 Chunyu Zhao1,2,3
Chunyu Zhao1,2,3 Shanshuo Liu
Shanshuo Liu Feng Gao
Feng Gao Yawen Zou
Yawen Zou Zhigang Ren
Zhigang Ren Zujiang Yu
Zujiang Yu- 1Department of Infectious Diseases, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Jinan Microecological Biomedicine Shandong Laboratory, Jinan, Shandong, China
- 3Gene Hospital of Henan Province; Precision Medicine Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Primary liver cancer (PLC), including hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC), and other rare tumours, is the second leading cause of cancer-related mortality. It has been a major contributor to the cancer burden worldwide. Of all primary liver cancer, HCC is the most common type. Over the past few decades, chemotherapy, immunotherapy and other therapies have been identified as applicable to the treatment of HCC. However, evidence suggests that chemotherapy resistance is associated with higher mortality rates in liver cancer. The tumour microenvironment (TME), which includes molecular, cellular, extracellular matrix(ECM), and vascular signalling pathways, is a complex ecosystem. It is now increasingly recognized that the tumour microenvironment plays a pivotal role in PLC prognosis, progression and treatment response. Cancer cells reprogram the tumour microenvironment to develop resistance to chemotherapy drugs distinct from normal differentiated tissues. Chemotherapy resistance mechanisms are reshaped during TME reprogramming. For this reason, TME reprogramming can provide a powerful tool to understand better both cancer-fate processes and regenerative, with the potential to develop a new treatment. This review discusses the recent progress of tumour drug resistance, particularly tumour microenvironment reprogramming in tumour chemotherapy resistance, and focuses on its potential application prospects.
Introduction
Over the past few years, cancer has always been a significant public health problem worldwide. According to the data the American Cancer Society reported in 2021, there are nearly 1.9 million new cancer cases and more than 600 thousand cancer deaths in the United States (1). Of these, liver cancer is the most common cause of cancer death worldwide and one of the fifth most common cancers in the United States, with its incidence rising yearly (2). The occurrence of PLC is mainly associated with chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections. However, other factors have also been implicated in the occurrence of HCC, including fatty liver disease, dietary aflatoxin exposure, alcohol-related cirrhosis, smoking, obesity, fatty liver disease, iron overload, Mellitus-related non-alcoholic fatty liver disease and type 2 diabetes (3, 4). Although ICC accounts for only a small proportion of primary liver cancer in most parts of the world, it is the most common subtype of primary liver cancer in Thailand due to the high incidence of chronic liver fluke infection. Cirrhosis is also one of its risk factors (5, 6).
The treatment of HCC has dramatically improved over the past decades. The Barcelona Clinic Liver Cancer (BCLC) staging system is the main basis for treatment. In principle, patients with early-stage are suitable for surgical resection, liver transplantation, and local ablation, while TACE is the first choice for patients with intermediate HCC. Due to the lack of effective chemoprevention strategies and early diagnosis, most patients are found at an advanced stage. However, the late prognosis is poor, and only systemic therapy can prolong survival time with a median survival time for advanced HCC of ~6 months (7). Sorafenib was the only systemic therapy approved for patients with advanced tumors in the last dozen years (8). The results of a global open-label randomized Phase-III trial (REFLECT) demonstrated that lenvatinib improved the overall survival of patients with advanced HCC, the first new drug to be approved in the first-line setting for advanced-stage HCC in more than 10 years, which represented a breakthrough in the clinical management of this cancer (7). Despite this, in advanced liver cancer, the survival benefit of these drugs is limited (9, 10). Despite this, in advanced liver cancer, the survival benefit of these drugs is limited.
The environment in which HCC tumor cells grow is called the liver tumor microenvironment, which is a complex mixture of tumor cells and stromal cells and the proteins they secrete (11). Normally, the stroma maintains the physiological homeostasis of normal tissues, and some stromal components act as a physical barrier to tumor formation (12, 13). However, neoplastic cells cause various changes, and stroma is inappropriately activated in cancer, transforming adjacent TME into pathological entities to support cancer development and make a contribution to the malignant characteristics of tumor cells (14). The elements of a typical TME are made up of surrounding blood vessels, cancer-associated fibroblasts(CAFs), immune and inflammatory cells, cytokines, chemokines or enzymes, and extracellular matrix (ECM) (15). There is increasing evidence that liver cancer progression and metastasis are influenced by the tumor microenvironment.
An adverse tumor microenvironment in a single tumor is recognized as common in most solid malignancies. It has been found that the degree of heterogeneity in the tumor microenvironment of normal and malignant cells is negatively correlated with the prognosis of patients (16). Some of the harmful features of the tumor microenvironment through reprogramming can act alone or in combination with cancer progression(via e.g. resistance to apoptosis, promotion of genetic instability and mutation, continuous angiogenesis, and distant metastasis), leading to chemotherapeutic resistance and ultimately poor patient outcomes (17). In this review, we mainly focus on the mechanism of tumor microenvironment reprogramming on chemotherapy resistance in PLC to spark new ideas for designing more specific therapies for cancer.
Cancer-associated fibroblasts
Among all the stromal cells that make up the tumor microenvironment, CAFs are the most abundant tumor stromal cell type and a key element of TME (18). CAFs predominantly arise from tissue-resident fibroblasts and mesenchymal stem cells (19–22). But studies have shown that even under limited conditions. Transformation of adipocytes, pericytes, and endothelial cells has also been observed (23). Normally, fibroblasts are usually quiescent, but fibroblasts can be activated under certain conditions, such as wound healing response, tissue fibrosis acute and chronic inflammation (24–26). Tumor cells recruit and secrete growth factors such as transforming growth factor β (TGFβ), platelet-derived growth factor (PDGF), and interleukin-6 (IL-6) to stimulate fibroblasts to convert to CAFs (23, 27). Although α-SMA, FAP, and PDGF receptor-α (PDGFRα)can recognize activated fibroblasts, there are still no comprehensive and specific biomarkers (23, 28, 29). Numerous studies have suggested that CAFs are associated with treatment resistance to colorectal cancer, ovarian cancer, breast cancer, and stomach cancer (30–33). Cancer-associated fibroblasts can mediate drug resistance by reprogramming the metabolic process of tumor cells (34).
Metastasis and metabolic reprogramming are known as two major features of cancer (35). The reason why liver tumorigenesis or tumor progression inevitably leads to metabolic reprogramming is that life is not only the largest metabolic organ in the human body but also is associated with almost all central metabolic processes. Tumor cells undergo metabolic reprogramming to meet bioenergy and biosynthesis requirements to maintain their abnormal proliferation and adapt to the tumor microenvironment (36). Unlike normal cells, tumor cells rely on glycolysis to produce energy even under aerobic conditions, also known as the Warburg effect (37). In this process, the rate of glucose uptake dramatically increases and produces more lactate. Cancer-associated fibroblasts have been observed to influence cancer drug response and play a positive metabolic role in tyrosine kinase inhibitor (TKIs) resistance. Specifically, increased lactate secretion leads to upregulation of hepatocyte growth factor (HGF) production by cancer-associated fibroblasts in a nuclear factor KB-dependent manner. MET-dependent signal transduction in cancer is activated by increased HGF and endows sustained resistance to TKIs (38). Studies have shown that in HCC patients with high c-MET expression, HGF reduces the anti-proliferation, pro-apoptotic and anti-invasion effects of lenvatinib on HCC cells. In addition, the activation of the HGF/c−MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells (39). Moreover, after long-term sorafenib treatment, HGF upregulation induces the autocrine activation of the HGF ⁄c-Met signaling pathway, increasing the anti-apoptotic and invasion ability of HCC cells, and leading to resistance to sorafenib (40). It can be seen that the HGF/c-Met axis plays an important role in the chemotherapy resistance of liver cancer. Several HGF/c-MET inhibitor drugs have been e assessed in clinical trials (Table 1).
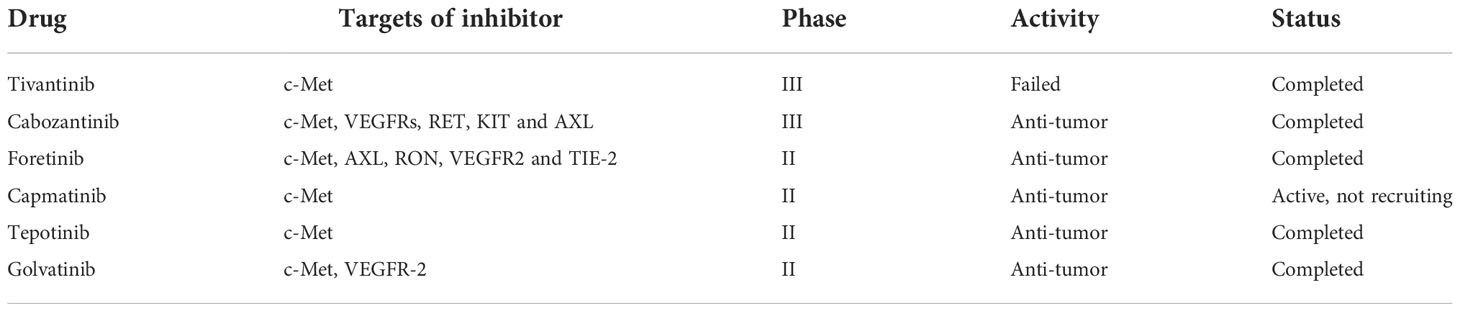
Table 1 Clinical trials of some HGF/c-Met signaling inhibitors in hepatocellular carcinoma patients.
Although the Warburg effect has been widely accepted as a distinctive character of tumor cells, accumulating evidence has revealed that other metabolic features, particularly the reverse Warburg effect (41), metabolic symbiosis, and glutamine metabolism, create challenges to antitumor therapy due to adaptive or acquired chemotherapy resistance (42). Lisanti et al. proposed the “reverse Warburg effect” in 2009: cancer cells induce glycolysis in cancer-associated fibroblasts which in turn produce lactate and pyruvate that are used by adjacent epithelial cancer cells as sources of the mitochondrial tricarboxylic acid cycle (TCA cycle), oxidative phosphorylation, and ATP production (43, 44) (Figure 1). The study of Migneco et al. showed that nutrients derived from glycolytic cancer-associated fibroblasts can effectively reduce the dependence of cancer cells on vascular blood supply, thus promoting the escape of tumor cells during antiangiogenic drugs (45).
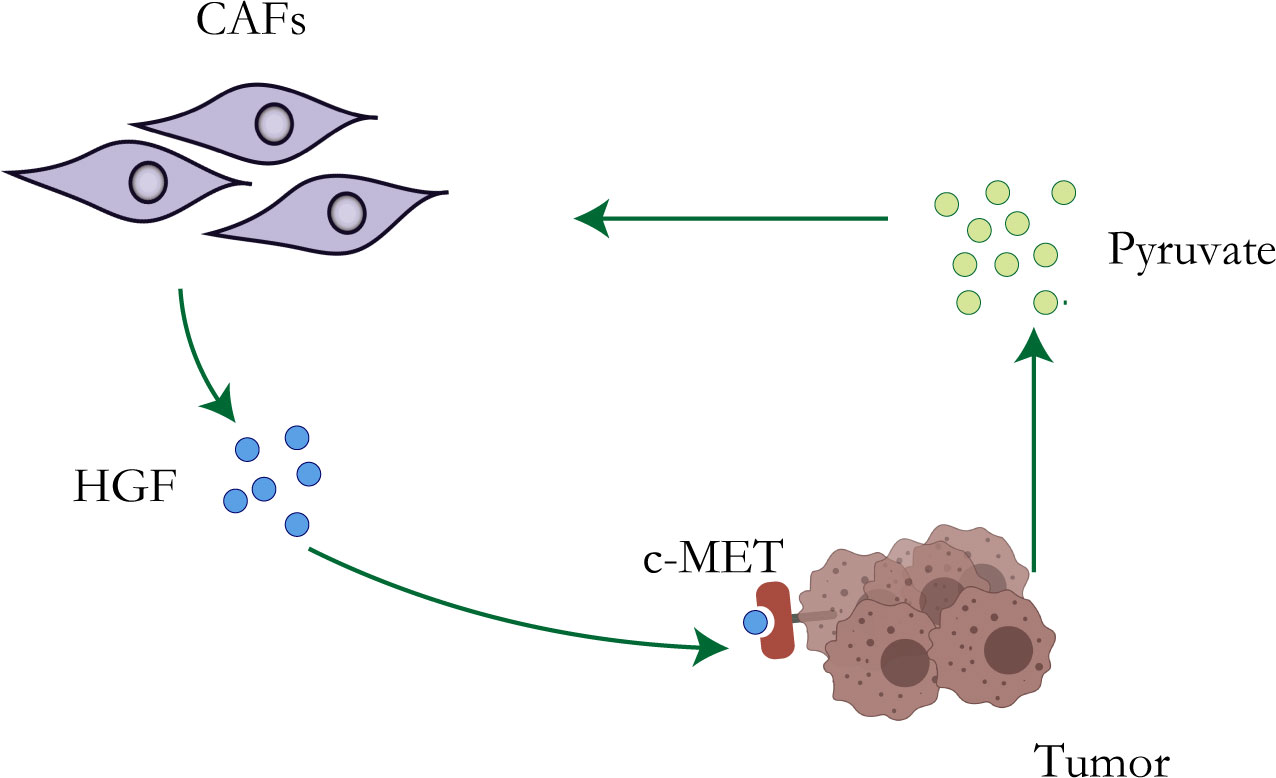
Figure 1 Resistance mechanisms of CAFs. Metabolic reprogramming of tumor cells leads to increased lactate secretion, which stimulates CAFs to up-regulate the production of HGF, leading to chemotherapy resistance of tumor cells.
In CAA, epidermal growth factor receptor (EGFR) overexpression often indicates a poor prognosis (46). It can be activated by heparin-binding epidermal growth factor (HB-EGF) secreted by CAFs. The HB-EGF/EGFR axis promotes CCA cell proliferation, migration, and invasion by activating the signal transducer and activator of transcription (STAT)-3. Interestingly, TGF-β1 secreted by tumor cells can enhance HB-EGF expression in CAFs, and TGF-β1 production is triggered by EGFR activation. Thus forming a continuous signaling loop (47).
In addition, c-Met expressed by tumor cells in CCA can be activated by HGF secreted by CAFs. After c-Met activation, its downstream pathways including mitogen-activated protein kinase (MAPK), extracellular signal-related kinase (ERK), and phosphatidylinositol 3-kinase (PI3K) are activated, which is conducive to tumor cells invasion and chemotherapy resistance (48, 49).
Mesenchymal stem cell
Mesenchymal stem cells also called mesenchymal stromal cells(MSCs), migrate to sites of inflammation, as well as migrate and fuse with tumors (50). MSCs are originally isolated from bone marrow. Besides, adipose tissue, peripheral blood, and umbilical cord are also important sources of MSCs (51). MSCs are a heterogeneous subpopulation of progenitor cells with the capability of self-renewal and multidirectional differentiation, which can transdifferentiate into osteocytes, chondrocytes, adipocytes, astrocytes, fibroblasts, and pericytes (52). Studies have shown that MSCs play a significant role in tumorigenesis and development, and participate in many steps of tumorigenesis, such as angiogenesis, invasion, metastasis, epithelial-mesenchymal transformation, anti-apoptosis, immunosuppression, and chemotherapeutic resistance (53). There is growing evidence showing the important role of MSCs in tumor resistance (54).
It has been proven that MSCs can secrete a variety of cytokines or growth factors that participate in multiple pathways and downstream mechanisms to activate a cascade of reactions, leading to drug resistance (55, 56). Inflammatory mediators are known to be an important part of the tumor microenvironment (57). TGF-β expressed by mesenchymal stem cells increased when exposed to tumor inflammatory microenvironment (58). It has been proved that autophagy can enhance the resistance of HCC cells to chemical drugs by affecting the apoptotic potential of HCC cells (59). Besides, Some studies have indicated that TGF-β plays an important role in inducing autophagy (60, 61). Taken together, MSCs promote the development of chemotherapy resistance of HCC cells by up-regulating the expression of TGF-β (62). MSCs, as mentioned before, have the potential to differentiate into multicellular lineages, so their transformation into CAFs may be another mechanism for the development of therapeutic resistance (Figure 2). Mishra et al. found that human bone marrow-derived mesenchymal stem cells (hMSCs) can transform into SDF-1-expressing CAFs when exposed to a tumor-conditioned medium (TCM) for a long time (63). As previously mentioned, CAFs play a critical role in tumor drug resistance.
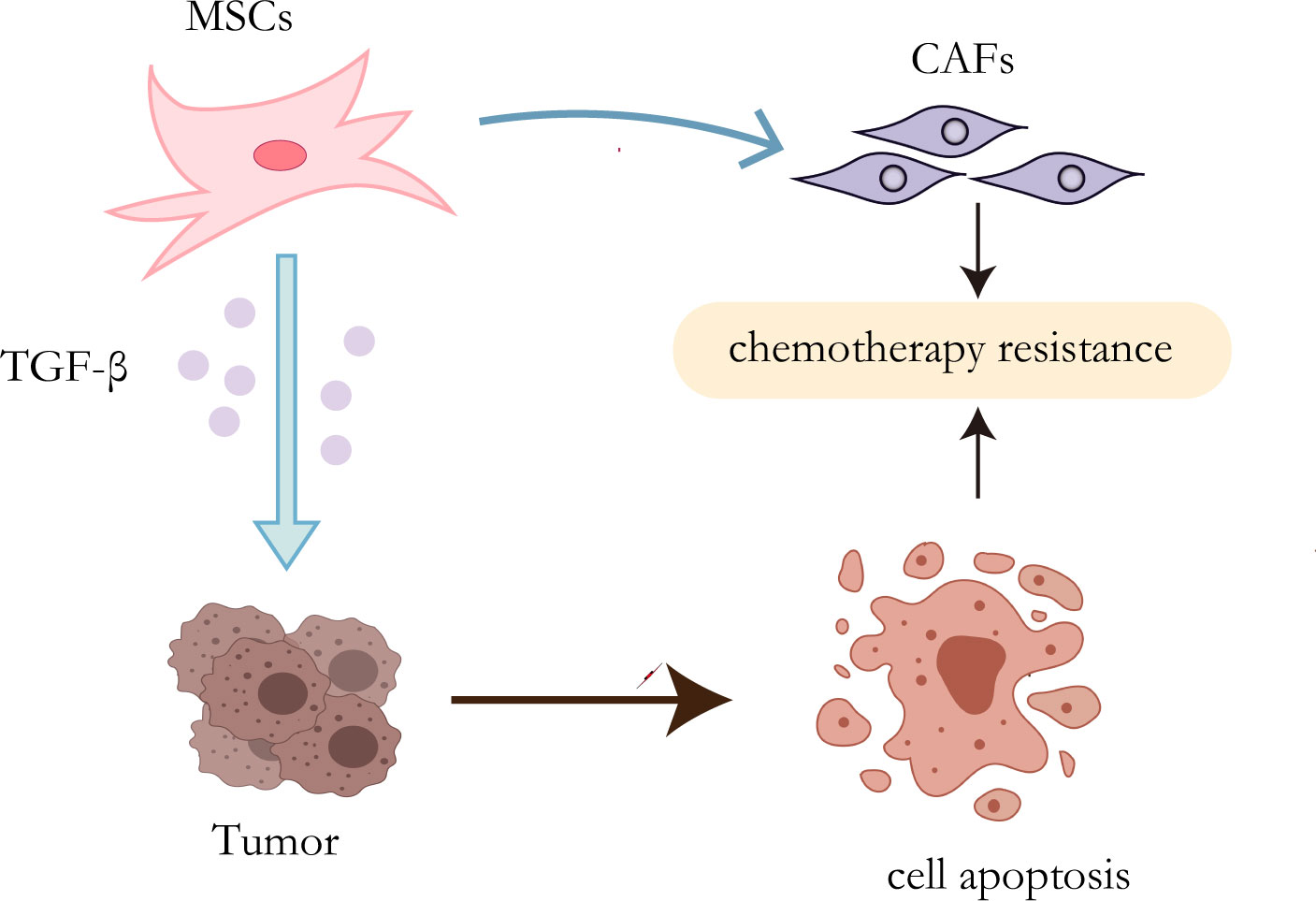
Figure 2 Resistance mechanisms of MSCs. On the one hand, TGF-β secreted by MSCs can induce autophagy of HCC cells, leading to chemotherapy resistance; on the other hand, MSCs can transform into CAFs to promote drug resistance.
Tumor-associated macrophages
Massive macrophage infiltration is a common characteristic of malignant tumors. Based on surface receptors and functional characteristics, macrophages can be divided into classical (M1) and alternative (M2) activated phenotypes. The macrophages surrounding the tumor area are called tumor-associated macrophages (TAM). Increased TAMs are known to be associated with poor prognosis after the surgical resection of hepatocellular carcinoma (64). The majority of macrophages located in intratumoral and peritumoral regions exhibit the M2 phenotype, and several studies have shown that cytokines found in peritumoral tissues, including vascular endothelial growth factor, macrophage colony-stimulating factor, and placental growth factor are significantly associated with HCC recurrence and poor survival outcomes (65–67).
Cancer-associated adipocytes
Epidemiological evidence suggests that obesity is associated with a combination of all cancers and a poor prognosis for multiple location-specific cancers (68). Systemic and local environmental changes caused by obesity can not only affect the occurrence and development of tumors but also induce chemotherapy resistance to tumors, especially in breast cancer, prostate cancer, ovarian cancer, and leukemia (69, 70). All of this suggests a potential role for adipocytes in tumor drug resistance. Adipocytes are one of the most important components of stromal cells in TME (71). It is noteworthy that co-cultured with cancer cells, adipocytes are observed to exhibit remarkable phenotypic changes and also exhibit an altered phenotype in terms of delipidation, which are designated as cancer-associated adipocytes (CAAs) (72). Bochet et al. demonstrated that tumor cells can secrete Wnt3a to promote changes in adipocyte phenotypes (73). In addition, the bidirectional cross-talk established between cancer cells and mature adipocytes may enhance the invasive capabilities of cancer cells by altering the adipocyte phenotype (72, 74). Accumulating recent evidence indicates that CAAs can induce chemoresistance through different mechanisms such as metabolic reprogramming, secretion of various factors, remodeling of the extracellular matrix, and altering chemotherapy pharmacokinetics (75–78).
The “Warburg effect” and “reverse Warburg effect” have been proved to be the cause of CAFs-mediated drug resistance, and this concept can also be applied to other cells in the tumor microenvironment, particularly adipocytes (79). For a long time, adipocytes were identified as a tremendous passive energy storage depot. Pérez de Heredia et al. showed that the release of lactate from adipocytes increased under hypoxic conditions (80). Previous studies have shown that lipids in adipocytes are the main source of tumor cells. CAAs release exogenous free fatty acids (FFAs) that can be taken up by CD36 on the surface of cancer cells (81). FFAs could yield sufficient energy for cancer cells through FAO (82), which contributes to therapy resistance. It has been proved that CD36 promotes epithelial-mesenchymal transition(EMT) in hepatocellular carcinoma by increased free fatty acid uptake (83). Notably, EMT is closely associated with the development of liver cancer and likely affects therapeutic responsiveness in HCC (84) (Figure 3).
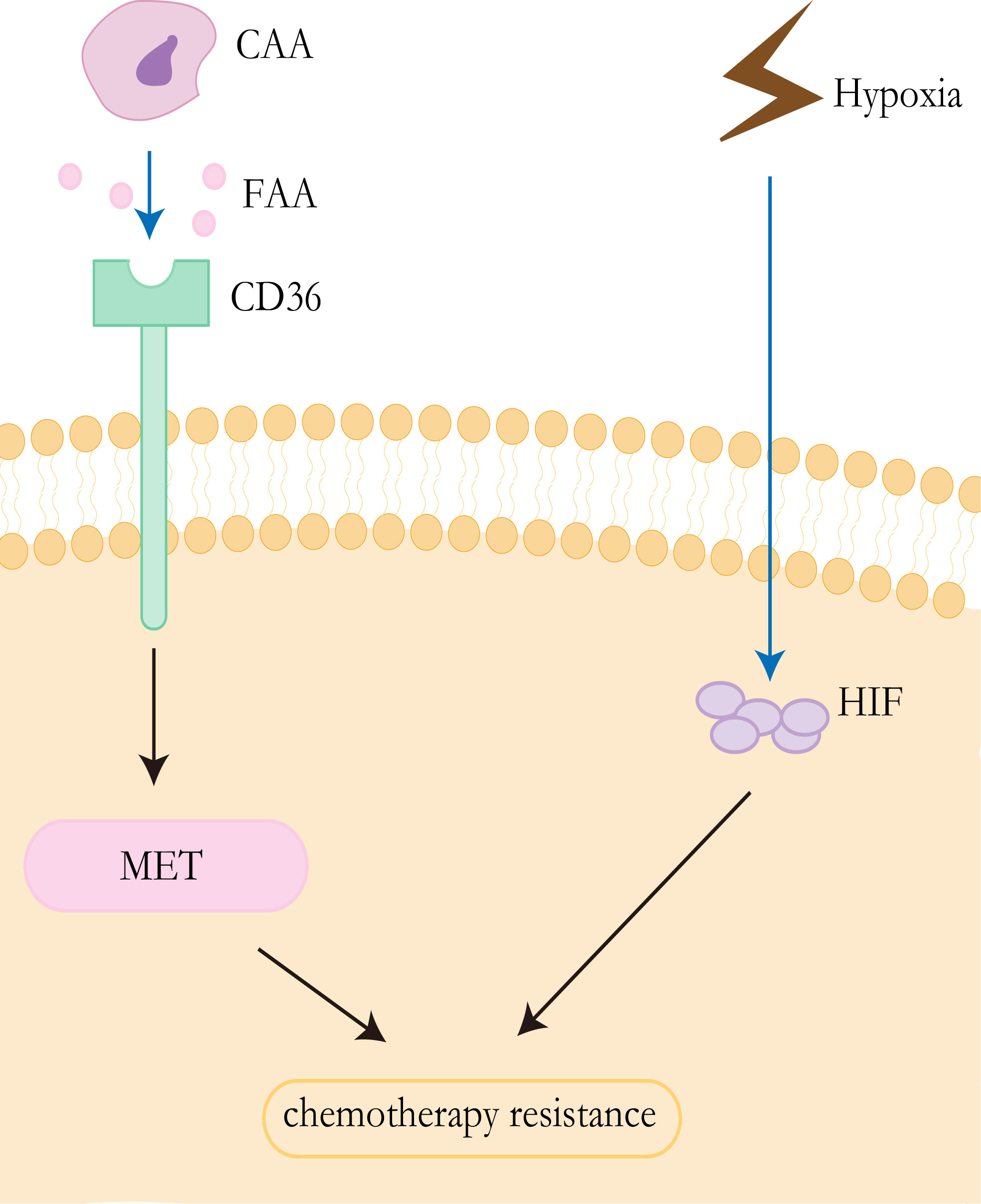
Figure 3 Resistance mechanisms of CAAs and hypoxia. CAAs release exogenous free fatty acids, which are absorbed by CD36 on the surface of tumor cells, to promote epithelial mesenchymal transformation and promote drug resistance of HCC. Tumor cells in anoxic environment lead to upregulated EXPRESSION of HIF, thus promoting drug resistance to chemotherapy.
Tumor-associated neutrophils
Traditionally, the recruitment mechanism and function of neutrophils have been studied mainly in inflammation. Neutrophils account for about 50-70% of myeloid-derived white blood cells in patients with infection or inflammation (85). The inflammatory cells in solid tumors are mainly neutrophils, and the intratumoral high density is correlated with lymph node metastasis, tumor stage axis, and tumor grade (86). Increasing clinical evidence shows that the tumor and the tumor microenvironment control neutrophil recruitment and in turn tumor-associated neutrophils (TANs) regulate tumor progression or growth control (87). Inflammatory neutrophils not only can phagocytose bacteria, activate and enhance the immunosuppressive system, but TANs also functions as immunosuppressive cells in tumors (88).
It has been well acknowledged that neutrophils have the capability to secret cytokine and chemokine (89). These cytokines enable them to interact directly with tumor cells as pro-tumor or anti-tumor effectors or indirectly by regulating angiogenesis, tumor growth, and anti-tumor immune responses (90). HGF, a heparin-binding factor, binds to a specific proto-oncogene tyrosine kinase receptor (C-MET) to stimulate hepatocytes by maintaining proliferation, promoting epithelial-mesenchymal transformation (EMT), and ultimately leading to invasion and metastasis during the malignant transformation of HCC (91, 92). In addition, other studies have shown that the HGF-Met axis plays an important role in chemotherapy resistance by coordinating liver cancer metabolism and autophagy. The combination of MET inhibitors and autophagy inhibitors significantly improved the efficacy of hepatocellular carcinoma therapy in mice (93). In liver cancer, however, cancer cells stimulate neutrophils to release hepatocyte growth factors, which in turn stimulates tumor cells to become more aggressive (94). CCL2 and CCL17 secreted by TANs can mediate intratumoral invasion of macrophages and Treg cells, thereby stimulating angiogenesis, promoting liver cancer growth and metastasis, and promoting sorafenib drug resistance. In addition, sorafenib also blocks tumor angiogenesis through targeted VEGF and PDGF receptor kinase activity, which also contributes to TAN accumulation and accompanying macrophage and Treg cell infiltration and promotes sorafenib resistance (95) (Figure 4).
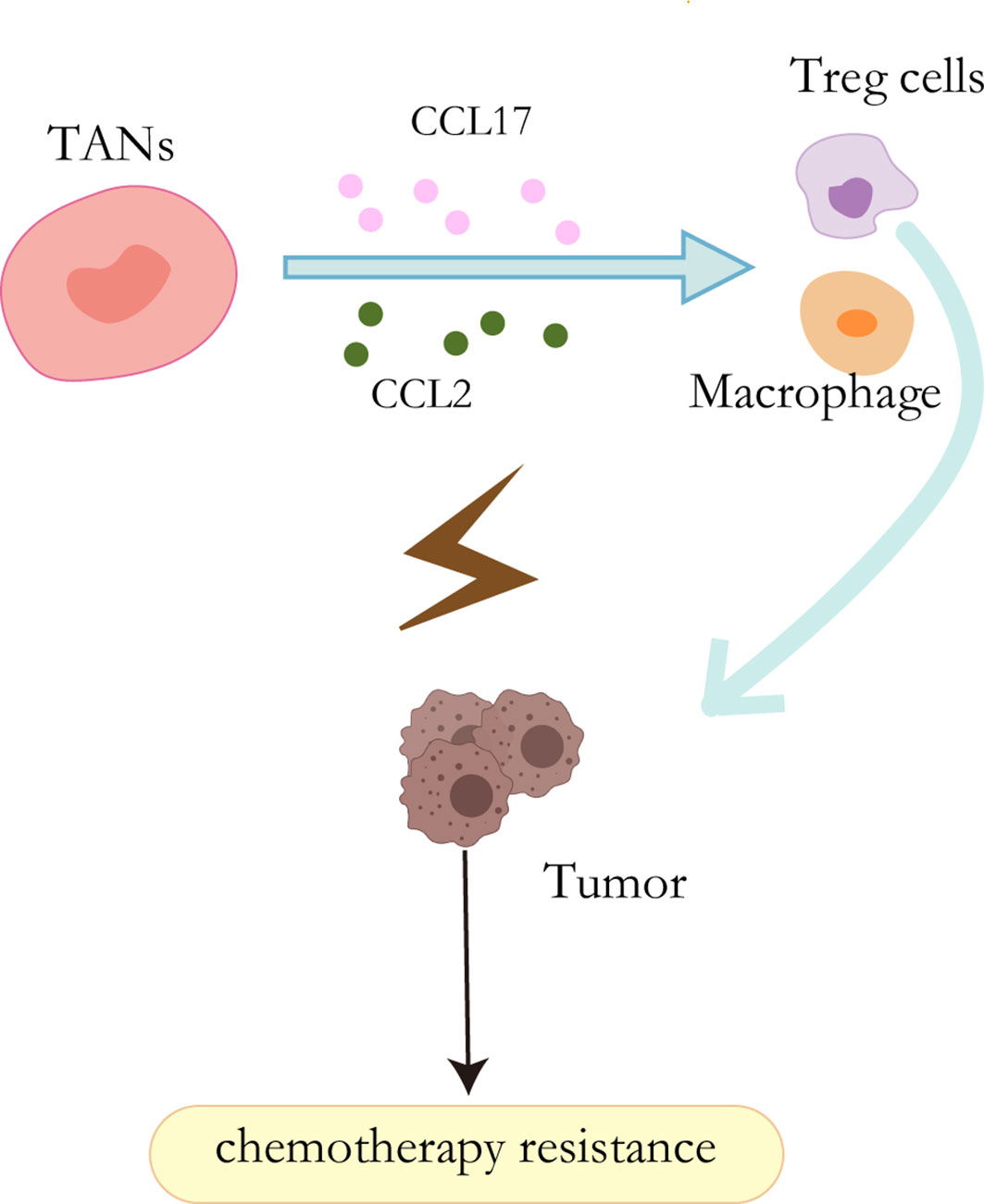
Figure 4 Resistance mechanisms of TANs. CCL2 and CCL17 secreted by TANs can mediate the intratumoral invasion of macrophages and Treg cells, thereby stimulating angiogenesis and promoting sorafenib resistance.
Extracellular matrix
Extracellular matrix (ECM), produced by stromal cells in the microenvironment, provides biochemical and mechanical clues to tumor cells and the surrounding tumor microenvironment. It is a highly dynamic structure that exists in all tissues and is constantly undergoing controlled remodeling. ECM remodeling is essential for regulating tissue morphogenesis (96). It has been demonstrated that some ECM components, directly and indirectly, interact with HCC and stromal cell types to alter the phenotype and function of HCC and stromal cells (97). These mechanical stresses and associated cellular strains could come from externally applied “outside-in” mechanical stimuli. To coordinate with outside-in stimuli, anchored cells also pull on the ECM by increasing ECM adhesions and focal adhesions, i.e. “inside-out” mechanical stimulation (98). This mutual stimulation results in a mechanically rigid microenvironment. The mechanical properties (stiffness) of the tumor cell niche affect both the differentiation of tumor cells and the characteristics of cancer stem cells. It has been shown that increased matrix hardness promotes tumor cell proliferation and chemotherapy resistance (99, 100). Therefore, ECM remodeling plays a crucial role in promoting tumor progression.
Hypoxia
Anti-angiogenic drugs can cause the tumor’s blood vessels to constrict, resulting in reduced blood flow. Inadequate blood supply affects both the effective delivery of antitumor drugs and the local concentration of oxygen and other nutrients (101). Hypoxia is a common feature of solid tumors, which has been demonstrated to be associated with chemotherapy failure and plays an important role in the selection of more aggressive and resistant clones, and poor clinical outcomes (102–104). The viability of hypoxic cells in solid tumors is increased by the adaptive response of cells to hypoxia, which is mainly controlled by hypoxia-inducible factors (HIFs) (105, 106). HIFs are transcription factors that mediate the adaptation of tumor cells to hypoxia by regulating genes involved in cell proliferation, angiogenesis, glucose metabolism, tumor invasion, and metastasis (103, 106). The HIFs factor consists of the HIF-α subunit and HIF-β subunit, where the α-subunit includes three subtypes(HIF-1α, HIF-2α, and HIF-3α) (107). Overexpression of HIF-1α and HIF-2α has been detected in nonalcoholic fatty liver disease, HCC, alcoholic liver disease, and radioactive liver injury (106). Studies have demonstrated that the multidrug resistance 1(MDR1/ABCB1) gene is hypoxia reactive and that HIF-1α can lead to the activation of the gene (108). Multidrug resistance (MDR1) gene product P-glycoprotein can reduce the intracellular concentration of sorafenib and other drugs, which is related to chemotherapy resistance (109). In addition, the stabilization of HIF1α can lead to metabolic reprogramming and enhance tumor growth (110) (Figure 3).
Conclusion and future development
The overall treatment outcome for liver cancer is far from satisfactory. The use of chemotherapeutic drugs has been hampered by drug resistance mechanisms in which the tumor microenvironment is an indispensable player. TME is a dynamic and constantly changing complex biological network whose diverse cellular and non-cellular components externally influence hallmarks and fates of tumor cells, which in part contributes to resistance to conventional therapeutic drugs. Therefore, TME would be an attractive target, both to sensitize tumors to traditional therapies and as a new option to fight the disease. Anyway, our current insights into tumor therapy indicate that the rapid elimination of therapeutic resistance in tumor cells is critical to reducing the incidence of adverse events. Although different combinations of therapies promise to achieve this goal, it is critical to find novel strategies to block primary crosstalk and reshape the microenvironment. It’s believed that in the foreseeable future, more and more preclinical and clinical research is expected to translate into novel, effective and safe clinical treatment options.
Author contributions
ZY and ZR designed the study. CZ, SL, FG, and YZ collected data and summarized the viewpoints. CZ and SL analyzed data and draw the figures. CZ wrote the manuscript. All authors reviewed and approved the manuscript.
Funding
This study was sponsored by grants from National Natural Science Foundation of China (U2004121, 82070643 and U1904164), and Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022015B).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistic. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Siegel RL, Miller KD, Jemal A. Cancer statistic. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
3. Center MM, Jemal A. International trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers Prev (2011) 20(11):2362–8. doi: 10.1158/1055-9965.EPI-11-0643
4. Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol (2014) 60(1):110–7. doi: 10.1016/j.jhep.2013.08.011
5. Sorensen HT, Friis S, Olsen JH, Thulstrup AM, Mellemkjaer L, Linet M, et al. Risk of liver and other types of cancer in patients with cirrhosis: A nationwide cohort study in Denmark. Hepatology (1998) 28(4):921–5. doi: 10.1002/hep.510280404
6. Islami F, Kamangar F, Boffetta P. Grand challenges in cancer epidemiology and prevention. Front Oncol (2011) 1:3. doi: 10.3389/fonc.2011.00003
7. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2021) 7(1):6. doi: 10.1038/s41572-020-00240-3
8. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology (2019) 156(2):477–491.e471. doi: 10.1053/j.gastro.2018.08.065
9. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
10. Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J Clin Oncol (2013) 31(32):4067–75. doi: 10.1200/JCO.2012.45.8372
11. Hernandez–Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology (2013) 144(3):512–27. doi: 10.1053/j.gastro.2013.01.002
12. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
13. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
14. Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med (2015) 13:45. doi: 10.1186/s12916-015-0278-7
15. Tahmasebi Birgani M, Carloni V. Tumor microenvironment, a paradigm in hepatocellular carcinoma progression and therapy. Int J Mol Sci (2017) 18(2):1–19. doi: 10.3390/ijms18020405
16. Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell (2019) 36(4):418–430.e416. doi: 10.1016/j.ccell.2019.08.007
17. Vaupel P, Multhoff G. Fatal alliance of hypoxia-/HIF-1alpha-Driven microenvironmental traits promoting cancer progression. Adv Exp Med Biol (2020) 1232:169–76. doi: 10.1007/978-3-030-34461-0_21
18. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov (2019) 18(2):99–115. doi: 10.1038/s41573-018-0004-1
19. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA (2010) 107(46):20009–14. doi: 10.1073/pnas.1013805107
20. Paunescu V, Bojin FM, Tatu CA, Gavriliuc OI, Rosca A, Gruia AT, et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med (2011) 15(3):635–46. doi: 10.1111/j.1582-4934.2010.01044.x
21. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell (2011) 19(2):257–72. doi: 10.1016/j.ccr.2011.01.020
22. Vicent S, Sayles LC, Vaka D, Khatri P, Gevaert O, Chen R, et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res (2012) 72(22):5744–56. doi: 10.1158/0008-5472.CAN-12-1097
23. Park D, Sahai E, Rullan A. SnapShot: Cancer-associated fibroblasts. Cell (2020) 181(2):486–486.e481. doi: 10.1016/j.cell.2020.03.013
24. Desmouliere A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: Role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest (2003) 83(12):1689–707. doi: 10.1097/01.lab.0000101911.53973.90
25. Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmoulière A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S5. doi: 10.1186/1755-1536-5-s1-s5
26. Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol (2014) 7:301–11. doi: 10.2147/CCID.S50046
27. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med (2014) 211(8):1503–23. doi: 10.1084/jem.20140692
28. Lazard D, Sastre X, Frid MG, Glukhova MA, Thiery JP, Koteliansky VE. Expression of smooth muscle-specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue. Proc Natl Acad Sci U.S.A. (1993) 90(3):999–1003. doi: 10.1073/pnas.90.3.999
29. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther (2006) 5(12):1640–6. doi: 10.4161/cbt.5.12.3354
30. Zhou B, Sun C, Li N, Shan W, Lu H, Guo L, et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int J Oncol (2016) 48(5):2087–97. doi: 10.3892/ijo.2016.3442
31. Wen X, He X, Jiao F, Wang C, Sun Y, Ren X, et al. Fibroblast activation protein-alpha-Positive fibroblasts promote gastric cancer progression and resistance to immune checkpoint blockade. Oncol Res (2017) 25(4):629–40. doi: 10.3727/096504016X14768383625385
32. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun (2018) 9(1):1–18 doi: 10.1038/s41467-018-05220-6
33. Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci USA (2018) 115(26):E5990–9. doi: 10.1073/pnas.1801348115
34. Ni Y, Zhou X, Yang J, Shi H, Li H, Zhao X, et al. The role of tumor-stroma interactions in drug resistance within tumor microenvironment. Front Cell Dev Biol (2021) 9:637675. doi: 10.3389/fcell.2021.637675
35. Zhu WW, Lu M, Wang XY, Zhou X, Gao C, Qin LX. The fuel and engine: The roles of reprogrammed metabolism in metastasis of primary liver cancer. Genes Dis (2020) 7(3):299–307. doi: 10.1016/j.gendis.2020.01.016
36. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
37. Warburg O, Wind F, Negelein E. THE METABOLISM OF TUMORS IN THE BODY. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
38. Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernandez-Perez D, Isella C, et al. Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab (2018) 28(6):848–865.e846. doi: 10.1016/j.cmet.2018.08.006
39. Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol (2020) 37(4):24. doi: 10.1007/s12032-020-01350-4
40. Firtina Karagonlar Z, Koc D, Iscan E, Erdal E, Atabey N. Elevated hepatocyte growth factor expression as an autocrine c-met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci (2016) 107(4):407–16. doi: 10.1111/cas.12891
41. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the "reverse warburg effect": a transcriptional informatics analysis with validation. Cell Cycle (2010) 9(11):2201–19. doi: 10.4161/cc.9.11.11848
42. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res (2015) 34(1):1–10. doi: 10.1186/s13046-015-0221-y
43. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
44. Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle (2011) 10(11):1772–83. doi: 10.4161/cc.10.11.15659
45. Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle (2010) 9(12):2412–22. doi: 10.4161/cc.9.12.11989
46. Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget (2016) 7(29):46750–67. doi: 10.18632/oncotarget.8775
47. Brivio S, Cadamuro M, Strazzabosco M, Fabris L. Tumor reactive stroma in cholangiocarcinoma: The fuel behind cancer aggressiveness. World J Hepatol (2017) 9(9):455–68. doi: 10.4254/wjh.v9.i9.455
48. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res (2013) 19(9):2310–8. doi: 10.1158/1078-0432.Ccr-12-2791
49. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell (2021) 39(6):883. doi: 10.1016/j.ccell.2021.05.010
50. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer (2017) 16(1):1–10. doi: 10.1186/s12943-017-0597-8
51. Orbay H, Tobita M, Mizuno H. Mesenchymal stem cells isolated from adipose and other tissues: basic biological properties and clinical applications. Stem Cells Int (2012) 2012:461718. doi: 10.1155/2012/461718
52. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8(9):726–36. doi: 10.1038/nri2395
53. Papaccio F, Paino F, Regad T, Papaccio G, Desiderio V, Tirino V. Concise review: Cancer cells, cancer stem cells, and mesenchymal stem cells: Influence in cancer development. Stem Cells Transl Med (2017) 6(12):2115–25. doi: 10.1002/sctm.17-0138
54. Shi Y, Du L, Lin L, Wang Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat Rev Drug Discov (2017) 16(1):35–52. doi: 10.1038/nrd.2016.193
55. Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene (2014) 33(21):2768–78. doi: 10.1038/onc.2013.233
56. Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int J Mol Sci (2017) 18(7):1–30. doi: 10.3390/ijms18071586
57. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
58. Nasef A, Chapel A, Mazurier C, Bouchet S, Lopez M, Mathieu N, et al. Identification of IL-10 and TGF-beta transcripts involved in the inhibition of T-lymphocyte proliferation during cell contact with human mesenchymal stem cells. Gene Expr (2007) 13(4-5):217–26. doi: 10.3727/000000006780666957
59. Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, et al. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy (2009) 5(8):1131–44. doi: 10.4161/auto.5.8.9996
60. Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee SJ, et al. TGF-{beta}1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem (2010) 285(48):37909–19. doi: 10.1074/jbc.M109.093724
61. Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy (2010) 6(5):645–7. doi: 10.4161/auto.6.5.12046
62. Han Z, Jing Y, Xia Y, Zhang S, Hou J, Meng Y, et al. Mesenchymal stem cells contribute to the chemoresistance of hepatocellular carcinoma cells in inflammatory environment by inducing autophagy. Cell Biosci (2014) 4:22. doi: 10.1186/2045-3701-4-22
63. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast–like differentiation of human mesenchymal stem cells. Cancer Res (2008) 68(11):4331–9. doi: 10.1158/0008-5472.Can-08-0943
64. Zhou J, Ding T, Pan W, Zhu LY, Li L, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer (2009) 125(7):1640–8. doi: 10.1002/ijc.24556
65. Jia J-B, Zhuang P-Y, Sun H-C, Zhang J-B, Zhang W, Zhu X-D, et al. Protein expression profiling of vascular endothelial growth factor and its receptors identifies subclasses of hepatocellular carcinoma and predicts survival. J Cancer Res Clin Oncol (2008) 135(6):847–54. doi: 10.1007/s00432-008-0521-0
66. Zhu X-D, Zhang J-B, Zhuang P-Y, Zhu H-G, Zhang W, Xiong Y-Q, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol (2008) 26(16):2707–16. doi: 10.1200/jco.2007.15.6521
67. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol (2016) 65:798–808. doi: 10.1016/j.jhep.2016.05.007
68. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med (2003) 348(17):1625–38. doi: 10.1056/NEJMoa021423
69. Ewertz M, Jensen M-B, Gunnarsdóttir K.Á., Højris I, Jakobsen EH, Nielsen D, et al. Effect of obesity on prognosis after early-stage breast cancer. J Clin Oncol (2011) 29(1):25–31. doi: 10.1200/jco.2010.29.7614
70. Lashinger LM, Rossi EL, Hursting SD. Obesity and resistance to cancer chemotherapy: Interacting roles of inflammation and metabolic dysregulation. Clin Pharmacol Ther (2014) 96(4):458–63. doi: 10.1038/clpt.2014.136
71. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest (2019) 129(8):3006–17. doi: 10.1172/JCI127201
72. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
73. Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res (2013) 73(18):5657–68. doi: 10.1158/0008-5472.CAN-13-0530
74. Motrescu ER, Rio MC. Cancer cells, adipocytes and matrix metalloproteinase 11: A vicious tumor progression cycle. Biol Chem (2008) 389(8):1037–41. doi: 10.1515/BC.2008.110
75. Muller C. Tumour-surrounding adipocytes are active players in breast cancer progression. Ann Endocrinol (Paris) (2013) 74(2):108–10. doi: 10.1016/j.ando.2013.02.007
76. Choi J, Cha YJ, Koo JS. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog Lipid Res (2018) 69:11–20. doi: 10.1016/j.plipres.2017.11.002
77. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discovery (2018) 8(8):1006–25. doi: 10.1158/2159-8290.CD-17-1371
78. Lehuede C, Li X, Dauvillier S, Vaysse C, Franchet C, Clement E, et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res (2019) 21(1):7. doi: 10.1186/s13058-018-1088-6
79. Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget (2017) 8(34):57622–41. doi: 10.18632/oncotarget.18038
80. Perez de Heredia F, Wood IS, Trayhurn P. Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflugers Arch (2010) 459(3):509–18. doi: 10.1007/s00424-009-0750-3
81. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
82. Yang E, Wang X, Gong Z, Yu M, Wu H, Zhang D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther (2020) 5(1):242. doi: 10.1038/s41392-020-00359-5
83. Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep (2015) 5:14752. doi: 10.1038/srep14752
84. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol (2016) 65(4):798–808. doi: 10.1016/j.jhep.2016.05.007
85. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33(5):657–70. doi: 10.1016/j.immuni.2010.11.011
86. Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-associated neutrophils as a new prognostic factor in cancer: a systematic review and meta-analysis. PloS One (2014) 9(6):e98259. doi: 10.1371/journal.pone.0098259
87. Uribe-Querol E, Rosales C. Neutrophils in cancer: Two sides of the same coin. J Immunol Res (2015) 2015:983698. doi: 10.1155/2015/983698
88. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol (2010) 184(6):3106–16. doi: 10.4049/jimmunol.0902661
89. Cassatella MA. Neutrophil-derived proteins: Selling cytokines by the pound. Adv Immunol (1999) 73:369–509. doi: 10.1016/s0065-2776(08)60791-9
90. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
91. Nakamura T, Sakai K, Nakamura T, Matsumoto K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol (2011) 26 Suppl 1:188–202. doi: 10.1111/j.1440-1746.2010.06549.x
92. Ogunwobi OO, Puszyk W, Dong HJ, Liu C. Epigenetic upregulation of HGF and c-met drives metastasis in hepatocellular carcinoma. PloS One (2013) 8(5):e63765. doi: 10.1371/journal.pone.0063765
93. Huang X, Gan G, Wang X, Xu T, Xie W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy (2019) 15(7):1258–79. doi: 10.1080/15548627.2019.1580105
94. Imai Y, Kubota Y, Yamamoto S, Tsuji K, Shimatani M, Shibatani N, et al. Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J Gastroenterol Hepatol (2005) 20(2):287–93. doi: 10.1111/j.1440-1746.2004.03575.x
95. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology (2016) 150(7):1646–1658.e1617. doi: 10.1053/j.gastro.2016.02.040
96. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
97. Rombouts K, Carloni V. The fibrotic microenvironment as a heterogeneity facet of hepatocellular carcinoma. Fibrogenesis Tissue Repair (2013) 6(1):17. doi: 10.1186/1755-1536-6-17
98. Provenzano PP, Keely PJ. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci (2011) 124(8):1195–1205. doi: 10.1242/jcs.067009
99. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology (2011) 54(4):1192–1205. doi: 10.1002/hep.24108
100. Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int (2014) 34(6):834–843. doi: 10.1111/liv.12465
101. Tang HW, Feng HL, Wang M, Zhu QL, Liu YQ, Jiang YX. In vivo longitudinal and multimodal imaging of hypoxia-inducible factor 1alpha and angiogenesis in breast cancer. Chin Med J (Engl) (2020) 133(2):205–11. doi: 10.1097/CM9.0000000000000616
102. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst (2007) 99(19):1441–54. doi: 10.1093/jnci/djm135
103. Wilson GK, Tennant DA, McKeating JA. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J Hepatol (2014) 61(6):1397–406. doi: 10.1016/j.jhep.2014.08.025
104. Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: A therapeutic target. World J Gastroenterol (2015) 21(42):12171–8. doi: 10.3748/wjg.v21.i42.12171
105. Powis G, Kirkpatrick L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther (2004) 3(5):647–54.
106. Ju C, Colgan SP, Eltzschig HK. Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (2016) 94(6):613–27. doi: 10.1007/s00109-016-1408-1
107. Chen C, Lou T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget (2017) 8(28):46691–703. doi: 10.18632/oncotarget.17358
108. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res (2002) 62(12):3387–94.
109. Poller B, Wagenaar E, Tang SC, Schinkel AH. Double-transduced MDCKII cells to study human p-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) interplay in drug transport across the blood–brain barrier. Mol Pharmaceutics (2011) 8(2):571–82. doi: 10.1021/mp1003898
Keywords: tumor microenvironment, reprogramming, primary liver cancer, chemotherapy resistance, hepatocellular carcinoma
Citation: Zhao C, Liu S, Gao F, Zou Y, Ren Z and Yu Z (2022) The role of tumor microenvironment reprogramming in primary liver cancer chemotherapy resistance. Front. Oncol. 12:1008902. doi: 10.3389/fonc.2022.1008902
Received: 01 August 2022; Accepted: 01 November 2022;
Published: 24 November 2022.
Edited by:
Fan Feng, The 302th Hospital of PLA, ChinaReviewed by:
Massimiliano Cadamuro, University of Padua, ItalyLindsay Jones Talbot, St. Jude Children’s Research Hospital, United States
Jose J. G. Marin, University of Salamanca, Spain
Copyright © 2022 Zhao, Liu, Gao, Zou, Ren and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang Ren, ZmNjcmVuemdAenp1LmVkdS5jbg==; Zujiang Yu, am9obnl1ZW1Aenp1LmVkdS5jbg==