 Xiaoyu Ma
Xiaoyu Ma Hongtao Zhu1
Hongtao Zhu1 Lidong Cheng
Lidong Cheng Xin Chen
Xin Chen Kai Shu
Kai Shu Suojun Zhang
Suojun Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 13 October 2022
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1004700
This article is part of the Research TopicTissue Resident Immune Cells in Tumorigenesis and Cancer TreatmentView all 6 articles
Glioblastoma (GBM) is the most malignant type of glioma with the worst prognosis. Traditional therapies (surgery combined with radiotherapy and chemotherapy) have limited therapeutic effects. As a novel therapy emerging in recent years, immunotherapy is increasingly used in glioblastoma (GBM), so we expect to discover more effective immune targets. FGL2, a member of the thrombospondin family, plays an essential role in regulating the activity of immune cells and tumor cells in GBM. Elucidating the role of FGL2 in GBM can help improve immunotherapy efficacy and design treatment protocols. This review discusses the immunosuppressive role of FGL2 in the GBM tumor microenvironment and its ability to promote malignant tumor progression while considering FGL2-targeted therapeutic strategies. Also, we summarize the molecular mechanisms of FGL2 expression on various immune cell types and discuss the possibility of FGL2 and its related mechanisms as new GBM immunotherapy.
Gliomas are the most universal malignant central nervous system (CNS) tumors, accounting for approximately 80% of all brain malignancies (1, 2). According to the classification of central nervous system tumors in the 2021 World Health Organization (WHO), Adult-type diffuse gliomas are classified into three types: Astrocytoma, IDH-mutant; Oligodendroglioma, IDH-mutant, and 1p/19q-co deleted; Glioblastoma, IDH-wildtype (3, 4). Among these, glioblastoma (GBM) is one of the most lethal gliomas, accounting for 70% of all widespread glioma diagnoses, with a median survival time of 15 months (5). Currently, conventional treatments for gliomas include surgery, chemotherapy, and radiotherapy. However, the 5-year survival rate of GBM patients is rarely 6.8% due to the infiltrative growth, aggressiveness, and recurrence of the malignant gliomas (6–8). The extremely low survival rate of GBM patients has prompted a search for more effective drugs and treatments (9).
Immunotherapy is an innovative treatment method in tumors today (10). The innate and adaptive immune system in the host recognizes and kills tumor cells in the initial stage of tumorigenesis, some tumor cells survive into the equilibrium phase, and in the later stages, tumor cells become resistant to the body’s immune response and immune cells escape. It is based on the study of tumor immune cell escape mechanisms that immunotherapy has developed. Immunotherapy attempts to activate immune cells by reactivating the tumor immune system and blocking immune escape pathways (11, 12).Targeted cytokines, immune checkpoint inhibitors (ICIs), pericyte therapy, oncolytic viruses, and cancer vaccines are currently used in clinical treatment and have been verified to be helpful in fighting tumor cells (13). Due to the success of immunotherapy in melanoma and hematologic tumors, attempts have been made to understand GBM in terms of the immune microenvironment and immune response and to improve its prognosis (14).Unfortunately, CTLA-4 monoclonal antibody Ipilimumab, PD-1 monoclonal antibody Nivolumab and so on have shown in clinical trials limited efficacy against GBM (15).
Due to the complex immune microenvironment of glioma, immunotherapy of glioma has been increasingly studied in recent years, and some promising results have been achieved. (16). In brain tumors, tumor cells secrete a large number of cytokines, growth factors and chemokines that promote the entry of numerous non-tumor cells such as infiltrating immune cells, pericytes, astrocytes, oligodendrocytes, and endothelial cells into the tumor (16–18). These cells and the various factors constitute the tumor microenvironment, which plays a crucial role in promoting tumor development, metastasis, and resistance to cancer therapy. The complex immune microenvironment of glioma makes it an excellent challenge for immunotherapy (19). In order to evade immune surveillance and clearance, tumor cells can suppress anti-tumor immunity through cancer immune editing, by which the immune response shifts from preventing the development of cancer to promoting the growth of tumor cells, thus evading immune surveillance (20, 21).
FGL2 is an important pleiotropic immunomodulatory cytokine discovered in recent years, which in cancer achieves immunosuppression by inhibiting antigen-presenting cells (APCs), suppressing T cell proliferation, inducing macrophage polarization to M2 and inducing regulatory T cell (TREG) activity (22, 23). However, the role of FGL2 in TME and the therapeutic potential of targeting this cytokine in gliomas is unclear. Here, we review the role of FGL2 in brain cancer and discuss its role in immunosuppression and tumor progression. We examined the molecular mechanisms responsible for regulating FGL2 expression in various cell types and considered the possibility that immunotherapies developed against this cytokine may improve the prognosis of GBM patients.
Fibrinogen-like protein 2 (Fgl2), as a number of the fibrinogen-associated protein superfamily, has a molecular weight of approximately 64 KD (24). It was first identified in the 1990s and was initially thought to be secreted by a constitutively expressed cytotoxic T lymphocyte. The protein encoded by FGL2 is homologous to the β and γ chains of fibrinogen at its carboxy terminus and carboxy terminus, with a FRED structure (25). Numerous studies surrounding FGL2 have revealed many functions of it. For example, Th1 and Th2 cytokines trigger the coagulation system by acting on FGL2 on endothelial cells, FGL2 can directly activate prothrombin to produce thrombin. However, FGL2, which is secreted by peripheral blood CD4+ and CD8+ T cells, has no coagulation activity (26). It can inhibit the maturation of dendritic cells and the proliferation of T cells and has immunosuppressive activity. Due to the two activities exhibited by FGL2, it is now more uniform to categorize FGL2 as a membrane-bound protein and a secretory protein, with the membrane-bound type have coagulation activity, and the secretory type with immunomodulatory function (27, 28). Previous studies have shown that FGL2 plays an important role in inflammatory diseases such as severe viral hepatitis, rheumatoid arthritis, chronic obstructive pulmonary disease (COPD) (28),and inflammatory bowel disease (IBD) (29). At the same time, FGL2 is implicated in the malignant progression of tumors in hepatocellular carcinoma, central nervous system tumors (22), breast cancer, ovarian cancer and so on.
Recent studies have shown that FGL2 can contribute to the growth of gliomas by inducing multiple immune mechanisms (30). However, the role of FGL2 in gliomas and the therapeutic potential to target this protein in cancer patients remains unclear. Therefore, this paper reviews the role of FGL2 in glioma and discusses its role in immunosuppression and malignant progression. We summarize the molecular mechanisms by which FGL2 may be expressed in various cell types and explore the potential prognostic value and immunotherapeutic targeting of FGL2 in gliomas.
Yan et al. were the first to identify the strong expression of FGL2 in gliomas and conducted experiments to prove it (30). They found that 83.8% of GBM patients had FGL2 gene amplification or copy increase, and 72.5% of LGG patients had diploidy of the FGL2 gene. They divided GBM patients into FGL2 High and FGL2 Low groups according to FGL2 mRNA expression levels. The results showed that the overall survival rate of patients in the FGL2 High group was lower than that of patients in the FGL2 Low group. The median survival times for low and high expression of FGL2 were 394 and 357 days. The 5-year survival rate was estimated to be 4.98% for patients with low FGL2 expression and 0.99% for patients with high FGL2 expression. Song et al. found by immunohistochemistry that the expression of FGL2 was significantly higher in glioma tissues than in normal tissues (31). And we also found that FGL2 mRNA expression levels were higher in high-grade gliomas than in low-grade gliomas, and low expression of FGL2 increased patient survival and prolonged patient survival time based on the Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/) and the Cancer Genome Atlas (TCGA)(http://cancergenome.nih.gov/) databases.(Figure 1) These data suggest that FGL2 expression in glioma is positively correlated with tumor malignancy and patient survival.
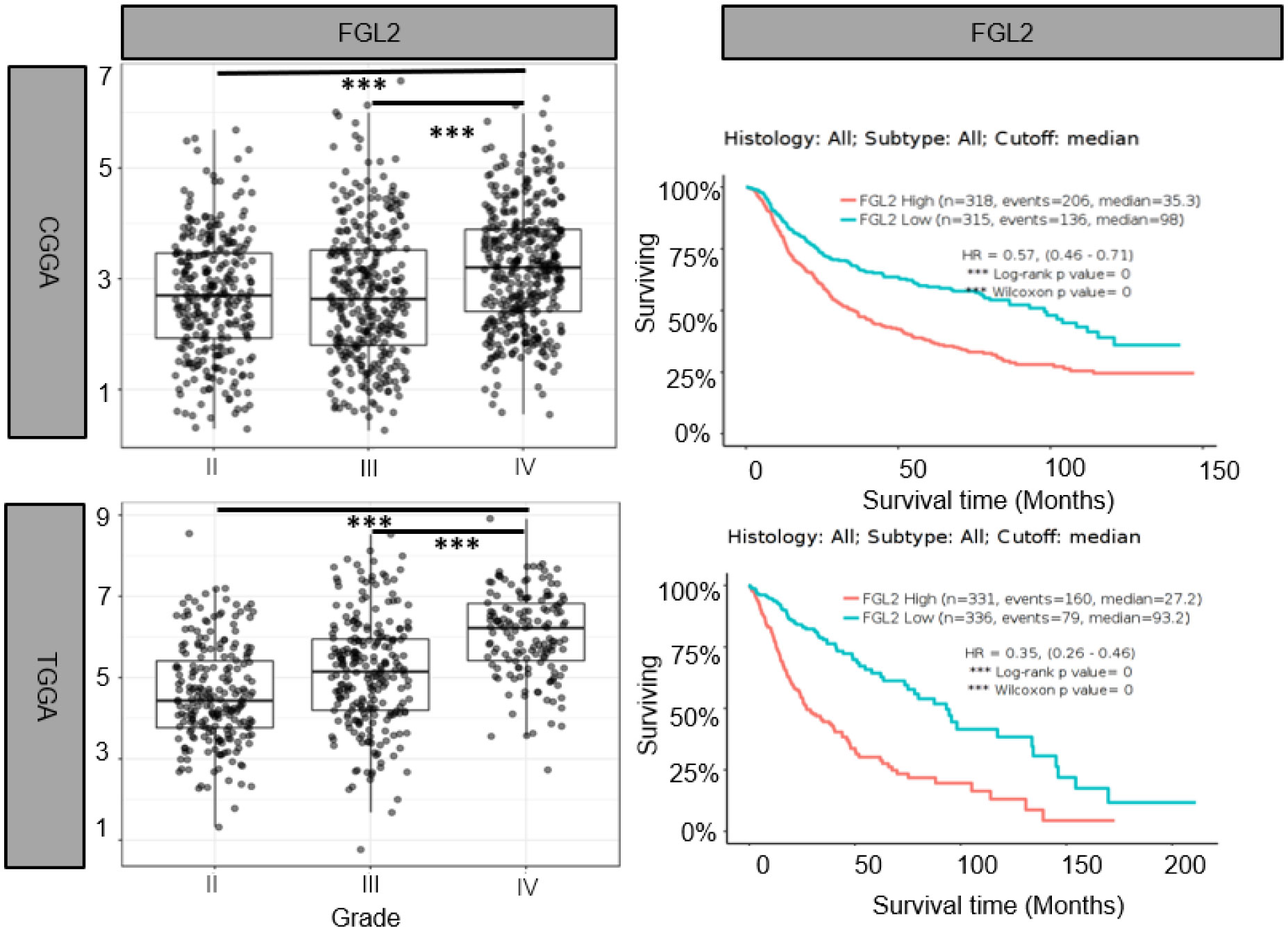
Figure 1 Elevated FGL2 mRNA levels in samples from patients with glioma and associated with poor patient prognosis. Analysis of glioma patient samples from The Chinese Glioma Genome Atlas (CCGA) and The Cancer Genome Atlas (TCGA) cohorts. FGL2 mRNA expression levels increased with glioma grade (II, III, IV). The figure was excerpted from GlioVis (http://gliovis.bioinfo.cnio.es/).
Although the cellular origin of FGL2 in gliomas has not been determined, it has been shown that FGL2 is higher expressed in glioblastoma and glioblastoma stem cells (GSCs) compared to fibroblast cells. In contrast, there is no significant difference in FGL2 expression between glioblastoma and GSC line (22, 31).Using murine glioma models, Yan et al. verified that FGL2 might promote glioma growth in murine models by inhibiting the infiltration of immunosuppressive cells in the tumor microenvironment and that neutralization of FGL2 protein using anti-FGL2 antibody prolonged the survival time of mic (30). FGL2 secreted by GSCs is thought to activate tumor-infiltrating immune cells. For example, Fgl2 can immunomodulate the organism by inhibiting T cell differentiation and proliferation, DCs maturation, CD8+ T cell function, complement activation and promoting B cell apoptosis, etc. (22, 23, 30, 31). In addition, FGL2 overexpression can skew macrophages and other activated antigen-presenting cells (APCs) in the tumor microenvironment from an inflammatory (M 1) or neoplastic (M0) phenotype to an immunosuppressive (M2) phenotype (23). The limitation of the current study is that the exact mechanism of FGL2-induced immunity is unclear and needs further investigation.
As well as its immunomodulatory functions of immunosuppressive and immunostimulatory activities, FGL2 can promote tumor progression by increasing cancer cell proliferation (32).In the mouse model of lung cancer, FGL2 promotes tumor growth by stimulating angiogenesis, immunosuppression, and tumor cell proliferation (33).Similarly, in vitro studies showed a dose-dependent increase in FGL2-mediated cell proliferation in LOVO and SW620 colorectal cancer cells, and knockdown of FGL2 resulted in reduced proliferation, migration and invasion (34, 35).Overexpression of FGL2 can eliminate the decrease in cell proliferation, migration, and aggression caused by the MAPK signaling inhibitor U0126 (34).In hepatocellular carcinoma studies, investigators used recombinant hFGL2 protein to stimulate HCCLM6 cells with thrombospondin and Ca2+, then found that phosphorylated p38 and ERK were significantly upregulated, while this upregulation could be abrogated by hirudin (36). Stable downregulation of FGL2 expression in the FGL2 knockdown HCCLM6 cell line was found to result in delayed tumor growth and reduced angiogenesis, along with decreased VEGF I and L-8 expression. These findings suggest that FGL2 may regulate HCCLM6 tumor cell growth by affecting angiogenesis (36). However, this phenomenon was not reproduced in vivo.
Although the origin of FGL2 in glioma is not fully understood, it has been shown to be expressed mainly in immune cells such as endothelial cells, macrophages, NK cells, T cells and tumor cells (27, 37). Low levels of FGL2 expression are associated with high granulocyte-macrophage (GM-CSF) colony-stimulating factor expression (38). Overexpression of FGL2 in a mouse glioma model was observed to increase CD4+FoxP3tregs cells and induced macrophages toward M2 phenotype shift. Ultimately, T cell initiation capacity was decreased (30).Yan et al. found that knockdown of FGL2 in immunoreactive mice reversed immune dysfunction in dendritic cells (DCs) and induced differentiation of CD103+ DCs in the brain (37). In addition, FGL2 has also been shown to play an important role in regulating myeloid-derived suppressor cells (MDSCs) (39, 40).FGL2 also was found to have a strong correlation with both immune cells in the TIMER database. (Figure 2) These findings suggest that FGL2 exerts its immunosuppressive effects through a variety of tumor-mediated immunosuppressive mechanisms (Figure 3).
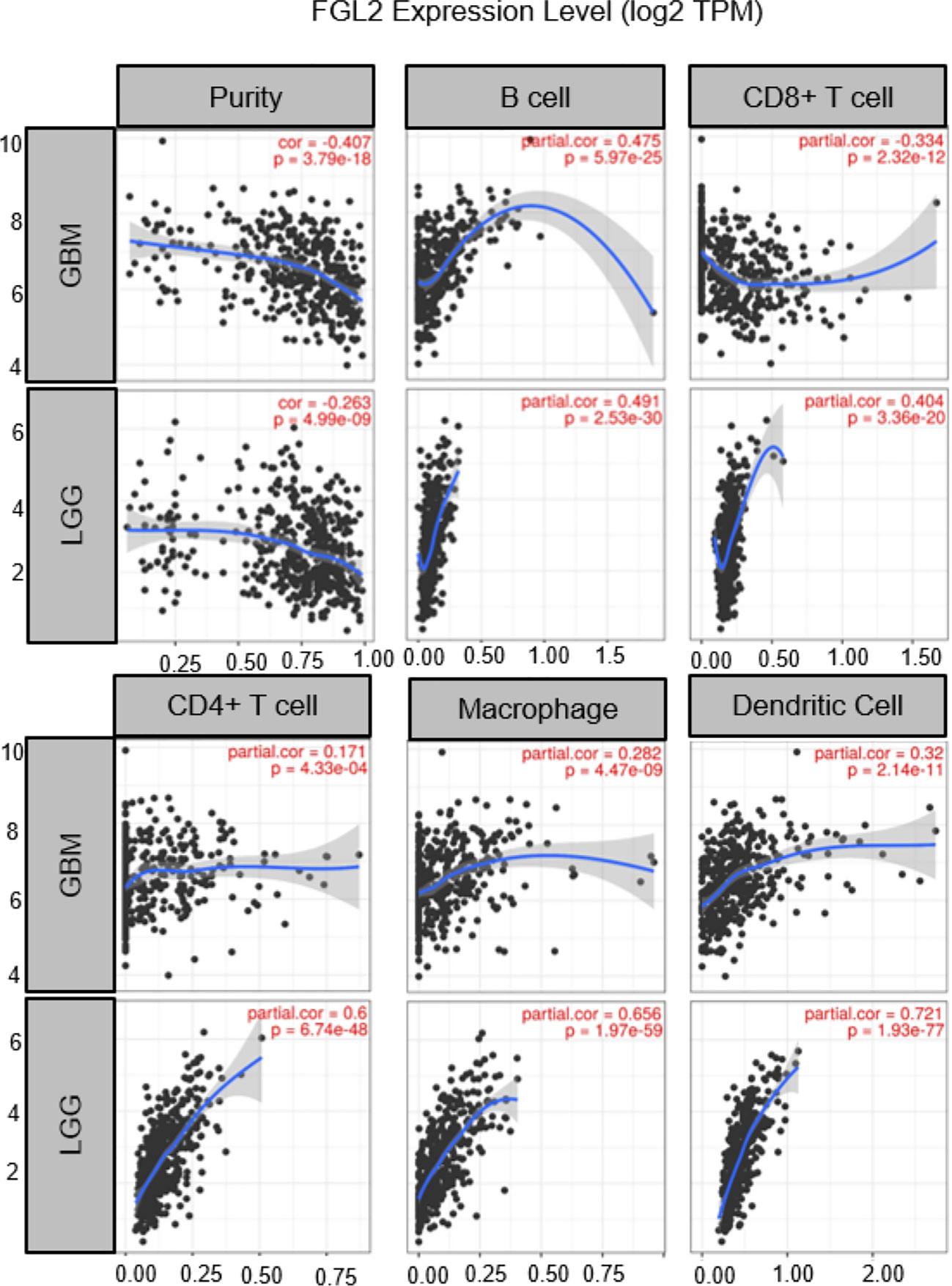
Figure 2 Correlation of FGL2 with immune cells. The figure was excerpted from TIMER (https://cistrome.shinyapps.io/timer/).
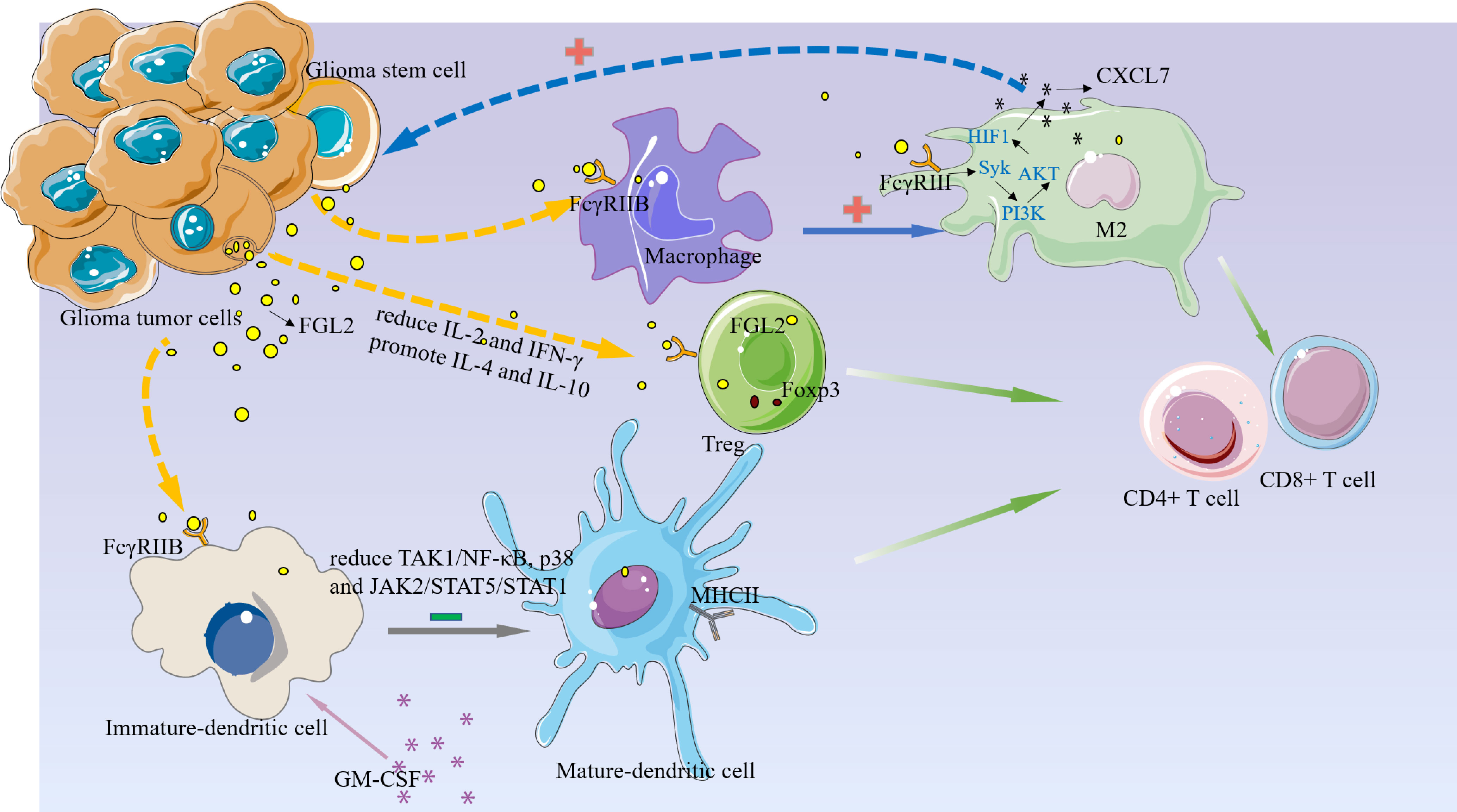
Figure 3 FGL2 plays an immunosuppressive function in the glioma microenvironment.1, FGL2 secreted by tumor cells binds to FCGR2B on immature DC cells and inhibits DC cell maturation and thus T cell proliferation by blocking the TAK1/NF-κB, p38, and JAK2/STAT5/STAT1 pathways.2, FGL2 inhibits the immune activators IL-2 and IFN-γ production, promotes immunosuppressive factors IL-4 and IL-10, increases Treg, and inhibits T-cell activation.3, FGL2 induces macrophages toward the M2 phenotype and produces CXCL7 in M2 cells via the FCGR2B/Syk/PI3K/AKT/HIF1α pathway, and CXCL7, in turn, acts on GSC to promote glioma cell stemness maintenance.
Among the immune cells involved in the immune response, dendritic cells (DC) are the most functional antigen-presenting cells known (41–44). It can effectively extract, process, treat, and present antigens to T and B lymphocytes to activate the body’s specific immunity (45). DC presents a large number of tumor antigen peptides so that the corresponding T cell receptors are fully occupied; binds to T cells, facilitating T cell clearance of tumors; promotes T cell enrichment and enhances activation of T cells; fully activates T cells (46). Immature DCs can be released into tissues via blood vessels and are constantly searching for invading antigens and mutated cells. Once found, DCs immediately function to engulf these antigens and cells while digesting them with digestive enzymes from intracellular lysosomes. Digested foreign bodies and cells leave behind fragments of biomolecules that DCs will put on MHC molecules on the cell surface and enter the lymphoid tissue for T cell recognition (47–49).. In this process, DC cells are gradually transformed into mature antigen-presenting cells, a process often referred to as “antigen presentation”. And studies in recent years have shown that DC cells are closely linked to the prognosis of tumors (50). In solid tumors, patients with a high number of DC cell infiltrations tend to have a better prognosis (51).Therefore, DC cells are also used in research for tumor treatment, such as DC vaccines (52).
The most abundant and intrinsically stimulatory migrating cell type is the CD103+ DC population. In melanoma, CD103+ DCs transport tumor antigens to Draining lymph nodes (dLN) in a CCR7-dependent manner, resulting in increased numbers of tumor-specific CD8+ T cells (53). FGL2 promotes glioma progression by inhibiting CD103+ dendritic cell differentiation (22). In tumors, tumor cells release antigen to bind with antigen-presenting cells APCs, which further activate T cells, and activated CD8+ T lymphocytes exert the ability to recognize and kill tumor cells (54). APCs express major histocompatibility class II (MHCII) molecules, including DCs, macrophages, monocytes, B lymphocytes, and microglia in the brain (55). Batf3 is a key transcription factor that regulates CD103+/CD8a+ DC differentiation (56).The study by Quintana et al. shows that in the absence of Batf3, there is an impairment in the recognition of tumor cells by T cells. GM-CSF promotes the differentiation of myeloid cells and plays an important role in the differentiation, proliferation and activation of DCs (57). Yan Jun et al. found that GM-CSF induced TAK1/NF-κB, p38 and JAK2/STAT5/STAT1 signaling to induce CD103 expression. And a small amount of FGL2 expression in tumor tissues could significantly reduce the intensity of these CD103+DC-induced signals (22).This discovery may shed new light on the development of DC immunotherapy.
T cells can shape immune responses in tumors, autoimmunity and infections, where CD4+ T (Th) cells and CD8+ T cells mediate effector responses that are suppressed by regulatory T (Treg) cells (58, 59). The balance between the effector T cell and Treg cell functions coordinates immune homeostasis and regulation of immune function. It has a vital role in tumorigenesis and development. Depending on the information the environment delivers, T cell metabolism dynamically changes and determines various aspects of its functional differentiation. CD4+ or CD8+ T cells recognize tumor antigens and autoantigens by expressing αβ T cell receptors (TCRs) and therefore play a crucial role in shaping the immune response in the formation of cancer or autoimmune diseases (60, 61). Following stimulation by cognate antigens, T cells are activated, proliferate, and undergo functional differentiation in response to environmental information. Initial CD8+ T cells without antigen stimulation differentiate into cytotoxic effector cells and long-term memory cells (62, 63). Initial CD4+ T cells differentiate into Th1, Th2, Th17, and Th effector cells, which can also form long-term memory cells, and immunosuppressive Treg cells expressing Foxp3 (64, 65). Cancer development is often associated with an immunosuppressive tumor microenvironment (TME), in which tumor-specific cytotoxic CD8+ T cells are often inadequate or dysfunctional and unable to eradicate malignant cells. T cell activation requires APCs such as CD103+ DCs to present antigens to T cells, while FGL2 can inhibit APC maturation and subsequently reduce T cell activation, the use of anti-FGL2 antibodies to antagonize FGL2 can activate T cells. Therefore, the development of FGL2 inhibitors may play an important role in the immunotherapy of gliomas (37).
Regulatory cells (Tregs) are a subpopulation of T cells that controls autoimmune reactivity in vivo, first reported by Sakakuchi et al. in 1995 (66, 67). Tregs can be divided into nTregs and aTregs. nTregs are mainly composed of Foxp3+CD25+CD4+ Treg cells, which function through cell-to-cell contacts (68). In recent years, many scholars have conducted numerous studies on the immunological aspects of Tregs in tumors. These studies confirm the large amounts of Tregs infiltration in tumor tissues such as liver (69), ovarian (70), lung (71), breast (72), and gastrointestinal tumors (73), and their significant presence is often associated with poor tumor prognosis. Attempts have been made to explain the mechanisms by which Tregs suppress the immune response at various cellular and molecular levels (74). They include: 1, through cell-to-cell contact-dependent inhibition, Tregs cells are inhibited by CTLA-4 and TGF-β etc. directly binding to the corresponding receptors on target cells, reducing the responsiveness of target cells to IL-2 and thus inhibiting the proliferation of effector T cells. 2, relying on suppressive cytokines such as IL-4 and IL-10. 3, modulating the body’s immunity by interacting with antigen-presenting cells, suppressing effector T cells by downregulating the function of APC cells or competing for co-stimulatory molecules on APC cells (75–82).
FGL2, which has immunomodulatory activity, has been reported to be highly expressed in Treg by many studies (83–85). To verify the importance of FGL2 on Treg function, Itay Shalev et al. used an anti-FGL2 monoclonal antibody to antagonize Treg in vitro. The experimental results showed that the FGL2 antibody could completely inhibit Treg’s function. This result further supports the previous finding that FGL2 may inhibit T cell proliferation by suppressing the production of immune- activating cytokines IL-2 and IFN-γ and promoting the production of suppressive cytokines IL-4 and IL-1 (25, 26).
Tumor-associated macrophages (TAM) are infiltrating macrophages in tumor tissues, which are mainly differentiated from monocytes (86, 87). Chemokines such as CSF1 and CCL2 secreted by tumor cells can recruit monocytes from peripheral circulating blood to the tumor microenvironment (TME), and then monocytes differentiate into macrophage (88, 89). A growing number of studies have shown that TAM has a series of tumor-promoting functions such as supporting tumor cell proliferation, invasion, and metastasis, and is highly correlated with poor prognosis of tumor patients (23, 90, 91). Based on phenotype and function, macrophages can be divided into two main types, M1 (pro-inflammatory, classically activated macrophages) and M2 (anti-inflammatory, alternatively activated macrophages (92–94). In addition, M1-type macrophages kill tumor cells and defend against pathogen invasion, while M2-type macrophages mainly play a role in promoting tumor growth, invasion and metastasis (95, 96).
In gliomas, TAMs include brain-resident microglia and bone marrow-derived macrophages, which account for 50% of GBM tumors (97).TAMs and glioma cells are important components in promoting glioma growth, and their interactions are critical for glioma proliferation, treatment resistance, and tumor recurrence (98). FGL2 may be a central effector molecule in this interaction. Yan et al. found differences in macrophage infiltration between FGL2hi and FGL2KO gliomas by flow cytometry analysis, demonstrating that FGL2 can increase the M2 macrophage population. Macrophage migration assays were performed by using FGL2hi and FGL2KO glioma cells conditioned medium (CM). It was found that CM from FGL2hi attracted more macrophage aggregation than that from FGL2KO. The migratory effect of macrophages was also significantly suppressed after blocking FGL2 or using antibody blockers of the FGL2 receptor FcγRIII (CD16) (23).These findings suggest that FGL2 acts as a potent chemokine that recruits macrophages into the tumor microenvironment (TME) of gliomas, and CD16 is the receptor that mediates this chemotactic effect. They also found that FGL2 secreted on glioma cells could bind to the receptor CD16 on TAMs and promote the Syk/PI3K/AKT/HIF1α signaling pathway, then induce the release of CXCL7, which in turn could act on glioma cells and facilitate their Stem-like transition (23). However, the relationship between FGL2, TAMs and gliomas cells has not been sufficiently studied, and more experiments are needed to verify the relationship in the future.
The current treatment of the new GBM diagnosis is still focused on maximal resection and combined radiation and chemotherapy. Unfortunately, almost all treated patients relapse within a short period, and there are currently no effective treatments to prolong their survival for relapsed patients (99, 100). So, it is urgent to find a more efficient treatment. Over the past few years, immune checkpoint inhibition has gradually received more attention in treating tumors, providing a ray of hope for some tumors with limited traditional treatment options (101, 102). For example, PD-1 (Programmed cell death protein 1) checkpoint inhibitor can bind to PD-1 or its ligand PD-L1 and block PD-1 from blocking T cell (103).But the current effect of immune checkpoint inhibitors represented by PD-1 in the treatment of glioma is not very satisfactory (104, 105). Therefore, it is essential to find more effective immune targets. In a review of previous studies, we found that FGL2 acts more on antigen-presenting cells APCs upstream of T cells. These cells are present in the glioma microenvironment (37), and play a key role in immunosuppression by suppressing T cell proliferation. As mentioned repeatedly in the literature, targeting a single molecule or pathway is not sufficient to inhibit the malignant progression of gliomas, so if we can find a key point that can inhibit multiple immunosuppressive pathways or mechanisms, it is possible to achieve good therapeutic results. As a molecule with such a role, the development of inhibitors of FGL2 may help reverse immunosuppression in the tumor microenvironment and may play an essential role in targeting glioma-mediated immunosuppressive therapy. Combined with previous findings, PD-1 can elevate FGL2 expression by inhibiting IFN-r and TNF-a. It is not difficult to infer that simultaneous inhibition of IL-10 and PD-1 can increase T cell proliferation, which will be verified in the future (Figure 4). Ntv-a mice injected with RCA-PDGFB+FGL2 were treated with anti-FGL2 antibodies, and the control mice were treated with IgG antibodies. Statistical analysis of the survival of the mice revealed that the median asymptomatic survival of the anti-FGL2 antibody-treated mice was significantly longer than the control. The tumors treated with anti-FGL2 antibody were also found to have lower CD44 expression and higher Olig2 expression than the control group, indirectly demonstrating that high expression of FGL2 is associated with the mesenchymal subtype of glioma. Comparing the number of Treg in the two groups after 20 days of treatment, it was found that the anti-FGL2 antibody-treated mice showed fewer Treg along with a small number of arginase1+/Iba1+ macrophages. These suggest that anti-FGL2 antibody treatment can effectively prevent immunosuppression of the tumor microenvironment (106).
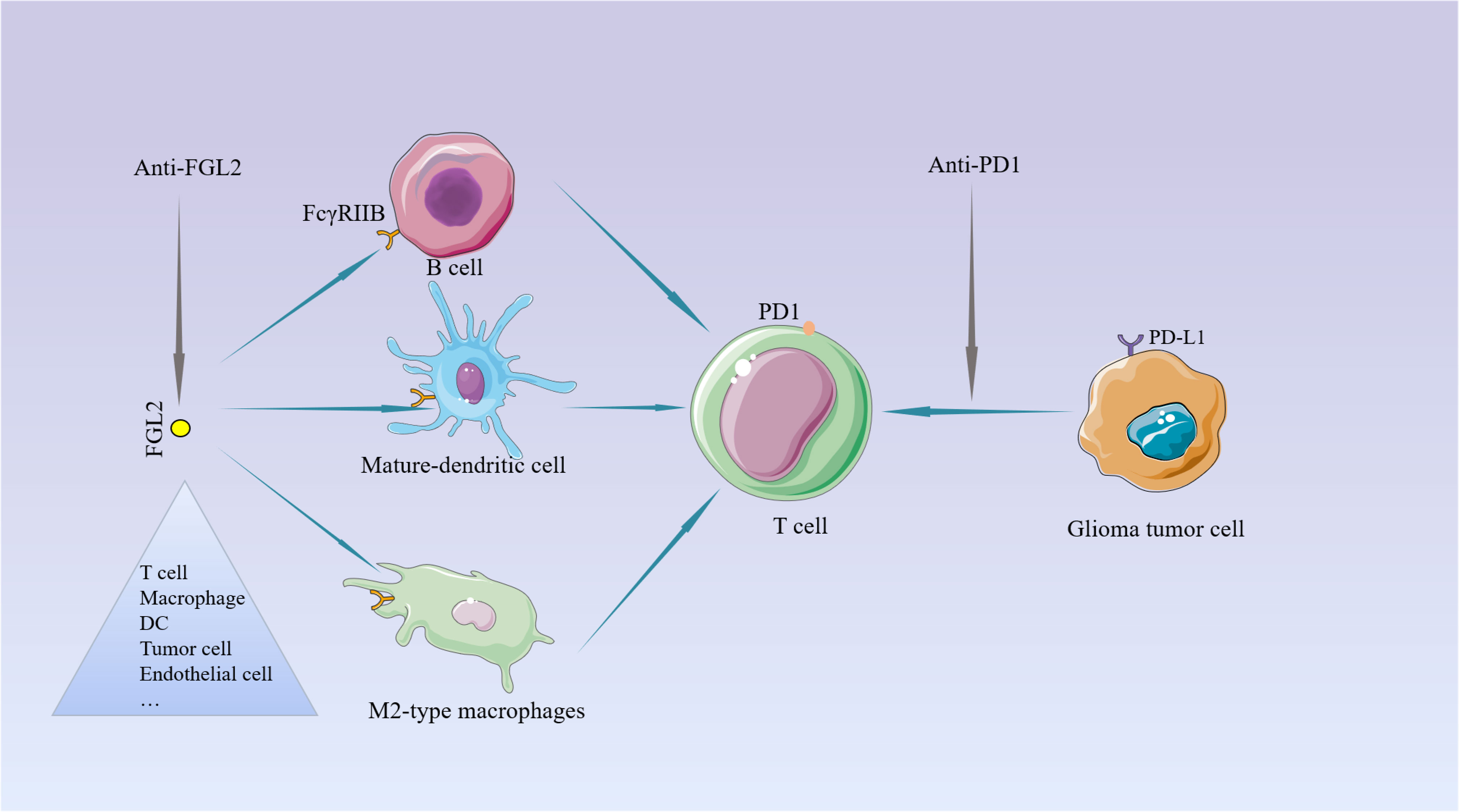
Figure 4 The potential for targeting FGL2. Unlike PD1 on T cells, FGL2 primarily targets the APC. PD-1 induces FGL2 expression by inhibiting IFN-γ and TNF-α.
Liu et al. established a stable knockdown of FGL2 in the HCCLM6 cell line and found that downregulation of FGL2 reduced tumor angiogenesis in HCCLM6 nude mouse xenograft. hFGL2 expression in HCC tumor cells promotes tumor growth and angiogenesis through activation of ERK and JNK pathways, and FGL2 protein secreted by tumor cells promotes angiogenesis and tumor growth through activation of the thrombin-dependent MAPK pathway. Extensive studies have shown that IL8 and VEGF are the most important activators of tumor-associated angiogenesis (107–109). Activation of multiple VEGF/VEGF receptor signaling pathways leads to endothelial cell survival, mitosis, migration, differentiation, vascular permeability, and endothelial progenitor cell mobilization (110, 111). IL-8 exerts its powerful angiogenic properties on endothelial cells through interaction with its receptors CXCR1 and CXCR2 (112, 113). In the nude mouse subcutaneous tumor model with stable downregulation of FGL2, FGL2 was found to be downregulated along with reduced expression of VEGF and IL-8 (28). At the same time, downregulation of FGL2 was found to affect angiogenesis in corneal microcapsule analysis in nude mice. However, FGL2 is also expressed in endothelial cells and macrophages, and FGL2 may also promote tumor angiogenesis by inducing value-added endothelial cells and recruiting inflammatory cells, this part needs to be further investigated. The correlation of FGL2 expression with enhanced phosphorylation of ERK and JNK strongly suggests that FGL2 plays an important role in tumor growth in HCC by regulating the activation of the MAPK pathway (36). FGL2 can lead to phosphorylation of ERK and p38 through thrombin production and subsequent activation of PAR1 and PAR3, or JNK through activation of PAR2, and these late cellular activities promote survival and proliferation, tumor growth and angiogenesis in hepatocellular carcinoma. These observations suggest that FGL2 prothrombinase, in conjunction with thrombin and tissue factor, may contribute to tumor hypercoagulability and possibly to angiogenesis and metastasis. In turn, FGL2 may serve as a novel target for the intervention of tumor development. Nevertheless, there are no relevant literature reports on whether FGL2 has an effect on tumor angiogenesis in glioma, and more experimental validation may be needed at a later stage.
FGL2 is widely expressed in gliomas and also plays an essential immunosuppressive role, thereby promoting tumor immune escape and malignant progression. Since FGL2 can act on antigen-presenting cells and inhibit the anti-tumor activity of T cells, it may be a future direction to try to apply FGL2 targeting therapy to GBM treatment. In-depth understanding and elucidation of the transcriptional and signaling mechanisms of FGL2 in different cell types of gliomas are essential for developing new specific targeted drugs.
SZ designed and led the project. XM wrote the manuscript. HZ, LC, XC, and KS provided valuable revisions to the manuscript. All authors contributed to this article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China, No. 82072805 and Hubei Natural Science Foundation, No. 2020CFB678.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet (2012) 205(12):613–21. doi: 10.1016/j.cancergen.2012.10.009
2. Pop S, Enciu AM, Necula LG, Tanase CP. Long non-coding RNAs in brain tumours: Focus on recent epigenetic findings in glioma. J Cell Mol Med (2018) 22(10):4597–610. doi: 10.1111/jcmm.13781
3. Louis DN, Perry A, Reifenberger G, Deimling AV, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathologica (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
4. Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol (2006) 2(9):494–503. doi: 10.1038/ncpneuro0289
5. Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol (2019) 15(7):405–17. doi: 10.1038/s41582-019-0220-2
6. Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H, et al. Hypoxic induction of vasorin regulates Notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell (2018) 22(1):104–118 e6. doi: 10.1016/j.stem.2017.10.005
7. Gusyatiner O, Hegi ME. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol (2018) 51:50–8. doi: 10.1016/j.semcancer.2017.11.010
8. Cruceru ML, Enciu AM, Popa AC, Albulescu R, Neagu M, Tanase CP, et al. Signal transduction molecule patterns indicating potential glioblastoma therapy approaches. Onco Targets Ther (2013) 6:1737–49. doi: 10.2147/OTT.S52365
9. Tanase C, Enciu AM, Codrici E, Popescu ID, Dudau M, Dobri AM, et al. Fatty acids, CD36, thrombospondin-1, and CD47 in glioblastoma: Together and/or separately? Int J Mol Sci (2022) 23(2):604. doi: 10.3390/ijms23020604
10. Lamplugh Z, Fan Y. Vascular microenvironment, tumor immunity and immunotherapy. Front Immunol (2021) 12:811485. doi: 10.3389/fimmu.2021.811485
11. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin (2020) 70(2):86–104. doi: 10.3322/caac.21596
12. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
13. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell (2018) 175(2):313–26. doi: 10.1016/j.cell.2018.09.035
14. Ghouzlani A, Kandoussi S, Tall M, Reddy KP, Rafii S, Badou A, et al. Immune checkpoint inhibitors in human glioma microenvironment. Front Immunol (2021) 12:679425. doi: 10.3389/fimmu.2021.679425
15. Kurz SC, Cabrera LP, Hastie D, Huang R, Unadkat P, Rinne M, et al. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology (2018) 91(14):e1355–9. doi: 10.1212/WNL.0000000000006283
16. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest (2017) 97(5):498–518. doi: 10.1038/labinvest.2017.19
17. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res (2006) 66(6):3294–302. doi: 10.1158/0008-5472.CAN-05-3773
18. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res (2011) 17(13):4296–308. doi: 10.1158/1078-0432.CCR-10-2557
19. Lim M, Xia Y, Bettegowda C, Weller M, et al. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol (2018) 15(7):422–42. doi: 10.1038/s41571-018-0003-5
20. Maire CL, Mohme M, Bockmayr M, Fita KD, Riecken K, Bornigen D, et al. Glioma escape signature and clonal development under immune pressure. J Clin Invest (2020) 130(10):5257–71. doi: 10.1172/JCI138760
21. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
22. Yan J, Zhao Q, Gabrusiewicz K, Kong LY, Xia X, Wang J, et al. FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nat Commun (2019) 10(1):448. doi: 10.1038/s41467-018-08271-x
23. Yan J, Zhao Q, Wang J, Tian X, Wang J, Xia X, et al. FGL2-wired macrophages secrete CXCL7 to regulate the stem-like functionality of glioma cells. Cancer Lett (2021) 506:83–94. doi: 10.1016/j.canlet.2021.02.021
24. Liu H, Yang PS, Zhu T, Manuel J, Zhang J, He W, et al. Characterization of fibrinogen-like protein 2 (FGL2): monomeric FGL2 has enhanced immunosuppressive activity in comparison to oligomeric FGL2. Int J Biochem Cell Biol (2013) 45(2):408–18. doi: 10.1016/j.biocel.2012.10.014
25. Chan CW, Kay LS, Khadaroo RG, Chan MW, Lakatoo S, Young KJ, et al. Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J Immunol (2003) 170(8):4036–44. doi: 10.4049/jimmunol.170.8.4036
26. Shalev I, Liu H, Koscik C, Bartczak A, Javadi M, Wong KM, et al. Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J Immunol (2008) 180(1):249–60. doi: 10.4049/jimmunol.180.1.249
27. Hu J, Yan J, Rao G, Latha K, Overwijk WW, Heimberger AB, et al. The duality of Fgl2 - secreted immune checkpoint regulator versus membrane-associated procoagulant: Therapeutic potential and implications. Int Rev Immunol (2016) 35(4):325–39. doi: 10.3109/08830185.2014.956360
28. Liu Y, Xu S, Xiao F, Xiong Y, Wang X, Gao S, et al. The FGL2/fibroleukin prothrombinase is involved in alveolar macrophage activation in COPD through the MAPK pathway. Biochem Biophys Res Commun (2010) 396(2):555–61. doi: 10.1016/j.bbrc.2010.04.145
29. Dong X, Ye X, Chen X, Chen T, Xie S, Li Q, et al. Intestinal and peripheral fibrinogen-like protein 2 expression in inflammatory bowel disease. Dig Dis Sci (2014) 59(4):769–77. doi: 10.1007/s10620-013-2962-9
30. Yan J, Kong LY, Hu J, Gabrusiewicz K, Dibra D, Xia X, et al. FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J Natl Cancer Inst (2015) 107(8):djv137. doi: 10.1093/jnci/djv137
31. Song Z, Wang Y, Du Y, Zhang Z, Yuan Y. Identification of integrative molecular and clinical profiles of fibrinogen-like protein 2 in gliomas using 1323 samples. Int Immunopharmacol (2020) 88:106894. doi: 10.1016/j.intimp.2020.106894
32. Yu J, Li J, Shen J, Du F, Wu X, Li M, et al. The role of fibrinogen-like proteins in cancer. Int J Biol Sci (2021) 17(4):1079–87. doi: 10.7150/ijbs.56748
33. Zhu Y, Zhang L, Zha H, Yang F, Hu C, Chen L, et al. Stroma-derived fibrinogen-like protein 2 activates cancer-associated fibroblasts to promote tumor growth in lung cancer. Int J Biol Sci (2017) 13(6):804–14. doi: 10.7150/ijbs.19398
34. Liu X, Chu Y, Wang D, Weng Y, Jia Z. MAPK-mediated upregulation of fibrinogen-like protein 2 promotes proliferation, migration, and invasion of colorectal cancer cells. Cell Biol Int (2019) 43(13):1483–91. doi: 10.1002/cbin.11198
35. Qin WZ, Li QL, Chen WF, Xu MD, Zhang YQ, Zhong YS, et al. Overexpression of fibrinogen-like protein 2 induces epithelial-to-mesenchymal transition and promotes tumor progression in colorectal carcinoma. Med Oncol (2014) 31(9):181. doi: 10.1007/s12032-014-0181-7
36. Liu Y, Xu L, Zeng Q, Wang J, Wang M, Xi D, et al. Downregulation of FGL2/prothrombinase delays HCCLM6 xenograft tumour growth and decreases tumour angiogenesis. Liver Int (2012) 32(10):1585–95. doi: 10.1111/j.1478-3231.2012.02865.x
37. Patel R, Traylor JI, Latha K, Heimberger AB, Li S, Rao G. Fibrinogen-like protein 2: a potential molecular target for glioblastoma treatment. Expert Opin Ther Targets (2019) 23(8):647–9. doi: 10.1080/14728222.2019.1628220
38. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood (2012) 119(15):3383–93. doi: 10.1182/blood-2011-11-370130
39. Liu BQ, Bao ZY, Zhu JY, Liu H. Fibrinogen-like protein 2 promotes the accumulation of myeloid-derived suppressor cells in the hepatocellular carcinoma tumor microenvironment. Oncol Lett (2021) 21(1):47. doi: 10.3892/ol.2020.12308
40. Wang M, Liu J, Xi D, Luo X, Ning Q. Adenovirus-mediated artificial microRNA against human fibrinogen like protein 2 inhibits hepatocellular carcinoma growth. J Gene Med (2016) 18(7):102–11. doi: 10.1002/jgm.2883
41. Lee YS, Radford KJ. The role of dendritic cells in cancer. Int Rev Cell Mol Biol (2019) 348:123–78. doi: 10.1016/bs.ircmb.2019.07.006
42. Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer (2019) 7(1):109. doi: 10.1186/s40425-019-0580-6
43. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
44. Pitt JM, André F, Amigorena S, Soria JC, Eggermont A, Kroeme RG, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest (2016) 126(4):1224–32. doi: 10.1172/JCI81137
45. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
46. Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: State of the art and beyond. Clin Cancer Res (2016) 22(8):1897–906. doi: 10.1158/1078-0432.CCR-15-1399
47. Del Balzo D, Capmany A, Cebrian I, Damiani MT. Chlamydia trachomatis infection impairs MHC-I intracellular trafficking and antigen cross-presentation by dendritic cells. Front Immunol (2021) 12:662096. doi: 10.3389/fimmu.2021.662096
48. Horrevorts SK, Duinkerken S, Bloem K, Secades P, Kalay H, Musters RJ, et al. Toll-like receptor 4 triggering promotes cytosolic routing of DC-SIGN-Targeted antigens for presentation on MHC class I. Front Immunol (2018) 9:1231. doi: 10.3389/fimmu.2018.01231
49. van Niel G, Wubbolts R, Stoorvogel W. Endosomal sorting of MHC class II determines antigen presentation by dendritic cells. Curr Opin Cell Biol (2008) 20(4):437–44. doi: 10.1016/j.ceb.2008.05.011
50. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12(4):265–77. doi: 10.1038/nrc3258
51. Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol (2014) 27:26–32. doi: 10.1016/j.coi.2014.01.005
52. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
53. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell (2016) 30(2):324–36. doi: 10.1016/j.ccell.2016.06.003
54. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol (2020) 17(12):725–41. doi: 10.1038/s41571-020-0413-z
55. Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: Parallels at non-CNS sites. Front Oncol (2015) 5:153. doi: 10.3389/fonc.2015.00153
56. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol (2012) 12(2):101–13. doi: 10.1038/nri3149
57. Zhan Y, Xu Y, Lew AM. The regulation of the development and function of dendritic cell subsets by GM-CSF: more than a hematopoietic growth factor. Mol Immunol (2012) 52(1):30–7. doi: 10.1016/j.molimm.2012.04.009
58. Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab (2017) 26(1):94–109. doi: 10.1016/j.cmet.2017.06.016
59. Schwartz RH. T Cell anergy. Annu Rev Immunol (2003) 21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110
60. Choi Y, Shi Y, Haymaker CL, Naing A, Ciliberto G, Hajjar J, et al. T-Cell agonists in cancer immunotherapy. J Immunother Cancer (2020) 8(2):e000966. doi: 10.1136/jitc-2020-000966
61. van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20(4):218–32. doi: 10.1038/s41568-019-0235-4
62. Reina-Campos M, Scharping NE, Goldrath AW. CD8(+) T cell metabolism in infection and cancer. Nat Rev Immunol (2021) 21(11):718–38. doi: 10.1038/s41577-021-00537-8
63. Han J, Khatwani N, Searles TG, Turk MJ, Angeles CV. Memory CD8(+) T cell responses to cancer. Semin Immunol (2020) 49:101435. doi: 10.1016/j.smim.2020.101435
64. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol (2018) 18(10):635–47. doi: 10.1038/s41577-018-0044-0
65. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity (2019) 51(6):1028–1042 e4. doi: 10.1016/j.immuni.2019.10.009
66. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
67. Tanaka A, Sakaguchi S. Targeting treg cells in cancer immunotherapy. Eur J Immunol (2019) 49(8):1140–6. doi: 10.1002/eji.201847659
68. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
69. Aghabi YO, Yasin A, Kennedy JI, Davies SP, Butler AE, Stamataki Z, et al. Targeting enclysis in liver autoimmunity, transplantation, viral infection and cancer. Front Immunol (2021) 12:662134. doi: 10.3389/fimmu.2021.662134
70. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol Res (2018) 6(12):1578–92. doi: 10.1158/2326-6066.CIR-17-0479
71. Xie M, Wei J, Xu J. Inducers, attractors and modulators of CD4(+) treg cells in non-Small-Cell lung cancer. Front Immunol (2020) 11:676. doi: 10.3389/fimmu.2020.00676
72. Bai F, Zhang P, Fu Y, Chen H, Zhang M, Huang Q, et al. Targeting ANXA1 abrogates treg-mediated immune suppression in triple-negative breast cancer. J Immunother Cancer (2020) 8(1):e000169. doi: 10.1136/jitc-2019-000169
73. Ji L, Qian W, Gui L, Ji Z, Yin P, Lin GN, et al. Blockade of beta-Catenin-Induced CCL28 suppresses gastric cancer progression via inhibition of treg cell infiltration. Cancer Res (2020) 80(10):2004–16. doi: 10.1158/0008-5472.CAN-19-3074
74. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
75. Barbi J, Pardoll D, Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev (2014) 259(1):115–39. doi: 10.1111/imr.12172
76. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
77. Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med (2007) 204(8):1765–74. doi: 10.1084/jem.20070719
78. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178(11):6725–9. doi: 10.4049/jimmunol.178.11.6725
79. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity (2008) 28(4):546–58. doi: 10.1016/j.immuni.2008.02.017
80. Zhang L, Jarvis LB, Baek HJ, Gaston JS. Regulatory IL4+CD8+ T cells in patients with ankylosing spondylitis and healthy controls. Ann Rheum Dis (2009) 68(8):1345–51. doi: 10.1136/ard.2008.088120
81. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118(30):e2023739118. doi: 10.1073/pnas.2023739118
82. Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS, et al. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer (2021) 20(1):171. doi: 10.1186/s12943-021-01464-x
83. Shalev I, Wong KM, Foerster K, Zhu Y, Chan C, Maknojia A, et al. The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen-like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology (2009) 49(2):387–97. doi: 10.1002/hep.22684
84. Foerster K, Helmy A, Zhu Y, Khattar R, Adeyi OA, Wong KM, et al. The novel immunoregulatory molecule FGL2: a potential biomarker for severity of chronic hepatitis c virus infection. J Hepatol (2010) 53(4):608–15. doi: 10.1016/j.jhep.2010.04.020
85. Zhao Z, Yang C, Wang L, Li L, Zhao T, Hu L, et al. The regulatory T cell effector soluble fibrinogen-like protein 2 induces tubular epithelial cell apoptosis in renal transplantation. Exp Biol Med (Maywood) (2014) 239(2):193–201. doi: 10.1177/1535370213514921
86. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab (2019) 30(1):36–50. doi: 10.1016/j.cmet.2019.06.001
87. Schulz M, Sevenich L. TAMs in brain metastasis: Molecular signatures in mouse and man. Front Immunol (2021) 12:716504. doi: 10.3389/fimmu.2021.716504
88. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
89. Yang Q, Guo N, Zhou Y, Chen J, Wei Q, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B (2020) 10(11):2156–70. doi: 10.1016/j.apsb.2020.04.004
90. Qiu SQ, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schroder CP, et al. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat Rev (2018) 70:178–89. doi: 10.1016/j.ctrv.2018.08.010
91. Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab (2016) 24(5):701–15. doi: 10.1016/j.cmet.2016.09.008
92. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
93. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater (2020) 32(40):e2002054. doi: 10.1002/adma.202002054
94. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
95. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J BioMed Sci (2019) 26(1):78. doi: 10.1186/s12929-019-0568-z
96. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol (2019) 12(1):76. doi: 10.1186/s13045-019-0760-3
97. Widodo SS, Dinevska M, Furst LM, Stylli SS, Mantamadiotis T. IL-10 in glioma. Br J Cancer (2021) 125(11):1466–76. doi: 10.1038/s41416-021-01515-6
98. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol (2015) 17(2):170–82. doi: 10.1038/ncb3090
99. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med (2018) 379(8):722–30. doi: 10.1056/NEJMoa1805453
100. Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol (2014) 15(9):e395–403. doi: 10.1016/S1470-2045(14)70011-7
101. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
102. O'Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol (2010) 21(8):1712–7. doi: 10.1093/annonc/mdq013
103. Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol (2015) 11(9):504–14. doi: 10.1038/nrneurol.2015.139
104. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
105. Lu Y, Ng AHC, Chow FE, Everson RG, Helmink BA, Tetzlaff MT, et al. Resolution of tissue signatures of therapy response in patients with recurrent GBM treated with neoadjuvant anti-PD1. Nat Commun (2021) 12(1):4031. doi: 10.1038/s41467-021-24293-4
106. Latha K, Yan J, Yang Y, Gressot LV, Kong LY, Manyam G, et al. The role of fibrinogen-like protein 2 on immunosuppression and malignant progression in glioma. J Natl Cancer Inst (2019) 111(3):292–300. doi: 10.1093/jnci/djy107
107. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem (2009) 284(10):6038–42. doi: 10.1074/jbc.C800207200
108. Funakoshi T, Lee CH, Hsieh JJ. A systematic review of predictive and prognostic biomarkers for VEGF-targeted therapy in renal cell carcinoma. Cancer Treat Rev (2014) 40(4):533–47. doi: 10.1016/j.ctrv.2013.11.008
109. Urbantat RM, Blank A, Kremenetskaia I, Vajkoczy P, Acker G, Brandenburg S, et al. The CXCL2/IL8/CXCR2 pathway is relevant for brain tumor malignancy and endothelial cell function. Int J Mol Sci (2021) 22(5):2634. doi: 10.3390/ijms22052634
110. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: Beyond discovery and development. Cell (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
111. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer (2013) 13(12):871–82. doi: 10.1038/nrc3627
112. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res (2008) 14(21):6735–41. doi: 10.1158/1078-0432.CCR-07-4843
Keywords: FGL2, resident immune cell, glioma microenvironment, immunotherapy, immune checkpoint molecules
Citation: Ma X, Zhu H, Cheng L, Chen X, Shu K and Zhang S (2022) Targeting FGL2 in glioma immunosuppression and malignant progression. Front. Oncol. 12:1004700. doi: 10.3389/fonc.2022.1004700
Received: 27 July 2022; Accepted: 26 September 2022;
Published: 13 October 2022.
Edited by:
Wenchao Zhou, University of Science and Technology of China, ChinaReviewed by:
Cristiana Tanase, Victor Babes National Institute of Pathology (INCDVB), RomaniaCopyright © 2022 Ma, Zhu, Cheng, Chen, Shu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suojun Zhang, emhhbmdzdW9qdW5AdGpoLnRqbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.