 Cheng-Jiang Wei1†
Cheng-Jiang Wei1† Zhi-Chao Wang
Zhi-Chao Wang Bin Wang
Bin Wang- 1Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Shanghai Universal Medical Imaging Diagnostic Center, Huaxin Business Center, Shanghai, China
Tuberous sclerosis complex (TSC) is an inherited disorder that typically presents with seizures, developmental delay, cutaneous lesions, and facial angiomas. Clinical diagnosis of TSC based on symptoms is sometimes challenging due to its clinical similarities with neurofibromatosis type 1 (NF1), another type of neurogenetic tumor syndrome. Differential diagnosis should be carefully performed on the basis of clinical presentations, imaging, laboratory, and genetic testing. Here, we presented a case of a patient with an aggressively enlarged right upper limb in the NF1 clinic, who was initially suspected of a giant plexiform neurofibroma. However, differential diagnosis revealed TSC as the final diagnosis. The treatments for NF1 and TSC vary significantly, and misdiagnoses can lead to serious threat to the patients’ health. We also systematically reviewed all previous cases regarding differential diagnoses between NF1 and TSC. This case report can help clinicians make more accurate diagnoses and benefit the potential patient community.
Introduction
Tuberous sclerosis complex (TSC) is a rare genetic disease characterized by seizures, developmental delay, and facial angiomas (Vogt’s triad) (1). Neurofibromatosis type 1 (NF1) is another neurogenetic tumor syndrome caused by the mutation of the NF1 gene (2). There are similarities in clinical symptoms between TSC with NF1, which might cause misdiagnosis. Here, we described a case of a young patient with TSC with an enlarged right arm who was first diagnosed as NF1 during first visit to the clinic. Genetic testing revealed the TSC1 mutation, and TSC was confirmed as the final diagnosis. Furthermore, the radiological imaging showed lung lymphangioleiomyoma and poor blood supply in the right arm. Last, we discussed the clinical treatment and follow-up recommendation for this patient (Figure 1).
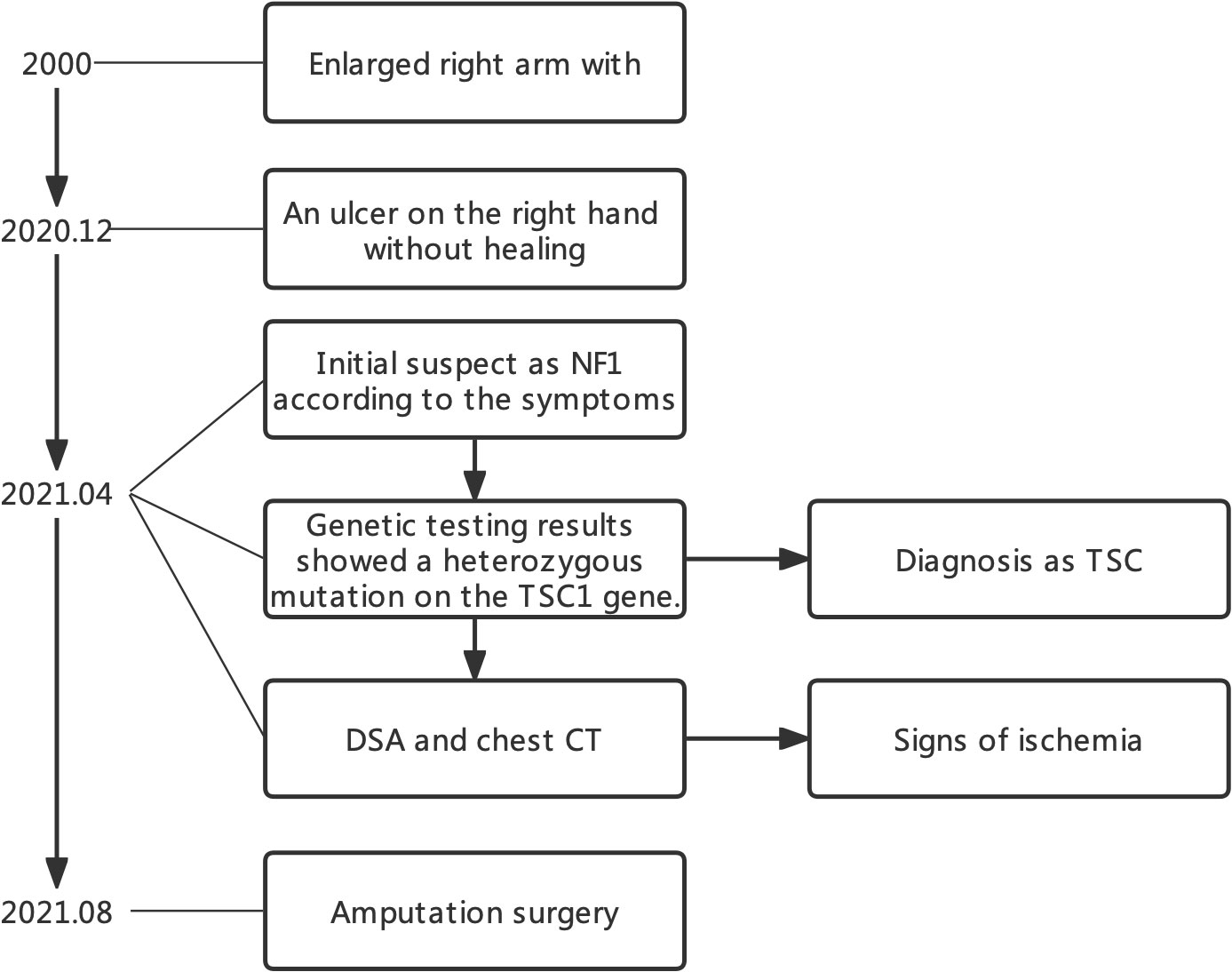
Figure 1 Timeline of this case.
Case presentation
A 20-year-old man presented to our institution, complaining that the right upper limb was much thicker than the left from birth, along with the enlargement of his left index finger and middle finger (Figures 2A–C). The volume of the right upper limb increased significantly during puberty. Five months before this visit, the patient suffered from an ulcer on the right hand that had failed to heal. He did not pay much attention to the enlarged right arm as he has got used to the condition for over 20 years. The main complaint for this visit was the non-healing right-hand ulcer, which he thought could be solved after simple debridement. He showed little awareness and had limited knowledge regarding the disease. Upon questioning, the patient declared no history of seizures and showed no signs of amentia. His parents also reported that the patient had no history of autism.
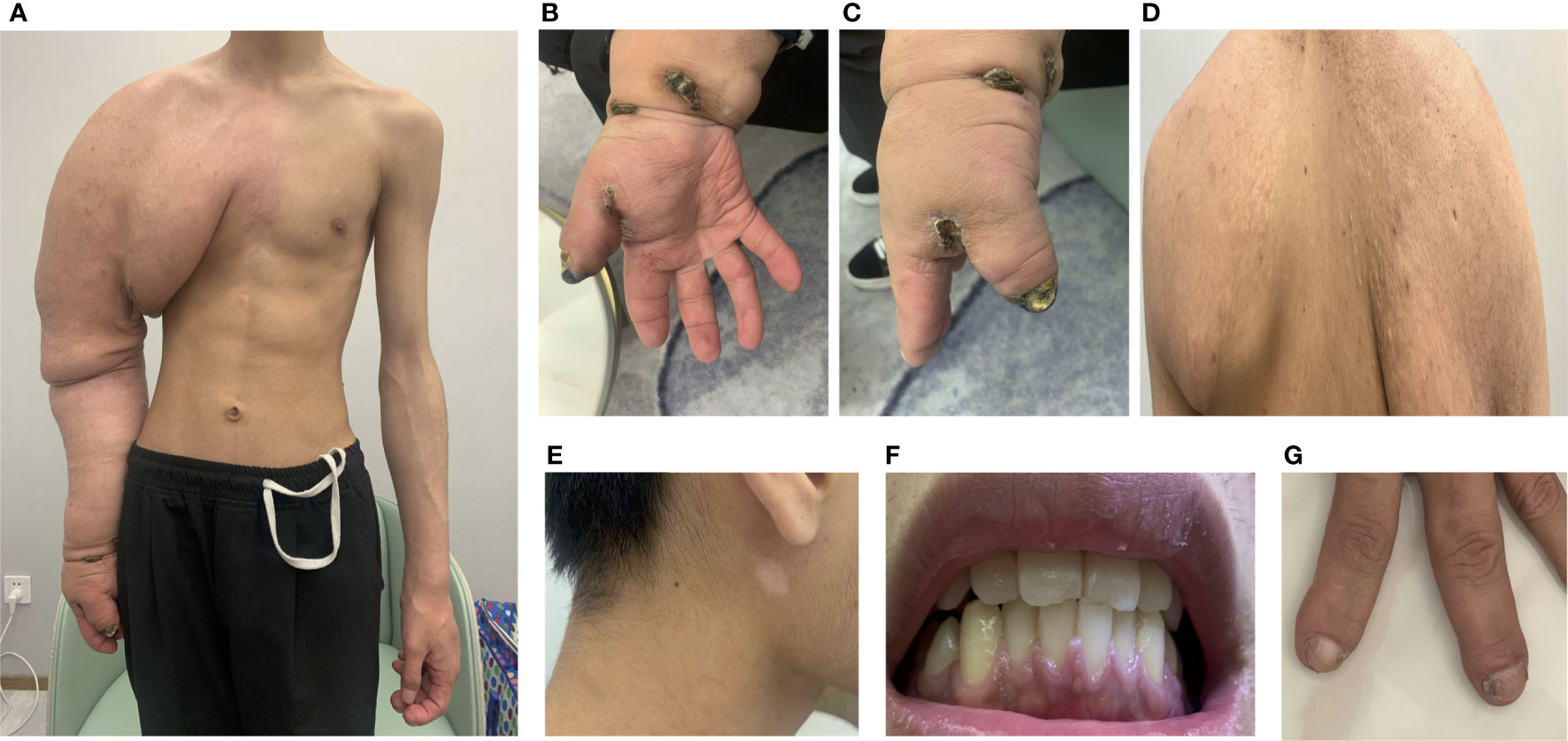
Figure 2 Clinical symptoms. (A) Enlarged right arm; (B, C) unhealing ulcer; (D) adenoma sebaceous; (E) depigmented macule; (F) gingiva; (G) periungal fibromas.
On clinical examination, the right arm was significantly more prominent than the left, with the soft tissue being much thicker than the opposite side. Meanwhile, there were sebaceous adenomas on the back, depigmented macules on the face, and fibromas under the nail bed (periungual fibromas) and gingiva (Figures 2D–G). Other skin lesions of tuberous sclerosis such as ash-leaf spots and shagreen patches were not evident. The physical examinations of cardiovascular and central nervous systems were also normal.
We initially suspected the case as plexiform neurofibroma with NF1 due to the enlargement of the right arm. However, the facial spots described by the patient as “café’ au lait spots” are actually depigmented macules. Whole-body imaging was then conducted for the patient. The head CT revealed subependymal, cortical, and subcortical nodules (Figure 3A). Meanwhile, we found lymphangioleiomyomatosis on the chest CT, focal sclerosis, and bone cysts in multiple bone regions such as the spine (Figures 3B, C). Although MRI might help clarify the nature of the enlarged right arm, it could not be conducted as the volume of the patient’s right arm exceeded the maximum size limit of our MRI machine. No hemorrhage and retinal hamartomas were found on ophthalmic examination, and mild mitral regurgitation was found on echocardiography.
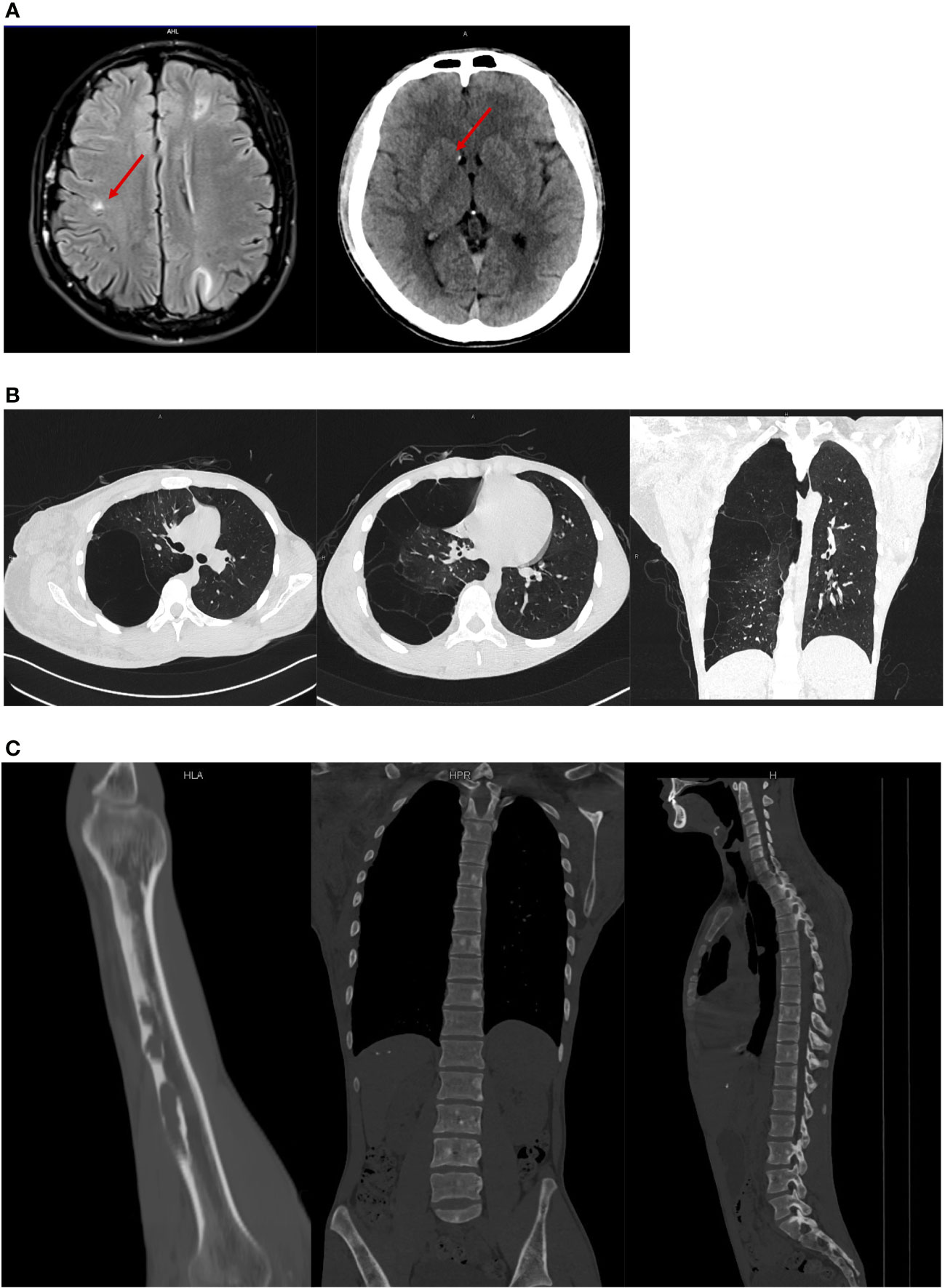
Figure 3 Medical imaging results. (A) Nodules showed on the head CT; (B) lymphangioleiomyomatosis on the chest CT and bone cysts in the different regions; (C) lymphangioleiomyomatosis on the chest CT and bone cysts in the different regions.
Genetic testing was conducted for further differential diagnosis. No NF1 mutation was found. Furthermore, there was a heterozygous mutation on the TSC1 gene.
On the basis of these features, a diagnosis of TSC was established, and the non-healing ulcer was considered to be associated with the vascular blockage caused by the thickened soft tissue. Digital subtraction angiography (DSA) was used to analyze the blood flow condition of the right arm (Supplementary File 1). The result showed insufficient blood supply in the distal right upper limb, and amputation appeared to be the only treatment plan for the patient. During consultation, the patient showed difficulties accepting this treatment plan and stated that he needed time for consideration. Approximately 4 months later, he finally accepted the amputation surgery of the right arm as the ulcer condition barely improved. After the surgery, we recommended life-long follow-up and monitoring of potentially fatal complications. Annual ophthalmology examination was also essential for the risk of hemorrhage. As the patient also presented with lung lymphangioleiomyoma and cyst, high-resolution CT should be performed every year and the annual pulmonary function examination was necessary.
Discussion and conclusions
TSC is caused by the pathogenic variants in either the TSC1 or TSC2 tumor suppressor genes, located on chromosomes 9q34 and 16p13, respectively (3). TSC1 and TSC2 encode the protein hamartin and tuberin, which together suppress the activity of the mammalian target of rapamycin (mTOR) pathway (4). Loss of their function leads to continuous activation of the mTOR pathway, which results in cell proliferation (4). Typical clinical symptoms of TSC include periungual fibromas, renal angiomyolipoma, benign interstitial expansion of lung pulmonary smooth muscle cells, and neurological symptoms like seizures and neurodevelopmental delay (1).
Neurofibromatosis type 1 (NF1) is also an autosomal dominant genetic disorder caused by another tumor suppressor gene NF1 (2). The NF1 gene encodes neurofibromin, a negative regulator of the RAS–mitogen-activated protein kinase pathway. The loss of NF1 contributes to the activation of this pathway and finally leads to tumor growth and development (5). Classic clinical features include café-au-lait macules, skinfold freckling, benign neurofibromas like cutaneous neurofibroma and plexiform neurofibroma, brain tumors, iris hamartomas, and typical bony lesions. Meanwhile, there are also reports of neurological manifestations such as epilepsy (6).
Comparing these two diseases, both may have vague and overlapping clinical presentations that can lead to missed diagnosis and finally cause delayed treatment. Coincidentally, both NF1 and TSC were first described by Von Recklinghausen, which proved from the side that these two diseases have some similar symptoms (7, 8). The major clinical diagnostic criteria of TSC such as hypomelanotic macule, angiofibroma, and shagreen patch are sometimes difficult to distinguish from typical NF1 phenomenon like café-au-lait macules, skinfold freckling, and benign neurofibroma (Table 1). Although NF1 and TSC are the first and second most common neurogenetic tumor syndromes, NF1 only has a prevalence of approximately 1:2,500 to 1:3,500, whereas TSC is even rarer and affects 1 in 5,500 to 1 in 10,000 live births (9). The limited amount of clinical cases further caused difficulties in clinical differential diagnosis, especially for doctors in remote areas. Moreover, it was shown that, in some cases, both NF1 and TSC could occur in a single individual, which might further confuse the diagnosis. We performed an extensive review of the literature using PubMed search including the words (NF1 and tuberous sclerosis) from the year 2000 until 2021 (Table 2) (10–14). Interestingly, Wheeler and Sadeghi-Nejad reported a case of a 4-month girl that suffered from both NF1 and TSC but reported a family history of NF1 only (12). After a definite diagnosis of TSC was made for this girl, her parents were carefully re-examined and also confirmed the diagnosis of TSC (12). This case further indicates the similarities of some clinical symptoms, and one definite diagnosis might further inhibit the recognition of another. Genetic testing and pathological biopsy may be necessary for a definite diagnosis of TSC. Moreover, as TSC and NF1 are both hereditary diseases, first-degree relatives should undergo a clinical assessment and a three-generation family history is required.

Table 1 Comparison of diagnosis criteria or neurofibromatosis type 1 and tuberous sclerosis complex.

Table 2 List of published case reports of patients with both NF-1 and TSC from 2000 to 2021.
Current clinical management of TSC is insufficient as most of them are symptomatic treatments, especially for seizures. Early therapeutic intervention for children who present with infantile spasms correlated with TSC is essential, and the first-line treatment is vigabatrin (15). Furthermore, the mTOR inhibitors such as everolimus have provided great therapeutic promise. A clinical study showed that treatment with everolimus in patients with TSC resulted in a sustained decrease in seizure frequency in children and adolescents (16). Nonetheless, this patient did not have any history of seizures. As he reached the age of 20 and presented with vascular blockage indicated by the non-healing ulcer, surgical approaches were considered according to the 2021 updated TSC international recommendations (17). DSA examination further showed signs of ischemia. As a result, amputation was the only surgical choice for him. Compared to the limited drug choices for patients with TSC, the development of drug therapies for patients with NF1 progressed rapidly. Selumetinib, a Mitogen-activated extracellularsignal-regulated kinase (MEK) inhibitor, was reported effective for plexiform neurofibroma and was approved for clinical usage by FDA (18). Early distinguishment of these two diseases is essential for further proper clinical treatment.
Another essential issue for the treatment of patients with TSC is life-long follow-up recommendations. TSC influences multiple organs in the body, and some manifestations are life-threatening. For example, our patient has lymphangioleiomyoma, which might cause respiratory dysfunction. According to the 2021 updated TSC international recommendations, we recommended routine serial pulmonary function test annually on chest CT (17). Meanwhile, we had several recommendations regarding other possible TSC features, including continuous sun protection for depigmented macules, detailed dental examination every 6 months, electrocardiography every 3 years for mild mitral regurgitation, annual ophthalmic evaluation, and renal function assessment. For patients with NF1, life-long follow-up is also essential, but they are less concerned with respiratory dysfunction. A definite diagnosis is essential for further proper follow-up.
Definite and accurate diagnosis is the basis for proper and effective treatment, and misdiagnosis might cause further damage to the patients and waste medical resources. It is far more essential for relatively rare diseases as most doctors might have relatively less experience with them. This case introduced the possible pitfall and similarities of clinical phenomena between TSC and NF1, which might be helpful for future clinical diagnosis and management of these diseases in this area.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the institutional review board of Shanghai Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
C-JW wrote the manuscript and was involved in the diagnostic and therapeutic clinical processes. L-LP analyzed radiology medical images and contributed to the diagnostic and therapeutic processes. M-HC made substantial contribution during revision. Z-CW and BW helped in the diagnostic process and critically revised the manuscript and were responsible for the diagnosis and treatment of the patient. All authors read and approved the final manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (82102344; 82172228; 81772115); Shanghai Rising Star Program supported by Science and Technology Commission of Shanghai Municipality (20QA1405600); Science and Technology Commission of Shanghai Municipality (19JC1413, 22MC1940300) ; Natural Science Foundation of Shanghai (22ZR1422300); “Chenguang Program” supported by Shanghai Education Development Foundation (SHEDF) (19CG18); Innovative research team of high-level local universities in Shanghai (SSMU-ZDCX20180700); the Project of Biobank (YBKA201901) from Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine; Research Project of Multi-Disciplinary Team of Shanghai Ninth People’s Hospital (201907).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1007651/full#supplementary-material
References
1. Randle SC. Tuberous sclerosis complex: A review. Pediatr Ann (2017) 46:e166–71. doi: 10.3928/19382359-20170320-01
2. Huson SM, Compston DA, Harper PS. A genetic study of von recklinghausen neurofibromatosis in south east wales. II. guidelines for genetic counselling. J Med Genet (1989) 26:712–21. doi: 10.1136/jmg.26.11.712
3. Portocarrero LKL, Quental KN, Samorano LP, Oliveira ZNP, Rivitti-Machado M. Tuberous sclerosis complex: review based on new diagnostic criteria. Bras Dermatol (2018) 93:323–31. doi: 10.1590/abd1806-4841.20186972
4. Kwiatkowski DJ. Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biol Ther (2003) 2:471–6. doi: 10.4161/cbt.2.5.446
5. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer (2015) 15:290–301. doi: 10.1038/nrc3911
6. Nix JS, Blakeley J, Rodriguez FJ. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol (2020) 139:625–41. doi: 10.1007/s00401-019-02002-2
7. Anderson JL, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol (2015) 132:75–86. doi: 10.1016/B978-0-444-62702-5.00004-4
8. Gomez MR. History of the tuberous sclerosis complex. Brain Dev (1995) 17 Suppl:55–7. doi: 10.1016/0387-7604(94)00130-8
9. Strowd RE 3rd, Plotkin SR. Familial nervous system tumor syndromes. Continuum (Minneap Minn) (2020) 26:1523–52. doi: 10.1212/CON.0000000000000950
10. Murakami Y, Wataya-Kaneda M, Tanaka M, Katayama I. Case of tuberous sclerosis complex complicated by mosaic localized neurofibromatosis type 1. J Dermatol (2013) 40:413–4. doi: 10.1111/1346-8138.12074
11. Erbay SH, Oljeski SA, Bhadelia R. Rapid development of optic glioma in a patient with hybrid phakomatosis: neurofibromatosis type 1 and tuberous sclerosis. AJNR Am J Neuroradiol (2004) 25:36–8. doi: 10.1016/S1076-6332(03)00599-3
12. Wheeler PG, Sadeghi-Nejad A. Simultaneous occurrence of neurofibromatosis type 1 and tuberous sclerosis in a young girl. Am J Med Genet A 133A (2005) 133A(1):78–81. doi: 10.1002/ajmg.a.30530
13. Suttur MS, Mysore SR, Krishnamurthy B, Nallur RB. Rare association of turner syndrome with neurofibromatosis type 1 and tuberous sclerosis complex. Indian J Hum Genet (2009) 15:75–7. doi: 10.4103/0971-6866.55220
14. Alaraj AM, Valyi-Nagy T, Roitberg B. Double phakomatosis; neurofibromatosis type-1 and tuberous sclerosis. Acta Neurochir (Wien) (2007) 149:505–9; discussion 509. doi: 10.1007/s00701-007-1140-2
15. Strzelczyk A, Grau J, Bast T, Bertsche A, Bettendorf U, Hahn A, et al. Prescription patterns of antiseizure drugs in tuberous sclerosis complex (TSC)-associated epilepsy: a multicenter cohort study from Germany and review of the literature. Expert Rev Clin Pharmacol (2021) 14:749–60. doi: 10.1080/17512433.2021.1911643
16. Franz DN, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Everolimus for treatment-refractory seizures in TSC: Extension of a randomized controlled trial. Neurol Clin Pract (2018) 8:412–20. doi: 10.1212/CPJ.0000000000000514
17. Northrup H, Aronow ME, Bebin EM, Bissler J, Darling TN, de Vries PJ, et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol (2021) 123:50–66. doi: 10.1016/j.pediatrneurol.2021.07.011
Keywords: tuberous sclerosis complex (TSC), neurofibromatosis type 1 (NF1), differential diagnosis, treatment, follow-up recommendation, case report
Citation: Wei C-J, Peng L-L, Chuang M-H, Wang Z-C and Wang B (2022) Case Report: Differential diagnosis for tuberous sclerosis and neurofibromatosis type 1 diagnostic pitfall of aggressively enlarged right upper limb. Front. Oncol. 12:1007651. doi: 10.3389/fonc.2022.1007651
Received: 30 July 2022; Accepted: 02 November 2022;
Published: 24 November 2022.
Edited by:
Ignazio Gaspare Vetrano, IRCCS Carlo Besta Neurological Institute Foundation, ItalyReviewed by:
Maria Guadalupe Moreno Treviño, University of Monterrey, MexicoDebora Garozzo, Mediclinic Parkview Hospital, United Arab Emirates
Copyright © 2022 Wei, Peng, Chuang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Chao Wang, c2htdXd6Y0AxNjMuY29t; Bin Wang, V2FuZ2IxNDM1QHNoOWhvc3BpdGFsLm9yZy5jbg==
†These authors have contributed equally to this work and share first authorship