 Tracey Immanuel1
Tracey Immanuel1 Jixia Li
Jixia Li Anna Bogdanova
Anna Bogdanova Maggie L. Kalev-Zylinska
Maggie L. Kalev-Zylinska- 1Blood and Cancer Biology Laboratory, Department of Molecular Medicine and Pathology, University of Auckland, Auckland, New Zealand
- 2Department of Laboratory Medicine, School of Medicine, Foshan University, Foshan City, China
- 3Red Blood Cell Research Group, Institute of Veterinary Physiology, Vetsuisse Faculty, University of Zurich, Zürich, Switzerland
- 4Zurich Center for Integrative Human Physiology, University of Zurich, Zürich, Switzerland
- 5Haematology Laboratory, Department of Pathology and Laboratory Medicine, Auckland City Hospital, Auckland, New Zealand
Intracellular calcium signaling regulates diverse physiological and pathological processes. In solid tumors, changes to calcium channels and effectors via mutations or changes in expression affect all cancer hallmarks. Such changes often disrupt transport of calcium ions (Ca2+) in the endoplasmic reticulum (ER) or mitochondria, impacting apoptosis. Evidence rapidly accumulates that this is similar in blood cancer. Principles of intracellular Ca2+ signaling are outlined in the introduction. We describe different Ca2+-toolkit components and summarize the unique relationship between extracellular Ca2+ in the endosteal niche and hematopoietic stem cells. The foundational data on Ca2+ homeostasis in red blood cells is discussed, with the demonstration of changes in red blood cell disorders. This leads to the role of Ca2+ in neoplastic erythropoiesis. Then we expand onto the neoplastic impact of deregulated plasma membrane Ca2+ channels, ER Ca2+ channels, Ca2+ pumps and exchangers, as well as Ca2+ sensor and effector proteins across all types of hematologic neoplasms. This includes an overview of genetic variants in the Ca2+-toolkit encoding genes in lymphoid and myeloid cancers as recorded in publically available cancer databases. The data we compiled demonstrate that multiple Ca2+ homeostatic mechanisms and Ca2+ responsive pathways are altered in hematologic cancers. Some of these alterations may have genetic basis but this requires further investigation. Most changes in the Ca2+-toolkit do not appear to define/associate with specific disease entities but may influence disease grade, prognosis, treatment response, and certain complications. Further elucidation of the underlying mechanisms may lead to novel treatments, with the aim to tailor drugs to different patterns of deregulation. To our knowledge this is the first review of its type in the published literature. We hope that the evidence we compiled increases awareness of the calcium signaling deregulation in hematologic neoplasms and triggers more clinical studies to help advance this field.
1 Introduction
The deregulation of signaling by calcium ions (Ca2+) has been extensively studied in solid tumors (1, 2). Changes to Ca2+ channels and effectors via mutations or changes in expression affect many functional capabilities responsible for cancer growth, invasion, and metastasis (2–5). The function of the endoplasmic reticulum (ER), the main site of Ca2+ storage in a cell, and Ca2+ transfer from the ER to mitochondria, the main regulation point for apoptotic cell death, are often deregulated in solid tumors (6, 7). Our review presents the rapidly accumulating data that this deregulation appears similar in many types of blood cancer. Therapeutic opportunities targeting Ca2+ signaling are emerging for disorders such as leukemia, lymphoma, and myeloproliferative neoplasms (MPN) (8–11), but this information is not yet widely known. Therefore, to increase awareness, we provide an outline of core findings that demonstrate deregulation of Ca2+ signaling in blood cancer. Research in this field has accelerated enormously in recent years, therefore, despite our great efforts, this review is unlikely to be complete. Nevertheless, we hope our compilation of data makes the subject of abnormal Ca2+ signaling in blood cancer more widely known. To our knowledge, this is the first review of this type in the published literature.
1.1 Unique relationship between extracellular Ca2+ and hematopoietic stem cells
Ca2+ signaling regulates many cellular processes, including gene expression, cell proliferation, motility, apoptosis, enzyme activity, and cytoskeletal dynamics, all of which are crucial to supporting normal cell differentiation including of hematopoietic stem cells (HSCs) (12–14). Specific effects of Ca2+ signaling are achieved through a tight control of intracellular Ca2+ homeostasis. At the resting state, cytosolic Ca2+ concentrations are maintained at very low levels: ∼50–100 nM in most cells and reported to be as low as 20–30 nM in HSCs (15). This contrasts with high extracellular Ca2+ concentrations of ~1.5 mM in most fluids, including in blood plasma and bone marrow interstitial space (16). On the background of this high extracellular-intracellular Ca2+ gradient, precisely regulated spatio-temporal increases in cytosolic Ca2+ levels trigger signaling events (17).
The bone marrow environment provides a unique extracellular context for Ca2+ signaling. High Ca2+ levels in the endosteal niche have been shown to assist homing of HSCs through their calcium-sensing receptor (18). Nevertheless, it remains unclear if low or high Ca2+ concentrations are required to support HSC quiescence, both were shown to apply (14, 15, 19, 20). A recent study demonstrates that there is heterogeneity in Ca2+ levels between bone marrow cavities, depending on the level of bone resorption, but unexpectedly, no sharp gradient towards the endosteal niche was observed (16). HSCs reside in locations with higher extracellular Ca2+ levels compared to the serum and to the overall Ca2+ levels in the bone marrow. With aging, there is a significant increase in extracellular Ca2+ levels in the bone marrow associated with clonal expansion of activated HSCs. It has been proposed that deregulated Ca2+ homeostasis may be involved in leukemic transformation of HSCs, but experimental validation is required (21). In support, changes in Ca2+ homeostasis influence cancer stem cell properties in other cancer types (22).
1.2 Principles of intracellular calcium signaling
Cytoplasmic free Ca2+ levels are maintained by Ca2+ buffer systems (23) and modulated by a system of molecules re-distributing Ca2+ between the intracellular stores (the ER, mitochondria, Golgi apparatus and lysosomes), taking Ca2+ in from the extracellular space, or extruding it from the cell (12). Various channels, exchangers and pumps regulate Ca2+ levels in cells, including in blood cells. The collective involvement of these molecules, often referred to as a Ca2+-signaling toolkit (13, 24), is shown in Figure 1 (with molecular details described in the figure legend).
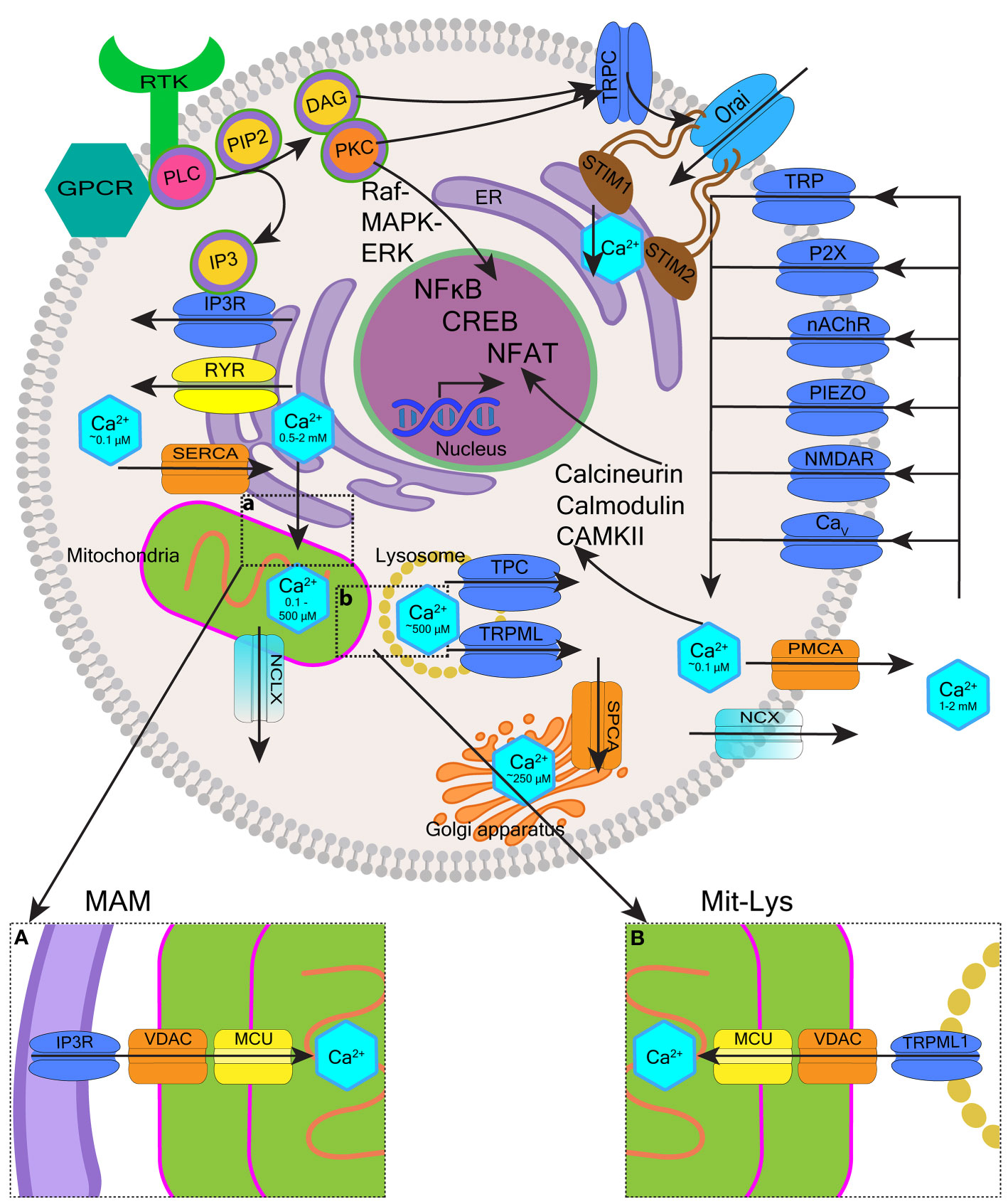
Figure 1 Overview of intracellular calcium homeostasis. Calcium homeostasis is maintained by the influx and efflux of Ca2+ through calcium channels and pumps located on the plasma membrane, as well as membranes of organelles such as the endoplasmic reticulum (ER), the Golgi apparatus, mitochondria, and endo-lysosomes. The cytoplasm, extracellular space, and each organelle have unique resting Ca2+ concentrations that have been indicated. Extracellular Ca2+ is transported into the cytosol through different channels such as transient receptor potential (TRP) channels, purinergic receptor (P2X) channels, nicotinic acetylcholine receptor (nAChR) channels, Piezo mechanosensitive channels, ionotropic glutamate receptor channels (e.g. N-methyl-D-aspartate receptors, NMDARs), and voltage-gated calcium (Cav) channels. Ca2+ is removed from the cytosol to the extracellular space by plasma membrane calcium ATPase (PMCA) efflux pumps and sodium-calcium exchangers (NCX). Activation of cell surface transmembrane receptors with tyrosine-based activation motifs (RTK e.g. B-cell and T-cell receptors) or G-protein coupled receptors (GPCR e.g. neurokinin-1 receptor) activate phospholipase C (PLC). PLC hydrolyses phosphatidylinositol-4,5-bisphosphate (PIP2) located in the plasma membrane which generates two second messengers inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG). IP3 binds to IP3 receptors (IP3Rs) located on the ER membrane leading to the release of Ca2+ from the ER and DAG activates protein kinase C (PKC). Depletion of ER Ca2+ activates stromal interaction molecules STIM1-STIM2 (located in the ER membrane), which then activates Orai1-Orai3 channels (located in the plasma membrane) to induce Ca2+ influx into the cytosol. This mechanism is called store-operated calcium entry (SOCE). Ryanodine receptors (RYRs) represent an alternative pathway for Ca2+ release from the ER regulated by Ca2+, Mg2+ and other molecules including ATP, calmodulin and CaMKII. The Ca2+ concentration in the ER is replenished via sarco-endoplasmic reticulum calcium ATPase 2b (SERCA2b) pump. The influx of Ca2+ from the ER to mitochondria occurs through voltage-dependent anion channels (VDAC) and mitochondrial calcium uniport (MCU) located in high numbers within mitochondria-associated ER membranes (MAMs) (insert a). Ca2+ leaves mitochondria mostly through Na+/Ca2+/Li+ exchanger (NCLX). Ca2+ stored in the endo-lysosomes is mobilized mostly by two-pore channels (TPC) and transient receptor potential mucolipin (TRPML) channels in response to nicotinic acid adenine dinucleotide phosphate (NAADP.) TRPML1 is involved in the mitochondrial-lysosomal contact sites (Mit-Lys), facilitating Ca2+ transfer to mitochondria through VDAC and MCU (insert b). Multiple effector molecules mediate effects of Ca2+ signaling including PKC, Raf-MAPK (mitogen-activated protein kinase)-ERK (extracellular signal-regulated kinase), calmodulin, calcium/calmodulin-dependent protein kinases (e.g. CaMKII), and calcineurin. These signaling molecules influence gene expression through transcription factors such as nuclear factor kappa B (NF-κB), cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB), and nuclear factor of activated T-cells (NFAT).
In this review, we wish to highlight the role of ER as the main site of Ca2+ storage in almost any cell, as this functionality is often deregulated in cancer including blood cancer (25). The concentration of free Ca2+ in the ER is ~500 μM and ~2 mM for total ER Ca2+, most of which is bound to Ca2+-binding proteins such as calreticulin (CALR) (26). Many pathways of cell activation converge on the efflux of Ca2+ from the ER that occurs through channels called inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) (27) (Figure 1). IP3Rs induce the release of Ca2+ from the ER upon binding of IP3 generated by phospholipase C (PLC) (28). PLC operates downstream of G-protein coupled receptors (GPCRs) and tyrosine kinase receptors located in the plasma membrane (29, 30). When ER Ca2+ becomes depleted, extracellular Ca2+ influx is initiated to maintain signaling. In non-neuronal cells most extracellular Ca2+ enters the cell through the mechanism called store-operated calcium entry (SOCE) (31, 32). SOCE is triggered by stromal interaction molecules (STIM1-STIM2) located in the ER membrane. Upon sensing ER Ca2+ depletion, STIM proteins oligomerize and redistribute to the plasma membrane where they interact with Orai1-Orai3 channels to activate Ca2+ influx into the cytosol (26, 32) STIM2 has low affinity for Ca2+ and activates when ER Ca2+ stores are <500 μM. In contrast, STIM1 has high affinity for Ca2+ and only activates when Ca2+ stores are <300 μM (33). Loss of STIM2 occuring in certain cancers is thought to reduce ER Ca2+ content (7). PLC also generates 1,2-diacylglycerol (DAG) that performs its signaling functions by binding and activating other proteins, including protein kinases C (PKC) and certain transient receptor potential (TRP) canonical (TRPC) channels, in particular TRPC1, that can interact with Orai1 and STIM1 to support SOCE (29, 34) (Figure 1).
Ca2+ transfer from the ER to mitochondria is another important mechanism often hijacked in solid tumors and of emerging importance in blood cancer (21, 35). ER and mitochondria interact through specialized ER-mitochondrial contact sites called mitochondria-associated ER membranes (MAMs) (36) (Figure 1, insert a). Within MAMs, IP3Rs on the ER interact with voltage-dependent anion channels (VDACs) located in the outer mitochondrial membrane allowing unrestricted Ca2+ entry into the inter-membrane space (37). The passage of Ca2+ through the inner mitochondrial membrane is restricted by the mitochondrial calcium uniport (MCU) and the membrane potential (ΔΨm ∼ −150 mV) (38). Small amounts of mitochondrial Ca2+ support mitochondrial metabolism, providing a mechanism that couples cellular activity with the generation of adenosine triphosphate (ATP). Ca2+ uptake into mitochondria activates pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase, thereby stimulating the tricyclic acid cycle and energy generation (39, 40). In contrast, high levels of Ca2+ in the mitochondria induce apoptosis (41, 42). Prolonged accumulation of Ca2+ in the mitochondria leads to the opening of the mitochondrial permeability transition pore (mPTP) formed when VDAC1 clusters with adenine nucleotide translocase (on the inner mitochondrial membrane) and cyclophilin D (in the mitochondrial matrix). The mPTP opening causes depolarization of the inner mitochondrial membrane, which uncouples the respiratory chain leading to increased mitochondrial membrane permeability and the release of cytochrome c (21, 35, 36).
Oncogenic effects have also been shown for certain endo-lysosomal Ca2+ storage and release mechanisms (43). Endo-lysosomes are heterogenous and dynamic acidic organelles that in addition to other roles, act as intracellular Ca2+ stores (44, 45). Endo-lysosomes sequester and release Ca2+ to the cytosol mainly through two-pore channels (TPC1-TPC2) and TRP mucolipin channels (TRPML1-TRPML3) activated by second messengers such as nicotinic acid adenine dinucleotide phosphate (NAADP), the most potent Ca2+-mobilizing second messenger known (46, 47). Effects of endo-lysosomal Ca2+ release may be both local and global. The latter occur when endo-lysosomal mechanisms act in conjuction with the ER to induce or inhibit ER Ca2+ release (48). Endo-lysosomal Ca2+ signaling regulates processes such as membrane trafficking, vesicle fusion and secretion which impacts a range of cellular behavious e.g. immune responses, autophagy, cell proliferation, and migration (43, 49). In analogy to MAMs, mitochondrial membrane contact sites have also been shown to involve lysosomes (50, 51) (Figure 1, insert b). The release of lysosomal Ca2+ through TRPML1 supports Ca2+ transfer to mitochondria, providing an additional mechanism through which intracellular Ca2+ signaling, mitochondrial bioenergetics and lysosomal effects can be regulated (51).
This review emphasizes importance of abnormal Ca2+ signaling in hematologic cancers. We begin by presenting the long-standing foundational data on Ca2+ homeostasis in red blood cells (RBCs) as historically, this work provided guidance for research into Ca2+ signaling in selected blood cancers. We then focus on the neoplastic impact of deregulated Ca2+ influx through the plasma membrane and the ER, Ca2+ efflux via Ca2+ pumps and exchangers, and the impact of deregulated Ca2+ sensor and effector proteins in blood cancer. Throughout the review we highlight potential therapeutic strategies being developed to abrogate this deregulation.
2 Foundational research into calcium signaling in red cells with an outline of the toolkit components
Research into Ca2+ homeostasis in RBCs has a long history and has been regularly reviewed (52–54). While reticulocytes and immature RBCs of patients with sickle cell disease retain some of the mitochondria (55), normal mammalian RBCs do not have true Ca2+ storage organelles. However, RBCs often contain inside-out vesicles that are formed in response to increased Ca2+ uptake. These vesicles contain plasma membrane calcium ATPases (PMCAs) that pump Ca2+ from the cytosol into vesicles and thus protect the cytosolic and membrane proteins from Ca2+-induced damage (oxidation, proteolysis, irreversible dehydration) (see Figure 2A and the corresponding legend for molecular details). Some of these Ca2+-filled vesicles are extruded, other inside-out endosomes are retained inside the cells (56–58). The resting concentration of cytosolic Ca2+ in RBCs are similar to nucleated cells, ranging from 30–60 nM in normal RBCs to the pathological 300 nM levels in patients with certain hereditary anemias (59). This compares with 1.2-1.8 mM in blood plasma (52, 53). Cytosolic Ca2+ concentrations in RBCs affect many aspects of red cell physiology including cell hydration, metabolic activity, redox state, and proteolysis. Regulation of Ca2+ concentrations translates into the control over the remodeling of the cytoskeletal elements and concomitant changes in cell shape, cell volume, rheological properties and ultimately, RBC longevity and clearance (52) (Figure 2A).
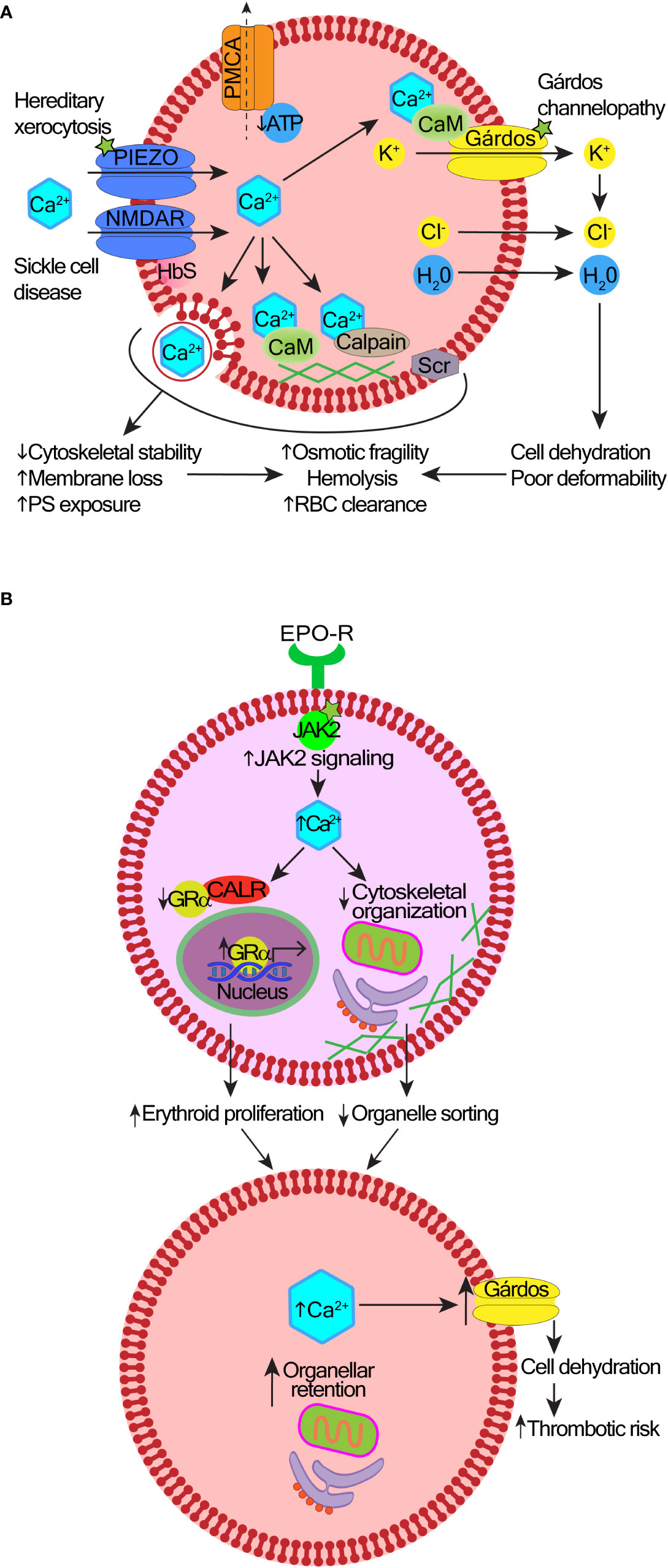
Figure 2 Mechanisms and consequences of deregulated calcium signaling in red cells and erythroid precursors. (A) In hereditary stomatocytosis/xerocytosis, heterogenous gain-of-function mutations in the Piezo1 or Gárdos channels cause excessive Ca2+ entry into red blood cells (RBCs). In sickle cell disease, there is an abnormally high abundance and activity of NMDA receptor channels, and probably other Ca2+-transporting ion channels contributing to the increased permeability of the RBC membrane to Ca2+. A layer of aggregated hemoglobin S (HbS) interferes with the shedding of NMDA receptor channels from the cell surface. Reduced levels of adenosine triphosphate (ATP) impair the function of the plasma membrane calcium pump (PMCA); as a result, Ca2+ uptake exceeds its efflux. Excess intracellular Ca2+ can be sequestered into vesicles and extruded, protecting the cytosolic and membrane proteins from Ca2+-induced damage. However, over time, increased cytosolic Ca2+ overactivates the Gárdos channel leading to cell dehydration. Membrane and cytoskeletal instability are induced by the overactive calcium/calmodulin (CaM) complexes, calpain, or scramblase (Scr). This leads to premature RBC clearance, hemolysis, and anemia. The exact contribution of these mechanisms to different types of anemia remains under investigation. (B) Effects of high intracellular Ca2+ in polycythemia vera (PV). Hyperactive JAK2 V617F mutation increases Ca2+ levels in erythroid precursors. Ca2+ overload impairs the nuclear export function of calreticulin (CALR), which results in nuclear retention of the glucocorticoid receptor α (GRα) responsible for stress response and erythroid proliferation. Defective organelle sorting and extrusion from erythroblasts leaves organellar remnants in reticulocytes.
Multiple types of channels permeable for Ca2+ are present in the RBC membrane supporting versatility and plasticity of intracellular Ca2+ signaling (53) (Figure 2A). These channels are present in RBCs in very low copy numbers to keep the basal Ca2+ permeability of the plasma membrane low. Each channel type responds to its own stimulus (e.g. mechanical, electrical or chemical) to induce Ca2+ oscillations under specific conditions. Due to the broad variance in channel copy number per cell, there is variation in RBC responses to stimulation, and the numbers of “responding cells” typically range from 10% to 30% (60–62).
One of the first Ca2+ signaling processes identified in RBCs was the function of the Gárdos channel (potassium calcium-activated channel subfamily N member 4, KCNN4) (63) (Figure 2A). KCNN4 is activated by Ca2+ that enters through any of the non-selective cation channels [e.g. piezo type mechanosensitive ion channel component 1, Piezo1 (64)]. Piezo channels are the largest plasma membrane Ca2+ channels known containing a three-bladed propeller-shaped structure that spans the lipid bilayer sensing membrane stretch (65–67). The activation of Piezo links mechanical forces applied to RBCs with the control of cell volume and lifespan (64, 68). The KCNN4 activation leads to K+ efflux and water loss (69), which reduces RBC volume and facilitates cell shape change. Activation of KCNN4 in RBCs of healthy people most likely enables better passage of RBCs through narrow capillaries (70), while its overactivation causes Ca2+-overload and RBC dehydration (71) (Figure 2A). Hereditary stomatocytosis/xerocytosis are caused by gain of function mutations in genes encoding either KCNN4 (58, 72, 73) or Piezo1 channels (74–76). Different mutations cause distinctive clinical phenotypes, including some with syndromic features (72). The increasing use of next-generation-sequencing will help characterize the scope of genetic variants that are clinically relevant.
Other Ca2+ channels in RBCs include selected TRP and voltage-gated Ca2+ channels (Cav), N-methyl-D-aspartate (NMDA) receptors, and VDACs (52, 53). The TRP channels are a large family of approximately 30 structurally related but diverse members, the majority of which function as non-selective cation channels with variable Ca2+ permeability (77, 78). TRP channels can be activated by multiple external ligands including inflammatory and pain mediators, certain spices (e.g. garlic, mint, camphor and chili), metabolites, or physical stimuli such as temperature and stretch. TRP channels act as environmental sensors and transduction channels that regulate intracellular Ca2+ levels in response to the depletion of internal Ca2+ stores with or without simultaneous activation by PLC (79–81). The importance of TRP and Piezo channels in human physiology and pathology is underscored by the award of the Nobel Prize in Physiology or Medicine in 2021 to David Julius and Ardem Patapoutian “for their discoveries of receptors for temperature and touch” (82–84). Based on the amino acid sequence homology, activation mode and function, TRP channels are divided into six subfamilies: TRPC (canonical, TRPC1-TRPC7), TRPV (vanilloid, TRPV1-TRPV6), TRPM (melastatin, TRPM1-TRPM8), TRPA (ankyrin, TRPA1), TRPML (mucolipin, TRPML1-TRPML3), and TRPP (polycystin, TRPP1-TRPP2) (77). All TRP channel types are tetrameric assemblies of subunits containing six transmembrane domains arranged around a central ion permeation pore (79).
All TRP channels mediate receptor-operated Ca2+ entry but some also function as components or regulators of SOCE (85, 86). The latter applies mostly to TRPC1 and TRPC4 as they can interact with and be activated by STIM1 upon depletion of the ER Ca2+ stores; in turn, TRPC1, TRPC3 and TRPC6 can interact and activate Orai1 channels to support ER Ca2+ store refilling (87). Other TRPC channels do not interact with STIM1 directly, however heteromeric assemblies combining TRPC1 with TRPC4/5 or TRPC3 with TRPC6/7 contribute to SOCE, implying single TRPC components can provide SOCE regulation (88). Other TRP channel types (e.g. TRPV4 and TRPV6) and other proteins also interact with TRPC channels, which influences diversity of their functioning (89).
TRPC6 is abundant in human RBCs and contributes to stress-stimulated Ca2+ entry but its specific function in RBCs remains elusive (90). The discovery of TRPV2 in RBCs is relatively recent (91). Similar to Piezo1, TRPV2 mediates Ca2+ influx into RBCs in response to mechanical activation, which modulates RBC osmotic fragility and may contribute to the RBC storage lesion (92).
NMDA receptors are ligand-gated non-specific cation channels with high Ca2+ permeability activated by glutamate and glycine (93). NMDA receptors play critical functions in the brain but are also expressed in non-neuronal cells, including all types of blood cells: red cells (60, 94, 95), platelets (96–98), neutrophils (99), monocytes (60), and lymphocytes (100, 101). In RBCs, NMDA receptor regulates hemoglobin oxygen affinity, nitric oxide production, cell hydration status, and proliferation of erythroid precursors (95). RBCs from patients with sickle cell disease carry higher numbers of NMDA receptors than in healthy donors (Figure 2A). NMDA receptor overactivity leads to Ca2+ overload, K+ loss, cell dehydration, and oxidative stress, which may contribute to sickle cell crises (94). The efficacy of NMDA receptor inhibitor memantine for symptomatic treatment of sickle cell disease is currently being explored (102, 103).
VDACs are the major components of the outer mitochondrial membrane but they are also present in the plasma membrane including in RBCs (104, 105). VDACs conductance and selectivity are voltage-dependent. In the plasma membrane, VDACs may be involved in the transmembrane electron transport (37, 104). VDACs permeability for Ca2+ is low but considering the large intracellular-extracellular Ca2+ gradient, their activation may still contribute significant Ca2+ influx (53).
Finally, voltage-gated Ca2+ (CaV) channels transduce changes in the plasma membrane potential to intracellular Ca2+ transients that initiate many crucial physiological processes (106). In neurons and muscle cells CaV channels primarily regulate synaptic transmission and contraction respectively but these channels also regulate secretion and biochemical processes such as enzyme activity, protein phosphorylation/dephosphorylation, and gene expression in other cell types. CaV channels are subdivided into CaV1, CaV2, and CaV3 (107). CaV2.1 is epressed in RBCs but its function is poorly defined (108).
Overall, Ca2+ channels play important roles in RBC membrane transport, metabolism, volume, shape and lifespan regulation, although many specific functions remain unknown (53). It has been proposed that increased Ca2+ levels in RBCs due to abnormal function of Ca2+ channels represents a common mechanism underlying an accelerated clearance of RBCs from the bloodstream and pathological hemolysis in a range of anemias, which is a new area for investigation (59).
3 Calcium signaling in normal and neoplastic erythropoiesis
The role of intracellular Ca2+ signaling during erythropoiesis has been recently reviewed (54). Ca2+ signaling regulates erythroid progenitor proliferation, differentiation, survival, and terminal enucleation. Changes in Ca2+ homeostasis are seen in reactive ineffective erythropoiesis (e.g. in β-thalassemia) (109) or in neoplastic erythropoiesis driven by Janus kinase 2 (JAK2) V617F mutation in polycythemia vera (PV) (110, 111). CALR is an ER-resident protein that regulates functions of other proteins by chaperoning them to their active sites in response to changing intracellular Ca2+ levels (112). In normal erythroid precursors, CALR promotes the nuclear export of glucocorticoid receptor α, which resets precursor proliferation to differentiation (110). In contrast, hyperactive JAK2 signaling in PV increases free intracellular Ca2+ levels, which impairs the nuclear export function of CALR (Figure 2B). Glucocorticoid receptor α is retained in the nucleus maintaining the expression of stress genes that increase proliferation of erythroblasts (110). Elevated levels of Ca2+ may also impair actin reorganization required to extrude organelles during enucleation (111). Consequently, PV reticulocytes have a high content of organellar remnants e.g. mitochondria, ER and ER-associated proteins including CALR. In mature RBCs from PV patients, high Ca2+ levels increase the activity of the Gárdos channel leading to cell dehydration (111) (Figure 2B).
Increased levels of cytoplasmic Ca2+, cell dehydration and the presence of organelle remnants in RBCs have the potential to promote thrombosis in PV (111). Dehydrated RBCs are more rigid, thus less amenable to shape changes required to pass through narrow capillaries, and also more susceptible to hemolysis under high-shear rates that occur in arterial circulation (113, 114). Higher cytoplasmic Ca2+ levels are known to increase adhesion between RBCs (115), and of RBCs to the endothelium (116, 117). Most previous work into PV-associated thrombosis focused on the role of a high hematocrit, white cell and platelet activation, coagulation factors and inflammation (118). However, a recent study used a laser-assisted optical rotational red cell analyzer to demonstrate abnormal RBC morphodynamics in 48 patients with PV (119). The deformability and stability of RBCs were reduced and RBC aggregation was increased. These alterations correlated with the incidence of ischemic stroke in 13 of these patients, suggesting a link between abnormal RBC morphodynamics and the increased risk of arterial thrombosis in PV, although this requires confirmation in larger studies (119).
Collectively, emerging data highlight a possible connection between the JAK2 V617F mutation and deregulated Ca2+ signaling in PV RBCs and precursors, with the potential to contribute to autonomous erythropoiesis and thrombosis. Therefore, strategies to modulate Ca2+ signaling may be useful for PV treatment.
4 Calcium signaling deregulation in blood cancer
Similar to solid tumors (3, 120), many blood cancers remodel Ca2+ signaling to promote their cancerous properties. Altered expression or activity of Ca2+ channels, pumps, and effectors can lead to the activation of transcription factors involved in the control of cell survival and proliferation.
4.1 Plasma membrane calcium-permeable channels
A number of Ca2+ influx channels located on the plasma membrane have been reported to impact on leukemic cells. These include the non-selective cation channels such as the TRP family, purinoreceptors (P2X7), nicotinic acetylcholine receptor (nAChR), Piezo1, NMDA receptor, and the Ca2+ selective Orai1 channels (Figure 1). Table 1 provides a summary of such changes in different blood cancers, and an explanation of their functional effects follows.

Table 1 The differential expression of plasma membrane calcium channels and their relative contribution to the malignant phenotype in different blood cancers.
4.1.1 Transient receptor potential channels
It is thought that the primary physiological roles of TRP channels are perception of various sensations ranging from pain, pressure, temperature, taste and vision. However, evidence accumulates that TRP channels also regulate proliferation, differentiation, invasion, metastasis, autophagy and apoptosis of malignant cells (80, 81, 166–170). TRP channels have been shown to contribute crucial oncogenic functions in a number of hematologic malignancies (166, 167). Leukemia, lymphoma, myeloma and Waldenström macroglobulinaemia patient cells and cell lines have altered expression of TRP channels that has been linked with changes in cell proliferation, cell death and cell migration (165–167) (Table 1).
TRPM2 is overexpressed in cells from patients with acute myeloid leukemia (AML) and in AML cell lines (e.g. Kasumi-1, U937, KG-1, MV-4-11, SKNO1, THP-1, MonoMac-6, AML-193, MOLM13 and SHSY5Y) (129). TRPM2 depletion in AML cells and xenograft mouse models has anti-leukemic effects. TRPV2, TRPM7 and TRPC1 have been studied in chronic myeloid leukemia (CML) cell lines (K-562, KU812, MOLM-6 and 32D-p210) (127, 146, 147, 171). The silencing of TRPV2 induces significant apoptosis in K-562 cells (127), while inhibition of TRPM7 reduces cell proliferation and increases differentiation (146). In BCR::ABL1-expressing murine myeloid progenitor cells (32D-p210), TRPC1 expression is reduced and may be one of the factors associated with SOCE reduction in these cells (171).
TRPV1, TRPV6 and TRPM2 contribute to the growth of cells derived from acute lymphoblastic leukemia (ALL) (142, 172). TRPV1 activation by resiniferatoxin (an analog of capsaicin, a vanilloid agonist) induces apoptosis, interferes with cell cycle progression and decreases proliferation in both Jurkat T-cells and patient-derived T-ALL lymphoblasts; however, the affect of resiniferatoxin on non-leukemic cells was not tested (172). TRPV6 is one of the necessary elements for migration and oncogenic signaling in Jurkat T-cells (142). TRPM2 is crucial for cell cycle arrest and decreases apoptosis of irradiated Jurkat T-cells and Bcl-2-overexpressing T-lymphoblasts (143).
In chronic lymphocytic leukemia (CLL) cells, patient-derived and the Jok-1 cell line, TRPC1 plays a role in promoting cell survival. It does so by contributing to the production of anti-inflammatory cytokines and the activation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathways triggered by CD5 activation (150, 168). TRPML2 is associated with the sensitivity of plasma cell myeloma cell lines to ibrutinib and/or bortezomib treatment. TRPML2 expression is low in ibrutinib-resistant U266 cells but high in ibrutinib-sensitive RPMI8226 cells (163). Upon TRPML2 RNA-silencing, RPMI8226 cells show worse response to ibrutinib than controls (163). These data raise the possibility that TRPML2 expression levels may help predict ibrutinib sensitivity in patients with myeloma (163).
Most recently, somatic mutations and copy number variations in TRP genes have been reported in 33 cancer types including hematologic malignancies, in particular diffuse large B-cell lymphoma (DLBCL) and AML cells (173). TRP mutations in the transmembrane regions were concluded to be likely deleterious and these genetic alterations were possibly linked to transcripitional deregulation of TRP genes and the consequent change in expression of TRP channels (173). The frequency of mutations in TRP channels was higher in DLBCL than in AML cells, with TRPM2, TRPM3 and TRPM6 showing the greatest mutation frequency (173). However, it is not clear what significance these genetic alterations have in the pathogenesis of cancer. Further work is required to uncover how these mutations contribute to cancer initiation and progression, and whether they can serve as markers for diagnosis, prognosis, or as treatment targets (173). Ex vivo studies with patient-derived cells demonstrate that targeting of TRP channels offers potential to inhibit malignant cell proliferation and improve chemotherapy effects (129, 172).
4.1.2 Purinoreceptor channels
P2X receptors are a family of ATP-dependent cation channels that have seven members (P2X1–7). An increase in extracellular ATP, often due to damage to the plasma membrane or exocytosis of ATP-containing granules, is the principal physiological stimulus for P2X receptor activation (174). Altered expression or function of P2X7 has been reported in a number of hematologic cancers (175). P2X7 is upregulated in cells from patients with AML and CLL and downregulated in B-ALL (124). Reports have differed on whether P2X7 in CML cells is up- or down-regulated (121, 124). When P2X7 activation is prolonged, and the receptor is exposed to high ATP levels, P2X7 opens an unselective membrane macropore and can trigger cell death (175, 176). P2X7RB is a splice variant that is unable to form this macropore (176). Both full-length P2X7RA and truncated variant P2X7RB are overexpressed in AML cells; whereas in relapsed AML patients, P2X7RB is increased and P2X7RA is decreased (126). AML blasts with high levels of P2X7RB have higher viability and much lower Ca2+ uptake than those expressing high levels of P2X7RA (126). AML development is slower and overall survival is extended in mice transplanted with P2X7-null AML cells compared to mice transplanted with control AML cells (125). Ca2+ influx is decreased in murine P2X7-null leukemia-initiating cells (LICs) and bulk AML cells compared to wild-type. The transcription factor cAMP-response element binding protein (CREB), which is involved in calcium signaling, is decreased in P2X7-null LICs and upregulated in AML patients. When CREB is overexpressed in P2X7-null AML cells, the development of leukemia is similar to wild-type AML cells (125). These results suggest that CREB-mediated Ca2+ signaling is required for the leukemogenic activities of P2X7.
4.1.3 Nicotinic acetylcholine receptor
Upon binding acetylcholine, nAChR channels assist the movement of cations into the cell, which causes membrane depolarization (177) and triggers the opening of voltage-gated Cav channels leading to Ca2+ influx (151). Homomeric α7-nAChRs are more permeable to Ca2+ and desensitize faster than heteromeric nAChRs (177). Primary CLL cells express α7-nAChR at a higher level than normal B-cells, and inhibiting α7-nAChRs in a range of leukemic cell lines reduces cell migration (151). Conversely, protein expression levels of α7-nAChRs in AML, CML and ALL patient peripheral blood or bone marrow-derived mononuclear cells was lower than in healthy subjects (178). Acetylcholine causes an increase in intracellular Ca2+ levels in CML-derived K-562 cells, and the α7-nAChR antagonist methyllycaconitine citrate inhibits K-562 cell proliferation as well as reduces the intracellular Ca2+ levels (177). The opposite was observed in Jurkat T-ALL cells, with methyllycaconitine causing intracellular Ca2+ levels to rise but this did not require extracellular Ca2+ (179).
4.1.4 N-methyl-D-aspartate receptor
Typical neuronal NMDA receptors are ligand-gated non-specific cation channels with high Ca2+ permeability activated by glutamate and glycine (93). In non-neuronal cells, including in megakaryocytes, NMDA receptors may also function in a metabotropic-like (i.e. flux independent) manner (97, 98, 180, 181) (see Figure 3A and the corresponding legend for molecular details). In leukemic cell lines with megakaryocytic features Meg-01, K-562, and Set-2, NMDA receptor supports cell proliferation (182). Deletion of NMDA receptor in Meg-01 cells shifts cell differentiation toward the erythroid lineage, suggesting NMDA receptor function at the level of a bipotential megakaryocyte-erythroid progenitor (183). NMDA receptor inhibitor memantine enhances cytotoxic effects of cytarabine in Meg-01 cells, thus this drug combination warrants testing on patient cells (183). In non-leukemic mice, the NMDA receptor regulates proplatelet formation through a mechanism that involves megakaryocyte interaction with the extracellular matrix and cytoskeletal reorganization (180). NMDA receptor exerts these effects by influencing Ca2+ and adenosine diphosphate (ADP) signaling, and the expression of transcripts involved in extracellular matrix remodeling (180) (Figure 3B). These mechanisms are relevant to the pathophysiology of primary myelofibrosis (PMF); therefore, NMDA receptor inhibitors should be tested in PMF models.
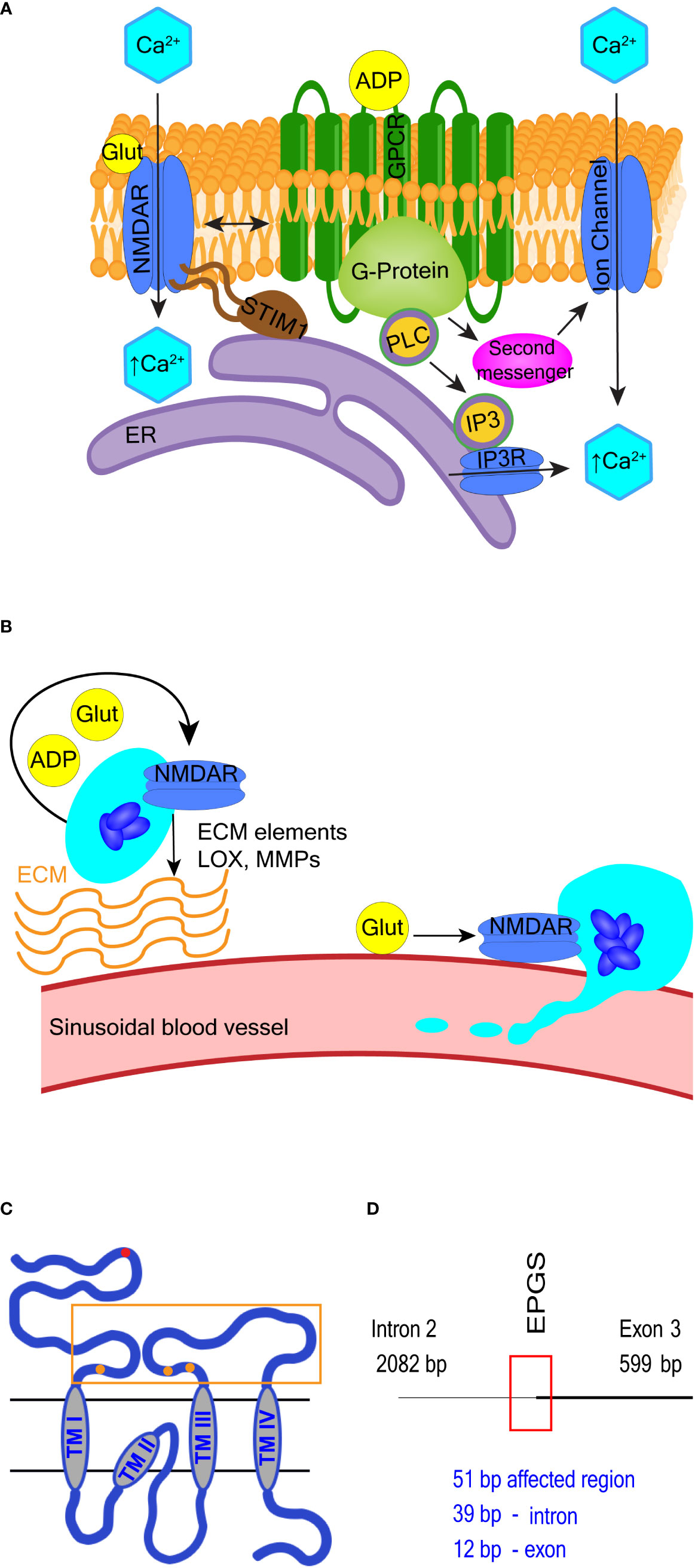
Figure 3 Selected NMDA receptor effects in hematopoietic cells. (A) Overview of NMDA receptor-induced calcium signaling. NMDA receptor directly facilitates Ca2+ entry into cells but may also operate in a metabotropic manner to induce Ca2+ release from the ER or via secondary messenger activation of ion channels such as transient receptor potential (TRP) channels. Adenosine diphosphate (ADP) and glutamate are both released from maturing megakaryocytes. ADP binds G protein-coupled receptors (GPCR) and activates PLC-β to increase intracellular Ca2+ levels. NMDA receptor modulates GPCR function in neuronal cells, so potentially may do so in hematopoietic cells. (B) Overview of NMDA receptor-associated effects in megakaryocytes. NMDA receptor assists proplatelet formation by regulating the expression of extracellular matrix (ECM) elements (e.g. collagen) and ECM remodeling enzymes (e.g. lysyl oxidase, LOX and matrix metalloproteinases, MMPs). (C, D) Schematics of NMDA receptor subunit GluN2C and the GRIN2C gene variants discovered in B-ALL. In (C), the glutamate-binding domain (400-539; 659-800 aa) is enclosed in an orange rectangle, and glutamate binding sites are represented by orange dots (at 509-511, 516, 687-688, and 729 aa respectively). Location of GRIN2C variants found in B-ALL is marked by a red dot in (C) and red rectangle in (D) The affected region is 51 base pairs long; the EPGS sequence is translated (134-137 aa).
In a random survival forest model, variants in the GRIN2C gene encoding the GluN2C subunit of NMDA receptor were part of a group of 7 variant genes found to predict shorter event-free survival in high-risk pediatric patients with B-ALL (145). The mutated GRIN2C region in ALL covers an intron/exon boundary located in the GluN2C protein’s N-terminal domain (see Figures 3C, D for further details). The presence of GRIN2C mutations was associated with accelerated relapse in children with high-risk B-ALL, but their functional impact is not known. These findings call for experimental studies to determine the NMDA receptor role in normal and leukemic B-cell precursors.
4.1.5 Voltage-dependent anion channels
VDAC has three isoforms in mammals with VDAC1 being the most abundant (37, 104). VDAC1 is a key regulator of metabolite transfer between the mitochondria and cytosol including of ATP, ADP, and of small ions such as Ca2+ and Na+. These functions are crucial for normal mitochondrial bioenergetics (37, 184). In its open state, VDAC1 facilitates metabolite exchange but is lowly permeable to Ca2+. In contrast, in the “closed” state VDAC1 is highly permeable to Ca2+ providing a proapoptotic signal (37, 185).
VDAC1 is overexpressed in U266 myeloma cells, which together with CD45 expression enhances the cells sensitivity to apoptosis via mitochondrial pathways (186). VDAC1 is also overexpressed in CLL patient cells compared to healthy controls (187). VDAC1-derived decoy sequences (Antp-LP4 and N-terminal-Antp) induce cell death in peripheral blood mononuclear cells from patients with CLL but not from healthy donors (187). Similarly, in a large panel of leukemic cell lines including from CLL (MEC-1, MO1043, and CLL), T-ALL (MOLT4, Jurkat), and AML (U-937, THP-1, K-562), VDAC1-targeting peptides induce cell death (188).
VDAC1 associating with Bcl-2, Bcl-xL or hexokinase prevents apoptosis in cancer cells. VDAC1 peptides disrupt this association leading to VDAC1 oligomerization, mitochondrial Ca2+ overload, cytochrome c release and apoptosis (187, 188). Combined treatment of acute promyelocytic leukemia (APL) cell line HL-60 with melatonin and retinoic acid decreases VDAC1 expression, suggesting its leukemia-promoting role (189). In B-ALL cell lines, VDAC1 was upregulated after prednisolone treatment in three steroid-sensitive cell lines (697, Sup-B15, RS4;11) but unchanged in the steroid-resistant cell line (REH), suggesting that VDAC1 has a role in steroid-induced apoptosis (190). Similar was seen in T-ALL cells. Cell death occurred in Jurkat T-cells when either rice or human VDAC proteins were overexpressed, and the effect was blocked by ectopically expressed Bcl-2 (191). Overall, VDAC1 interactions with pro-survival proteins support anabolic metabolism and inhibit apoptosis thus maintaining leukemia growth. Strategies that target these interactions are being explored for treatment of leukemia, with T-ALL cells emerging as the most vulnerable to this form of manipulation (192, 193).
4.1.6 Voltage-gated ion channels (CaV channels)
Voltage-gated Ca2+ channels are coded by CACNA genes (calcium voltage-gated channel subunit alpha) and are subdivided into three families CaV1, CaV2 and CaV3 (194). CaV1 and CaV3 channels are expressed in many cell types while CaV2 are mostly expressed in neurons (106). CaV channels mediate Ca2+ influx in response to plasma membrane depolarization, influencing muscle contraction and neurotransmission, as well as secretion and gene expression in may cell types (137). CaV1 channels are activated by high voltage (> −40 mV with a peak at 0 mV) and mediate long-lasting (L-type) currents with slow inactivation. In contrast, CaV3 channels are activated by low voltage (around −60 mV with a peak at −20 mV) and mediate transient currents (T-type) with faster kinetics than the L-type currents. CaV2 channels are activated by high voltage and mediate P/Q-type, N-type and R-type Ca2+ currents (106).
Bioinformatic analysis of Oncomine, a web-based cancer microarray database of patient tissue revealed increased expression of CACNA transcripts in diverse cancer types including of CACNA1S and CACNA1C (coding for CaV1.1 and CaV1.2 channels respectively) in AML and B-ALL samples and of CACNA1A (coding for CaV2.1) in samples from patients with CLL, marginal zone lymphoma and monoclonal gammopathy of unknown significance (136, 137). On the other hand, downregulation of CACNA1C transcripts (coding for CaV1.2) was seen in centroblastic lymphoma, CACNA1F (coding for CaV1.4) in anaplastic large cell lymphoma, and CACNA1G (coding for CaV3.1) in mantle cell lymphoma (195). A recent study confirmed distinct expression of CaV channels in a range of lymphoma cell lines and patient-derived samples (156). CaV1.2 (CACNA1C) expression was increased in classical Hodgkin lymphoma cell lines when compared to other B-cell lymphoma cell lines. CaV1.3 (CACNA1D) showed higher expression in samples from patients with activated B-cell-like DLBCL (ABC-DLBCL), whereas expression of CaV1.1 (CACNA1S), CaV1.2 (CACNA1C), and CaV1.4 (CACNA1F) were higher in germinal centre B-cell like DLBCL (GCB-DLBCL) (156). Therapeutic potential of inhibiting CaV1.2 in AML was revealed in an elegant 3D-culture model that mimicked the human bone marrow niche and utilized AML-derived mesenchymal stromal cells (AML-MSCs) from pediatric patients (138). Inhibition of CaV1.2 channel in AML-MSCs using lercanidipine (an anti-hypertensive drug) interfered with leukemia growth ex vivo and in vivo, with no toxic effects on normal MSCs or healthy CD34-positive HSCs (138). Further work is required to determine the mechanism through which CaV channels influence blood cancer growth.
4.1.7 Orai1 channels
Multimers of Orai1 proteins form an ion pore in the plasma membrane that is highly selective for Ca2+. SOCE is triggered when Orai1 and STIM1/STIM2 proteins interact in response to ER Ca2+ store depletion. Increased expression of Orai1 or STIM1/STIM2 has been recorded in cell lines derived from AML (132, 133), T-ALL (134), CLL (11) and various lymphoma cell lines (134, 156). Mantle cell lymphoma Rec-1 cell line does not have high expression of Orai1 and STIM1 but Rec-1 and patient cells have significantly higher cytoplasmic Ca2+ concentrations under physiological conditions compared to normal cells suggesting “leaky SOCE” (196). High expression of Orai1 and STIM1 in CLL patients is associated with worse treatment- and progression-free survival (11). In mice models of T-ALL, deletion of STIM1 and STIM2 abolishes SOCE and results in prolonged survival (134). The underlying mechanism is intriguing, as the absence of SOCE does not change leukemic cells proliferation; instead, prolonged survival is associated with reduced inflammation in organs infiltrated by leukemia (134). In the HL-60 APL cell line, silencing of Orai1 and Orai2 reduces cell migration and proliferation (132). In the KG-1 and U937 AML cell lines, Orai1 contributes to cell proliferation and cell cycle progression (133). ORAI1 gene expression was increased in peripheral blood mononuclear cells from 9 patients with AML compared with normal cells and correlated with adverse risk in the cohort of 439 AML patients (133).
Orai1 and STIM1 function is also linked with the CD20 molecule in B-cells and required for the efficacy of anti-CD20 antibody therapy in B-cell cancers. CD20 (MS4A1) is part of the membrane-spanning 4-domain family, subfamily A (MS4A) (197). The exact biological role of CD20 is unknown but it may act as an amplifier of Ca2+ signals transmitted through the B-cell receptor (BCR) in immature and mature B-cells to modulate cell proliferation and differentiation (198). CD20 has been reported to be physically coupled to or affect the phosphorylation of BCR and BCR-associated kinases; which are upstream regulators of the signaling cascade that activates SOCE (199–201).
Monoclonal anti-CD20 antibodies such as rituximab and obinutuzumab activate Ca2+ influx in patient CLL cells and cell lines such as SUDHL-4, BL2, Ramos, BL60, Raji, Daudi, and normal B-cells (202–206). Using genetically encoded Ca2+ indicator GCaMP-CD20 as a precise method to measure Ca2+ concentration changes around CD20, it was determined that anti-CD20 antibodies do not cause Ca2+ influx through or near CD20 (207). Instead, obinutuzumab activates intracellular Ca2+ efflux from either lysosomes or the ER into the cytosol (206) (see Figure 4A and the corresponding legend for molecular details). Inhibition of this intracellular Ca2+ movement reduces obinutuzumab-induced cell death (206, 207). Binding of rituximab to CD20 induces co-clustering of CD20 with Orai1 and STIM1 in SUDHL-4 cells, leading to extracellular Ca2+ influx and internal Ca2+ store release (205). CD20 overexpression in HEK293 cells increases Ca2+ influx, which is abolished when Orai1 and STIM1 are knocked down (207). CD20 strongly interacts with STIM1 but only when Orai1 is present (207). Influx of Ca2+ induced by rituximab or obinutuzumab is significantly reduced in Orai1 knockdown cells (205, 206). In B-CLL cells expressing high levels of STIM1, a combination of an anti-STIM1 monoclonal antibody and rituximab significantly reduces cell viability compared to rituximab alone (11). Thus, CD20 interactions with Orai1/STIM1 are important for the therapeutic efficacy of anti-CD20 antibodies. Manipulation of these interactions may help develop more effective therapeutic combinations for B-cell malignancies (Figure 4A).
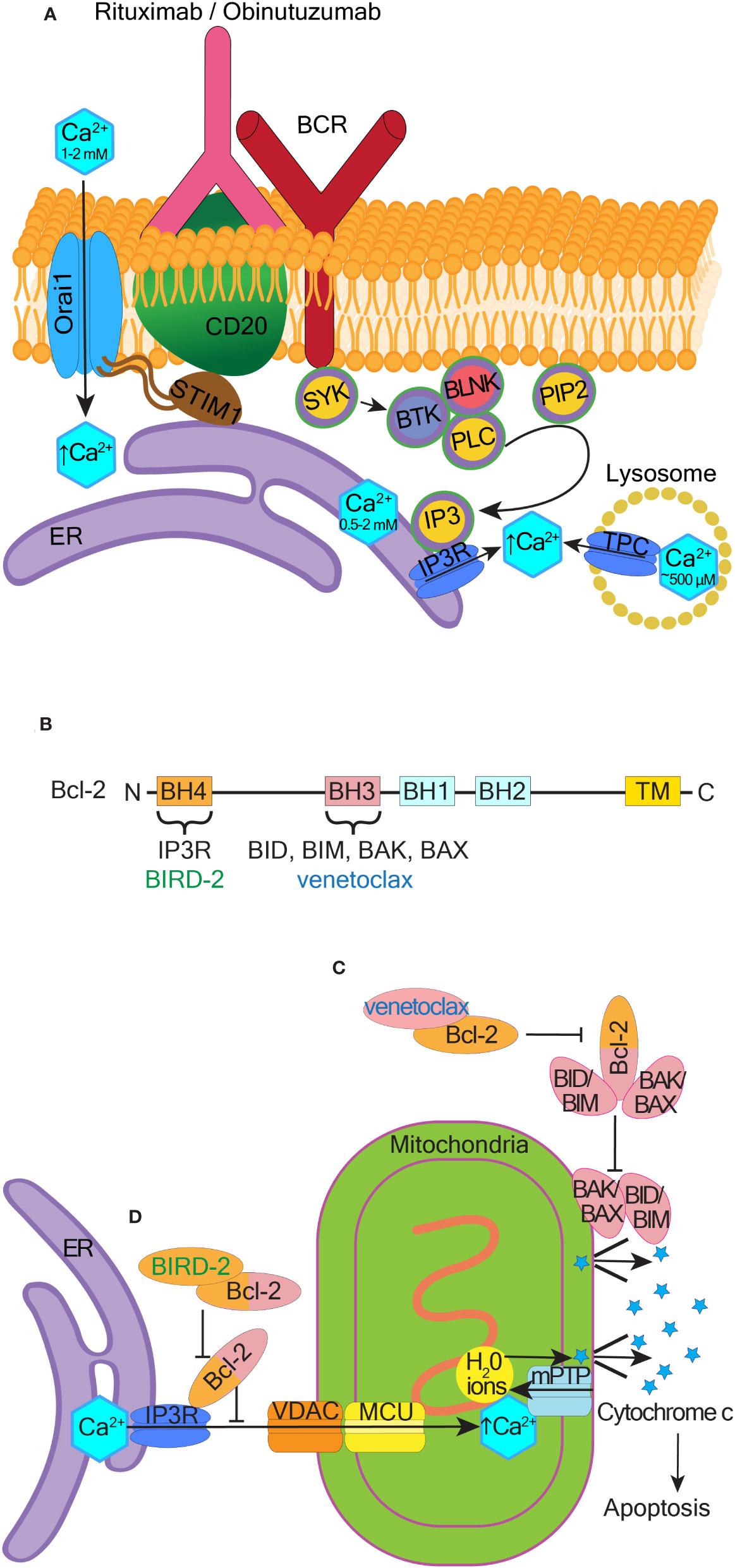
Figure 4 Therapeutic potential of pro-apoptotic calcium signaling at the ER and mitochondria. (A) Mechanism of BCR activated Ca2+ influx in response to rituximab and obinutuzumab. The membrane-spanning 4-domain protein CD20 is physically-coupled to the BCR. Rituximab and obinutuzumab induce phosphorylation of several proteins involved in BCR signaling, including BLNK (B-cell linker kinase), BTK (Bruton’s tyrosine kinase), and PLC-γ (phospholipase C-γ). CD20 binds STIM1 and this binding is dependent on the presence of Orai1. Upon binding of rituximab/obinutuzumab, Ca2+ is released from lysosomes, the ER, and/or extracellularly via activation of store-operated calcium entry, which assists cell killing. (B) Schematic showing the four Bcl-2 homology (BH) domains. Venetoclax binds to the hydrophobic cleft located in the BH3 domain, and BIRD-2 binds to the BH4 domain. Transmembrane domain (TM), N- and C- termini are indicated. (C) Canonical BAX and BAK dependent pathway of apoptosis and the mechanism through which venetoclax inhibits this pathway. (D) Non-canonical ER Ca2+-dependent pathway of apoptosis and the mechanism through which BIRD-2 inhibits this pathway.
4.2 IP3 signaling cascade and Ca2+ release from the ER
IP3 is a major regulator of Ca2+ signaling; it binds to IP3Rs on the ER to release Ca2+ into the cytosol (27, 28) (Figure 1). IP3 and another second messenger DAG are generated when PLC, activated downstream of G-protein coupled or tyrosine kinase receptors, hydrolyses PIP2 located in the plasma membrane (26).
4.2.1 Phospholipase C
PLC has six isoforms; β, γ, δ, ϵ, ζ, and η. PLC-β operates downstream of GPCRs and PLC-γ downstream of tyrosine kinase receptors (29, 30). Important activators of PLC-γ in hematologic cells are BCR and T-cell receptors (TCR), which are transmembrane tyrosine kinase receptors (208). T-cells mainly express the PLC-γ1 isoform, and B-cells mainly express the PLC-γ2 isoform. PLC-γ1 is essential for IP3 production and Ca2+ release in normal T-cells, whereas PLC-β3 is the main regulator of these responses in Jurkat T-cells and patient-derived T-ALL blasts (209).
Leukemic stem cells (LSCs) are multipotent, proliferative, and self-renewing cells that propagate leukemia (210). In AML LSCs, oxysterol-binding protein-related protein 4L (ORP4L) acts as a scaffold protein that facilitates PIP2 presentation to PLC-β3 for cleavage into IP3 (211). ORP4L is expressed in LSCs, but not in normal HSCs (211). Knocking down or inhibiting ORP4L decreases the survival of T-ALL cells and AML LSCs and reduces spontaneous cytosolic and mitochondrial Ca2+ oscillations (209, 211, 212). In T-ALL cells, ORP4L also interacts with PLC-β3 and IP3R1, which enhances Ca2+ release from the ER by facilitating the binding of IP3 to IP3R1 (212). Overall, the ORP4L regulated Ca2+ release into the mitochondria helps sustain mitochondrial oxidative respiration required for survival of T-ALL cells and AML LSCs (209, 211, 212)
BCR and TCR recruit kinases such as SYK (spleen tyrosine kinase), BTK (Bruton’s tyrosine kinase), BLNK (B-cell linker kinase), and ZAP70 (zeta chain of T-cell receptor-associated protein kinase 7) to phosphorylate and activate PLC-γ. PLC-γ2 plays a role in CLL, DLBCL, Hodgkin lymphoma, endemic Burkitt lymphoma, MALT-associated gastric lymphoma, and plasma cell myeloma (208). For example, B-CLL cells showing high responsiveness after BCR engagement have higher PLC-γ2 activity and calcium signaling compared to non-responding cells (213). Patients with such hyperresponsive B-cells have a poorer prognosis than non-responders (213). Ibrutinib, which inhibits BTK and thus blocks PLC-γ2 signaling, has become an important and effective treatment for CLL and other B-cell lymphomas (208).
4.2.2 Inositol 1,4,5-trisphosphate receptors
Three isoforms of IP3Rs exist and most cell types express more than one isoform (214). Mice with all three IP3R isoforms deleted develop T-cell malignancies throughout the body that resemble T-ALL (215). Cytogenetically normal AML cells have higher expression of IP3R2 than healthy progenitors and patients with high IP3R2 expression have shorter overall and event-free survival (135). DLBCL SU-DHL-4 cells also have high levels of IP3R2 and constitutive IP3 signaling, which leads to elevated basal levels of cytoplasmic Ca2+ and increased cell survival (154). Inhibition of IP3 production via inhibiting PLC reverses the prosurvival effect and increases cell death in SU-DHL-4 cells (154).
Several oncogenes and tumor suppressors directly interact with IP3Rs and regulate their activities to control Ca2+ influx into the mitochondria. Among such IP3R regulators are the Bcl-2 family proteins, which consist of different anti-apoptotic members (e.g. Bcl-2, Bcl-xL, Mcl-1, and Bcl-10) and pro-apoptotic members (e.g. BIM, BID, BAX, and BAK) (216) (Figure 4B). Bcl-2 overexpression is common in blood cancer, including in DLBCL, AML and CLL (154, 217, 218). Bcl-2 binds to and prevents the activation of pro-apoptotic proteins through their BH3 domains (219, 220). To overcome Bcl-2 effects in cancer cells, BH3 mimetics like venetoclax were developed that target the hydrophobic BH3-binding groove of Bcl-2 (Figure 4B). Venetoclax binding to Bcl-2 liberates BIM, which activates BAX or BAK, leading to apoptosis (221) (Figure 4C).
In addition to the canonical BAX/BAK-dependent pathway of apoptosis, Bcl-2 also directly binds to IP3Rs on the ER through its BH4 domain. The binding of Bcl-2 to IP3R inhibits Ca2+ release from the ER and prevents cell apoptosis triggered by mitochondrial Ca2+ overload (222). Venetoclax does not interfere with this BH4-dependent mechanism of cell death (223). In contrast, a designer peptide Bcl-2 IP3R Disruptor-2 (BIRD-2) targets the BH4 domain of Bcl-2 (224) (Figure 4B). BIRD-2 binding to Bcl-2 unleashes IP3R activation and cytotoxic Ca2+ levels are released from the ER (155, 225) (Figure 4D).
BIRD-2 induces apoptosis in multiple blood cancer cell lines and/or patient-derived cells, including from DLBCL, CLL, plasma cell myeloma, and follicular lymphoma (153, 226, 227). DLBCL cells with high levels of IP3R2 and constitutive IP3 signaling are particularly sensitive to BIRD-2 (154). In DLBCL cell lines, BIRD-2 induces cell death in a caspase-dependent manner, however in contrast to venetoclax, BIRD-2-induced cell death is independent of BAX/BAK (228). In both DLBCL cell lines and primary CLL patient samples, BIRD-2 triggers mitochondrial Ca2+ overload to induce caspase-dependent cell death. Cyclosporine A, which desensitizes mPTP to excessive Ca2+-induced opening, and ruthenium265 (Ru265), which inhibits mitochondrial Ca2+ uptake, both counteract BIRD-2-induced cell death (228). Combining venetoclax with BIRD-2 enhances cell death of DLBCL cell lines, however DLBCL cells with acquired resistance to venetoclax were not sensitized to BIRD-2 (155, 229). BIRD-2 may be useful in combination with venetoclax as a therapeutic strategy in DLBCL, however the clinical relevance of these approaches remains to be determined.
Another therapeutic target that can induce mitochondrial Ca2+ overload is the GPCR neurokinin-1 receptor (NK-R1), expression of which is elevated in patient-derived AML cells and cell lines (230). Targeting NK-R1 with the antagonists SR140333 or aprepitant in AML and CML cell lines increases cytosolic and mitochondrial Ca2+ concentrations, resulting in increased production of reactive oxygen specied (ROS) and apoptosis (230). When IP3R or VDAC1 are inhibited, ROS production and apoptosis are decreased (230), but neither antagonist inhibits proliferation of normal CD34-positive HSCs. Aprepitant has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting (230). Therefore, this and other NK-R1 antagonists could be repurposed for testing efficacy in AML (230).
4.3 Endo-lysosomal Ca2+ channels
The role of endo-lysosomal Ca2+ signaling in blood cancer has not been systematically studied but there are a number of observations pointing towards its importance. TRPML3, TPC1 and TPC2 endo-lysosomal Ca2+ efflux channels are expressed in the megakaryoblastic leukemia cell line Meg-01 (231). NAADP releases Ca2+ from the lysosomal-like Ca2+ stores in Meg-01 cells and TPC knockdown reduces this response (231). TPC2 is localized to platelet dense granules that are lysosome-related organelles and is involved in their maturation and function (232). TPC2 mediates Ca2+ release and formation of perigranular Ca2+ nanodomains in Meg-01 cells that mark “kiss-and-run” events mediating material transfer between different granules (232). Upon genetic deletion of NMDA receptors in Meg-01 cells, accumulation of lysosomal organelles and upregulation of MCOLN3 transcripts (coding for TRPML3) were observed. This suggests a link between lysosomal biogenesis, NMDA receptor function and Meg-01 cell proliferation (183). TPC1 and TPC2 inhibitor tetrandrine suppresses growth and increases cell death in several AML cell lines (U937, NB4, K-562, HL-60 and THP-1) (233–236). A recent study also demonstrates that TPC2 inhibition and its genetic deletion sensitizes ALL cells (cell lines and patient-derived) to cytotoxic drugs (237). Upon TPC2 deletion, leukemic cells are not able to sequester cytotoxic drugs within lysosomes, which increases drug concentration in the cytoplasm and enhances its cytotoxic effectiveness. Therefore, targeting lysosomal TPC2 may help overcome chemoresistance in ALL cells (237). Similar may apply in AML, although different mechanisms may contribute to lysosomal deregulation in different types of leukemia (238–240).
4.4 Calcium ATPases and secondary-active Ca2+ transporters
Several types of Ca2+ ATPases are involved in the maintenance of transmembrane Ca2+ gradients between the cytosol and the blood plasma as well as between the cytosol and the inner compartments of the organelles. Plasma membranes are equipped with several isoforms of the plasma membrane calcium ATPases (PMCAs) (241, 242) (Figure 1). Human monocytes and macrophages express plasma membrane Na+/Ca2+ exchanger NCX that actively extrudes Ca2+ while taking in Na+ transported passively using the energy of transmembrane Na+ gradient generated by the Na+/K+ ATPase (243). Mitochondrial membrane contains its own Na+(Li+)/Ca2+ exchanger (NCLX) that controls Ca2+ levels in the mitochondria (244). Cells also pump Ca2+ out of the cytoplasm into the Golgi apparatus through secretory pathway Ca2+ ATPases (SPCA), and to the ER through SERCA (245) (Figure 1). The activity of SERCA reflects the activation state of T-cells (246), B-cells (247), Th1 and Th2 lymphocytes (248). Of these molecules, SERCA has been reported to be dysregulated in a number of hematologic malignancies.
4.4.1 Sarco-endoplasmic reticulum calcium ATPase
There are three SERCA genes in humans and alternative splicing can give rise to many isoforms (10). In response to differentiation, SERCA3 expression changes in leukemic megakaryocytic cell line Meg-01, precursor B-ALL cell lines (Kasumi-2 and RCH-ACV), and APL cell lines (NB4 and HL-60) (249–251). All-trans retinoic acid-induced differentiation of APL cells results in increasing SERCA3 expression and SERCA3-dependent Ca2+ accumulation (249). When SERCA activity is inhibited, lower concentrations of retinoic acid can induce differentiation of NB4 and HL-60 cell lines (252).
In a human T-ALL xenograft mouse model, SERCA inhibition with thapsigargin reduces tumor growth (253). Thapsigargin prevents the activation of the transmembrane receptor, NOTCH1, which often contains activating mutations in T-ALL, CLL, mantle cell lymphoma, and a subset of DLBCL (10, 253). The reduction of Ca2+ entering the ER upon SERCA inhibition changes the folding and trafficking of NOTCH1 (10, 253). As reviewed by Pagliaro et al, other SERCA inhibitors have been developed that induce apoptosis in a range of leukemic cell lines and xenograft models including T-ALL, B-ALL, mantle cell lymphoma, and AML (10). In contrast, when SERCA expression is reduced or its activity is inhibited by oncogenes such as Bcl-2 and Ras, the decrease in ER Ca2+ concentrations reduces the potential for a Ca2+ overload and initiation of apoptosis in response to ER stress (219, 254).
4.5 Calcium sensor and effector proteins
Some of the downstream Ca2+ sensor proteins implicated in blood cancer include the S100 family, calcium/calmodulin-stimulated protein kinases (CaMKs), calcineurin, CALR, and PKC. Dysregulation of these Ca2+ sensors alters gene regulation associated with cell apoptosis, proliferation, and migration (Table 2).
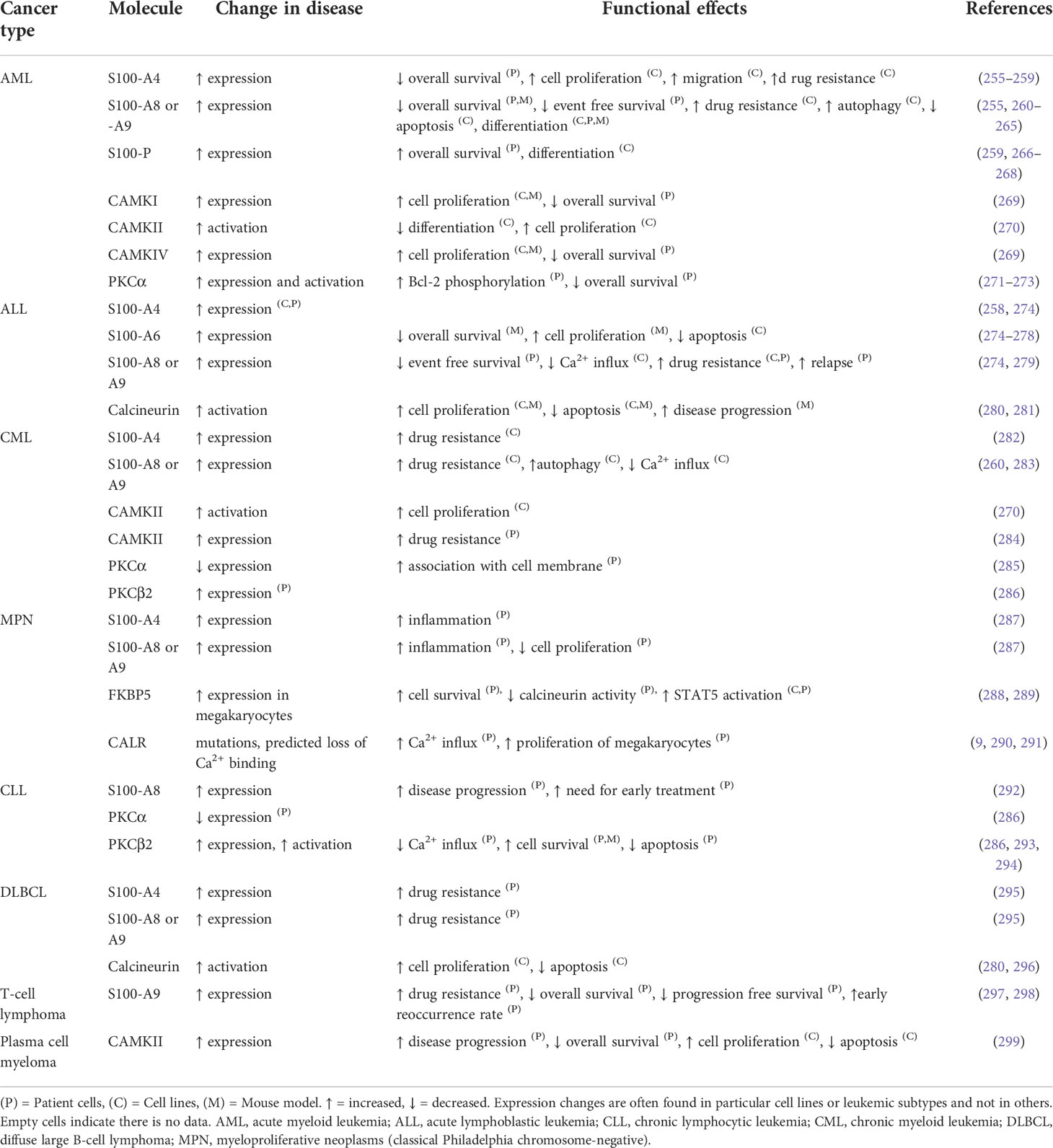
Table 2 The differential expression of calcium sensor and effector proteins and their relative contribution to the malignant phenotype of different blood cancers.
4.5.1 S100 family
The S100 family are Ca2+ binding proteins of which many have been reported to be dysregulated in AML, ALL, CLL, MPN, and lymphomas (292, 295, 300, 301). S100- A8 and A9 are the most well-studied members of the S100 family in leukemia. Dysregulation of their expression, and changes in plasma levels, or secretion levels in the bone marrow microenvironment have been reported in AML (300, 301). S100- A8 and A9 are dose-dependent regulators of myeloid differentiation and leukemic cell proliferation and can play contradictory roles, depending on their expression levels as monomers, homodimers, or heterodimers (300, 301). S100- A8 and A9 are expressed constitutively in the cytoplasm of myeloid cells, including myeloid precursors, but are absent from lymphocytes (301). Increased activity of multiple S100 family members is associated with increased drug resistance in hematologic malignancies including AML, CML, ALL, and B-cell lymphomas (302, 303). For example, S100-A8/A9 contribute to gilteritinib resistance in FLT3- internal tandem duplications- (FLT3-ITD)-positive AML primary cells and cell lines (304). Particularly, S100-A9 expression has been found to be more consistently and remarkably altered than S100-A8 in human FLT3-ITD-positive AML cell lines (MOLM13 and MOLM13-RES) after gilteritinib treatment. The potential mechanism is gilteritinib promotes Bcl-6 dissociation from the S100-A9 promoter, which leads to upregulation of S100-A9 (304).
4.5.2 Calcium/calmodulin-stimulated protein kinase family
Calmodulin is a Ca2+-binding protein that regulates a wide variety of cellular processes via interaction with multiple target proteins (305). The CaMK family members are activated when bound to Ca2+-saturated calmodulin (306). CaMK family members are overexpressed or aberrantly activated in CML, AML, and plasma cell myeloma (269, 270, 284, 299, 307). Inhibition or knockdown of CaMKI, CaMKII, or CaMKIV reduces proliferation in different myeloid leukemia cells and multiple CAMK or calmodulin antagonists have been used to inhibit leukemic cell growth and proliferation (269, 270, 305–307). High expression of CaMKs is associated with a poor overall survival probability in patients with plasma cell myeloma or AML (269, 299). Deletion of CaMKII suppresses T-cell lymphomagenesis in mice, and T-cell lymphoma cell line growth (comprising H9, JB6, Jurkat, and SU-DHL-1) is suppressed when CaMKII activity is inhibited (308). CaMKII is also activated by the constitutively active tyrosine kinase BCR::ABL1 in CML cells. The tyrosine kinase inhibitor (TKI) imatinib, inhibits proliferation of BCR::ABL1 expressing cells and is accompanied by a rapid decrease in activated (autophosphorylated) CaMKII (270). In an inducible BCR::ABL1 cell line (TonB210.1), decreased BCR::ABL1 expression is also accompanied by a reduction of autophosphorylated CaMKII, and inducing BCR::ABL1 expression restores CaMKII autophosphorylation (270). CAMKII-γ is highly activated in CML LSCs and its aberrant activation accelerates CML blast crisis (309). In a mouse xenograft model of patient-derived CML cells, LSCs were eliminated by an ATP-competitive CAMKII-γ inhibitor berbamine (310).
4.5.3 Calcineurin
Calcineurin is a calcium-calmodulin-dependent phosphatase, that when activated by Ca2+ and calmodulin, dephosphorylates its substrates including nuclear factor of activated T-cells (NFAT) (311). Dephosphorylated NFAT proteins translocate into the nucleus to regulate the transcription of genes important for cell proliferation, growth, migration, differentiation, and survival (311). The calcineurin-NFAT pathway negatively regulates megakaryopoiesis (312). Inappropriate inhibition of this pathway may contribute to the pathological expansion of megakaryocytes and their precursors, in particular in the context of Down syndrome (313, 314) (see Figure 5A and the corresponding legend for molecular details). The calcineurin inhibitor FKBP5 (FK506 binding protein) is overexpressed in megakaryocytes from patients with PMF. FKBP5 overexpression in UT-7 cells (a factor-dependent human cell line with a megakaryocytic phenotype) promotes cell survival after cytokine deprivation, suggesting a pathway to disease development through the inhibition of calcineurin (288).
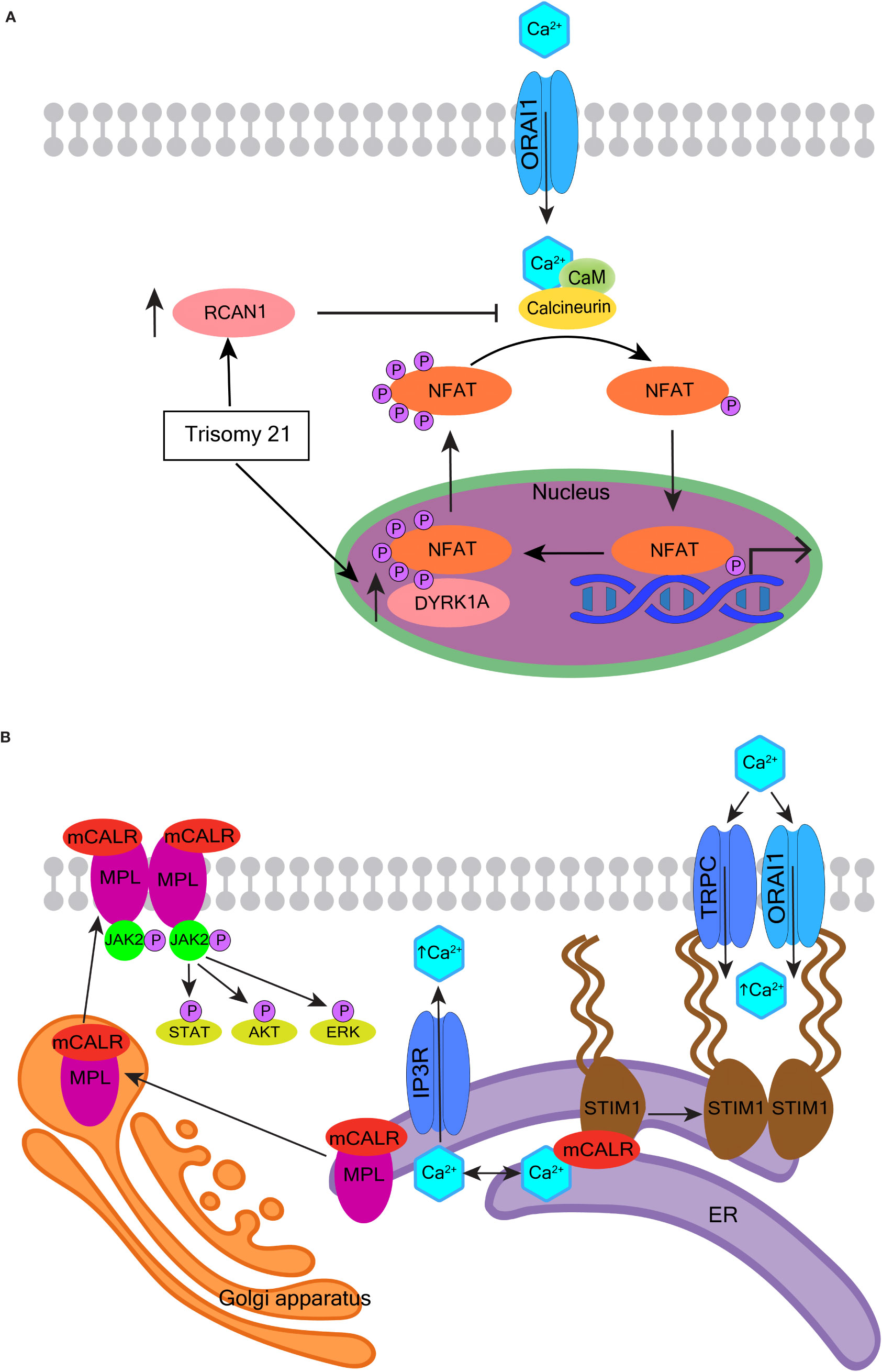
Figure 5 Oncogenic effects of calcineurin and calreticulin. (A) The role of calcineurin-NFAT signaling in the pathogenesis of myeloid proliferation associated with Down syndrome. Human chromosome 21 encodes two important regulators of nuclear factor of activated T-cells (NFAT) - regulator of calcineurin 1 (RCAN1) and dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A). When calcineurin is activated by Ca2+ and calmodulin (CaM), it dephosphorylates NFAT. Dephosphorylated NFAT translocates to the nucleus and transcriptionally regulates numerous genes involved in cell proliferation, growth, migration, differentiation, and survival. NFAT is re-phosphorylated by DYRK1A and is exported back to the cytoplasm. RCAN1 inhibits calcineurin, and so also inhibits the dephosphorylation and translocation of NFAT. RCAN1 and DYRK1A are overexpressed in Down syndrome and are suspected to contribute to the development of transient abnormal myeloproliferation and megakaryoblastic leukemia in Down syndrome children. (B) The role of mutated calreticulin (CALR) in myeloproliferative neoplasms. More than 50 mutations have been reported in exon 9 of the CALR gene; most generate a +1-frameshift causing the mutated CALR protein (mCALR) to stably associate with the thrombopoietin receptor MPL protein in the ER. The mCALR-MPL complex is transported from the ER through the Golgi apparatus and secretory system to the plasma membrane. The binding of mCALR to MPL constitutively activates signaling through JAK2 and its downstream targets such as STAT, AKT, and ERK (left). The most common mCALR variants are type 1 that also impair the Ca2+ binding activity of mCALR more than type 2. The type 1 mCALR with reduced Ca2+ binding dissociates from STIM1 in the ER. This allows STIM1 to dimerize and bind Orai1 and TRPC, which leads to constitutive activation of SOCE (right).
Calcineurin and NFAT have also been implicated in other hematologic malignancies, including Burkitt lymphoma, T-cell lymphoma, T-ALL, DLBCL, CML, CLL, and AML (311, 315, 316). The calcineurin inhibitors, cyclosporin A, and tacrolimus (FK506), have anti-leukemic effects in mice T-ALL models, and deletion of calcineurin specifically in T-ALL leukemic cells results in impaired leukemia progression (280, 281). Inhibition of calcineurin by cyclosporin A or FK506 is also selectively cytotoxic against the ABC-DLBCL (296). This response to calcineurin inhibitors is associated with reduced NFAT-mediated expression of critical genes, including c-Jun, signal transducer and activator of transcription 3 (STAT3), interleukin-6 and interleukin-10 that are crucial for survival of ABC-DLBCL cells (296).
4.5.4 Calreticulin
CALR, amongst its other functions, is an ER-resident Ca2+-buffering protein that helps maintain intracellular Ca2+ homeostasis and assists the folding of proteins destined for secretion or insertion into the plasma membrane (317). CALR is mutated in approximately one-quarter of the Philadelphia chromosome-negative MPNs, PMF and essential thrombocythemia (290, 318). The mutations in CALR, as well as JAK2 and MPL (Figure 5B, left), converge to constitutively activate JAK2-STAT signaling, which drives deregulated expansion of HSCs and megakaryocytes (317, 318). CALR mutations have two main variants, type 1 and type 2. Type 1 or type 1-like mutations are mostly large deletions of which a 52-bp deletion is the most common; while type 2 or type 2-like are mostly small insertions of which a 5-bp insertion is the most common. Type 1 mutations are predicted to impair the Ca2+-binding activity of CALR more than type 2 (319). Congruently, type 1 mutations associate with higher ER Ca2+ release, higher SOCE, and spontaneous cytosolic Ca2+ oscillations in cultured megakaryocytes (9, 290).
CALR assists the folding of STIM1, and whilst they are bound, STIM1 is in an inactive configuration on the ER membrane inhibiting SOCE (9). Megakaryocytes with mutated CALR have a decreased association between CALR and STIM1 (Figure 5B, right), and the CALR type 1 variant has the highest level of dissociation from STIM1. Defective interaction between mutant CALR and STIM1 activates SOCE and generates spontaneous cytosolic Ca2+ influx. This, in turn, increases megakaryocyte proliferation, which can be reversed using a specific SOCE inhibitor (9). Thus, the impact of mutated CALR on Ca2+ homeostasis may be influencing the course of MPN in combination with its aberrant activation of JAK2-STAT signaling. Further elucidation of these mechanisms may inform the development of new drugs to improve the effects of JAK2 inhibition.
4.5.5 Protein kinase C
PKC is activated by the second messenger DAG, which is hydrolyzed from PIP2 following receptor engagement and PLC activation (320, 321). The PKC family has many isoforms that can be categorized into three groups: conventional PKC isoforms, novel PKC isoforms, and atypical PKC isoforms (320, 321). A range of PKC isoforms are up- or down-regulated which can affect cell growth and survival in AML, CML, CLL, and plasma cell myeloma (286, 322, 323). Only the conventional PKC isoforms (α, β, and γ) are activated by Ca2+ as well as DAG (320, 321). In CML, the BCR::ABL1 phosphorylates a range of PKC isoforms leading to altered activity (323). ER Ca2+ release and SOCE are reduced in cell lines that express BCR::ABL1 (171, 324). These abnormal Ca2+ responses are Bcl-2 independent but PKC dependent. PKC-β overexpression is significantly associated with resistance to TKIs such as imatinib (323, 325). Suppressing PKC-β activity or expression in TKI-resistant CML patient cells and cell lines increases the sensitivity to imatinib (325). Inhibition of PKC-β increases the effect of imatinib on reducing leukemic cell proliferation in a CML mouse model and also prolongs survival (325). Outside of CML, PKC-β was found to be essential for CLL development in a mouse model and promotes cell growth and survival of CLL cells (294, 322, 326).
5 Mutational landscape in the calcium-toolkit encoding genes recorded in publically accessible blood cancers databases
We reviewed publically available genetic cancer databases for mutations affecting molecules described in this review across the main blood cancer types. Figure 6 demonstrates the mutational landscape in lymphoid cancers and Figure 7 in myeloid cancers. We obtained this data using cBioPortal for Cancer Genomics platform (https://www.cbioportal.org/) (354, 355). The Ca2+-toolkit encoding genes were queried as gene sets grouped according to function (see Supplementary Table S1). Results are observational only and require validation but are useful to generate hypotheses for future research and to assist discussion. The datasets available for interrogation and the samples reviewed are listed in Supplementary Tables S2 and S3. The lymphoid neoplasms reviewed included B-ALL (n = 234-269 patients depending on the gene), DLBCL (n = 1288), low-grade B-cell neoplasms (n= 1542) including CLL (n = 1254), monoclonal B-cell lymphocytosis (n = 54), mantle cell lymphoma (n = 29), plasma cell myeloma (n= 205) and low-grade T-cell neoplasms (n = 43) including Sezary syndrome (n = 26), primary cutaneous CD8/CD30 positive lymphomas (n = 14) and mycosis fungoides (n = 3) (Figure 6, Table 3 and Supplementary Table 2). Of these lymphoid cancers, patients with B-ALL had the lowest frequency of variants in the Ca2+-toolkit encoding genes (5.9%) and patients with low-grade T-cell neoplasms had the highest frequency (48.8%), most carrying multiple variants (Figure 6 and Table 3). The GRIN genes encoding NMDA receptor subunits and the ITPR genes encoding IP3Rs were mutated in 1.3% of B-ALL patients each, other variants were present in <1% of B-ALL patients. The cBioPortal data did not yet contain results of Bohannan et al. published earlier this year that reported the presence of GRIN2C mutations in high-risk B-ALL patients (145) (Figure 3C). It would be interesting to review these data when it becomes publically available, and pursue similar analysis in larger studies in the future.
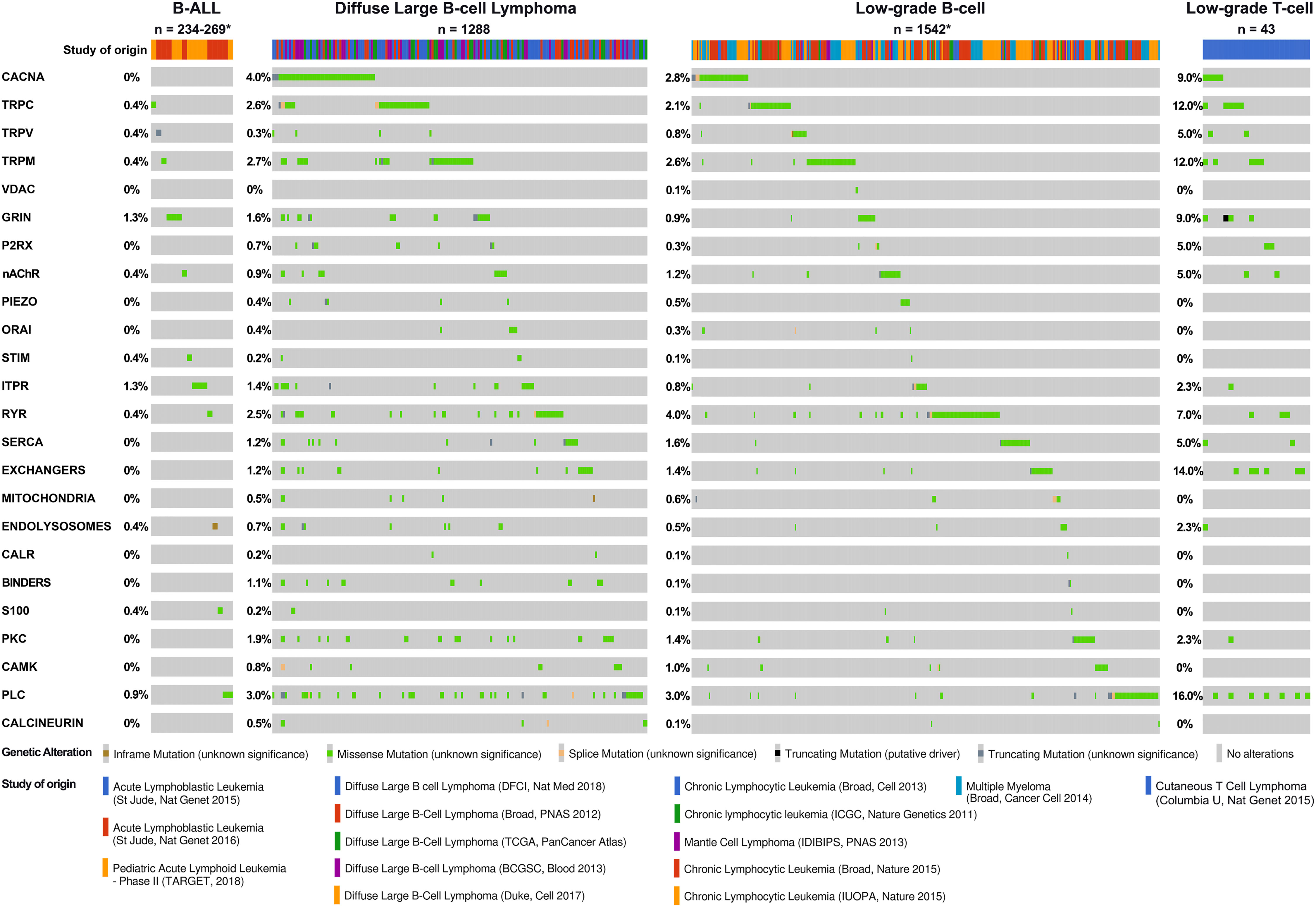
Figure 6 Calcium-toolkit mutations in lymphoid neoplasms. Oncoprints are shown generated using cBioportal for Cancer Genomics platform (https://www.cbioportal.org/). Events were analyzed per patient, frequencies are listed. Unaltered columns and whitespaces between columns are not shown. The Ca2+-toolkit genes were grouped according to function (see Supplemental Table 1). The molecular profiles queried included mutations but excluded copy number variations and structural variants as these were not available for most patients. The databases analyzed are indicated in the figure and referenced below: 1) For B-ALL: Acute Lymphoblastic Leukemia databases St Jude Nat Genet 2015 (327), St Jude Nat Genet 2016 (328) and Pediatric Acute Lymphoid Leukemia Phase II TARGET 2018. TARGET data was generated by the Therapeutically Applicable Research to Generate Effective Treatments initiative and is available at https://portal.gdc.cancer.gov/projects. The St Jude database also contained 8 T-ALL, 10 AML, and 5 unspecified leukemias - none had relevant mutations and these cases were excluded from the total. There were no other T-ALL cases with mutational data available for analysis so this cancer type could not be analyzed further. 2) For DLBCL: Diffuse Large B cell Lymphoma databases DFCI Nat Med 2018 (329), Duke Cell 2017 (330), Broad PNAS 2012 (331), TCGA PanCancer Atlas (332–340), and BCGSC Blood 2013 (341). 3) For low-grade B-cell neoplasms: Chronic Lymphocytic Leukemia databases Broad Cell 2013 (342), Broad Nature 2015 (343), IUOPA Nature 2015 (344), ICGC Nature Genetics 2011 (345), Mantle Cell Lymphoma database IDIBIPS PNAS 2013 (346), and Multiple Myeloma database Broad Cancer Cell 2014 (347). 4) For low-grade T-cell neoplasms: Cutaneous T Cell Lymphoma database Columbia U Nat Genet 2015 (348). Only patients with the appropriate diagnoses were selected. Specific cases analyzed are listed in Supplemental Table 2. The cBioPortal queries can be retrieved at the following links: B-ALL https://bit.ly/3Q9ZGvv; DLBCL https://bit.ly/3CZJpq8; low-grade B-cell neoplasms https://bit.ly/3AP87Xx; low-grade T-cell neoplasms https://bit.ly/3TvDZsT. Specific genetic variants can be found through these links, all were of unknown significance. *Numbers of patients analyzed and disease groups are clarified in Table 3.

Figure 7 Calcium-toolkit mutations in myeloid neoplasms. Oncoprints are shown generated using cBioportal for Cancer Genomics platform (https://www.cbioportal.org/). Events were analyzed per patient, frequencies are listed. Unaltered columns and whitespaces between columns are not shown. The Ca2+-toolkit genes were grouped according to function (see Supplemental Table 1). The molecular profiles queried included mutations but excluded copy number variations and structural variants as these were not available for most patients. The databases analyzed are indicated in the figure and referenced below: 1) Acute Myeloid Leukemia databases OHSU Nature 2018 (349); TCGA PanCancer Atlas (332–340), and Pediatric Acute Myeloid Leukemia TARGET 2018. TARGET data was generated by the Therapeutically Applicable Research to Generate Effective Treatments initiative and is available at https://portal.gdc.cancer.gov/projects. 2) Myelodysplasia databases UTokyo Nature 2011 (350)), MSKCC 2020 (349, 351, 352). 466 MDS patients were excluded from the analysis as they were not profiled for any queried genes. 3) Myeloproliferative Neoplasms database CIMR NEJM 2013 (353). Only patients with the appropriate diagnoses were selected. Specific cases analyzed are listed in Supplemental Table 3. The cBioPortal queries can be retrieved at the following links: AML https://bit.ly/3Ts6NlX; MDS https://bit.ly/3AuvJkc; MPN https://bit.ly/3q1TZ8C. Specific genetic variants can be found through these links, apart from CALR mutations all were of unknown significance. *Numbers of patients analyzed are clarified in Table 3.
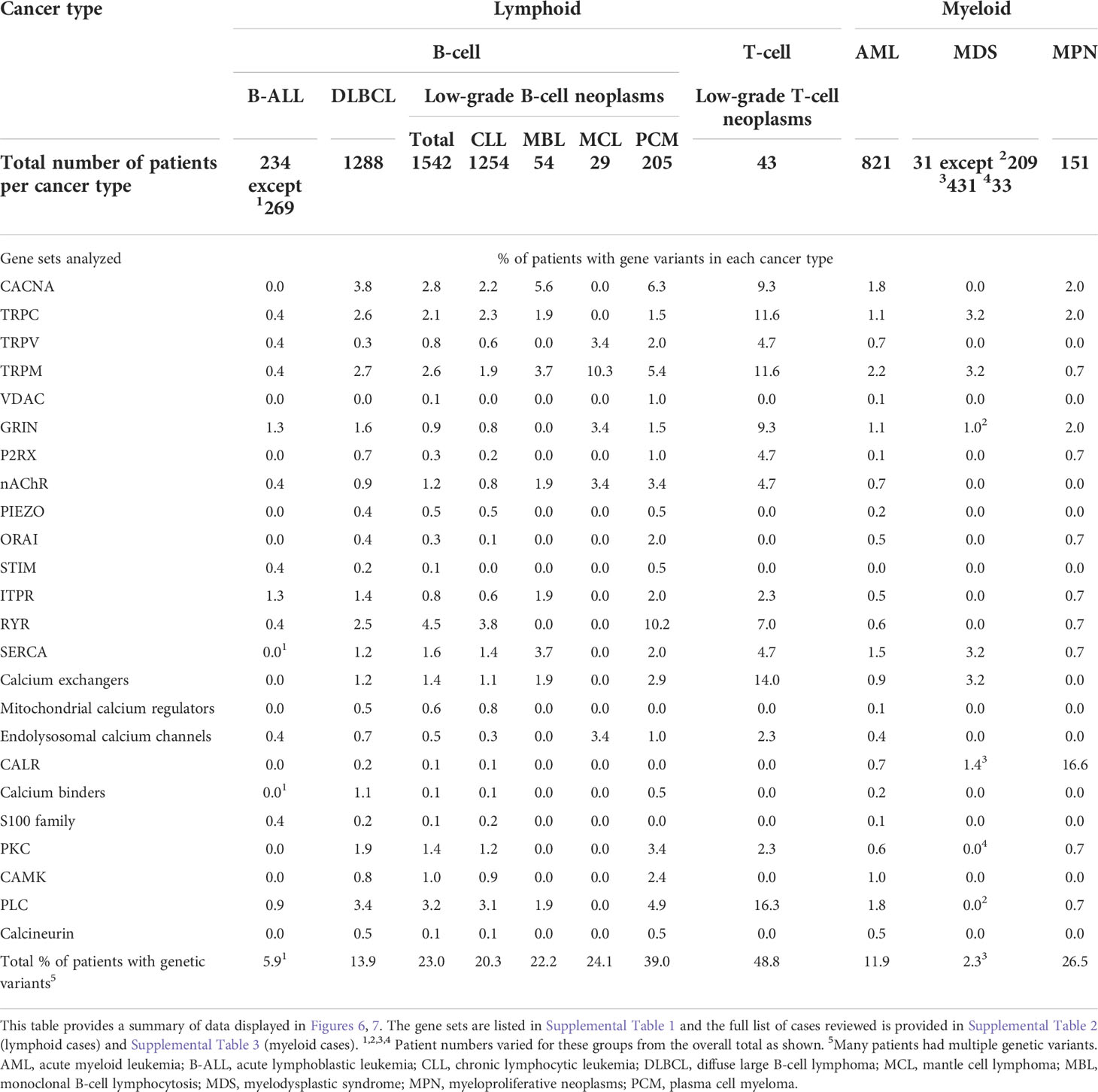
Table 3 Frequencies of genetic variants in the calcium-toolkit encoding genes documented in lymphoid and myeloid cancer databases.
In mature T-cell neoplasms, the most affected genes were those coding for PLC (16.3%), Ca2+/Na+/K+ exchangers (14.0%), TRPC and TRPM channels (11.6% each), CaV and NMDA receptor channels (9.3% each), ryanodine receptors (RYR) (7.0%), and a few others at 4.7% each (TRPV, P2RX, and SERCA) (Figure 6 and Table 3). This particularly high frequency of variants in cutaneous T-cell neoplasms requires confirmation in larger cohorts. Unfortunately, no other mature or precursor T-cell malignancies could be reliably reviewed. Less than 10 T-ALL samples with mutational data were identified and none had relevant mutations. Ca2+ signaling is critical for T-cell activation downstream of TCR and linked closely with the regulation of T-cell metabolism (356). Multiple studies highlighted the role of Orai, CaV, TRP, NMDA receptors and other Ca2+ regulators in normal and malignant T-cells (134, 192, 357–361). Further studies are required to examine potential contribution of these changes in T-cell cancers.
In mature B-cell neoplasms, mutated genes were broadly similar between the low-grade and high-grade cancers (Figure 6 and Table 3). The frequency of variants was higher in patients with low-grade B-cell neoplasms, 23.0% compared with 13.9% in DLBCL, but many patients with DLBCL had multiple variants. The types of variants were similar between patients with CLL, monoclonal B-cell lymphocytosis, mantle cell lymphoma and plasma cell myeloma. In low-grade B-cell lymphoproliferative disorders, variants in RYR1-3 were the most common (4.5%), followed by PLC (3.2%), CACNA (2.8%), TRPM (2.6%) and TRPC genes (2.1%), with others present in <2% of patients. Some of these mutations appeared exclusive between each other. In DLBCL, RYR mutations were less common (2.5%) but CACNA variants were more common (3.8%) (Figure 6 and Table 3). One could hypothesize that the acquisition of multiple mutations associates with a higher grade. Further bioinformatics analysis and laboratory studies to determine the role of gene variants in lymphoid cancers appears warranted. For example, experimental studies to examine the contribution of mutations in plasma membrane Ca2+ channels and RYRs would be of interest. The existing literature in this area is limited. RYRs facilitate the release of Ca2+ from the ER in addition to IP3Rs (362) (Figure 1). They are the largest ion channels known regulated by CaV1.1/CaV1.2-mediated Ca2+ entry, as well as other small molecules including ATP, calmodulin, calsequestrin and CaMKII (363). RYR1 is primarily expressed in skeletal muscles but is also present in B-lymphocytes (364, 365) and Burkitt lymphoma-derived Namalwa cells (366). The RYR role in B-cell function remains unclear but its activity downregulates CD38 expression (366). High levels of CD38 associate with poor risk CLL (367). Studies to determine the mechanism of the RYR contribution to the regulation of CD38 and the impact of RYR mutations on CD38 expression and B-cell activation may be helpful.
In the group of myeloid neoplasms, we reviewed 821 patients with AML, 151 with MPN and 31-431 patients with myelodysplastic syndrome (MDS) (depending on the gene) (Figure 7, Table 3 and Supplementary Table S3). Compared with B-ALL, AML patients carried more variants (11.9% versus 5.5% in B-ALL patients). Variants in CACNA (1.8%) and TRP genes (2.2% in TRPM, 1.1% in TRPC, 0.7% in TRPV) were the most common in AML but no variants exceeded 3% frequency. MDS patients had a lower frequency of variants at 2.3% overall, although data for these patients was limited. In the MPN group, 17% had CALR mutations, consistent with it being a known MPN driver (353, 368). In addition, a further 9.5% of MPN patients had variants in other Ca2+-toolkit genes (including 2.0% in CACNA, TRPC and GRIN genes each). It was intriguing that these variants were exclusive with CALR mutations and also appeared exclusive with each other, raising the possibility of their independent effects in MPN (Figure 7 and Table 3). Evidence slowly accumulates that Ca2+ signaling is aberrant in CALR-mutated MPN (9, 290, 291, 319). The pattern of the mutational landscape revealed by our review argues that further work into Ca2+ signaling in MPN is warranted, including in patients without CALR mutations.
6 Concluding remarks
In all cell types, including hematopoietic, Ca2+ is an essential second messenger controlling a wide range of cellular functions, including activation of gene transcription, protein kinase signaling, cell cycle, cell survival, proliferation, differentiation, migration, and apoptosis (2–4). Others have reviewed deregulation of Ca2+ signaling in specific cancer subtypes such as AML, CLL and plasma cell myeloma, and summarized the influence of Ca2+ remodeling on cell proliferation and differentiation (369–371). Our review is the first one, to our knowledge, that highlights the extent and intricacies of alterations in the Ca2+-toolkit in a wide range of hematologic cancers. We also provide a review of blood cancer databases for genetic variants in the Ca2+-toolkit encoding genes.
It has been well documented that in solid tumors, cancer cells remodel Ca2+ signaling to enhance cancer hallmarks (1, 2). Our review emphasizes that similar alterations occur in blood cancer, driven by changes in expression, function and possibly mutations in the Ca2+-toolkit components. Figure 8 provides a schematic summary of the underlying mechanisms and their consequences in blood cancer cells. Normal cells in response to activation mostly use Ca2+ released from the ER to support signaling. However, prolonged IP3R-mediated Ca2+ release in chronically activated cancer cells (e.g. due to oncogenic mutations) may lead to mitochondrial Ca2+ overload triggering apoptosis. Therefore, to escape apoptosis, cancer cells shift to preferentially utilize extracellular Ca2+ transferred into cells by overexpressed or overactive plasma membrane Ca2+ channels. On the other hand, mechanisms that utilize ER-derived Ca2+ may be reduced, and overactive SERCA2 pumps Ca2+ back into the ER. This remodeling of Ca2+ signaling helps cells maintain heightened oncogenic signaling while shielding mitochondria from Ca2+ overload. The examples of mechanisms providing anti-apoptotic effects include Bcl-2-mediated inhibition of IP3R in DLBCL and reduced ER Ca2+ binding by mutated CALR in MPN. In addition, intracellular mediators of Ca2+ signaling may be overexpressed or overactive e.g. PLC, PKC, ORP4L, calcineurin and CaMK. Understanding how Ca2+ signaling remodels in cancer cells creates therapeutic opportunities, with the potential to spare normal cells and tailor therapies according to the underlying mechanism in different cancers/patients (Figure 8).
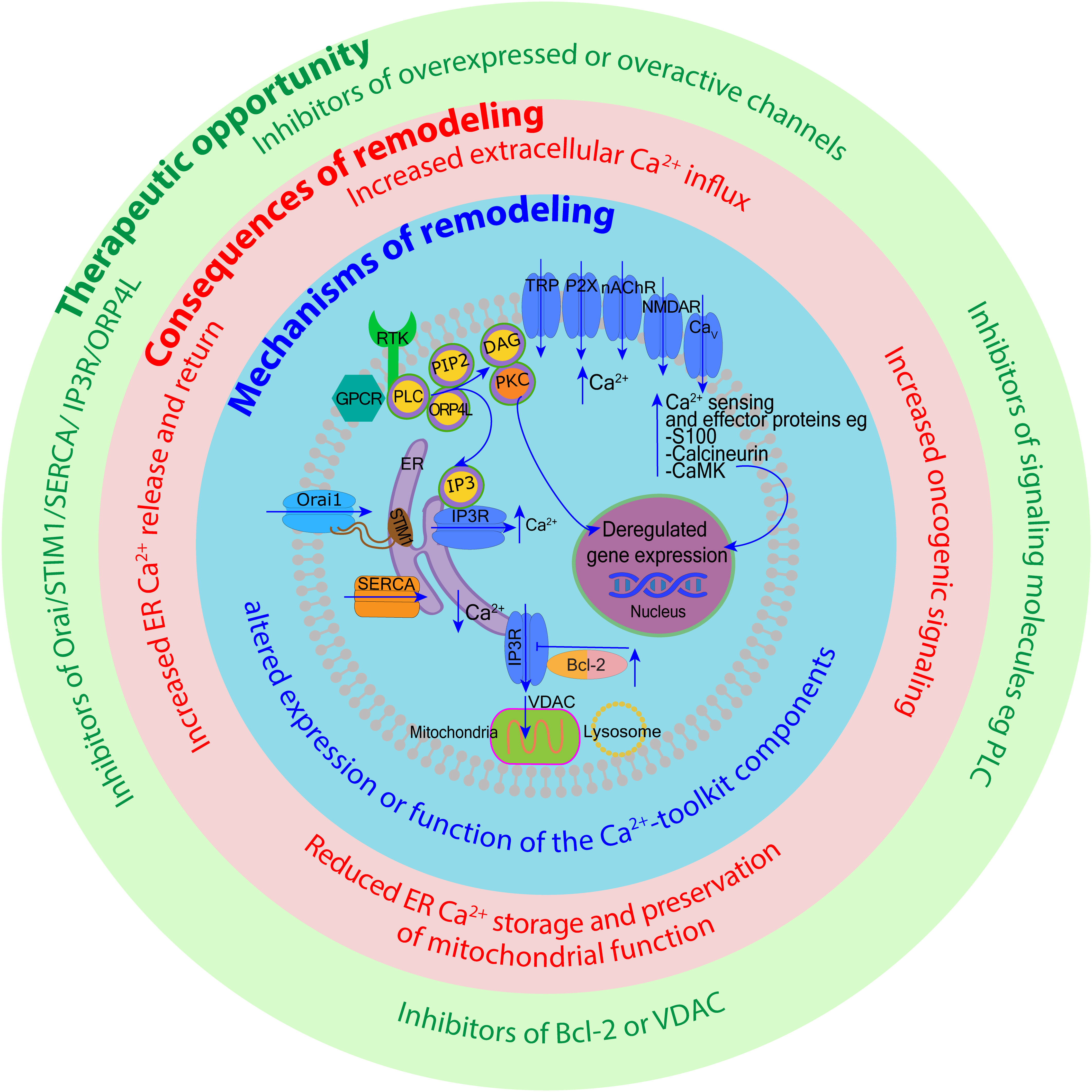
Figure 8 Remodeling of calcium signaling in hematologic cancers and therapeutic opportunities this presents. Examples of mechanisms responsible for the remodeling of Ca2+ signaling in blood cancer are depicted within the central blue circle. Their direct consequences are listed in the middle red circle, and therapeutic opportunities arising from these changes are highlighted in the outer green circle. For example, the increased expression of the plasma membrane Ca2+ channels (a mechanism of remodeling) leads to increased extracellular Ca2+ influx supporting oncogenic signaling (a consequence). Such changes could be counteracted by specific Ca2+ channel inhibitors (a therapeutic opportunity). In another example, mechanisms that spare cancer cells from mitochondrial Ca2+ overload (e.g. through the overexpression of Bcl-2 or VDAC) could be counteracted by inhibitors of these molecules. The design of novel therapies heavily relies on our understanding of the Ca2+-toolkit remodeling in different blood cancers.
Research into Ca2+ signaling in blood cancer has become very active in recent years. Many critical discoveries have been made but multiple challenges remain. We highlighted some areas for future investigation throughout this review, including the need to characterize diverse mechanisms of Ca2+ remodeling and determine the significance of the mutational landscape affecting the Ca2+-toolkit genes in different cancer types. Such work is not easy. Ca2+ signaling is a complex network of intertwining pathways and ubiquitous for cellular functioning. It can be difficult to identify the causes and consequences of the changes found in blood cancer. Most of the previous studies focused on a specific mechanism of Ca2+ signaling in isolation and used cell lines to characterize it. Moving forward, we should consider changes to the entire Ca2+ toolkit and use multiple disease models ex vivo and in vivo to study effects of multiple gene networks in cancer cells and stromal cells. This will require innovative approaches and collaboration between experts in Ca2+ signaling, hematological sciences and clinical hematologists.
The ultimate aim of pursuing research in this area is to improve the treatment of patients. Calcium pathways are amenable to modulation and may offer novel points for therapeutic intervention. The main targets/pathways were recently summarized for AML (371). Our Table 4 provides a range of examples of compounds/drugs that have been found to exert an impact on the functional outcome in vitro or in vivo in diverse blood cancer types. Some of these compounds are in clinical use for other applications or undergo pre-clinical/clinical testing in solid tumors (394, 395). If their targets are found to be pathogenic in blood cancer, these drugs could be rapidly transitioned to hematologic applications.
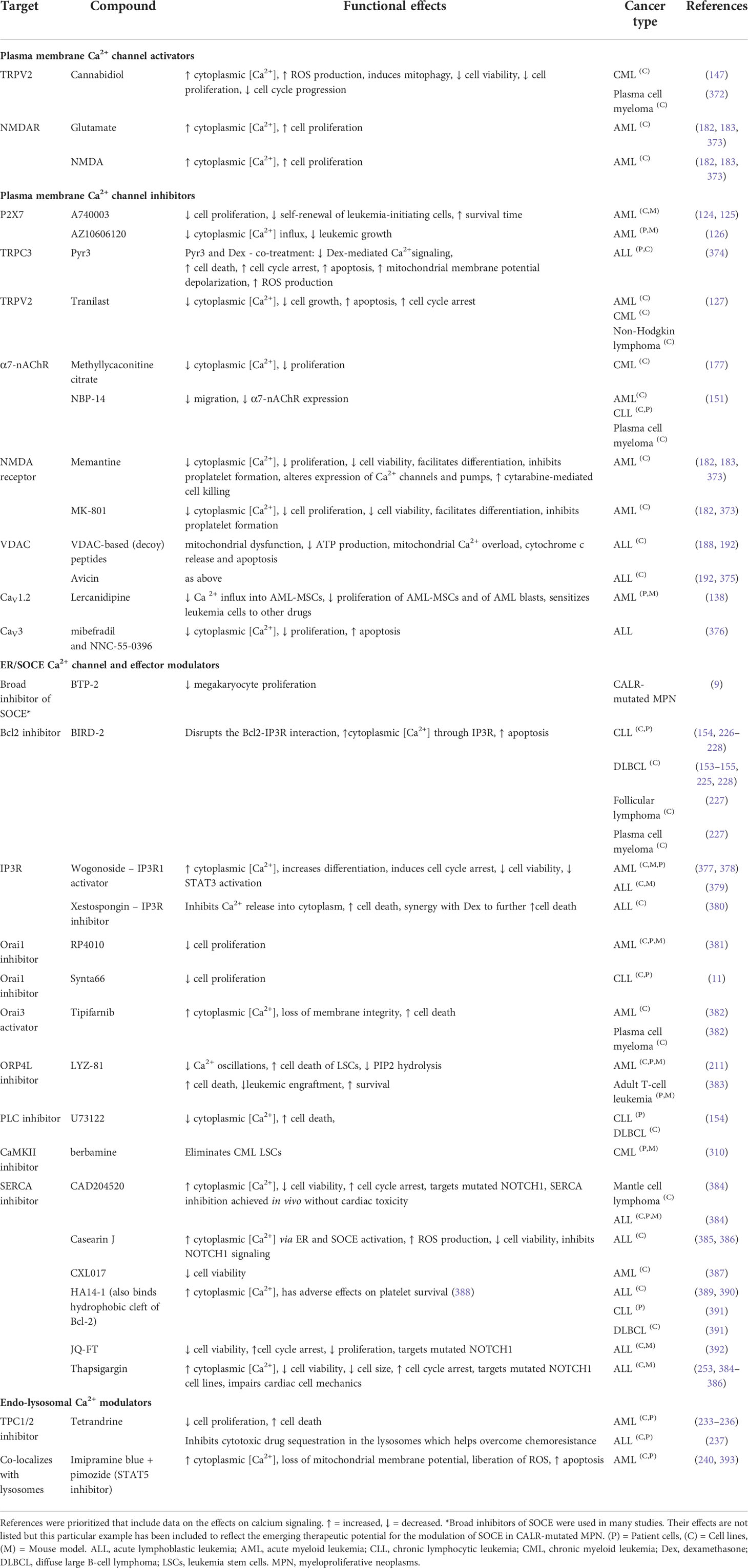
Table 4 Demonstrative examples of calcium-related compounds/drugs (specific inhibitors or activators of calcium channels and receptors) that have been found to exert an impact on the functional outcome in vivo or in vitro in diverse blood cancer types.
In conclusion, multiple Ca2+ homeostatic mechanisms and Ca2+ responsive pathways are altered in hematologic cancers. Some of these alterations may have genetic basis, including in MPN, B-cell and T-cell lymphoproliferative disorders, but studies are limited. Most changes in the Ca2+-toolkit do not appear to define or associate with specific cancer types but may influence variables such as grade (e.g. in mature B-cell neoplasms), prognosis including responsiveness to chemotherapy (e.g. in ALL, AML and CLL), and complications (e.g. thrombosis and bone marow fibrosis in MPN). Deregulation of Ca2+signaling provides an opportunity to design novel therapeutic interventions. Some options are currently investigated mostly at the pre-clinical level in various cancer models (e.g. of AML, ALL and DLBCL). Similar opportunities are being considered in solid tumours, which may facilitate faster clinical translation to blood cancer. Future research to define the role of specific Ca2+ regulatory mechanisms in different blood cancer types will be challenging but such work is likely to advance therapies.
Author contributions
MLK-Z conceived and designed the work. TI and MLK-Z wrote most of the paper. JL and TNG helped with the literature review, figure preparation, and writing of specific sections; JL wrote the TRP section, TNG wrote about VDACs and helped analyze the cBioPortal data. AB provided advice and wrote about calcium homeostasis in mature red cells. All authors contributed to the article and approved the submitted version.
Funding
Bone Marrow Cancer Research Trust (Christchurch) (UoA 9102-3720536) and Auckland Medical Research Foundation (Funder reference 1119009) provided salary funding to staff working on this project but had no influence over any aspects of the work or the decision to publish.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1010506/full#supplementary-material
References
1. Alharbi A, Zhang Y, Parrington J. Deciphering the role of Ca(2+) signalling in cancer metastasis: From the bench to the bedside. Cancers (Basel) (2021) 13(2):179. doi: 10.3390/cancers13020179
2. Patergnani S, Danese A, Bouhamida E, Aguiari G, Previati M, Pinton P, et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int J Mol Sci (2020) 21(21):8323. doi: 10.3390/ijms21218323
3. Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer (2017) 17(6):367–80. doi: 10.1038/nrc.2017.18
4. Tiffner A, Derler I. Molecular choreography and structure of Ca(2+) release-activated Ca(2+) (Crac) and Kca2+ channels and their relevance in disease with special focus on cancer. Membranes (Basel) (2020) 10(12):425. doi: 10.3390/membranes10120425
5. Audero MM, Prevarskaya N, Fiorio Pla A. Ca(2+) signalling and Hypoxia/Acidic tumour microenvironment interplay in tumour progression. Int J Mol Sci (2022) 23(13):7377. doi: 10.3390/ijms23137377
6. Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, et al. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (Mams). Biochim Biophys Acta Bioenerg (2017) 1858(8):615–27. doi: 10.1016/j.bbabio.2017.01.003
7. Villalobos C, Sobradillo D, Hernandez-Morales M, Nunez L. Remodeling of calcium entry pathways in cancer. Adv Exp Med Biol (2016) 898:449–66. doi: 10.1007/978-3-319-26974-0_19
8. de Ridder I, Kerkhofs M, Veettil SP, Dehaen W, Bultynck G. Cancer cell death strategies by targeting bcl-2's Bh4 domain. Biochim Biophys Acta Mol Cell Res (2021) 1868(5):118983. doi: 10.1016/j.bbamcr.2021.118983
9. Di Buduo CA, Abbonante V, Marty C, Moccia F, Rumi E, Pietra D, et al. Defective interaction of mutant calreticulin and soce in megakaryocytes from patients with myeloproliferative neoplasms. Blood (2020) 135(2):133–44. doi: 10.1182/blood.2019001103
10. Pagliaro L, Marchesini M, Roti G. Targeting oncogenic notch signaling with serca inhibitors. J Hematol Oncol (2021) 14(1):8. doi: 10.1186/s13045-020-01015-9
11. Debant M, Burgos M, Hemon P, Buscaglia P, Fali T, Melayah S, et al. Stim1 at the plasma membrane as a new target in progressive chronic lymphocytic leukemia. J Immunother Cancer (2019) 7(1):111. doi: 10.1186/s40425-019-0591-3
12. Bootman MD, Bultynck G. Fundamentals of cellular calcium signaling: A primer. Cold Spring Harb Perspect Biol (2020) 12(1):a038802. doi: 10.1101/cshperspect.a038802
13. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi: 10.1038/35036035
14. Snoeck HW. Calcium regulation of stem cells. EMBO Rep (2020) 21(6):e50028. doi: 10.15252/embr.202050028
15. Luchsinger LL, Strikoudis A, Danzl NM, Bush EC, Finlayson MO, Satwani P, et al. Harnessing hematopoietic stem cell low intracellular calcium improves their maintenance in vitro. Cell Stem Cell (2019) 25(2):225–40.e7. doi: 10.1016/j.stem.2019.05.002
16. Yeh SA, Hou J, Wu JW, Yu S, Zhang Y, Belfield KD, et al. Quantification of bone marrow interstitial ph and calcium concentration by intravital ratiometric imaging. Nat Commun (2022) 13(1):393. doi: 10.1038/s41467-022-27973-x
17. Samanta K, Parekh AB. Spatial Ca(2+) profiling: Decrypting the universal cytosolic Ca(2+) oscillation. J Physiol (2017) 595(10):3053–62. doi: 10.1113/JP272860
18. Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature (2006) 439(7076):599–603. doi: 10.1038/nature04247
19. Fukushima T, Tanaka Y, Hamey FK, Chang CH, Oki T, Asada S, et al. Discrimination of dormant and active hematopoietic stem cells by G0 marker reveals dormancy regulation by cytoplasmic calcium. Cell Rep (2019) 29(12):4144–58.e7. doi: 10.1016/j.celrep.2019.11.061
20. Umemoto T, Hashimoto M, Matsumura T, Nakamura-Ishizu A, Suda T. Ca(2+)-mitochondria axis drives cell division in hematopoietic stem cells. J Exp Med (2018) 215(8):2097–113. doi: 10.1084/jem.20180421
21. Bonora M, Kahsay A, Pinton P. Mitochondrial calcium homeostasis in hematopoietic stem cell: Molecular regulation of quiescence, function, and differentiation. Int Rev Cell Mol Biol (2021) 362:111–40. doi: 10.1016/bs.ircmb.2021.05.003
22. Jardin I, Lopez JJ, Sanchez-Collado J, Gomez LJ, Salido GM, Rosado JA. Store-operated calcium entry and its implications in cancer stem cells. Cells (2022) 11(8):1332. doi: 10.3390/cells11081332
23. Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harb Perspect Biol (2011) 3(3):a004069. doi: 10.1101/cshperspect.a004069
24. Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, et al. Calcium signalling–an overview. Semin Cell Dev Biol (2001) 12(1):3–10. doi: 10.1006/scdb.2000.0211
25. Roberts-Thomson SJ, Chalmers SB, Monteith GR. The calcium-signaling toolkit in cancer: Remodeling and targeting. Cold Spring Harb Perspect Biol (2019) 11(8):a035204. doi: 10.1101/cshperspect.a035204
26. Berridge MJ. The inositol Trisphosphate/Calcium signaling pathway in health and disease. Physiol Rev (2016) 96(4):1261–96. doi: 10.1152/physrev.00006.2016
27. Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta (2009) 1793(6):933–40. doi: 10.1016/j.bbamcr.2008.10.005
28. Berridge MJ. Inositol trisphosphate and diacylglycerol as second messengers. Biochem J (1984) 220(2):345–60. doi: 10.1042/bj2200345
29. Wang H, Cheng X, Tian J, Xiao Y, Tian T, Xu F, et al. Trpc channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol Ther (2020) 209:107497. doi: 10.1016/j.pharmthera.2020.107497
30. Kadamur G, Ross EM. Mammalian phospholipase c. Annu Rev Physiol (2013) 75:127–54. doi: 10.1146/annurev-physiol-030212-183750
31. Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium (1986) 7(1):1–12. doi: 10.1016/0143-4160(86)90026-6
32. Putney JW. Forms and functions of store-operated calcium entry mediators, stim and orai. Adv Biol Regul (2018) 68:88–96. doi: 10.1016/j.jbior.2017.11.006
33. Brandman O, Liou J, Park WS, Meyer T. Stim2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell (2007) 131(7):1327–39. doi: 10.1016/j.cell.2007.11.039
34. Brose N, Betz A, Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol (2004) 14(3):328–40. doi: 10.1016/j.conb.2004.05.006
35. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium (2018) 69:62–72. doi: 10.1016/j.ceca.2017.05.003
36. Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res (2019) 1866(7):1068–78. doi: 10.1016/j.bbamcr.2018.10.016
37. Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM. Mitochondrial Vdac1: A key gatekeeper as potential therapeutic target. Front Physiol (2017) 8:460. doi: 10.3389/fphys.2017.00460
38. Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev (2022) 102(2):893–992. doi: 10.1152/physrev.00041.2020
39. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry (2012) 51(14):2959–73. doi: 10.1021/bi2018909
40. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (2009) 1787(11):1309–16. doi: 10.1016/j.bbabio.2009.01.005
41. Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium (2012) 52(1):36–43. doi: 10.1016/j.ceca.2012.02.008
42. Zhang L, Qi J, Zhang X, Zhao X, An P, Luo Y, et al. The regulatory roles of mitochondrial calcium and the mitochondrial calcium uniporter in tumor cells. Int J Mol Sci (2022) 23(12):6667. doi: 10.3390/ijms23126667
43. Faris P, Shekha M, Montagna D, Guerra G, Moccia F. Endolysosomal Ca(2+) signalling and cancer hallmarks: Two-pore channels on the move, Trpml1 lags behind! Cancers (Basel) (2018) 11(1):27. doi: 10.3390/cancers11010027
44. Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J (2011) 439(3):349–74. doi: 10.1042/BJ20110949
45. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal biology and function: Modern view of cellular debris bin. Cells (2020) 9(5):1131. doi: 10.3390/cells9051131
46. Rosato AS, Tang R, Grimm C. Two-pore and trpml cation channels: Regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol Ther (2021) 220:107713. doi: 10.1016/j.pharmthera.2020.107713
47. Yuan Y, Jaslan D, Rahman T, Bolsover SR, Arige V, Wagner LE 2nd, et al. Segregated cation flux by Tpc2 biases Ca(2+) signaling through lysosomes. Nat Commun (2022) 13(1):4481. doi: 10.1038/s41467-022-31959-0
48. Cremer T, Neefjes J, Berlin I. The journey of Ca(2+) through the cell - pulsing through the network of er membrane contact sites. J Cell Sci (2020) 133(24):jcs249136. doi: 10.1242/jcs.249136
49. Davis LC, Morgan AJ, Galione A. Acidic Ca(2+) stores and immune-cell function. Cell Calcium (2022) 101:102516. doi: 10.1016/j.ceca.2021.102516
50. Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission Via Rab7 gtp hydrolysis. Nature (2018) 554(7692):382–6. doi: 10.1038/nature25486
51. Peng W, Wong YC, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics Via lysosomal Trpml1. Proc Natl Acad Sci U.S.A. (2020) 117(32):19266–75. doi: 10.1073/pnas.2003236117
52. Bogdanova A, Makhro A, Wang J, Lipp P, Kaestner L. Calcium in red blood cells-a perilous balance. Int J Mol Sci (2013) 14(5):9848–72. doi: 10.3390/ijms14059848
53. Kaestner L, Bogdanova A, Egee S. Calcium channels and calcium-regulated channels in human red blood cells. Adv Exp Med Biol (2020) 1131:625–48. doi: 10.1007/978-3-030-12457-1_25
54. Zhang Y, Xu Y, Zhang S, Lu Z, Li Y, Zhao B. The regulation roles of Ca(2+) in erythropoiesis: What have we learned? Exp Hematol (2022) 106:19–30. doi: 10.1016/j.exphem.2021.12.192
55. Martino S, Arlet JB, Odievre MH, Jullien V, Moras M, Hattab C, et al. Deficient mitophagy pathways in sickle cell disease. Br J Haematol (2021) 193(5):988–93. doi: 10.1111/bjh.17416
56. Nguyen DB, Ly TB, Wesseling MC, Hittinger M, Torge A, Devitt A, et al. Characterization of microvesicles released from human red blood cells. Cell Physiol Biochem (2016) 38(3):1085–99. doi: 10.1159/000443059
57. Lew VL, Hockaday A, Sepulveda MI, Somlyo AP, Somlyo AV, Ortiz OE, et al. Compartmentalization of sickle-cell calcium in endocytic inside-out vesicles. Nature (1985) 315(6020):586–9. doi: 10.1038/315586a0
58. Jansen J, Qiao M, Hertz L, Wang X, Fermo E, Zaninoni A, et al. Mechanistic ion channel interactions in red cells of patients with gardos channelopathy. Blood Adv (2021) 5(17):3303–8. doi: 10.1182/bloodadvances.2020003823
59. Hertz L, Huisjes R, Llaudet-Planas E, Petkova-Kirova P, Makhro A, Danielczok JG, et al. Is increased intracellular calcium in red blood cells a common component in the molecular mechanism causing anemia? Front Physiol (2017) 8:673. doi: 10.3389/fphys.2017.00673
60. Makhro A, Hanggi P, Goede JS, Wang J, Bruggemann A, Gassmann M, et al. N-Methyl-D-Aspartate receptors in human erythroid precursor cells and in circulating red blood cells contribute to the intracellular calcium regulation. Am J Physiol Cell Physiol (2013) 305(11):C1123–38. doi: 10.1152/ajpcell.00031.2013
61. Wang J, Wagner-Britz L, Bogdanova A, Ruppenthal S, Wiesen K, Kaiser E, et al. Morphologically homogeneous red blood cells present a heterogeneous response to hormonal stimulation. PloS One (2013) 8(6):e67697. doi: 10.1371/journal.pone.0067697
62. Rotordam MG, Fermo E, Becker N, Barcellini W, Bruggemann A, Fertig N, et al. A novel gain-of-Function mutation of Piezo1 is functionally affirmed in red blood cells by high-throughput patch clamp. Haematologica (2019) 104(5):e179–e83. doi: 10.3324/haematol.2018.201160
63. Gardos G. The function of calcium in the potassium permeability of human erythrocytes. Biochim Biophys Acta (1958) 30(3):653–4. doi: 10.1016/0006-3002(58)90124-0
64. Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, Patapoutian A. Piezo1 links mechanical forces to red blood cell volume. Elife (2015) 4:e07370. doi: 10.7554/eLife.07370
65. Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, et al. Architecture of the mammalian mechanosensitive Piezo1 channel. Nature (2015) 527(7576):64–9. doi: 10.1038/nature15247
66. Wang L, Zhou H, Zhang M, Liu W, Deng T, Zhao Q, et al. Structure and mechanogating of the mammalian tactile channel Piezo2. Nature (2019) 573(7773):225–9. doi: 10.1038/s41586-019-1505-8
67. Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature (2018) 554(7693):487–92. doi: 10.1038/nature25743
68. Rogers S, Lew VL. Piezo1 and the mechanism of the long circulatory longevity of human red blood cells. PloS Comput Biol (2021) 17(3):e1008496. doi: 10.1371/journal.pcbi.1008496
69. Grygorczyk R, Schwarz W, Passow H. Ca2+-activated k+ channels in human red cells. comparison of single-channel currents with ion fluxes. Biophys J (1984) 45(4):693–8. doi: 10.1016/S0006-3495(84)84211-3
70. Danielczok JG, Terriac E, Hertz L, Petkova-Kirova P, Lautenschlager F, Laschke MW, et al. Red blood cell passage of small capillaries is associated with transient Ca(2+)-mediated adaptations. Front Physiol (2017) 8:979. doi: 10.3389/fphys.2017.00979
71. Brugnara C, de Franceschi L, Alper SL. Inhibition of Ca(2+)-dependent k+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest (1993) 92(1):520–6. doi: 10.1172/JCI116597
72. Fermo E, Monedero-Alonso D, Petkova-Kirova P, Makhro A, Peres L, Bouyer G, et al. Gardos channelopathy: Functional analysis of a novel Kcnn4 variant. Blood Adv (2020) 4(24):6336–41. doi: 10.1182/bloodadvances.2020003285
73. Lew VL, Tiffert T. On the mechanism of human red blood cell longevity: Roles of calcium, the sodium pump, Piezo1, and gardos channels. Front Physiol (2017) 8:977. doi: 10.3389/fphys.2017.00977
74. Knight T, Zaidi AU, Wu S, Gadgeel M, Buck S, Ravindranath Y. Mild erythrocytosis as a presenting manifestation of Piezo1 associated erythrocyte volume disorders. Pediatr Hematol Oncol (2019) 36(5):317–26. doi: 10.1080/08880018.2019.1637984
75. Fermo E, Bogdanova A, Petkova-Kirova P, Zaninoni A, Marcello AP, Makhro A, et al. 'Gardos channelopathy': A variant of hereditary stomatocytosis with complex molecular regulation. Sci Rep (2017) 7(1):1744. doi: 10.1038/s41598-017-01591-w
76. Picard V, Guitton C, Thuret I, Rose C, Bendelac L, Ghazal K, et al. Clinical and biological features in Piezo1-hereditary xerocytosis and gardos channelopathy: A retrospective series of 126 patients. Haematologica (2019) 104(8):1554–64. doi: 10.3324/haematol.2018.205328
77. Birnbaumer L. The trpc class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol (2009) 49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928
78. Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient receptor potential canonical (Trpc) channels: Then and now. Cells (2020) 9(9):1983. doi: 10.3390/cells9091983
79. Gees M, Owsianik G, Nilius B, Voets T. Trp channels. Compr Physiol (2012) 2(1):563–608. doi: 10.1002/cphy.c110026
80. Nielsen N, Lindemann O, Schwab A. Trp channels and Stim/Orai proteins: Sensors and effectors of cancer and stroma cell migration. Br J Pharmacol (2014) 171(24):5524–40. doi: 10.1111/bph.12721
81. Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev (2007) 87(1):165–217. doi: 10.1152/physrev.00021.2006
82. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature (1997) 389(6653):816–24. doi: 10.1038/39807
83. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science (2010) 330(6000):55–60. doi: 10.1126/science.1193270
84. Cheng Y. Trpv1 and piezo: The 2021 Nobel prize in physiology or medicine. IUCrJ (2022) 9(Pt 1):4–5. doi: 10.1107/S2052252521013488
85. Smani T, Shapovalov G, Skryma R, Prevarskaya N, Rosado JA. Functional and physiopathological implications of trp channels. Biochim Biophys Acta (2015) 1853(8):1772–82. doi: 10.1016/j.bbamcr.2015.04.016
86. Ong HL, de Souza LB, Ambudkar IS. Role of trpc channels in store-operated calcium entry. Adv Exp Med Biol (2016) 898:87–109. doi: 10.1007/978-3-319-26974-0_5
87. Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, et al. Stim1 carboxyl-terminus activates native soc, I(Crac) and Trpc1 channels. Nat Cell Biol (2006) 8(9):1003–10. doi: 10.1038/ncb1454
88. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. Stim1 heteromultimerizes trpc channels to determine their function as store-operated channels. Nat Cell Biol (2007) 9(6):636–45. doi: 10.1038/ncb1590
89. Kiselyov K, Shin DM, Kim JY, Yuan JP, Muallem S. Trpc channels: Interacting proteins. Handb Exp Pharmacol (2007) 179):559–74. doi: 10.1007/978-3-540-34891-7_33
90. Foller M, Kasinathan RS, Koka S, Lang C, Shumilina E, Birnbaumer L, et al. Trpc6 contributes to the Ca(2+) leak of human erythrocytes. Cell Physiol Biochem (2008) 21(1-3):183–92. doi: 10.1159/000113760
91. Belkacemi A, Trost CF, Tinschert R, Flormann D, Malihpour M, Wagner C, et al. The Trpv2 channel mediates Ca2+ influx and the Delta9-Thc-Dependent decrease in osmotic fragility in red blood cells. Haematologica (2021) 106(8):2246–50. doi: 10.3324/haematol.2020.274951
92. Egee S, Kaestner L. The transient receptor potential vanilloid type 2 (Trpv2) channel-a new druggable Ca(2+) pathway in red cells, implications for red cell ion homeostasis. Front Physiol (2021) 12:677573. doi: 10.3389/fphys.2021.677573
93. Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, et al. Structure, function, and allosteric modulation of nmda receptors. J Gen Physiol (2018) 150(8):1081–105. doi: 10.1085/jgp.201812032
94. Hanggi P, Makhro A, Gassmann M, Schmugge M, Goede JS, Speer O, et al. Red blood cells of sickle cell disease patients exhibit abnormally high abundance of n-methyl d-aspartate receptors mediating excessive calcium uptake. Br J Haematol (2014) 167(2):252–64. doi: 10.1111/bjh.13028
95. Hanggi P, Telezhkin V, Kemp PJ, Schmugge M, Gassmann M, Goede JS, et al. Functional plasticity of the n-Methyl-D-Aspartate receptor in differentiating human erythroid precursor cells. Am J Physiol Cell Physiol (2015) 308(12):C993–C1007. doi: 10.1152/ajpcell.00395.2014
96. Kalev-Zylinska ML, Green TN, Morel-Kopp MC, Sun PP, Park YE, Lasham A, et al. N-Methyl-D-Aspartate receptors amplify activation and aggregation of human platelets. Thromb Res (2014) 133(5):837–47. doi: 10.1016/j.thromres.2014.02.011
97. Kalev-Zylinska ML, Morel-Kopp MC, Ward CM, Hearn JI, Hamilton JR, Bogdanova AY. Ionotropic glutamate receptors in platelets: Opposing effects and a unifying hypothesis. Platelets (2021) 32(8):998–1008. doi: 10.1080/09537104.2020.1852542
98. Kalev-Zylinska ML, Hearn JI, Makhro A, Bogdanova A. N-Methyl-D-Aspartate receptors in hematopoietic cells: What have we learned? Front Physiol (2020) 11:577. doi: 10.3389/fphys.2020.00577
99. Del Arroyo AG, Hadjihambi A, Sanchez J, Turovsky E, Kasymov V, Cain D, et al. Nmda receptor modulation of glutamate release in activated neutrophils. EBioMedicine (2019) 47:457–69. doi: 10.1016/j.ebiom.2019.08.004
100. Miglio G, Varsaldi F, Lombardi G. Human T lymphocytes express n-Methyl-D-Aspartate receptors functionally active in controlling T cell activation. Biochem Biophys Res Commun (2005) 338(4):1875–83. doi: 10.1016/j.bbrc.2005.10.164
101. Sadat-Shirazi MS, Vousooghi N, Alizadeh B, Makki SM, Zarei SZ, Nazari S, et al. Expression of nmda receptor subunits in human blood lymphocytes: A peripheral biomarker in online computer game addiction. J Behav Addict (2018) 7(2):260–8. doi: 10.1556/2006.7.2018.35
102. Hegemann I, Sasselli C, Valeri F, Makhro A, Muller R, Bogdanova A, et al. Memsid: Results from a phase 2 pilot study on memantine treatment for sickle cell disease. Hemasphere (2020) 4(4):e452. doi: 10.1097/HS9.0000000000000452
103. Makhro A, Hegemann I, Seiler E, Simionato G, Claveria V, Bogdanov N, et al. A pilot clinical phase ii trial memsid: Acute and durable changes of red blood cells of sickle cell disease patients on memantine treatment. eJHaem (2020) 1(1):23–34. doi: 10.1002/jha2.11
104. Najbauer EE, Becker S, Giller K, Zweckstetter M, Lange A, Steinem C, et al. Structure, gating and interactions of the voltage-dependent anion channel. Eur Biophys J (2021) 50(2):159–72. doi: 10.1007/s00249-021-01515-7
105. Bouyer G, Cueff A, Egee S, Kmiecik J, Maksimova Y, Glogowska E, et al. Erythrocyte peripheral type benzodiazepine Receptor/Voltage-dependent anion channels are upregulated by plasmodium falciparum. Blood (2011) 118(8):2305–12. doi: 10.1182/blood-2011-01-329300
106. Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol (2011) 3(8):a003947. doi: 10.1101/cshperspect.a003947
107. Dolphin AC. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci Adv (2018) 2:2398212818794805. doi: 10.1177/2398212818794805
108. Kaestner L, Wang X, Hertz L, Bernhardt I. Voltage-activated ion channels in non-excitable cells-a viewpoint regarding their physiological justification. Front Physiol (2018) 9:450. doi: 10.3389/fphys.2018.00450
109. Wannatung T, Lithanatudom P, Leecharoenkiat A, Svasti S, Fucharoen S, Smith DR. Increased erythropoiesis of beta-Thalassaemia/Hb e proerythroblasts is mediated by high basal levels of Erk1/2 activation. Br J Haematol (2009) 146(5):557–68. doi: 10.1111/j.1365-2141.2009.07794.x
110. Falchi M, Varricchio L, Martelli F, Marra M, Picconi O, Tafuri A, et al. The calreticulin control of human stress erythropoiesis is impaired by Jak2v617f in polycythemia Vera. Exp Hematol (2017) 50:53–76. doi: 10.1016/j.exphem.2017.02.001
111. Buks R, Dagher T, Rotordam MG, Monedero Alonso D, Cochet S, Gautier EF, et al. Altered Ca(2+) homeostasis in red blood cells of polycythemia Vera patients following disturbed organelle sorting during terminal erythropoiesis. Cells (2021) 11(1):49. doi: 10.3390/cells11010049
112. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J (2009) 417(3):651–66. doi: 10.1042/BJ20081847
113. Weisel JW, Litvinov RI. Red blood cells: The forgotten player in hemostasis and thrombosis. J Thromb Haemost (2019) 17(2):271–82. doi: 10.1111/jth.14360
114. Alexy T, Detterich J, Connes P, Toth K, Nader E, Kenyeres P, et al. Physical properties of blood and their relationship to clinical conditions. Front Physiol (2022) 13:906768. doi: 10.3389/fphys.2022.906768
115. Steffen P, Jung A, Nguyen DB, Muller T, Bernhardt I, Kaestner L, et al. Stimulation of human red blood cells leads to Ca2+-mediated intercellular adhesion. Cell Calcium (2011) 50(1):54–61. doi: 10.1016/j.ceca.2011.05.002
116. Borst O, Abed M, Alesutan I, Towhid ST, Qadri SM, Foller M, et al. Dynamic adhesion of eryptotic erythrocytes to endothelial cells Via Cxcl16/Sr-psox. Am J Physiol Cell Physiol (2012) 302(4):C644–51. doi: 10.1152/ajpcell.00340.2011
117. Smeets MW, Bierings R, Meems H, Mul FP, Geerts D, Vlaar AP, et al. Platelet-independent adhesion of calcium-loaded erythrocytes to Von willebrand factor. PloS One (2017) 12(3):e0173077. doi: 10.1371/journal.pone.0173077
118. Kroll MH, Michaelis LC, Verstovsek S. Mechanisms of thrombogenesis in polycythemia Vera. Blood Rev (2015) 29(4):215–21. doi: 10.1016/j.blre.2014.12.002
119. Kuznetsova PI, Raskurazhev AA, Shabalina AA, Melikhyan AL, Subortseva IN, Tanashyan MM. Red blood cell morphodynamics in patients with polycythemia Vera and stroke. Int J Mol Sci (2022) 23(4):2247. doi: 10.3390/ijms23042247
120. Tajada S, Villalobos C. Calcium permeable channels in cancer hallmarks. Front Pharmacol (2020) 11:968. doi: 10.3389/fphar.2020.00968
121. Zhang XJ, Zheng GG, Ma XT, Yang YH, Li G, Rao Q, et al. Expression of P2x7 in human hematopoietic cell lines and leukemia patients. Leukemia Res (2004) 28(12):1313–22. doi: 10.1016/j.leukres.2004.04.001
122. Salvestrini V, Zini R, Rossi L, Gulinelli S, Manfredini R, Bianchi E, et al. Purinergic signaling inhibits human acute myeloblastic leukemia cell proliferation, migration, and engraftment in immunodeficient mice. Blood (2012) 119(1):217–26. doi: 10.1182/blood-2011-07-370775
123. Chong JH, Zheng GG, Zhu XF, Guo Y, Wang L, Ma CH, et al. Abnormal expression of P2x family receptors in Chinese pediatric acute leukemias. Biochem Biophys Res Commun (2010) 391(1):498–504. doi: 10.1016/j.bbrc.2009.11.087
124. Feng WL, Yang X, Wang LN, Wang R, Yang FF, Wang H, et al. P2x7 promotes the progression of mll-Af9 induced acute myeloid leukemia by upregulation of Pbx3. Haematologica (2021) 106(5):1278–89. doi: 10.3324/haematol.2019.243360
125. He XX, Wan JB, Yang XN, Zhang XZ, Huang D, Li X, et al. Bone marrow niche atp levels determine leukemia-initiating cell activity Via P2x7 in leukemic models. J Clin Invest (2021) 131(4):e140242. doi: 10.1172/JCI140242
126. Pegoraro A, Orioli E, De Marchi E, Salvestrini V, Milani A, Di Virgilio F, et al. Differential sensitivity of acute myeloid leukemia cells to daunorubicin depends on P2x7a versus P2x7b receptor expression. Cell Death Dis (2020) 11(10):876. doi: 10.1038/s41419-020-03058-9
127. Siveen KS, Prabhu KS, Parray AS, Merhi M, Arredouani A, Chikri M, et al. Evaluation of cationic channel Trpv2 as a novel biomarker and therapeutic target in leukemia-implications concerning the resolution of pulmonary inflammation. Sci Rep (2019) 9(1):1554. doi: 10.1038/s41598-018-37469-8
128. Zatkova A, Merk S, Wendehack M, Bilban M, Muzik EM, Muradyan A, et al. Aml/Mds with 11q/Mll amplification show characteristic gene expression signature and interplay of DNA copy number changes. Genes Chromosomes Cancer (2009) 48(6):510–20. doi: 10.1002/gcc.20658
129. Chen SJ, Bao L, Keefer K, Shanmughapriya S, Chen L, Lee J, et al. Transient receptor potential ion channel Trpm2 promotes aml proliferation and survival through modulation of mitochondrial function, ros, and autophagy. Cell Death Dis (2020) 11(4):247. doi: 10.1038/s41419-020-2454-8
130. Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, et al. A novel Trpm2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem (2003) 278(18):16222–9. doi: 10.1074/jbc.M300298200
131. Wang F, Wu P, Gong S, Chen Y, Gao J, Wang S, et al. Aberrant Trpm4 expression in mll-rearranged acute myeloid leukemia and its blockade induces cell cycle arrest Via Akt/Gli1/Cyclin D1 pathway. Cell Signal (2020) 72:109643. doi: 10.1016/j.cellsig.2020.109643
132. Diez-Bello R, Jardin I, Salido GM, Rosado JA. Orai1 and Orai2 mediate store-operated calcium entry that regulates Hl60 cell migration and fak phosphorylation. Biochim Biophys Acta Mol Cell Res (2017) 1864(6):1064–70. doi: 10.1016/j.bbamcr.2016.11.014
133. Lewuillon C, Guillemette A, Titah S, Shaik FA, Jouy N, Labiad O, et al. Involvement of Orai1/Soce in human aml cell lines and primary cells according to Abcb1 activity, lsc compartment and potential resistance to ara-c exposure. Int J Mol Sci (2022) 23(10):5555. doi: 10.3390/ijms23105555
134. Saint Fleur-Lominy S, Maus M, Vaeth M, Lange I, Zee I, Suh D, et al. Stim1 and Stim2 mediate cancer-induced inflammation in T cell acute lymphoblastic leukemia. Cell Rep (2018) 24(11):3045–60.e5. doi: 10.1016/j.celrep.2018.08.030
135. Shi JL, Fu L, Wang WD. High expression of inositol 1,4,5-trisphosphate receptor, type 2 (Itpr2) as a novel biomarker for worse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget (2015) 6(7):5299–309. doi: 10.18632/oncotarget.3024
136. Wang CY, Lai MD, Phan NN, Sun Z, Lin YC. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PloS One (2015) 10(7):e0125766. doi: 10.1371/journal.pone.0125766
137. Buchanan PJ, McCloskey KD. Cav channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur Biophys J (2016) 45(7):621–33. doi: 10.1007/s00249-016-1144-z
138. Borella G, Da Ros A, Borile G, Porcu E, Tregnago C, Benetton M, et al. Targeting the plasticity of mesenchymal stromal cells to reroute the course of acute myeloid leukemia. Blood (2021) 138(7):557–70. doi: 10.1182/blood.2020009845
139. Wenning AS, Neblung K, Strauss B, Wolfs MJ, Sappok A, Hoth M, et al. Trp expression pattern and the functional importance of Trpc3 in primary human T-cells. Biochim Biophys Acta (2011) 1813(3):412–23. doi: 10.1016/j.bbamcr.2010.12.022
140. Vassilieva IO, Tomilin VN, Marakhova II, Shatrova AN, Negulyaev YA, Semenova SB. Expression of transient receptor potential vanilloid channels Trpv5 and Trpv6 in human blood lymphocytes and jurkat leukemia T cells. J Membr Biol (2013) 246(2):131–40. doi: 10.1007/s00232-012-9511-x
141. Tomilin VN, Cherezova AL, Negulyaev YA, Semenova SB. Trpv5/V6 channels mediate Ca(2+) influx in jurkat T cells under the control of extracellular ph. J Cell Biochem (2016) 117(1):197–206. doi: 10.1002/jcb.25264
142. Bobkov D, Yudintceva N, Lomert E, Shatrova A, Kever L, Semenova S. Lipid raft integrity is required for human leukemia jurkat T-cell migratory activity. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(6):158917. doi: 10.1016/j.bbalip.2021.158917
143. Klumpp D, Misovic M, Szteyn K, Shumilina E, Rudner J, Huber SM. Targeting Trpm2 channels impairs radiation-induced cell cycle arrest and fosters cell death of T cell leukemia cells in a bcl-2-Dependent manner. Oxid Med Cell Longev (2016) 2016:8026702. doi: 10.1155/2016/8026702
144. Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. Trpm4 regulates calcium oscillations after T cell activation. Science (2004) 306(5700):1374–7. doi: 10.1126/science.1098845
145. Bohannan ZS, Coffman F, Mitrofanova A. Random survival forest model identifies novel biomarkers of event-free survival in high-risk pediatric acute lymphoblastic leukemia. Comput Struct Biotechnol J (2022) 20:583–97. doi: 10.1016/j.csbj.2022.01.003
146. Takahashi K, Umebayashi C, Numata T, Honda A, Ichikawa J, Hu Y, et al. Trpm7-mediated spontaneous Ca(2+) entry regulates the proliferation and differentiation of human leukemia cell line K562. Physiol Rep (2018) 6(14):e13796. doi: 10.14814/phy2.13796
147. Maggi F, Morelli MB, Tomassoni D, Marinelli O, Aguzzi C, Zeppa L, et al. The effects of cannabidiol Via Trpv2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci (2022) 113(4):1235–49. doi: 10.1111/cas.15257
148. Vasileva V, Morachevskaya E, Sudarikova A, Negulyaev Y, Chubinskiy-Nadezhdin V. Selective chemical activation of Piezo1 in leukemia cell membrane: Single channel analysis. Int J Mol Sci (2021) 22(15):7839. doi: 10.3390/ijms22157839
149. Adinolfi E, Melchiorri L, Falzoni S, Chiozzi P, Morelli A, Tieghi A, et al. P2x7 receptor expression in evolutive and indolent forms of chronic b lymphocytic leukemia. Blood (2002) 99(2):706–8. doi: 10.1182/blood.v99.2.706
150. Garaud S, Taher TE, Debant M, Burgos M, Melayah S, Berthou C, et al. Cd5 expression promotes il-10 production through activation of the Mapk/Erk pathway and upregulation of Trpc1 channels in b lymphocytes. Cell Mol Immunol (2018) 15(2):158–70. doi: 10.1038/cmi.2016.42
151. Pepper C, Tu H, Morrill P, Garcia-Rates S, Fegan C, Greenfield S. Tumor cell migration is inhibited by a novel therapeutic strategy antagonizing the alpha-7 receptor. Oncotarget (2017) 8(7):11414–24. doi: 10.18632/oncotarget.14545
152. Loo SK, Ch'ng ES, Md Salleh MS, Banham AH, Pedersen LM, Moller MB, et al. Trpm4 expression is associated with activated b cell subtype and poor survival in diffuse Large b cell lymphoma. Histopathology (2017) 71(1):98–111. doi: 10.1111/his.13204
153. Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. Ip3r2 levels dictate the apoptotic sensitivity of diffuse Large b-cell lymphoma cells to an Ip3r-derived peptide targeting the Bh4 domain of bcl-2. Cell Death Dis (2013) 4:e632. doi: 10.1038/cddis.2013.140
154. Bittremieux M, La Rovere RM, Akl H, Martines C, Welkenhuyzen K, Dubron K, et al. Constitutive Ip3 signaling underlies the sensitivity of b-cell cancers to the bcl-2/Ip3 receptor disruptor bird-2. Cell Death Differ (2019) 26(3):531–47. doi: 10.1038/s41418-018-0142-3
155. Vervloessem T, Akl H, Tousseyn T, De Smedt H, Parys JB, Bultynck G. Reciprocal sensitivity of diffuse Large b-cell lymphoma cells to bcl-2 inhibitors bird-2 versus venetoclax. Oncotarget (2017) 8(67):111656–71. doi: 10.18632/oncotarget.22898
156. Stanwood SR, Chong LC, Steidl C, Jefferies WA. Distinct gene expression patterns of calcium channels and related signaling pathways discovered in lymphomas. Front Pharmacol (2022) 13:795176. doi: 10.3389/fphar.2022.795176
157. Beider K, Rosenberg E, Dimenshtein-Voevoda V, Sirovsky Y, Vladimirsky J, Magen H, et al. Blocking of transient receptor potential vanilloid 1 (Trpv1) promotes terminal mitophagy in multiple myeloma, disturbing calcium homeostasis and targeting ubiquitin pathway and bortezomib-induced unfolded protein response. J Hematol Oncol (2020) 13(1):158. doi: 10.1186/s13045-020-00993-0
158. Amachi R, Hiasa M, Teramachi J, Harada T, Oda A, Nakamura S, et al. A vicious cycle between acid sensing and survival signaling in myeloma cells: Acid-induced epigenetic alteration. Oncotarget (2016) 7(43):70447–61. doi: 10.18632/oncotarget.11927
159. Bai H, Zhu H, Yan Q, Shen X, Lu X, Wang J, et al. Trpv2-induced Ca(2+)-Calcineurin-Nfat signaling regulates differentiation of osteoclast in multiple myeloma. Cell Commun Signal (2018) 16(1):68. doi: 10.1186/s12964-018-0280-8
160. Fabris S, Todoerti K, Mosca L, Agnelli L, Intini D, Lionetti M, et al. Molecular and transcriptional characterization of the novel 17p11.2-P12 amplicon in multiple myeloma. Genes Chromosomes Cancer (2007) 46(12):1109–18. doi: 10.1002/gcc.20494
161. Samart P, Luanpitpong S, Rojanasakul Y, Issaragrisil S. O-Glcnacylation homeostasis controlled by calcium influx channels regulates multiple myeloma dissemination. J Exp Clin Cancer Res (2021) 40(1):100. doi: 10.1186/s13046-021-01876-z
162. Hirai A, Aung NY, Ohe R, Nishida A, Kato T, Meng H, et al. Expression of Trpm8 in human reactive lymphoid tissues and mature b-cell neoplasms. Oncol Lett (2018) 16(5):5930–8. doi: 10.3892/ol.2018.9386
163. Santoni G, Amantini C, Maggi F, Marinelli O, Santoni M, Morelli MB. The mucolipin Trpml2 channel enhances the sensitivity of multiple myeloma cell lines to ibrutinib and/or bortezomib treatment. Biomolecules (2022) 12(1):107. doi: 10.3390/biom12010107
164. Wang W, Ren Y, Wang L, Zhao W, Dong X, Pan J, et al. Orai1 and Stim1 mediate the majority of store-operated calcium entry in multiple myeloma and have strong implications for adverse prognosis. Cell Physiol Biochem (2018) 48(6):2273–85. doi: 10.1159/000492645
165. Trojani A, Di Camillo B, Bossi LE, Leuzzi L, Greco A, Tedeschi A, et al. Identification of a candidate gene set signature for the risk of progression in igm mgus to Smoldering/Symptomatic waldenstrom macroglobulinemia (Wm) by a comparative transcriptome analysis of b cells and plasma cells. Cancers (Basel) (2021) 13(8):1837. doi: 10.3390/cancers13081837
166. Maggi F, Morelli MB, Nabissi M, Marinelli O, Zeppa L, Aguzzi C, et al. Transient receptor potential (Trp) channels in haematological malignancies: An update. Biomolecules (2021) 11(5):765. doi: 10.3390/biom11050765
167. Morelli MB, Liberati S, Amantini C, Nabiss M, Santoni M, Farfariello V, et al. Expression and function of the transient receptor potential ion channel family in the hematologic malignancies. Curr Mol Pharmacol (2013) 6(3):137–48. doi: 10.2174/187446720603140415215431
168. Morelli MB, Amantini C. Transient receptor potential (Trp) channels: Markers and therapeutic targets for cancer? Biomolecules (2022) 12(4):547. doi: 10.3390/biom12040547
169. Qi J, Xing Y, Liu Y, Wang MM, Wei X, Sui Z, et al. Mcoln1/Trpml1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy (2021) 17(12):4401–22. doi: 10.1080/15548627.2021.1917132
170. Du W, Gu M, Hu M, Pinchi P, Chen W, Ryan M, et al. Lysosomal zn 2+ release triggers rapid, mitochondria-mediated, non-apoptotic cell death in metastatic melanoma. Cell Rep (2021) 37(3):109848. doi: 10.1016/j.celrep.2021.109848
171. Cabanas H, Harnois T, Magaud C, Cousin L, Constantin B, Bourmeyster N, et al. Deregulation of calcium homeostasis in bcr-Abl-Dependent chronic myeloid leukemia. Oncotarget (2018) 9(41):26309–27. doi: 10.18632/oncotarget.25241
172. Punzo F, Manzo I, Tortora C, Pota E, Angelo V, Bellini G, et al. Effects of Cb2 and Trpv1 receptors' stimulation in pediatric acute T-lymphoblastic leukemia. Oncotarget (2018) 9(30):21244–58. doi: 10.18632/oncotarget.25052
173. Pan T, Gao Y, Xu G, Zhou P, Li S, Guo J, et al. Pan-cancer analyses reveal the genetic and pharmacogenomic landscape of transient receptor potential channels. NPJ Genom Med (2022) 7(1):32. doi: 10.1038/s41525-022-00304-1
174. North RA. Molecular physiology of P2x receptors. Physiol Rev (2002) 82(4):1013–67. doi: 10.1152/physrev.00015.2002
175. De Marchi E, Pegoraro A, Adinolfi E. P2x7 receptor in hematological malignancies. Front Cell Dev Biol (2021) 9:645605. doi: 10.3389/fcell.2021.645605
176. Di Virgilio F, Schmalzing G, Markwardt F. The elusive P2x7 macropore. Trends Cell Biol (2018) 28(5):392–404. doi: 10.1016/j.tcb.2018.01.005
177. Narin GO, Aydin B, Cabadak H. Studies on the role of alpha 7 nicotinic acetylcholine receptors in K562 cell proliferation and signaling. Mol Biol Rep (2021) 48(6):5045–55. doi: 10.1007/s11033-021-06498-4
178. Suriyo T, Chotirat S, Auewarakul CU, Chaiyot K, Promsuwicha O, Satayavivad J. Variation of nicotinic subtype Alpha7 and muscarinic subtype M3 acetylcholine receptor expression in three main types of leukemia. Oncol Lett (2019) 17(1):1357–62. doi: 10.3892/ol.2018.9663
179. Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, Nandi JS, Mishra NC, Singh SP, et al. T Cells express Alpha7-nicotinic acetylcholine receptor subunits that require a functional tcr and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol (2007) 179(5):2889–98. doi: 10.4049/jimmunol.179.5.2889
180. Hearn JI, Green TN, Hisey CL, Bender M, Josefsson EC, Knowlton N, et al. Deletion of Grin1 in mouse megakaryocytes reveals nmda receptor role in platelet function and proplatelet formation. Blood (2022) 139(17):2673–90. doi: 10.1182/blood.2021014000
181. Montes de Oca Balderas P. Flux-independent nmdar signaling: Molecular mediators, cellular functions, and complexities. Int J Mol Sci (2018) 19(12):3800. doi: 10.3390/ijms19123800
182. Kamal T, Green TN, Morel-Kopp MC, Ward CM, McGregor AL, McGlashan SR, et al. Inhibition of glutamate regulated calcium entry into leukemic megakaryoblasts reduces cell proliferation and supports differentiation. Cell Signal (2015) 27(9):1860–72. doi: 10.1016/j.cellsig.2015.05.004
183. Hearn JI, Green TN, Chopra M, Nursalim YNS, Ladvanszky L, Knowlton N, et al. N -Methyl-D-Aspartate receptor hypofunction in Meg-01 cells reveals a role for intracellular calcium homeostasis in balancing megakaryocytic-erythroid differentiation. Thromb Haemostasis (2020) 120(4):671–86. doi: 10.1055/s-0040-1708483
184. Lemeshko VV. Vdac electronics: 3. vdac-creatine kinase-dependent generation of the outer membrane potential in respiring mitochondria. Biochim Biophys Acta (2016) 1858(7 Pt A):1411–8. doi: 10.1016/j.bbamem.2016.04.005
185. Tan W, Colombini M. Vdac closure increases calcium ion flux. Biochim Biophys Acta (2007) 1768(10):2510–5. doi: 10.1016/j.bbamem.2007.06.002
186. Liu S, Ishikawa H, Tsuyama N, Li FJ, Abroun S, Otsuyama KI, et al. Increased susceptibility to apoptosis in Cd45(+) myeloma cells accompanied by the increased expression of Vdac1. Oncogene (2006) 25(3):419–29. doi: 10.1038/sj.onc.1208982
187. Prezma T, Shteinfer A, Admoni L, Raviv Z, Sela I, Levi I, et al. Vdac1-based peptides: Novel pro-apoptotic agents and potential therapeutics for b-cell chronic lymphocytic leukemia. Cell Death Dis (2013) 4:e809. doi: 10.1038/cddis.2013.316
188. Shteinfer-Kuzmine A, Amsalem Z, Arif T, Zooravlov A, Shoshan-Barmatz V. Selective induction of cancer cell death by Vdac1-based peptides and their potential use in cancer therapy. Mol Oncol (2018) 12(7):1077–103. doi: 10.1002/1878-0261.12313
189. Krestinina O, Fadeev R, Lomovsky A, Baburina Y, Kobyakova M, Akatov V. Melatonin can strengthen the effect of retinoic acid in hl-60 cells. Int J Mol Sci (2018) 19(10):2873. doi: 10.3390/ijms19102873
190. Jiang N, Kham SK, Koh GS, Suang Lim JY, Ariffin H, Chew FT, et al. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (All). J Proteomics (2011) 74(6):843–57. doi: 10.1016/j.jprot.2011.02.034
191. Godbole A, Varghese J, Sarin A, Mathew MK. Vdac is a conserved element of death pathways in plant and animal systems. Biochim Et Biophys Acta-Molecular Cell Res (2003) 1642(1-2):87–96. doi: 10.1016/S0167-4889(03)00102-2
192. Olivas-Aguirre M, Pottosin I, Dobrovinskaya O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J Leukoc Biol (2019) 105(5):935–46. doi: 10.1002/JLB.5VMR0818-330RR
193. Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A, Arif T. Voltage-dependent anion channel 1 as an emerging drug target for novel anti-cancer therapeutics. Front Oncol (2017) 7:154. doi: 10.3389/fonc.2017.00154
194. Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, et al. Nomenclature of voltage-gated calcium channels. Neuron (2000) 25(3):533–5. doi: 10.1016/s0896-6273(00)81057-0
195. Phan NN, Wang CY, Chen CF, Sun Z, Lai MD, Lin YC. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol Lett (2017) 14(2):2059–74. doi: 10.3892/ol.2017.6457
196. Doignon I, Fayol O, Dellis O. Improvement of the rituximab-induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Oncotarget (2019) 10(43):4466–78. doi: 10.18632/oncotarget.27063
197. Liang Y, Tedder TF. Identification of a Cd20-, fcepsilonribeta-, and Htm4-related gene family: Sixteen new Ms4a family members expressed in human and mouse. Genomics (2001) 72(2):119–27. doi: 10.1006/geno.2000.6472
198. Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ. Cd20 as a target for therapeutic type I and ii monoclonal antibodies. Semin Hematol (2010) 47(2):107–14. doi: 10.1053/j.seminhematol.2010.01.001
199. Pavlasova G, Mraz M. The regulation and function of Cd20: An "Enigma" of b-cell biology and targeted therapy. Haematologica (2020) 105(6):1494–506. doi: 10.3324/haematol.2019.243543
200. Thomsen EA, Rovsing AB, Anderson MV, Due H, Huang J, Luo Y, et al. Identification of blnk and btk as mediators of rituximab-induced programmed cell death by crispr screens in gcb-subtype diffuse Large b-cell lymphoma. Mol Oncol (2020) 14(9):1978–97. doi: 10.1002/1878-0261.12753
201. Polyak MJ, Li H, Shariat N, Deans JP. Cd20 homo-oligomers physically associate with the b cell antigen receptor. dissociation upon receptor engagement and recruitment of phosphoproteins and calmodulin-binding proteins. J Biol Chem (2008) 283(27):18545–52. doi: 10.1074/jbc.M800784200
202. Walshe CA, Beers SA, French RR, Chan CH, Johnson PW, Packham GK, et al. Induction of cytosolic calcium flux by Cd20 is dependent upon b cell antigen receptor signaling. J Biol Chem (2008) 283(25):16971–84. doi: 10.1074/jbc.M708459200
203. Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF. Transfection of the Cd20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in b lymphocytes. J Cell Biol (1993) 121(5):1121–32. doi: 10.1083/jcb.121.5.1121
204. Janas E, Priest R, Wilde JI, White JH, Malhotra R. Rituxan (Anti-Cd20 antibody)-induced translocation of Cd20 into lipid rafts is crucial for calcium influx and apoptosis. Clin Exp Immunol (2005) 139(3):439–46. doi: 10.1111/j.1365-2249.2005.02720.x
205. Vacher P, Vacher AM, Pineau R, Latour S, Soubeyran I, Pangault C, et al. Localized store-operated calcium influx represses Cd95-dependent apoptotic effects of rituximab in non-Hodgkin b lymphomas. J Immunol (2015) 195(5):2207–15. doi: 10.4049/jimmunol.1402942
206. Latour S, Zanese M, Le Morvan V, Vacher AM, Menard N, Bijou F, et al. Role of calcium signaling in Ga101-induced cell death in malignant human b cells. Cancers (Basel) (2019) 11(3):291. doi: 10.3390/cancers11030291
207. Heo W, Jin N, Park MS, Kim HY, Yoon SM, Lee J, et al. Stim1 knock-down decreases the affinity of obinutuzumab for Cd20 by altering Cd20 localization to triton-soluble membrane. Clin Exp Immunol (2020) 200(3):260–71. doi: 10.1111/cei.13427
208. Jackson JT, Mulazzani E, Nutt SL, Masters SL. The role of plc gamma 2 in immunological disorders, cancer, and neurodegeneration. J Biol Chem (2021) 297(2):100905. doi: 10.1016/j.jbc.2021.100905
209. Zhong WB, Yi Q, Xu B, Li SQ, Wang T, Liu FP, et al. Orp4l is essential for T-cell acute lymphoblastic leukemia cell survival. Nat Commun (2016) 7:12702. doi: 10.1038/ncomms12702
210. Marchand T, Pinho S. Leukemic stem cells: From leukemic niche biology to treatment opportunities. Front Immunol (2021) 12:775128. doi: 10.3389/fimmu.2021.775128
211. Zhong WB, Xu MY, Li CJ, Zhu BY, Cao XY, Li D, et al. Orp4l extracts and presents Pip2 from plasma membrane for plc beta 3 catalysis: Targeting it eradicates leukemia stem cells. Cell Rep (2019) 26(8):2166. doi: 10.1016/j.celrep.2019.01.082
212. Cao X, Chen J, Li D, Xie P, Xu M, Lin W, et al. Orp4l couples Ip3 to Itpr1 in control of endoplasmic reticulum calcium release. FASEB J (2019) 33(12):13852–65. doi: 10.1096/fj.201900933RR
213. Le Roy C, Deglesne PA, Chevallier N, Beitar T, Eclache V, Quettier M, et al. The degree of bcr and nfat activation predicts clinical outcomes in chronic lymphocytic leukemia. Blood (2012) 120(2):356–65. doi: 10.1182/blood-2011-12-397158
214. Parys JB, De Smedt H. Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol (2012) 740:255–79. doi: 10.1007/978-94-007-2888-2_11
215. Ouyang K, Leandro Gomez-Amaro R, Stachura DL, Tang H, Peng X, Fang X, et al. Loss of Ip3r-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun (2014) 5:4814. doi: 10.1038/ncomms5814
216. Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene (2016) 35(39):5079–92. doi: 10.1038/onc.2016.31
217. Nagy B, Lundan T, Larramendy ML, Aalto Y, Zhu Y, Niini T, et al. Abnormal expression of apoptosis-related genes in haematological malignancies: Overexpression of myc is poor prognostic sign in mantle cell lymphoma. Br J Haematol (2003) 120(3):434–41. doi: 10.1046/j.1365-2141.2003.04121.x
218. Scarfo L, Ghia P. Reprogramming cell death: Bcl2 family inhibition in hematological malignancies. Immunol Lett (2013) 155(1-2):36–9. doi: 10.1016/j.imlet.2013.09.015
219. Bittremieux M, Parys JB, Pinton P, Bultynck G. Er functions of oncogenes and tumor suppressors: Modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta (2016) 1863(6 Pt B):1364–78. doi: 10.1016/j.bbamcr.2016.01.002
220. Balatti V, Pekarky Y, Rizzotto L, Croce CM. Mir deregulation in cll. Adv Exp Med Biol (2013) 792:309–25. doi: 10.1007/978-1-4614-8051-8_14
221. Perini GF, Ribeiro GN, Pinto Neto JV, Campos LT, Hamerschlak N. Bcl-2 as therapeutic target for hematological malignancies. J Hematol Oncol (2018) 11(1):65. doi: 10.1186/s13045-018-0608-2
222. Distelhorst CW, Bootman MD. Creating a new cancer therapeutic agent by targeting the interaction between bcl-2 and Ip3 receptors. Cold Spring Harb Perspect Biol (2019) 11(9):a035196. doi: 10.1101/cshperspect.a035196
223. Vervloessem T, Ivanova H, Luyten T, Parys JB, Bultynck G. The selective bcl-2 inhibitor venetoclax, a Bh3 mimetic, does not dysregulate intracellular Ca(2+) signaling. Biochim Biophys Acta Mol Cell Res (2017) 1864(6):968–76. doi: 10.1016/j.bbamcr.2016.11.024
224. Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting bcl-2-Ip3 receptor interaction to reverse bcl-2's inhibition of apoptotic calcium signals. Mol Cell (2008) 31(2):255–65. doi: 10.1016/j.molcel.2008.06.014
225. Bittremieux M, La Rovere RM, Schuermans M, Luyten T, Mikoshiba K, Vangheluwe P, et al. Extracellular and er-stored Ca(2+) contribute to bird-2-Induced cell death in diffuse Large b-cell lymphoma cells. Cell Death Discovery (2018) 4:101. doi: 10.1038/s41420-018-0118-6
226. Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of bcl-2-Ip3 receptor interaction. Blood (2011) 117(10):2924–34. doi: 10.1182/blood-2010-09-307405
227. Lavik AR, Zhong F, Chang MJ, Greenberg E, Choudhary Y, Smith MR, et al. A synthetic peptide targeting the Bh4 domain of bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the Bh3-binding pocket of bcl-2. Oncotarget (2015) 6(29):27388–402. doi: 10.18632/oncotarget.4489
228. Kerkhofs M, La Rovere R, Welkenhuysen K, Janssens A, Vandenberghe P, Madesh M, et al. Bird-2, a Bh4-Domain-Targeting peptide of bcl-2, provokes Bax/Bak-independent cell death in b-cell cancers through mitochondrial Ca(2+)-dependent mptp opening. Cell Calcium (2021) 94:102333. doi: 10.1016/j.ceca.2020.102333
229. Kerkhofs M, Vervloessem T, Stopa KB, Smith VM, Vogler M, Bultynck G. Dlbcl cells with acquired resistance to venetoclax are not sensitized to bird-2 but can be resensitized to venetoclax through bcl-xl inhibition. Biomolecules (2020) 10(7):1081. doi: 10.3390/biom10071081
230. Ge C, Huang H, Huang F, Yang T, Zhang T, Wu H, et al. Neurokinin-1 receptor is an effective target for treating leukemia by inducing oxidative stress through mitochondrial calcium overload. Proc Natl Acad Sci U.S.A. (2019) 116(39):19635–45. doi: 10.1073/pnas.1908998116
231. Dionisio N, Albarran L, Lopez JJ, Berna-Erro A, Salido GM, Bobe R, et al. Acidic naadp-releasable Ca(2+) compartments in the megakaryoblastic cell line Meg01. Biochim Biophys Acta (2011) 1813(8):1483–94. doi: 10.1016/j.bbamcr.2011.05.005
232. Ambrosio AL, Boyle JA, Di Pietro SM. Tpc2 mediates new mechanisms of platelet dense granule membrane dynamics through regulation of Ca2+ release. Mol Biol Cell (2015) 26(18):3263–74. doi: 10.1091/mbc.E15-01-0058
233. Jang BC, Lim KJ, Paik JH, Cho JW, Baek WK, Suh MH, et al. Tetrandrine-induced apoptosis is mediated by activation of caspases and pkc-delta in U937 cells. Biochem Pharmacol (2004) 67(10):1819–29. doi: 10.1016/j.bcp.2004.01.018
234. Liu T, Men Q, Wu G, Yu C, Huang Z, Liu X, et al. Tetrandrine induces autophagy and differentiation by activating ros and Notch1 signaling in leukemia cells. Oncotarget (2015) 6(10):7992–8006. doi: 10.18632/oncotarget.3505
235. Liu T, Zhang Z, Yu C, Zeng C, Xu X, Wu G, et al. Tetrandrine antagonizes acute megakaryoblastic leukaemia growth by forcing autophagy-mediated differentiation. Br J Pharmacol (2017) 174(23):4308–28. doi: 10.1111/bph.14031
236. Wu G, Liu T, Li H, Li Y, Li D, Li W. C-myc and reactive oxygen species play roles in tetrandrine-induced leukemia differentiation. Cell Death Dis (2018) 9(5):473. doi: 10.1038/s41419-018-0498-9
237. Geisslinger F, Muller M, Chao YK, Grimm C, Vollmar AM, Bartel K. Targeting Tpc2 sensitizes acute lymphoblastic leukemia cells to chemotherapeutics by impairing lysosomal function. Cell Death Dis (2022) 13(8):668. doi: 10.1038/s41419-022-05105-z
238. Rafiq S, McKenna SL, Muller S, Tschan MP, Humbert M. Lysosomes in acute myeloid leukemia: Potential therapeutic targets? Leukemia (2021) 35(10):2759–70. doi: 10.1038/s41375-021-01388-x
239. Jain V, Bose S, Arya AK, Arif T. Lysosomes in stem cell quiescence: A potential therapeutic target in acute myeloid leukemia. Cancers (Basel) (2022) 14(7):1618. doi: 10.3390/cancers14071618
240. Metts J, Bradley HL, Wang Z, Shah NP, Kapur R, Arbiser JL, et al. Imipramine blue sensitively and selectively targets Flt3-itd positive acute myeloid leukemia cells. Sci Rep (2017) 7(1):4447. doi: 10.1038/s41598-017-04796-1
241. Wu X, Weng L, Zhang J, Liu X, Huang J. The plasma membrane calcium atpases in calcium signaling network. Curr Protein Pept Sci (2018) 19(8):813–22. doi: 10.2174/1389203719666180416122745
242. Brini M, Cali T, Ottolini D, Carafoli E. The plasma membrane calcium pump in health and disease. FEBS J (2013) 280(21):5385–97. doi: 10.1111/febs.12193
243. Staiano RI, Granata F, Secondo A, Petraroli A, Loffredo S, Annunziato L, et al. Human macrophages and monocytes express functional Na(+)/Ca (2+) exchangers 1 and 3. Adv Exp Med Biol (2013) 961:317–26. doi: 10.1007/978-1-4614-4756-6_27
244. Katoshevski T, Ben-Kasus Nissim T, Sekler I. Recent studies on nclx in health and diseases. Cell Calcium (2021) 94:102345. doi: 10.1016/j.ceca.2020.102345
245. Papp B, Launay S, Gelebart P, Arbabian A, Enyedi A, Brouland JP, et al. Endoplasmic reticulum calcium pumps and tumor cell differentiation. Int J Mol Sci (2020) 21(9):3351. doi: 10.3390/ijms21093351
246. Gil D, Guse AH, Dupont G. Three-dimensional model of Sub-plasmalemmal Ca(2+) microdomains evoked by the interplay between Orai1 and Insp3 receptors. Front Immunol (2021) 12:659790. doi: 10.3389/fimmu.2021.659790
247. Dellis O, Arbabian A, Brouland JP, Kovacs T, Rowe M, Chomienne C, et al. Modulation of b-cell endoplasmic reticulum calcium homeostasis by Epstein-Barr virus latent membrane protein-1. Mol Cancer (2009) 8:59. doi: 10.1186/1476-4598-8-59
248. Toldi G, Kaposi A, Zsembery A, Treszl A, Tulassay T, Vasarhelyi B. Human Th1 and Th2 lymphocytes are distinguished by calcium flux regulation during the first 10 Min of lymphocyte activation. Immunobiology (2012) 217(1):37–43. doi: 10.1016/j.imbio.2011.08.007
249. Launay S, Gianni M, Kovacs T, Bredoux R, Bruel A, Gelebart P, et al. Lineage-specific modulation of calcium pump expression during myeloid differentiation. Blood (1999) 93(12):4395–405. doi: 10.1182/blood.V93.12.4395
250. Lacabaratz-Porret C, Launay S, Corvazier E, Bredoux R, Papp B, Enouf J. Biogenesis of endoplasmic reticulum proteins involved in Ca2+ signalling during megakaryocytic differentiation: An in vitro study. (2000) Biochem J 350(Pt 3):723–34. doi: 10.1042/bj3500723
251. Ait Ghezali L, Arbabian A, Roudot H, Brouland JP, Baran-Marszak F, Salvaris E, et al. Induction of endoplasmic reticulum calcium pump expression during early leukemic b cell differentiation. J Exp Clin Cancer Res (2017) 36(1):87. doi: 10.1186/s13046-017-0556-7
252. Launay S, Gianni M, Diomede L, Machesky LM, Enouf J, Papp B. Enhancement of atra-induced cell differentiation by inhibition of calcium accumulation into the endoplasmic reticulum: Cross-talk between rar alpha and calcium-dependent signaling. Blood (2003) 101(8):3220–8. doi: 10.1182/blood-2002-09-2730
253. Roti G, Carlton A, Ross KN, Markstein M, Pajcini K, Su AH, et al. Complementary genomic screens identify serca as a therapeutic target in Notch1 mutated cancer. Cancer Cell (2013) 23(3):390–405. doi: 10.1016/j.ccr.2013.01.015
254. Zhai X, Sterea AM, Hiani YE. Lessons from the endoplasmic reticulum Ca(2+) transporters-a cancer connection. Cells (2020) 9(6):1536. doi: 10.3390/cells9061536
255. Karjalainen R, Liu M, Kumar A, He L, Malani D, Parsons A, et al. Elevated expression of S100a8 and S100a9 correlates with resistance to the bcl-2 inhibitor venetoclax in aml. Leukemia (2019) 33(10):2548–53. doi: 10.1038/s41375-019-0504-y
256. Alanazi B, Munje CR, Rastogi N, Williamson AJK, Taylor S, Hole PS, et al. Integrated nuclear proteomics and transcriptomics identifies S100a4 as a therapeutic target in acute myeloid leukemia. Leukemia (2020) 34(2):427–40. doi: 10.1038/s41375-019-0596-4
257. Lyu T, Wang Y, Li D, Yang H, Qin B, Zhang W, et al. Exosomes from bm-mscs promote acute myeloid leukemia cell proliferation, invasion and chemoresistance Via upregulation of S100a4. Exp Hematol Oncol (2021) 10(1):24. doi: 10.1186/s40164-021-00220-7
258. Guo L, Liu J, Yang Y, Zeng Y, Yuan F, Zhong F, et al. Purple sweet potato anthocyanins elicit calcium overload-induced cell death by inhibiting the calcium-binding protein S100a4 in acute lymphoblastic leukemia. Food Bioscience (2021) 42:101214. doi: 10.1016/j.fbio.2021.101214
259. Yang XY, Jin J, Huang J, Li P, Xue JW, Wu XJ, et al. Expression and clinical significance profile analysis of S100 family members in human acute myeloid leukemia. Eur Rev Med Pharmacol Sci (2020) 24(13):7324–34. doi: 10.26355/eurrev_202007_21896
260. Yang L, Yang M, Zhang H, Wang Z, Yu Y, Xie M, et al. S100a8-targeting sirna enhances arsenic trioxide-induced myeloid leukemia cell death by down-regulating autophagy. Int J Mol Med (2012) 29(1):65–72. doi: 10.3892/ijmm.2011.806
261. Yang XY, Zhang MY, Zhou Q, Wu SY, Zhao Y, Gu WY, et al. High expression of S100a8 gene is associated with drug resistance to etoposide and poor prognosis in acute myeloid leukemia through influencing the apoptosis pathway. Onco Targets Ther (2016) 9:4887–99. doi: 10.2147/OTT.S101594
262. Nicolas E, Ramus C, Berthier S, Arlotto M, Bouamrani A, Lefebvre C, et al. Expression of S100a8 in leukemic cells predicts poor survival in De novo aml patients. Leukemia (2011) 25(1):57–65. doi: 10.1038/leu.2010.251
263. Laouedj M, Tardif MR, Gil L, Raquil MA, Lachhab A, Pelletier M, et al. S100a9 induces differentiation of acute myeloid leukemia cells through Tlr4. Blood (2017) 129(14):1980–90. doi: 10.1182/blood-2016-09-738005
264. Mondet J, Laurin D, Lo Presti C, Jacob MC, Meunier M, Giraudon E, et al. Increased S100a8 expression in bone marrow plasma by monocytic cells from acute myeloid leukemia patients. Hematol Oncol (2020) 38(1):114–8. doi: 10.1002/hon.2707
265. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult De novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
266. Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med (2004) 350(16):1605–16. doi: 10.1056/NEJMoa031046
267. Tsumura H, Akimoto M, Kiyota H, Ishii Y, Ishikura H, Honma Y. Gene expression profiles in differentiating leukemia cells induced by methyl jasmonate are similar to those of cytokinins and methyl jasmonate analogs induce the differentiation of human leukemia cells in primary culture. Leukemia (2009) 23(4):753–60. doi: 10.1038/leu.2008.347
268. Ishii Y, Kasukabe T, Honma Y. Immediate up-regulation of the calcium-binding protein S100p and its involvement in the cytokinin-induced differentiation of human myeloid leukemia cells. Biochim Biophys Acta (2005) 1745(2):156–65. doi: 10.1016/j.bbamcr.2005.01.005
269. Kang X, Cui C, Wang C, Wu G, Chen H, Lu Z, et al. Camks support development of acute myeloid leukemia. J Hematol Oncol (2018) 11(1):30. doi: 10.1186/s13045-018-0574-8
270. Si J, Collins SJ. Activated Ca2+/Calmodulin-dependent protein kinase iigamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res (2008) 68(10):3733–42. doi: 10.1158/0008-5472.CAN-07-2509
271. Kornblau SM, Vu HT, Ruvolo P, Estrov Z, O'Brien S, Cortes J, et al. Bax and pkcalpha modulate the prognostic impact of Bcl2 expression in acute myelogenous leukemia. Clin Cancer Res (2000) 6(4):1401–9.
272. Kurinna S, Konopleva M, Palla SL, Chen W, Kornblau S, Contractor R, et al. Bcl2 phosphorylation and active pkc alpha are associated with poor survival in aml. Leukemia (2006) 20(7):1316–9. doi: 10.1038/sj.leu.2404248
273. Ruvolo PP, Zhou L, Watt JC, Ruvolo VR, Burks JK, Jiffar T, et al. Targeting pkc-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J Cell Biochem (2011) 112(6):1696–707. doi: 10.1002/jcb.23090
274. Spijkers-Hagelstein JA, Schneider P, Hulleman E, de Boer J, Williams O, Pieters R, et al. Elevated S100a8/S100a9 expression causes glucocorticoid resistance in mll-rearranged infant acute lymphoblastic leukemia. Leukemia (2012) 26(6):1255–65. doi: 10.1038/leu.2011.388
275. Yamaguchi H, Hanawa H, Uchida N, Inamai M, Sawaguchi K, Mitamura Y, et al. Multistep pathogenesis of leukemia Via the mll-Af4 chimeric Gene/Flt3 gene tyrosine kinase domain (Tkd) mutation-related enhancement of S100a6 expression. Exp Hematol (2009) 37(6):701–14. doi: 10.1016/j.exphem.2009.02.007
276. Tamai H, Miyake K, Yamaguchi H, Takatori M, Dan K, Inokuchi K, et al. Resistance of mll-Aff1-Positive acute lymphoblastic leukemia to tumor necrosis factor-alpha is mediated by S100a6 upregulation. Blood Cancer J (2011) 1(11):e38. doi: 10.1038/bcj.2011.37
277. Tamai H, Miyake K, Yamaguchi H, Shimada T, Dan K, Inokuchi K. Inhibition of S100a6 induces gvl effects in Mll/Af4-positive all in human pbmc-scid mice. Bone Marrow Transplant (2014) 49(5):699–703. doi: 10.1038/bmt.2014.18
278. Tamai H, Yamaguchi H, Miyake K, Takatori M, Kitano T, Yamanaka S, et al. Amlexanox downregulates S100a6 to sensitize Kmt2a/Aff1-positive acute lymphoblastic leukemia to tnfalpha treatment. Cancer Res (2017) 77(16):4426–33. doi: 10.1158/0008-5472.CAN-16-2974
279. Chow YP, Alias H, Jamal R. Meta-analysis of gene expression in relapsed childhood b-acute lymphoblastic leukemia. BMC Cancer (2017) 17(1):120. doi: 10.1186/s12885-017-3103-1
280. Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, et al. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat Med (2007) 13(6):736–41. doi: 10.1038/nm1588
281. Gachet S, Genesca E, Passaro D, Irigoyen M, Alcalde H, Clemenson C, et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia (2013) 27(12):2289–300. doi: 10.1038/leu.2013.156
282. He X, Deng Y, Yue W. Investigating critical genes and gene interaction networks that mediate cyclophosphamide sensitivity in chronic myelogenous leukemia. Mol Med Rep (2017) 16(1):523–32. doi: 10.3892/mmr.2017.6636
283. Yang L, Li D, Tang P, Zuo Y. Curcumin increases the sensitivity of K562/Dox cells to doxorubicin by targeting S100 calcium-binding protein A8 and p-glycoprotein. Oncol Lett (2020) 19(1):83–92. doi: 10.3892/ol.2019.11083
284. Gonzalez M, De Brasi C, Ferri C, Bengio R, Bianchini M, Larripa I. Camkiigamma, Hsp70 and Hsp90 transcripts are differentially expressed in chronic myeloid leukemia cells from patients with resistant mutated disease. Leuk Lymphoma (2014) 55(9):2101–8. doi: 10.3109/10428194.2013.861070
285. Balasubramanian N, Advani SH, Zingde SM. Protein kinase c isoforms in normal and leukemic neutrophils: Altered levels in leukemic neutrophils and changes during myeloid maturation in chronic myeloid leukemia. Leuk Res (2002) 26(1):67–81. doi: 10.1016/s0145-2126(01)00098-4
286. Kabir NN, Ronnstrand L, Kazi JU. Protein kinase c expression is deregulated in chronic lymphocytic leukemia. Leuk Lymphoma (2013) 54(10):2288–90. doi: 10.3109/10428194.2013.769220
287. Kovacic M, Mitrovic-Ajtic O, Beleslin-Cokic B, Djikic D, Suboticki T, Diklic M, et al. Tlr4 and rage conversely mediate pro-inflammatory S100a8/9-mediated inhibition of proliferation-linked signaling in myeloproliferative neoplasms. Cell Oncol (Dordr) (2018) 41(5):541–53. doi: 10.1007/s13402-018-0392-6
288. Giraudier S, Chagraoui H, Komura E, Barnache S, Blanchet B, LeCouedic JP, et al. Overexpression of Fkbp51 in idiopathic myelofibrosis regulates the growth factor independence of megakaryocyte progenitors. Blood (2002) 100(8):2932–40. doi: 10.1182/blood-2002-02-0485
289. Komura E, Chagraoui H, Mansat de Mas V, Blanchet B, de Sepulveda P, Larbret F, et al. Spontaneous Stat5 activation induces growth factor independence in idiopathic myelofibrosis: Possible relationship with Fkbp51 overexpression. Exp Hematol (2003) 31(7):622–30. doi: 10.1016/s0301-472x(03)00085-7
290. Pietra D, Rumi E, Ferretti VV, Di Buduo CA, Milanesi C, Cavalloni C, et al. Differential clinical effects of different mutation subtypes in calr-mutant myeloproliferative neoplasms. Leukemia (2016) 30(2):431–8. doi: 10.1038/leu.2015.277
291. Jaiswal A, Wang Z, Zhu X, Ju Z. Unraveling the connections between calreticulin and myeloproliferative neoplasms Via calcium signalling. bioRxiv (2021) 2021.08.05.455248. doi: 10.1101/2021.08.05.455248
292. Alsagaby SA, Khanna S, Hart KW, Pratt G, Fegan C, Pepper C, et al. Proteomics-based strategies to identify proteins relevant to chronic lymphocytic leukemia. J Proteome Res (2014) 13(11):5051–62. doi: 10.1021/pr5002803
293. Abrams ST, Lakum T, Lin K, Jones GM, Treweeke AT, Farahani M, et al. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase cbetaii. Blood (2007) 109(3):1193–201. doi: 10.1182/blood-2006-03-012021
294. Lutzny G, Kocher T, Schmidt-Supprian M, Rudelius M, Klein-Hitpass L, Finch AJ, et al. Protein kinase c-Beta-Dependent activation of nf-kappab in stromal cells is indispensable for the survival of chronic lymphocytic leukemia b cells in vivo. Cancer Cell (2013) 23(1):77–92. doi: 10.1016/j.ccr.2012.12.003
295. Fornecker LM, Muller L, Bertrand F, Paul N, Pichot A, Herbrecht R, et al. Multi-omics dataset to decipher the complexity of drug resistance in diffuse Large b-cell lymphoma. Sci Rep (2019) 9(1):895. doi: 10.1038/s41598-018-37273-4
296. Bucher P, Erdmann T, Grondona P, Xu W, Schmitt A, Schurch C, et al. Targeting chronic nfat activation with calcineurin inhibitors in diffuse Large b-cell lymphoma. Blood (2020) 135(2):121–32. doi: 10.1182/blood.2019001866
297. Zhou Z, Li Z, Sun Z, Zhang X, Lu L, Wang Y, et al. S100a9 and Orm1 serve as predictors of therapeutic response and prognostic factors in advanced extranodal Nk/T cell lymphoma patients treated with Pegaspargase/Gemcitabine. Sci Rep (2016) 6:23695. doi: 10.1038/srep23695
298. Zhou Z, Chen X, Li Z, Wang X, Zhang M. Overexpression of S100a9 in tumor stroma contribute to immune evasion of Nk/T cell lymphoma and predict poor response rate. Sci Rep (2021) 11(1):11220. doi: 10.1038/s41598-021-90794-3
299. Yang L, Wu B, Wu Z, Xu Y, Wang P, Li M, et al. Camkiiγ is a targetable driver of multiple myeloma through camkiiγ/ Stat3 axis. Aging (2020) 12(13):13668–83. doi: 10.18632/aging.103490
300. Brenner AK, Bruserud O. S100 proteins in acute myeloid leukemia. Neoplasia (2018) 20(12):1175–86. doi: 10.1016/j.neo.2018.09.007
301. Mondet J, Chevalier S, Mossuz P. Pathogenic roles of S100a8 and S100a9 proteins in acute myeloid and lymphoid leukemia: Clinical and therapeutic impacts. Molecules (2021) 26(5):1323. doi: 10.3390/molecules26051323
302. Hua X, Zhang H, Jia J, Chen S, Sun Y, Zhu X. Roles of S100 family members in drug resistance in tumors: Status and prospects. BioMed Pharmacother (2020) 127:110156. doi: 10.1016/j.biopha.2020.110156
303. Zhang L, Zhou S, Zhou T, Yuan K, Li X, Tang J. S100a8 promotes chemoresistance Via augmenting autophagy in bcell lymphoma cells. Oncol Rep (2021) 45(1):151–8. doi: 10.3892/or.2020.7841
304. Zavorka Thomas ME, Jeon JY, Talebi Z, Buelow DR, Silvaroli J, Campbell MJ, et al. Gilteritinib-induced upregulation of S100a9 is mediated through Bcl6 in acute myeloid leukemia. Blood Adv (2021) 5(23):5041–6. doi: 10.1182/bloodadvances.2021005614
305. Berchtold MW, Villalobo A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta (2014) 1843(2):398–435. doi: 10.1016/j.bbamcr.2013.10.021
306. Brzozowski JS, Skelding KA. The multi-functional Calcium/Calmodulin stimulated protein kinase (Camk) family: Emerging targets for anti-cancer therapeutic intervention. Pharm (Basel) (2019) 12(1):8. doi: 10.3390/ph12010008
307. Monaco S, Rusciano MR, Maione AS, Soprano M, Gomathinayagam R, Todd LR, et al. A novel crosstalk between Calcium/Calmodulin kinases ii and iv regulates cell proliferation in myeloid leukemia cells. Cell Signal (2015) 27(2):204–14. doi: 10.1016/j.cellsig.2014.11.007
308. Gu Y, Zhang J, Ma X, Kim BW, Wang H, Li J, et al. Stabilization of the c-myc protein by camkiigamma promotes T cell lymphoma. Cancer Cell (2017) 32(1):115–28.e7. doi: 10.1016/j.ccell.2017.06.001
309. Terrie E, Coronas V, Constantin B. Role of the calcium toolkit in cancer stem cells. Cell Calcium (2019) 80:141–51. doi: 10.1016/j.ceca.2019.05.001
310. Gu Y, Chen T, Meng Z, Gan Y, Xu X, Lou G, et al. Camkii gamma, a critical regulator of cml Stem/Progenitor cells, is a target of the natural product berbamine. Blood (2012) 120(24):4829–39. doi: 10.1182/blood-2012-06-434894
311. Medyouf H, Ghysdael J. The Calcineurin/Nfat signaling pathway: A novel therapeutic target in leukemia and solid tumors. Cell Cycle (2008) 7(3):297–303. doi: 10.4161/cc.7.3.5357
312. Zaslavsky A, Chou ST, Schadler K, Lieberman A, Pimkin M, Kim YJ, et al. The calcineurin-nfat pathway negatively regulates megakaryopoiesis. Blood (2013) 121(16):3205–15. doi: 10.1182/blood-2012-04-421172
313. Kyttala S, Habermann I, Minami T, Ehninger G, Kiani A. Regulation of down syndrome critical region 1 expression by nuclear factor of activated T cells in megakaryocytes. Br J Haematol (2009) 144(3):395–408. doi: 10.1111/j.1365-2141.2008.07490.x
314. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of down syndrome. J Clin Invest (2012) 122(3):948–62. doi: 10.1172/JCI60455
315. Qin JJ, Nag S, Wang W, Zhou J, Zhang WD, Wang H, et al. Nfat as cancer target: Mission possible? Biochim Biophys Acta (2014) 1846(2):297–311. doi: 10.1016/j.bbcan.2014.07.009
316. Gao R, Zhang Y, Zeng C, Li Y. The role of nfat in the pathogenesis and targeted therapy of hematological malignancies. Eur J Pharmacol (2022) 921:174889. doi: 10.1016/j.ejphar.2022.174889
317. Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res (2021) 31(1):5–16. doi: 10.1038/s41422-020-0383-9
318. Szybinski J, Meyer SC. Genetics of myeloproliferative neoplasms. Hematol Oncol Clin North Am (2021) 35(2):217–36. doi: 10.1016/j.hoc.2020.12.002
319. Shivarov V, Ivanova M, Tiu RV. Mutated calreticulin retains structurally disordered c terminus that cannot bind Ca(2+): Some mechanistic and therapeutic implications. Blood Cancer J (2014) 4:e185. doi: 10.1038/bcj.2014.7
320. Isakov N, Protein Kinase C. (Pkc) isoforms in cancer, tumor promotion and tumor suppression. Semin Cancer Biol (2018) 48:36–52. doi: 10.1016/j.semcancer.2017.04.012
321. Newton AC. Protein kinase c: Perfectly balanced. Crit Rev Biochem Mol Biol (2018) 53(2):208–30. doi: 10.1080/10409238.2018.1442408
322. Kazi JU, Kabir NN, Ronnstrand L. (Pkc) as a drug target in chronic lymphocytic leukemia. Med Oncol (2013) 30(4):757. doi: 10.1007/s12032-013-0757-7
323. Mencalha AL, Correa S, Abdelhay E. Role of calcium-dependent protein kinases in chronic myeloid leukemia: Combined effects of pkc and bcr-abl signaling on cellular alterations during leukemia development. Onco Targets Ther (2014) 7:1247–54. doi: 10.2147/OTT.S64303
324. Piwocka K, Vejda S, Cotter TG, O'Sullivan GC, McKenna SL. Bcr-abl reduces endoplasmic reticulum releasable calcium levels by a bcl-2-Independent mechanism and inhibits calcium-dependent apoptotic signaling. Blood (2006) 107(10):4003–10. doi: 10.1182/blood-2005-04-1523
325. Ma D, Liu P, Wang P, Zhou Z, Fang Q, Wang J. Pkc-Beta/Alox5 axis activation promotes bcr-Abl-Independent tki-resistance in chronic myeloid leukemia. J Cell Physiol (2021) 236(9):6312–27. doi: 10.1002/jcp.30301
326. Holler C, Pinon JD, Denk U, Heyder C, Hofbauer S, Greil R, et al. Pkcbeta is essential for the development of chronic lymphocytic leukemia in the Tcl1 transgenic mouse model: Validation of pkcbeta as a therapeutic target in chronic lymphocytic leukemia. Blood (2009) 113(12):2791–4. doi: 10.1182/blood-2008-06-160713
327. Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant mll-rearranged acute lymphoblastic leukemias. Nat Genet (2015) 47(4):330–7. doi: 10.1038/ng.3230
328. Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of Dux4 and erg in acute lymphoblastic leukemia. Nat Genet (2016) 48(12):1481–9. doi: 10.1038/ng.3691
329. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse Large b cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
330. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse Large b cell lymphoma. Cell (2017) 171(2):481–94.e15. doi: 10.1016/j.cell.2017.09.027
331. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse Large b-cell lymphoma (Dlbcl) by whole-exome sequencing. Proc Natl Acad Sci USA (2012) 109(10):3879–84. doi: 10.1073/pnas.1121343109
332. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell (2018) 173(2):291–304.e6. doi: 10.1016/j.cell.2018.03.022
333. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst (2018) 6(3):271–81.e7. doi: 10.1016/j.cels.2018.03.002
334. Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell (2018) 33(4):676–89.e3. doi: 10.1016/j.ccell.2018.03.007
335. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated tcga pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell (2018) 173(2):400–16.e11. doi: 10.1016/j.cell.2018.02.052
336. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell (2018) 173(2):321–37.e10. doi: 10.1016/j.cell.2018.03.035
337. Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep (2018) 23(1):227–38.e3. doi: 10.1016/j.celrep.2018.03.050
338. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature (2020) 579(7800):567–74. doi: 10.1038/s41586-020-2095-1
339. Ding L, Bailey MH, Porta-Pardo E, Thorsson V, Colaprico A, Bertrand D, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell (2018) 173(2):305–20.e10. doi: 10.1016/j.cell.2018.03.033
340. Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol (2017) 2017:PO.17.00073. doi: 10.1200/PO.17.00073
341. Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, et al. Mutational and structural analysis of diffuse Large b-cell lymphoma using whole-genome sequencing. Blood (2013) 122(7):1256–65. doi: 10.1182/blood-2013-02-483727
342. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell (2013) 152(4):714–26. doi: 10.1016/j.cell.2013.01.019
343. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving cll and their evolution in progression and relapse. Nature (2015) 526(7574):525–30. doi: 10.1038/nature15395
344. Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature (2015) 526(7574):519–24. doi: 10.1038/nature14666
345. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor Sf3b1 gene in chronic lymphocytic leukemia. Nat Genet (2011) 44(1):47–52. doi: 10.1038/ng.1032
346. Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U.S.A. (2013) 110(45):18250–5. doi: 10.1073/pnas.1314608110
347. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell (2014) 25(1):91–101. doi: 10.1016/j.ccr.2013.12.015
348. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and sezary syndrome. Nat Genet (2015) 47(12):1465–70. doi: 10.1038/ng.3442
349. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature (2018) 562(7728):526–31. doi: 10.1038/s41586-018-0623-z
350. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature (2011) 478(7367):64–9. doi: 10.1038/nature10496
351. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
352. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood (2013) 122(22):3616–27. doi: 10.1182/blood-2013-08-518886
353. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic calr mutations in myeloproliferative neoplasms with nonmutated Jak2. N Engl J Med (2013) 369(25):2391–405. doi: 10.1056/NEJMoa1312542
354. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
355. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signal (2013) 6(269):l1. doi: 10.1126/scisignal.2004088
356. Fracchia KM, Pai CY, Walsh CM. Modulation of T cell metabolism and function through calcium signaling. Front Immunol (2013) 4:324. doi: 10.3389/fimmu.2013.00324
357. Kotturi MF, Carlow DA, Lee JC, Ziltener HJ, Jefferies WA. Identification and functional characterization of voltage-dependent calcium channels in T lymphocytes. J Biol Chem (2003) 278(47):46949–60. doi: 10.1074/jbc.M309268200
358. Pelletier L, Savignac M. Ca(2+) signaling in T-cell subsets with a focus on the role of Cav1 channels: Possible implications in therapeutics. Front Immunol (2013) 4:150. doi: 10.3389/fimmu.2013.00150
359. Pelletier L, Moreau M. Cav1 channels is also a story of non excitable cells: Application to calcium signalling in two different non related models. Biochim Biophys Acta Mol Cell Res (2021) 1868(6):118996. doi: 10.1016/j.bbamcr.2021.118996
360. Fenninger F, Han J, Stanwood SR, Nohara LL, Arora H, Choi KB, et al. Mutation of an l-type calcium channel gene leads to T lymphocyte dysfunction. Front Immunol (2019) 10:2473. doi: 10.3389/fimmu.2019.02473
361. Nohara LL, Stanwood SR, Omilusik KD, Jefferies WA. Tweeters, woofers and horns: The complex orchestration of calcium currents in T lymphocytes. Front Immunol (2015) 6:234. doi: 10.3389/fimmu.2015.00234
362. Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH. Molecular cloning of cdna encoding the Ca2+ release channel (Ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem (1990) 265(23):13472–83. doi: 10.1016/S0021-9258(18)77371-7
363. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol (2010) 2(11):a003996. doi: 10.1101/cshperspect.a003996
364. Sei Y, Gallagher KL, Basile AS. Skeletal muscle type ryanodine receptor is involved in calcium signaling in human b lymphocytes. J Biol Chem (1999) 274(9):5995–6002. doi: 10.1074/jbc.274.9.5995
365. Hosoi E, Nishizaki C, Gallagher KL, Wyre HW, Matsuo Y, Sei Y. Expression of the ryanodine receptor isoforms in immune cells. J Immunol (2001) 167(9):4887–94. doi: 10.4049/jimmunol.167.9.4887
366. McCarthy TV, Datar S, Mackrill JJ. Activation of ryanodine Receptor/Ca2+ release channels downregulates Cd38 in the namalwa b lymphoma. FEBS Lett (2003) 554(1-2):133–7. doi: 10.1016/s0014-5793(03)01122-0
367. Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. Cd38 and chronic lymphocytic leukemia: A decade later. Blood (2011) 118(13):3470–8. doi: 10.1182/blood-2011-06-275610
368. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369(25):2379–90. doi: 10.1056/NEJMoa1311347
369. Debant M, Hemon P, Brigaudeau C, Renaudineau Y, Mignen O. Calcium signaling and cell fate: How can Ca2+ signals contribute to wrong decisions for chronic lymphocytic leukemic b lymphocyte outcome? Int J Dev Biol (2015) 59(7-9):379–89. doi: 10.1387/ijdb.150204om
370. Li T, Chen J, Zeng Z. Pathophysiological role of calcium channels and transporters in the multiple myeloma. Cell Commun Signal (2021) 19(1):99. doi: 10.1186/s12964-021-00781-4
371. Lewuillon C, Laguillaumie MO, Quesnel B, Idziorek T, Touil Y, Lemonnier L. Put in a "Ca(2+)Ll" to acute myeloid leukemia. Cells (2022) 11(3):543. doi: 10.3390/cells11030543
372. Morelli MB, Offidani M, Alesiani F, Discepoli G, Liberati S, Olivieri A, et al. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. a role for transient receptor potential vanilloid type-2. Int J Cancer (2014) 134(11):2534–46. doi: 10.1002/ijc.28591
373. Kamal T, Green TN, Hearn JI, Josefsson EC, Morel-Kopp MC, Ward CM, et al. N-Methyl-D-Aspartate receptor mediated calcium influx supports in vitro differentiation of normal mouse megakaryocytes but proliferation of leukemic cell lines. Res Pract Thromb Hae (2018) 2(1):125–38. doi: 10.1002/rth2.12068
374. Abdoul-Azize S, Buquet C, Vannier JP, Dubus I. Pyr3, a Trpc3 channel blocker, potentiates dexamethasone sensitivity and apoptosis in acute lymphoblastic leukemia cells by disturbing Ca(2+) signaling, mitochondrial membrane potential changes and reactive oxygen species production. Eur J Pharmacol (2016) 784:90–8. doi: 10.1016/j.ejphar.2016.05.014
375. Haridas V, Li X, Mizumachi T, Higuchi M, Lemeshko VV, Colombini M, et al. Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion (2007) 7(3):234–40. doi: 10.1016/j.mito.2006.12.005
376. Huang W, Lu C, Wu Y, Ouyang S, Chen Y. T-Type calcium channel antagonists, mibefradil and nnc-55-0396 inhibit cell proliferation and induce cell apoptosis in leukemia cell lines. J Exp Clin Cancer Res (2015) 34:54. doi: 10.1186/s13046-015-0171-4
377. Li H, Xu J, Zhou Y, Liu X, Shen LE, Zhu YU, et al. Plscr1/Ip3r1/Ca(2+) axis contributes to differentiation of primary aml cells induced by wogonoside. Cell Death Dis (2017) 8(5):e2768. doi: 10.1038/cddis.2017.175
378. Chen Y, Hui H, Yang H, Zhao K, Qin Y, Gu C, et al. Wogonoside induces cell cycle arrest and differentiation by affecting expression and subcellular localization of Plscr1 in aml cells. Blood (2013) 121(18):3682–91. doi: 10.1182/blood-2012-11-466219
379. Xiao R, Gan M, Jiang T. Wogonoside exerts growth-suppressive effects against T acute lymphoblastic leukemia through the Stat3 pathway. Hum Exp Toxicol (2017) 36(11):1169–76. doi: 10.1177/0960327116679716
380. Cruz P, Ahumada-Castro U, Bustos G, Molgo J, Sauma D, Lovy A, et al. Inhibition of Insp3r with xestospongin b reduces mitochondrial respiration and induces selective cell death in T cell acute lymphoblastic leukemia cells. Int J Mol Sci (2021) 22(2):651. doi: 10.3390/ijms22020651
381. Carmichael B, Lehman A, Elgamal OA, Orwick SJ, Truxall J, Beaver L, et al. Abstract 1938: Store-operated calcium signaling is an effective therapeutic target in aml. Cancer Res (2018) 78(13_Supplement):1938. doi: 10.1158/1538-7445.AM2018-1938
382. Yanamandra N, Buzzeo RW, Gabriel M, Hazlehurst LA, Mari Y, Beaupre DM, et al. Tipifarnib-induced apoptosis in acute myeloid leukemia and multiple myeloma cells depends on Ca2+ influx through plasma membrane Ca2+ channels. J Pharmacol Exp Ther (2011) 337(3):636–43. doi: 10.1124/jpet.110.172809
383. Zhong W, Cao X, Pan G, Niu Q, Feng X, Xu M, et al. Orp4l is a prerequisite for the induction of T-cell leukemogenesis associated with human T-cell leukemia virus 1. Blood (2022) 139(7):1052–65. doi: 10.1182/blood.2021013579
384. Marchesini M, Gherli A, Montanaro A, Patrizi L, Sorrentino C, Pagliaro L, et al. Blockade of oncogenic Notch1 with the serca inhibitor Cad204520 in T cell acute lymphoblastic leukemia. Cell Chem Biol (2020) 27(6):678–97.e13. doi: 10.1016/j.chembiol.2020.04.002
385. De Ford C, Heidersdorf B, Haun F, Murillo R, Friedrich T, Borner C, et al. The clerodane diterpene casearin J induces apoptosis of T-all cells through serca inhibition, oxidative stress, and interference with Notch1 signaling. Cell Death Dis (2016) 7:e2070. doi: 10.1038/cddis.2015.413
386. De Ford C, Calderon C, Sehgal P, Fedosova NU, Murillo R, Olesen C, et al. Discovery of tricyclic clerodane diterpenes as Sarco/Endoplasmic reticulum Ca(2+)-atpase inhibitors and structure-activity relationships. J Nat Prod (2015) 78(6):1262–70. doi: 10.1021/acs.jnatprod.5b00062
387. Bleeker NP, Cornea RL, Thomas DD, Xing C. A novel serca inhibitor demonstrates synergy with classic serca inhibitors and targets multidrug-resistant aml. Mol Pharm (2013) 10(11):4358–66. doi: 10.1021/mp400458u
388. Akl H, Vandecaetsbeek I, Monaco G, Kauskot A, Luyten T, Welkenhuyzen K, et al. Ha14-1, but not the Bh3 mimetic abt-737, causes Ca2+ dysregulation in platelets and human cell lines. Haematologica (2013) 98(4):e49–51. doi: 10.3324/haematol.2012.080598
389. Hermanson D, Addo SN, Bajer AA, Marchant JS, Das SG, Srinivasan B, et al. Dual mechanisms of sha 14-1 in inducing cell death through endoplasmic reticulum and mitochondria. Mol Pharmacol (2009) 76(3):667–78. doi: 10.1124/mol.109.055830
390. Tian D, Das SG, Doshi JM, Peng J, Lin J, Xing C. Sha 14-1, a stable and ros-free antagonist against anti-apoptotic bcl-2 proteins, bypasses drug resistances and synergizes cancer therapies in human leukemia cell. Cancer Lett (2008) 259(2):198–208. doi: 10.1016/j.canlet.2007.10.012
391. Akl H, La Rovere RM, Janssens A, Vandenberghe P, Parys JB, Bultynck G. Ha14-1 potentiates apoptosis in b-cell cancer cells sensitive to a peptide disrupting ip 3 receptor / bcl-2 complexes. Int J Dev Biol (2015) 59(7-9):391–8. doi: 10.1387/ijdb.150213gb
392. Roti G, Qi J, Kitara S, Sanchez-Martin M, Saur Conway A, Varca AC, et al. Leukemia-specific delivery of mutant Notch1 targeted therapy. J Exp Med (2018) 215(1):197–216. doi: 10.1084/jem.20151778
393. Wang Z, Mi T, Bradley HL, Metts J, Sabnis H, Zhu W, et al. Pimozide and imipramine blue exploit mitochondrial vulnerabilities and reactive oxygen species to cooperatively target high risk acute myeloid leukemia. Antioxidants (Basel) (2021) 10(6):956. doi: 10.3390/antiox10060956
394. Gross S, Mallu P, Joshi H, Schultz B, Go C, Soboloff J. Ca(2+) as a therapeutic target in cancer. Adv Cancer Res (2020) 148:233–317. doi: 10.1016/bs.acr.2020.05.003
Keywords: Calcium signaling, calcium homeostasis, blood cells, lymphoma, myeloproliferative neoplasms, red cell abnormalities, leukaemia, cancer biological pathways
Citation: Immanuel T, Li J, Green TN, Bogdanova A and Kalev-Zylinska ML (2022) Deregulated calcium signaling in blood cancer: Underlying mechanisms and therapeutic potential. Front. Oncol. 12:1010506. doi: 10.3389/fonc.2022.1010506
Received: 03 August 2022; Accepted: 21 September 2022;
Published: 18 October 2022.
Edited by:
Giuseppe Maurizio Campo, University of Messina, ItalyReviewed by:
Pawan Faris, University of Pavia, ItalyDavid Lawrence Prole, University of Cambridge, United Kingdom
Carlos Villalobos, Spanish National Research Council (CSIC), Spain
Giovanni Monaco, Centre for Drug Design and Discovery, Belgium
Copyright © 2022 Immanuel, Li, Green, Bogdanova and Kalev-Zylinska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maggie L. Kalev-Zylinska, bS5rYWxldkBhdWNrbGFuZC5hYy5ueg==