 Xiangwan Miao1,2,3†
Xiangwan Miao1,2,3† Beilei Wang1,2,3†Kaili Chen1,2,3
Beilei Wang1,2,3†Kaili Chen1,2,3 Rui Ding1,2,3
Rui Ding1,2,3 Jichang Wu1,2,3Yi Pan1,2,3
Jichang Wu1,2,3Yi Pan1,2,3 Peilin Ji1,2,3
Peilin Ji1,2,3 Bin Ye1,2,3*
Bin Ye1,2,3* Mingliang Xiang1,2,3*
Mingliang Xiang1,2,3*- 1Department of Otolaryngology & Head and Neck Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Shanghai Key Laboratory of Translational Medicine on Ear and Nose Diseases, Shanghai, China
- 3Ear Institute, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Recent studies showed that lipid metabolism reprogramming contributes to tumorigenicity and malignancy by interfering energy production, membrane formation, and signal transduction in cancers. HNSCCs are highly reliant on aerobic glycolysis and glutamine metabolism. However, the mechanisms underlying lipid metabolism reprogramming in HNSCCs remains obscure. The present review summarizes and discusses the “vital” cellular signaling roles of the lipid metabolism reprogramming in HNSCCs. We also address the differences between HNSCCs regions caused by anatomical heterogeneity. We enumerate these recent findings into our current understanding of lipid metabolism reprogramming in HNSCCs and introduce the new and exciting therapeutic implications of targeting the lipid metabolism.
Introduction
Over 850,000 people are diagnosed with head and neck squamous cell carcinomas (HNSCCs) worldwide, and 440,000 people die of it (1, 2). Although human papillomavirus (HPV)-positive HNSCCs patients have better outcomes with overall survival (OS) rate of 70% (3, 4), patients with stage III–IV disease still suffer from local invasion and therapeutic failure, with a poor prognosis and OS of approximately 40% at 5 years (5). The treatment for HNSCCs is individualized, with either surgery or combined with radiotherapy, chemotherapy, target therapy or immunotherapy, as indicated by the pathological or clinical features and anatomical regions (6). Extensive tissue resection, reconstruction, and side effects of radiotherapy and chemotherapy seriously affect the life quality and survival rate of HNSCCs patients, primarily due to impaired swallowing, speaking and breathing functions (7). Because of inadequate nutrient intake, half of the HNSCCs patients are malnourished and about 80% of them lose weight during treatment (8, 9), whereas some lose up to 20% of body weight (10). After exposure to treatments, several metabolic changes occur because of wound repairing and immune response (7), accompanied with other existed metabolism reprogramming in tumors (11).
It’s well known that HNSCCs are highly reliant on glucose metabolism, known as Warburg effect (12, 13). However, nutritional limitation of the total calorie intake in HNSCCs patients promote cancer cell proliferation (14, 15), indicating that not only glucose metabolism, but also other metabolic processes, such as glutamine and lipid metabolism, are vital. As a newly discovered cancer characteristic (16), studies have found that lipid metabolism is reprogrammed in cancers, too (17). Lipid metabolism could support survival, proliferation, invasion, and metastasis in cancer cells by contributing to membrane formation, energy production and signal transduction, and even mediate drug resistance (18, 19). Due to the rapid proliferation rates and high metabolic energy requirements, cancer cells have tremendous demand of lipids (20, 21). Moreover, a variety of intermediate substrates produced by glucose and glutamine metabolism could participate in lipid metabolism, forming a “shortcut” cycling (22). Thus, lipid metabolism reprogramming plays a “vital” role in HNSCCs.
However, it should be noted that the anatomical HNSCCs regions, especially in the neck and supraclavicular regions, mainly contain brown adipose tissue and beige adipose cells (23), which promote energy consumption and help improve the glucose and lipid metabolic disorders (24). And this could partially explain the marked heterogeneity among different head and neck regions (25), especially nasopharyngeal carcinoma (NPC). In NPC, the most common manifestation is cervical lymph node metastasis, which is riches in brown adipose tissue (23). Distant metastasis occurs in about 20% of NPC cases, and half of them are bone/bone marrow metastasis (26), a region where adipocytes predominate (27). Latest studies found that activated brown adipose tissue can reduce glucose around cancers and inhibit cancer growth (28). Because of these differences in adipose tissue distribution, the mechanisms underlying lipid metabolism in different HNSCCs, and other solid carcinomas may vary.
Up to date, most studies were working on the key enzymes involved in lipid uptake and synthesis in HNSCCs, and the upregulation of these enzymes indicates the therapeutic potentials of lipid uptake and synthesis inhibitors in HNSCCs. In this review, we summarize the current studies working on lipid metabolism enzymes and signal transduction molecules and introduce the advancements for lipid metabolism disruption in HNSCCs. Lipids are composed of fat (triglyceride, TG) and lipoid (phospholipid, cholesterol, and cholesterol esters) and both are involved in lipid uptake, synthesis, storage, and catabolism (20). Thus, this review introduces the reprogramming of lipid metabolism in HNSCCs by FA and cholesterol, which are the main substrate for fat and lipoid.
Lipid uptake in HNSCCs
Cholesterol uptake
Cholesterol, which plays a crucial role in membrane structure, is absorbed by intestinal enterocytes (29) and used to synthesize very low-density lipoprotein (VLDL) in the liver (30). VLDL is released into the blood and processed into low-density lipoprotein (LDL), which is taken up by low-density lipoprotein receptors (LDLR) on peripheral cells (31). Nicotine in tobacco can induce an increase in LDLR expression in oral epithelial cells, while smoking is an important risk factor for HNSCCS (32). But the blood cholesterol and LDL levels are significantly decreased in oral carcinoma patients (33, 34). These results indicate that HNSCCs require more cholesterol and LDL than normal cells. Daker et al. also found that Epstein-Barr virus encoded RNA (EBERs) up-regulated LDLR and FA synthase (FASN) in NPC cells (35). Besides, experiments on head and neck cancer (HNC) cell lines revealed that the expression of CD36 and LOX-1, another two LDL membrane receptors, were significantly upregulated after exposure to oxidized LDL (oxLDL) (36), which also suggested that the uptake of cholesterol increased in HNSCCSs. However, lipid metabolism varies according to different tumor microenvironment (TME) and progression stages (37, 38). When oxLDL upregulated CD36 in HNC cell lines, the migration of cancer cells were reduced after oxLDL exposure (36). Thus, the regulation of LDL receptors needs further exploration in order to guide the administration of cholesterol uptake inhibitor. The mechanisms underlying cholesterol efflux proteins, such as LXR or ABCA1, in HNSCCs are still lacking, which worth more attention since they affect the total concentration of cholesterol inside the cells, too.
FA uptake
FA is another essential molecule involved in lipid biosynthesis and serves as a substrate for energy production metabolism. Mammals produce only a few endogenous FAs, which carry a double bond at δ 9 in the hydrocarbon chain. Other necessary FAs, especially polyunsaturated FAs, need to be obtained from food (20, 39). FA are taken up by simple diffusion through the lipid bilayer or by FA transporters on the membrane (22). The currently known FA transporters include differentiated cluster 36 (CD36, also known as FA translocation enzyme), FA transporter family (FATPs, also known as SLC27), and FA binding proteins on the plasma membrane (also known as FABPs). Abnormal elevation of these three proteins occurs in a variety of cancers (20, 40). Among them, CD36 has been studied most comprehensively in HNSCCs. In oral squamous cell carcinoma (SCC), CD36 upregulation promotes tumor metastasis, while its inhibition leads to complete remission or elimination of lymph node and lung metastases in in vivo oral carcinoma models (40). These findings suggest the therapeutical use of CD36 inhibitors in advanced HNSCCs patients. What’s more, CD36 inhibitors could reduce the growth of oral SCC cells and inhibited lipid droplet (LD) formation, tumor progression, and metastasis (41–43). And it is important to know that our daily dietary intake may affect the expression of CD36 as Pascual et al. found that dietary palmitic acid (PA) activated CD36 in oropharyngeal carcinoma and stimulated metastasis of cancer cells, which was promoted by a specialized proregenerative extracellular matrix secreted from cancer-associated Schwann cells (44). Thus, nutritional interventions should be considered together with lipid metabolism inhibitors for cancer treatment. Similar to CD36, Rauch et al. found that FABP protein expression was significantly increased in HNSCCs compared to normal tissues (45). Then, Ohyama et al. further found abnormal expressions of FABP4 and FABP5 in tongue carcinoma, whereas only FABP5 was expressed in normal tongue epithelial cells, which showed a higher expression level in injured and cancer tissues (46). Although few studies have evaluated the role of FA transporter family in HNSCCs, these studies revealed that HNSCCs require more FAs than normal cells. However, the killing efficiency of the FA uptake inhibitors should be researched more specifically, along with the optimal duration of use, usefulness and efficiency of nutritional interventions, and long-term side effects.
Lipid synthesis and storage in HNSCCs
Citric acid, produced by the citric acid cycle or glutamine metabolism, is the starting molecule involved in intracellular lipid synthesis. ATP-citric acid lyase (ACLY) converts citric acid to acetyl-CoA and oxaloacetate, which are used to synthesize different lipids in the cells (Figure 1). Although there is no direct evidence of ACLY expression in HNSCCs, Zheng et al. found that the long non-coding RNA TINCR could bind to ACLY and protect it from degradation in NPC, which maintained the total acetyl-CoA level in cells (47). In addition, Sur et al. reported that bitter gourd extract could significantly reduce the expression of ACLY, acetyl-CoA carboxylase (ACC), and FASN genes in oral carcinoma, and promote cell apoptosis (48). These results suggest that the ACLY expression is increased in HNSCCs, which may contribute to the survival of cancer cells and ACLY inhibition may be used as a new anticancer treatment in HNSCCs.
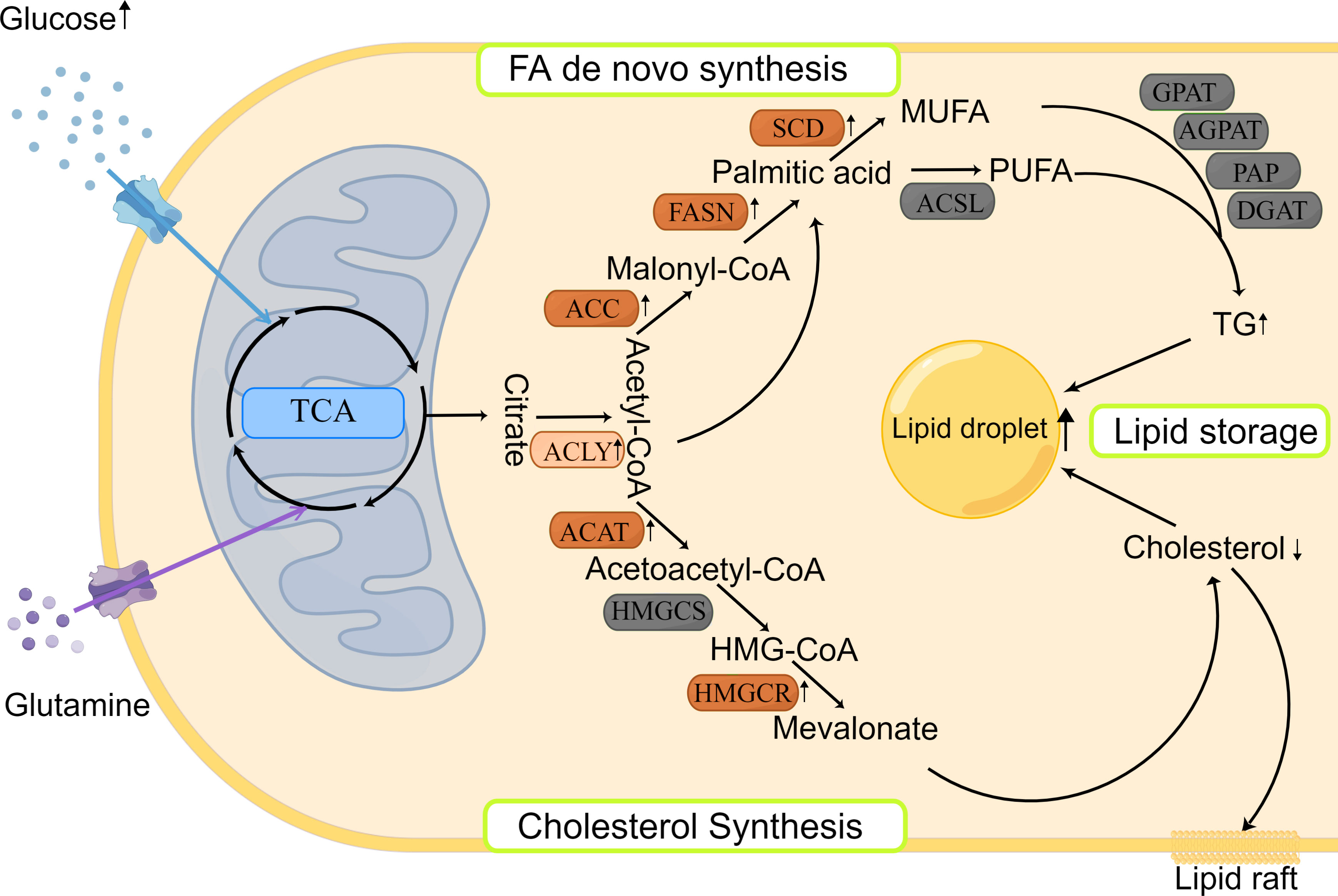
Figure 1 Lipid synthesis and storage in HNSCCs. Lipid synthesis begins with citric acid, produced from the TCA cycle, which is used to synthesize different lipids in the cytoplasm. There are two main pathways involved: FA de novo synthesis and cholesterol synthesis. The produced lipids are stored as LDs. Most enzymes involved in lipid synthesis are upregulated in HNSCCs.
Cholesterol synthesis
Cholesterol biosynthesis begins with the conversion of two molecules of acetyl-CoA to acetoacetyl-CoA by acetyl-CoA acetyltransferase (ACAT). Subsequently, a third acetyl-CoA molecule is synthesized into HMG-CoA by HMG-CoA synthase (HMGCS). HMG-COA reductase (HMGCR) is the next rate-limiting step in cholesterol synthesis and produces mevalonate (Figure 1). Mevalonate can be modified to produce different cholesterols with various physiological functions, such as lipid raft in cell membrane (29). Using genetic variation assessment, Gormley et al. reported that there was limited evidence regarding LDL reduction by HMGCR, Niemann-Pick type C1-like 1 (NPC1L1), CETP, or other circulating lipid trait genes on the risk of oral or oropharyngeal carcinoma (49). However, ACAT1 was reported to be associated with poor prognosis of oral SCC (50). This may be explained by the lack of consideration of cholesterol efflux in the previous study, which affects the total quantity of cholesterol inside the cancer cells. Although previous findings related to cholesterol synthesis are controversial and there are limited reports about the expression and prognostic role of cholesterol synthesis-related enzymes in HNSCCs, statins, which are the cholesterol-lowering drugs that act by HMGCR inhibition (51), could induce apoptosis of cancer cells by consuming non-steroidal mevalanoic acid metabolites in HNSCCs (52). Furthermore, statins could enhance the effects of cisplatin with concomitant use and potentiate the efficacy of immunotherapy in HNSCCs (53). These results highlight the potential therapeutic use of statins in HNSCCs, which should be further studied to clarify the mechanisms behind.
FA de novo synthesis
FA de novo synthesis begins with the conversion of acetyl-CoA to malonyl-CoA by ACC. Then, acetyl-CoA and malonyl-CoA are catalyzed by FASN to form palmitate, which is further modified by elongase of very long chain fatty acids (ELOVL) enzymes to elongate the length of FA chains. Finally, polysaturated FAs, such as palmitic acid, are desaturated to produce unsaturated FAs by stearoyl-CoA desaturase (SCD) and/or other fatty acyl-CoA desaturases (Figure 1). The expression of various rate-limiting enzymes involved in FA de novo synthesis was increased in HNSCCs. In HNSCCs with lymph node metastasis, highly phosphorylated ACC expression was found to be associated with poor survival outcomes (54). And ACC2 serves as a vital prognostic indicator and potential therapeutic target in HNSCCs (55). As another key rate-limiting enzyme in FA synthesis, FASN expression was found to be increased in HNSCCs, too. Epstein-Barr virus could promote FASN expression in NPC cells (35, 56) and FASN transcription was increased in cisplatin-resistant SCCs and played a role in cisplatin resistance (57). Furthermore, FASN siRNA inhibited the growth of in vivo oral SCC and lymph node metastasis (58), and FASN inhibitors increased the sensitivity to radiotherapy (59). The aforementioned results suggest that inhibition of FA synthesis would be a novel and exciting treatment for HNSCCs. Clinical trials evaluating the efficiency of the FASN inhibitors are currently ongoing on variety of cancers, including oral cancers (NCT02223247) (www.clinicaltrails.gov). Besides, SCD inhibitors could hinder cancer cell proliferation and invasion in oral carcinoma (60, 61), but need more in-depth and long-term studies.
Total lipid synthesis and storage
After synthesis, FAs bind to different backbones to produce different classes of fat in the body, such as phospholipids and TGs with glycerol is the most common backbone, except phospholipids. FAs produce TGs through several enzymes, including Gly3P phosphate acyltransferase (GPAT), 1-acyl-sn-Gly3P acyltransferase (AGPAT), PA phosphatase (PAP), and DAG acyltransferase (DGAT). TGs are then encapsulated in LDs, which is the main storage form of lipids (Figure 1). LD accumulation serves as a phenotype for metastasis initiation, energy storage, and regulatory mechanism of reactive oxygen species in carcinomas (62). HNSCCs show increased LD accumulation, too (63, 64). However, the distribution and mechanism of key enzymes and molecules, such as lipins, in HNSCCs have not been reported previously, and merit further exploration.
Lipid catabolism in HNSCCs
Lipolysis
In response to the requirements of rapid growth and invasion, intracellular lipolytic enzyme activity is also increased (65). In mitochondria, long chain FAs are transformed into acetyl-CoA through lipid catabolism (20), thereby providing ATP and substrates for lipid synthesis (66). The initial step of lipolysis is the hydrolysis of TG into diacylglycerol (DAG) by lipases. Two main lipases are involved in this process, namely, hormone-sensitive lipase (HSL) and fatty triglyceride lipase (ATGL, also known as phospholipase A2, PNPLA2, or PLA2). Rather than TG, HSL hydrolyzes DAG to monoacylglycerol (MAG), while ATGL almost completely hydrolyzes TGs to release DAG (67, 68). DAG is derived from TGs via ATGL, and DAG is hydrolyzed by HSL to 2-MAG. Then, 2-MAG is hydrolyzed by MAG lipase (MGL) to free FAs and glycerol, which is then secreted extracellularly (22) (Figure 2).
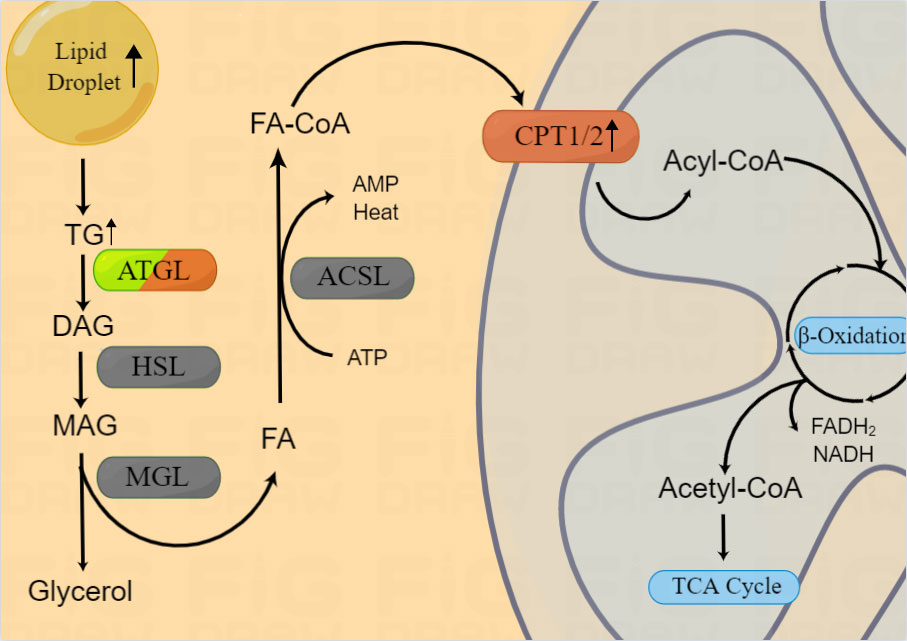
Figure 2 Lipid catabolism in HNSCCs. Enzymes involved in lipid catabolism are shown in the figure. After release from the LD, triacylglycerol is broken into FA-CoA by lipolysis-related enzymes and catabolized by FAO to produce energy and substrates for the mitochondrial TCA cycle. β-oxidization are reported to be upregulated in HNSCCs, however, the expression of ATGL in HNSCCs is still controversial.
In 2012, Tripathi et al. found that, along with the Warburg effect, the phosphatidylcholin/lysophosphatidylcholine and phosphatidylcholine/glycerophosphatidylcholine ratios were significantly increased and the activity of ATGL in HNSCCs (oral, tongue, and larynx) was enhanced (69). However, Zhou et al. found that in NPC, ATGL expression was inhibited, lipolysis was reduced, and LD accumulation was increased (63). In addition, they found that low ATGL expression was associated with poor prognosis of patients and ATGL inhibition was regulated by Epstein-Barr virus-encoded membrane latent protein 2A (LMP2A). LMP2A not only promoted lipid accumulation by inhibiting ATGL, but also enhanced migration in vitro (64). Thus, the expression and mechanisms of ATGL varied according to the anatomical regions of HNSCCs. As we mentioned above, this gene heterogeneity may be related with the different adipose tissue distribution and lipid metabolism in HNSCCs.
FA catabolism
In addition to being a metabolic intermediate in lipid anabolism, FAs are an important energy source. FAs are catabolized by fatty acid oxidation (FAO), also known as β oxidation. FA-CoA was transformed into FA-carnitine by carnitine palmityl transferase (CPT) and transported from the cytosol across the outer mitochondrial membrane. Within the mitochondria, FAs are repeatedly cleaved to produce acetyl-CoA, which is recycled into the citric acid cycle to produce the reductive equivalent of oxidative phosphorylation (Figure 2). Du et al. found that increased CPT1A-mediated FAO was significantly associated with radiotherapy resistance in NPC (70), suggesting the potential use of combination treatment of FAO inhibitors and radiotherapy. However, the mechanism underlying the role of CPT1/2 in other HNSCCs as well as its other functions require further evaluation.
Regulation pathways of lipid metabolism reprogramming in HNSCCs
In addition to the key lipid metabolic steps mentioned previously, there are also many important signaling pathways involved in lipid metabolism regulation, such as PI3K/AKT, mTOR, and AMPK pathways, which have been discussed previously and are not included in this review (22, 71). In addition to the above pathways, there is a star lipid regulation pathway, which is involved in the regulation of synthesis of multiple lipids, namely, INSIG/SCAP/SREBPs, and requires further attention. The INSIG/SCAP/SREBPs complex is located on the endoplasmic reticulum but does not have any regulatory activity. After cholesterol or glucose stimulates INSIG, the SCAP/SREBPs complex is transported to the Golgi and cleaved into the activated form. Then, SREBP-1c is released into the cell nucleus and regulates the downstream genes as a transcriptional factor. SREBPs has three main forms, namely, SREBP1a, SREBP1c, and SREBP2. SREBP1 mainly regulates the expression of FA synthesis genes and LDLR, while SREBP2 preferentially regulates the expression of cholesterol biosynthesis genes (20). In NPC, SREBP1 activation mediated lipid synthesis and promoted tumor proliferation and progression (72). However, the distributions, expression levels, and specific mechanisms of INSIG/SCAP/SREBPs in different HNSCCs are still unclear.
Effects of high-risk factors on lipid metabolism reprogramming in HNSCCs tumor microenvironment
Tobacco and alcohol
Compared with non-smoker who never drank, those who drank and smoked every day had a 14-fold higher risk for head and neck squamous cell carcinoma (73). Alcohol consumption alone increases the risk for head and neck squamous cell carcinoma (74, 75). Ethanol is oxidized into acetaldehyde after absorption, which forms various proteins and DNA adducts that promote DNA repair failure, lipid peroxidation and metabolism (76). In HNSCCs, there is a significant positive dose-response relationship between prediagnosis alcohol intake and worse OS, especially associated with the fast ADH1B and the slow/nonfunctional ALDH2 genotype combination (77), two dehydrogenase for alcohol and aldehyde. Chronic alcohol exposure decreases the DNA binding ability of PPARα, a nuclear hormone receptor involved in mitochondrial β-oxidation regulation (78, 79), and impairs cholesterol synthesis (80, 81), which may promote cancer progression, and may also occur in head and neck epithelial cells. Another risk factor that HNSCCs patients are frequently exposed to is tobacco. Difference in lipidome signatures can be found between smokers and non-smokers across a number of lipid species (82, 83). Compared with unexposed, active or passive smokers have higher LDL (84–87) and lower HDL (88). Nicotine in tobacco can induce up-regulation of LDLR expression in oral epithelial cells (32). However, the serum levels of total lipids, cholesterol and HDL in patients with oral cancer are significantly reduced, while triglycerides and VLDL are increased (33, 34). Above results support that lipid metabolism reprogramming has a significant relationship with HNSCCs development, although the specific mechanisms of alcohol and tobacco regulation is still unclear.
Virus infection
As a part of the upper aerodigestive tract, HNSCCs are often affected by viral or bacterial microbes, such as HPV and EBV. Viruses require lipid-mediated endocytosis to enter the cell and HPV proteins L1 and L2 could activate lipid-raft mediated endocytosis to increase its infection (89). The HPV16 E5 protein even can change the lipid composition in cells to help establishing an immune suppressed TME that favors HPV long-term infection (90). HPV16 E6 and E7 could up-regulate lipid synthesis by activating PI3K/AKT/mTOR (91) and SREBPs lipid synthesis signaling pathways (92–94). HPV-positive HNSCCs patients had higher levels of gene expression in TCA cycle, oxidative phosphorylation and β-oxidation, compared with HPV-negative patients (95). However, due to the different adipose tissue distribution, the lipid metabolism regulated by HPV may also varied in different cancers. For example, although HPV is involved in the regulation of cell metabolism in both cervical cancer and HNSCCs, its functions varied. In HPV-associated HNSCCs, it mainly promotes oxidative phosphorylation to obtain energy (96, 97), while in cervical cancer, HPV E6 protein up-regulates lipolysis and down-regulates oxidative phosphorylation (98).
Another well-known virus risk factor in HNSCCs is Epstein-Barr virus, which is also involved in lipid metabolism reprogramming in HNSCCs TME. EBV encoded LMP1 has been reported to regulate glycolysis and lipogenesis in NPC (56, 99, 100). EBV-mediated reprogramming of lipid biosynthesis promotes B-cell activation and differentiation surrounding TME (101, 102), which help shaping a tumor favored TME. At the same time, EBV can also release inflammatory factors, such as IL6, IL-10 and leptin, which promote fat consumption (103–105) and help cancer cells to evade immune surveillance (106). Therefore, virus associated HNSCCs show differences in lipid metabolism compared with non-infectious HNSCCs, which worth more study in the future.
Dietary interventions
Dietary interventions alter the metabolic substrates concentrations in the TME, which will reprogram the cancer cell metabolism and induce cancer development and progression (107–111). Caloric restriction inhibits the growth of pancreatic cancers and helps limit cancer progression (112). However, the total calory intake restriction intervention does not improve the survival prognosis in HNSCCs, but improves the cancer cells proliferation (14, 15). Whether a hypoglycemic diet will inhibit cancer growth may be determined by the mismatch between the fatty acid desaturation degree and the available specific fatty acid types in the cancer (112). Therefore, the role of lipid metabolism in HNSCCs deserves further investigation.
Ferroptosis is an iron-mediated lipid peroxidation that causes non-apoptotic cell death, which is associated with cancer development and therapy response. Inhibition of GPX4, an important ferroptosis regulation molecule, can sensitize drug-resistant cancer cells in HNSCCs (113). During ferroptosis, polyunsaturated fatty acids (PUFAs) are most susceptible to peroxidation, which can cause the destruction of the lipid bilayer and affect membrane function (114). In oral cancer, glutathione can regulate lipid oxidation by binding to PTGS2 which promotes ferroptosis (115). What’s more, high fat-soluble vitamins, such as Vitamin D is associated with lower risk of cancer (116). Thus essential nutrients such as glutathione (GSH), fat-soluble vitamins A, D and K, which help remove ROS (98) and regulate lipid peroxidation and ferroptosis (117), have potential anticancer application in HNSCCs by promoting ferroptosis.
In addition to essential nutrients, there are many exogenous lipid nutrients with potential cancer killing effects in HNSCCs. Reports have shown that docosahexaenoic acid, a ω-3 fatty acid, can induce the degradation of HPV E6/E7 oncoprotein and promote apoptosis (118). Ergosterol Peroxide extracted from mushroom can increase radiotherapy sensitization in cervical cancer cells (119). Salvianolic acid B extracted from salvia miltiorrhiza, which could can also inhibit the malignant transformation of oral premalignant lesion (120), which has been reported had the protective effect on metabolic homeostasis by regulating PPARγ, FASN, SCD1 and CD36 (121). These results suggest that exogenous unsaturated fatty acids and lipid nutrients extracted from plants may have therapeutical potential in HNSCCs.
Regulation of lipid metabolism in HNSCCs by cancer associated cells in TME
Apart from cancer cells, cancer-associated cells in the TME also play an important role in the occurrence and development in cancers. Among them, the one that has been studied the most is cancer-associated fibroblasts (CAFs), which can be derived from normal fibroblasts around cancers, mesenchymal stem cells and cancer cells undergoing EMT transformation (27). It interacts with cancer cells and other components in the TME which help forming a tumor-supporting TME (122–124). HPV-negative oropharyngeal cancer cells can stimulate normal fibroblasts to produce HGF and IL-6 (125), and senescent CAFs will secrete more IL-6, COX2 and PGE2 (126). And then, IL-6 further promotes cancer cell invasion, lipid depletion and immunosuppression (103–106). Although there is evidence that CAFs in HNSCCs exhibit similar metabolic characteristics with cancer cells (127, 128), studies on lipid metabolism in CAFs are still lacking. What’s more, Pascual et al. found that dietary PA could induce the cancer associated Schwann cells to secrete a specialized extracellular matrix to promote metastasis (44). All these results support that cancer associated cells help reprogramming the lipid metabolism in HNSCCs TME.
Adipocytes also can differentiate into CAFs (27). Cancer cells could regulate lipid metabolism of adipocytes to produce cancer-associated adipocytes, which are morphologically and functionally different from normal adipocytes (129). Cancer-associated adipocytes then further release fatty acids, mitogens and proinflammatory adipokines to promote the occurrence and development of cancers (130–134). Adipokines such as leptin and adiponectin are lower in HNSCCs patients (135–138), whereas visfatin and chemerinze are higher (28, 139).However, adipocytes distribution in different regions of HNSCCs varies, which may be the reason for the distinct metabolism CAFs subtypes in HNSCCs (128, 140). All these evidences support that cancer associated adipocytes and CAFs play a vital role in lipid metabolism in HNSCCs, but needs more exploration.
Summary and future prospects
The mask of lipid metabolic reprogramming in HNSCCs is gradually being revealed. Previous studies reported that a variety of lipid metabolic enzymes are upregulated in HNSCCs, but heterogeneous was also existed according to different TME and anatomical regions. Cancer cells are constantly reprogramming their lipid metabolisms in response to the TME and/or metastasis/colonization needs in HNSCCs. In this article, we summarized the previous research on lipid metabolism reprogramming in HNSCCs. However, as shown in Table 1, only few lipid metabolism enzymes have been researched and there are still a lot of vacancy in this area which need further exploration in the future. Importantly, HNSCCs comprise a diverse group of cancers that affect the upper aerodigestive tract. The differences in reprogramming of lipid metabolism under different TMEs in HNSCCs require additional studies. Lipid metabolism reprogramming not only shows extensive interaction with other metabolic mechanisms, but also has various crosstalk with surrounding cells, cytokines, growth factors, and even nutrient molecules within the malignant cancer cells. Therefore, the role of lipid metabolism reprogramming in HNSCCs needs additional studies, including, but not limited to, its effects on the immune microenvironment and angiogenesis. Further understanding of the lipid metabolism reprogramming mechanisms, key rate-limiting enzyme functions, and regulatory pathways in HNSCCs may help to develop the potential use of lipid metabolism pathways as targets for anti-tumor therapy, as well as the use of dietary/nutritional interventions to improve the prognosis and life quality of HNSCCs patients.
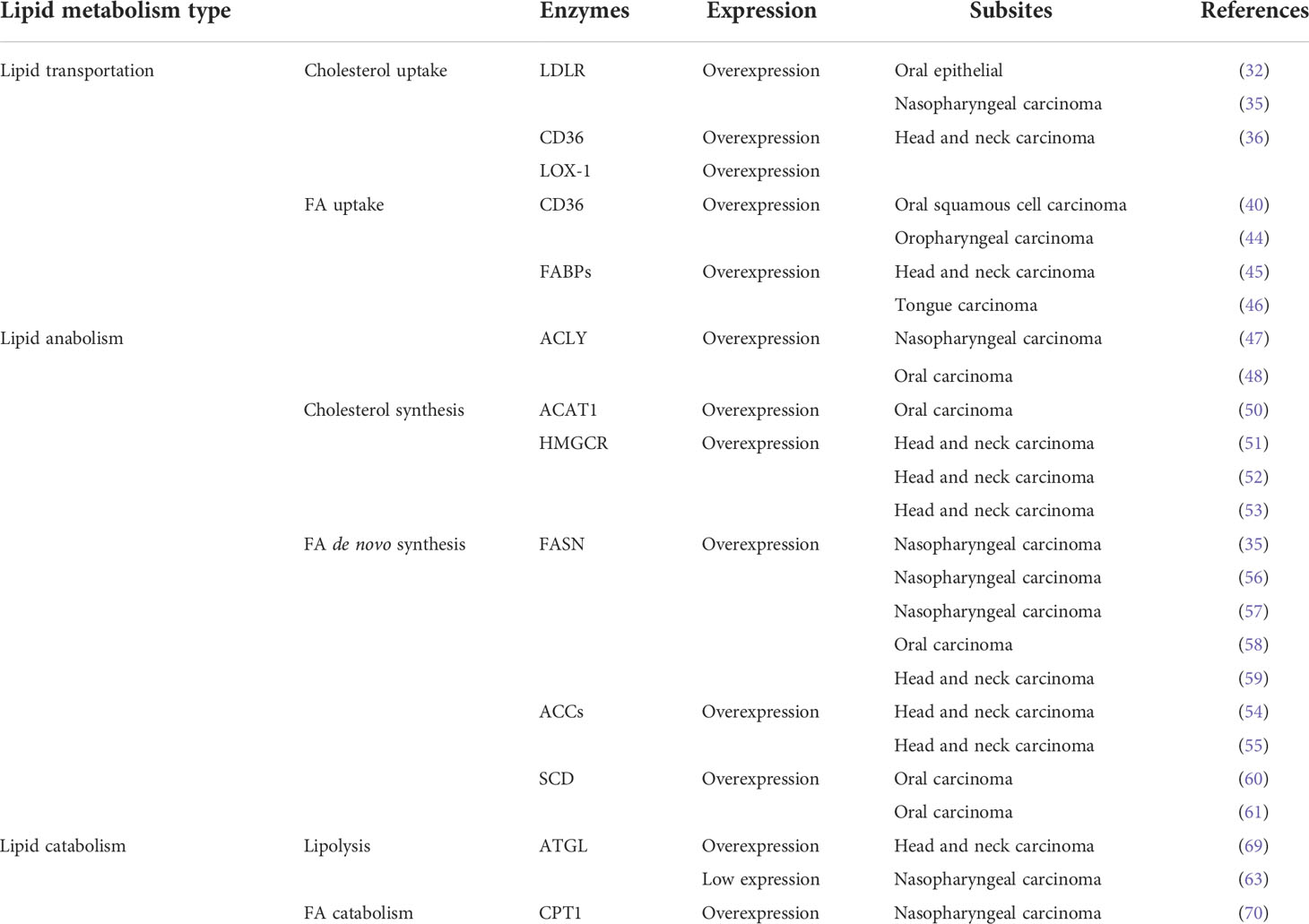
Table 1 Expression of key lipid metabolism enzymes in HNSCCs.
Author contributions
Conceptualization, XM, BY, and MX; methodology, YP; software, KC and PJ; validation, XM, BW, and BY; investigation, RD; resources, JW; data curation, XM and BW; writing—original draft preparation, XM; writing—review and editing, BY; visualization, BW; supervision, MX; project administration, MX; funding acquisition, XM, BY, and MX. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by grants from the Cultivation Project of the Major Research Plan of the National Natural Science Foundation of China (grant No. 91949119), National Natural Science Foundation of China (No.82101209 and No.82101212), Science and Technology Commission of Shanghai Municipality (grant No.21ZR1440200), the Shanghai Sailing Program (20YF1426400, 19YF1430300), and Ruijin Youth NSFC Cultivation Fund.
Acknowledgments
Figures were generated by using Figdraw (www.figdraw.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. New Engl J Med (2010) 363:24–35. doi: 10.1056/NEJMoa0912217
4. Nguyen-Tan PF, Zhang Q, Ang KK, Weber RS, Rosenthal DI, Soulieres D, et al. Randomized phase III trial to test accelerated versus standard fractionation in combination with concurrent cisplatin for head and neck carcinomas in the radiation therapy oncology group 0129 trial: long-term report of efficacy and toxicity. J Clin oncol: Off J Am Soc Clin Oncol (2014) 32:3858–66. doi: 10.1200/JCO.2014.55.3925
5. Cohen N, Fedewa S, Chen AY. Epidemiology and demographics of the head and neck cancer population. Oral Maxillofac Surg Clinics North America (2018) 30:381–95. doi: 10.1016/j.coms.2018.06.001
6. David G, Pfister SS, Douglas A, Birkeland A, Brizel D, Busse P, et al. National comprehensive cancer network. head and neck cancers version 2. (2022). p. ed2022. Available at: http://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf
7. Muller-Richter U, Betz C, Hartmann S, Brands RC. Nutrition management for head and neck cancer patients improves clinical outcome and survival. Nutr Res (2017) 48:1–8. doi: 10.1016/j.nutres.2017.08.007
8. Orell-Kotikangas H, Osterlund P, Makitie O, Saarilahti K, Ravasco P, Schwab U, et al. Cachexia at diagnosis is associated with poor survival in head and neck cancer patients. Acta Otolaryngol (2017) 137:778–85. doi: 10.1080/00016489.2016.1277263
9. Gorenc M, Kozjek NR, Strojan P. Malnutrition and cachexia in patients with head and neck cancer treated with (chemo)radiotherapy. Rep Pract Oncol radiother: J Greatpoland Cancer Center Poznan Polish Soc Radiat Oncol (2015) 20:249–58. doi: 10.1016/j.rpor.2015.03.001
10. Ehrsson YT, Langius-Eklof A, Laurell G. Nutritional surveillance and weight loss in head and neck cancer patients. Supportive Care cancer: Off J Multinational Assoc Supportive Care Cancer (2012) 20:757–65. doi: 10.1007/s00520-011-1146-4
11. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Sci (2020) 368(6487):1–10. doi: 10.1126/science.aaw5473.
12. Bozzetti F, Stanga Z. Does nutrition for cancer patients feed the tumour? a clinical perspective. Crit Rev Oncology/Hematol (2020) 153:103061. doi: 10.1016/j.critrevonc.2020.103061
13. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys (2001) 51:349–53. doi: 10.1016/S0360-3016(01)01630-3
14. Edstrom S, Westin T, Delle U, Lundholm K. Cell cycle distribution and ornithine decarboxylase activity in head and neck cancer in response to enteral nutrition. Eur J Cancer Clin Oncol (1989) 25:227–32. doi: 10.1016/0277-5379(89)90013-8
15. Baron PL, Lawrence W Jr., Chan WM, White FK, Banks WL Jr. Effects of parenteral nutrition on cell cycle kinetics of head and neck cancer. Arch Surg (1986) 121:1282–6. doi: 10.1001/archsurg.1986.01400110072012
16. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
17. Ocana MC, Martinez-Poveda B, Quesada AR, Medina MA. Glucose favors lipid anabolic metabolism in the invasive breast cancer cell line MDA-MB-231. Biol (2020) 9(1):1–12. doi: 10.3390/biology9010016
18. Chen X, Chen S, Yu D. Metabolic reprogramming of chemoresistant cancer cells and the potential significance of metabolic regulation in the reversal of cancer chemoresistance. Metabolites (2020) 10(7):1–15. doi: 10.3390/metabo10070289
19. Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P. Lipid metabolism and resistance to anticancer treatment. Biol (2020) 9(12):1–21. doi: 10.3390/biology9120474
20. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med (2021) 218(1):1–17. doi: 10.1084/jem.20201606
21. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer (2016) 16:732–49. doi: 10.1038/nrc.2016.89
22. Prentki M, Madiraju SR. Glycerolipid metabolism and signaling in health and disease. Endocr Rev (2008) 29:647–76. doi: 10.1210/er.2008-0007
23. Zhang F, Hao G, Shao M, Nham K, An Y, Wang Q, et al. An adipose tissue atlas: An image-guided identification of human-like BAT and beige depots in rodents. Cell Metab (2018) 27:252–62.e3. doi: 10.1016/j.cmet.2017.12.004
24. White JD, Dewal RS, Stanford KI. The beneficial effects of brown adipose tissue transplantation. Mol Aspects Med (2019) 68:74–81. doi: 10.1016/j.mam.2019.06.004
26. Bensouda Y, Kaikani W, Ahbeddou N, Rahhali R, Jabri M, Mrabti H, et al. Treatment for metastatic nasopharyngeal carcinoma. Eur Ann otorhinolaryngol Head Neck Diseases (2011) 128:79–85. doi: 10.1016/j.anorl.2010.10.003
27. Liu SC, Tsang NM, Lee PJ, Sui YH, Huang CH, Liu TT. Epstein-Barr Virus induces adipocyte dedifferentiation to modulate the tumor microenvironment. Cancer Res (2021) 81:3283–94. doi: 10.1158/0008-5472.CAN-20-3121
28. Wang N, Wang QJ, Feng YY, Shang W, Cai M. Overexpression of chemerin was associated with tumor angiogenesis and poor clinical outcome in squamous cell carcinoma of the oral tongue. Clin Oral Investigations (2014) 18:997–1004. doi: 10.1007/s00784-013-1046-8
29. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell (2021) 56:1363–93. doi: 10.1016/j.devcel.2021.04.013
30. Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol hepatol (2020) 17:169–83. doi: 10.1038/s41575-019-0250-7
31. Goldstein JL, Brown MS. The LDL receptor. Arteriosclerosis thrombosis Vasc Biol (2009) 29:431–8. doi: 10.1161/ATVBAHA.108.179564
32. Ito S, Gojoubori T, Tsunoda K, Yamaguchi Y, Asano M, Goke E, et al. Nicotine-induced expression of low-density lipoprotein receptor in oral epithelial cells. PloS One (2013) 8:e82563. doi: 10.1371/journal.pone.0082563
33. Srinivas GV, Namala S, Ananthaneni A, Puneeth HK, Devi BS. Evaluation and correlation of serum lipid profile in oral and gastrointestinal cancer patients. J Int Oral Health: JIOH (2013) 5:72–7. doi: 10.1084/jem.20201606
34. Chawda JG, Jain SS, Patel HR, Chaduvula N, Patel K. The relationship between serum lipid levels and the risk of oral cancer. Indian J Med paediatric oncol: Off J Indian Soc Med Paediatric Oncol (2011) 32:34–7. doi: 10.4103/0971-5851.81888
35. Daker M, Bhuvanendran S, Ahmad M, Takada K, Khoo AS. Deregulation of lipid metabolism pathway genes in nasopharyngeal carcinoma cells. Mol Med Rep (2013) 7:731–41. doi: 10.3892/mmr.2012.1253
36. Kindt N, Journe F, Carlier S, Trelcat A, Scalia A, Saussez S. Effect of oxidized low-density lipoprotein on head and neck squamous cell carcinomas. Biomedicines (2021) 9(5):1–11. doi: 10.3390/biomedicines9050513
37. Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab (2016) 24:311–23. doi: 10.1016/j.cmet.2016.07.003
38. Wang MD, Wu H, Huang S, Zhang HL, Qin CJ, Zhao LH, et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget (2016) 7:6711–26. doi: 10.18632/oncotarget.6817
39. Nakamura MT, Nara TY. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu Rev Nutr (2004) 24:345–76. doi: 10.1146/annurev.nutr.24.121803.063211
40. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541:41–5. doi: 10.1038/nature20791
41. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S, et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics (2019) 9:5359–73. doi: 10.7150/thno.34024
42. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J Exp Clin Cancer Research: CR. (2019) 38:52. doi: 10.1186/s13046-019-1049-7
43. Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C, et al. TGFbeta2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun (2020) 11:454. doi: 10.1038/s41467-019-14262-3
44. Pascual G, Dominguez D, Elosua-Bayes M, Beckedorff F, Laudanna C, Bigas C, et al. Dietary palmitic acid promotes a prometastatic memory via schwann cells. Nature (2021) 599:485–90. doi: 10.1038/s41586-021-04075-0
45. Rauch J, Ahlemann M, Schaffrik M, Mack B, Ertongur S, Andratschke M, et al. Allogenic antibody-mediated identification of head and neck cancer antigens. Biochem Biophys Res Commun (2004) 323:156–62. doi: 10.1016/j.bbrc.2004.08.071
46. Ohyama Y, Kawamoto Y, Chiba T, Kikuchi K, Sakashita H, Imai K. Differential expression of fatty acid-binding proteins and pathological implications in the progression of tongue carcinoma. Mol Clin Oncol (2014) 2:19–25. doi: 10.3892/mco.2013.198
47. Zheng ZQ, Li ZX, Guan JL, Liu X, Li JY, Chen Y, et al. Long noncoding RNA TINCR-mediated regulation of acetyl-CoA metabolism promotes nasopharyngeal carcinoma progression and chemoresistance. Cancer Res (2020) 80:5174–88. doi: 10.1158/0008-5472.CAN-19-3626
48. Sur S, Nakanishi H, Flaveny C, Ippolito JE, McHowat J, Ford DA, et al. Inhibition of the key metabolic pathways, glycolysis and lipogenesis, of oral cancer by bitter melon extract. Cell Commun Signaling: CCS (2019) 17:131. doi: 10.1186/s12964-019-0447-y
49. Gormley M, Yarmolinsky J, Dudding T, Burrows K, Martin RM, Thomas S, et al. Using genetic variants to evaluate the causal effect of cholesterol lowering on head and neck cancer risk: A mendelian randomization study. PloS Genet (2021) 17:e1009525. doi: 10.1371/journal.pgen.1009525
50. Zhang L, Zhao S, Liu Y, Lv F, Geng X. Identification and validation of transcription factor-driven enhancers of genes related to lipid metabolism in metastatic oral squamous cell carcinomas. BMC Oral Health (2022) 22:126. doi: 10.1186/s12903-022-02157-7
51. Ahmadi M, Amiri S, Pecic S, Machaj F, Rosik J, Los MJ, et al. Pleiotropic effects of statins: A focus on cancer. Biochim Biophys Acta Mol basis disease (2020) 1866:165968. doi: 10.1016/j.bbadis.2020.165968
52. Dimitroulakos J, Marhin WH, Tokunaga J, Irish J, Gullane P, Penn LZ, et al. Microarray and biochemical analysis of lovastatin-induced apoptosis of squamous cell carcinomas. Neoplasia (2002) 4:337–46. doi: 10.1038/sj.neo.7900247
53. Kwon M, Nam GH, Jung H, Kim SA, Kim S, Choi Y, et al. Statin in combination with cisplatin makes favorable tumor-immune microenvironment for immunotherapy of head and neck squamous cell carcinoma. Cancer Letters (2021) 522:198–210. doi: 10.1016/j.canlet.2021.09.029
54. Su YW, Lin YH, Pai MH, Lo AC, Lee YC, Fang IC, et al. Association between phosphorylated AMP-activated protein kinase and acetyl-CoA carboxylase expression and outcome in patients with squamous cell carcinoma of the head and neck. PloS One (2014) 9:e96183. doi: 10.1371/journal.pone.0096183
55. Li K, Zhang C, Chen L, Wang P, Fang Y, Zhu J, et al. The role of acetyl-coA carboxylase2 in head and neck squamous cell carcinoma. PeerJ (2019) 7:e7037. doi: 10.7717/peerj.7037
56. Lo AK, Lung RW, Dawson CW, Young LS, Ko CW, Yeung WW, et al. Activation of sterol regulatory element-binding protein 1 (SREBP1)-mediated lipogenesis by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) promotes cell proliferation and progression of nasopharyngeal carcinoma. J pathol (2018) 246:180–90. doi: 10.1002/path.5130
57. Huang Y, Bell LN, Okamura J, Kim MS, Mohney RP, Guerrero-Preston R, et al. Phospho-DeltaNp63alpha/SREBF1 protein interactions: bridging cell metabolism and cisplatin chemoresistance. Cell Cycle (2012) 11:3810–27. doi: 10.4161/cc.22022
58. Boelcke WP, Teixeira IF, Aquino IG, Mazzaro AR, Cuadra-Zelaya FJM, de Souza AP, et al. Pharmacological fatty acid synthase inhibitors differently affect the malignant phenotype of oral cancer cells. Arch Oral Biol (2022) 135:105343. doi: 10.1016/j.archoralbio.2021.105343
59. Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW, et al. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res (2015) 183:291–304. doi: 10.1667/RR13828.1
60. Jain P, Nattakom M, Holowka D, Wang DH, Thomas Brenna J, Ku AT, et al. Runx1 role in epithelial and cancer cell proliferation implicates lipid metabolism and Scd1 and Soat1 activity. Stem Cells (2018) 36:1603–16. doi: 10.1002/stem.2868
61. Nanjappa V, Renuse S, Sathe GJ, Raja R, Syed N, Radhakrishnan A, et al. Chronic exposure to chewing tobacco selects for overexpression of stearoyl-CoA desaturase in normal oral keratinocytes. Cancer Biol Ther (2015) 16:1593–603. doi: 10.1080/15384047.2015.1078022
62. Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death disease (2020) 11:105. doi: 10.1038/s41419-020-2297-3
63. Zhou X, Wei J, Chen F, Xiao X, Huang T, He Q, et al. Epigenetic downregulation of the ISG15-conjugating enzyme UbcH8 impairs lipolysis and correlates with poor prognosis in nasopharyngeal carcinoma. Oncotarget (2015) 6:41077–91. doi: 10.18632/oncotarget.6218
64. Zheng S, Matskova L, Zhou X, Xiao X, Huang G, Zhang Z, et al. Downregulation of adipose triglyceride lipase by EB viral-encoded LMP2A links lipid accumulation to increased migration in nasopharyngeal carcinoma. Mol Oncol (2020) 14:3234–52. doi: 10.1002/1878-0261.12824
65. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol cancer (2017) 16:76. doi: 10.1186/s12943-017-0646-3
66. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large b cell lymphoma. Cancer Cell (2012) 22:547–60. doi: 10.1016/j.ccr.2012.08.014
67. Zechner R, Strauss JG, Haemmerle G, Lass A, Zimmermann R. Lipolysis: pathway under construction. Curr Opin lipidol (2005) 16:333–40. doi: 10.1097/01.mol.0000169354.20395.1c
68. Mairal A, Langin D, Arner P, Hoffstedt J. Human adipose triglyceride lipase (PNPLA2) is not regulated by obesity and exhibits low in vitro triglyceride hydrolase activity. Diabetologia (2006) 49:1629–36. doi: 10.1007/s00125-006-0272-x
69. Tripathi P, Kamarajan P, Somashekar BS, MacKinnon N, Chinnaiyan AM, Kapila YL, et al. Delineating metabolic signatures of head and neck squamous cell carcinoma: phospholipase A2, a potential therapeutic target. Int J Biochem Cell Biol (2012) 44:1852–61. doi: 10.1016/j.biocel.2012.06.025
70. Du Q, Tan Z, Shi F, Tang M, Xie L, Zhao L, et al. PGC1alpha/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer sci (2019) 110:2050–62. doi: 10.1111/cas.14011
71. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
72. Liu F, Wei J, Hao Y, Lan J, Li W, Weng J, et al. Long intergenic non-protein coding RNA 02570 promotes nasopharyngeal carcinoma progression by adsorbing microRNA miR-4649-3p thereby upregulating both sterol regulatory element binding protein 1, and fatty acid synthase. Bioengineered (2021) 12:7119–30. doi: 10.1080/21655979.2021.1979317
73. Hashibe M, Brennan P, Chuang SC, Boccia S, Castellsague X, Chen C, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the international head and neck cancer epidemiology consortium. Cancer epidemiol Biomarkers prevention: Publ Am Assoc Cancer Research cosponsored by Am Soc Prev Oncol (2009) 18:541–50. doi: 10.1158/1055-9965.EPI-08-0347
74. Di Credico G, Polesel J, Dal Maso L, Pauli F, Torelli N, Luce D, et al. Alcohol drinking and head and neck cancer risk: the joint effect of intensity and duration. Br J cancer (2020) 123:1456–63. doi: 10.1038/s41416-020-01031-z
75. Koo HY, Han K, Shin DW, Yoo JE, Cho MH, Jeon KH, et al. Alcohol drinking pattern and risk of head and neck cancer: A nationwide cohort study. Int J Environ Res Public Health (2021) 18(21):1–14. doi: 10.3390/ijerph182111204
76. Ganne-Carrie N, Nahon P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J hepatol (2019) 70:284–93. doi: 10.1016/j.jhep.2018.10.008
77. Lee WT, Hsiao JR, Ou CY, Huang CC, Chang CC, Tsai ST, et al. The influence of prediagnosis alcohol consumption and the polymorphisms of ethanol-metabolizing genes on the survival of head and neck cancer patients. Cancer epidemiol Biomarkers prevention: Publ Am Assoc Cancer Research cosponsored by Am Soc Prev Oncol (2019) 28:248–57. doi: 10.1158/1055-9965.EPI-18-0425
78. Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem (2003) 278:27997–8004. doi: 10.1074/jbc.M302140200
79. Salaspuro MP, Shaw S, Jayatilleke E, Ross WA, Lieber CS. Attenuation of the ethanol-induced hepatic redox change after chronic alcohol consumption in baboons: metabolic consequences in vivo and in vitro. Hepatology (1981) 1:33–8. doi: 10.1002/hep.1840010106
80. Tomita K, Azuma T, Kitamura N, Nishida J, Tamiya G, Oka A, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-met. Gastroenterology (2004) 126:873–85. doi: 10.1053/j.gastro.2003.12.008
81. Li Q, Zhong W, Qiu Y, Kang X, Sun X, Tan X, et al. Preservation of hepatocyte nuclear factor-4alpha contributes to the beneficial effect of dietary medium chain triglyceride on alcohol-induced hepatic lipid dyshomeostasis in rats. Alcoholism Clin Exp Res (2013) 37:587–98. doi: 10.1111/acer.12013
82. Middlekauff HR, William KJ, Su B, Haptonstall K, Araujo JA, Wu X, et al. Changes in lipid composition associated with electronic cigarette use. J Trans Med (2020) 18:379. doi: 10.1186/s12967-020-02557-9
83. Chelland Campbell S, Moffatt RJ, Stamford BA. Smoking and smoking cessation – the relationship between cardiovascular disease and lipoprotein metabolism: a review. Atherosclerosis (2008) 201:225–35. doi: 10.1016/j.atherosclerosis.2008.04.046
84. Azizi F, Raiszadeh F, Salehi P, Rahmani M, Emami H, Ghanbarian A, et al. Determinants of serum HDL-c level in a Tehran urban population: the Tehran lipid and glucose study. Nutrition metabolism Cardiovasc diseases: NMCD (2002) 12:80–9.
85. Mizoue T, Ueda R, Hino Y, Yoshimura T. Workplace exposure to environmental tobacco smoke and high density lipoprotein cholesterol among nonsmokers. Am J Epidemiol (1999) 150:1068–72. doi: 10.1093/oxfordjournals.aje.a009930
86. Merianos AL, Jandarov RA, Khoury JC, Mahabee-Gittens EM. Tobacco smoke exposure association with lipid profiles and adiposity among U.S. adolescents. J Adolesc health: Off Publ Soc Adolesc Med (2018) 62:463–70. doi: 10.1016/j.jadohealth.2017.10.001
87. Zakhar J, Amrock SM, Weitzman M. Passive and active tobacco exposure and children’s lipid profiles. Nicotine tobacco research: Off J Soc Res Nicotine Tobacco (2016) 18:982–7. doi: 10.1093/ntr/ntv158
88. Steenland K, Sieber K, Etzel RA, Pechacek T, Maurer K. Exposure to environmental tobacco smoke and risk factors for heart disease among never smokers in the third national health and nutrition examination survey. Am J Epidemiol (1998) 147:932–9. doi: 10.1093/oxfordjournals.aje.a009383
89. Bousarghin L, Touze A, Sizaret PY, Coursaget P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J virol (2003) 77:3846–50. doi: 10.1128/JVI.77.6.3846-3850.2003
90. Bravo IG, Crusius K, Alonso A. The E5 protein of the human papillomavirus type 16 modulates composition and dynamics of membrane lipids in keratinocytes. Arch virol (2005) 150:231–46. doi: 10.1007/s00705-004-0420-x
91. Zhang L, Wu J, Ling MT, Zhao L, Zhao KN. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol Cancer (2015) 14:87. doi: 10.1186/s12943-015-0361-x
92. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (2018) 38:27. doi: 10.1186/s40880-018-0301-4
93. Chopjitt P, Pientong C, Bumrungthai S, Kongyingyoes B, Ekalaksananan T. Activities of E6 protein of human papillomavirus 16 Asian variant on miR-21 up-regulation and expression of human immune response genes. Asian Pacific J Cancer prevention: APJCP (2015) 16:3961–8. doi: 10.7314/APJCP.2015.16.9.3961
94. Ni K, Wang D, Xu H, Mei F, Wu C, Liu Z, et al. miR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int (2019) 19:219. doi: 10.1186/s12935-019-0941-8
95. Prusinkiewicz MA, Gameiro SF, Ghasemi F, Dodge MJ, Zeng PYF, Maekebay H, et al. Survival-associated metabolic genes in human papillomavirus-positive head and neck cancers. Cancers (2020) 12(1):1–17. doi: 10.3390/cancers12010253
96. Martinez-Ramirez I, Carrillo-Garcia A, Contreras-Paredes A, Ortiz-Sanchez E, Cruz-Gregorio A, Lizano M. Regulation of cellular metabolism by high-risk human papillomaviruses. Int J Mol Sci (2018) 19(7):1–17. doi: 10.3390/ijms19071839
97. Cruz-Gregorio A, Martinez-Ramirez I, Pedraza-Chaverri J, Lizano M. Reprogramming of energy metabolism in response to radiotherapy in head and neck squamous cell carcinoma. Cancers (2019) 11(2):1–17. doi: 10.3390/cancers11020182
98. Cruz-Gregorio A, Aranda-Rivera AK, Ortega-Lozano AJ, Pedraza-Chaverri J, Mendoza-Hoffmann F. Lipid metabolism and oxidative stress in HPV-related cancers. Free Radical Biol Med (2021) 172:226–36. doi: 10.1016/j.freeradbiomed.2021.06.009
99. Cai TT, Ye SB, Liu YN, He J, Chen QY, Mai HQ, et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PloS pathog (2017) 13:e1006503. doi: 10.1371/journal.ppat.1006503
100. Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang M, et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene (2014) 33:4568–78. doi: 10.1038/onc.2014.32
101. Petras G, Adam MM. Phases of the serological response in pseudomonas aeruginosa infections. Acta microbiol Hungarica (1987) 34:147–57.
102. Wang LW, Wang Z, Ersing I, Nobre L, Guo R, Jiang S, et al. Epstein-Barr Virus subverts mevalonate and fatty acid pathways to promote infected b-cell proliferation and survival. PloS Pathog (2019) 15:e1008030. doi: 10.1371/journal.ppat.1008030
103. Arora GK, Gupta A, Narayanan S, Guo T, Iyengar P, Infante RE. Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. JCI Insight (2018) 3(14):1–18. doi: 10.1172/jci.insight.121221
104. Strassmann G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest (1992) 89:1681–4. doi: 10.1172/JCI115767
105. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab (2016) 24:672–84. doi: 10.1016/j.cmet.2016.10.010
106. Middeldorp JM, Pegtel DM. Multiple roles of LMP1 in Epstein-Barr virus induced immune escape. Semin Cancer Biol (2008) 18:388–96. doi: 10.1016/j.semcancer.2008.10.004
107. Lien EC, Vander Heiden MG. A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer (2019) 19:651–61. doi: 10.1038/s41568-019-0198-5
108. Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab (2019) 29:1410–21 e4. doi: 10.1016/j.cmet.2019.02.015
109. Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife (2019) 8:1–27. doi: 10.7554/eLife.44235
110. Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature (2017) 544:372–6. doi: 10.1038/nature22056
111. Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature (2019) 572:397–401. doi: 10.1038/s41586-019-1437-3
112. Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, Lau AN, et al. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature (2021) 599:302–7. doi: 10.1038/s41586-021-04049-2
113. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radical Biol Med (2018) 129:454–62. doi: 10.1016/j.freeradbiomed.2018.10.426
114. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
115. Huang C, Zhan L. Network pharmacology identifies therapeutic targets and the mechanisms of glutathione action in ferroptosis occurring in oral cancer. Front Pharmacol (2022) 13:851540. doi: 10.3389/fphar.2022.851540
116. Budhathoki S, Hidaka A, Yamaji T, Sawada N, Tanaka-Mizuno S, Kuchiba A, et al. Plasma 25-hydroxyvitamin d concentration and subsequent risk of total and site specific cancers in Japanese population: large case-cohort study within Japan public health center-based prospective study cohort. Bmj (2018) 360:k671. doi: 10.1136/bmj.k671
117. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature (2022) 608(7924):778–83. doi: 10.1038/s41586-022-05022-3
118. Jing K, Shin S, Jeong S, Kim S, Song KS, Park JH, et al. Docosahexaenoic acid induces the degradation of HPV E6/E7 oncoproteins by activating the ubiquitin-proteasome system. Cell Death disease (2014) 5:e1524. doi: 10.1038/cddis.2014.477
119. Meza-Menchaca T, Poblete-Naredo I, Albores-Medina A, Pedraza-Chaverri J, Quiroz-Figueroa FR, Cruz-Gregorio A, et al. Ergosterol peroxide isolated from oyster medicinal mushroom, pleurotus ostreatus (Agaricomycetes), potentially induces radiosensitivity in cervical cancer. Int J med mushrooms (2020) 22:1109–19. doi: 10.1615/IntJMedMushrooms.2020036673
120. Zhou ZT, Yang Y, Ge JP. The preventive effect of salvianolic acid b on malignant transformation of DMBA-induced oral premalignant lesion in hamsters. Carcinogenesis (2006) 27:826–32. doi: 10.1093/carcin/bgi271
121. Meng LC, Zheng JY, Qiu YH, Zheng L, Zheng JY, Liu YQ, et al. Salvianolic acid b ameliorates non-alcoholic fatty liver disease by inhibiting hepatic lipid accumulation and NLRP3 inflammasome in ob/ob mice. Int immunopharmacol (2022) 111:109099. doi: 10.1016/j.intimp.2022.109099
122. Buchsbaum RJ, Oh SY. Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers (2016) 8(2):1–19. doi: 10.3390/cancers8020019
123. Sun DY, Wu JQ, He ZH, He MF, Sun HB. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-beta signaling pathway. Life Sci (2019) 235:116791. doi: 10.1016/j.lfs.2019.116791
124. Ham IH, Lee D, Hur H. Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J Oncol (2019) 2019:6270784. doi: 10.1155/2019/6270784
125. Bolt R, Foran B, Murdoch C, Lambert DW, Thomas S, Hunter KD. HPV-negative, but not HPV-positive, oropharyngeal carcinomas induce fibroblasts to support tumour invasion through micro-environmental release of HGF and IL-6. Carcinogenesis (2018) 39:170–9. doi: 10.1093/carcin/bgx130
126. Kabir TD, Leigh RJ, Tasena H, Mellone M, Coletta RD, Parkinson EK, et al. A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. Aging (2016) 8:1608–35. doi: 10.18632/aging.100987
127. Jiang E, Xu Z, Wang M, Yan T, Huang C, Zhou X, et al. Tumoral microvesicle-activated glycometabolic reprogramming in fibroblasts promotes the progression of oral squamous cell carcinoma. FASEB journal: Off Publ Fed Am Societies Exp Biol (2019) 33:5690–703. doi: 10.1096/fj.201802226R
128. Zhang Z, Gao Z, Rajthala S, Sapkota D, Dongre H, Parajuli H, et al. Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell Mol Life Sci: CMLS (2020) 77:1115–33. doi: 10.1007/s00018-019-03209-y
129. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol (2011) 12:722–34. doi: 10.1038/nrm3198
130. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res (2011) 71:2455–65. doi: 10.1158/0008-5472.CAN-10-3323
131. James RF, Lake SP, Chamberlain J, Thirdborough S, Bassett PD, Mistry N, et al. Gamma irradiation of isolated rat islets pretransplantation produces indefinite allograft survival in cyclosporine-treated recipients. Transplantation (1989) 47:929–33. doi: 10.1097/00007890-198906000-00001
132. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140:197–208. doi: 10.1016/j.cell.2009.12.052
133. Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev (2011) 32:550–70. doi: 10.1210/er.2010-0030
134. Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med (2013) 64:45–57. doi: 10.1146/annurev-med-121211-091527
135. Young MRI, Levingston C, Johnson SD. Cytokine and adipokine levels in patients with premalignant oral lesions or in patients with oral cancer who did or did not receive 1 alpha,25-dihydroxyvitamin d-3 treatment upon cancer diagnosis. Cancers (2015) 7:1109–24. doi: 10.3390/cancers7030827
136. Gharote HP, Mody RN. Estimation of serum leptin in oral squamous cell carcinoma. J Oral Pathol med: Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol (2010) 39:69–73. doi: 10.1111/j.1600-0714.2009.00808.x
137. Lo HC, Yang CS, Tsai LJ. Simultaneous measurements of serum insulin-like growth factor-I and leptin reflect the postoperative nutrition status of oral tumor patients. Nutrition (2003) 19:327–31. doi: 10.1016/S0899-9007(02)01012-2
138. Guo XH, Wang JY, Gao Y, Gao M, Yu GY, Xiang RL, et al. Decreased adiponectin level is associated with aggressive phenotype of tongue squamous cell carcinoma. Cancer sci (2013) 104:206–13. doi: 10.1111/cas.12077
139. Yu-Duan T, Chao-Ping W, Chih-Yu C, Li-Wen L, Tsun-Mei L, Chia-Chang H, et al. Elevated plasma level of visfatin/pre-b cell colony-enhancing factor in male oral squamous cell carcinoma patients. Med oral patol Oral y cirugia bucal (2013) 18:e180–6. doi: 10.4317/medoral.18574
Keywords: lipid metabolism reprogramming, lipid catabolism, lipid synthesis, lipid uptake, HNSCCs
Citation: Miao X, Wang B, Chen K, Ding R, Wu J, Pan Y, Ji P, Ye B and Xiang M (2022) Perspectives of lipid metabolism reprogramming in head and neck squamous cell carcinoma: An overview. Front. Oncol. 12:1008361. doi: 10.3389/fonc.2022.1008361
Received: 31 July 2022; Accepted: 31 August 2022;
Published: 16 September 2022.
Edited by:
Donglai Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Jian-Fei Pei, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaBo Chu, Shandong University, China
Copyright © 2022 Miao, Wang, Chen, Ding, Wu, Pan, Ji, Ye and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingliang Xiang, bWluZ2xpYW5neGlhbmdAMTYzLmNvbQ==; Bin Ye, YXlkeWViaW5AMTI2LmNvbQ==
†These authors have contributed equally to this work