 Daniel M. Freed
Daniel M. Freed Josh Sommer
Josh Sommer Nindo Punturi
Nindo Punturi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 October 2022
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1009193
This article is part of the Research TopicChordoma: Advances in Biology and Clinical ManagementView all 14 articles
The development of effective and personalized treatment options for patients with rare cancers like chordoma is hampered by numerous challenges. Biomarker-guided repurposing of therapies approved in other indications remains the fastest path to redefining the treatment paradigm, but chordoma’s low mutation burden limits the impact of genomics in target discovery and precision oncology efforts. As our knowledge of oncogenic mechanisms across various malignancies has matured, it’s become increasingly clear that numerous properties of tumors transcend their genomes – leading to new and uncharted frontiers of therapeutic opportunity. In this review, we discuss how the implementation of cutting-edge tools and approaches is opening new windows into chordoma’s vulnerabilities. We also note how a convergence of emerging observations in chordoma and other cancers is leading to the identification and evaluation of new therapeutic hypotheses for this rare cancer.
Chordoma is an ultra-rare bone cancer that arises in the skull base or spine of pediatrics and adults, and originates from vestigial remnants of the embryonic notochord. Normally a low-grade but locally invasive disease, current standard of care for chordoma involves maximum surgical resection and/or radiotherapy (1). Despite significant advances in surgical techniques and radiotherapy strategies, the majority of chordoma patients eventually develop recurrent and/or metastatic disease and ultimately require systemic therapy to control further progression (2). To date, no drugs are approved for the treatment of advanced chordoma and conventional chemotherapy is generally ineffective (1, 2), resulting in a poor prognosis in the advanced disease setting. Research efforts over the past two decades have focused intensively on evaluating drug repurposing opportunities, primarily guided by detection of activated signaling pathways (3–5), focused drug screens (6–8), or anecdotal clinical responses to therapies (9, 10). These investigations inspired several Phase II clinical trials primarily involving multi-kinase inhibition with agents such as imatinib (11), sorafenib (12), lapatinib (13), or everolimus plus imatinib (14), for example, although modest efficacy and low objective response rates were observed in each study. In parallel, efforts to discover novel drug targets indicate that chordoma relies on the lineage-specific transcription factor brachyury (15–17), positioning it as arguably the most attractive – though, as of yet undruggable – target in chordoma.
Over the same time period, the continued growth of genome-guided precision oncology prompted an explosion of drug repurposing efforts for molecularly-defined tumor types – a trend that also extended into the realm of rare cancers. For example, following its approval in chronic myelogenous leukemia, imatinib was successfully repurposed for KIT-mutant gastrointestinal stromal tumors (18), and dabrafenib plus trametinib was repositioned for BRAF V600-mutated anaplastic thyroid cancer (19) after the approval of this combination in non-small cell lung cancer (NSCLC) and melanoma. These and other success stories motivated a series of genomic profiling efforts in chordoma (20–27), with the hope that lifting the veil on chordoma genomes might reveal actionable therapeutic opportunities. Instead, these studies revealed that, similar to most sarcomas and pediatric cancers (28–30), chordoma appears to be characterized by a low and infrequently-actionable mutation burden – with only ~14% of chordomas harboring genomic biomarkers predictive of response to FDA-approved or investigational therapies in other indications (Table 1) (31).
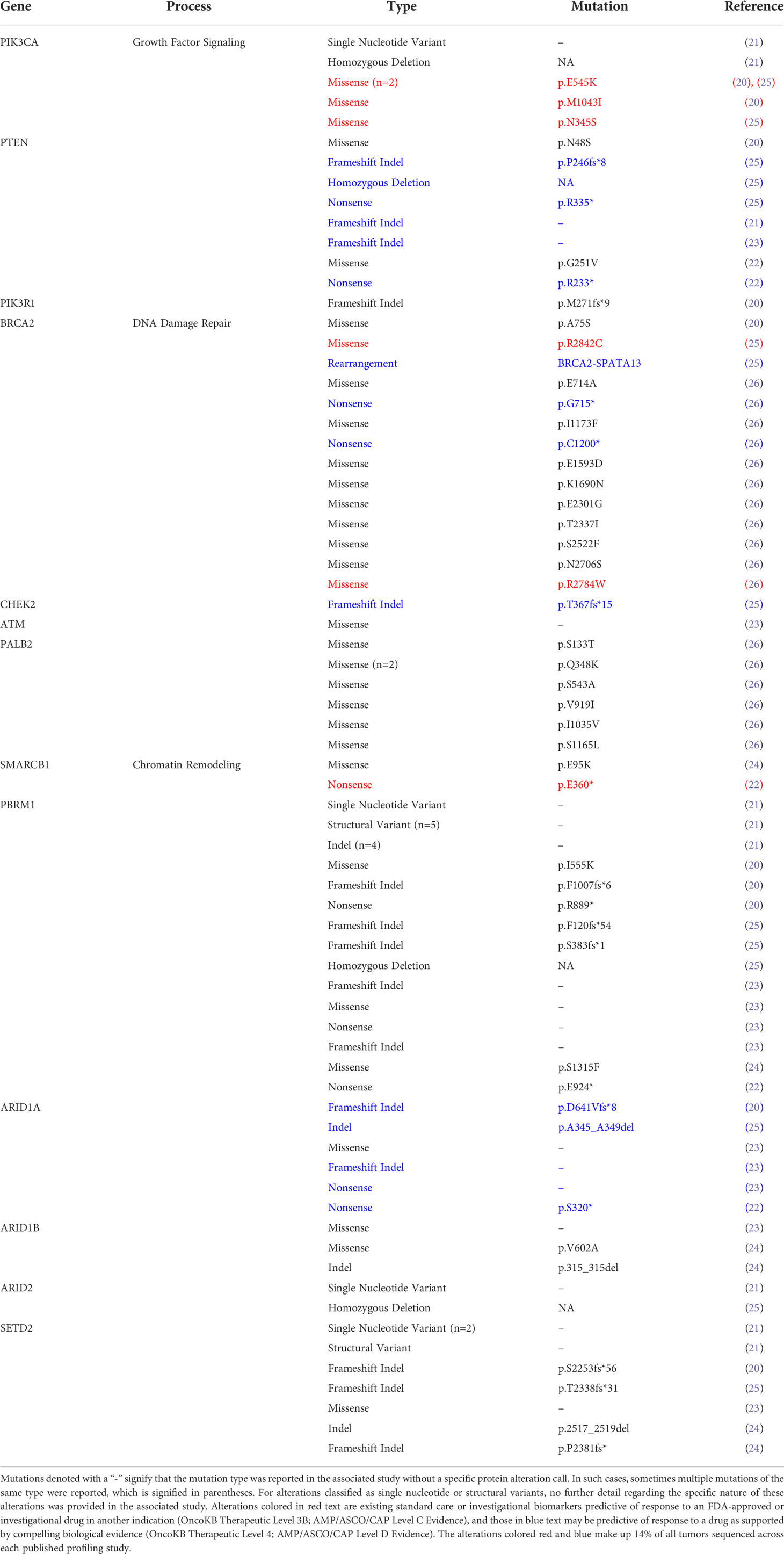
Table 1 Chordoma mutations reported in genes of potential therapeutic significance.
Although this observation limits the current impact of traditional genomic profiling on drug repurposing campaigns and precision oncology efforts in chordoma, it does not mean that chordoma is devoid of exploitable alterations per se (32). Indeed, genomic profiling studies have identified several potentially actionable alterations based on emerging science – many of which we discuss further below – and validating these therapeutic opportunities may increase the number of advanced-stage chordoma patients that can benefit from genomics-guided precision oncology. Moreover, systematic functional studies in other rare cancers argue that multiple therapeutically actionable vulnerabilities nonetheless exist in the context of a genomically “quiet” background (33–35). In this review, we provide a snapshot of the emerging drug repurposing landscape in chordoma, while highlighting state-of-the-art approaches that can open new windows into chordoma biology to extend our view beyond that provided by genomics. We also discuss opportunities to repurpose lessons learned in other cancers to catalyze the identification of novel therapeutic hypotheses in chordoma. The synthesis of this emerging knowledge may lead to the discovery of new targets and the development of personalized drug repurposing opportunities for chordoma.
Although ~95-97% of chordomas belong to a single histological subtype, multiple observations suggest that its biology and disease mechanisms are heterogeneous. For example, over half of patients experience disease recurrence following complete tumor resection (2), and exhibit vastly different responses to systemic therapies in the advanced disease setting (36). Additionally, many chordomas are defined by complex genomic rearrangements (20, 37) or recurrent copy number losses (38), whereas other tumors harbor no detectable alterations. This molecular heterogeneity is also reflected in recent chordoma tumor profiling studies, which have utilized next-generation sequencing to identify potentially actionable alterations in chordoma (Table 1) (20–23). These studies indicate that only ~14% of chordomas have biomarkers predictive of response to FDA-approved or investigational therapies in other indications. However, several opportunities for molecularly-guided drug repurposing are emerging based on recent scientific advances in chordoma and other cancers, and validation of these therapeutic hypotheses may increase the number of chordoma patients that can benefit from precision oncology (Figure 1A).
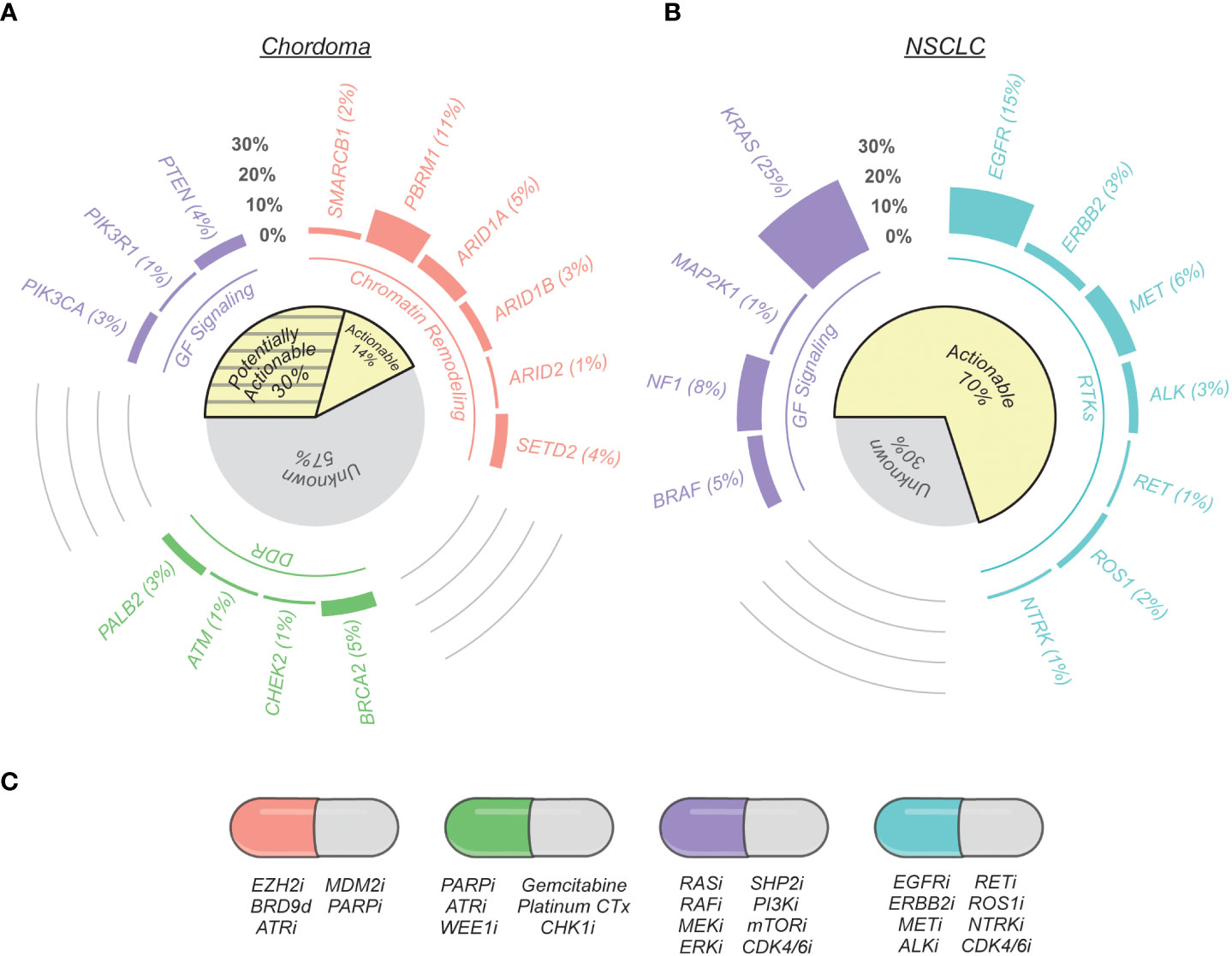
Figure 1 Therapeutically relevant genomic alterations found in chordoma and non-small cell lung cancer (NSCLC). (A) Nonsynonymous mutation call data from chordoma tumors were compiled from seven published genomic profiling studies (20–26). Genes are grouped by their protein functionality (chromatin remodeling, DNA damage repair (DDR), growth factor (GF) signaling, and receptor tyrosine kinases (RTKs)) and were selected by on their association with potential therapeutic opportunities based on current scientific literature, as reviewed here. The cohort of tumors analyzed for potentially targetable alterations varied on a per gene basis to account for variation between sequencing techniques and data presentation across studies: SMARCB1 (n = 2 altered/123 total), PBRM1 (22/203), ARID1A (6/123), ARID1B (3/104), ARID2 (2/203), SETD2 (8/203), PTEN (8/179), PIK3CA (6/203), PIK3R1 (1/123), BRCA2 (12/260), CHEK2 (1/123), ATM (1/123), PALB2 (7/260). The subset of actionable gene alterations are existing standard care or investigational biomarkers predictive of response to an FDA-approved or investigational drug in another indication (OncoKB Therapeutic Level 3B; AMP/ASCO/CAP Level C Evidence) or are predictive of response to a drug as supported by compelling biological evidence (OncoKB Therapeutic Level 4; AMP/ASCO/CAP Level D Evidence). Potentially actionable alterations are variants of currently-unknown significance (31). (B) Actionable gene alterations in metastatic NSCLC (39). (C) Examples of potential therapeutic opportunities indicated by specific gene alterations. The letter “i” signifies an inhibitor, whereas “d” denotes a degrader.
In one large cohort (20), PI3K pathway alterations were observed in 16% of cases (n = 17/104), indicating an opportunity to explore repurposing of inhibitors targeting PI3K or its downstream effector mTOR. One of the most frequently altered genes in chordoma is PTEN (Figure 1A); the resulting potential dependence on PI3Kβ signaling (40) suggests an opportunity to evaluate PI3Kβ inhibitors in chordoma (41). Indeed, a recent preclinical study revealed significant tumor growth inhibition by the pan-PI3K inhibitor buparlisib (BKM120) in patient-derived xenograft models (42). Downstream of PI3K, clinical trials involving mTOR inhibitor combinations have demonstrated modest clinical benefit in chordoma patients, particularly in tumors with mTOR effector activation (14, 43). Intriguingly, chordoma sometimes occurs in patients with tuberous sclerosis complex (44–46), which is characterized by loss of the mTOR negative regulators TSC1/2, further hinting at a role for the PI3K/mTOR pathway in chordoma pathogenesis. Moreover, PI3K and mTOR are regulated by receptor tyrosine kinases (RTKs), of which several appear to be activated in most chordoma tumors (4). Several studies have analyzed the activation state or effects of targeting RTKs including MET (47, 48), IGF1R (49, 50), and the FGFR family (51), though PDGFRβ (5, 10) and EGFR (3, 52) have received the most attention, primarily owing to evidence of some clinical benefit from agents targeting these RTKs (9–11, 13). Since RTK mutations are not frequently seen in chordoma, these receptors are presumably activated through alternative mechanisms such as aberrant growth factor production, which may be directly regulated by brachyury (53). The frequent activation of RTKs observed in chordoma may be related to the role of the notochord in regulating embryonic tissue patterning; in this context RTKs are thought to dictate proliferation and differentiation through the interpretation of morphogen gradients (54, 55). Inhibitors of wild-type EGFR, such as afatinib and cetuximab, have reproducibly shown promising activity against chordoma cell lines (3, 6, 7) and xenograft models (39, 47), which has motivated two Phase II clinical trials (NCT03083678 and NCT05041127). Since these strategies rely on inhibition of wild-type EGFR, it remains to be seen whether skin and gastrointestinal toxicities will limit their efficacy in the clinic (56).
Growth factor signaling drives cell proliferation by upregulation of cyclin D, CDK4/6 activation, and progression through the G1/S cell cycle checkpoint. RTK activation along with frequent loss of the cell cycle tumor suppressor CDKN2A in chordomas (24, 38, 57, 58) has motivated preclinical repurposing studies with CDK4/6 inhibitors (39, 59, 60) and a Phase II trial involving palbociclib in CDKN2A-null chordoma patients (NCT03110744). It remains unclear, however, whether CDKN2A loss is a faithful predictor of sensitivity to CDK4/6 inhibition (61, 62) – possibly because, in addition to p16INK4A, CDKN2A encodes p14ARF, whose loss results in CDK2 deregulation and compensatory G1/S cell cycle progression. Nevertheless, tumors with co-deletion of the CDKN2A-proximal MTAP gene may present an opportunity for combinations involving CDK4/6 inhibitors and antagonists of the PRMT5 axis (63–65). Notably, CDK4/6 inhibition has been reported to potentiate T cell immunity in several contexts (66, 67), and we discuss opportunities for evaluating CDK4/6 inhibitor combinations in this context further below.
Genomic profiling studies have also revealed potential synthetic lethality strategies in chordoma. Several deleterious alterations have been reported in genes involved in DNA damage repair and response, including BRCA2, CHEK2, PALB2 and ATM (20, 23, 25, 26). In one recent study, a novel defective homologous recombination signature was identified in advanced chordomas that appears to impart a “BRCAness” phenotype and sensitivity to PARP inhibition (22). This strategy is being explored further in a Phase 2 clinical trial combining olaparib plus trabectedin for solid tumors with this defective homologous recombination signature (NCT03127215). Future studies aimed at examining the potential link between DNA damage repair defects and complex genomic rearrangements in chordoma may provide further mechanistic insight into this therapeutic opportunity.
Other studies have identified alterations in chromatin remodeling genes such as SETD2 and SWI/SNF complex members SMARCB1, ARID1A, and PBRM1 (20, 21, 24). Notably, biallelic loss of SMARCB1 defines an aggressive, poorly differentiated histopathological subtype of chordoma (<5% of cases) that most commonly afflicts the pediatric patient population (68, 69). Based on the apparent EZH2 dependence bestowed by SWI/SNF alterations (70, 71), a Phase II study is underway to explore repurposing of tazemetostat for SMARCB1-null chordoma (NCT02601950). The presence of SWI/SNF alterations also suggests opportunities for therapeutically exploiting aberrant SWI/SNF function, for example through resulting synthetic lethality with BRD9 antagonists (72–74), inhibitors of DNA repair (75–77), or p53 activation (78). Implementation of functional genomics screens may lead to the discovery of additional chordoma-specific synthetic lethal strategies in this context, which we discuss in more detail below.
An interesting connection appears to exist between SWI/SNF, the Hippo pathway, and brachyury, chordoma’s main Achilles’ heel (79). Hippo transcriptional effectors YAP and TEAD are critical for notochord differentiation during embryonic development (80). Indeed, a YAP/TEAD motif is one of the top brachyury binding sites in chordoma cells (81), and reports have linked brachyury-mediated YAP upregulation to stemness and growth (82) – suggesting convergence between the Hippo and brachyury signaling networks. Intriguingly, SWI/SNF appears to sequester YAP, preventing its association with TEAD and thus antagonizing oncogenic Hippo transcriptional outputs (83). A key role of loss-of-function SWI/SNF alterations in chordoma may therefore be de-sequestration of YAP, which, when augmented by brachyury-mediated upregulation of YAP synthesis and stability, drives Hippo pathway flux to an oncogenic level. These observations suggest opportunities for evaluating an emerging class of TEAD palmitoylation inhibitors (84–86) in chordoma.
Although genomic profiling studies have informed our understanding of chordoma biology and expanded the list of potentially actionable therapeutic targets, chordoma nevertheless remains largely devoid of the recurring, actionable genomic alterations that define other solid tumors. For example, therapeutic biomarkers guide care for over two-thirds of metastatic NSCLC patients, with response rates to targeted therapies often approaching 70-80%, while chordoma profiling studies indicate ~14% of cases have potentially actionable genomic alterations (Figure 1). As highlighted in the previous section, our developing understanding of cancer biology suggests up to an additional ~30% of chordomas might have actionable genomic alterations; nevertheless, a majority of advanced-stage patients lack clear or effective treatment options.
This creates a need to open new windows into chordoma biology that extend our view beyond the “single oncogenic driver” perspective of cancer’s dependencies. To this end, studies across several cancers have revealed new categories of therapeutic targets, called “non-oncogene dependencies”, that mediate epigenetic changes, dysregulated signal transduction, metabolic rewiring, immune evasion, and other hallmarks of cancer (32). Multiple efforts are underway to analyze and integrate data layers derived from different aspects of cell biology, with a view to providing a more detailed molecular-resolution view of chordoma pathogenesis. For example, a recent investigation of methylation signatures in circulating tumor DNA revealed the existence of two distinct epigenetic subtypes in chordoma with prognostic relevance (87). A gene-set enrichment analysis pointed to dysregulated signaling pathways operating within each subtype, uncovering potential therapeutic opportunities that prompt further evaluation in functional studies. The exploration of additional data layers may further elucidate chordoma’s molecular subtypes, including their association with specific therapeutic vulnerabilities, risk of recurrence, and other features of the disease. Such multi-omics studies may also lead to the identification of tumor-specific or lineage-restricted cell surface proteins that can serve as targets for antibody-drug conjugates, bispecific antibodies, chimeric antigen receptor T cells, or other surface antigen-targeted modalities.
In addition to tumor cell intrinsic targets, therapeutic opportunities may exist within the tumor microenvironment, where crosstalk with various immune and stromal cell subsets can profoundly influence chordoma progression and therapy response (88, 89). Studies of the chordoma immune microenvironment to date have focused on the PD-1 axis (90, 91), as well as other potentially important immune checkpoints such as B7-H3 and HHLA2 (92, 93). A recent single-cell transcriptomic analysis of six chordoma tumors identified putative immunosuppressive contributions from regulatory T cells, tumor-associated macrophages, and TGFβ signaling (94). Notably, TGFβ pathway genes are upregulated by brachyury (81). These results point to a repurposing opportunity for antagonists of TGFβ signaling in combination with immune checkpoint blockade (95, 96). Interestingly, a chordoma patient treated with a bifunctional fusion protein targeting TGFβ and PD-L1 experienced late-onset tumor shrinkage in a Phase 1 trial (97). The set of factors that govern antitumor immunity is complex, and more comprehensive phenotyping of the chordoma immune microenvironment – through single-cell sequencing, digital spatial profiling, multispectral immunofluorescence and other approaches – will be important for creating an atlas of the various lineage states in chordoma and revealing therapeutically-reversible defects in the cancer-immunity cycle (98).
Other important tumor cell extrinsic features extend beyond the microenvironment, highlighting the need to study chordoma biology at various resolutions – including contributions from host physiology. For example, germline genetics are now understood to play a role in cancer predisposition (99) and tumor immunity (100). Additionally, the gut microbiome impacts immunotherapy efficacy in several solid tumor types (101–103), and recent data indicate that certain dietary habits can modulate the composition of the gut microbiome and influence immunotherapy response (104). Though it remains unclear how these factors contribute to the biology or treatment response of chordoma tumors, some studies are beginning to explore these questions. For example, MD Anderson’s Patient Mosaic initiative aims to collect genetic, immune, and microbiome profiles from thousands of cancer patients to inform treatment strategies. Biospecimens collected from chordoma patients enrolled on the cetuximab Phase II study at MD Anderson will be included in the Patient Mosaic protocol, shedding light on how host (and other tumor extrinsic) factors shape chordoma tumor biology.
The functional validation of new therapeutic targets and strategies resulting from multi-omics studies requires appropriate patient-derived samples and preclinical models. To this end, a variety of chordoma models have been developed by several groups (105, 106). In addition, the Chordoma Foundation has built a tumor biobank of over 500 biospecimens and a model repository currently consisting of 26 cell lines, 12 patient-derived xenograft (PDX) models, and a PBMC-humanized mouse model (www.chordoma.org/research). The majority of these models have been characterized by whole-exome and whole-transcriptome sequencing and will undergo additional multi-omics characterization in the future, with a view to facilitating hypothesis testing through the establishment of models representing the full diversity of chordoma. Moreover, these models are available to the research community, as are in-kind drug testing services offered through the Chordoma Foundation’s Drug Screening Program. As emerging drug repurposing concepts are evaluated in the Drug Screening Program, resulting data are publicly shared, whenever possible (107), to provide justification for further evaluation of the most promising therapeutic opportunities.
In translational cancer research, PDX models have been the gold standard for preclinical drug testing because they accurately recapitulate features of the patient’s tumor (108, 109); this has motivated the development of over two dozen chordoma PDXs by the Chordoma Foundation and others (47, 105) that represent the anatomical, age, histopathological, and known molecular diversity of chordoma. More recently, patient-derived organoids (PDOs) have generated significant interest as functional models because they provide faithful representations of patient tumors, while improving on initiation time, cost, and efficiency scales compared to PDXs (110). This technology is now being actively explored in chordoma; one recent proof-of-concept study reportedly developed chordoma PDOs from five different patients and screened them against various drugs to nominate personalized repurposing opportunities (111). In other cancer types, PDOs accurately mimic patient drug response (112–114) and have been utilized for personalized therapy (115, 116). The slow growth rate of chordoma tumors provides a large window of opportunity to develop protocols for establishing, validating, screening chordoma PDOs from high-risk or relapsing patients to enable identification of effective drug repurposing opportunities within the timeframe required to make treatment decisions.
Patient avatars like PDXs, PDOs and cell lines also serve as key platforms for target discovery because they allow functional studies capable of revealing or validating non-oncogene dependencies in chordoma. Genome-scale loss-of-function screens in various cancer cell lines have enabled the creation of “dependency maps” (33, 117), and this cutting-edge approach has recently been applied to chordoma to identify selective genetic dependencies (79). Perhaps unsurprisingly, T (or TBXT), the gene encoding brachyury, appears to be the most selectively essential gene in chordoma. Since brachyury (like most transcription factors) is a challenging drug target, the authors performed a drug repurposing screen and found that inhibitors of CDK9 or CDK7/12/13 (118) downregulate TBXT transcription and suppress chordoma cell proliferation. These results have motivated further in vivo testing of transcriptional CDK inhibitors, including KB-0742 (119), in the Chordoma Foundation’s Drug Screening Program (120).
Ongoing systematic screening of genetic and chemical vulnerabilities in chordoma is facilitating the development of new therapeutic hypotheses. For example, CDK6 – but not CDK4 – appears to be a genetic essentiality in some chordoma cell lines (79). Outside of their common cell-cycle target RB1, CDK6 possesses a much broader substrate repertoire than does CDK4 (121) – suggesting that one or more non-RB1 targets may be mechanistically linked to chordoma’s CDK6 dependence. One interesting possibility relates to the observation that chordoma cells are sensitive to the lipid hydroperoxidase inhibitor RSL3 (79), which is known to promote ferroptotic cell death via antagonism of GPX4. CDK6 can upregulate glutathione and NADPH via phosphorylation of two glycolytic enzymes (122); depletion of these antioxidants can prime cells for ferroptosis (123). CDK6 may therefore be crucial for maintaining redox homeostasis in chordoma to safeguard against ferroptosis, providing rationale for evaluation of CDK4/6 inhibitors in combination with ferroptosis inducers.
Chordoma’s apparent CDK6 dependence and potential ferroptosis susceptibility raises intriguing and unexpected parallels with clear-cell carcinomas (124, 125). Histologically, clear-cell renal cell carcinoma (ccRCC) is almost indistinguishable from chordoma, owing to morphological similarities between ccRCC’s characteristic lipid droplets and the physaliferous cells that define conventional chordoma (126). Notably, ferroptosis susceptibility in clear-cell carcinomas has been linked to HIF-1/2α-dependent accumulation of polyunsaturated lipids within the intracellular droplets that give rise to the clear-cell morphology (124). Both brachyury and mTOR are known to upregulate HIF-1α (81, 127–129), suggesting a possible connection between dysregulated hypoxia signaling, physaliferous morphology, and establishment of a ferroptosis-susceptible state in chordoma (Figure 2A). Although the precise composition of physaliferous vacuoles remains unclear (130–132), chordoma and ccRCC share additional similarities, including resistance to chemotherapy and modest mutational burdens enriched in chromatin modifier and PI3K/mTOR pathway alterations (133). Collectively, these observations suggest these cancers of different tissue origins share a similar cellular context, and potentially associated therapeutic vulnerabilities – providing opportunities for repurposing lessons learned from a well-studied and common cancer.
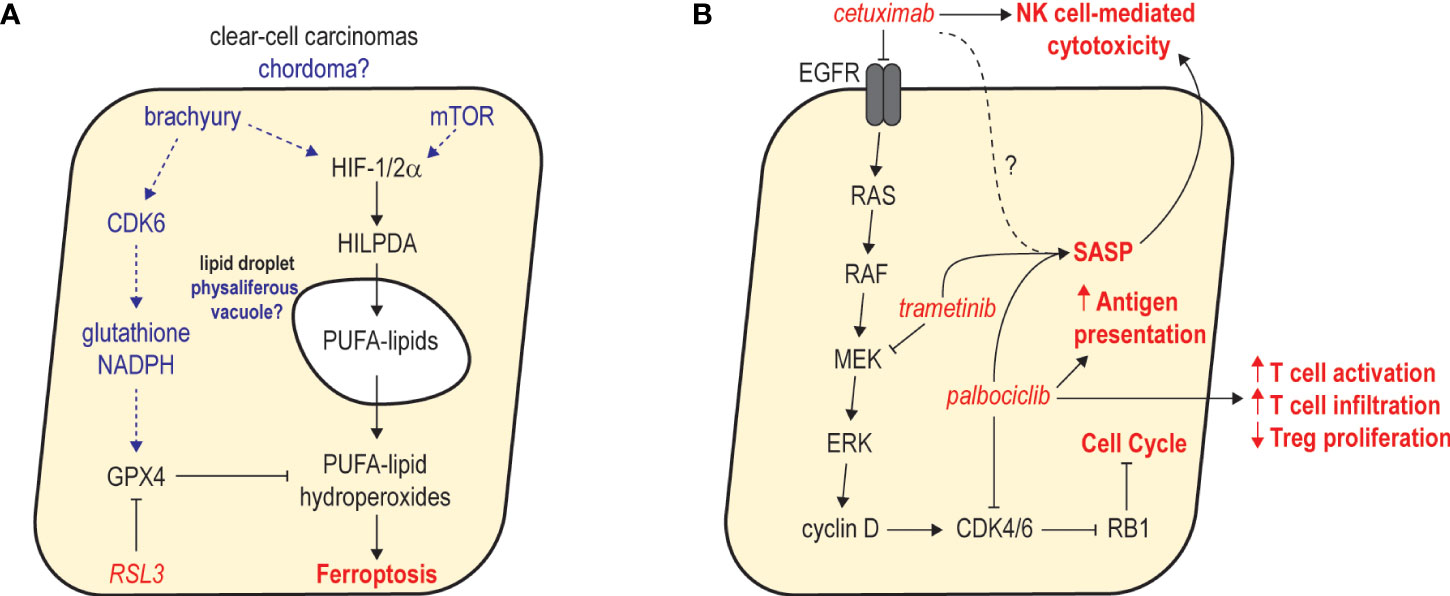
Figure 2 Examples of emerging therapeutic hypotheses in chordoma. (A) Potential mechanisms of ferroptosis susceptibility in chordoma and parallels with clear-cell carcinomas. In clear-cell carcinomas, dysregulated HIF-1/2α functions through HILPDA to promote deposition of polyunsaturated fatty acid (PUFA) lipids in intracellular lipid droplets. GPX4 activity counteracts PUFA-lipid oxidation and protects cells from ferroptosis. In chordoma, brachyury and mTOR activity may upregulate HIF-1/2α, resulting in deposition of PUFA lipids in physaliferous vacuoles. CDK6 activity may be critical for antioxidant production to protect chordoma cells against ferroptosis. (B) Combined inhibition of MAPK signaling and CDK4/6 activity promotes robust cell cycle arrest and antitumor immunity. Studies in other cancers revealed that combined inhibition of MEK and CDK4/6 with trametinib and palbociclib leads to sustained proliferative arrest and a senescence-associated secretory phenotype (SASP) that promotes NK and T cell immunity. As single agents, cetuximab and palbociclib reportedly augment NK and T cell immunity, respectively. Thus, combining cetuximab with palbociclib may synergistically inhibit tumor growth while stimulating a SASP that augments the activation of NK and T cell-based immunity promoted by each agent.
Another key implementation of systematic functional screens involves the discovery of synthetic lethal and combination therapy strategies. Loss-of-function screens in large cell line panels have led to identification of new synthetic lethal interactions (117, 134, 135), including PRMT5 dependence in cells with MTAP loss (63, 64). As noted above, the CDKN2A/MTAP locus is frequently deleted in chordoma (20, 21), suggesting an opportunity for repurposing PRMT5 or MAT2A inhibitors (136, 137). Exploiting such synthetic lethalities not only provides an avenue for targeting tumor suppressor loss in cancer, but is a particularly important approach to explore in genomically quiet malignancies. In addition to potential vulnerabilities created by loss of MTAP, SWI/SNF, or homologous recombination repair (as noted above), an intriguing candidate for synthetic lethality screening is LYST – a lysosomal trafficking protein of unknown function that’s lost in 10% of chordomas (20). Functional genomics screens in chordoma cell lines with LYST loss may reveal targetable vulnerabilities created by this unique alteration.
Preclinical and clinical research has yet to identify a therapy capable of producing frequent responses in chordoma (36), motivating the development of combination strategies aimed at increasing the magnitude and duration of therapeutic benefit. One approach with this goal in mind involves performing unbiased anchor screens, in which genome-wide CRISPR screening is utilized to identify genes whose loss sensitizes cells to a given targeted therapy ‘anchor’ (136). Such genes – if druggable – may serve as attractive targets for combination therapy regimens. A similar approach can also be employed to identify candidate resistance mechanisms – that is, genes whose loss (or gain) reduce sensitivity to the anchor drug.
As one of the most well-validated therapeutic targets in chordoma, inhibitors of wild-type EGFR are arguably the best ‘anchors’ to initially explore in unbiased screens or rational combination studies. Indeed, combination therapy investigations with afatinib (47) or cetuximab (138) have yielded encouraging results. One interesting hypothesis involves combining cetuximab with a CDK4/6 inhibitor (Figure 2B). Since CDK4/6-mediated G1/S cell cycle progression is highly dependent on RTK/MAPK signaling, concomitant antagonism of EGFR-mediated cyclin D upregulation and CDK4/6 kinase activity may cause a more complete cell cycle arrest. This effect has been observed in lung (139) and pancreatic cancers (140), where combined MEK and CDK4/6 inhibition induced a profound G1/S arrest, resulting in a senescence-associated secretory phenotype (SASP) that promoted increased NK cell-mediated cytotoxicity and infiltration of CD8+ T cells, respectively. Importantly, as an IgG1 antibody, cetuximab monotherapy appears to promote antibody-dependent NK cell-mediated cytotoxicity in several cancers including chordoma (141). A cetuximab/CDK4/6 inhibitor combination may therefore act synergistically to halt the growth of chordoma tumor cells and provoke a strong NK- and T-cell based antitumor response. As a result, further exploration of this concept may be warranted, particularly once the single-agent activity of cetuximab (NCT05041127) and palbociclib (NCT03110744) in chordoma is benchmarked in the clinic. Notably, similar combination immunotherapy approaches aiming to enhance NK cell-mediated killing have recently been described in chordoma (138), and these strategies were reported to selectively target the reservoir of cancer stem-like cells that promote recurrence and therapy resistance.
Achieving deep and durable responses in chordoma will likely require identification of therapeutic concepts capable of invigorating antitumor immunity. Despite a low tumor mutational burden, a significant proportion of chordomas appear to be characterized by complex genomic rearrangements (20, 37), which may lead to high neoantigen expression. In addition to the examples noted above, documented patient responses to vaccines (142) and PD-1 inhibitors (143–146) provide important proof-of-concept for the use of immunotherapies in chordoma, and prompt evaluation of different immune checkpoints and combinations thereof. For example, strong scientific rationale exists for co-blockade of the PD-1 and TIGIT checkpoints in cancer (147), and a new clinical study enrolling chordoma patients is testing this concept with atezolizumab plus tiragolumab (NCT05286801). Another promising approach involves the cell-surface protein CD24, which is frequently expressed in chordomas and – along with brachyury and low molecular weight cytokeratins – has been used as a diagnostic marker for chordoma in some cases (148). Intriguingly, tumor-derived CD24 was recently identified as a key anti-phagocytic “don’t eat me” signal in other solid tumors (149), making it a promising immunotherapeutic target and prompting evaluation of CD24 blockade in chordoma. The evaluation of immunotherapy combinations in chordoma, such as PD-1 antagonism plus inhibition of TIGIT or TGFβ signaling as noted above, may reinvigorate the tumor-immunity cycle at multiple points. Multi-omics studies of chordoma may be valuable in guiding these efforts and revealing key molecular details governing the chordoma immune microenvironment.
Tumorigenesis appears to require three main ingredients: an oncogenic signaling input, deregulation of the signal through tumor suppressor loss (150, 151), and a permissive transcriptional environment for interpretation of oncogenic signaling (152, 153). Lineage-specific transcription factors – such as brachyury in chordoma – are essential for creating a permissive environment (79), and thus represent attractive drug targets. While oncogenic signaling and tumor suppressor loss can be targeted by kinase inhibitors and synthetic lethal strategies, respectively, transcription factors like brachyury are inherently challenging drug targets (Figure 3A). However, advances in drug discovery and the development of new targeted protein degradation technologies, such as proteolysis targeting chimeras (PROTACs) and molecular glues, provide opportunities to redefine this paradigm (154). To this end, numerous projects have recently been launched to develop novel compounds that bind brachyury with high affinity, which can either serve as functional inhibitors, molecular glues, or warheads for PROTACS. Notably, an open-source project through the Structural Genomics Consortium is focusing on the development of high-quality probes that bind pockets identified in brachyury crystal structures (Figure 3B) to induce industry investment in further brachyury drug discovery.
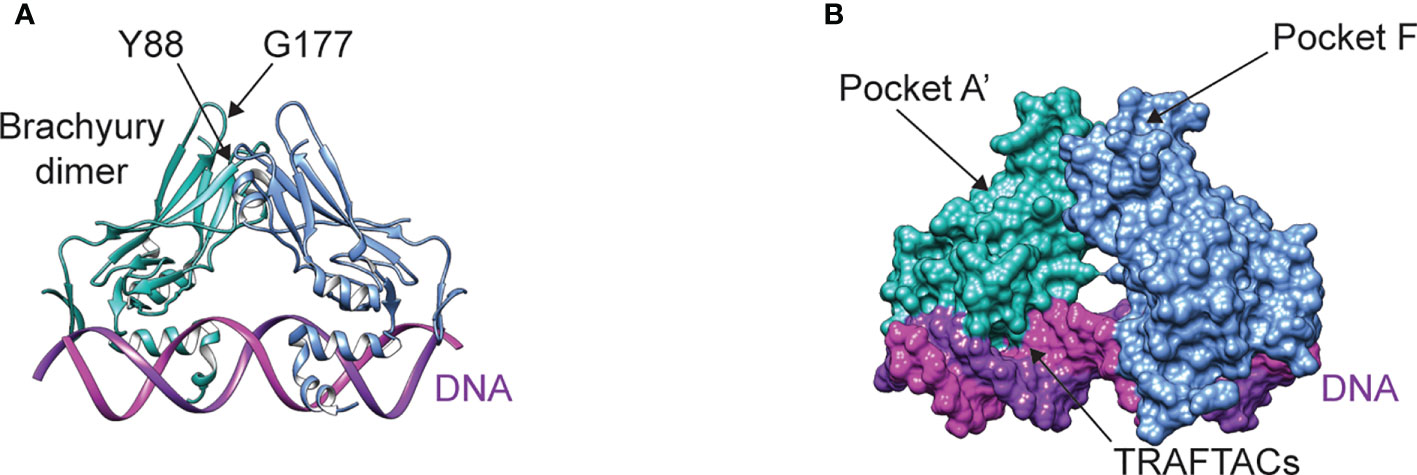
Figure 3 (A) Structure of the brachyury dimer bound to DNA (PDB ID 6F58). The alpha carbons of residues Y88 and G177 are separated by a distance of ~9 Å (B) Brachyury rendered in surface representation, showing the two main pockets being targeted by open-source drug discovery efforts and the DNA binding interface targeted by TRAFTACs.
The development of functional inhibitors is a challenging endeavor, given that brachyury lacks the deep binding pockets commonly associated with enzymatic activity. Yet, transcription factors like brachyury are often involved in multiprotein complexes, pointing to the development of compounds that modulate protein-protein interactions as an attractive strategy. For example, brachyury associates with the histone acetyltransferase p300 using an interface involving amino acid residue Y88 (Figure 3A) (155). The proximity of Y88 to residue G177 (Figure 3A) is interesting, as a G177D germline variant is strongly associated with chordoma (15). Because residue G177 is on a flexible, solvent-exposed loop, the G177D mutation is unlikely to affect brachyury structure – however this substitution may stabilize intermolecular contacts with p300 or other binding partners, thus modulating brachyury function. Indeed, the interaction between brachyury and p300 appears to regulate histone 3 lysine 27 acetylation (155) – a modification associated with active enhancers. Since association of brachyury with super-enhancers appears to be crucial to its role in chordoma (79, 81), designing compounds that can block or allosterically modulate this protein-protein interaction – for example, by targeting pocket A’ or F (Figure 3B) – may represent an attractive therapeutic strategy. Another novel approach to functionally modulating brachyury involves the development of Transcription Factor Targeting Chimeras (TRAFTACs) (156). In contrast to PROTACs, TRAFTACs utilize a transcription factor-specific DNA sequence to achieve target specificity, which is linked to an E3 ligase-recruiting moiety that directs brachyury to the proteasome for degradation.
Although new drug discovery in an ultra-rare indication presents numerous challenges, the concept of “reverse” drug repurposing – that is, repurposing drugs initially developed in a rare cancer to more common indications – represents a promising path. Further highlighting the intriguing parallels between chordoma and kidney cancer, brachyury expression is associated with poor survival in ccRCC and papillary RCC (Figure 4) (133, 157). Interestingly, expression of CDK6 – an apparent dependency in both chordoma and ccRCC (125), as noted above – appears to be regulated by brachyury (79). Numerous additional studies indicate brachyury is associated with poor prognosis and implicated in driving recurrence, metastasis, and/or resistance to standard of care therapy in several more common cancers including breast (158–161), lung (162–166), and colon (167). Thus, chordoma represents a “pure” and target-rich setting for the initial development of brachyury antagonists, which can then be expanded into larger indications where brachyury plays a role in disease progression.
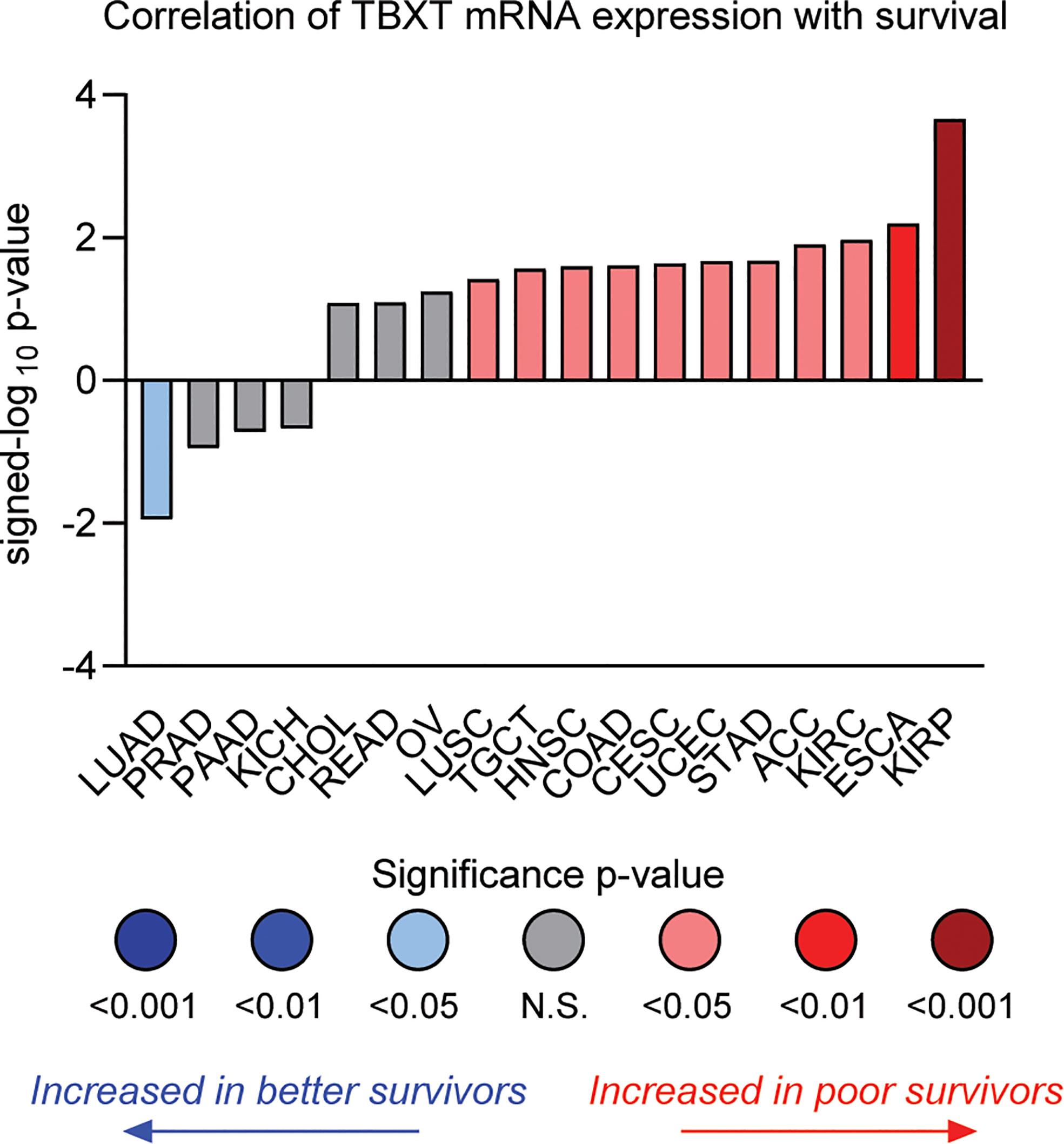
Figure 4 Correlation of TBXT (brachyury) mRNA expression with overall survival by tumor type in TCGA datasets. The individual values are sign-corrected log10 p-values of correlation. Negative signed-log10 p-values (y-axis) indicate that high TBXT expression is associated with better survival, whereas positive values indicate high TBXT expression predicts poor survival. Cancer types are listed by their respective TCGA study abbreviations (e.g., KIRP, kidney renal papillary cell carcinoma; ESCA, esophageal carcinoma; KIRC, kidney renal clear-cell carcinoma; LUAD, lung adenocarcinoma). The results shown in this figure are in whole or part based upon data generated through the Lumin Bioinformatics Software of Champions Oncology, Inc.
Even when macroscopic complete resection is achieved using cutting-edge surgical approaches, the majority of chordoma patients experience disease recurrence and are unlikely to be cured (1, 2). At some point, local therapies such as surgery and/or radiation are no longer safe or feasible, and treatment options become limited due to a lack of effective systemic therapies. This has motivated intensive research to identify effective therapeutic strategies in chordoma, but drug repurposing efforts have been hampered by chordoma’s resistance to conventional chemotherapy and a paucity of actionable genomic alterations.
In this review, we highlight several therapeutic hypotheses inspired by developing knowledge of chordoma biology and its parallels with other cancer types. In particular, we focus on emerging therapeutic opportunities based on emerging knowledge linking drug sensitivity to specific biomarkers. Nevertheless, through the lens of genomic sequencing, most chordomas still lack actionable alterations – underscoring the need to implement more sophisticated multi-omics approaches. Indeed, genomics is only one piece of the puzzle; tumor growth is controlled by multiple integrated systems, with each contributing uniquely to chordoma’s biology. Therapeutic opportunities exist within each of these systems, and efforts focused on elucidating and integrating them will provide a fuller view of chordoma’s biology. A key goal of multi-omics profiling efforts will be the identification of molecular subtypes, stratified by risk and therapeutic vulnerabilities, as has been demonstrated in other cancers (168–170).
In parallel, unbiased functional assays, utilizing genome-wide CRISPR or high-throughput drug screening, may reveal non-oncogene dependencies or combination therapy strategies that would otherwise be difficult to detect through multi-omics profiling approaches. The identification of additive or synergistic therapeutic combinations is of particular interest, given the low historical response rates in chordoma (36). In addition to guiding target discovery campaigns, functional assays can provide personalized medicine opportunities. For example, if multi-omics studies identify patients at high risk for recurrence, the ability to establish and profile drug sensitivity of PDXs or PDOs at time of initial surgery may allow nomination of potential therapeutic options upon disease recurrence, as successfully demonstrated recently in breast cancer (116). Due to the intrinsically slow growth of chordoma tumors, such an approach could also be considered at the time of recurrence.
Finally, we highlight how technological advances are opening the door to targeting the transcription factor brachyury, the main Achilles heel of chordoma. The identification of binding pockets on brachyury can serve as target sites for PROTAC warheads or molecular glues, but they may also be functionally important. One such potential site is pocket A’ or F (Figure 3B), near the putative p300 interface and residue G177, which is the site of a germline variant strongly associated with chordoma development. If efforts to target brachyury are ultimately successful, these drugs can be repurposed for more common cancers in circumstances where brachyury drives resistance to standard of care therapy.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was funded by the generous donors to Chordoma Foundation.
We acknowledge and thank our patient community for giving us constant motivation to help identify and develop better treatments for chordoma. We are also grateful for our generous donors who provide the necessary funding – and to our colleagues at Chordoma Foundation and the research community for their efforts – to work towards this goal. Finally, we thank Champions Oncology for their assistance with figure generation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stacchiotti S, Sommer J, G. Chordoma Global Consensus. Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol (2015) 16(2):e71–83. doi: 10.1016/S1470-2045(14)71190-8
2. Stacchiotti S, Gronchi A, Fossati P, Akiyama T, Alapetite C, Baumann M, et al. Best practices for the management of local-regional recurrent chordoma: A position paper by the chordoma global consensus group. Ann Oncol (2017) 28(6):1230–42. doi: 10.1093/annonc/mdx054
3. Shalaby A, Presneau N, Ye H, Halai D, Berisha F, Idowu B, et al. The role of epidermal growth factor receptor in chordoma pathogenesis: A potential therapeutic target. J Pathol (2011) 223(3):336–46. doi: 10.1002/path.2818
4. Tamborini E, Virdis E, Negri T, Orsenigo M, Brich S, Conca E, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro Oncol (2010) 12(8):776–89. doi: 10.1093/neuonc/noq003
5. Tamborini E, Miselli F, Negri T, Lagonigro MS, Staurengo S, Dagrada GP, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) b, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res (2006) 12(23):6920–8. doi: 10.1158/1078-0432.CCR-06-1584
6. Scheipl S, Barnard M, Cottone L, Jorgensen M, Drewry DH, Zuercher WJ, et al. EGFR inhibitors identified as a potential treatment for chordoma in a focused compound screen. J Pathol (2016) 239(3):320–34. doi: 10.1002/path.4729
7. Magnaghi P, Salom B, Cozzi L, Amboldi N, Ballinari D, Tamborini E, et al. Afatinib is a new therapeutic approach in chordoma with a unique ability to target EGFR and brachyury. Mol Cancer Ther (2018) 17(3):603–13. doi: 10.1158/1535-7163.MCT-17-0324
8. Cottone L, Cribbs AP, Khandelwal G, Wells G, Ligammari L, Philpott M, et al. Inhibition of histone H3K27 demethylases inactivates brachyury (TBXT) and promotes chordoma cell death. Cancer Res (2020) 80(20):4540–51. doi: 10.1158/0008-5472.CAN-20-1387
9. Hof H, Welzel T, Debus J. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie (2006) 29(12):572–4. doi: 10.1159/000096283
10. Casali PG, Messina A, Stacchiotti S, Tamborini E, Crippa F, Gronchi A, et al. Imatinib mesylate in chordoma. Cancer (2004) 101(9):2086–97. doi: 10.1002/cncr.20618
11. Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol (2012) 30(9):914–20. doi: 10.1200/JCO.2011.35.3656
12. Bompas E, Le Cesne A, Tresch-Bruneel E, Lebellec L, Laurence V, Collard O, et al. Sorafenib in patients with locally advanced and metastatic chordomas: A phase II trial of the French sarcoma group (GSF/GETO). Ann Oncol (2015) 26(10):2168–73. doi: 10.1093/annonc/mdv300
13. Stacchiotti S, Tamborini E, Lo Vullo S, Bozzi F, Messina A, Morosi C, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol (2013) 24(7):1931–6. doi: 10.1093/annonc/mdt117
14. Stacchiotti S, Morosi C, Lo Vullo S, Casale A, Palassini E, Frezza AM, et al. Imatinib and everolimus in patients with progressing advanced chordoma: A phase 2 clinical study. Cancer (2018) 124(20):4056–63. doi: 10.1002/cncr.31685
15. Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet (2012) 44(11):1185–7. doi: 10.1038/ng.2419
16. Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: A genetic and functional-based study. J Pathol (2011) 223(3):327–35. doi: 10.1002/path.2816
17. Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol (2006) 209(2):157–65. doi: 10.1002/path.1969
18. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med (2002) 347(7):472–80. doi: 10.1056/NEJMoa020461
19. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol (2018) 36(1):7–13. doi: 10.1200/JCO.2017.73.6785
20. Tarpey PS, Behjati S, Young MD, Martincorena I, Alexandrov LB, Farndon SJ, et al. The driver landscape of sporadic chordoma. Nat Commun (2017) 8(1):890. doi: 10.1038/s41467-017-01026-0
21. Bai J, Shi J, Li C, Wang S, Zhang T, Hua X, et al. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat Commun (2021) 12(1):757. doi: 10.1038/s41467-021-21026-5
22. Groschel S, Hubschmann D, Raimondi F, Horak P, Warsow G, Frohlich M, et al. Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nat Commun (2019) 10(1):1635. doi: 10.1038/s41467-019-09633-9
23. Mattox AK, Yang B, Douville C, Lo SF, Sciubba D, Wolinsky JP, et al. The mutational landscape of spinal chordomas and their sensitive detection using circulating tumor DNA. Neurooncol Adv (2021) 3(1):vdaa173. doi: 10.1093/noajnl/vdaa173
24. Wang L, Zehir A, Nafa K, Zhou N, Berger MF, Casanova J, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosomes Cancer (2016) 55(7):591–600. doi: 10.1002/gcc.22362
25. Gounder MM, Agaram NP, Trabucco SE, Robinson V, Ferraro RA, Millis SZ, et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nat Commun (2022) 13(1):3406. doi: 10.1038/s41467-022-30496-0
26. Xia B, Biswas K, Foo TK, Torres T, Riedel-Topper M, Southon E, et al. Rare germline variants in PALB2 and BRCA2 in familial and sporadic chordoma. Hum Mutat (2022) 43(10):1396–407. doi: 10.1002/humu.24427
27. Liang WS, Dardis C, Helland A, Sekar S, Adkins J, Cuyugan L, et al. Identification of therapeutic targets in chordoma through comprehensive genomic and transcriptomic analyses. Cold Spring Harb Mol Case Stud (2018) 4(6):a003418. doi: 10.1101/mcs.a003418
28. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature (2018) 555(7696):371–6. doi: 10.1038/nature25795
29. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555(7696):321–7. doi: 10.1038/nature25480
30. Cancer Genome Atlas Research Network. Electronic address and N. Cancer Genome Atlas Research. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell (2017) 171(4):950–965 e28. doi: 10.1016/j.cell.2017.10.014
31. Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol (2017) PO.17.00011. doi: 10.1200/PO.17.00011
32. Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, Druker BJ, et al. An expanded universe of cancer targets. Cell (2021) 184(5):1142–55. doi: 10.1016/j.cell.2021.02.020
33. Dharia NV, Kugener G, Guenther LM, Malone CF, Durbin AD, Hong AL, et al. A first-generation pediatric cancer dependency map. Nat Genet (2021) 53(4):529–38. doi: 10.1038/s41588-021-00819-w
34. Hong AL, Tseng YY, Cowley GS, Jonas O, Cheah JH, Kynnap BD, et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun (2016) 7:11987. doi: 10.1038/ncomms11987
35. Oberlick EM, Rees MG, Seashore-Ludlow B, Vazquez F, Nelson GM, Dharia NV, et al. Small-molecule and CRISPR screening converge to reveal receptor tyrosine kinase dependencies in pediatric rhabdoid tumors. Cell Rep (2019) 28(9):2331–2344 e8. doi: 10.1016/j.celrep.2019.07.021
36. Wedekind MF, Widemann BC, Cote G. Chordoma: Current status, problems, and future directions. Curr Probl Cancer (2021) 45(4):100771. doi: 10.1016/j.currproblcancer.2021.100771
37. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell (2011) 144(1):27–40. doi: 10.1016/j.cell.2010.11.055
38. Le LP, Nielsen GP, Rosenberg AE, Thomas D, Batten JM, Deshpande V, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PloS One (2011) 6(5):e18846. doi: 10.1371/journal.pone.0018846
39. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet (2017) 49(12):1693–704. doi: 10.1038/ng.3990
40. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature (2008) 454(7205):776–9. doi: 10.1038/nature07091
41. Mateo J, Ganji G, Lemech C, Burris HA, Han SW, Swales K, et al. A first-Time-in-Human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin Cancer Res (2017) 23(19):5981–92. doi: 10.1158/1078-0432.CCR-17-0725
42. Michmerhuizen NL, Owen JH, Heft Neal ME, Mann JE, Leonard E, Wang J, et al. Rationale for the advancement of PI3K pathway inhibitors for personalized chordoma therapy. J Neurooncol (2020) 147(1):25–35. doi: 10.1007/s11060-020-03418-7
43. Stacchiotti S, Marrari A, Tamborini E, Palassini E, Virdis E, Messina A, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol (2009) 20(11):1886–94. doi: 10.1093/annonc/mdp210
44. Borgel J, Olschewski H, Reuter T, Miterski B, Epplen JT. Does the tuberous sclerosis complex include clivus chordoma? a case report. Eur J Pediatr (2001) 160(2):138. doi: 10.1007/s004310000645
45. Lee-Jones L, Aligianis I, Davies PA, Puga A, Farndon PA, Stemmer-Rachamimov A, et al. Sacrococcygeal chordomas in patients with tuberous sclerosis complex show somatic loss of TSC1 or TSC2. Genes Chromosomes Cancer (2004) 41(1):80–5. doi: 10.1002/gcc.20052
46. McMaster ML, Goldstein AM, Parry DM. Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J Med Genet (2011) 48(7):444–9. doi: 10.1136/jmg.2010.085092
47. Zhao T, Siu IM, Williamson T, Zhang H, Ji C, Burger PC, et al. AZD8055 enhances in vivo efficacy of afatinib in chordomas. J Pathol (2021) 255(1):72–83. doi: 10.1002/path.5739
48. Scheipl S, Barnard M, Lohberger B, Zettl R, Brcic I, Liegl-Atzwanger B, et al. Drug combination screening as a translational approach toward an improved drug therapy for chordoma. Cell Oncol (Dordr) (2021) 44(6):1231–42. doi: 10.1007/s13402-021-00632-x
49. Sommer J, Itani DM, Homlar KC, Keedy VL, Halpern JL, Holt GE, et al. Methylthioadenosine phosphorylase and activated insulin-like growth factor-1 receptor/insulin receptor: potential therapeutic targets in chordoma. J Pathol (2010) 220(5):608–17. doi: 10.1002/path.2679
50. Aleksic T, Browning L, Woodward M, Phillips R, Page S, Henderson S, et al. Durable response of spinal chordoma to combined inhibition of IGF-1R and EGFR. Front Oncol (2016) 6:98. doi: 10.3389/fonc.2016.00098
51. Hu Y, Mintz A, Shah SR, Quinones-Hinojosa A, Hsu W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis (2014) 35(7):1491–9. doi: 10.1093/carcin/bgu014
52. Dewaele B, Maggiani F, Floris G, Ampe M, Vanspauwen V, Wozniak A, et al. Frequent activation of EGFR in advanced chordomas. Clin Sarcoma Res (2011) 1(1):4. doi: 10.1186/2045-3329-1-4
53. Nelson AC, Pillay N, Henderson S, Presneau N, Tirabosco R, Halai D, et al. An integrated functional genomics approach identifies the regulatory network directed by brachyury (T) in chordoma. J Pathol (2012) 228(3):274–85. doi: 10.1002/path.4082
54. Shilo BZ. Regulating the dynamics of EGF receptor signaling in space and time. Development (2005) 132(18):4017–27. doi: 10.1242/dev.02006
55. Darras S, Nishida H. The BMP signaling pathway is required together with the FGF pathway for notochord induction in the ascidian embryo. Development (2001) 128(14):2629–38. doi: 10.1242/dev.128.14.2629
56. Politi K, Ayeni D, Lynch T. The next wave of EGFR tyrosine kinase inhibitors enter the clinic. Cancer Cell (2015) 27(6):751–3. doi: 10.1016/j.ccell.2015.05.012
57. Choy E, MacConaill LE, Cote GM, Le LP, Shen JK, Nielsen GP, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PloS One (2014) 9(7):e101283. doi: 10.1371/journal.pone.0101283
58. Cottone L, Eden N, Usher I, Lombard P, Ye H, Ligammari L, et al. Frequent alterations in p16/CDKN2A identified by immunohistochemistry and FISH in chordoma. J Pathol Clin Res (2020) 6(2):113–23. doi: 10.1002/cjp2.156
59. Anderson E, Havener TM, Zorn KM, Foil DH, Lane TR, Capuzzi SJ, et al. Synergistic drug combinations and machine learning for drug repurposing in chordoma. Sci Rep (2020) 10(1):12982. doi: 10.1038/s41598-020-70026-w
60. von Witzleben A, Goerttler LT, Marienfeld R, Barth H, Lechel A, Mellert K, et al. Preclinical characterization of novel chordoma cell systems and their targeting by pharmocological inhibitors of the CDK4/6 cell-cycle pathway. Cancer Res (2015) 75(18):3823–31. doi: 10.1158/0008-5472.CAN-14-3270
61. Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic aberrations that activate d-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell (2017) 32(6):761–776 e6. doi: 10.1016/j.ccell.2017.11.006
62. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol (2015) 16(1):25–35. doi: 10.1016/S1470-2045(14)71159-3
63. Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science (2016) 351(6278):1214–8. doi: 10.1126/science.aad5214
64. Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, et al. Sellers: Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science (2016) 351(6278):1208–13. doi: 10.1126/science.aad5944
65. Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell (2010) 18(4):329–40. doi: 10.1016/j.ccr.2010.08.012
66. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
67. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discovery (2018) 8(2):216–33. doi: 10.1158/2159-8290.CD-17-0915
68. Hasselblatt M, Thomas C, Hovestadt V, Schrimpf D, Johann P, Bens S, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol (2016) 132(1):149–51. doi: 10.1007/s00401-016-1574-9
69. Shih AR, Cote GM, Chebib I, Choy E, DeLaney T, Deshpande V, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol (2018) 31(8):1237–45. doi: 10.1038/s41379-018-0002-1
70. Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med (2015) 21(12):1491–6. doi: 10.1038/nm.3968
71. Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell (2010) 18(4):316–28. doi: 10.1016/j.ccr.2010.09.006
72. Michel BC, D'Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol (2018) 20(12):1410–20. doi: 10.1038/s41556-018-0221-1
73. Wang X, Wang S, Troisi EC, Howard TP, Haswell JR, Wolf BK, et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun (2019) 10(1):1881. doi: 10.1038/s41467-019-09891-7
74. Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife (2018) 7:e41305. doi: 10.7554/eLife.41305
75. Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, et al. PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Res (2021) 81(11):2888–902. doi: 10.1158/0008-5472.CAN-21-0628
76. Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, et al. ATR is a therapeutic target in synovial sarcoma. Cancer Res (2017) 77(24):7014–26. doi: 10.1158/0008-5472.CAN-17-2056
77. Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun (2016) 7:13837. doi: 10.1038/ncomms13837
78. Howard TP, Arnoff TE, Song MR, Giacomelli AO, Wang X, Hong AL, et al. MDM2 and MDM4 are therapeutic vulnerabilities in malignant rhabdoid tumors. Cancer Res (2019) 79(9):2404–14. doi: 10.1158/0008-5472.CAN-18-3066
79. Sharifnia T, Wawer MJ, Chen T, Huang QY, Weir BA, Sizemore A, et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat Med (2019) 25(2):292–300. doi: 10.1038/s41591-018-0312-3
80. Sawada A, Kiyonari H, Ukita K, Nishioka N, Imuta Y, Sasaki H. Redundant roles of Tead1 and Tead2 in notochord development and the regulation of cell proliferation and survival. Mol Cell Biol (2008) 28(10):3177–89. doi: 10.1128/MCB.01759-07
81. Sheppard HE, Dall'Agnese A, Park WD, Shamim MH, Dubrulle J, Johnson HL, et al. Targeted brachyury degradation disrupts a highly specific autoregulatory program controlling chordoma cell identity. Cell Rep Med (2021) 2(1):100188. doi: 10.1016/j.xcrm.2020.100188
82. Shah SR, David JM, Tippens ND, Mohyeldin A, Martinez-Gutierrez JC, Ganaha S, et al. Brachyury-YAP regulatory axis drives stemness and growth in cancer. Cell Rep (2017) 21(2):495–507. doi: 10.1016/j.celrep.2017.09.057
83. Chang L, Azzolin L, Di Biagio D, Zanconato F, Battilana G, Lucon Xiccato R, et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature (2018) 563(7730):265–9. doi: 10.1038/s41586-018-0658-1
84. Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, et al. Palmitoylation of TEAD transcription factors is required for their stability and function in hippo pathway signaling. Structure (2016) 24(1):179–86. doi: 10.1016/j.str.2015.11.005
85. Holden JK, Crawford JJ, Noland CL, Schmidt S, Zbieg JR, Lacap JA, et al. Small molecule dysregulation of TEAD lipidation induces a dominant-negative inhibition of hippo pathway signaling. Cell Rep (2020) 31(12):107809. doi: 10.1016/j.celrep.2020.107809
86. Tang TT, Konradi AW, Feng Y, Peng X, Ma M, Li J, et al. Small molecule inhibitors of TEAD auto-palmitoylation selectively inhibit proliferation and tumor growth of NF2-deficient mesothelioma. Mol Cancer Ther (2021) 20(6):986–98. doi: 10.1158/1535-7163.MCT-20-0717
87. Zuccato JA, Patil V, Mansouri S, Liu JC, Nassiri F, Mamatjan Y, et al. DNA Methylation-based prognostic subtypes of chordoma tumors in tissue and plasma. Neuro Oncol (2022) 24(3):442–54. doi: 10.1093/neuonc/noab235
88. Zou MX, Lv GH, Wang XB, Huang W, Li J, Jiang Y, et al. Clinical impact of the immune microenvironment in spinal chordoma: Immunoscore as an independent favorable prognostic factor. Neurosurgery (2019) 84(6):E318–33. doi: 10.1093/neuros/nyy274
89. Zou MX, Zheng BW, Liu FS, Wang XB, Hu JR, Huang W, et al. The relationship between tumor-stroma ratio, the immune microenvironment, and survival in patients with spinal chordoma. Neurosurgery (2019) 85(6):E1095–110. doi: 10.1093/neuros/nyz333
90. Feng Y, Shen J, Gao Y, Liao Y, Cote G, Choy E, et al. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget (2015) 6(13):11139–49. doi: 10.18632/oncotarget.3576
91. Mathios D, Ruzevick J, Jackson CM, Xu H, Shah SR, Taube JM, et al. PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J Neurooncol (2015) 121(2):251–9. doi: 10.1007/s11060-014-1637-5
92. Xia C, Huang W, Chen YL, Fu HB, Tang M, Zhang TL, et al. Coexpression of HHLA2 and PD-L1 on tumor cells independently predicts the survival of spinal chordoma patients. Front Immunol (2021) 12:797407. doi: 10.3389/fimmu.2021.797407
93. Long C, Li G, Zhang C, Jiang T, Li Y, Duan X, et al. B7-H3 as a target for CAR-T cell therapy in skull base chordoma. Front Oncol (2021) 11:659662. doi: 10.3389/fonc.2021.659662
94. Duan W, Zhang B, Li X, Chen W, Jia S, Xin Z, et al. Single-cell transcriptome profiling reveals intra-tumoral heterogeneity in human chordomas. Cancer Immunol Immunother (2022) 71(9):2185–95. doi: 10.1007/s00262-022-03152-1
95. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Powles: TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
96. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
97. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin Cancer Res (2018) 24(6):1287–95. doi: 10.1158/1078-0432.CCR-17-2653
98. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
99. Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell (2018) 173(2):355–370 e14. doi: 10.1016/j.cell.2018.03.039
100. Sayaman RW, Saad M, Thorsson V, Hu D, Hendrickx W, Roelands J, et al. Germline genetic contribution to the immune landscape of cancer. Immunity (2021) 54(2):367–386 e8. doi: 10.1016/j.immuni.2021.01.011
101. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (2018) 359(6371):104–8. doi: 10.1126/science.aao3290
102. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
103. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
104. Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science (2021) 374(6575):1632–40. doi: 10.1126/science.aaz7015
105. Passeri T, Dahmani A, Masliah-Planchon J, Naguez A, Michou M, El Botty R, et al. Dramatic In vivo efficacy of the EZH2-inhibitor tazemetostat in PBRM1-mutated human chordoma xenograft. Cancers (Basel) (2022) 14(6):1486. doi: 10.3390/cancers14061486
106. Walker RL, Hornicek FJ, Duan Z. Advances in the development of chordoma models for drug discovery and precision medicine. Biochim Biophys Acta Rev Cancer (2022), 1877(6):188812. doi: 10.1016/j.bbcan.2022.188812
107. Chordoma FoundationChordoma foundation in vivo drug screening program data. figshare. Available at: https://figshare.com/projects/Chordoma_Foundation_In_Vivo_Drug_Screening_Program/25948.
108. Woo XY, Giordano J, Srivastava A, Zhao ZM, Lloyd MW, de Bruijn R, et al. Eur: Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat Genet (2021) 53(1):86–99. doi: 10.1038/s41588-020-00750-6
109. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med (2015) 21(11):1318–25. doi: 10.1038/nm.3954
110. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer (2018) 18(7):407–18. doi: 10.1038/s41568-018-0007-6
111. Al Shihabi A, Davarifar A, Nguyen HTL, Tavanaie N, Nelson SD, Yanagawa J, et al. Personalized chordoma organoids for drug discovery studies. Sci Adv (2022) 8(7):eabl3674. doi: 10.1126/sciadv.abl3674
112. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science (2018) 359(6378):920–6. doi: 10.1126/science.aao2774
113. Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discovery (2018) 8(9):1112–29. doi: 10.1158/2159-8290.CD-18-0349
114. Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med (2019) 11(513):eaay2574. doi: 10.1126/scitranslmed.aay2574
115. Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized In vitro and In vivo cancer models to guide precision medicine. Cancer Discovery (2017) 7(5):462–77. doi: 10.1158/2159-8290.CD-16-1154
116. Guillen KP, Fujita M, Butterfield AJ, Scherer SD, Bailey MH, Chu Z, et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat Cancer (2022) 3(2):232–50. doi: 10.1038/s43018-022-00337-6
117. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell (2017) 170(3):564–576 e16. doi: 10.1016/j.cell.2017.06.010
118. Vervoort SJ, Devlin JR, Kwiatkowski N, Teng M, Gray NS, Johnstone RW. Targeting transcription cycles in cancer. Nat Rev Cancer (2022) 22(1):5–24. doi: 10.1038/s41568-021-00411-8
119. Richters A, Doyle SK, Freeman DB, Lee C, Leifer BS, Jagannathan S, et al. Modulating androgen receptor-driven transcription in prostate cancer with selective CDK9 inhibitors. Cell Chem Biol (2021) 28(2):134–147 e14. doi: 10.1016/j.chembiol.2020.10.001
120. Day MAS, Hood T, Obholzer N, Pandey A, Lin CY, Kumar P, et al. CDK9 inhibition via KB-0742 is a potential strategy to treat transcriptionally addicted cancers. Cancer Res (2022) 82(12_Supplement):2564. doi: 10.1158/1538-7445.AM2022-2564
121. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell (2011) 20(5):620–34. doi: 10.1016/j.ccr.2011.10.001
122. Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature (2017) 546(7658):426–30. doi: 10.1038/nature22797
123. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
124. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10(1):1617. doi: 10.1038/s41467-019-09277-9
125. Nicholson HE, Tariq Z, Housden BE, Jennings RB, Stransky LA, Perrimon N, et al. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Sci Signal (2019) 12(601):eaay0482. doi: 10.1126/scisignal.aay0482
126. Coffin CM, Swanson PE, Wick MR, Dehner LP. An immunohistochemical comparison of chordoma with renal cell carcinoma, colorectal adenocarcinoma, and myxopapillary ependymoma: a potential diagnostic dilemma in the diminutive biopsy. Mod Pathol (1993) 6(5):531–8.
127. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
128. Dodd KM, Yang J, Shen MH, Sampson JR, Tee AR. mTORC1 drives HIF-1alpha and VEGF-a signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene (2015) 34(17):2239–50. doi: 10.1038/onc.2014.164
129. Li X, Ji Z, Ma Y, Qiu X, Fan Q, Ma B. Expression of hypoxia-inducible factor-1alpha, vascular endothelial growth factor and matrix metalloproteinase-2 in sacral chordomas. Oncol Lett (2012) 3(6):1268–74. doi: 10.3892/ol.2012.645
130. Lam R. The nature of cytoplasmic vacuoles in chordoma cells. a correlative enzyme and electron microscopic histochemical study. Pathol Res Pract (1990) 186(5):642–50. doi: 10.1016/S0344-0338(11)80228-1
131. Pandiar D, Thammaiah S. Physaliphorous cells. J Oral Maxillofac Pathol (2018) 22(3):296–7. doi: 10.4103/jomfp.JOMFP_265_18
132. Ellis K, Bagwell J, Bagnat M. Notochord vacuoles are lysosome-related organelles that function in axis and spine morphogenesis. J Cell Biol (2013) 200(5):667–79. doi: 10.1083/jcb.201212095
133. N. Cancer Genome Atlas Research. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) 499(7456):43–9. doi: 10.1038/nature12222
134. McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, et al. Sellers: Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by Large-scale, deep RNAi screening. Cell (2017) 170(3):577–592 e10. doi: 10.1016/j.cell.2017.07.005
135. Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature (2019) 568(7753):511–6. doi: 10.1038/s41586-019-1103-9
136. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery (2020) 19(1):23–38. doi: 10.1038/s41573-019-0046-z
137. Kalev P, Hyer ML, Gross S, Konteatis Z, Chen CC, Fletcher M, et al. MAT2A inhibition blocks the growth of MTAP-deleted cancer cells by reducing PRMT5-dependent mRNA splicing and inducing DNA damage. Cancer Cell (2021) 39(2):209–224 e11. doi: 10.1016/j.ccell.2020.12.010
138. Hoke ATK, Padget MR, Fabian KP, Nandal A, Gallia GL, Bilusic M, et al. Combinatorial natural killer cell-based immunotherapy approaches selectively target chordoma cancer stem cells. Cancer Res Commun (2021) 1(3):127–39. doi: 10.1158/2767-9764.crc-21-0020
139. Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science (2018) 362(6421):1416–22. doi: 10.1126/science.aas9090
140. Ruscetti M, t. Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell (2020) 181(2):424–441 e21. doi: 10.1016/j.cell.2020.03.008
141. Fujii R, Schlom J, Hodge JW. A potential therapy for chordoma via antibody-dependent cell-mediated cytotoxicity employing NK or high-affinity NK cells in combination with cetuximab. J Neurosurg (2018) 128(5):1419–27. doi: 10.3171/2017.1.JNS162610
142. DeMaria PJ, Lee-Wisdom K, Donahue RN, Madan RA, Karzai F, Schwab A, et al. Phase 1 open-label trial of intravenous administration of MVA-BN-brachyury-TRICOM vaccine in patients with advanced cancer. J Immunother Cancer (2021) 9(9):e003238. doi: 10.1136/jitc-2021-003238
143. Migliorini D, Mach N, Aguiar D, Vernet R, Landis BN, Becker M, et al. First report of clinical responses to immunotherapy in 3 relapsing cases of chordoma after failure of standard therapies. Oncoimmunology (2017) 6(8):e1338235. doi: 10.1080/2162402X.2017.1338235
144. Williamson LM, Rive CM, Di Francesco D, Titmuss E, Chun HE, Brown SD, et al. Clinical response to nivolumab in an INI1-deficient pediatric chordoma correlates with immunogenic recognition of brachyury. NPJ Precis Oncol (2021) 5(1):103. doi: 10.1038/s41698-021-00238-4
145. Wu X, Lin X, Chen Y, Kong W, Xu J, Yu Z. Response of metastatic chordoma to the immune checkpoint inhibitor pembrolizumab: A case report. Front Oncol (2020) 10:565945. doi: 10.3389/fonc.2020.565945
146. Blay JY, Penel N, Ray-Coquard IL, Cousin S, Bertucci F, Bompas E, et al. High clinical activity of pembrolizumab in chordoma, alveolar soft part sarcoma (ASPS) and other rare sarcoma histotypes: The French AcSe pembrolizumab study from unicancer. J Clin Oncol (2021) 39(no. 15_suppl):11520–0. doi: 10.1200/JCO.2021.39.15_suppl.11520
147. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity (2022) 55(3):512–526 e9. doi: 10.1016/j.immuni.2022.02.005
148. Fujita N, Miyamoto T, Imai J, Hosogane N, Suzuki T, Yagi M, et al. CD24 is expressed specifically in the nucleus pulposus of intervertebral discs. Biochem Biophys Res Commun (2005) 338(4):1890–6. doi: 10.1016/j.bbrc.2005.10.166
149. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0
150. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9
151. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature (2005) 436(7051):720–4. doi: 10.1038/nature03890
152. Baggiolini A, Callahan SJ, Montal E, Weiss JM, Trieu T, Tagore MM, et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science (2021) 373(6559):eabc1048. doi: 10.1126/science.abc1048
153. Patel SA, Hirosue S, Rodrigues P, Vojtasova E, Richardson EK, Ge J, et al. The renal lineage factor PAX8 controls oncogenic signalling in kidney cancer. Nature (2022) 606(7916):999–1006. doi: 10.1038/s41586-022-04809-8
154. Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer (2021) 21(10):638–54. doi: 10.1038/s41568-021-00365-x
155. Beisaw A, Tsaytler P, Koch F, Schmitz SU, Melissari MT, Senft AD, et al. BRACHYURY directs histone acetylation to target loci during mesoderm development. EMBO Rep (2018) 19(1):118–34. doi: 10.15252/embr.201744201
156. Samarasinghe KTG, Jaime-Figueroa S, Burgess M, Nalawansha DA, Dai K, Hu Z, et al. Targeted degradation of transcription factors by TRAFTACs: TRAnscription factor TArgeting chimeras. Cell Chem Biol (2021) 28(5):648–661 e5. doi: 10.1016/j.chembiol.2021.03.011
157. N. Cancer Genome Atlas Research, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med (2016) 374(2):135–45. doi: 10.1056/NEJMoa1505917
158. Palena C, Roselli M, Litzinger MT, Ferroni P, Costarelli L, Spila A, et al. Overexpression of the EMT driver brachyury in breast carcinomas: association with poor prognosis. J Natl Cancer Inst (2014) 106(5):dju054. doi: 10.1093/jnci/dju054
159. Hamilton DH, Roselli M, Ferroni P, Costarelli L, Cavaliere F, Taffuri M, et al. Brachyury, a vaccine target, is overexpressed in triple-negative breast cancer. Endocr Relat Cancer (2016) 23(10):783–96. doi: 10.1530/ERC-16-0037
160. Li K, Ying M, Feng D, Du J, Chen S, Dan B, et al. Brachyury promotes tamoxifen resistance in breast cancer by targeting SIRT1. BioMed Pharmacother (2016) 84:28–33. doi: 10.1016/j.biopha.2016.09.011
161. Lee KH, Kim EY, Yun JS, Park YL, Do SI, Chae SW, et al. Prognostic significance of expression of epithelial-mesenchymal transition driver brachyury in breast cancer and its association with subtype and characteristics. Oncol Lett (2018) 15(1):1037–45. doi: 10.3892/ol.2017.7402
162. Miettinen M, Wang Z, Lasota J, Heery C, Schlom J, Palena C. Nuclear brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: An immunohistochemical study of 5229 cases. Am J Surg Pathol (2015) 39(10):1305–12. doi: 10.1097/PAS.0000000000000462
163. Roselli M, Fernando RI, Guadagni F, Spila A, Alessandroni J, Palmirotta R, et al. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res (2012) 18(14):3868–79. doi: 10.1158/1078-0432.CCR-11-3211
164. Huang B, Cohen JR, Fernando RI, Hamilton DH, Litzinger MT, Hodge JW, et al. The embryonic transcription factor brachyury blocks cell cycle progression and mediates tumor resistance to conventional antitumor therapies. Cell Death Dis (2013) 4:e682. doi: 10.1038/cddis.2013.208
165. Xu K, Liu B, Liu Y. Impact of brachyury on epithelial-mesenchymal transitions and chemosensitivity in non-small cell lung cancer. Mol Med Rep (2015) 12(1):995–1001. doi: 10.3892/mmr.2015.3348
166. Shimamatsu S, Okamoto T, Haro A, Kitahara H, Kohno M, Morodomi Y, et al. Prognostic significance of expression of the epithelial-mesenchymal transition-related factor brachyury in intrathoracic lymphatic spread of non-small cell lung cancer. Ann Surg Oncol (2016) 23(Suppl 5):1012–20. doi: 10.1245/s10434-016-5530-7
167. Kilic N, Feldhaus S, Kilic E, Tennstedt P, Wicklein D, Wasielewski R, et al. Brachyury expression predicts poor prognosis at early stages of colorectal cancer. Eur J Cancer (2011) 47(7):1080–5. doi: 10.1016/j.ejca.2010.11.015
168. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21(11):1350–6. doi: 10.1038/nm.3967
169. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol (2009) 27(8):1160–7. doi: 10.1200/JCO.2008.18.1370
Keywords: chordoma, rare cancer, drug repurposing, target discovery, multi-omics, functional genomics, synthetic lethality, precision oncology
Citation: Freed DM, Sommer J and Punturi N (2022) Emerging target discovery and drug repurposing opportunities in chordoma. Front. Oncol. 12:1009193. doi: 10.3389/fonc.2022.1009193
Received: 01 August 2022; Accepted: 11 October 2022;
Published: 27 October 2022.
Edited by:
Jiwei Bai, Beijing Tiantan Hospital, Capital Medical University, ChinaReviewed by:
Christopher Asquith, University of Eastern Finland, FinlandCopyright © 2022 Freed, Sommer and Punturi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel M. Freed, ZGFuQGNob3Jkb21hLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.