 Ashlynn Ai Li Ler
Ashlynn Ai Li Ler Michael P. Carty
Michael P. Carty
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 07 February 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.822500
This article is part of the Research Topic Targeting DNA Repair and the DNA Damage Response: Beyond the Standard PI3 Kinase-like Kinases View all 16 articles
DNA lesions arising from both exogenous and endogenous sources occur frequently in DNA. During DNA replication, the presence of unrepaired DNA damage in the template can arrest replication fork progression, leading to fork collapse, double-strand break formation, and to genome instability. To facilitate completion of replication and prevent the generation of strand breaks, DNA damage tolerance (DDT) pathways play a key role in allowing replication to proceed in the presence of lesions in the template. The two main DDT pathways are translesion synthesis (TLS), which involves the recruitment of specialized TLS polymerases to the site of replication arrest to bypass lesions, and homology-directed damage tolerance, which includes the template switching and fork reversal pathways. With some exceptions, lesion bypass by TLS polymerases is a source of mutagenesis, potentially contributing to the development of cancer. The capacity of TLS polymerases to bypass replication-blocking lesions induced by anti-cancer drugs such as cisplatin can also contribute to tumor chemoresistance. On the other hand, during homology-directed DDT the nascent sister strand is transiently utilised as a template for replication, allowing for error-free lesion bypass. Given the role of DNA damage tolerance pathways in replication, mutagenesis and chemoresistance, a more complete understanding of these pathways can provide avenues for therapeutic exploitation. A number of small molecule inhibitors of TLS polymerase activity have been identified that show synergy with conventional chemotherapeutic agents in killing cancer cells. In this review, we will summarize the major DDT pathways, explore the relationship between damage tolerance and carcinogenesis, and discuss the potential of targeting TLS polymerases as a therapeutic approach.
It is estimated that up to 50,000 DNA lesions can occur per cell in a single day, an average of around 2,000 DNA lesions per cell per hour (1). While the majority of DNA damage is removed by repair pathways, including nucleotide excision repair and base excision repair, prior to cells entering S-phase, lesions can remain in the DNA template during DNA replication. The main DNA polymerases that carry out genomic DNA replication, polymerase δ (Pol δ) on the lagging strand and polymerase ϵ (Pol ϵ) on the leading strand, can both be blocked by DNA damage in the template strand, leading to replication fork stalling, fork collapse, chromosome breakage and genomic instability. To resolve this problem, DNA damage tolerance (DDT) pathways that allow replication of damaged DNA to continue while reducing genomic instability, are present in virtually all organisms (1–4).
There are two main DDT pathways described in eukaryotic cells, namely (i) translesion synthesis (TLS) and (ii) homology-directed DDT (Figure 1). TLS involves the recruitment of specialized TLS DNA polymerases to the arrested replication fork to facilitate lesion bypass, which can take place either directly at the replication fork, or behind the fork by repriming DNA synthesis at daughter strand gaps (DSGs) (Figure 1) (5). In response to DNA damage, monoubiquitination of the clamp protein proliferating cell nuclear antigen (PCNA) results in recruitment of the specialized TLS polymerases required to bypass the DNA lesion. Lesion bypass takes place either directly at the site of the arrested fork, or during gap-filling subsequent to replication restart away from the lesion site (6, 7) (Figure 1). However, despite some exceptions discussed below, bypass by TLS polymerases contributes to mutagenesis owing to the tendency for base misincorporation opposite lesions (7–9). In fact, the error-prone nature of TLS polymerases has been implicated both in the development of cancer and in promoting chemoresistance in cancer cells (10–12). Hence, TLS is considered an error-prone DDT pathway (13).
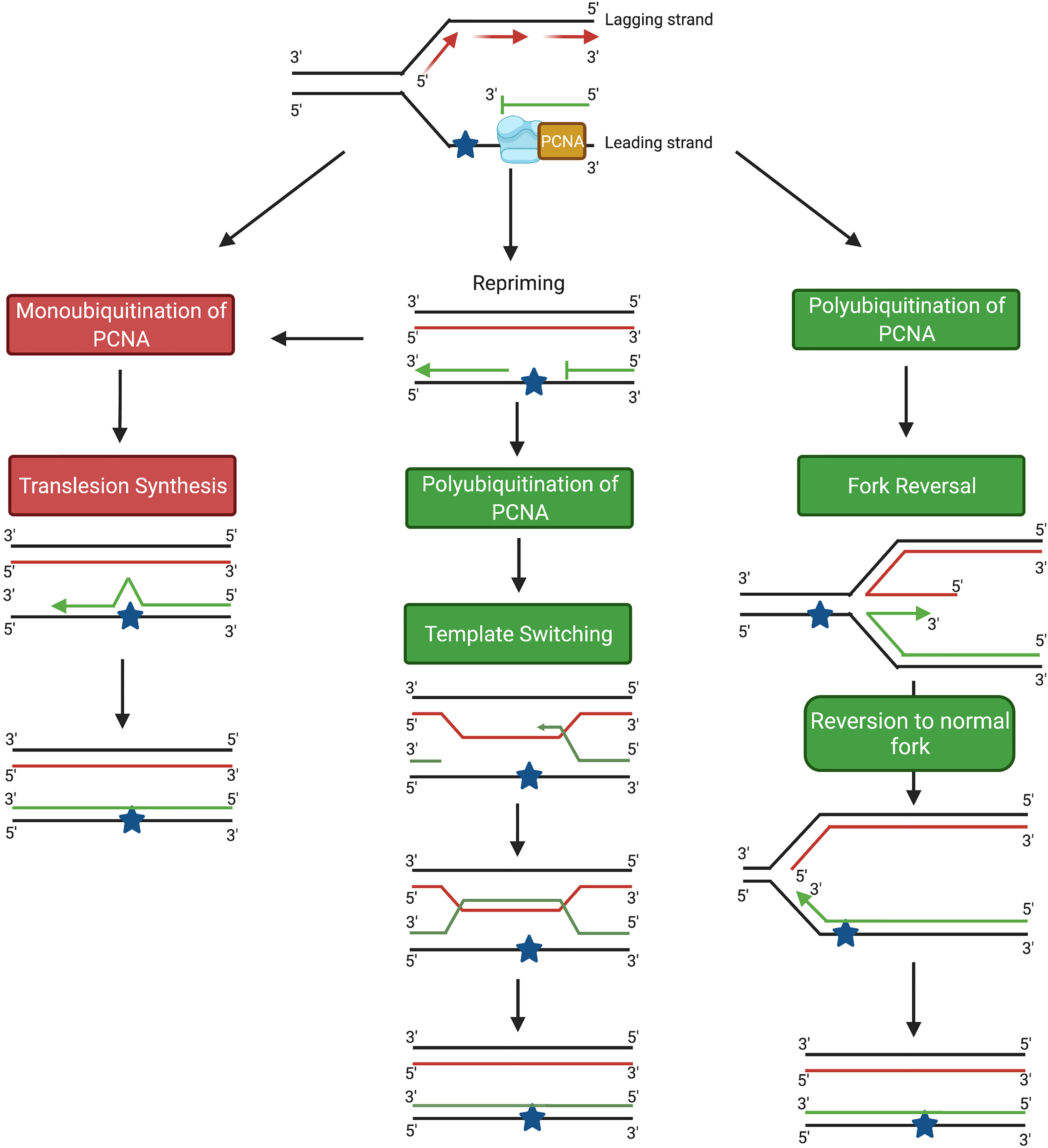
Figure 1 Schematic diagram showing the main DDT pathways in eukaryotic cells. TLS pathways are highlighted in red; homology-based damage tolerance pathways are highlighted in green. Image generated with BioRender.com.
In addition to TLS, damage tolerance can also take place through homology-directed DDT, which consists of two pathways, fork reversal (FR) and template switching (TS) (Figure 1) (5). Both FR and TS are initiated through the polyubiquitination of PCNA, and involve a temporary switch from the damaged template strand to using the newly-synthesized copy of the complementary strand on a homologous sister chromatid as the template for DNA synthesis. Because an undamaged template is copied, FR and TS are error-free lesion bypass pathways (5). Fork reversal involves formation of a ‘chicken foot’-like DNA structure, allowing the replisome on the arrested nascent strand to gain access to the homologous sister template (14, 15) (Figure 1). In contrast, TS occurs following repriming at DSGs generated at lesion sites behind the replication fork (16, 17). TS involves strand invasion, where the newly-synthesized strand from the homologous sister chromatid transiently serves as a template for nascent strand synthesis to allow the replication machinery to bypass the lesion (13, 18) (Figure 1).
On a biochemical level, the process of damage tolerance is complex, requiring multiple proteins. While these proteins are potential targets for development of novel cancer therapeutics, a more complete understanding of the molecular genetics, cell biology and biochemistry of damage tolerance is necessary to advance this potential. The present review provides an overview of the main DDT pathways in human cells, and discusses recent advances in targeting these pathways to develop cancer therapeutics.
Both the TLS and TS pathways share common initial steps. Stalling of the replicative DNA polymerase at a DNA lesion site together with ongoing helicase activity at the replication fork generates a region of single-stranded DNA (ssDNA) on the template strand which is bound by replication protein A (RPA). The ssDNA-RPA complex recruits ATRIP, and activates the ataxia telangiectasia and RAD3-related protein (ATR)-dependent replication checkpoint (18–20). At the same time, the chromatin remodelling protein INO80 binds to the stalled replication fork (18, 21–23). This, in conjunction with the RPA-ssDNA complex facilitates the recruitment of the RAD18 E3 ubiquitin ligase to the site of DNA damage (18, 23–26). At the lesion site, RAD18 recruits the E2 ligase RAD6, leading to the formation of the E2-E3 ubiquitinase (18, 25, 27–30) which monoubiquitinates PCNA on K164 (18, 25, 31–33). Monoubiquitination can be facilitated by other E3 ligases such as ring finger protein 8 (RNF8) in conjunction with the E2 ligase, Ubiquitin-conjugating Enzyme H5c (UbcH5c) (34). At this step, the two DDT pathways diverge, with monoubiquitination of PCNA on K164 resulting in the induction of TLS, while polyubiquitination at K164 leads to homology-directed DDT (Figure 2).

Figure 2 Schematic diagram showing key proteins involved in the initial steps of DNA damage tolerance pathway activation at an arrested replication fork. PCNA, proliferating cell nuclear antigen; RPA, replication protein A; UBC13, Ubiquitin-conjugating enzyme E2 13; MMS2, Ubiquitin-conjugating enzyme variant MMS2. Image generated with BioRender.com.
Following monoubiquitination of PCNA, one or more TLS polymerases are recruited to the stalled replication fork. Human TLS polymerases comprise proteins belonging to 4 families: the Y-family (Rev 1, Pol η, Pol ι and Pol κ), the A-family (Pol θ), the B-family (Pol ζ) and the archaeo-eukaryotic primase (AEP) family (PrimPol) (6, 35–38). In Y-family TLS polymerases, the active site that catalyses lesion bypass is located within the conserved N-terminal domain (39, 40), while the variable C-terminal region facilitates recruitment of the protein to stalled forks (39, 40). Y-family TLS polymerases can bind directly to K164-ubiquitinated PCNA through ubiquitin-binding zinc fingers (UBZ) found in Pol η and κ, or to ubiquitin-binding motifs (UBM) present in Pol ι and Rev1 (39). The PCNA-interacting peptide (PIP) boxes on Pol ι, η and κ, and the BRCA1 C-terminus (BRCT) domain in the N-terminal of Rev1, also facilitate the binding of TLS polymerases to PCNA (39, 41, 42).
TLS generally occurs by either a ‘one-polymerase’ mechanism or a ‘multiple-polymerase’ mechanism (35). Upon replication fork stalling in the presence of DNA damage, the replicative polymerase (δ or ϵ) is replaced by a TLS polymerase. Following this step, in the one-polymerase mechanism, a single TLS polymerase inserts nucleotides at the lesion site and continues to extend the replicated DNA strand past the lesion site, and is then replaced again by the replicative polymerase (35). The multiple-polymerase mechanism usually involves two TLS polymerases working in concert, such that one polymerase inserts a nucleotide opposite the lesion site, while the other extends the primer beyond the lesion site (35, 40, 43). Rev1 incorporates a single dCTP opposite a lesion site, but does not carry out subsequent polymerization (44–48). During bypass by S. cerevisiae Rev1, the lesion on the template strand is flipped into an extra-helical position and stabilised inside a hydrophobic pocket of Rev1, where it remains during incorporation of the incoming cytosine (49). The R324 side-chain of Rev1 displaces the DNA lesion, acting as an alternative template for Watson-Crick base pairing with the incoming cytosine (49). Following phosphodiester bond formation coupled with the hydrolysis of pyrophosphate, hydrogen bonding between the cytosine and R324 is broken (49). Rev1 then dissociates from the DNA and the lesion is reincorporated into the double helix (49).
Y-family DNA polymerases are characterised by a more open active site that can accommodate altered bases, and by the absence of 3’->5’ proofreading exonuclease activity (9, 40, 50, 51). For example, the active site of human Pol η can accommodate the two covalently-linked thymine bases in a UV-induced cis-syn thymine-thymine CPD lesion (50, 51). A β-strand in the little finger (LF) domain of the protein provides a molecular splint that stabilises the newly-synthesized double-stranded DNA into a B-form structure, preventing CPD-induced duplex distortion and frameshift formation, which facilitates efficient and accurate Pol η-mediated bypass of thymine-thymine CPDs (40, 50, 51).
As noted above, the capacity of TLS polymerases to accommodate altered bases in the active site, and the absence of 3’ to 5’ exonuclease activity facilitate lesion bypass (Table 1). Bypass is often at the cost of replication fidelity (70). The accuracy of TLS polymerases is lesion-dependent, such that specific TLS polymerases are more accurate than others when encountering particular lesions (70). For example, Pol θ predominantly incorporates the correct base when replicating across a 1,N6-ethenodeoxyadenosine lesion in human cells (66). However, Pol θ also plays an important role in the error-prone bypass of UV-induced cis-syn thymine-thymine CPDs and (6–4) PP lesions (66). By preventing the collapse of arrested replication forks and thereby reducing genome instability, error-prone lesion bypass by Pol θ protects against UV-induced skin cancer in mice (66). The overall fidelity of lesion bypass during TLS results from a combination of the biochemical properties of the individual TLS polymerases, the affinity of the polymerases for the lesion, as well as the sequence context of the lesion (9, 71).
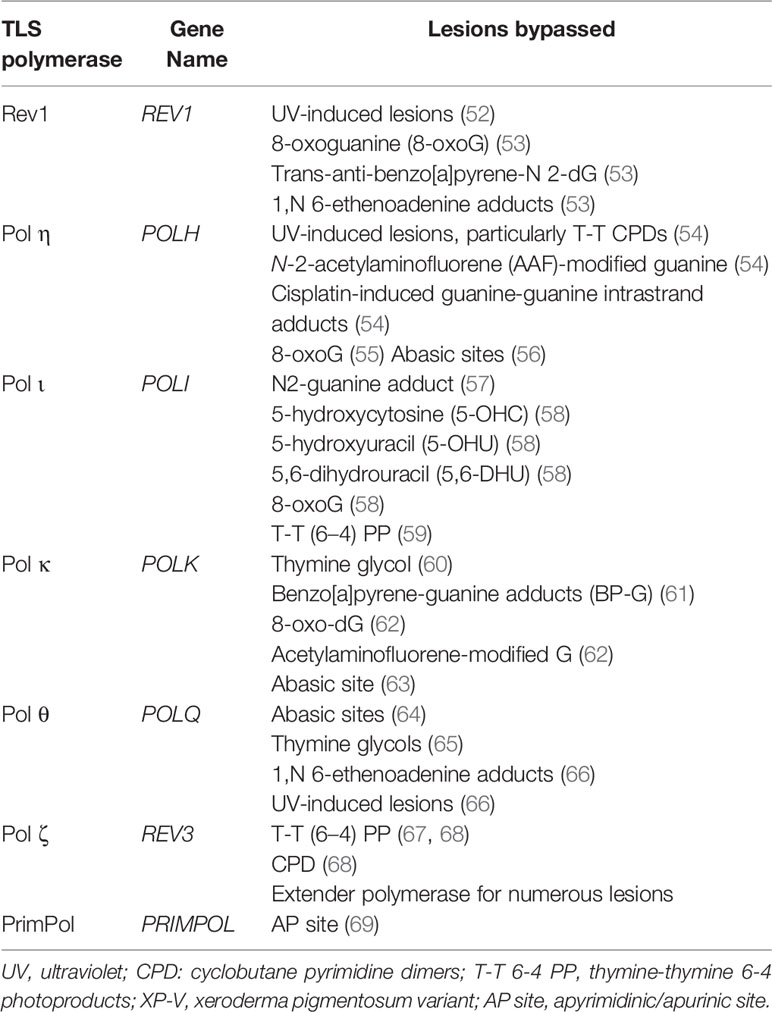
Table 1 Examples of lesion bypass by human TLS polymerases.
In addition to the error-prone TLS pathway, lesion bypass during S-phase can occur through the error-free homology-directed DDT pathways, FR and TS. Error-free DNA damage tolerance requires PCNA polyubiquitination, mediated by the recruitment of one of the yeast RAD5 homologues, SNF2 histone linker PHD RING helicase (SHPRH) or helicase-like transcription factor (HLTF), to the RAD6/RAD18 complex (2). In the FR pathway, remodeling of the stalled replication fork into the characteristic ‘chicken-foot’ structure (Figure 1) is initiated by the recruitment of the helicase protein SMARCAL1, which binds directly to ssDNA and removes bound RPA (5, 72–74). Following the removal of RPA, translocase zinc finger RANBP2-type containing 3 (ZRANB3) then promotes further fork reversal (5, 75–77). Binding of the Fanconi anemia complementation group M (FANCM) helicase to the protein-DNA complex leads to the formation of a four-way junction (78, 79). The reversed fork is stabilized by BRCA1, BRCA2 and RAD51, which bind to the exposed ends of the nascent leading and lagging strands and prevent MRE-11-mediated exonucleolytic degradation (5, 80–83). Following successful lesion bypass, regression of the reversed fork from a four-way junction into the original three-way junction (Figure 1) is catalysed by RecQ-like helicase (RECQ1), Werner syndrome RECQ-like helicase (WRN) and DNA replication helicase/nuclease 2 (DNA2) (5, 84, 85).
In the TS pathway (Figure 1), following polyubiquitination of PCNA, the 9-1-1 complex binds to the 5’ end of the gap on the nascent DNA strand and recruits exonuclease 1 (EXO1) (18, 86). A RAD51-ssDNA presynaptic filament, stabilized by RAD55/RAD57, then forms on the ssDNA region of the template strand (87–89). ATP-dependent DNA helicase SRS2 (SRS2) disrupts the nucleofilament and opposes the action of RAD55/RAD57; the balance between these processes determines the overall stability of the RAD51-ssDNA presynaptic filament (18, 90, 91). The nucleofilament, with RAD52 and RAD54, carries out both the homology search and strand invasion of the sister chromatid (18, 89, 92). After complementary base-pairing between the invading strand and the homologous template, DNA pol δ is recruited and continues DNA replication, generating a D-loop and subsequently a sister-chromatid junction (SCJ) (18, 89, 93–95). D-loop formation is negatively regulated by SRS2 (18, 96). Finally, the slow growth suppressor 1 (SGS1)/DNA topoisomerase 3 (TOP3)/RECQ-mediated genome instability protein 1 (RMI1) complex pries the SCJ apart, regenerating the normal double-helical DNA structure (16, 18, 97, 98) (Figure 1).
The type of PCNA ubiquitination plays a key role in the choice of DDT pathway between either TLS or homology-directed DDT (5, 9, 99). PCNA monoubiquitination leads to TLS, while polyubiquitination results in the initiation of homology-directed DDT. The overall level of ubiquitinated PCNA is also regulated by ubiquitin-specific processing protease 7 (USP7), by the USP1/upstream activation factor (UAF1) complex, and by enhanced level of genomic instability 1 (ELG1) (99–103). Following UV irradiation, USP1 undergoes auto-cleavage and degradation, increasing the level of modified PCNA (104). It has been proposed that the extent of replication arrest is a factor in determining the type of PCNA ubiquitination, such that prolonged replication arrest leads to polyubiquitination of PCNA molecules that remain bound at the arrest site, promoting a switch to homology-directed DDT (99). Alternatively, homology-directed DDT could be activated first where HLTF is recruited together with the RAD6/RAD18 complex, resulting in the immediate polyubiquitination of PCNA (99). In addition to ubiquitination, PCNA undergoes other related modifications. Protein inhibitor of STAT (PIAS1 and PIAS4)-mediated SUMOylation of PCNA on K164 promotes template switching rather than TLS (105). After TLS is completed, monoubiquitinated PCNA is modified by the addition of interferon-stimulated gene 15 (ISG15) molecules, leading to recruitment of USP10 and PCNA deubiquitination (99, 106, 107). Understanding the interplay between PCNA modification and the choice of DNA damage tolerance pathway is an important area for further study.
Since TLS polymerases are generally error-prone it is critical that TLS activity is tightly regulated. The main points of regulation of TLS involve the interactions between TLS polymerases, accessory proteins, RAD18 and PCNA (108). CHK1 and CLASPIN are essential for binding of RAD18 to chromatin (18, 109). SIVA1, TIMELESS and HLTF play important roles in PCNA monoubiquitination (18, 109–111), while protein with SprT-like domain at the N-terminus (SPARTAN) is crucial both for binding of RAD18 to chromatin and for monoubiquitination of PCNA (112, 113).
Regulation of the TLS pathway also occurs at the level of the individual polymerases, where TLS polymerases undergo post-translational modification including ubiquitination, SUMOylation and phosphorylation (as shown in Table 2 for human Pol η). In the absence of DNA damage in the template, or when DNA lesions have been bypassed, TLS polymerases are monoubiquitinated, switching the protein from an open conformation, where the C-terminal ubiquitin-binding domain is available to interact with monoubiquitinated PCNA, to a closed conformation, where this domain is bound in cis to ubiquitin and is unable to interact with PCNA (128). For human Pol η, in the closed conformation ubiquitination of one of the four lysines K682, K686, K694 and K709 results in interaction between the ubiquitin moiety and the UBZ domain of the polymerase which competes with ubiquitinated PCNA for UBZ binding, thereby abrogating the PCNA interaction (118, 128). Following UV-induced DNA damage in non-small cell lung and colon carcinoma cell lines, ubiquitinated Pol η is polyubiquitinated by mouse double minute 2 homologue (MDM2) resulting in degradation by the proteasome by 24 hours post-irradiation (121, 128). The other Y family TLS polymerases Rev 1, Pol ι and Pol κ are also ubiquitinated (51, 128). In addition to ubiquitination, Pol η undergoes SUMOylation (126, 127). PIAS1-dependent SUMOylation on K163 targets Pol η to difficult-to-replicate regions of the genome such as fragile sites even in the absence of exogenous DNA damage (127). Following completion of TLS, SUMOylation of Pol η on multiple lysine residues prevents ongoing interaction with ubiquitinated PCNA, leading to SUMO-targeted ubiquitin ligase (STUbL)-mediated ubiquitination of Pol η and its’ exclusion from damage sites (127).

Table 2 Proteins regulating Pol η function in TLS.
TLS polymerase activity is also modulated by phosphorylation. Pol η is phosphorylated at a number of sites in the C-terminus by protein kinases including ATR, CDK2 and PKC. Following DNA damage, ATR-mediated phosphorylation of Pol η on serine 601 (114) releases it from sequestration by Pol δ-interacting protein of 38 kDa (PDIP38), freeing Pol η to bind to monoubiquitinated PCNA (115). This links ATR activation by replication arrest-induced single-stranded DNA, with recruitment of TLS polymerases to the arrested fork (115). Pol η is additionally phosphorylated by PKC on S587 and T617 (129), and on serine 687 by CDK2, which increases the stability of the polymerase in late-S and G2/M (125).
In addition to post-translational modification of specific proteins, TLS is regulated at the transcriptional level. Following DNA damage, POLH expression is p53-dependent (130), while POLK expression is regulated by the aryl hydrocarbon receptor (AhR) (131, 132). A recent report shows that TLS is negatively regulated by Pumilio RNA Binding Family Member 1 (PUM1), a protein that mediates mRNA decay (133). miRNAs have also been identified which modulate expression of DNA damage tolerance proteins. Examples include MiR-145 and miR-630 which downregulate RAD18 expression, and miR-93 and miR-619 which downregulate Pol η expression (39, 134). Furthermore, alternative polyadenylation of the POLH mRNA transcript in lung and bladder cancer cells generates three transcripts having 3’-UTR sequences of 427, 2516 or 6245 nucleotides, respectively (135). Of note, miR-619 only targets the longer transcript, while the shortest transcript is resistant to miR-619, and is responsible for increased Pol η expression and cisplatin resistance in cancer cell lines (135).
Interplay between fork-protective and fork-degradative factors plays a key role in modulating fork reversal (104). BRCA1, BRCA2 and RAD51 shield the nascent DNA strands at the reversed fork from degradation by the exonuclease action of MRE-11 (5, 80, 81). WRN helicase interacting protein 1 (WRNIP1) also protects reversed forks from structure-specific endonuclease subunit SLX4 (SLX4)-mediated fork cleavage and subsequent DNA2-mediated fork degradation (136–138). The interaction of polyubiquitinated PCNA with ZRANB3 slows fork progression, promoting fork reversal through the translocase activity of ZRANB3 (75, 76). Recruitment of SMARCAL1 to the stalled fork is regulated by ATR-mediated phosphorylation, thereby limiting the extent of fork reversal (136, 139). Poly(ADP-ribose) polymerase 1 (PARP1) modulates fork reversal and fork restart by inhibiting RECQ1 helicase, and prolongs FR by preventing RECQ1-mediated regression of reversed forks to three-way junctions (15, 85, 136).
Template switching is regulated at a number of points including PCNA polyubiquitination, the formation of the RAD51-ssDNA presynaptic filament and SCJ formation [reviewed in (18)]. The chromatin remodeling protein INO80, and the human Rad5 orthologues, HLTF and SHPRH are important for PCNA polyubiquitination (140). Chromatin remodeling by INO80 facilitates the addition of K63-linked polyubiquitin chains to PCNA by HLTF and SHRPH (5, 23, 141–143). The stability of the RAD51-ssDNA filament involved in homology searching is negatively regulated by SRS2 (18, 144, 145), while exonuclease 1 (EXO1), INO80 and high mobility group protein 1 (HMO1) facilitate SCJ formation (18, 23, 89, 146).
The role of TLS in preventing cancer is clearly demonstrated in the sun-sensitive skin cancer-prone disease xeroderma pigmentosum variant (XP-V), where the absence of Pol η as a result of inactivating mutations in POLH (147, 148) leads to prolonged replication arrest at the sites of UV-induced lesions in the template. In the absence of error-free bypass of UV-induced CPDs by Pol η in XP-V cells, error-prone lesion bypass is carried out by polymerases including Pol ι and Pol ζ, resulting in increased mutagenesis that contributes to skin carcinogenesis in XP-V patients (9, 149). However, error-prone TLS can also play an anti-carcinogenic role. As noted, error-prone bypass of UV-induced lesions by Pol θ protects against skin cancer in mice, by allowing ongoing DNA synthesis to proceed thereby preventing strand break formation and the resulting genomic rearrangements (66). As a source of spontaneous mutagenesis, low-fidelity TLS polymerases may play a role in driving carcinogenesis. Y-family TLS polymerases in particular have been implicated as a source of somatic mutations in tumors (150). For example, Pol η mutational signatures are found in the genome of cancer cells from patients with malignant B-cell lymphoma and chronic lymphocytic leukemia (151).
Polymorphisms in genes encoding TLS polymerases are also associated with increased cancer risk. Polymorphisms in REV1 and POLI, leading to single amino acid substitutions in Rev1 and Pol ι, were associated with increased risk of squamous cell carcinoma and adenocarcinoma, respectively (152), while POLH polymorphisms are associated with increased risk of malignant melanoma (153). In addition to polymorphic variants, sequencing of tumor DNA has revealed somatic mutations in TLS polymerase genes in a number of tumor types (10, 154). While the functional significance of most of these mutations remains to be determined experimentally, mutations in TLS genes that affect protein function could in principle lead to genome instability and contribute to tumor development, or alter the response of tumor cells to chemotherapeutic DNA damaging agents (10).
TLS polymerases are overexpressed in a number of different cancers. It has been proposed that overexpression of TLS polymerases can facilitate error-prone replication and adaptation of the cancer cells to targeted therapy (155). For example, expression of TLS polymerases ι, κ, λ, μ and Rev1 was upregulated in colorectal cancer cells following treatment with inhibitors of B-RAF or EGFR signalling (155). However, whether increased levels of TLS polymerases directly contribute to the acquisition of adaptive mutations requires further investigation. In non-small-cell lung tumors increased expression of Pol η is associated with poorer prognosis (156, 157), while increased expression of Pol ι is associated with oesophageal squamous cell cancer and directly correlates with the degree of metastasis (158). Pol ι expression also correlates with the grade of bladder tumors (159), while high expression of Pol κ in glioblastoma tumors is associated with poor prognosis (160).
From the perspective of cancer treatment, TLS can increase the tolerance of cancer cells to DNA damage induced by chemotherapeutic anti-cancer agents, thus promoting cancer cell survival, and increasing the mutational burden as result of error-prone lesion bypass. Pol κ plays a role in the response to the alkylating agent temozolomide used in the treatment of glioblastoma. Increased expression of Pol κ enhanced the resistance of human glioblastoma cell lines to temozolomide while down-regulation sensitised the cells to the drug (135). Pol η can bypass cisplatin-induced intrastrand lesions (161–163), and also plays a role in interstrand crosslink repair (164, 165). Human cells lacking Pol η are more sensitive to platinum-based chemotherapeutic agents (162, 163, 166–168). Overexpression of Pol η and Pol ζ contributes to cisplatin resistance in ovarian cancer stem cells and human glioma cells (169, 170). It was recently shown that PrimPol enhances survival of cisplatin-treated BRCA-deficient human ovarian cancer and osteosarcoma cells (171). PrimPol promotes repriming by reinitiating DNA synthesis downstream of blocking lesions in the template, thereby preventing fork reversal and degradation (171). In addition to promoting resistance to direct DNA-damaging agents, TLS polymerase levels also affect the response to signalling pathway inhibitors. Pol κ increased the resistance of melanoma cells to the B-RAF inhibitor vemurafenib (70). Although the mechanisms of TLS polymerase overexpression in cancer cells remain to be elucidated, overexpression of Pol κ is regulated through activation of the aryl hydrocarbon receptor (AhR) by the endogenous tryptophan-derived ligand kyneurin, as well as by DNA damaging agents such as benzo[a]pyrene (B[a]P) (131, 132, 172, 173). In the case of Pol η, expression is regulated in a p53-dependent manner after exposure of cells to DNA damage (130).
Other than DNA polymerases, alterations to regulatory proteins that play a role in TLS may also contribute to cancer development. For example, RAD18 deletions were identified in 5% of pancreatic tumors and 11% of renal cell carcinoma tumors examined (174). Increased expression of RAD18 in a variety of human cancer cell lines (including H1299 non-small cell lung carcinoma cells, H157 and H650 adenocarcinoma cells and U2OS osteosarcoma cells) leads to excessive activation of the TLS pathway, contributing significantly to hypermutability (150). RAD18 protein levels can be increased by upregulation of melanoma antigen-A4 (MAGE-A4), which binds to and stabilises RAD18, activating the TLS pathway (175). The RAD5 ortholog HLTF, important in PCNA polyubiquitination, is downregulated through promoter methylation in colon cancer cell lines and in primary tumors (176).
Given the role of DNA damage tolerance pathways in driving chemoresistance, there is potential to sensitize cancer cells to chemotherapy by inhibiting these pathways (177). To date, the major focus has been on identification of TLS inhibitors. A number of inhibitors of TLS-mediated lesion bypass have been reported (Table 3) and are discussed below. The inhibitors fall broadly into two categories: (i) inhibitors that directly interfere with TLS polymerase catalytic function and (ii) inhibitors that interfere with protein-protein interactions (PPIs) to inhibit TLS indirectly.
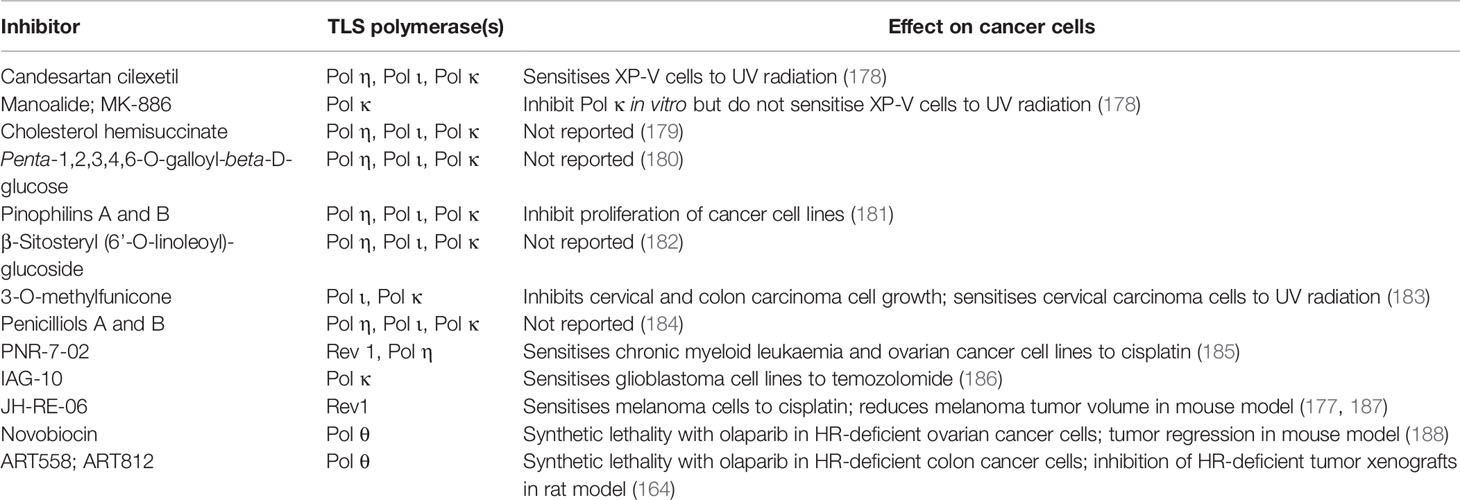
Table 3 Inhibitors of TLS polymerases.
In recent years, several TLS polymerase inhibitors have been reported (187). Examples include indole thiobarbituric acid (ITBA) and its’ derivatives (185, 186). ITBA binds directly to the finger and LF domains of Pol η, which may prevent the polymerase from binding to ssDNA and interfere with nucleotide incorporation (186). The ITBA derivative ITBA-12 inhibits Pol η and Pol κ activity (186), while ITBA-16 and ITBA-19, containing N-1-naphthoyl and N-2-naphthoyl Ar-substituents have increased specificity towards Pol η (186). The ITBA derivative, PNR-7-02 which binds to the little finger domain and inhibits Pol η function, acts synergistically with cisplatin to kill chronic myeloid leukaemia and ovarian cancer cell lines (185). An indole-aminoguanidine analogue, IAG-10, binds human Pol κ preventing the N-clasp domain from holding the LF domain in place, triggering a conformational change that decreases the contact between the protein and the DNA template (190). IAG-10 synergises with temozolomide to kill glioblastoma cell lines in culture (190). This supports the concept that direct inhibitors of TLS polymerases can increase the cytotoxic effects of conventional chemotherapeutic agents (190).
In addition to the identification of novel compounds that inhibit TLS polymerases, certain existing small molecules in clinical use have been reported to inhibit TLS. These include candesartan cilexetil, used clinically as an angiotensin-II receptor antagonist in the treatment of hypertension (191); manoalide, a phospholipase A2 inhibitor with both analgesic and anti-inflammatory properties (192), and MK-886, a leukotriene antagonist (193). Candesartan cilexetil, manoalide and MK-886 were shown to inhibit the in vitro activity of purified human Pol κ on undamaged DNA templates and to inhibit bypass of a γ-HOPdG lesion by Pol κ (178). Candesartan cilexetil, but not the other two compounds, sensitised Pol η-deficient XP-V cells to UV radiation (178). The fungal-derived molecules 3-O-methylfunicone, and Penicilliols A and B have been identified by screening for natural products that inhibit TLS polymerases (183). 3-O-methylfunicone, isolated from an Australian sea salt fungal strain, inhibits Y-polymerases κ, ι and η (183), competing directly with the DNA template-primer for interaction with the DNA binding domain of Pol κ (183). 3-O-methylfunicone decreased the growth of two cervical carcinoma and colon carcinoma cell lines, while having little effect on the growth and proliferation of normal human cells (183). Penicilliols A and B, isolated from a strain of Penicillium daleae, also inhibit mammalian Y-family polymerases, in particular Pol ι (184).
Recent reports (188, 189) demonstrating that novel inhibitors of human Pol θ synergise with HR-deficiency or resistance to PARP inhibition to kill cancer cells provides strong support for the strategy of targeting specialised DNA polymerases in cancer therapy. The Pol θ inhibitors include the antibiotic novobiocin (188) and the synthetic small molecule allosteric inhibitor ART558 (189). When used in conjunction with PARP inhibitors, Pol θ inhibitors induce synthetic lethality in HR-deficient cancer cells. PARP is required for repair of single strand breaks and inhibition of PARP-dependent single-strand break repair increases the level of double-strand breaks in the genome. Novobiocin synergistically increased the cytotoxic effects of the PARP inhibitors rucaparib and olaparib in BRCA1-deficient human retinal pigment epithelial cells and ovarian cancer cell lines, respectively (188). In mouse studies, novobiocin sensitized tumors arising from PARPi-resistant ovarian carcinoma cells to treatment with olaparib, resulting in tumor regression (188). The novel small molecule ART558 induced synthetic lethality in PARPi-resistant BRCA2-deficient human colon cancer cells treated with olaparib (189). ART812, a more bioavailable derivative of ART558, inhibits tumor xenograft growth in a rat model (189). Mechanistically, the cytotoxic effects of the Pol θ inhibitors are synergistic with HR-deficiency and PARP inhibitor resistance due to the effect of the molecules on Pol θ activity in the Theta-mediated end-joining pathway of DNA double-strand break repair (188, 189).
Protein-protein interactions (PPIs) play a critical role in lesion bypass. PPIs include the key interactions between Ub-PCNA and TLS polymerases, as well as interactions between inserter and extender TLS polymerases, for example between Rev1 and other TLS polymerases, and between the Rev7 and Rev3 subunits of the Pol ζ complex. A number of PPI inhibitors have been developed based on detailed structural information on the interaction domains of the target proteins (194).
Indirect inhibitors of TLS can inhibit the recruitment of TLS polymerases to PCNA, thereby preventing lesion bypass. Small molecule inhibitors of the PCNA/PIP-box interaction compete with the PIP-box sequence of TLS polymerases for binding to PCNA during the initiation phase of TLS. The compound 3,3’,5-triiodothyronine (T3) and its synthetic derivative T2 amino alcohol (T2AA) were reported to inhibit the PCNA/PIP-box PPI (195). T2AA and its analogues suppressed TLS in NER-deficient human cells, decreased cell division in osteosarcoma cells treated with cisplatin (196), and inhibited interstrand DNA cross-link (ICL) repair, slowing proliferation of cervical cancer cells (196). Consistent with the importance of the PCNA-PIP box interaction, a novel compound which specifically targets the L126 to Y133 region of the PIP-interaction loop of PCNA sensitises cancer cells to cisplatin (197). Other inhibitors of the PCNA/TLS polymerase interaction specifically prevent the recruitment of Rev1 to PCNA. One small molecule inhibitor, compound 1, binds Rev1 directly via the UBM2 motif and prevents interaction with K164-ubiquitinated PCNA (198). Compound 1 increased the cytotoxicity of both 4-hydroxycyclophosphamide and cisplatin by up to 10-fold in cultured cells (198).
Inhibitors of essential PPIs between inserter and extender TLS polymerases have also been identified that suppress TLS and enhance the cytotoxicity of chemotherapeutic agents. Among these are small molecule inhibitors of the interaction between the C-terminal domain of Rev1 (Rev1-CT) and the Rev1-interacting region (RIR) of other Y-family TLS polymerases. Two such compounds, 4 and 5, have been reported (199) that bind to the Rev1-CT, preventing the recruitment of Pol ζ to Rev1, and sensitising fibrosarcoma cell lines to cisplatin and to UV radiation. Both compounds decreased the level of cisplatin-induced HPRT gene mutations, indicating the molecules can attenuate the mutagenicity associated with error-prone replication of platinum-induced DNA lesions (199). Additional derivatives have been developed that also compete with TLS polymerases for binding to the Rev1-CT domain, and increase the cytotoxicity of cisplatin (200, 201). Of note, inhibition of the Rev1-CT/RIR interaction was synergistic with the ATM and ATR inhibitor VE-821 and the Wee1 inhibitor MK-1775, leading to the formation of daughter strand gaps (DSGs) in replicating DNA, and sensitising bone osteosarcoma and colon cancer cells to these agents (201). Consistent with this, Pol η-deficient cells are significantly more sensitive to ATR inhibitors than normal cells (129, 202, 203). Since daughter strand gaps can also result from replication stress in oncogene-mutated cancer cells, it is proposed that in addition to direct lesion bypass, TLS polymerases contribute to cancer cell survival by carrying out DNA synthesis at DSGs. This limits the accumulation of single-stranded DNA in the genome (201), a process termed gap suppression (201). TLS inhibitors could therefore be used to achieve synthetic lethality in combination with cell cycle checkpoint inhibitors that induce DSGs, such as inhibitors of ATR and Wee1 (201).
Aside from inhibitors that interrupt Rev1-CT-RIR interactions, a small molecule inhibitor that disrupts the interaction between Rev1 and the Rev7 subunit of Pol ζ has also been identified. In both human and mouse cell lines, JH-RE-06, a small molecule that binds to the C-terminal domain of Rev1 and blocks interaction with the Rev7 subunit of Pol ζ, sensitised melanoma cells to cisplatin, and reduced drug-induced mutagenesis (204). Combination treatment with JH-RE-06 and cisplatin reduced tumor volume and improved survival in a mouse xenograft model of A375 melanoma cells, demonstrating the potential of targeting key PPIs as a therapeutic strategy (204, 205). Treatment of fibrosarcoma and melanoma cells with JH-RE-06 leads to senescence following cisplatin-induced DNA damage (204, 205). The impact of chemical inhibition of Pol ζ activity on overall genome stability should also be considered, since genetic ablation of REV3L encoding the catalytic subunit of Pol ζ increased genome instability in REV3L-null mouse embryo fibroblasts (206), and contributed to development of lymphomas and mammary tumors in mice where REV3L was conditionally deleted (207).
Recent evidence indicates that the interaction between Rev7 and Rev3 proteins required to form active Pol ζ is actively regulated in cells (208). The ATPase thyroid receptor-interacting protein 13, (TRIP13), modulates the conformation of Rev7, preventing both its’ interaction with Rev3 to form of active Pol ζ which is required for TLS, and interaction with the Shieldin complex which activates NHEJ (208). TRIP13 therefore mediates pathway choice, promoting error-free HDR over mutagenic TLS or NHEJ (208). TRIP13 overexpression correlates with BRCA1-deficiency in breast cancer cells and contributes to chemoresistance towards PARP inhibitors (208). The interaction of Rev7 with the Shieldin complex and with Rev3 is also inhibited by expression of the p31comet HORMA-like protein (209, 210), identifying Rev7 as a key modulator of pathway choice after DNA damage. Overall, inhibition of key interactions between TLS polymerases and partner proteins represents a promising approach to sensitising cancer cells to chemotherapy.
While targeting the catalytic activity and protein-protein interactions of TLS polymerases has been shown to be effective in overcoming chemoresistance in cancer cells, there may also be potential in targeting upstream regulators of TLS to inhibit the action of multiple TLS polymerases for therapeutic effect. For example, targeting the TLS pathway regulator RAD6 and PCNA monoubiquitination could be a more potent method to inhibit TLS than targeting individual TLS polymerases. However, the effects of inhibiting RAD6 on cytotoxicity and genome stability after DNA damage should be directly compared with the effects of targeting individual TLS polymerases using specific inhibitors. Inhibition of RAD6 by a small-molecule inhibitor, SMI#9, attenuated cisplatin resistance in triple-negative breast cancer cells, and enhanced the cytotoxicity of oxaliplatin towards the oxaliplatin-resistant colorectal carcinoma cell line HCT116-OxR (211, 212). Co-administration of SMI#9 with cisplatin decreased the growth of tumors arising from triple-negative breast cancer cells and lymph node metastasis (211). Molecules that modulate the extent of PCNA monoubiquitination and therefore the recruitment of TLS polymerases to DNA damage sites have also been described. C11 and G8, two inhibitors of the protein kinase AKT, inhibit damage-induced PCNA monoubiquitination and show synthetic lethality with UV-irradiation in BRCA1-deficient triple-negative breast cancer and colon cancer cell lines (213). The specific targets of AKT that modulate PCNA monoubiquitination are of interest (213).
Additional potential targets for inhibition of TLS include the USP1/UAF1 deubiquitinase complex. Two small molecule USP1/UAF1 inhibitors, pimozide and GW7647, enhanced the cytotoxicity of cisplatin and decreased cell division in non-small cell lung cancer cell lines (214). A third USP1/UAF1 inhibitor, ML323, was found to increase the cytotoxic effect of cisplatin on osteosarcoma and non-small cell lung cancer cells (215). As well as affecting TLS, USP1/UAF1 inhibitors increase cisplatin sensitivity by disrupting deubiquitination of the FANCD2/FANCI complex, preventing repair of drug-induced interstrand adducts by the FA crosslink repair pathway (215–217). The level of the chaperone protein Hsp90 is also important in the stability of TLS polymerases, including Rev1 (218) and Pol η (219). Tanespimycin (17-AAG), which promotes proteasomal degradation of Hsp90, decreased the amount of Rev1 in human prostate and bone osteosarcoma cancer cells (218), and downregulated the recruitment of Rev 1 to sites of UV-induced DNA damage in the nucleus (218). The proteasome inhibitors, lactacystin and MG-132, prevented the reduction in Rev1 levels induced by 17-AAG (218), indicating that Hsp90 normally protects Rev1 from proteasomal degradation. Following UV-induced DNA damage, Pol η undergoes direct PIAS1-mediated poly-SUMOylation upon recruitment to PCNA. The protein is then modified by SUMO-targeted ubiquitin ligases (STUbLs), which is crucial for clearance of the polymerase from damage sites following lesion bypass. Targeting the human STUbLs RNF4 and RNF11 could potentially enhance the turnover rate of mutagenic TLS polymerases and decrease lesion bypass in cancer cells (127). Given the extensive ubiquitination of proteins involved in DNA damage tolerance, the response to general inhibitors of the proteasome is complex. Of interest, it has been reported that the proteasome inhibitors MG-132, lactacystin, and MG-262 inhibited TLS in human cancer cell lines, but not in normal cells (220), indicating that targeting the degradation of specific proteins could represent another approach to modulation of the DNA damage tolerance pathway in cancer cells.
Alternative approaches of TLS inhibition include the use of novel non-natural nucleotides to inhibit bypass synthesis, and down-regulation of DDT protein expression using miRNAs. Two synthetic nucleotide analogues, 5-nitro-indolyl-2′deoxyriboside triphosphate (5-NITP) and 5-phenyl-indolyl-2′deoxyriboside triphosphate (5-PhITP), are preferentially incorporated opposite abasic sites during DNA replication, while resisting both excision by proofreading exonuclease activity and subsequent elongation, preventing further DNA replication past the damage sites (221). TLS polymerases, specifically Pol η and Pol ι, preferentially inserted 5-NITP over dATP opposite temozolomide-induced abasic sites, preventing lesion bypass at the damage sites (222). The synthetic nucleoside potentiated the cytotoxic effects of temozolomide in glioblastoma cancer cell lines, and led to tumor regression in mouse models of tumor growth (222).
Identification of miRNAs that regulate expression of key genes represents another potential mechanism of DDT inhibition. For example, miR-96 regulates expression of RAD51 and REV1 (223). Knockdown of REV1 expression using miR-96 contributed to cisplatin sensitization in bone osteosarcoma cells with intact HR repair, as well as in BRCA1-deficient breast cancer and BRCA2-deficient ovarian cancer cell lines with compromised HR pathways (223). Direct inhibition of RAD51 and REV1 expression by overexpression of miR-96 ultimately slowed growth of tumors from triple-negative breast cancer cells in mice (223).
There is increasing evidence that inhibition of DNA damage tolerance pathways can sensitize cancer cells to conventional chemotherapeutic agents. Ongoing research into the molecular basis of DNA damage tolerance will provide opportunities for further advances. The factors that influence pathway choice in specific circumstances require further investigation. For the TLS pathway, understanding the contribution of individual DNA polymerases to lesion bypass in cancer and normal cells will be important, and will further inform the design of specific inhibitors that target polymerases or accessory proteins. In addition to polymerases, other DDT proteins could also represent therapeutic targets, using agents that directly block protein function, or interfere with specific PPIs that are essential for DNA damage tolerance. The major focus to date has been on inhibition of TLS, but given the recent advances in elucidating the genetics and biochemistry of fork reversal and template switching, there is potential to identify new inhibitors targeting these pathways. The risk of directing replication intermediates into other more error-prone pathways, increasing genetic instability and further contributing to the development of resistant cancer cells, also has to be considered in this context. Further challenges include the development of specific inhibitors, proving that cellular phenotypes are due to inhibition of the proposed target, and demonstrating clinical utility for small molecule inhibitors. Ongoing research will advance the potential to target DNA tolerance pathways as a therapeutic approach for cancer.
DDT pathways are critical to allow cells to tolerate DNA lesions and facilitate the completion of DNA replication. However, imbalances in these pathways in cancer cells can lead to significant mutagenesis, contributing to chemoresistance and increased cancer cell survival. Considering the evidence that inhibiting DDT pathways can sensitize cancer cells to chemotherapy, more research into novel therapeutics in this area could eventually lead to the development of a new class of cancer therapeutic agents that enhance the response to treatment with conventional chemotherapy.
AL wrote the initial draft of the review and MC revised and edited the review. Both authors approved the final version.
Open access publication fees were supported by the Discipline of Biochemistry, The National University of Ireland Galway.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors apologise to colleagues for any work not cited in this review.
ATR, ataxia telangiectasia and RAD3-related protein; ATRIP, ATR-interacting protein; DDT, DNA damage tolerance; FA, Fanconi anemia; FR, fork reversal; HTLF, helicase-like transcription factor; PARP, Poly(ADP-ribose) polymerase; PCNA, proliferating cell nuclear antigen; PIP, PCNA-interacting peptide; PPI, protein-protein interactions; Rev1-CT, Rev1 C-terminal; RIR, Rev1-interacting region; RPA, Replication protein A; SHPRH, SNF2 histone linker PHD RING helicase; SMARCAL1, SWI/SNF-related matrix associated actin-dependent regulator of chromatin subfamily A-like protein 1; SPARTAN, SprT-like domain at the N-terminus; ssDNA, single-stranded DNA; TLS, translesion synthesis; TLSi, translesion synthesis inhibitor; TS, template switching; UBM, ubiquitin-binding motif; UBZ, ubiquitin-binding zinc finger; USP1, ubiquitin carboxyl-terminal hydroxylase 1; USP7, ubiquitin carboxyl-terminal hydroxylase 7; XP-V, xeroderma pigmentosum variant; ZRANB3, Translocase zinc finger RANBP2 type –containing 3.
1. Livneh Z, Cohen IS, Paz-Elizur T, Davidovsky D, Carmi D, Swain U, et al. High-Resolution Genomic Assays Provide Insight Into the Division of Labor Between TLS and HDR in Mammalian Replication of Damaged DNA. DNA Repair (2016) 44:59–67. doi: 10.1016/j.dnarep.2016.05.007
2. Xu X, Blackwell S, Lin A, Li F, Qin Z, Xiao W. Error-Free DNA-Damage Tolerance in Saccharomyces Cerevisiae. Mutat Research/Reviews Mutat Res (2015) 764:43–50. doi: 10.1016/j.mrrev.2015.02.001
3. Ulrich H. Conservation of DNA Damage Tolerance Pathways From Yeast to Humans. Biochem Soc Trans (2007) 35(5):1334–7. doi: 10.1042/BST0351334
4. Kunz BA, Xiao W. DNA Damage Tolerance in Plants via Translesion Synthesis. Genes Genomes Genomics (2007) 1(1):89–99.
5. Pilzecker B, Buoninfante OA, Jacobs H. DNA Damage Tolerance in Stem Cells, Ageing, Mutagenesis, Disease and Cancer Therapy. Nucleic Acids Res (2019) 47(14):7163–81. doi: 10.1093/nar/gkz531
6. Yang W. An Overview of Y-Family DNA Polymerases and a Case Study of Human DNA Polymerase η. Biochemistry (2014) 53(17):2793–803. doi: 10.1021/bi500019s
7. Makarova AV, Burgers PM. Eukaryotic DNA Polymerase ζ. DNA Repair (2015) 29:47–55. doi: 10.1016/j.dnarep.2015.02.012
8. Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol Mol Biol Rev (2009) 73(1):134–54. doi: 10.1128/MMBR.00034-08
9. McCulloch SD, Kunkel TA. The Fidelity of DNA Synthesis by Eukaryotic Replicative and Translesion Synthesis Polymerases. Cell Res (2008) 18(1):148–61. doi: 10.1038/cr.2008.4
10. Makridakis N, Reichardt J. Translesion DNA Polymerases and Cancer. Front Genet (2012) 3:174. doi: 10.3389/fgene.2012.00174
11. Basu A, Broyde S, Iwai S, Kisker C. DNA Damage, Mutagenesis, and DNA Repair. J Nucleic Acids (2010) 182894.
12. Knobel PA, Marti TM. Translesion DNA Synthesis in the Context of Cancer Research. Cancer Cell Int (2011) 11(1):1–19. doi: 10.1186/1475-2867-11-39
13. Branzei D, Psakhye I. DNA Damage Tolerance. Curr Opin Cell Biol (2016) 40:137–44. doi: 10.1016/j.ceb.2016.03.015
14. Sogo JM, Lopes M, Foiani M. Fork Reversal and ssDNA Accumulation at Stalled Replication Forks Owing to Checkpoint Defects. Science (2002) 297(5581):599–602. doi: 10.1126/science.1074023
15. Chaudhuri AR, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, et al. Topoisomerase I Poisoning Results in PARP-Mediated Replication Fork Reversal. Nat Struct Mol Biol (2012) 19(4):417–23. doi: 10.1038/nsmb.2258
16. Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D. Visualization of Recombination-Mediated Damage Bypass by Template Switching. Nat Struct Mol Biol (2014) 21(10):884–92. doi: 10.1038/nsmb.2888
17. Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity During DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol Cell (2015) 57(5):812–23. doi: 10.1016/j.molcel.2014.12.038
18. Bi X. Mechanism of DNA Damage Tolerance. World J Biol Chem (2015) 6(3):48–56. doi: 10.4331/wjbc.v6.i3.48
19. Binz SK, Sheehan AM, Wold MS. Replication Protein A Phosphorylation and the Cellular Response to DNA Damage. DNA repair (2004) 3(8-9):1015–24. doi: 10.1016/j.dnarep.2004.03.028
20. Ünsal-Kaçmaz K, Makhov AM, Griffith JD, Sancar A. Preferential Binding of ATR Protein to UV-Damaged DNA. Proc Natl Acad Sci (2002) 99(10):6673–8. doi: 10.1073/pnas.102167799
21. Papamichos-Chronakis M, Peterson CL. The Ino80 Chromatin-Remodeling Enzyme Regulates Replisome Function and Stability. Nat Struct Mol Biol (2008) 15(4):338–45. doi: 10.1038/nsmb.1413
22. Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, et al. Ino80 Chromatin Remodeling Complex Promotes Recovery of Stalled Replication Forks. Curr Biol (2008) 18(8):566–75. doi: 10.1016/j.cub.2008.03.049
23. Falbo KB, Alabert C, Katou Y, Wu S, Han J, Wehr T, et al. Involvement of a Chromatin Remodeling Complex in Damage Tolerance During DNA Replication. Nat Struct Mol Biol (2009) 16(11):1167–72. doi: 10.1038/nsmb.1686
24. Huttner D, Ulrich HD. Cooperation of Replication Protein A With the Ubiquitin Ligase Rad18 in DNA Damage Bypass. Cell Cycle (2008) 7(23):3629–33. doi: 10.4161/cc.7.23.7166
25. Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 Activity During DNA Damage Tolerance. Annu Rev Biophysics (2015) 44:207–28. doi: 10.1146/annurev-biophys-060414-033841
26. Hedglin M, Aitha M, Pedley A, Benkovic SJ. Replication Protein A Dynamically Regulates Monoubiquitination of Proliferating Cell Nuclear Antigen. J Biol Chem (2019) 294(13):5157–68. doi: 10.1074/jbc.RA118.005297
27. Notenboom V, Hibbert RG, van Rossum-Fikkert SE, Olsen JV, Mann M, Sixma TK. Functional Characterization of Rad18 Domains for Rad6, Ubiquitin, DNA Binding and PCNA Modification. Nucleic Acids Res (2007) 35(17):5819–30. doi: 10.1093/nar/gkm615
28. Ulrich HD, Jentsch S. Two RING Finger Proteins Mediate Cooperation Between Ubiquitin-Conjugating Enzymes in DNA Repair. EMBO J (2000) 19(13):3388–97. doi: 10.1093/emboj/19.13.3388
29. Bailly V, Lamb J, Sung P, Prakash S, Prakash L. Specific Complex Formation Between Yeast RAD6 and RAD18 Proteins: A Potential Mechanism for Targeting RAD6 Ubiquitin-Conjugating Activity to DNA Damage Sites. Genes Dev (1994) 8(7):811–20. doi: 10.1101/gad.8.7.811
30. Bailly V, Lauder S, Prakash S, Prakash L. Yeast DNA Repair Proteins Rad6 and Rad18 Form a Heterodimer That has Ubiquitin Conjugating, DNA Binding, and ATP Hydrolytic Activities. J Biol Chem (1997) 272(37):23360–5. doi: 10.1074/jbc.272.37.23360
31. Finley D, Ulrich HD, Sommer T, Kaiser P. The Ubiquitin–Proteasome System of Saccharomyces Cerevisiae. Genetics (2012) 192(2):319–60. doi: 10.1534/genetics.112.140467
32. Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S. RAD6-Dependent DNA Repair is Linked to Modification of PCNA by Ubiquitin and SUMO. Nature (2002) 419(6903):135–41. doi: 10.1038/nature00991
33. Yoon J-H, Prakash S, Prakash L. Requirement of Rad18 Protein for Replication Through DNA Lesions in Mouse and Human Cells. Proc Natl Acad Sci (2012) 109(20):7799–804. doi: 10.1073/pnas.1204105109
34. Zhang S, Chea J, Meng X, Zhou Y, Lee EY, Lee MY. PCNA is Ubiquitinated by RNF8. Cell Cycle (2008) 7(21):3399–404. doi: 10.4161/cc.7.21.6949
35. Quinet A, Lerner LK, Martins DJ, Menck CF. Filling Gaps in Translesion DNA Synthesis in Human Cells. Mutat Research/Genetic Toxicol Environ Mutagenesis (2018) 836:127–42. doi: 10.1016/j.mrgentox.2018.02.004
36. Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, et al. Translesion Synthesis: Y-Family Polymerases and the Polymerase Switch. DNA Repair (2007) 6(7):891–9. doi: 10.1016/j.dnarep.2007.02.003
37. Guo C, Kosarek-Stancel JN, Tang T-S, Friedberg EC. Y-Family DNA Polymerases in Mammalian Cells. Cell Mol Life Sci (2009) 66(14):2363–81. doi: 10.1007/s00018-009-0024-4
38. Friedberg EC, Wagner R, Radman M. Specialized DNA Polymerases, Cellular Survival, and the Genesis of Mutations. Science (2002) 296(5573):1627–30. doi: 10.1126/science.1070236
39. Ma X, Tang TS, Guo C. Regulation of Translesion DNA Synthesis in Mammalian Cells. Environ Mol Mutagen (2020) 61(7):680–92. doi: 10.1002/em.22359
40. Yang W, Gao Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem (2018) 87:239–61. doi: 10.1146/annurev-biochem-062917-012405
41. Guo C, Sonoda E, Tang T-S, Parker JL, Bielen AB, Takeda S, et al. REV1 Protein Interacts With PCNA: Significance of the REV1 BRCT Domain In Vitro and In Vivo. Mol Cell (2006) 23(2):265–71. doi: 10.1016/j.molcel.2006.05.038
42. Pustovalova Y, Maciejewski MW, Korzhnev DM. NMR Mapping of PCNA Interaction With Translesion Synthesis DNA Polymerase Rev1 Mediated by Rev1-BRCT Domain. J Mol Biol (2013) 425(17):3091–105. doi: 10.1016/j.jmb.2013.05.029
43. Livneh Z, Shachar S. Multiple Two-Polymerase Mechanisms in Mammalian Translesion DNA Synthesis. Cell Cycle (2010) 9(4):729–35. doi: 10.4161/cc.9.4.10727
44. Yoon J-H, Park J, Conde J, Wakamiya M, Prakash L, Prakash S. Rev1 Promotes Replication Through UV Lesions in Conjunction With DNA Polymerases η, ι, and κ But Not DNA Polymerase ζ. Genes Dev (2015) 29(24):2588–602. doi: 10.1101/gad.272229.115
45. Pustovalova Y, Magalhães MT, D’Souza S, Rizzo AA, Korza G, Walker GC, et al. Interaction Between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polζ Suggests a Mechanism of Polymerase Exchange Upon Rev1/Polζ-Dependent Translesion Synthesis. Biochemistry (2016) 55(13):2043–53. doi: 10.1021/acs.biochem.5b01282
46. Kikuchi S, Hara K, Shimizu T, Sato M, Hashimoto H. Structural Basis of Recruitment of DNA Polymerase ζ by Interaction Between REV1 and REV7 Proteins. J Biol Chem (2012) 287(40):33847–52. doi: 10.1074/jbc.M112.396838
47. Pozhidaeva A, Pustovalova Y, D’Souza S, Bezsonova I, Walker GC, Korzhnev DM. NMR Structure and Dynamics of the C-Terminal Domain From Human Rev1 and its Complex With Rev1 Interacting Region of DNA Polymerase η. Biochemistry (2012) 51(27):5506–20. doi: 10.1021/bi300566z
48. Washington MT, Minko IG, Johnson RE, Haracska L, Harris TM, Lloyd RS, et al. Efficient and Error-Free Replication Past a Minor-Groove N2-Guanine Adduct by the Sequential Action of Yeast Rev1 and DNA Polymerase ζ. Mol Cell Biol (2004) 24(16):6900–6. doi: 10.1128/MCB.24.16.6900-6906.2004
49. Weaver TM, Cortez LM, Khoang TH, Washington MT, Agarwal PK, Freudenthal BD. Visualizing Rev1 Catalyze Protein-Template DNA Synthesis. Proc Natl Acad Sci (2020) 117(41):25494–504. doi: 10.1073/pnas.2010484117
50. Biertümpfel C, Zhao Y, Kondo Y, Ramón-Maiques S, Gregory M, Lee JY, et al. Structure and Mechanism of Human DNA Polymerase η. Nature (2010) 465(7301):1044–8. doi: 10.1038/nature09196
51. Sale JE, Lehmann AR, Woodgate R. Y-Family DNA Polymerases and Their Role in Tolerance of Cellular DNA Damage. Nat Rev Mol Cell Biol (2012) 13(3):141–52. doi: 10.1038/nrm3289
52. Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, et al. Translesion Synthesis Mechanisms Depend on the Nature of DNA Damage in UV-Irradiated Human Cells. Nucleic Acids Res (2016) 44(12):5717–31. doi: 10.1093/nar/gkw280
53. Zhang Y, Wu X, Rechkoblit O, Geacintov NE, Taylor J-S, Wang Z. Response of Human REV1 to Different DNA Damage: Preferential dCMP Insertion Opposite the Lesion. Nucleic Acids Res (2002) 30(7):1630–8. doi: 10.1093/nar/30.7.1630
54. Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of Accurate Translesion Synthesis by Human DNA Polymerase η. EMBO J (2000) 19(12):3100–9. doi: 10.1093/emboj/19.12.3100
55. Haracska L, Yu S-L, Johnson RE, Prakash L, Prakash S. Efficient and Accurate Replication in the Presence of 7, 8-Dihydro-8-Oxoguanine by DNA Polymerase η. Nat Genet (2000) 25(4):458–61. doi: 10.1038/78169
56. Choi J-Y, Lim S, Kim E-J, Jo A, Guengerich FP. Translesion Synthesis Across Abasic Lesions by Human B-Family and Y-Family DNA Polymerases α, δ, η, ι, κ, and REV1. J Mol Biol (2010) 404(1):34–44. doi: 10.1016/j.jmb.2010.09.015
57. Choi J-Y, Guengerich FP. Kinetic Evidence for Inefficient and Error-Prone Bypass Across Bulky N2-Guanine DNA Adducts by Human DNA Polymerase ι. J Biol Chem (2006) 281(18):12315–24. doi: 10.1074/jbc.M600112200
58. Vaisman A, Woodgate R. Unique Misinsertion Specificity of Polι may Decrease the Mutagenic Potential of Deaminated Cytosines. EMBO J (2001) 20(22):6520–9. doi: 10.1093/emboj/20.22.6520
59. Vaisman A, Frank EG, Iwai S, Ohashi E, Ohmori H, Hanaoka F, et al. Sequence Context-Dependent Replication of DNA Templates Containing UV-Induced Lesions by Human DNA Polymerase ι. DNA Repair (2003) 2(9):991–1006. doi: 10.1016/S1568-7864(03)00094-6
60. Fischhaber PL, Gerlach VL, Feaver WJ, Hatahet Z, Wallace SS, Friedberg EC. Human DNA Polymerase κ Bypasses and Extends Beyond Thymine Glycols During Translesion Synthesis In Vitro, Preferentially Incorporating Correct Nucleotides. J Biol Chem (2002) 277(40):37604–11. doi: 10.1074/jbc.M206027200
61. Ogi T, Shinkai Y, Tanaka K, Ohmori H. Polκ Protects Mammalian Cells Against the Lethal and Mutagenic Effects of Benzo [a] Pyrene. Proc Natl Acad Sci (2002) 99(24):15548–53. doi: 10.1073/pnas.222377899
62. Zhang Y, Yuan F, Wu X, Wang M, Rechkoblit O, Taylor J-S, et al. Error-Free and Error-Prone Lesion Bypass by Human DNA Polymerase κ In Vitro. Nucleic Acids Res (2000) 28(21):4138–46. doi: 10.1093/nar/28.21.4138
63. Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, et al. Error-Prone Bypass of Certain DNA Lesions by the Human DNA Polymerase κ. Genes Dev (2000) 14(13):1589–94. doi: 10.1101/gad.14.13.1589
64. Seki M, Masutani C, Yang LW, Schuffert A, Iwai S, Bahar I, et al. High-Efficiency Bypass of DNA Damage by Human DNA Polymerase Q. EMBO J (2004) 23(22):4484–94. doi: 10.1038/sj.emboj.7600424
65. Yoon J-H, Choudhury JR, Park J, Prakash S, Prakash L. A Role for DNA Polymerase θ in Promoting Replication Through Oxidative DNA Lesion, Thymine Glycol, in Human Cells. J Biol Chem (2014) 289(19):13177–85. doi: 10.1074/jbc.M114.556977
66. Yoon J-H, McArthur MJ, Park J, Basu D, Wakamiya M, Prakash L, et al. Error-Prone Replication Through UV Lesions by DNA Polymerase θ Protects Against Skin Cancers. Cell (2019) 176(6):1295–309.e15. doi: 10.1016/j.cell.2019.01.023
67. Yoon J-H, Prakash L, Prakash S. Error-Free Replicative Bypass of (6–4) Photoproducts by DNA Polymerase ζ in Mouse and Human Cells. Genes Dev (2010) 24(2):123–8. doi: 10.1101/gad.1872810
68. Yoon J-H, Basu D, Sellamuthu K, Johnson RE, Prakash S, Prakash L. A Novel Role of DNA Polymerase λ in Translesion Synthesis in Conjunction With DNA Polymerase ζ. Life Sci alliance (2021) 4(4). doi: 10.26508/lsa.202000900
69. García-Gómez S, Reyes A, Martínez-Jiménez MI, Chocrón ES, Mourón S, Terrados G, et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol Cell (2013) 52(4):541–53. doi: 10.1016/j.molcel.2013.09.025
70. Temprine K, Campbell NR, Huang R, Langdon EM, Simon-Vermot T, Mehta K, et al. Regulation of the Error-Prone DNA Polymerase Polκ by Oncogenic Signaling and its Contribution to Drug Resistance. Sci Signaling (2020) 13(629). doi: 10.1126/scisignal.aau1453
71. Chatterjee N, Walker GC. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ Mol Mutagenesis (2017) 58(5):235–63. doi: 10.1002/em.22087
72. Yusufzai T, Kadonaga JT. HARP is an ATP-Driven Annealing Helicase. Science (2008) 322(5902):748–50. doi: 10.1126/science.1161233
73. Poole LA, Cortez D. Functions of SMARCAL1, ZRANB3, and HLTF in Maintaining Genome Stability. Crit Rev Biochem Mol Biol (2017) 52(6):696–714. doi: 10.1080/10409238.2017.1380597
74. Bétous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, et al. SMARCAL1 Catalyzes Fork Regression and Holliday Junction Migration to Maintain Genome Stability During DNA Replication. Genes Dev (2012) 26(2):151–62. doi: 10.1101/gad.178459.111
75. Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, et al. Replication Fork Slowing and Reversal Upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell (2017) 67(5):882–90.e5. doi: 10.1016/j.molcel.2017.08.010
76. Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity After Replication Stress. Mol Cell (2012) 47(3):396–409. doi: 10.1016/j.molcel.2012.05.024
77. Tian T, Bu M, Chen X, Ding L, Yang Y, Han J, et al. The ZATT-TOP2A-PICH Axis Drives Extensive Replication Fork Reversal to Promote Genome Stability. Mol Cell (2021) 81(1):198–211.e6. doi: 10.1016/j.molcel.2020.11.007
78. Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA Replication Structures by the Branch Point Translocase FANCM. Proc Natl Acad Sci (2008) 105(42):16107–12. doi: 10.1073/pnas.0804777105
79. Gari K, Décaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi Anemia Protein FANCM can Promote Branch Migration of Holliday Junctions and Replication Forks. Mol Cell (2008) 29(1):141–8. doi: 10.1016/j.molcel.2007.11.032
80. Lemaçon D, Jackson J, Quinet A, Brickner J, Li S. MRE11 and EXO1 Nucleases Degrade Reversed Forks and Elicit MUS81-Dependent Fork Rescue in BRCA2-Deficient Cells. Nat Commun (2017) 8:860. doi: 10.1038/s41467-017-01180-5
81. Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell (2017) 67(5):867–81.e7. doi: 10.1016/j.molcel.2017.07.001
82. Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, et al. Replication Fork Reversal Triggers Fork Degradation in BRCA2-Defective Cells. Nat Commun (2017) 8(1):1–11. doi: 10.1038/s41467-017-01164-5
83. Mason JM, Chan Y-L, Weichselbaum RW, Bishop DK. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat Commun (2019) 10(1):1–11. doi: 10.1038/s41467-019-12297-0
84. Quinet A, Lemaçon D, Vindigni A. Replication Fork Reversal: Players and Guardians. Mol Cell (2017) 68(5):830–3. doi: 10.1016/j.molcel.2017.11.022
85. Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, et al. Human RECQ1 Promotes Restart of Replication Forks Reversed by DNA Topoisomerase I Inhibition. Nat Struct Mol Biol (2013) 20(3):347–54. doi: 10.1038/nsmb.2501
86. Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S. Noncanonical Role of the 9-1-1 Clamp in the Error-Free DNA Damage Tolerance Pathway. Mol Cell (2013) 49(3):536–46. doi: 10.1016/j.molcel.2012.11.016
87. Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer W-D. Rad51 Paralogues Rad55–Rad57 Balance the Antirecombinase Srs2 in Rad51 Filament Formation. Nature (2011) 479(7372):245–8. doi: 10.1038/nature10522
88. Godin S, Wier A, Kabbinavar F, Bratton-Palmer DS, Ghodke H, Van Houten B, et al. The Shu Complex Interacts With Rad51 Through the Rad51 Paralogues Rad55–Rad57 to Mediate Error-Free Recombination. Nucleic Acids Res (2013) 41(8):4525–34. doi: 10.1093/nar/gkt138
89. Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. Replication and Recombination Factors Contributing to Recombination-Dependent Bypass of DNA Lesions by Template Switch. PloS Genet (2010) 6(11):e1001205. doi: 10.1371/journal.pgen.1001205
90. Bernstein KA, Reid RJ, Sunjevaric I, Demuth K, Burgess RC, Rothstein R. The Shu Complex, Which Contains Rad51 Paralogues, Promotes DNA Repair Through Inhibition of the Srs2 Anti-Recombinase. Mol Biol Cell (2011) 22(9):1599–607. doi: 10.1091/mbc.e10-08-0691
91. Sasanuma H, Tawaramoto M, Lao J, Hosaka H, Sanda E, Suzuki M, et al. A New Protein Complex Promoting the Assembly of Rad51 Filaments. Nat Commun (2013) 4:1676. doi: 10.1038/ncomms2678
92. Zhang H, Lawrence CW. The Error-Free Component of the RAD6/RAD18 DNA Damage Tolerance Pathway of Budding Yeast Employs Sister-Strand Recombination. Proc Natl Acad Sci (2005) 102(44):15954–9. doi: 10.1073/pnas.0504586102
93. Branzei D. Ubiquitin Family Modifications and Template Switching. FEBS Lett (2011) 585(18):2810–7. doi: 10.1016/j.febslet.2011.04.053
94. San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem (2008) 77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255
95. Symington LS. Role of RAD52 Epistasis Group Genes in Homologous Recombination and Double-Strand Break Repair. Microbiol Mol Biol Rev (2002) 66(4):630–70. doi: 10.1128/MMBR.66.4.630-670.2002
96. Burkovics P, Sebesta M, Sisakova A, Plault N, Szukacsov V, Robert T, et al. Srs2 Mediates PCNA-SUMO-Dependent Inhibition of DNA Repair Synthesis. EMBO J (2013) 32(5):742–55. doi: 10.1038/emboj.2013.9
97. Cejka P, Plank JL, Dombrowski CC, Kowalczykowski SC. Decatenation of DNA by the S. Cerevisiae Sgs1-Top3-Rmi1 and RPA Complex: A Mechanism for Disentangling Chromosomes. Mol Cell (2012) 47(6):886–96. doi: 10.1016/j.molcel.2012.06.032
98. Bernstein KA, Shor E, Sunjevaric I, Fumasoni M, Burgess RC, Foiani M, et al. Sgs1 Function in the Repair of DNA Replication Intermediates Is Separable From its Role in Homologous Recombinational Repair. EMBO J (2009) 28(7):915–25. doi: 10.1038/emboj.2009.28
99. Masuda Y, Masutani C. Spatiotemporal Regulation of PCNA Ubiquitination in Damage Tolerance Pathways. Crit Rev Biochem Mol Biol (2019) 54(5):418–42. doi: 10.1080/10409238.2019.1687420
100. Kashiwaba S-I, Kanao R, Masuda Y, Kusumoto-Matsuo R, Hanaoka F, Masutani C. USP7 is a Suppressor of PCNA Ubiquitination and Oxidative-Stress-Induced Mutagenesis in Human Cells. Cell Rep (2015) 13(10):2072–80. doi: 10.1016/j.celrep.2015.11.014
101. Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, et al. A UAF1-Containing Multisubunit Protein Complex Regulates the Fanconi Anemia Pathway. Mol Cell (2007) 28(5):786–97. doi: 10.1016/j.molcel.2007.09.031
102. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, et al. Inactivation of Murine Usp1 Results in Genomic Instability and a Fanconi Anemia Phenotype. Dev Cell (2009) 16(2):314–20. doi: 10.1016/j.devcel.2009.01.001
103. Lee K-Y, Yang K, Cohn MA, Sikdar N, D’Andrea AD, Myung K. Human ELG1 Regulates the Level of Ubiquitinated Proliferating Cell Nuclear Antigen (PCNA) Through Its Interactions With PCNA and USP1. J Biol Chem (2010) 285(14):10362–9. doi: 10.1074/jbc.M109.092544
104. Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, et al. Regulation of Monoubiquitinated PCNA by DUB Autocleavage. Nat Cell Biol (2006) 8(4):341–7. doi: 10.1038/ncb1378
105. Mohiuddin M, Evans TJ, Rahman MM, Keka IS, Tsuda M, Sasanuma H, et al. SUMOylation of PCNA by PIAS1 and PIAS4 Promotes Template Switch in the Chicken and Human B Cell Lines. Proc Natl Acad Sci (2018) 115(50):12793–8. doi: 10.1073/pnas.1716349115
106. Park JM, Yang SW, Yu KR, Ka SH, Lee SW, Seol JH, et al. Modification of PCNA by ISG15 Plays a Crucial Role in Termination of Error-Prone Translesion DNA Synthesis. Mol Cell (2014) 54(4):626–38. doi: 10.1016/j.molcel.2014.03.031
107. Leung W, Baxley RM, Moldovan G-L, Bielinsky A-K. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes (2019) 10(1):10. doi: 10.3390/genes10010010
108. Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 Guides Polη to Replication Stalling Sites Through Physical Interaction and PCNA Monoubiquitination. EMBO J (2004) 23(19):3886–96. doi: 10.1038/sj.emboj.7600383
109. Yang XH, Shiotani B, Classon M, Zou L. Chk1 and Claspin Potentiate PCNA Ubiquitination. Genes Dev (2008) 22(9):1147–52. doi: 10.1101/gad.1632808
110. Han J, Liu T, Huen MS, Hu L, Chen Z, Huang J. SIVA1 Directs the E3 Ubiquitin Ligase RAD18 for PCNA Monoubiquitination. J Cell Biol (2014) 205(6):811–27. doi: 10.1083/jcb.201311007
111. Lin J-R, Zeman MK, Chen J-Y, Yee M-C, Cimprich KA. SHPRH and HLTF Act in a Damage-Specific Manner to Coordinate Different Forms of Postreplication Repair and Prevent Mutagenesis. Mol Cell (2011) 42(2):237–49. doi: 10.1016/j.molcel.2011.02.026
112. Centore RC, Yazinski SA, Tse A, Zou L. Spartan/C1orf124, a Reader of PCNA Ubiquitylation and a Regulator of UV-Induced DNA Damage Response. Mol Cell (2012) 46(5):625–35. doi: 10.1016/j.molcel.2012.05.020
113. Juhasz S, Balogh D, Hajdu I, Burkovics P, Villamil MA, Zhuang Z, et al. Characterization of Human Spartan/C1orf124, an Ubiquitin-PCNA Interacting Regulator of DNA Damage Tolerance. Nucleic Acids Res (2012) 40(21):10795–808. doi: 10.1093/nar/gks850
114. Göhler T, Sabbioneda S, Green CM, Lehmann AR. ATR-Mediated Phosphorylation of DNA Polymerase η Is Needed for Efficient Recovery From UV Damage. J Cell Biol (2011) 192(2):219–27. doi: 10.1083/jcb.201008076
115. Peddu C, Zhang S, Zhao H, Wong A, Lee EY, Lee MY, et al. Phosphorylation Alters the Properties of Pol η: Implications for Translesion Synthesis. iScience (2018) 6:52–67. doi: 10.1016/j.isci.2018.07.009
116. Yanagihara H, Kobayashi J, Tateishi S, Kato A, Matsuura S, Tauchi H, et al. NBS1 Recruits RAD18 via a RAD6-Like Domain and Regulates Pol η-Dependent Translesion DNA Synthesis. Mol Cell (2011) 43(5):788–97. doi: 10.1016/j.molcel.2011.07.026
117. Saito Y, Komatsu K. Functional Role of NBS1 in Radiation Damage Response and Translesion DNA Synthesis. Biomolecules (2015) 5(3):1990–2002. doi: 10.3390/biom5031990
118. Jung Y-S, Hakem A, Hakem R, Chen X. Pirh2 E3 Ubiquitin Ligase Monoubiquitinates DNA Polymerase Eta to Suppress Translesion DNA Synthesis. Mol Cell Biol (2011) 31(19):3997–4006. doi: 10.1128/MCB.05808-11
119. Jung Y-S, Liu G, Chen X. Pirh2 E3 Ubiquitin Ligase Targets DNA Polymerase Eta for 20S Proteasomal Degradation. Mol Cell Biol (2010) 30(4):1041–8. doi: 10.1128/MCB.01198-09
120. Qian J, Pentz K, Zhu Q, Wang Q, He J, Srivastava AK, et al. USP7 Modulates UV-Induced PCNA Monoubiquitination by Regulating DNA Polymerase Eta Stability. Oncogene (2015) 34(36):4791–6. doi: 10.1038/onc.2014.394
121. Jung Y-S, Qian Y, Chen X. DNA Polymerase Eta is Targeted by Mdm2 for Polyubiquitination and Proteasomal Degradation in Response to Ultraviolet Irradiation. DNA Repair (2012) 11(2):177–84. doi: 10.1016/j.dnarep.2011.10.017
122. Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, Poulsen JW, et al. Systems-Wide Analysis of Ubiquitylation Dynamics Reveals a Key Role for PAF15 Ubiquitylation in DNA-Damage Bypass. Nat Cell Biol (2012) 14(10):1089–98. doi: 10.1038/ncb2579
123. Nicolae CM, Aho ER, Vlahos AH, Choe KN, De S, Karras GI, et al. The ADP-Ribosyltransferase PARP10/ARTD10 Interacts With Proliferating Cell Nuclear Antigen (PCNA) and Is Required for DNA Damage Tolerance. J Biol Chem (2014) 289(19):13627–37. doi: 10.1074/jbc.M114.556340
124. Huang M, Zhou B, Gong J, Xing L, Ma X, Wang F, et al. RNA-Splicing Factor SART3 Regulates Translesion DNA Synthesis. Nucleic Acids Res (2018) 46(9):4560–74. doi: 10.1093/nar/gky220
125. Bertoletti F, Cea V, Liang CC, Lanati T, Maffia A, Avarello MDM, et al. Phosphorylation Regulates Human Polη Stability and Damage Bypass Throughout the Cell Cycle. Nucleic Acids Res (2017) 45(16):9441–54. doi: 10.1093/nar/gkx619
126. Despras E, Sittewelle M, Pouvelle C, Delrieu N, Cordonnier AM, Kannouche PL. Rad18-Dependent SUMOylation of Human Specialized DNA Polymerase Eta is Required to Prevent Under-Replicated DNA. Nat Commun (2016) 7(1):1–15. doi: 10.1038/ncomms13326
127. Guérillon C, Smedegaard S, Hendriks IA, Nielsen ML, Mailand N. Multisite SUMOylation Restrains DNA Polymerase η Interactions With DNA Damage Sites. J Biol Chem (2020) 295(25):8350–62. doi: 10.1074/jbc.RA120.013780
128. Cipolla L, Maffia A, Bertoletti F, Sabbioneda S. The Regulation of DNA Damage Tolerance by Ubiquitin and Ubiquitin-Like Modifiers. Front Genet (2016) 7:105. doi: 10.3389/fgene.2016.00105
129. Chen Y-W, Cleaver JE, Hatahet Z, Honkanen RE, Chang J-Y, Yen Y, et al. Human DNA Polymerase η Activity and Translocation Is Regulated by Phosphorylation. Proc Natl Acad Sci (2008) 105(43):16578–83. doi: 10.1073/pnas.0808589105
130. Lerner LK, Francisco G, Soltys DT, Rocha CR, Quinet A, Vessoni AT, et al. Predominant Role of DNA Polymerase Eta and P53-Dependent Translesion Synthesis in the Survival of Ultraviolet-Irradiated Human Cells. Nucleic Acids Res (2017) 45(3):1270–80. doi: 10.1093/nar/gkw1196
131. Bostian AC, Maddukuri L, Reed MR, Savenka T, Hartman JH, Davis L, et al. Kynurenine Signaling Increases DNA Polymerase Kappa Expression and Promotes Genomic Instability in Glioblastoma Cells. Chem Res Toxicol (2016) 29(1):101–8. doi: 10.1021/acs.chemrestox.5b00452
132. Brauze D, Rawłuszko AA. The Effect of Aryl Hydrocarbon Receptor Ligands on the Expression of Polymerase (DNA Directed) Kappa (Polκ), Polymerase RNA II (DNA Directed) Polypeptide A (PolR2a), CYP1B1 and CYP1A1 Genes in Rat Liver. Environ Toxicol Pharmacol (2012) 34(3):819–25. doi: 10.1016/j.etap.2012.09.004
133. Yamada T, Imamachi N, Imamura K, Taniue K, Kawamura T, Suzuki Y, et al. Systematic Analysis of Targets of Pumilio-Mediated mRNA Decay Reveals That PUM1 Repression by DNA Damage Activates Translesion Synthesis. Cell Rep (2020) 31(5):107542. doi: 10.1016/j.celrep.2020.107542
134. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced Expression of DNA Polymerase Eta Contributes to Cisplatin Resistance of Ovarian Cancer Stem Cells. Proc Natl Acad Sci (2015) 112(14):4411–6. doi: 10.1073/pnas.1421365112
135. Zhang J, Sun W, Ren C, Kong X, Yan W, Chen X. A PolH Transcript With a Short 3′ UTR Enhances PolH Expression and Mediates Cisplatin Resistance. Cancer Res (2019) 79(14):3714–24. doi: 10.1158/0008-5472.CAN-18-3928
136. Neelsen KJ, Lopes M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat Rev Mol Cell Biol (2015) 16(4):207–20. doi: 10.1038/nrm3935
137. Porebski B, Wild S, Kummer S, Scaglione S, Gaillard P-HL, Gari K. WRNIP1 Protects Reversed DNA Replication Forks From SLX4-Dependent Nucleolytic Cleavage. iScience (2019) 21:31–41. doi: 10.1016/j.isci.2019.10.010
138. Leuzzi G, Marabitti V, Pichierri P, Franchitto A. WRNIP 1 Protects Stalled Forks From Degradation and Promotes Fork Restart After Replication Stress. EMBO J (2016) 35(13):1437–51. doi: 10.15252/embj.201593265
139. Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, et al. ATR Phosphorylates SMARCAL1 to Prevent Replication Fork Collapse. Genes Dev (2013) 27(14):1610–23. doi: 10.1101/gad.214080.113
140. Unk I, Hajdú I, Blastyák A, Haracska L. Role of Yeast Rad5 and its Human Orthologs, HLTF and SHPRH in DNA Damage Tolerance. DNA Repair (2010) 9(3):257–67. doi: 10.1016/j.dnarep.2009.12.013
141. MacKay C, Toth R, Rouse J. Biochemical Characterisation of the SWI/SNF Family Member HLTF. Biochem Biophys Res Commun (2009) 390(2):187–91. doi: 10.1016/j.bbrc.2009.08.151
142. Motegi A, Sood R, Moinova H, Markowitz SD, Liu PP, Myung K. Human SHPRH Suppresses Genomic Instability Through Proliferating Cell Nuclear Antigen Polyubiquitination. J Cell Biol (2006) 175(5):703–8. doi: 10.1083/jcb.200606145
143. Unk I, Hajdú I, Fátyol K, Szakál B, Blastyák A, Bermudez V, et al. Human SHPRH is a Ubiquitin Ligase for Mms2–Ubc13-Dependent Polyubiquitylation of Proliferating Cell Nuclear Antigen. Proc Natl Acad Sci (2006) 103(48):18107–12. doi: 10.1073/pnas.0608595103
144. Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 Helicase Prevents Recombination by Disrupting Rad51 Nucleoprotein Filaments. Nature (2003) 423(6937):309–12. doi: 10.1038/nature01585
145. Antony E, Tomko EJ, Xiao Q, Krejci L, Lohman TM, Ellenberger T. Srs2 Disassembles Rad51 Filaments by a Protein-Protein Interaction Triggering ATP Turnover and Dissociation of Rad51 From DNA. Mol Cell (2009) 35(1):105–15. doi: 10.1016/j.molcel.2009.05.026
146. Gonzalez-Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, et al. DNA Bending Facilitates the Error-Free DNA Damage Tolerance Pathway and Upholds Genome Integrity. EMBO J (2014) 33(4):327–40. doi: 10.1002/embj.201387425
147. Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, et al. The XPV (Xeroderma Pigmentosum Variant) Gene Encodes Human DNA Polymerase η. Nature (1999) 399(6737):700–4. doi: 10.1038/21447
148. Johnson RE, Kondratick CM, Prakash S, Prakash L. Hrad30 Mutations in the Variant Form of Xeroderma Pigmentosum. Science (1999) 285(5425):263–5. doi: 10.1126/science.285.5425.263
149. Ziv O, Geacintov N, Nakajima S, Yasui A, Livneh Z. DNA Polymerase ζ Cooperates With Polymerases κ and ι in Translesion DNA Synthesis Across Pyrimidine Photodimers in Cells From XPV Patients. Proc Natl Acad Sci (2009) 106(28):11552–7. doi: 10.1073/pnas.0812548106
150. Yang Y, Gao Y, Zlatanou A, Tateishi S, Yurchenko V, Rogozin IB, et al. Diverse Roles of RAD18 and Y-Family DNA Polymerases in Tumorigenesis. Cell Cycle (2018) 17(7):833–43. doi: 10.1080/15384101.2018.1456296
151. Rogozin IB, Pavlov YI, Goncearenco A, De S, Lada AG, Poliakov E, et al. Mutational Signatures and Mutable Motifs in Cancer Genomes. Briefings Bioinf (2018) 19(6):1085–101. doi: 10.1093/bib/bbx049
152. Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, Sobue T, et al. Association of Amino Acid Substitution Polymorphisms in DNA Repair Genes TP53, POLI, REV1 and LIG4 With Lung Cancer Risk. Int J Cancer (2005) 114(5):730–7. doi: 10.1002/ijc.20790
153. Di Lucca J, Guedj M, Lacapère J-J, Fargnoli MC, Bourillon A, Dieudé P, et al. Variants of the Xeroderma Pigmentosum Variant Gene (POLH) are Associated With Melanoma Risk. Eur J Cancer (2009) 45(18):3228–36. doi: 10.1016/j.ejca.2009.04.034
154. Yadav S, Anbalagan M, Baddoo M, Chellamuthu VK, Mukhopadhyay S, Woods C, et al. Somatic Mutations in the DNA Repairome in Prostate Cancers in African Americans and Caucasians. Oncogene (2020) 39(21):4299–311. doi: 10.1038/s41388-020-1280-x
155. Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, et al. Adaptive Mutability of Colorectal Cancers in Response to Targeted Therapies. Science (2019) 366(6472):1473–80. doi: 10.1126/science.aav4474
156. Ceppi P, Novello S, Cambieri A, Longo M, Monica V, Iacono ML, et al. Polymerase η mRNA Expression Predicts Survival of Non–Small Cell Lung Cancer Patients Treated With Platinum-Based Chemotherapy. Clin Cancer Res (2009) 15(3):1039–45. doi: 10.1158/1078-0432.CCR-08-1227
157. Berdis AJ. Inhibiting DNA Polymerases as a Therapeutic Intervention Against Cancer. Front Mol Biosci (2017) 4:78. doi: 10.3389/fmolb.2017.00078
158. Sun H, Zou S, Zhang S, Liu B, Meng X, Li X, et al. Elevated DNA Polymerase Iota (Poli) is Involved in the Acquisition of Aggressive Phenotypes of Human Esophageal Squamous Cell Cancer. Int J Clin Exp Pathol (2015) 8(4):3591.
159. Yuan F, Xu Z, Yang M, Wei Q, Zhang Y, Yu J, et al. Overexpressed DNA Polymerase Iota Regulated by JNK/c-Jun Contributes to Hypermutagenesis in Bladder Cancer. PloS One (2013) 8(7):e69317. doi: 10.1371/journal.pone.0069317
160. Peng C, Chen Z, Wang S, Wang H-W, Qiu W, Zhao L, et al. The Error-Prone DNA Polymerase κ Promotes Temozolomide Resistance in Glioblastoma Through Rad17-Dependent Activation of ATR-Chk1 Signaling. Cancer Res (2016) 76(8):2340–53. doi: 10.1158/0008-5472.CAN-15-1884
161. Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Guéranger Q, et al. Differential Roles for DNA Polymerases Eta, Zeta, and REV1 in Lesion Bypass of Intrastrand Versus Interstrand DNA Cross-Links. Mol Cell Biol (2010) 30(5):1217–30. doi: 10.1128/MCB.00993-09
162. Albertella MR, Green CM, Lehmann AR, O’Connor MJ. A Role for Polymerase η in the Cellular Tolerance to Cisplatin-Induced Damage. Cancer Res (2005) 65(21):9799–806. doi: 10.1158/0008-5472.CAN-05-1095
163. Sokol AM, Cruet-Hennequart S, Pasero P, Carty MP. DNA Polymerase η Modulates Replication Fork Progression and DNA Damage Responses in Platinum-Treated Human Cells. Sci Rep (2013) 3(1):1–9. doi: 10.1038/srep03277
164. Roy U, Mukherjee S, Sharma A, Frank EG, Schärer OD. The Structure and Duplex Context of DNA Interstrand Crosslinks Affects the Activity of DNA Polymerase η. Nucleic Acids Res (2016) 44(15):7281–91. doi: 10.1093/nar/gkw485
165. Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, et al. Nucleotide Excision Repair-and Polymerase η-Mediated Error-Prone Removal of Mitomycin C Interstrand Cross-Links. Mol Cell Biol (2003) 23(2):754–61. doi: 10.1128/MCB.23.2.754-761.2003
166. Chen Y-W, Cleaver JE, Hanaoka F, Chang C-F, Chou K-M. A Novel Role of DNA Polymerase η in Modulating Cellular Sensitivity to Chemotherapeutic Agents. Mol Cancer Res (2006) 4(4):257–65. doi: 10.1158/1541-7786.MCR-05-0118
167. Cruet-Hennequart S, Villalan S, Kaczmarczyk A, O’Meara E, Sokol AM, Carty MP. Characterization of the Effects of Cisplatin and Carboplatin on Cell Cycle Progression and DNA Damage Response Activation in DNA Polymerase Eta-Deficient Human Cells. Cell Cycle (2009) 8(18):3043–54. doi: 10.4161/cc.8.18.9624
168. Cruet-Hennequart S, Glynn MT, Murillo LS, Coyne S, Carty MP. Enhanced DNA-PK-Mediated RPA2 Hyperphosphorylation in DNA Polymerase Eta-Deficient Human Cells Treated With Cisplatin and Oxaliplatin. DNA Repair (2008) 7(4):582–96. doi: 10.1016/j.dnarep.2007.12.012
169. Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced Expression of DNA Polymerase eta Contributes to Cisplatin Resistance of Ovarian Cancer Stem Cells. PNAS (2015) 112(14):4411–6. doi: 10.1073/pnas.1421365112
170. Wang H, Zhang S-Y, Wang S, Lu J, Wu W, Weng L, et al. REV3L Confers Chemoresistance to Cisplatin in Human Gliomas: The Potential of Its RNAi for Synergistic Therapy. Neuro-Oncology (2009) 11(6):790–802. doi: 10.1215/15228517-2009-015
171. Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell (2020) 77(3):461–74.e9. doi: 10.1016/j.molcel.2019.10.008
172. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature (2011) 478(7368):197–203. doi: 10.1038/nature10491
173. Cheong JE, Sun L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy–Challenges and Opportunities. Trends Pharmacol Sci (2018) 39(3):307–25. doi: 10.1016/j.tips.2017.11.007
174. Buoninfante OA, Pilzecker B, Aslam MA, Zavrakidis I, van der Wiel R, van de Ven M, et al. Precision Cancer Therapy: Profiting From Tumor Specific Defects in the DNA Damage Tolerance System. Oncotarget (2018) 9(27):18832. doi: 10.18632/oncotarget.24777
175. Gao Y, Mutter-Rottmayer E, Greenwalt AM, Goldfarb D, Yan F, Yang Y, et al. A Neomorphic Cancer Cell-Specific Role of MAGE-A4 in Trans-Lesion Synthesis. Nat Commun (2016) 7(1):1–14. doi: 10.1038/ncomms12105
176. Moinova HR, Chen W-D, Shen L, Smiraglia D, Olechnowicz J, Ravi L, et al. HLTF Gene Silencing in Human Colon Cancer. Proc Natl Acad Sci (2002) 99(7):4562–7. doi: 10.1073/pnas.062459899
177. Yamanaka K, Chatterjee N, Hemann MT, Walker GC. Inhibition of Mutagenic Translesion Synthesis: A Possible Strategy for Improving Chemotherapy? PloS Genet (2017) 13(8):e1006842. doi: 10.1371/journal.pgen.1006842
178. Yamanaka K, Dorjsuren D, Eoff RL, Egli M, Maloney DJ, Jadhav A, et al. A Comprehensive Strategy to Discover Inhibitors of the Translesion Synthesis DNA Polymerase κ. PLoS One (2012) 7(10):1–8. doi: 10.1371/journal.pone.0045032
179. Ishimaru C, Kuriyama I, Shimazaki N, Koiwai O, Sakaguchi K, Kato I, et al. Cholesterol Hemisuccinate: A Selective Inhibitor of Family X DNA Polymerases. Biochem Biophys Res Commun (2007) 354(2):619–25. doi: 10.1016/j.bbrc.2007.01.034
180. Mizushina Y, Zhang J, Pugliese A, Kim S-H, Lü J. Anti-Cancer Gallotannin Penta-O-Galloyl-Beta-D-Glucose is a Nanomolar Inhibitor of Select Mammalian DNA Polymerases. Biochem Pharmacol (2010) 80(8):1125–32. doi: 10.1016/j.bcp.2010.06.031
181. Myobatake Y, Takeuchi T, Kuramochi K, Kuriyama I, Ishido T, Hirano K, et al. Inhibitors of Mammalian A-, B-, and Y-Family DNA Polymerases and Human Cancer Cell Proliferation. J Natural products (2012) 75(2):135–41. doi: 10.1021/np200523b
182. Horie S, Okuda C, Yamashita T, Watanabe K, Sato Y, Yamaguchi Y, et al. β-Sitosteryl (6’-O-Linoleoyl)-Glucoside of Soybean (Glycine Max L.) Crude Extract Inhibits Y-Family DNA Polymerases. J oleo Sci (2010) 59(11):621–30. doi: 10.5650/jos.59.621
183. Mizushina Y, Motoshima H, Yamaguchi Y, Takeuchi T, Hirano K, Sugawara F, et al. 3-O-Methylfunicone, a Selective Inhibitor of Mammalian Y-Family DNA Polymerases From an Australian Sea Salt Fungal Strain. Marine Drugs (2009) 7(4):624–39. doi: 10.3390/md7040624
184. Kimura T, Takeuchi T, Kumamoto-Yonezawa Y, Ohashi E, Ohmori H, Masutani C, et al. Penicilliols A and B, Novel Inhibitors Specific to Mammalian Y-Family DNA Polymerases. Bioorganic medicinal Chem (2009) 17(5):1811–6. doi: 10.1016/j.bmc.2009.01.064
185. Zafar MK, Maddukuri L, Ketkar A, Penthala NR, Reed MR, Eddy S, et al. A Small-Molecule Inhibitor of Human DNA Polymerase η Potentiates the Effects of Cisplatin in Tumor Cells. Biochemistry (2018) 57(7):1262–73. doi: 10.1021/acs.biochem.7b01176
186. Coggins GE, Maddukuri L, Penthala NR, Hartman JH, Eddy S, Ketkar A, et al. N-Aroyl Indole Thiobarbituric Acids as Inhibitors of DNA Repair and Replication Stress Response Polymerases. ACS Chem Biol (2013) 8(8):1722–9. doi: 10.1021/cb400305r
187. Saha P, Mandal T, Talukdar AD, Kumar D, Kumar S, Tripathi PP, et al. DNA Polymerase Eta: A Potential Pharmacological Target for Cancer Therapy. J Cell Physiol (2021) 236(6):4106–20. doi: 10.1002/jcp.30155
188. Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A First in Class Polymerase Theta Inhibitor Selectively Targets Homologous Recombination-Deficient Tumors. Nat Cancer (2021) 2(6):598–620. doi: 10.1038/s43018-021-00203-x
189. Zatreanu D, Robinson HM, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-23463-8
190. Ketkar A, Maddukuri L, Penthala NR, Reed MR, Zafar MK, Crooks PA, et al. Inhibition of Human DNA Polymerases Eta and Kappa by Indole-Derived Molecules Occurs Through Distinct Mechanisms. ACS Chem Biol (2019) 14(6):1337–51. doi: 10.1021/acschembio.9b00304
191. Ardiana F, Lestari ML, Indrayanto G. Candesartan Cilexetil. Profiles Drug Substances Excipients Related Method (2012) 37:79–112. doi: 10.1016/B978-0-12-397220-0.00003-9
192. Soriente A, De Rosa M, Scettri A, Sodano G, Terencio M, Paya M, et al. Manoalide. Curr medicinal Chem (1999) 6(5):415–31. doi: 10.1021/jm980027h
193. Jawien J, Gajda M, Rudling M, Mateuszuk L, Olszanecki R, Guzik T, et al. Inhibition of Five Lipoxygenase Activating Protein (FLAP) by MK-886 Decreases Atherosclerosis in Apoe/LDLR-Double Knockout Mice. Eur J Clin Invest (2006) 36(3):141–6. doi: 10.1111/j.1365-2362.2006.01606.x
194. Patel SM, Dash RC, Hadden MK. Translesion Synthesis Inhibitors as a New Class of Cancer Chemotherapeutics. Expert Opin Investigational Drugs (2021) 30(1):13–24. doi: 10.1080/13543784.2021.1850692
195. Punchihewa C, Inoue A, Hishiki A, Fujikawa Y, Connelly M, Evison B, et al. Identification of Small Molecule Proliferating Cell Nuclear Antigen (PCNA) Inhibitor That Disrupts Interactions With PIP-Box Proteins and Inhibits DNA Replication. J Biol Chem (2012) 287(17):14289–300. doi: 10.1074/jbc.M112.353201
196. Actis M, Inoue A, Evison B, Perry S, Punchihewa C, Fujii N. Small Molecule Inhibitors of PCNA/PIP-Box Interaction Suppress Translesion DNA Synthesis. Bioorganic medicinal Chem (2013) 21(7):1972–7. doi: 10.1016/j.bmc.2013.01.022
197. Gu L, Lingeman R, Yakushijin F, Sun E, Cui Q, Chao J, et al. The Anticancer Activity of a First-in-Class Small-Molecule Targeting PCNA. Clin Cancer Res (2018) 24(23):6053–65. doi: 10.1158/1078-0432.CCR-18-0592
198. Vanarotti M, Evison BJ, Actis ML, Inoue A, McDonald ET, Shao Y, et al. Small-Molecules That Bind to the Ubiquitin-Binding Motif of REV1 Inhibit REV1 Interaction With K164-Monoubiquitinated PCNA and Suppress DNA Damage Tolerance. Bioorganic medicinal Chem (2018) 26(9):2345–53. doi: 10.1016/j.bmc.2018.03.028
199. Sail V, Rizzo AA, Chatterjee N, Dash RC, Ozen Z, Walker GC, et al. Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein–Protein Interaction. ACS Chem Biol (2017) 12(7):1903–12. doi: 10.1021/acschembio.6b01144
200. Dash RC, Ozen Z, McCarthy KR, Chatterjee N, Harris CA, Rizzo AA, et al. Virtual Pharmacophore Screening Identifies Small-Molecule Inhibitors of the Rev1-CT/RIR Protein–Protein Interaction. ChemMedChem (2019) 14(17):1610–7. doi: 10.1002/cmdc.201900307
201. Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, et al. Inhibition of the Translesion Synthesis Polymerase REV1 Exploits Replication Gaps as a Cancer Vulnerability. Sci Adv (2020) 6(24):eaaz7808. doi: 10.1126/sciadv.aaz7808
202. Quinet A, Vessoni AT, Rocha CR, Gottifredi V, Biard D, Sarasin A, et al. Gap-Filling and Bypass at the Replication Fork are Both Active Mechanisms for Tolerance of Low-Dose Ultraviolet-Induced DNA Damage in the Human Genome. DNA Repair (2014) 14:27–38. doi: 10.1016/j.dnarep.2013.12.005
203. Barnes RP, Tsao W-C, Moldovan G-L, Eckert KA. DNA Polymerase Eta Prevents Tumor Cell-Cycle Arrest and Cell Death During Recovery From Replication Stress. Cancer Res (2018) 78(23):6549–60. doi: 10.1158/0008-5472.CAN-17-3931
204. Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell (2019) 178(1):152–9.e11. doi: 10.1016/j.cell.2019.05.028
205. Chatterjee N, Whitman MA, Harris CA, Min SM, Jonas O, Lien EC, et al. REV1 Inhibitor JH-RE-06 Enhances Tumor Cell Response to Chemotherapy by Triggering Senescence Hallmarks. Proc Natl Acad Sci (2020) 117(46):28918–21. doi: 10.1073/pnas.2016064117
206. Wittschieben JP, Reshmi SC, Gollin SM, Wood RD. Loss of DNA Polymerase ζ Causes Chromosomal Instability in Mammalian Cells. Cancer Res (2006) 66(1):134–42. doi: 10.1158/0008-5472.CAN-05-2982
207. Wittschieben JP, Patil V, Glushets V, Robinson LJ, Kusewitt DF, Wood RD. Loss of DNA Polymerase ζ Enhances Spontaneous Tumorigenesis. Cancer Res (2010) 70(7):2770–8. doi: 10.1158/0008-5472.CAN-09-4267
208. Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, et al. TRIP13 Regulates DNA Repair Pathway Choice Through REV7 Conformational Change. Nat Cell Biol (2020) 22(1):87–96. doi: 10.1038/s41556-019-0442-y
209. Alfieri C, Chang L, Barford D. Mechanism for Remodelling of the Cell Cycle Checkpoint Protein MAD2 by the ATPase Trip13. Nature (2018) 559(7713):274–8. doi: 10.1038/s41586-018-0281-1
210. Sarangi P, Clairmont CS, Galli LD, Moreau LA, D’Andrea AD. P31comet Promotes Homologous Recombination by Inactivating REV7 Through the TRIP13 ATPase. Proc Natl Acad Sci (2020) 117(43):26795–803. doi: 10.1073/pnas.2008830117
211. Sanders MA, Haynes B, Nangia-Makker P, Polin LA, Shekhar MP. Pharmacological Targeting of RAD6 Enzyme-Mediated Translesion Synthesis Overcomes Resistance to Platinum-Based Drugs. J Biol Chem (2017) 292(25):10347–63. doi: 10.1074/jbc.M117.792192
212. Haynes B, Gajan A, Nangia-Makker P, Shekhar MP. RAD6B is a Major Mediator of Triple Negative Breast Cancer Cisplatin Resistance: Regulation of Translesion Synthesis/Fanconi Anemia Crosstalk and BRCA1 Independence. Biochim Biophys Acta (BBA)-Molecular Basis Dis (2020) 1866(1):165561. doi: 10.1016/j.bbadis.2019.165561
213. Villafañez F, García IA, Carbajosa S, Pansa MF, Mansilla S, Llorens MC, et al. AKT Inhibition Impairs PCNA Ubiquitylation and Triggers Synthetic Lethality in Homologous Recombination-Deficient Cells Submitted to Replication Stress. Oncogene (2019) 38(22):4310–24. doi: 10.1038/s41388-019-0724-7
214. Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, et al. Selective and Cell-Active Inhibitors of the USP1/UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-Small Cell Lung Cancer Cells. Chem Biol (2011) 18(11):1390–400. doi: 10.1016/j.chembiol.2011.08.014
215. Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, et al. A Selective USP1–UAF1 Inhibitor Links Deubiquitination to DNA Damage Responses. Nat Chem Biol (2014) 10(4):298–304. doi: 10.1038/nchembio.1455
216. Kim H, D’Andrea AD. Regulation of DNA Cross-Link Repair by the Fanconi Anemia/BRCA Pathway. Genes Dev (2012) 26(13):1393–408. doi: 10.1101/gad.195248.112
217. Wang W. Emergence of a DNA-Damage Response Network Consisting of Fanconi Anaemia and BRCA Proteins. Nat Rev Genet (2007) 8(10):735–48. doi: 10.1038/nrg2159
218. Mayca Pozo F, Oda T, Sekimoto T, Murakumo Y, Masutani C, Hanaoka F, et al. Molecular Chaperone Hsp90 Regulates REV1-Mediated Mutagenesis. Mol Cell Biol (2011) 31(16):3396–409. doi: 10.1128/MCB.05117-11
219. Sekimoto T, Oda T, Pozo FM, Murakumo Y, Masutani C, Hanaoka F, et al. The Molecular Chaperone Hsp90 Regulates Accumulation of DNA Polymerase η at Replication Stalling Sites in UV-Irradiated Cells. Mol Cell (2010) 37(1):79–89. doi: 10.1016/j.molcel.2009.12.015
220. Takezawa J, Ishimi Y, Yamada K. Proteasome Inhibitors Remarkably Prevent Translesion Replication in Cancer Cells But Not Normal Cells. Cancer Sci (2008) 99(5):863–71. doi: 10.1111/j.1349-7006.2008.00764.x
221. Zhang X, Lee I, Berdis AJ. A Potential Chemotherapeutic Strategy for the Selective Inhibition of Promutagenic DNA Synthesis by Nonnatural Nucleotides. Biochemistry (2005) 44(39):13111–21. doi: 10.1021/bi050584n
222. Choi J-S, Kim CS, Berdis A. Inhibition of Translesion DNA Synthesis as a Novel Therapeutic Strategy to Treat Brain Cancer. Cancer Res (2018) 78(4):1083–96. doi: 10.1158/0008-5472.CAN-17-2464
Keywords: DNA damage, DNA damage tolerance pathways, DNA replication, translesion synthesis (TLS), TLS inhibitors, cancer therapeutics
Citation: Ler AAL and Carty MP (2022) DNA Damage Tolerance Pathways in Human Cells: A Potential Therapeutic Target. Front. Oncol. 11:822500. doi: 10.3389/fonc.2021.822500
Received: 25 November 2021; Accepted: 30 December 2021;
Published: 07 February 2022.
Edited by:
John Turchi, Indiana University Bloomington, United StatesReviewed by:
Christine Canman, University of Michigan, United StatesCopyright © 2022 Ler and Carty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael P. Carty, bWljaGFlbC5jYXJ0eUBudWlnYWx3YXkuaWU=
†Present address: Ashlynn Ai Li Ler, National University of Singapore
Yong Loo Lin School of Medicine, Singapore, Singapore
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.