 Ye Yang
Ye Yang Min Wang
Min Wang Ying-Ying Zhang
Ying-Ying Zhang Shu-Zhi Zhao4,5*†
Shu-Zhi Zhao4,5*†- 1Obstetrics and Gynecology Department, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2General Surgery Department, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Respiratory and Critical Care Medicine Department, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 4Department of Ophthalmology, Shanghai General Hospital (Shanghai First People’s Hospital), Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 5National Clinical Research Center for Eye Diseases, Shanghai Key Laboratory of Ocular Fundus Diseases, Shanghai Engineering Center for Visual Science and Photomedicine, Shanghai Engineering Center for Precise Diagnosis and Treatment of Eye Diseases, Shanghai, China
- 6Trauma Center, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
The endosomal sorting complex required for transport (ESCRT) machinery plays a key role in the repair of damaged plasma membranes with puncta form and removes pores from the plasma membrane in regulated cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy. ESCRT-I overexpression and ESCRT-III-associated charged multivesicular body protein (CHMP) 4B participate in apoptosis, and the ESCRT-1 protein TSG 101 maintains low levels of ALIX and ALG-2 and prevents predisposition to apoptosis. The ESCRT-III components CHMP2A and CHMP4B are recruited to broken membrane bubble sites with the requirement of extracellular Ca2+, remove membrane vesicles from cells, and delay the time required for active MLKL to mediate necroptosis, thus preserving cell survival. CHMP4B disturbed pyroptosis by recruiting around the plasma membrane neck to remove the GSDMD pores and preserve plasma membrane integrity depending on Ca2+ influx. The accumulation of the ESCRT-III subunits CHMP5 and CHMP6 in the plasma membrane is increased by the classical ferroptosis activators erastin-1 and ras-selective lethal small molecule 3 (RSL3) upon cytosolic calcium influx and repairs the ferroptotic plasma membrane. ESCRT-III- and VPS4-induced macroautophagy, ESCRT-0-initiated microautophagy. ESCRT-I, ESCRT-II, ESCRT-III, ALIX, and VPS4A are recruited to damaged lysosomes and precede lysophagy, indicating that ESCRT is a potential target to overcome drug resistance during tumor therapy.
Introduction
The cell plasma membrane (PM) made of glycerophospholipids separates the inner and outer parts of the cell. Under physiological conditions, it acts as a gatekeeper to protect cells from the environment (1). In pathological situations, it undergoes structural and functional changes, resulting in cell damage (2). According to the recommendations of the Nomenclature Committee on Cell Death, cell death can be divided into accidental cell death (ACD) and regulated cell death (RCD); the latter, also called “active” cell death, can be further classified into apoptotic and non-apoptotic cell death with different morphological, genetic, and biochemical characteristics (3). Non-apoptotic cell death, such as necroptosis (4), pyroptosis (5), and ferroptosis (6, 7), triggers various types of plasma membrane damage to suppress tumor growth and could be used in the treatment of cancer against apoptosis resistance. Apoptosis, necroptosis, and pyroptosis all feature a passive “suicide” form promoting molecular processes and form a few nanometers of plasma membrane pores, leading to a catastrophic bursting of the cell (8). A rise in cytosolic calcium (Ca2+) has been related to the execution of different cell death processes, including apoptosis, necroptosis, pyroptosis, and unregulated necrosis (9). Apoptosis involves fragmentation and margination of chromatin, as well as generation of apoptotic bodies and plasma membrane blebbing (10). Necroptosis has been demonstrated to involve swelling of cells and was shown to be associated with the generation of released plasma membrane broken pieces as vesicles. Necrotic cells lose the integrity of the plasma membrane and thereby release intracellular damage-associated molecular patterns. Necroptosis signaling can be switched from apoptosis through anti-caspase mechanisms such as genetic ablation of caspase-8 (11). Pyroptosis relies on the protein gasdermin D (GSDMD) to precede rapid necrotic cell death by intensive blebbing. However, such features have not been observed during ferroptosis. Correspondingly, ferroptosis damages the active cell integrity with the accumulation of lipid peroxides due to a defective antioxidant response of the cell (12).
However, cells undergoing necroptosis do not always die. The known repair mechanisms involving the endosomal sorting complex required for transport (ESCRT) machinery outward vesiculates or sheds damaged membranes (13) and plays a critical counterbalancing role in sorting and downregulating activated cell-surface receptors (14) and repairing damaged plasma membranes to maintain membrane integrity (15), thus delaying cell death pathways, including apoptosis (16), necroptosis (17), pyroptosis (18, 19), ferroptosis (20), autophagy (21), and endosomal processing (22). In this review, we summarized the relationship between the ESCRT pathway and regulated cell death, aiming to identify ESCRT as a potential target to overcome drug resistance during tumor therapy.
Summary of ESCRT function in cell death
ESCRT components are implicated in cellular events such as viral budding, cell signaling regulation (23), modulation of cytokine release, communication with immune cells (24), vesicle budding, and sustained cell viability and survival (25, 26). Four different multimeric protein complexes comprise ESCRT-0 to ESCRT-III machinery sequentially recruited from the cytoplasm to the endosomal membrane: ESCRT-0 (Vps27), ESCRT-I (Vps23, Vps28, Vps37, MVB12), ESCRT-II (Vps22, Vps25, Vps36), and ESCRT-III (Did2, Vps2, Vps20, Vps24, Snf7, Vps60, Chm7, Ist1). Another ESCRT machinery-functioning subcomplex, the AAA-ATPase complex vacuolar protein sorting-associated 4 (VPS4) and apoptosis-linked gene 2 (ALG-2)–interacting protein X (ALIX) protein, are potential decisive factors in the function of ESCRT-I and ESCRT-II. The ESCRT machinery mediates the “reverse-topology” cellular membrane scission mechanism (27) to process membrane remodeling, including cytokinesis, plasma membrane and lysosomal membrane repair, and nuclear envelope reformation (22). ESCRT-0 triggered by Ca2+ influx recruits other ESCRT machinery to endosomes during multivesicular body formation (MVB) sorting. Then, ESCRT-I binds ubiquitinated cargo and canonically activates ESCRT-II recruitment. ESCRT-III assembly starts with Vps20, followed by the core polymer subunits Vps2 and Vps24 binding Snf7 in tandem and finally recruiting VPS4, which dissociates the ESCRT structure from the endosomal membrane (28–31). Subsequently, the assembly of ESCRT-III machinery was recruited to wounds, assembled on the inner surface of the membrane neck to form single- or multiple-stranded polymorphic filaments shapes from spirals (32, 33) to conical spirals (34) and tubular helices (35), and then broken down within the lysosomal lumen to create plasma membrane blebs and intraluminal vesicles (ILVs) (36), mediate membrane scission, remove damaged parts of cell membranes away from the cytoplasm (27), and shed off the extracellular space.
ESCRT-I, ESCRT-II, ESCRT-III, VPS4A, and ALIX were recruited to damaged lysosomes and mediated lysosomal membrane repair. In mammalian cells, the ESCRT-III complex consists of 12 subunits: charged multivesicular body protein 1A (CHMP1A), CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4A, CHMP4B, CHMP4C, CHMP5, CHMP6, CHMP7, and increased sodium tolerance 1 (IST1) (37). ESCRT-III plays a key role in the repair of damaged plasma membranes in various types of regulated cell death, such as necroptosis, pyroptosis, and ferroptosis. The inhibition of ESCRT-III machinery through genetic depletion of its core components increases susceptibility to anticancer agent-induced cell death (1), indicating that ESCRT III is a potential target to overcome drug resistance during tumor therapy.
Apoptosis
Apoptosis is the most extensively investigated type of regulated cell death characterized by the sequential activation of cysteine-aspartic protease caspases (12). It is composed of the extrinsic and intrinsic major pathways that activate and cleave the downstream “executioner” caspase-3 and “initiator” caspase-7. Bcl-2 family effector proteins specific effector molecules B-cell/CLL lymphoma 2 (BCL2)-associated protein X (Bax) aBak and Bok (19, 38) regulated the integrity of the outer mitochondrial membrane permeabilization, releasing proteins of the mitochondrial intermembrane space into the cytosol. The intrinsic endolysosomal pathway in cellular stress (39) is enriched in phagosomes, and exosomes dominate the control of membrane budding and scission (36) to control apoptosis (40) in response to DNA damage (41). The extrinsic pathway is activated upon the binding of extracellular ligands to cell surface death receptors.
The ESCRT machinery components apoptosis-linked gene-2 (ALG-2) and exosomes interact with apoptosis-linked gene interacting protein X (ALIX) and are suggested to be a bridge between the endolysosomal system and apoptosis. ALG-2 is a penta-EF-hand protein enriched in phagosomes, also known as programmed cell death 6–interacting protein (PDCD6IP), which is regarded as apoptotic machinery in T-cell lines (42) and participates in T-cell receptor-, Fas-, and glucocorticoid-induced programmed cell death. ALIX interacts with Cbl-interacting protein of 85 kDa (CIN85)/SRC homology 3 (SH3) and sensitizes astrocytes to apoptosis in response to DNA damage (43, 44). The ALIX–ALG-2 complex undergoes apoptosis in a Ca2+/K+-dependent manner (45), while these events individually do not activate the downstream caspase cascade to eventually lead to apoptosis.
Tumor susceptibility gene 101 (TSG101) is an ESCRT-1 protein homologue of the yeast class E VPS protein complex ESCRT-III (46) that directly participates in mitigating ER stress-mediated apoptosis. The association of TSG 101 with ALIX prevents predisposition to apoptosis, but deregulating cytosolic Ca2+ and upregulating the levels of ALG-2 could disrupt this process. In healthy cells, when cytosolic Ca2+ is low, mahogunin RING finger 1 (MGRN1)-mediated ubiquitination of the ESCRT-I protein tumor susceptibility gene 101 (TSG101) promotes amphisomal–lysosomal and endolysosomal degradation pathways (47, 48) and helps maintain low levels of ALIX and ALG-2 as well as cell viability. MGRN1 depletion leads to cell surface glycoprotein mammalian PrP (CtmPrP)-mediated ER stress, and an increase in cytosolic Ca2+ results in the ALIX–ALG-2 protein interaction. Overexpression of TSG101 also increases ALIX and ALG-2 levels, eventually eliciting predisposition to death in selected brain regions or myocardial apoptosis during embryonic development (16).
Under pathological conditions, the ESCRT-III-associated protein CHMP4B could also interact with ALIX-ALG-2 and participate in the endolysosomal system or cell apoptosis. CHMP4B mutation prevented early programmed cell death caused by overexpression of ALIX (49). However, increasing CHMP4B levels were accompanied by the upregulation of Fas receptor (Fas), Fas ligand (FasL), active caspase-8, and caspase-3 in neurons, which implicated a proapoptotic function in neuronal cells induced by hemin stimulation following intracerebral hemorrhage (ICH) via the extrinsic apoptotic pathway (50) (Figure 1 and Table 1).
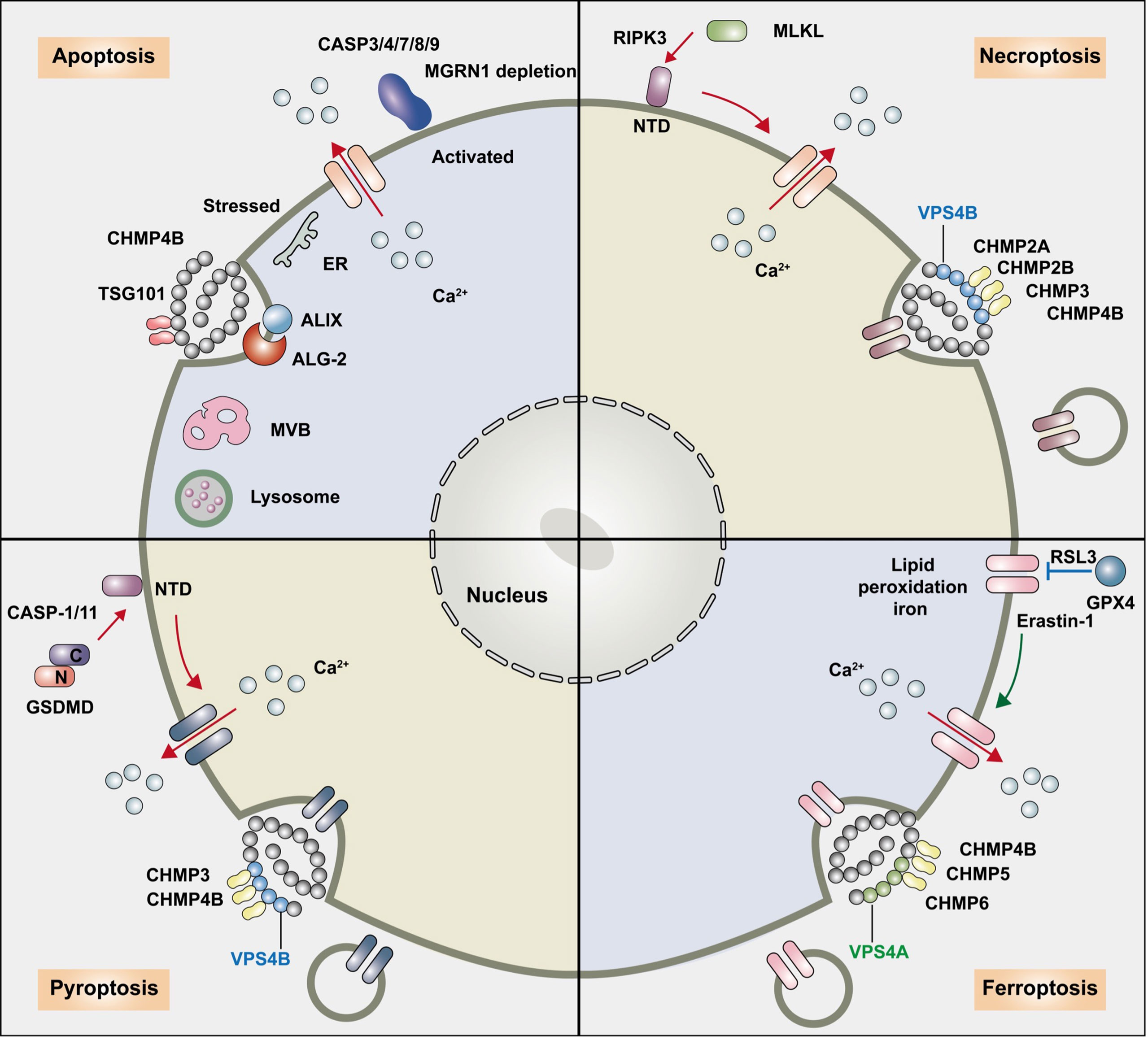
Figure 1 The relationship between the ESCRT pathway and regulated cell death. The endosomal sorting complex required for transport (ESCRT) machinery participates in the following regulated cell death: apoptosis, necroptosis, pyroptosis, ferroptosis and autophagy. ESCRT-I overexpression and ESCRT-III-associated charged multivesicular body protein (CHMP) 4B participate in apoptosis, and ESCRT-1 protein TSG 101 maintains low levels of ALIX and ALG-2 and prevents predisposition to apoptosis. The ESCRT-III components CHMP2A and CHMP4B are recruited to broken membrane bubble sites with the requirement of extracellular Ca2+, remove membrane vesicles from cells, and delay the time required for active MLKL to mediate necroptosis, thus preserving cell survival. CHMP4B disturbed pyroptosis by recruiting around the plasma membrane neck to remove the GSDMD pores and preserve plasma membrane integrity depending on Ca2+ influx. The accumulation of the ESCRT-III subunits CHMP5 and CHMP6 in the plasma membrane is increased by the classical ferroptosis activators erastin-1 and ras-selective lethal small molecule 3 (RSL3) upon cytosolic calcium influx and repairs the ferroptotic plasma membrane.
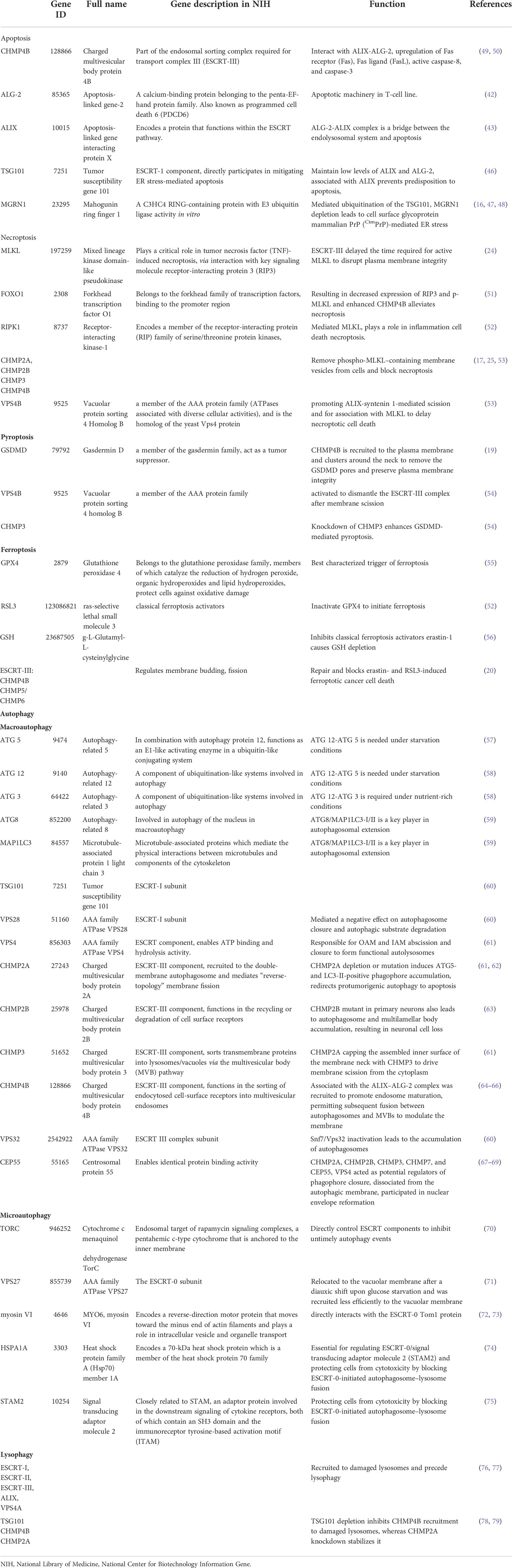
Table 1 The endosomal sorting complex required for transport (ESCRT) machinery plays a key role in the repair of damaged plasma membranes in regulated cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy.
Necroptosis
Necroptosis is characterized by permeability and finally plasma membrane rupturing in necrotic cells, different from shrinking and blebbing of plasma membranes in apoptotic cells (4, 19). Phosphatidylserine (PS) is localized to the inner leaflet of the plasma membrane of healthy cells, and exposure revealed small plasma membrane (PM) “bubbles” of broken plasma membrane released from the cell surface. Phospholipid scrambling (exposing PS) and disrupting plasma membrane integrity dominated the initial extracellular death signals and initiated necroptosis. Necroptosis can be mediated by tumor necrosis factor alpha (TNFα) and/or Fas or activated by the execution of necroptosis receptor-interacting kinase-1 (RIPK1)/RIPK3-mediated phosphorylation of mixed lineage kinase domains such as pseudokinase (MLKL) (24, 52). MLKL plays a critical role in tumor necrosis factor (TNF)-induced necroptosis via interaction with receptor-interacting protein 3 (RIP3). MLKL oligomerization-mediated PM disruption occurs prior to the loss of PM integrity, resulting in a rapid Ca2+ influx into the cell (17, 80) and induced bubbles, thus making the core machinery of the unique “membrane-explosive” necroptosis cell death pathway (80).
However, cells that are exposed to PS upon MLKL activation can be “resuscitated” and survive. The calcium-dependent ESCRT-III machinery plays a wider role in modulating various types of RCD by delaying cell membrane rupture. Sustained viability by ESCRT can either antagonize (pyroptosis) or enhance (necroptosis) the release of signaling events upstream of terminal effector activation (81). ESCRT-III greatly delayed the time required for active MLKL to disrupt plasma membrane integrity and the onset of membrane permeabilization, sustained the integrity of the plasma membrane, and enhanced necroptosis bubble formation, therefore sustaining survival of the cell (17, 82). When MLKL activation is subsequently halted, cells have sufficient time to permit surrounding cells to activate intracellular signaling pathways such as the cytokines C–X–C motif chemokine ligand 1 (CXCL1) and CXCL10 (80). Previous studies have indicated that the ESCRT pathway can remove phospho-MLKL–containing membrane vesicles from cells and block necroptosis, and CHMP4B decreases necroptosis through different transcriptional activators (24, 83). MLKL localizes to sites of broken membrane bubbles with the requirement of extracellular Ca2+, and ESCRT-III components CHMP2A or CHMP4B are then recruited and reduce cell membrane damage caused by p-MLKL, thus preserving survival despite MLKL activation in kidney transplantation (17, 25). The presence of the N-terminal ubiquitin-binding UEV domain in TSG101 and the disassembly complex VPS4B is critical in promoting ALIX-syntenin 1-mediated scission and for association with MLKL to delay necroptotic cell death (84). Forkhead transcription factor O1 (FOXO1) binds to the specific region on the CHMP4B promoter, and enhanced CHMP4B alleviates necroptosis in microglia by binding to the promoter region, resulting in decreased expression of RIP3 and p-MLKL and protecting against cell death after traumatic brain injury (TBI), thus improving neurological function recovery (51). On the other hand, the activation of necroptosis might alter the expression levels of ESCRT III proteins as a compensatory mechanism. CHMP2B is a marker for granulovacuolar degeneration (GVD) bodies in the Alzheimer’s disease (AD) brain (85). A significantly increased expression of CHMP2B, CHMP3, and VPS4B was shown in pMLKL+ neurons and counterbalanced necroptosis (53). CHMP2B mutation was associated with neurodegeneration in frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (86) (Figure 1 and Table 1).
Pyroptosis
Pyroptosis is a form of regulated necrosis induced by the pore-forming protein gasdermin D (GSDMD) that damages the plasma membrane (19). Caspase-1, caspase-4, caspase-5, and caspase-11 cleave GSDMD, release the N-terminal domain, and then translocate into the plasma membrane. While components of ESCRT-III mainly repaired the plasma membrane damage and function in preserving cell survival when the activity of the effectors is sufficiently low or the engaged pathway is disrupted prior to lysis (87). During pyroptosis, after cytosolic caspases cleave GSDMD to form nanoscale (10–15-nm) membrane pores, CHMP4B is recruited to the plasma membrane and clusters around the neck to remove the GSDMD pores and preserve plasma membrane integrity, thus limiting proinflammatory cytokine interleukin-1β (IL-1β) and IL-18 release through GSDMD pores (19), which rely on the influx of Ca2+. In contrast, VPS4B ATPase is activated to dismantle the ESCRT-III complex after membrane scission. Knockdown of CHMP3 enhances GSDMD-mediated pyroptosis. Recently, Kai et al. revealed that Ca2+ and K+ influx as well as activation of NLR family pyrin domain containing 3 (NLRP3)-dependent IL-1β release resulted in pyroptosis, and Mycobacterium tuberculosis (Mtb) infection spread to neighboring cells. Upon NLRP3 inflammasome activation, ESCRT composite ALG-2 and ALIX recruitment repaired plasma membrane damage in macrophages (54) (Figure 1 and Table 1).
Ferroptosis
Ferroptosis is a caspase-independent form of regulated pathological necroinflammation (56) and activation of the innate immune system, causing cell metabolic state changes involving cell enlargement, organelle swelling, membrane rupture, mitochondrial shrinkage, and increased outer membrane density (52, 88). Ferroptosis proceeds even in the absence of key effectors of apoptosis (Bax, Bak, and caspases) or necroptosis (MLKL, RIPK1, and RIPK3) (89). Cells dying by ferroptosis primarily exhibit shrunken and damaged mitochondria by electron microscopy, with few other morphological changes evident prior to the point of cell death (90, 91). The ferroptosis pathway occurs in cells involving targeting the amino acid antiporter system xc– or iron transport molecule shuttles such as transferrin (92) and lactotransferrin (93) or is activated after intracellular antioxidant enzymes are blocked (94). Glutathione peroxidase 4 (GPX4) is the best-characterized trigger of ferroptosis. Ferroptosis only occurs when the function of GPX4 is inactivated by ras-selective lethal small molecule 3 (RSL3) (55) or when it inhibits erastin-1, which causes g-L-glutamyl-L-cysteinylglycine (GSH) depletion (52, 56). Then, iron-dependent membrane phospholipid hydroperoxide accumulation precedes a sustained increase in cytosolic Ca2+ (95, 96), ultimately forming nanopores to trigger plasma membrane rupture and release of intracellular components (97).
ESCRT-III-dependent membrane repair blocks ferroptosis through association with the plasma membrane model and acts as a protective mechanism (20). The accumulation of the ESCRT-III subunits CHMP5 and CHMP6 in the plasma membrane is increased by the classical ferroptosis activators erastin-1 and RSL3, and the increase in cytosolic calcium influx relies on endoplasmic reticulum stress. CHMP5 or CHMP6 depletion increases erastin- and RSL3-induced ferroptosis (98). ESCRT-III is recruited to the plasma membrane to form CHMP4B puncta, removing pores from the plasma membrane, shedding them in ectosomes, and reducing lipid peroxidation as well as DAMP release, causing damaged membrane sections to be removed by endocytosis to delay ferroptosis membrane damage (81, 98–100). ESCRT-III also impacts cytokine secretion in ferroptotic cells (20). Apoptosis-inducing factor mitochondria-associated 2 (AIFM2)-dependent ESCRT-III recruitment regulates membrane budding, fission, and repair and blocks erastin- and RSL3-induced ferroptotic cancer cell death, which is responsible for ferroptosis resistance (101) (Figure 1 and Table 1).
Autophagy
Autophagy is a catabolic lysosomal degradation pathway responsible for nutrient recycling, protein and organelle quality control, and degradation and recycling of cellular material to maintain cell homeostasis and cope with stressful conditions (21). Autophagy is characterized by phagophores forming a small crescent-shaped membrane that stretches and seals cytoplasmic cargoes in double-membrane autophagosomes (21). Autophagosomes are nucleated from endoplasmic reticulum (ER) sites called omegasomes by phosphatidyl inositol 3 phosphate (PI3P) kinase complex class III and PI3P-binding proteins. Autophagosomes can fuse with early endosomes and MVBs to generate an intermediate compartment, the amphisome, which ultimately fuses with lysosomes (102). There are three most degradative systems types: the ubiquitin proteasome system, endocytosis, and autophagy vesicular processes converging on the lysosome (102). Lysosomes originate from endolysosomes or autolysosomes, act as single membrane-bound organelles, and recycle cellular nutrients through the outer membrane of mature autophagosomes and release acid hydrolases to degrade the autophagosomal content; they are also critical junctures between autophagy and endocytosis (103) as well as essential processes for maintaining intracellular homeostasis. ESCRT participates in four coexisting types of autophagy processes in a cell, namely, the recycling of cytosolic components by macroautophagy (often simply called autophagy), endosomal microautophagy, chaperone-mediated autophagy (CMA), and lysosomal and autophagic cell death pathways, ultimately directed to the lysosome for degradation (104). In macroautophagy, autophagic substrates are transported to the vacuole by autophagosomes with double-membrane structures; in microautophagy, substrates are directly engulfed by the vacuolar membrane.
Macroautophagy
Macroautophagy is the main cytosolic degradative system involved in the formation of preautophagosomal structures called omegasomes (105). Autophagosome formation fuses with the lysosome to form an autolysosome, including extending, closing, and fusioning the isolation membrane (IM) dependent on autophagy-related (ATG) proteins to enwrap cargoes, thus initiating the macroautophagy degradation pathway characterized by double-membrane vesicles, termed autophagosome maturation, and encompassing multiple lysosomal-dependent mechanisms. The ESCRT machinery rescued slightly damaged lysosomes, and the ESCRT machinery and ATG proteins interact between endocytosis and macroautophagy to form a bridge between the endolysosomal system and cell death (82, 106). ATG12-ATG5 is needed under starvation conditions, while ATG12-ATG3 is required under nutrient-rich conditions (57, 58). Autophagosome closure requires a similar membrane scission machinery as ESCRT-III (107). ATG8/microtubule-associated protein 1 light chain 3 (MAP1LC3)-I/II is a key player in autophagosomal extension. Upon closure, LC3-II on the outer autophagosomal membrane (OAM) is delipidated and released to the cytosol, while LC3-II associated with the inner autophagosomal membrane (IAM) is degraded upon autophagosome–lysosome fusion (59).
Autophagosomal membranes can serve as activation platforms for intracellular death-inducing signaling complexes (iDISCs) to initiate caspase-8-dependent apoptosis. Mutations in ESCRT I (TSG101 and VPS28), II (SNF8, VPS22, VPS25), and III (VPS32) affected fusion between endosome and autophagosome accumulation to produce amphisomes (48, 60). The ESCRT-I subunit VPS28 variant mediated a negative effect on autophagosome closure and autophagic substrate degradation (108). The ESCRT component AAA-ATPase VPS4 is responsible for OAM and IAM abscission and closure to form functional autolysosomes (61) as well as dissociate the ESCRT machinery from the endosomal membrane (60). Prior to lysosomal recruitment, the autophagosome closure regulator ESCRT-III component CHMP2A is recruited to the double-membrane autophagosome and mediates “reverse-topology” membrane fission (61, 62), capping the assembled inner surface of the membrane neck with CHMP3 to drive membrane scission from the cytoplasm, shape MVB formation, and cut the membrane, and nuclear envelope reformation and remodeling processes involve regulating membrane fission phagophore closure (22, 61, 63, 109). CHMP2A depletion or mutation induces iDISC-mediated non-canonical caspase-8 activation on immature autophagosomal membranes and leads to ATG5- and LC3-II-positive phagophore accumulation, and redirects protumorigenic autophagy to apoptosis in osteosarcoma and neuroblastoma cells, thus inhibiting mouse xenograft model tumor growth (109–111), which may open new avenues for therapeutic targeting of autophagy in cancer.
Prior to final fusion with lysosomes, CHMP4B associated with the ALIX–ALG-2 complex was recruited to promote endosome maturation, permitting subsequent fusion between autophagosomes and MVBs to modulate the membrane (49, 64, 65, 112). ESCRT-III Snf7/Vps32 inactivation leads to the accumulation of autophagosomes, probably due to a blockage of autophagic flux in HeLa cells in the late stage of autophagosome formation (63, 66). Starvation-induced ESCRT-III components (CHMP2A, CHMP2B, CHMP3, CHMP7, and CEP55) as well as VPS4 acted as potential regulators of phagophore closure, dissociated from the autophagic membrane, participated in nuclear envelope reformation, and directly mediated membrane scission in human bone osteosarcoma epithelial cells (U-2 OS), HeLa cells, and human retinal pigment ephitilial-1 cells under basal and starved conditions (61, 67–69). The CHMP2B mutant in primary neurons also leads to autophagosome and multilamellar body accumulation, resulting in neuronal cell loss (63). The accumulation of autophagosomes in plant ESCRT mutants may result from inefficient closure of autophagosomes. The ESCRT-II subunit VPS36 localizes to endosomes and the plasma membrane. In Arabidopsis, autophagic turnover of plastids decreased in the ESCRT-related CHMP1 (VPS46) mutant due to defects in phagophore maturation and transport (113) (Figure 2 and Table 1).
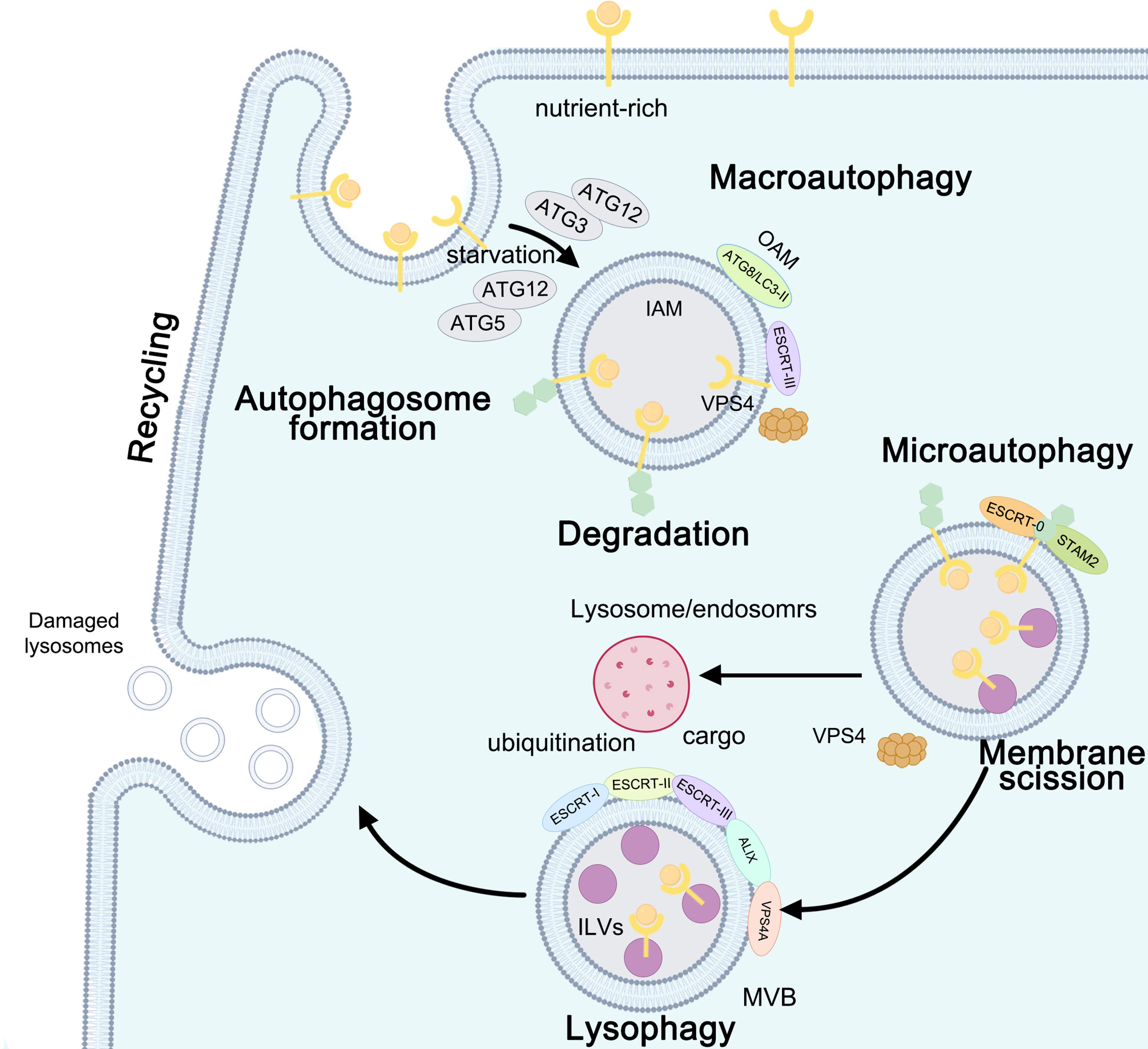
Figure 2 Autophagy: the function between ESCRT and macroautophagy, microautophagy, and lysophagy. ESCRT-III- and VPS4-induced macroautophagy is involved in the formation of autophagosomes: ATG12-ATG5 is needed under starvation conditions, while ATG12-ATG3 is required under nutrient-rich conditions. Upon closure, ATG8/light chain 3 (LC3)-II on the outer autophagosomal membrane (OAM) is delipidated and released to the cytosol, while LC3-II associated with the inner autophagosomal membrane (IAM) is degraded upon autophagosome-lysosome fusion. ESCRT-0- and signal transducingadaptor molecule 2 (STAM2)-initiated microautophagy requires membrane scission. ESCRT-I, ESCRT-II, ESCRT-III, ALIX, and VPS4A are recruited to damaged lysosomes and precede lysophagy.
Microautophagy
Microautophagy requires membrane scission at the vacuolar membrane, similar to ILV formation at the MVE. Microautophagy occurs in the following three different membrane dynamics: protrusion of the lysosomal membrane to engulf the cargo, invagination of the lysosomal membrane or endosome to entrap the cargo inside the lysosome or endosomes (114). The ESCRT machinery plays catabolic roles in cell starvation through the sorting and degradation of cytosolic proteins and lipids, similar to a hub-like system involved in the final maturation of both late endosomes and autophagosomes. ESCRT participates in plasma membrane scission as well as in cytosolic components, proteins, and lipids during starvation and affects the fusion of vesicles with lysosomes to form autolysosomes (115, 116). Upon ER stress-induced macrolipophagy in budding yeast, the whole ESCRT machinery is recruited to the scission site on the vacuolar membrane and lipid droplets to remove the ER whorls and Snf7, thus clearing the defective proteasomes (117). The ESCRT-0 subunit Vps27 relocated to the vacuolar membrane after a diauxic shift upon glucose starvation and was recruited less efficiently to the vacuolar membrane; endosomal target of rapamycin (TOR) signaling complexes (TORC) directly control ESCRT components to inhibit untimely autophagy events (70, 71). In mammalian cells, on the one hand, myosin VI directly interacts with the ESCRT-0 Tom1 protein; on the other hand, myosin VI interacts with autophagy adaptors and optineurin, which are involved in selective autophagy (72, 73). Heat shock protein family A (Hsp70) member 1A (HSPA1A) is essential for regulating ESCRT-0/signal transducing adaptor molecule 2 (STAM2) and protecting cells from cytotoxicity by blocking ESCRT-0-initiated autophagosome–lysosome fusion (74), abolishing autophagic flux in cellular thermoresistance, significantly reducing thermal cytotoxicity, and promoting cell survival (75) (Figure 2 and Table 1).
Lysophagy
Lysophagy occurs only when the repair mechanism fails to be initiated due to extensive damage to the lysosomal membrane and the inability to recruit ESCRT repair complexes. Damaged lysosomes are selectively eliminated by lysophagy when ESCRT-mediated membrane repair fails (118). Rescue of lysosomes mediated by ESCRT, removal of damaged lysosomes via lysophagy, and lysosome biogenesis can restore lysosome function and improve autophagic clearance (118). ESCRT-mediated membrane remodeling may contribute to both immediate and delayed responses to lysosomal damage by the multivesicular body pathway (76, 77). ESCRT-III is recruited to damaged lysosomes, which requires ESCRT-I and ALIX. ESCRT-I, ESCRT-II, ESCRT-III, ALIX, and VPS4A are recruited to damaged lysosomes and precede lysophagy. TSG101 depletion inhibits CHMP4B recruitment to damaged lysosomes, whereas CHMP2A knockdown stabilizes CHMP4B. ESCRT recruitment protects cells against cell death caused by lysosome damage (78, 79). After membrane damage, ubiquitination is delayed by 30 min compared to the recruitment of ESCRT components (77). Dampening ESCRT responses by depleting TSG101 and ALIX slows or completely blocks this rapid recovery and thereby implicates ESCRT function in lysosomal repair (119, 120) (Figure 2 and Table 1).
ESCRT as a potential target for tumor therapy
Tumor cells develop drug-resistance effects to escape cell death and cause treatment failure. ESCRT contributes to resistance to cell death and is generally regarded as a tumor-suppressor gene. The ESCRT-0 protein Hrs was upregulated in tumor specimens of the stomach, colon, liver, cervix, and melanoma (121). The ESCRT-I subunit Vps37A was significantly reduced in the hepatocellular carcinoma cell line (122). ESCRT-III subunit CHMP1A overexpression inhibited tumor xenograft growth of human pancreatic carcinoma cells (123). Upregulation of CHMP3 was detected in non-small cell lung carcinoma (124). CHMP5 or CHMP6 confers resistance to ferroptotic PANC1 and HepG2 human cancer cell death (98). CHMP4CT232 is associated with increased susceptibility to cancer tumorigenesis in ovarian cancer (125) and male genital tract, prostate, and skin cancers (126). Inhibition of these ESCRT proteins could block membrane remodeling and induce cancer cell death. In addition, observed from the uterine corpus endometrial carcinoma (UCEC) dataset of The Cancer Genome Atlas database (TCGA) database (https://portal.gdc.cancer.gov/) and GEPIA (http://gepia.cancer-pku.cn/), the ESCRT-III components CHMP2A, CHMP4B, CHMP4C, CHMP5, and CHMP6 were significantly related to tumor-infiltrating lymphocytes (TILs), revealing that a deregulated ESCRT pathway would offer a potential target or effective markers in cancer immunotherapy.
Discussion
Apoptosis is a non-inflammatory form of PCD mediated by activation of apoptotic caspases and can occur either via an extrinsic or an intrinsic pathway that converges on the activation of the executioner caspase-3, 6, and 7 (12). At the terminal stage of apoptosis, cells are phagocytosed in vivo by scavenger cells, such as macrophages or neutrophils. However, if these cells are not removed in a timely fashion, as is the case in vitro, they progress to a final phase called secondary necrosis characterized by cytoplasmic swelling and plasma membrane damage, similar to the phenotype of cells undergoing pyroptosis or necroptosis (8, 9). Necroptosis is triggered by the activation of receptor-interacting protein kinase-3 (RIPK3), which phosphorylates the pseudokinase MLKL, causing it to translocate to the plasma membrane to induce cell permeabilization (56). Pyroptosis is triggered primarily by activation of inflammatory caspases, which include caspase-1 and caspase-11 (caspase-4/-5 in humans) (19). Autophagy is a process of bulk protein degradation in which cytoplasmic components, including organelles, are enclosed in double-membrane structures called autophagosomes and delivered to lysosomes or vacuoles for degradation (21). The ESCRT machinery is involved in the above regulated cell death processes, such as apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy, in a Ca2+-dependent manner (47, 48). Ca2+ influx plays an important role in the activation and recruitment of the ESCRT-III complex, leading to the repair of damaged plasma membranes during cell death. The ESCRT-1 protein TSG 101 maintains low levels of ALIX and ALG-2 and prevents predisposition to apoptosis (47, 48). ESCRT-III components CHMP2A or CHMP4B are recruited to the broken membrane bubble sites with the requirement of extracellular Ca2+, delaying the time required for active MLKL to mediate necroptosis and thus preserving cell survival despite MLKL (17, 25). Upon Ca2+ and K+ influx activating the NLRP3 inflammasome, the ESCRT composites ALG-2, ALIX, and CHMP4B are recruited around the plasma membrane neck to remove the GSDMD pores and block pyroptosis and thus preserve plasma membrane integrity (19, 54). The accumulation of the ESCRT-III subunits CHMP5 and CHMP6 in the plasma membrane is increased by the classical ferroptosis activators erastin-1 and ras-selective lethal small molecule 3 (RSL3) upon cytosolic calcium influx and repairs the ferroptotic plasma membrane (98). ESCRT is also involved in membrane scission machinery on autophagosome closure, and mutations in ESCRT I (TSG101 and VPS28), II (SNF8, VPS22, VPS25), and III (VPS32) affect fusion between endosome and autophagosome accumulation to produce amphisomes (48, 60).
Conclusion
In summary, ESCRT provides time to the dying cell, and ESCRT-dependent membrane repair negatively regulates cell death: ESCRT-I and -III participate in apoptosis, ESCRT-III mediates necroptosis, pyroptosis, and ferroptosis, ESCRT-0 initiates microautophagy, ESCRT-III induces macroautophagy, and ESCRT-I, -II, and -III precede lysophagy. All these types of cell death can cause plasma membrane damage through different mechanisms; however, some essential mechanisms need to be clarified, such as the exact time point or target site ESCRT-rescued cell death, and it will be interesting to investigate the role of ESCRT as a potential target to overcome drug resistance in cancer cells.
Author contributions
YY and MW analyzed and interpreted the data. Y-YZ and SG collected information. YY, MW, Y-YZ, S-ZZ and SG worked equally as major contributors in writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81902628, 81900036), Clinical Research Project of Shanghai Health Commission (202040455), and Shanghai Songjiang District Science and Technology Commission for funding the project (20SJKJGG139, 20SJKJGG304)
Acknowledgments
We thank Elsevier Author Services (https://webshop.elsevier.com/language-editing-services/language-editing/) and for its linguistic assistance during the preparation of this manuscript. We thank American Journal Experts (AJE)(https://www.aje.com) for helping us revise the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liu J, Kang R, Tang D. ESCRT-III-mediated membrane repair in cell death and tumor resistance. Cancer Gene Ther (2021) 28(1-2):1–4. doi: 10.1038/s41417-020-0200-0
2. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol (2018) 19(5):281–96. doi: 10.1038/nrm.2017.138
3. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Kroemer G. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
4. Ros U, Peña-Blanco A, Hänggi K, Kunzendorf U, Krautwald S, Wong WW, et al. Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep (2017) 19(1):175–87. doi: 10.1016/j.celrep.2017.03.024
5. Vasconcelos NMD, Opdenbosch NV, Gorp HV, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ (2019) 26(1):146–61. doi: 10.1038/s41418-018-0106-7
6. Lglic A, Kulkarni CV, Rappolt M. Advances in biomembranes and lipid self-assembly (Academic Press), Vol. 28. (2018).
7. Andrews NW, Corrotte M. Plasma membrane repair. Curr Biol (2018) 28(8):R392–R7. doi: 10.1016/j.cub.2017.12.034
8. Espiritu RA. Repairing plasma membrane damage in regulated necrotic cell death. Mol Biol Rep (2021) 48(3):2751–9. doi: 10.1007/s11033-021-06252-w
9. Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol (2003) 5(12):1041–3. doi: 10.1038/ncb1203-1041
10. Elmore S. A review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi: 10.1080/01926230701320337
11. Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: The expanding network of nonapoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
12. Green DR. The coming decade of cell death research: Five riddles. Cell (2019) 177(5):1094–107. doi: 10.1016/j.cell.2019.04.024
13. Romero M, Keyel M, Shi G, Bhattacharjee P, Roth R, Heuser JE, et al. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ (2017) 24(5):798–808. doi: 10.1038/cdd.2017.11
14. Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature (2009) 458(7237):445–52. doi: 10.1038/nature07961
15. Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci (2017) 42(1):42–56. doi: 10.1016/j.tibs.2016.08.016
16. Kaul Z, Chakrabarti O. Tumor susceptibility gene 101 regulates predisposition to apoptosis via ESCRT machinery accessory proteins. Mol Biol Cell (2017) 28(15):2106–22. doi: 10.1091/mbc.e16-12-0855
17. Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell (2017) 169(2):286–300 e16. doi: 10.1016/j.cell.2017.03.020
18. Neggers JE, Paolella BR, Asfaw A, Rothberg MV, Skipper TA, Yang A, et al. Synthetic lethal interaction between the ESCRT paralogue enzymes VPS4A and VPS4B in cancers harboring loss of chromosome 18q or 16q. Cell Rep (2020) 33(11):108493. doi: 10.1016/j.celrep.2020.108493
19. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362(6417):956–60. doi: 10.1126/science.aar7607
20. Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ (2021) 28(5):1644–57. doi: 10.1038/s41418-020-00691-x
21. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature (2008) 451(7182):1069. doi: 10.1038/nature06639
22. Vietri M, Radulovic M, Stenmark H. The many functions of ESCRTs. Nat Rev Mol Cell Biol (2020) 21(1):25–42. doi: 10.1038/s41580-019-0177-4
23. Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell (2010) 143(7):1136–48. doi: 10.1016/j.cell.2010.11.034
24. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, additionally, regulates endosomal trafficking and extracellular vesicle generation. Immunity (2017) 47(1):51–65.e7. doi: 10.1016/j.immuni.2017.06.001
25. Gong YN, Guy C, Crawford JC, Green DR. Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle (2017) 1748:1748–60. doi: 10.1080/15384101.2017.1371889
26. Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol (2007) 8(5):355–68. doi: 10.1038/nrm2162
27. Schneberg J, Lee IH, Iwasa JH, Hurley JH. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol (2017) 18(1):5–17. doi: 10.1038/nrm.2016.121
28. Mierzwa BE, Chiaruttini N, Redondo-Morata L, Joachim MVF, KöNig J, Larios J, et al. Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat Cell Biol (2017) 19(7):787–98. doi: 10.1038/ncb3559
29. Saksena S, Wahlman J, Teis D, Johnson AE, Emr SD. Functional reconstitution of ESCRT-III assembly and disassembly. Cell (2009) 136(1):97–109. doi: 10.1016/j.cell.2008.11.013
30. Schoneberg J, Pavlin MR, Yan S, Righini M, Lee IH, Carlson LA, et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science (2018) 362(6421):1423–8. doi: 10.1126/science.aad8305
31. Maity S, Caillat C, Miguet N, Sulbaran G, Effantin G, Schoehn G, et al. VPS4 triggers constriction and cleavage of ESCRT-III helical filaments. Sci Adv (2019) 5(4):eaau7198–206. doi: 10.1126/sciadv.aau7198
32. Chiaruttini N, Redondo-Morata L, Colom A, Humbert F, Lenz M, Scheuring S, et al. Relaxation of loaded ESCRT-III spiral springs drives membrane deformation. Cell (2015) 163(4):866–79. doi: 10.1016/j.cell.2015.10.017
33. Shen QT, Schuh AL, Zheng Y, Quinney K, Wang L, Hanna M, et al. Structural analysis and modelling reveals new mechanisms governing ESCRT-III spiral filament assembly. J Cell Biol (2014) 206(6):763–77. doi: 10.1083/jcb.201403108
34. Mccullough J, Clippinger AK, Talledge N, Skowyra ML, Saunders MG, Naismith TV, et al. Structure and membrane remodelling activity of ESCRT-III helical polymers. Science (2015) 350(6267):1548. doi: 10.1126/science.aad8305
35. Effantin Grégory, Dordor Aurélien, Sandrin V, Martinelli N, Sundquist WI, Schoehn G, et al. ESCRT-III CHMP2A and CHMP3 form variable helical polymersin vitroand act synergistically during HIV-1 budding. Cell Microbiol (2013) 15(2):213–26. doi: 10.1111/cmi.12041
36. Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, et al. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun (2014) 5:5646. doi: 10.1038/ncomms6646
37. McCullough J, Frost A, Sundquist W. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodelling and fission complexes. Annu Rev Cell Dev Biol (2018) 34:85–109. doi: 10.1146/annurev-cellbio-100616-060600
38. Gatta AT, Carlton JG. The ESCRT-machinery: Closing holes and expanding roles. Curr Opin Cell Biol (2019) 59:121–32. doi: 10.1016/j.ceb.2019.04.005
39. Mcarthur K, Kile BT. Apoptotic caspases: Multiple or mistaken identities? Trends Cell Biol (2018) 28(6):475–93. doi: 10.1016/j.tcb.2018.02.003
40. Missotten M, Nichols A, Rieger K, Sadoul R. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ (1999) 6(2):124–9. doi: 10.1038/sj.cdd.4400456
41. Chen B. The glioma-associated protein SETA interacts with AIP1/Alix and ALG-2 and modulates apoptosis in astrocytes. Jbiolchem (2000) 275(25):19275–81. doi: 10.1074/jbc.M908994199
42. Vito P, Lacana E. Interfering with apoptosis: Ca2+-binding protein ALG-2 and alzheimer's disease gene ALG-3. Science (1996) 271(5248):521–5. doi: 10.1126/science.271.5248.521
43. Maki M, Maemoto Y, Osako Y, Shibata H. Evolutionary and physical linkage between calpains and penta-EF-hand Ca2+-binding proteins. FEBS J (2012) 279(8):1414–21. doi: 10.1111/j.1742-4658.2012.08560.x
44. Chen B. The glioma-associated protein SETA interacts with AIP1\/Alix and ALG-2 and modulates apoptosis in astrocytes. J Biol Chem (2000) 275(25):19275–81. doi: 10.1074/jbc.M908994199
45. Trioulier Y, Torch S, Blot B, Cristina N, Chatellard-Causse C, Verna JM, et al. Alix, a protein regulating endosomal trafficking, is involved in neuronal death. J Biol Chem (2004) 279(3):2046–52. doi: 10.1074/jbc.M309243200
46. Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell (2003) 114(6):689–99. doi: 10.1016/S0092-8674(03)00653-6
47. Martin-Serrano J, Yaravoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U.S.A. (2003) 100(21):12414–9. doi: 10.1073/pnas.2133846100
48. Majumder P, Chakrabarti O. Mahogunin regulates fusion between amphisomes/MVBs and lysosomes via ubiquitination of TSG101. Cell Death Dis (2015) 6(11):e1970. doi: 10.1038/cddis.2015.257
49. Sagona AP, Nezis IP, Stenmark H. Association of CHMP4B and autophagy with micronuclei: Implications for cataract formation. BioMed Res Int (2014) 2014:1–10. doi: 10.1155/2014/974393
50. Shen J, Liu Y, Song Y, Li L, Duan C, Zhou Y, et al. CHMP4B, ESCRT-III associating protein, associated with neuronal apoptosis following intracerebral hemorrhage. Brain Res (2015) 1597:1–13. doi: 10.1016/j.brainres.2014.11.043
51. Zhao P, Li C, Chen B, Sun G, Chao H, Tu Y, et al. Upregulation of CHMP4B alleviates microglial necroptosis induced by traumatic brain injury. J Cell Mol Med (2020) 24(15):8466–79. doi: 10.1111/jcmm.15406
52. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
53. Jayaraman A, Htike T, James R, Picon C, Reynolds R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in alzheimer's disease hippocampus. (2021) 9(1):159–79. doi: 10.1186/s40478-021-01264-w
54. Beckwith KS, Beckwith MS, Ullmann S, Saetra RS, Kim H, Marstad A, et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during mycobacterium tuberculosis infection. Nat Commun (2020) 11(1):2270. doi: 10.1038/s41467-020-16143-6
55. Dixon SJ, Patel DN, Welsch M, Skouta R, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife Sci (2014) 3:e02523. doi: 10.7554/eLife.02523
56. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
57. Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with alix to promote basal autophagic flux and late endosome function. Nat Cell Biol (2015) 11(6):961–2. doi: 10.1038/ncb3112
58. Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem (1998) 273(34):21883–92. doi: 10.1074/jbc.273.34.21883
59. Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
60. Rusten TE, Vaccari T, Lindmo K, Rodahl LM, Nezis IP, Sem-Jacobsen C, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol (2007) 17(20):1817–25. doi: 10.1016/j.cub.2007.09.032
61. Takahashi Y, He H, Tang Z, Hattori T, Wang HG. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun (2018) 9(1):2855–67. doi: 10.1038/s41467-018-05254-w
62. Hollenstein DM, Kraft C. Autophagosomes are formed at a distinct cellular structure. Curr Opin Cell Biol (2020) 65:50–7. doi: 10.1016/j.ceb.2020.02.012
63. Zhen Y, Spangenberg H, Munson MJ, Brech A, Schink KO, Tan KW, et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy (2020) 16(5):826–41. doi: 10.1080/15548627.2019.1639301
64. Zhu SY, Yao RQ, Li YX, Zhao PY, Yao YM. Lysosomal quality control of cell fate: a novel therapeutic target for human diseases. Cell Death Dis (2020) 11(9):817–29. doi: 10.1038/s41419-020-03032-5
65. Takahashi Y, Liang X, Hattori T, Tang Z, Wang HG. VPS37A directs ESCRT recruitment for phagophore closure. J Cell Biol (2019) 218(10):jcb.201902170. doi: 10.1083/jcb.201902170
66. Lu Y, Zhang Z, Sun D, Sweeney S, Gao FB. Syntaxin 13, a genetic modifier of mutant CHMP2B in frontotemporal dementia, is required for autophagosome maturation. Mol Cell (2013) 52(2):264–71. doi: 10.1016/j.molcel.2013.08.041
67. Gu M, Lajoie D, Chen OS, Von Appen A, Ladinsky MS, Redd MJ, et al. LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc Natl Acad ences (2017) 114(11):E2166. doi: 10.1073/pnas.1613916114
68. Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG. ESCRT-III controls nuclear envelope reformation. Nature (2015) 522(7555):236–9. doi: 10.1038/nature14503
69. Zhou F, Wu Z, Zhao M, Murtazina R, Cai J, Zhang A, et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Biol (2019) 218(6):1908–27. doi: 10.1083/jcb.201811173
70. Lahiri V, Klionsky D. Spatially distinct pools of TORC1 balance protein homeostasis. Autophagy (2019) 15(4):561–4. doi: 10.1080/15548627.2019.1575162
71. Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, et al. A Rab5 endosomal pathway mediates parkin-dependent mitochondrial clearance. Nat Commun (2017) 8:14050. doi: 10.1038/ncomms14050
72. Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol (2012) 14(10):1024–35. doi: 10.1038/ncb2589
73. Oku M, Maeda Y, Kagohashi Y, Kondo T, Yamada M, Fujimoto T, et al. Evidence for ESCRT- and clathrin-dependent microautophagy. J Cell Biol (2017) 3263:3263–74. doi: 10.1083/jcb.201611029
74. Olsvik HL, Svenning S, Abudu YP, Brech A, Stenmark H, Johansen T, et al Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy (2019) 15(1):182–3. doi: 10.1080/15548627.2018.1532265
75. Wu S, Pei Q, Ni W, Fu X, Zhang W, Song C, et al. HSPA1A protects cells from thermal stress by impeding ESCRT-0-Mediated autophagic flux in epidermal thermoresistance. J Invest Dermatol (2021) 141(1):48–58 e3. doi: 10.1016/j.jid.2020.05.105
76. Jia J, Claude-Taupin A, Gu Y, Choi SW, Deretic V. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell (2019) 52(1):69–87.e8. doi: 10.1016/j.devcel.2019.10.025
77. McNally KE, Brett LC. The intralumenal fragment pathway mediates ESCRT-independent surface transporter downregulation. Nat Commun (2018) 9(1):5358. doi: 10.1038/s41467-018-07734-5
78. Radulovic M, Schink K, Wenzel E, Nähse V, Bongiovanni A, Lafont F, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J (2018) 37(21):e99753. doi: 10.15252/embj.201899753
79. Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science (2018) 360(6384):eaar5078. doi: 10.1126/science.aar5078
80. Zhang J, Yang Y, He W, Sun L. Necrosome core machinery: MLKL. Cell Mol Life ences CMLS (2016) 73(11-12):2153–63. doi: 10.1007/s00018-016-2190-5
81. Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science (2014) 343(6174):1247136. doi: 10.1126/science.1247136
82. Rusten TE, Stenmark H. How do ESCRT proteins control autophagy? J Cell Sci (2009) 122(13):2179–83. doi: 10.1242/jcs.050021
83. Sefi Z, Inbar S, Ziv E, Aria H, Yifat OB, Croker BA, et al. Phosphatidylserine externalization, "necroptotic bodies" release, and phagocytosis during necroptosis. PloS Biol (2017) 15(6):e2002711. doi: 10.1371/journal.pbio.2002711
84. Fan W, Guo J, Gao B, Zhang W, Wang X. Flotillin-mediated endocytosis and ALIX–syntenin-1–mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci Signaling (2019) 12(583):eaaw3423. doi: 10.1126/scisignal.aaw3423
85. Yamazaki Y, Takahashi T, Hiji M, Kurashige T, Matsumoto M. Immunopositivity for ESCRT-III subunit CHMP2B in granulovacuolar degeneration of neurons in the alzheimer's disease hippocampus. Neurosci Lett (2010) 477(2):86–90. doi: 10.1016/j.neulet.2010.04.038
86. Cu A, Rjhwb C. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis - ScienceDirect. Neurobiol Dis (2021) 147:105144. doi: 10.1016/j.nbd.2020.105144
87. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting k+ efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
88. Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death differ (2019) 26(1):14–24. doi: 10.1038/s41418-018-0173-9
89. Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III–dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun (2020) 522(2):415–21. doi: 10.1016/j.bbrc.2019.11.110
90. Angeli JF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Free Radical Biol Med (2014) 76(12):1180–91. doi: 10.1038/ncb3064
91. Abrams RP, Carroll WL, Woerpel KA. Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol (2016) 11(5):1305–12. doi: 10.1021/acschembio.5b00900
92. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
93. Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4 l-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun (2020) 531(4):581–7. doi: 10.1016/j.bbrc.2020.07.032
94. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol (2019) 15(12):1137–47. doi: 10.1038/s41589-019-0408-1
95. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: Process and function. Cell Death Differ (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
96. Wan S, Yang K, Kim M, Patel M, Shchepinov MS, Stockwell BR, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci United States America (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
97. Wan SY, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol (2015) 26(3):165–76. doi: 10.1016/j.tcb.2015.10.014
98. Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun (2020) 522(2):415–21. doi: 10.1016/j.bbrc.2019.11.110
99. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: Molecular mechanisms and health implications. Cell Death Differ (2021) 28(5):1644–57. doi: 10.1038/s41422-020-00441-1
100. Yan HF, Zou T, Tuo QZ, Xu S, Lei P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct Targeted Ther (2021) 6(1):49. doi: 10.1038/s41392-020-00428-9
101. Dai E, Zhang W, Cong D, Kang R, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun (2020) 523(4):966–71. doi: 10.1016/j.bbrc.2020.01.066
102. Lefebvre C, Legouis R, Culetto E. ESCRT and autophagies: Endosomal functions and beyond. Semin Cell Dev Biol (2018) 74:21–8. doi: 10.1016/j.semcdb.2017.08.014
103. Mahapatra KK, Mishra SR, Behera BP, Patil S, Gewirtz DA, Bhutia SK. The lysosome as an imperative regulator of autophagy and cell death. Cell Mol Life Sci (2021) 78(23):7435–49. doi: 10.1007/s00018-021-03988-3
104. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi FC, et al. Molecular definitions of autophagy and related processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
105. Rubinsztein D, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol (2012) 22(1):R29–34. doi: 10.1016/j.cub.2011.11.034
106. Mcknight NC, Zhong Y, Wold MS, Gong S, Phillips GR, Dou Z, et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PloS Genet (2014) 10(10):e1004626. doi: 10.1371/journal.pgen.1004626
107. Kalinowska K, Nagel MK, Goodman K, Cuyas L, Isono E. Arabidopsis ALIX is required for the endosomal localization of the deubiquitinating enzyme AMSH3. Proc Natl Acad Sci United States America (2015) 112(40):E5543–51. doi: 10.1073/pnas.1510516112
108. Flower TG, Takahashi Y, Hudait A, Rose K, Hurley JH. A helical assembly of human ESCRT-I scaffolds reverse-topology membrane scission. Nat Struct Mol Biol (2020) 27(6):570–80. doi: 10.1038/s41594-020-0426-4
109. Hattori T, Takahashi Y, Chen L, Tang Z, Wills CA, Liang X, et al. Targeting the ESCRT-III component CHMP2A for noncanonical caspase-8 activation on autophagosomal membranes. Cell Death Differ (2021) 28(2):657–70. doi: 10.1038/s41418-020-00610-0
110. Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol Cb (2007) 17(18):1561–7. doi: 10.1016/j.cub.2007.07.029
111. Herz H-M. vps25 mosaics display nonautonomous cell survival andovergrowth, and autonomous apoptosis. Development (2006) 133(10):1871. doi: 10.1242/dev.02356
112. Papadopoulos C, Kravic B, Meyer H. Repair or lysophagy: Dealing with damaged lysosomes. J Mol Biol (2020) 432(1):231–39. doi: 10.1016/j.jmb.2019.08.010
113. Spitzer C, Li F, Buono R, Roschzttardtz H, Chung T, Zhang M, et al. The endosomal protein CHARGED MULTIVESICULAR BODY PROTEIN1 regulates the autophagic turnover of plastids in arabidopsis. Plant Cell (2015) 27(2):391. doi: 10.1105/tpc.114.135939
114. Oku M, Sakai Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays News Rev Mol Cell Dev Biol (2018) 40(6):e1800008. doi: 10.1002/bies.201800008
115. Müller M, Schmidt O, Angelova M, Faserl K, Weys S, Kremser L, et al. The coordinated action of the MVB pathway and autophagy ensures cell survival during starvation. eLife (2015) 4(4):e07736. doi: 10.7554/eLife.07736
116. Schfer JA, Schessner JP, Bircham PW, Tsuji T, Funaya C, Pa Jonk O, et al. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J (2020) 39(2):e102586. doi: 10.15252/embj.2019102586
117. Garcia EJ, Liao PC, Tan G, Vevea JD, Sing CN, Tsang CA, et al. Membrane dynamics and protein targets of lipid droplet microautophagy during ER stress-induced proteostasis in the budding yeast, saccharomyces cerevisiae. Autophagy (2021) 17(9):2363–83. doi: 10.1080/15548627.2020.1826691
118. Chen XC, Li ZH, Yang C, Tang JX, Lan HY, Liu HF. Lysosome depletion-triggered autophagy impairment in progressive kidney injury. Kidney Dis (Basel) (2021) 7(4):254–67. doi: 10.1159/000515035
119. Repnik U, Borg Distefano M, Speth MT, Ng MYW, Progida C, Hoflack B, et al. L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J Cell Sci (2017) 130(18):3124–40. doi: 10.1242/jcs.204529
120. López-Jiménez AT, Cardenal-Muñoz E, Leuba F, Gerstenmaier L, Barisch C, Hagedorn M, et al. The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the mycobacterium-containing vacuole but have opposite impact on containing the infection. PloS Pathog (2018) 14(12):e1007501. doi: 10.1371/journal.ppat.1007501
121. Toyoshima M, Tanaka N, Aoki J, Tanaka Y, Murata K, Kyuuma M, et al. Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein hrs: its regulatory role on e-cadherin and beta-catenin. Cancer Res (2007) 67(11):5162–71. doi: 10.1158/0008-5472.CAN-06-2756
122. Moh MC, Lee LH, Yang X, Shen S. HEPN1, a novel gene that is frequently down-regulated in hepatocellular carcinoma, suppresses cell growth and induces apoptosis in HepG2 cells. J Hepatol (2003) 39(4):580–6. doi: 10.1016/S0168-8278(03)00359-3
123. Li J, Belogortseva N, Porter D, Park M. Chmp1A functions as a novel tumor suppressor gene in human embryonic kidney and ductal pancreatic tumor cells. Cell Cycle (2008) 7(18):2886–93. doi: 10.4161/cc.7.18.6677
124. Walker GE, Antoniono RJ, Ross HJ, Paisley TE, Oh Y. Neuroendocrine-like differentiation of non-small cell lung carcinoma cells: Regulation by cAMP and the interaction of mac25/IGFBP-rP1 and 25.1. Oncogene (2006) 25(13):1943–54. doi: 10.1038/sj.onc.1209213
125. Nikolova DN, Doganov N, Dimitrov R, Angelov K, Low SK, Dimova I, et al. Genome-wide gene expression profiles of ovarian carcinoma: Identification of molecular targets for the treatment of ovarian carcinoma. Mol Med Rep (2009) 2(3):365–84. doi: 10.3892/mmr_00000109
126. Sadler JBA, Wenzel DM, Strohacker LK, Guindo-Martínez M, Alam SL, Mercader JM, et al. A cancer-associated polymorphism in ESCRT-III disrupts the abscission checkpoint and promotes genome instability. Proc Natl Acad Sci USA (2018) 115(38):E8900–8. doi: 10.1073/pnas.1805504115
Glossary

Keywords: the endosomal sorting complex required for transport (ESCRT), apoptosis, necroptosis, pyroptosis, ferroptosis, autophagy
Citation: Yang Y, Wang M, Zhang Y-Y, Zhao S-Z and Gu S (2022) The endosomal sorting complex required for transport repairs the membrane to delay cell death. Front. Oncol. 12:1007446. doi: 10.3389/fonc.2022.1007446
Received: 30 July 2022; Accepted: 20 September 2022;
Published: 18 October 2022.
Edited by:
Yanan Jiang, Harbin Medical University, ChinaReviewed by:
Glaucia Maria Machado-Santelli, Universidade de São Paulo, BrazilHai Feng, Shanghai University of Traditional Chinese Medicine, China
Copyright © 2022 Yang, Wang, Zhang, Zhao and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song Gu, U29uZ2d1MjAyMkAxNjMuY29t; Shu-Zhi Zhao, d2VsbHNub3cyMDA4QDE2My5jb20=; Ying-Ying Zhang, YWxwaGFmbHlAMTYzLmNvbQ==; Min Wang, d2FuZ21pbjA1MDFAaG90bWFpbC5jb20=
†These authors have contributed equally to this work