA for the treatment of NSCLC and melanoma (39). However, ICIs did not achieve similar success in PDAC, even in combinatorial approaches. PDAC is characterized by a desmoplastic tumor microenvironment (TME) with low numbers of tumor-infiltrating lymphocytes (TILs) and is largely considered to be immunosuppressive (40). Immune cells that are found within the PDAC TME include tumor-associated macrophages (TAMs), myeloid-derived suppressive cells (MDSCs), and regulatory T cells (Tregs) which contribute to immune evasion, PDAC progression, and resistance to immunotherapies (38, 41). These factors could explain the observed low response of PDAC to ICIs in clinical trials (42–44). The exception is a small subset of PDAC patients with high microsatellite instability (MSI-H) tumors or mismatch repair deficiency (<1%) who can benefit from anti-PD-1 therapy (45, 46). Nevertheless, attempts are being made to identify targets that could reinvigorate the immune suppressive tumor microenvironment to become more immunogenic and better responsive to immunotherapy.
Mutant KRAS modulates tumor immune microenvironment
Several studies have highlighted the role of oncogenic KRAS in establishing a pro-inflammatory microenvironment that enables PDAC tumorigenesis and progression (47–50). Oncogenic KRAS G12D signaling in tumor cells regulates the signaling of surrounding stromal cells and establishes reciprocal signaling between tumor and stromal cells (49, 51). PD-L1 expression is also suppressed by oncogenic KRAS G12D, and patients with G12D mutation have lower PD-L1 expression compared to other KRAS mutants (52, 53). Additional evidence for the role of oncogenic KRAS in modulating the TME comes from a recent study utilizing the anti-KRAS G12C agent AMG510 (19); where treatment with AMG510 increased the number of cytotoxic CD8+ T cells that infiltrated tumors in mice. Anti-KRAS therapy also synergized with anti-PD-1 treatment and produced durable anti-tumor responses (19). This provides strong evidence that KRAS signaling is involved in modulating the TME and maintaining an immune evasive environment, which hinders the development of ICIs as monotherapy in PDAC. Nevertheless, it also shows that using the right tools, the TME can be modulated and PDAC can potentially become responsive to immunotherapeutic approaches.
T cell therapy targeting KRAS G12D
Adoptive cell therapy utilizes the patient’s own lymphocytes which may be engineered to express receptors that specifically target tumor neoantigens (54). This immunotherapeutic approach may be suitable to target KRAS neoantigens for PDAC therapy and to address the challenge of targeting KRAS, as well as the challenge of implementation of immunotherapies in PDAC. A study identified HLA-A*11:01 to be able to present KRAS neoantigens, and then generated murine T cells that recognize G12D mutated PDAC in an HLA-A*11:01 restricted manner and could inhibit the growth of tumors in vivo. (55). An ongoing phase I/II clinical trial is investigating transfer of T-cells engineered to express a G12D specific murine T-cell receptor (TCR) in HLA-A*11:01 patients with solid tumors, including pancreatic cancer, harboring the KRAS G12D mutation (NCT03745326).
In a recent report, a patient with metastatic G12D-mutated PDAC received adoptive cell transfer therapy using engineered autologous T cells that target KRAS G12D mutant protein in the tumor, resulting in regression of metastases in the patient (Figure 2) (56). A patient with CRC had been previously reported to receive an infusion of ex-vivo expanded T cells with HLA-C*08:02 restriction and KRAS G12D reactivity, resulting in the regression of metastatic lung lesions (57). Based on the previous success of this approach, a heavily pretreated PDAC patient with lung metastases was treated with T-cells that were engineered to express HLA-C*08:02 restricted TCRs with specificity against KRAS G12D (56). A single infusion of 16.2×109 T-cells was given to the patient, which contained 85% CD8+ T-cells, and 15% CD4+ T cells, with high-dose interleukin-2 therapy to support T-cell expansion. One month follow-up revealed regression of metastatic lung lesions, which continued to regress at the 6-month follow-up with an overall objective partial response of 72% according to RECIST, version 1.1, criteria (56). Additionally, the infused T-cells were found to persist in circulation at 6 months, making up 2.4% of total circulating T cells.
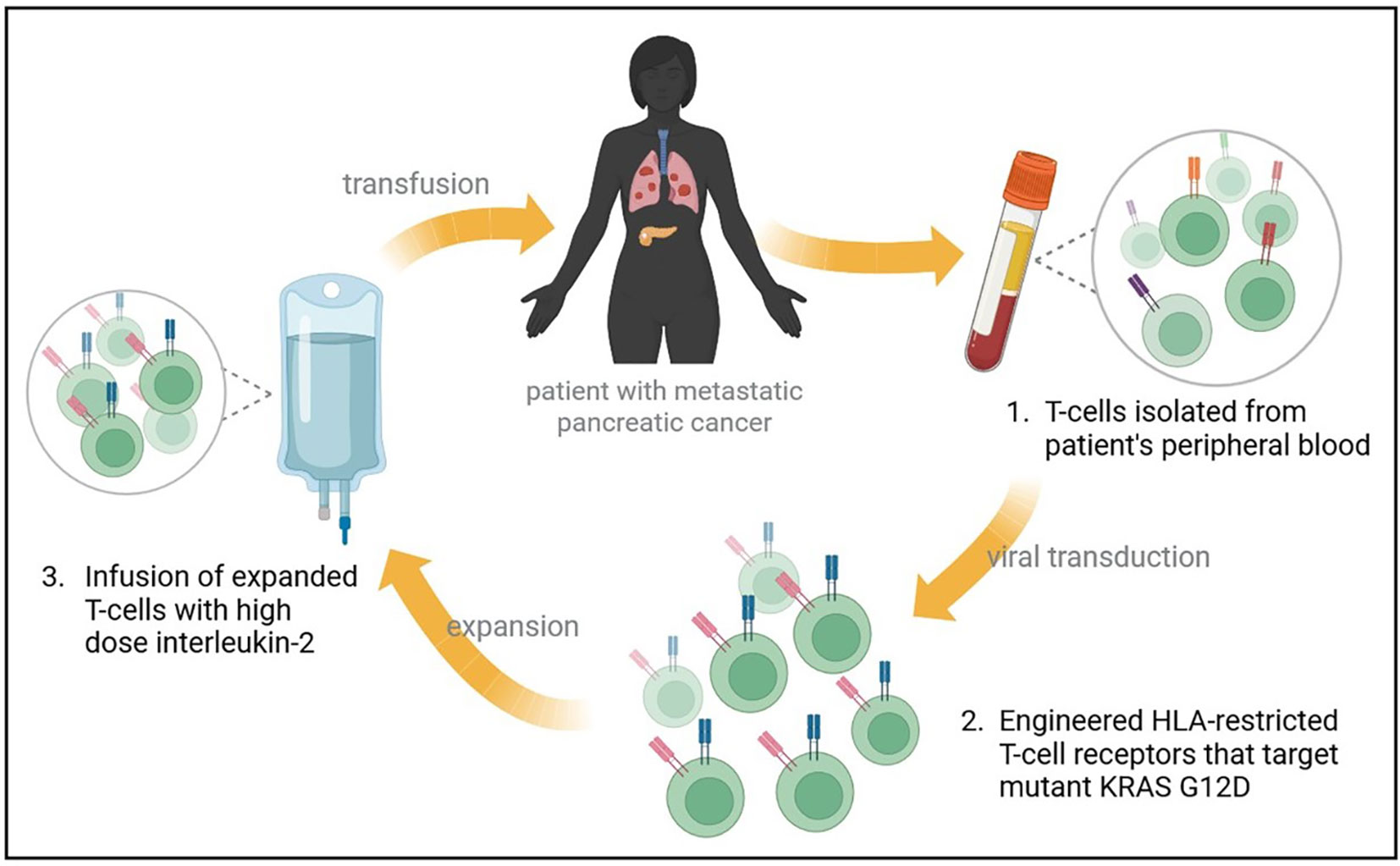
Figure 2 Treatment of metastatic PDAC with mutant KRAS-targeting genetically engineered T-cells. A heavily pretreated patient with metastatic PDAC to the lungs underwent an expiremental immunotherapy that targets mutant KRAS. Autologous T-cells were isolated and transfected with genes encoding two HLA-restricted T-cell receptors targeting KRAS G12D epitopes. The infusion product contained 16.2x109 T cells, and supportive high dose interleukin-2 therapy was administered. Metastatic lung lesions regressed at 1 month follow up, with continued regression at 6-months and an overall partial response rate of 72%.
This reported case shows that engineered TCRs could be beneficial to some patients with metastatic PDAC. The drawback of this therapy is that TCRs were restricted by a specific HLA-C*08:02, which is expressed by a small subset of patients. Further studies and clinical trials are needed to investigate this therapy on a larger scale, and to identify other KRAS-G12D-reactive TCRs which could be utilized for similar therapies (58, 59). Meanwhile, results from the ongoing clinical trial for HLA-A*11:01+ patients are eagerly awaited.
Targeting KRAS mutations beyond G12D
Since the discovery of G12C inhibitors, there has been growing interest in finding novel mutant-specific inhibitors to other common KRAS mutants. For a more detailed review, Nagasaka and colleagues have recently published an article highlighting novel strategies to target KRAS beyond G12C inhibitors including cancer vaccines, adoptive cell therapy, PROTACs and CRISPR/Cas9 (60).
KRAS G12R mutation is present in 17% of PDAC cases (Figure 1) and is therefore an important target for PDAC. Recent work has shown that the mutant arginine 12 in KRAS G12R can be targeted covalently with small molecule electrophiles (61). Although the molecules that target G12R have not shown activity in mutant cells, they could be further optimized to develop more potent molecules, and thus this presents an important proof of concept for targeting G12R. Less prevalent in PDAC, the G12S mutation in KRAS has also been targeted recently with compounds that acylate the noncatalytic mutant serine 12 residue (62). These compounds have shown selectivity for cells harboring the G12S mutation, but currently remain in vitro tools that require further optimization. Another class of drugs mentioned previously, is the tricomplex Ras(On) inhibitors, which are being developed against various KRAS mutants including G12C, G12D, and G13C, in addition to a G12X inhibitor that targets several G12 mutants (63).
Conclusion
RAS genes are the most commonly mutated oncogenes in cancers. A mutation in KRAS is the most common oncogenic driver in PDAC and is also a known driver of NSCLC and CRC. Until the recent discovery of KRAS G12C inhibitors, KRAS had been considered an undruggable target for decades (64). Given the success of translating this discovery to the clinic, research efforts are focusing on drugging KRAS G12D, the most common hotspot mutation. The discovery of the preclinical agent MRTX1133 is an exciting advance for pancreatic cancer research. MRTX1133 binds potently and reversibly in the switch II pocket of mutant KRAS G12D. We hope that this discovery will lead to precipitous efforts by various groups to introduce optimized agents that can be tested clinically for PDAC patients.
The switch-II pocket seems to be a druggable pocket in various KRAS mutants and is susceptible to reversible non-covalent inhibition. Additionally, this pocket could potentially be targeted in both the GDP as well as the GTP-bound states of KRAS (31, 65), meaning it is possible to develop inhibitors that target KRAS mutants with low intrinsic GTPase activity (28).
Cancer immunotherapy using ICIs and the observed durable responses with limited toxicities has generated a lot of excitement within the cancer research field. Despite many approvals for various cancers, immunotherapeutic approaches are yet to be approved for the treatment of PDAC. Targeting KRAS G12D using engineered T cells is an exciting development for immunotherapy in the PDAC KRAS G12D space.
To answer the question, has the KRAS G12D fortress been conquered? At this point in time, there are definitely exciting advances towards that goal, but G12D agents have a few more hurdles to overcome. The fortress may not have been conquered yet, but the walls we once thought were impervious have certainly been breached.
Author contributions
SB researched and drafted the article. HK and AA supervised the content. All authors contributed to the article and approved the submitted version.
Funding
Work in the lab of AA is supported by NIH NCI R01CA24060701 and R37CA215427
Acknowledgments
AA acknowledges support from SKY Foundation Inc and U CAN-CER VIVE foundation. SB is supported by WSU Thomas C. Rumble Fellowship.
Conflict of interest
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AA received funding from Karyopharm Therapeutics Inc and Purple Biotech. AA serves as a consultant for GLG and Guidepoint.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistic. CA Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
2. Torres C, Grippo PJ. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann Med (2018) 50:277–87. doi: 10.1080/07853890.2018.1453168
3. Moore AR, Rosenberg SC, Mccormick F, Malek S. RAS-targeted therapies: Is the undruggable drugged? Nat Rev Drug Discov (2020) 19:533–52. doi: 10.1038/s41573-020-0068-6
4. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology (2012) 142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042
5. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell (2003) 4:437–50. doi: 10.1016/s1535-6108(03)00309-x
6. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest (2012) 122:639–53. doi: 10.1172/JCI59227
7. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduction Targeted Ther (2021) 6:386. doi: 10.1038/s41392-021-00780-4
8. Arbour KC, Lito P. Expanding the arsenal of clinically active KRAS G12C inhibitors. J Clin Oncol (2022) 40:2609–11. doi: 10.1200/JCO.22.00562
9. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci (2016) 129:1287–92. doi: 10.1242/jcs.182873
10. Buday L, Downward J. Many faces of ras activation. Biochim Biophys Acta (BBA) - Rev Cancer (2008) 1786:178–87. doi: 10.1016/j.bbcan.2008.05.001
11. Hennig A, Markwart R, Esparza-Franco MA, Ladds G, Rubio I. Ras activation revisited: Role of GEF and GAP systems. Biol Chem (2015) 396:831–48. doi: 10.1515/hsz-2014-0257
12. Bannoura SF, Uddin MH, Nagasaka M, Fazili F, Al-Hallak MN, Philip PA, et al. Targeting KRAS in pancreatic cancer: New drugs on the horizon. Cancer Metastasis Rev (2021) 40:819–35. doi: 10.1007/s10555-021-09990-2
13. Nollmann FI, Ruess DA. Targeting mutant KRAS in pancreatic cancer: Futile or promising? Biomedicines (2020) 8(8):281. doi: 10.3390/biomedicines8080281
14. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503:548–51. doi: 10.1038/nature12796
15. Potashman MH, Duggan ME. Covalent modifiers: An orthogonal approach to drug design. J Med Chem (2009) 52:1231–46. doi: 10.1021/jm8008597
16. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science (2016) 351:604–8. doi: 10.1126/science.aad6204
17. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov (2016) 6:316–29. doi: 10.1158/2159-8290.CD-15-1105
18. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell (2018) 172:578–589.e17. doi: 10.1016/j.cell.2018.01.006
19. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575:217–23. doi: 10.1038/s41586-019-1694-1
20. Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem (2020) 63:52–65. doi: 10.1021/acs.jmedchem.9b01180
21. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167
22. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
23. Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non-Small-Cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med (2022) 387:120–31. doi: 10.1056/NEJMoa2204619
24. Liu J, Kang R, Tang D. The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther (2022) 29:875–8. doi: 10.1038/s41417-021-00383-9
25. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature (2020) 577:421–5. doi: 10.1038/s41586-019-1884-x
26. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med (2021) 384:2382–93. doi: 10.1056/NEJMoa2105281
27. Nakajima EC, Drezner N, Li X, Mishra-Kalyani PS, Liu Y, Zhao H, et al. FDA Approval summary: Sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin Cancer Res (2022) 28:1482–6. doi: 10.1158/1078-0432.CCR-21-3074
28. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res (2015) 13:1325–35. doi: 10.1158/1541-7786.MCR-15-0203
29. Zheng Q, Peacock DM, Shokat KM. Drugging the next undruggable KRAS allele-Gly12Asp. J Medicinal Chem (2022) 65:3119–22. doi: 10.1021/acs.jmedchem.2c00099
30. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J Med Chem (2022) 65:3123–33. doi: 10.1021/acs.jmedchem.1c01688
31. Mao Z, Xiao H, Shen P, Yang Y, Xue J, Yang Y, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov (2022) 8:5. doi: 10.1038/s41421-021-00368-w
32. Li L, Liu J, Yang Z, Zhao H, Deng B, Ren Y, et al. Discovery of Thieno[2,3-d]pyrimidine-based KRAS G12D inhibitors as potential anticancer agents via combinatorial virtual screening. Eur J Med Chem (2022) 233:114243. doi: 10.1016/j.ejmech.2022.114243
33. Knox J, Jiang J, Burnett L, Liu Y, Weller C, Wang Z, et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, tri-complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Res (2022) 82(12_Supplement):3596. doi: 10.1158/1538-7445.AM2022-3596
35. Khan I, Marelia-Bennet C, Lefler J, Zuberi M, Denbaum E, Koide A, et al. Targeting the KRAS α4-α5 allosteric interface inhibits pancreatic cancer tumorigenesis. Small GTPases (2021), 1–14. doi: 10.1080/21541248.2021.1906621
36. Khan I, Spencer-Smith R, O'bryan JP. Targeting the α4-α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene (2019) 38:2984–93. doi: 10.1038/s41388-018-0636-y
37. Spencer-Smith R, Li L, Prasad S, Koide A, Koide S, O'bryan JP. Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases (2019) 10:378–87. doi: 10.1080/21541248.2017.1333188
38. Leinwand J, Miller G. Regulation and modulation of antitumor immunity in pancreatic cancer. Nat Immunol (2020) 21:1152–9. doi: 10.1038/s41590-020-0761-y
39. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med (2018) 50:1–11. doi: 10.1038/s12276-018-0191-1
40. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
41. Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for increasing pancreatic tumor immunogenicity. Clin Cancer Res (2017) 23:1656–69. doi: 10.1158/1078-0432.CCR-16-2318
42. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33:828–33. doi: 10.1097/CJI.0b013e3181eec14c
43. O'reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol (2019) 5:1431–8. doi: 10.1001/jamaoncol.2019.1588
44. Kamath SD, Kalyan A, Kircher S, Nimeiri H, Fought AJ, Benson A 3rd, et al. Ipilimumab and gemcitabine for advanced pancreatic cancer: A phase ib study. Oncologist (2020) 25:e808–15. doi: 10.1634/theoncologist.2019-0473
45. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
46. Das S, Berlin J, Cardin D. Harnessing the immune system in pancreatic cancer. Curr Treat Options Oncol (2018) 19:48. doi: 10.1007/s11864-018-0566-5
47. Mcallister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-Epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
48. Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov (2020) 10:608–25. doi: 10.1158/2159-8290.CD-19-0297
49. Tape CJ, Ling S, Dimitriadi M, Mcmahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell (2016) 165:910–20. doi: 10.1016/j.cell.2016.03.029
50. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21:836–47. doi: 10.1016/j.ccr.2012.04.024
51. Wang HC, Lin YL, Hsu CC, Chao YJ, Hou YC, Chiu TJ, et al. Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics (2019) 9:7168–83. doi: 10.7150/thno.36830
52. Liu C, Zheng S, Wang Z, Wang S, Wang X, Yang L, et al. KRAS-G12D mutation drives immune suppression and the primary resistance of anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer Commun (Lond) (2022) 42(9):828–47. doi: 10.1002/cac2.12327
53. Falk AT, Yazbeck N, Guibert N, Chamorey E, Paquet A, Ribeyre L, et al. Effect of mutant variants of the KRAS gene on PD-L1 expression and on the immune microenvironment and association with clinical outcome in lung adenocarcinoma patients. Lung Cancer (2018) 121:70–5. doi: 10.1016/j.lungcan.2018.05.009
54. Wang Z, Cao YJ. Adoptive cell therapy targeting neoantigens: A frontier for cancer research. Front Immunol (2020) 11:176. doi: 10.3389/fimmu.2020.00176
55. Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res (2016) 4:204–14. doi: 10.1158/2326-6066.CIR-15-0188
56. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med (2022) 386:2112–9. doi: 10.1056/NEJMoa2119662
57. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
58. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun (2019) 10:449. doi: 10.1038/s41467-019-08304-z
59. Bear AS, Blanchard T, Cesare J, Ford MJ, Richman LP, Xu C, et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat Commun (2021) 12:4365. doi: 10.1038/s41467-021-24562-2
60. Nagasaka M, Potugari B, Nguyen A, Sukari A, Azmi AS, Ou SI. KRAS inhibitors- yes but what next? direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev (2021) 101:102309. doi: 10.1016/j.ctrv.2021.102309
61. Zhang Z, Morstein J, Ecker AK, Guiley KZ, Shokat KM. Chemoselective covalent modification of K-Ras(G12R) with a small molecule electrophile. J Am Chem Soc (2022) 144:15916–21. doi: 10.1021/jacs.2c05377
62. Zhang Z, Guiley KZ, Shokat KM. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat Chem Biol (2022) 18(11):1177–83. doi: 10.1038/s41589-022-01065-9
63. Koltun E, Rice M, Gustafson WC, Wilds D, Jiang J, Lee B, et al. Abstract 3597: Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res (2022) 82:3597–7. doi: 10.1158/1538-7445.AM2022-3597
64. Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat Rev Drug Discov (2016) 15:771–85. doi: 10.1038/nrd.2016.139
Keywords: KRAS, KRAS G12C, KRAS G12D, sotorasib, adagrasib, MRTX1133, adoptive cell therapy, immunotherapy
Citation: Bannoura SF, Khan HY and Azmi AS (2022) KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front. Oncol. 12:1013902. doi: 10.3389/fonc.2022.1013902
Received: 08 August 2022; Accepted: 08 November 2022;
Published: 29 November 2022.
Edited by:
Cyril Corbet, Fonds National de la Recherche Scientifique (FNRS), BelgiumReviewed by:
Aaron Hobbs, Medical University of South Carolina, United StatesBoshi Wang, Shanghai Cancer Institute, China
Copyright © 2022 Bannoura, Khan and Azmi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asfar S. Azmi, YXptaWFAa2FybWFub3Mub3Jn