
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 11 January 2022
Sec. Gynecological Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.812656
This article is part of the Research Topic Identification of Genetic Changes and Targeted Therapies in Breast and Ovarian Cancers View all 9 articles
 Jun Li1,2,3*†
Jun Li1,2,3*† Ping Wang4†
Ping Wang4† Cuiyun Zhang1,2,3
Cuiyun Zhang1,2,3 Sile Han1
Sile Han1 Han Xiao5Zhiyuan Liu6Xiaoyan Wang1,2,3Weiling Liu7Bing Wei1,2,3Jie Ma1,2,3Hongle Li1*
Han Xiao5Zhiyuan Liu6Xiaoyan Wang1,2,3Weiling Liu7Bing Wei1,2,3Jie Ma1,2,3Hongle Li1* Yongjun Guo1,2,3*
Yongjun Guo1,2,3*Breast cancer gene 1 (BRCA1) and BRCA2 are tumor suppressors involved in DNA damage response and repair. Carriers of germline pathogenic or likely pathogenic variants in BRCA1 or BRCA2 have significantly increased lifetime risks of breast cancer, ovarian cancer, and other cancer types; this phenomenon is known as hereditary breast and ovarian cancer (HBOC) syndrome. Accurate interpretation of BRCA1 and BRCA2 variants is important not only for disease management in patients, but also for determining preventative measures for their families. BRCA1:c.132C>T (p.Cys44=) is a synonymous variant recorded in the ClinVar database with “conflicting interpretations of its pathogenicity”. Here, we report our clinical tests in which we identified this variant in two unrelated patients, both of whom developed breast cancer at an early age with ovarian presentation a few years later and had a family history of relevant cancers. Minigene assay showed that this change caused a four-nucleotide loss at the end of exon 3, resulting in a truncated p.Cys44Tyrfs*5 protein. Reverse transcription-polymerase chain reaction identified two fragments (123 and 119 bp) using RNA isolated from patient blood samples, in consistency with the results of the minigene assay. Collectively, we classified BRCA1:c.132C>T (p.Cys44=) as a pathogenic variant, as evidenced by functional studies, RNA analysis, and the patients’ family histories. By analyzing variants recorded in the BRCA Exchange database, we found synonymous changes at the ends of exons could potentially influence splicing; meanwhile, current in silico tools could not predict splicing changes efficiently if the variants were in the middle of an exon, or in the deep intron region. Future studies should attempt to identify variants that influence gene expression and post-transcription modifications to improve our understanding of BRCA1 and BRCA2, as well as their related cancers.
Hereditary breast and ovarian cancer (HBOC) syndrome is an autosomal dominant genetic disorder that is caused by the predisposition of pathogenic variants in breast cancer gene 1 (BRCA1) and BRCA2. It is characterized by an increased lifetime risk of developing breast cancer, ovarian cancer (OC) and prostate cancer, as well as other cancers to a lower extent, such as pancreatic cancer (1). According to the National Comprehensive Cancer Network guidelines for 2021, identifying heterozygous pathogenic BRCA1/2 germline variants (gBRCAMUT) in affected individuals is sufficient for the diagnosis of HBOC. This test is of paramount importance for both patients and their families, as knowing the gBRCAMUT status can not only guide the treatment options for patients, but also help carriers to take preventive measures. The commonly used variant classification criteria were drafted by experts from different academic committees (2–4); in general, a five-tier classification system [Benign, Likely Benign, Variant of Uncertain Significance (VUS), Likely Pathogenic, and Pathogenic] is used to interpret detected variants to guide therapeutic decisions and disease management. However, it is particularly difficult for oncologists and genetic counselors to deal with variants classified as VUS (5).
Efforts to reduce the number of VUSs in BRCA1/2 genetic testing have been ongoing for decades, including classical function studies to evaluate the biological consequences of specific variants (6), clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-based saturation genome editing of all possible single nucleotide variants (SNVs) in a defined region to test their effects in the native genomic context (7), yeast 2-hybrid minigene assays to determine splicing changes (8), in silico predictions of changes in protein structure and function (9), and multifactorial likelihood quantitative analysis (10), among others. These methods have been systematically reviewed by several researchers (11–13).
However, approximately 7–10% of variants detected in BRCA1/2 are still classified as VUSs (14–16), depending on the criteria used for classification and the training of staff in different laboratories. Most VUSs in BRCA1/2 are missense variants, which have uncertain influences on the function of the protein product and the cancer risk of the carrier in question. Synonymous variants, meanwhile, alter the nucleotide without changing the encoded amino acid; they are usually considered to have a minimal impact on the function of the protein product, unless this change interferes with splicing regulation, such as the c.641A>G variant in BRCA1 and the c.859G>A variant in BRCA2.
Genomic DNA was isolated from 500 µl of peripheral blood using the QIAsymphony SP system (QIAGEN, Germany) following the manufacturer’s instructions. The concentration and purity of the resulting DNA was determined by Qubit dsDNA HS assay (Life Technologies, USA) and NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, USA), respectively. Library construction was performed as previously described using 200 ng of genomic DNA. The DNA was first fragmented into segments of approximately 300 bp by sonication (Diagenode, USA). Then appropriate sizing and quantification of the fragments was evaluated by the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The fragments were blunt-end-repaired and A-tailed to allow ligation of adapters, followed by PCR amplification. Enrichment of fragments covering 45 breast/ovarian cancer related genes was achieved using a probe set (Novogene, China) that captures a 0.26-Mb genomic region. The enriched library was sequenced on a NextSeq550 sequencer (Illumina, USA) generating paired end reads of 150 bp to a targeted coverage of over 500 unique reads.
Raw sequencing reads were cleaned and aligned to a human reference genome (GRCh37) using BWA (v0.7.17). SNVs and small insertions and deletions (indels) were called using the Genome Analysis Toolkit, Haplotype (v 4.1.7.0), freebayes (v 1.1.0.46), and SAMtools (v1.9). Then the called variants were annotated using snpEff (v4.3.1t). The pathogenicity of the detected variants was independently evaluated by two clinical geneticists according to the ACMG and ENIGMA (v 2.5.1) criteria. Annotations of the variants followed the Human Genome Variant Society recommendations.
Total RNA was extracted and purified from 500 µl of peripheral blood using the EZ-press RNA Purification Kit PLUS (EZBioscience, China) according to the manufacturer’s instructions. Quantification and qualification of the isolated RNA were carried out using the NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, USA). Reverse transcription was conducted to generate cDNA using 200ng of total RNA using HiScript II Q RT SuperMix (Vazyme, China), followed by PCR amplification using a pair of primers flanking the exon3 of BRCA1 (F′-AGAGTGTCCCATCTGTCTGGA, R′- AAAGGACACTGTGAAGGCCC). Fragment analysis of the amplicon was performed using the Agilent 2100 Bioanalyzer (Agilent Technologies, USA).
Wild type and mutant DNA fragments containing the entire exon and its flanking sequence of ±150 bp were synthesized by Sangon China. Then KpnI and BamHI sites were introduced at both ends of the sequence to insert into the pCAS2 vector as previously reported (8). The wild type and mutant vectors were transfected into HEK293T cells. After 24 hours of culturing, RNA was isolated from the transfected cells followed by RT-PCR using the following primers: F-TGACCCTGACCCCCCCT, R-TAAGGGCGATGCGAA. Then the amplicon was separated by gel electrophoresis for Sanger sequencing.
The first case was a 56-year-old female patient who was diagnosed with serous ovarian cancer (stage II) in the Affiliated Cancer Hospital of Zhengzhou University in 2018. The patient underwent a total abdominal hysterectomy with a lymph node dissection, followed by six cycles of carboplatin plus paclitaxel with a standard-dose regimen. Genetic testing identified a synonymous germline variant of NM_007294.4 (BRCA1):c.132C>T (p.Cys44=) (Figure 1A) in this patient, which was first interpreted as a VUS but could not exclude the possibility of being likely pathogenic. Genetic counseling revealed that the patient had undergone a mastectomy six years ago because of breast cancer, and that her mother experienced a similar history: breast cancer first followed by subsequent ovarian cancer (Figure 1B). Fortunately, the disease has been maintained in a stable state for several years, including at the latest review in May 2021.
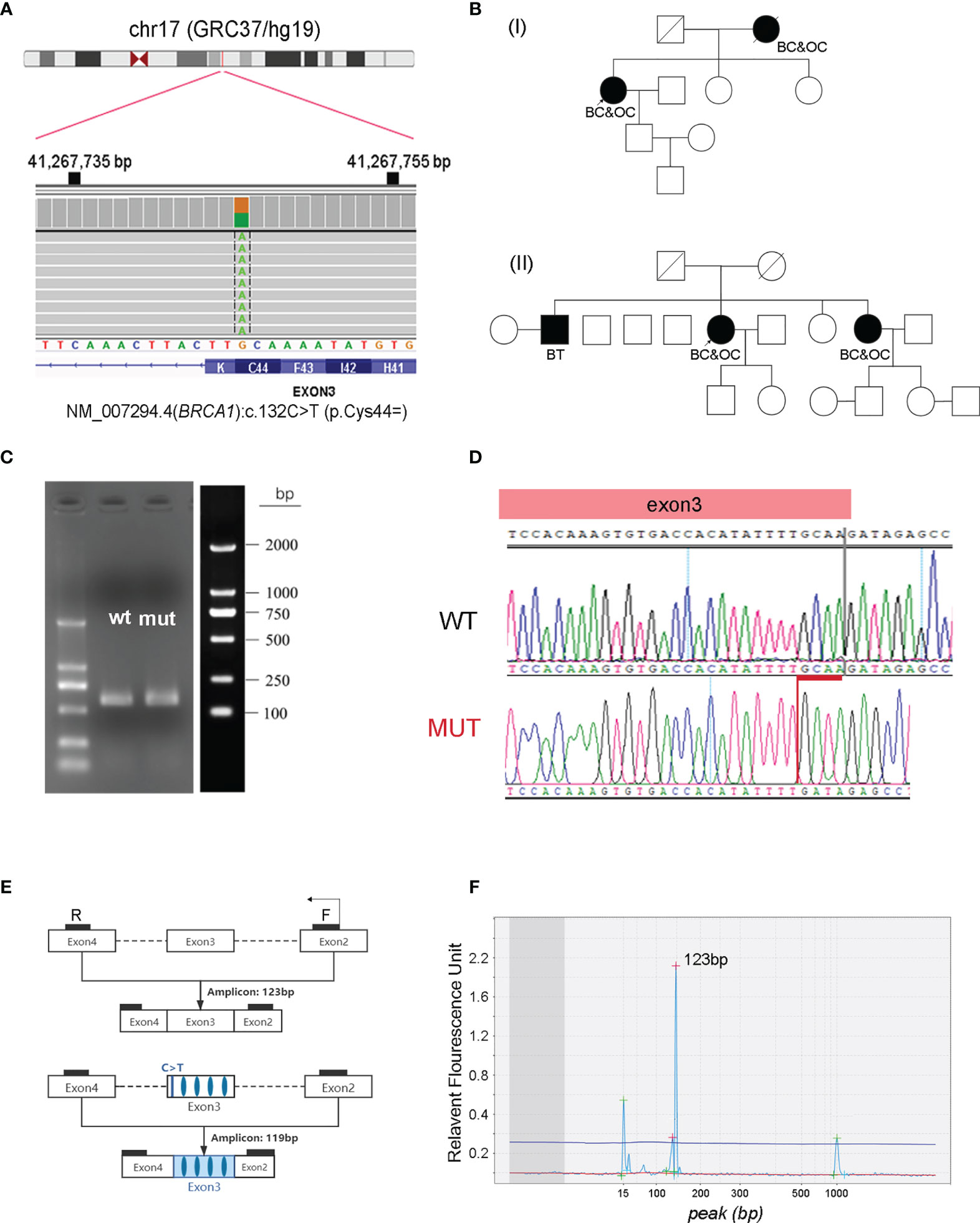
Figure 1 Characterization of NM_007294.4 (BRCA1):c.132C>T (p.Cys44=) as a pathogenic variant. (A) Identification of the heterozygous BRCA1:c.132C>T variant in one of the patients. The screen shot of this variant in the Integrative Genomics Viewer is presented. (B) Pedigrees of two unrelated patients carrying the synonymous BRCA1:c.132C>T variant. (C) PCR amplicons generated from wild-type (wt) and BRCA1:c.132C>T mutant (mut) constructs by a minigene assay, followed by agarose gel electrophoresis and band analysis. (D) Sanger sequencing determination of the deletion of GCAA (red) at the end of exon three in BRCA1:c.132C>T mutant cells. (E) Designed primers flanking exon three for RT-PCR amplification. (F) Fragment analysis of the RT-PCR product using RNA isolated from the blood cells of patients with BRCA1:c.132C>T variant. The amplicon size is evaluated by using the high-resolution automated electrophoresis on an Agilent 2100 Bioanalyzer. BC, breast cancer; OC, ovarian cancer; BT, brain tumor.
The second case was a 64-year-old female patient, who was first diagnosed with breast cancer at 60 years of age, and then presented with ovarian cancer (stage IIIc) 4 years later in 2018. The patient followed a similar treatment as the case presented above: surgical resection followed by six cycles of carboplatin plus paclitaxel and three cycles of pegylated liposomal doxorubicin (PLD) plus nedaplatin. The disease was maintained in a stable state until January 2021, and the patient survived after local treatment. This patient was found to harbor the same germline variant of NM_007294.4 (BRCA1):c.132C>T (p.Cys44=). It was confirmed that one of her sisters, who is a carrier of the same variant, also developed breast cancer and ovarian cancer in her sixties (Figure 1B).
This synonymous BRCA1:c.132C>T (p.Cys44=) variant is located in the 3rd coding exon. It features a C to T substitution that occurs at nucleotide position 132 of the BRCA1 gene. It has been recorded in the ClinVar database as having “conflicting interpretations of pathogenicity” (variation ID: 230061), with four submitted interpretations (two are of uncertain significance; the others are pathogenic). Observing these two independent carriers of BRCA1:c.132C>T prompted us to clarify the pathogenicity of this variant. This substitution occurs 3 bp from the end of exon two and does not change the amino acid at codon 44 (Figure 1A), in silico prediction (SSF, MaxEnt, NNSPLICE and GeneSplicer) suggests that this change may result in a truncated protein product of p.C44Yfs*5 (Supplementary Figures 1, 2). Therefore, we first used the minigene assay to clarify if splicing was influenced by this synonymous variant, which revealed that this change caused the loss of the last four nucleotides (GCAA) in exon three (Figures 1C, D), consistent with a previous report by Steffensen et al. (17). We then isolated ribonucleic acid (RNA) from blood cells, followed by reverse transcription-polymerase chain reaction (RT-PCR) amplification using a pair of primers flanking exon three (Figure 1E). Fragment analysis using the Agilent 2100 bioanalyzer confirmed two peaks next to each other: a strong peak that was 123 bp long and a weak peak that was a few bp shorter (Figure 1F). This weak peak was probably caused by nonsense-mediated mRNA decay. Collectively, functional studies, in combination with the family history of these two independent carriers, convinced us to classify this synonymous BRCA1:c.132C>T (p.Cys44=) as a pathogenic variant.
Female individuals carrying germline variants in BRCA1/2 (gBRCA1/2MUT) have been well documented to have increased lifetime risk of developing multiple cancers. For ovarian cancer, their cumulative risk up to an age of 80 years increases to 44% for gBRCA1MUT and to 17% for gBRCA2MUT carriers, respectively (18). The frequencies and spectrums of gBRCA1/2MUT vary dramatically between different geographical regions and ethnic groups, ranging from 41% in Ashkenazi Jews to 13.8% in Americans (19). Several studies have shown that approximately 23–28% of Chinese Han OC patients present pathogenic or likely pathogenic gBRCA1/2MUT. In addition, these variants are distributed throughout the whole coding sequence, as well as in the flanking splicing regions, without any “hot spot” (20–22).
Medical professionals have suggested that the gBRCA1/2 MUT status in OC patients should be clarified as early as possible following their first diagnosis (23) because of its profound impact on treatment decisions and disease management. Both the Food and Drug Administration and European Medicines Agency have approved PARP inhibitors for the treatment of germline BRCAMUT-associated ovarian cancer. However, it remains challenging to correctly classify a new BRCA1/2 variant in routine clinical practice. Approximately 10–20% of patients are reported to harbor BRCA1/2 variants of unknown significance, which introduces uncertainty to their clinical management (24). We analyzed all of the submitted variants in the BRCA Exchange database (accessed in August 2021, https://brcaexchange.org) and found that in total, 38.63% (8355/21627) of the variants were classified as being of unknown significance, of which 92.75% (7749/8355) were missense and 7.25% (606/8355) were synonymous. Plotting the locations of expert-reviewed missense variants revealed a preference for pathogenic or likely pathogenic aberrations in the functional domains of BRCA1 and BRCA2; almost all of the synonymous variants were benign or likely benign (Figure 2).
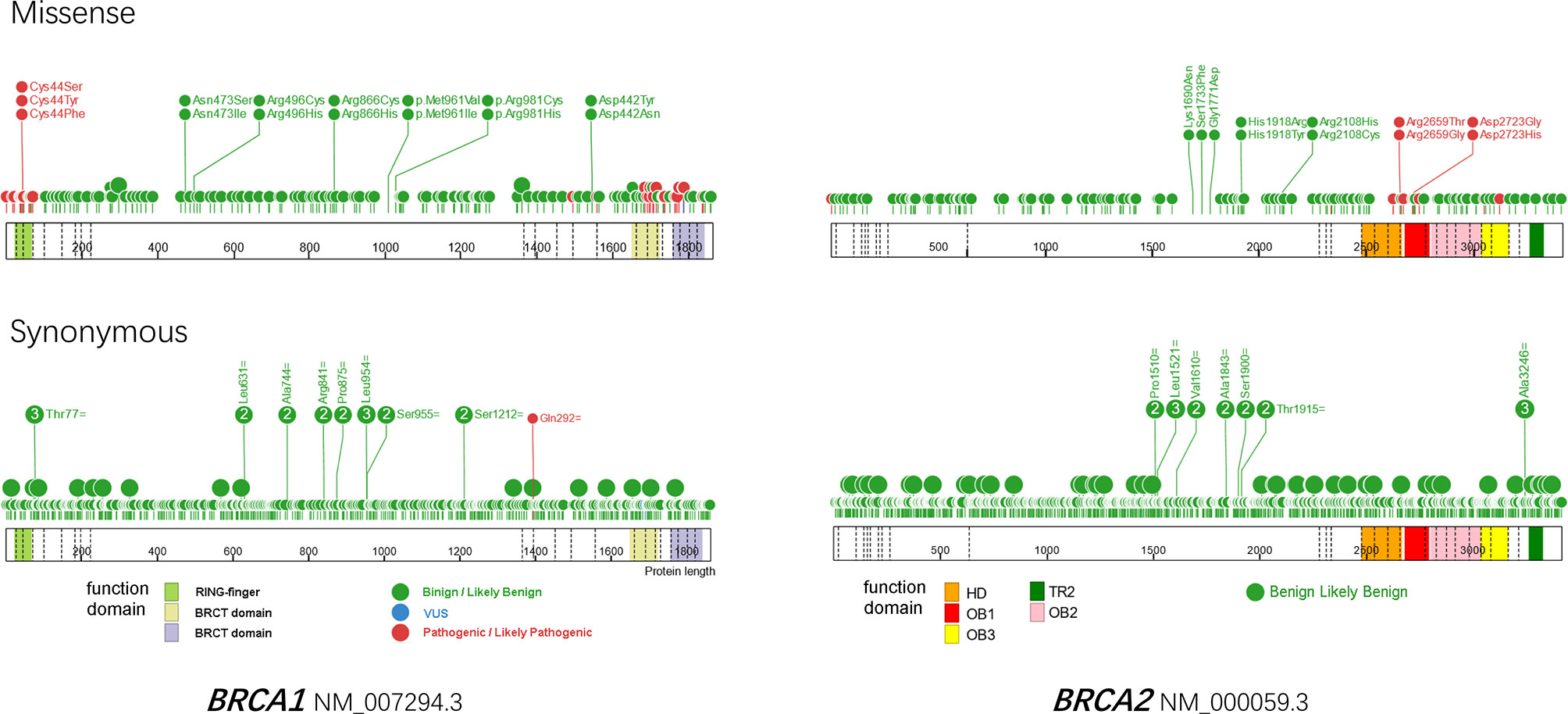
Figure 2 Protein paint of missense (upper) and synonymous variants reviewed by ENIGMA expert panel in BRCA1 (left) and BRCA2 (right). A total of 173 missense and 462 synonymous variants are plotted for BRCA1, as well as 143 missense and 782 synonymous variants for BRCA2. The reference transcripts of NM_007294.3 and NM_000059.3 are used for BRCA1 and BRCA2 respectively. Benign and likely benign variants are in green, VUS are in blue, pathogenic and likely pathogenic variants are in red. Dashed lines indicate exons. HD, helical domain; OB, oligosaccharide-binding folds; TR2, C-terminal RAD51 interaction domain.
Synonymous variants are changes in a DNA sequence that do not change the encoded amino acid. Therefore, they are considered to be “silent”. However, if a synonymous variant disrupts splicing, either by creating a cryptic splice site or by interrupting the splicing regulatory elements, it could potentially abrogate protein function (25). Synonymous variants, as the “sound of silence”, is one of the most mysterious fields in the interpretation of BRCA1/2 variants. Several in silico tools have been developed to predict the splicing changes caused by genetic variants (Supplementary Table 1), which is the most readily and commonly used approach (26–33).
Jian et al. compared the efficiency of these tools in predicting splicing defects by evaluating SNVs, without affecting the GT-AG dinucleotides at the 5′ and 3′ splice sites. They found that MaxEntScan and PWM outperformed other tools (9). This result was also confirmed by Houdayer et al. in BRCA1 and BRCA2 (34). The usDSM method, which uses 14-dimensional biology features and a random forest classifier, has been applied to achieve a superior performance in detecting deleterious synonymous variants. The deep learning model did not make a substantial contribution to the prediction, however, probably because of the limited training dataset used in the study (35). In addition to in silico prediction, the splicing reporter minigene assay is an efficient approach for evaluating the impact of an unclassified variant on mRNA splicing (36). It has been widely used in many laboratories worldwide. In this method, the variant of interest, along with its flanking intronic sequence (~150 bp) is PCR-amplified and cloned into the plasmid, followed by transient transfection into cultured cells. Then, RNA is isolated for reverse transcription, and the chimeric transcripts generated from both wild-type and mutant constructs are compared by PCR and sequencing to determine splicing changes. Several studies have shown that the impacts of variants on splicing patterns and protein functions are not always equivalent; they can vary depending on the proportions of truncated and functional isoforms (37–40).
We analyzed all synonymous variants in the BRCA exchange database. Unsurprisingly, 92.6% (4033/4356) of the variants were consistently classified as being benign or likely benign, 7.1% (311/4356) were VUS, and only 0.3% (12/4356) were pathogenic or likely pathogenic (Figure 3A). Furthermore, discordant interpretations were more frequently observed in benign/likely benign/VUS variants than in pathogenic/likely pathogenic/VUS ones (Figure 3B). In total, ten synonymous variants were classified as being pathogenic or likely pathogenic by at least one submitter (Supplementary Table 2). By reviewing these variants, we interpreted c.4992C>T (p.Leu1664=) in BRCA1 (NM_007294.4) as benign, BRCA1:c.5022C>T (p.Ile1674=) and BRCA2:c.7992T>A (p.Ile2664=) as likely benign, and c.5277G>A (p.Lys1759=) in BRCA1 and c.9117G>T (p.Pro3039=) in BRCA2 as VUS (due to insufficient evidence). The other variants were classified as being either pathogenic or likely pathogenic. One common feature of pathogenic or likely pathogenic variants with synonymous changes is that they always occur at the end of an exon. This suggests that particular caution is warranted regarding the variants in this region. If only in silico predictions of splicing changes are to be used as evidence, without functional and RNA confirmation, then these variants could only be classified as VUS. As mentioned above, the proportions of dysfunctional isoforms caused by aberrant splicing also determines the eventual biological consequences. For example, the minigene assay showed that BRCA1: c.557C>A results in an isoform with excluded exons. However, the carrier’s RNA analysis revealed only a minor proportion of this aberrant isoform; therefore, this variant was still considered to be a VUS. It is worth noting that BRCA1: c.557C>A was not predicted to cause aberrant splicing by several in silico tools, probably because of its location in the internal exon. Some synonymous variants that occur in exons, such as NM_000059.3 (BRCA2):c.9057A>G (p.Lys3019=), can induce aberrant splicing, but most bioinformatic tools cannot efficiently predict these culprits (41).
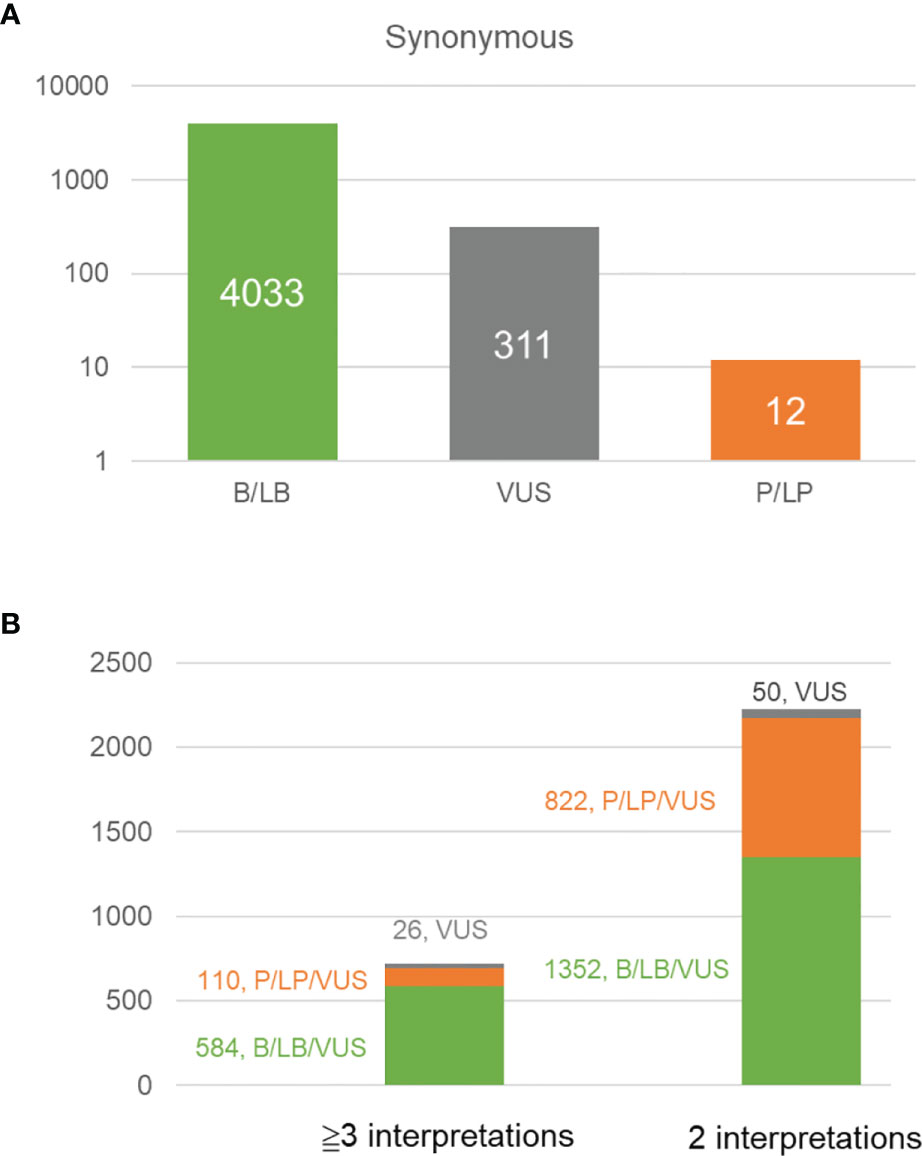
Figure 3 Analysis of the variants in different classes recorded in BRCA exchange database (accessed in Aug. 2021, https://brcaexchange.org). (A) Bar plot showing the number of benign/likely benign variants (4033, in green), VUS (311, in gray), and pathogenic/likely pathogenic variants (12, in orange) in BRCA exchange database. (B) Bar plot shows the number of variants with different interpretations in the BRCA exchange database. Green, the variant with different interpretations of benign, likely benign or VUS; orange, the variant with different interpretations of pathogenic, likely benign and VUS; gray, the variants of VUS.
In addition to the canonical approaches to interpreting BRCA1/2 variants, such as functional studies and multifactorial likelihood quantitative analysis, genome-wide association studies have identified many genetic variants associated with BRCA1/2 expression levels and post-translational modifications. These variants are thereby associated with the risk of developing breast and ovarian cancer (42–44). Expression quantitative trait locus (eQTL) analysis is commonly used to interpret the transcription regulatory mechanisms of genetic variants, which can be either in cis (<1 Mb) or trans (>5 Mb or on another chromosome) (45). The single nucleotide polymorphisms (SNPs) rs17742929 and rs12952924 have been identified as cis-eQTL and trans-eQTL, respectively, for BRCA1 expression in breast cancer. Furthermore, rs7988807 and rs277271 have been identified as cis-eQTL and trans-eQTL, respectively, for BRCA2 expression (46). It has previously been shown that rs57025206 alone can serve as an independent survival marker for estrogen receptor negative BRCA1MUT breast cancer patients, so it can predict unfavorable outcomes (47). The SNPs rs56187033 and rs56012641 are in the post-translational modification sites of BRCA1 and are associated with decreased phosphorylation and N-glycosylation, respectively. However, they were predicted by multiple in silico tools to be neutral, and they have been classified by an expert panel as benign variants (variation IDs 37661 and 37423, respectively). Other non-coding SNPs, such as rs799923, rs799916, and rs3092994, have also been predicted to affect transcription factor binding and have been shown to be circular RNA binding sites, leading to deleterious biological consequences (42). Future studies should pay more attention to these SNPs, as most of them are considered to be benign variants based on current American College of Medical Genetics and Genomics or ENIGMA guidelines but are likely to also cause deleterious clinical outcomes.
BRCA1 and BRCA2 were identified and isolated in the 1990s by researchers from the United States of America and the United Kingdom, respectively. They were identified as breast cancer susceptibility genes, and their essential roles in maintaining genome stability and integrity by regulating DNA damage response and repair were revealed (48). Although many studies have focused on BRCA1/2, as Aristotle famously wrote: “the more you know, the more you know you don’t know”. Future studies should aim to clarify the clinical significance of remaining VUS, investigate the regulatory mechanism of BRCA1/2 expression, and build connections between genetic variants with large-scale clinical data. These efforts could help to better understand these genes and related cancers.
The datasets presented in this study can be found in Genome Sequence Archive (GSA) database with the accession numbers of HRI102015 and HRI102067, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the ethics committee of Henan Cancer Hospital. The patients/participants provided their written informed consent to participate in this study.
JL, PW, and HX conceived the idea and drafted the manuscript. CZ performed the bioinformatics analysis. SH and PW collected the patients’ medical records and performed the RT-PCR experiment. ZL carried out the minigene assay. XW, BW, and JM joined manuscript editing. HL and YG supervised and supported the study. All authors contributed to the article and approved the submitted version.
JL was supported by the Henan provincial young researcher program. This work was financially supported by the funding from National Natural Science Foundation of China (grant number: 81802779), Henan Provincial Health Commission (grant number:SBGJ202002020), Henan science and technology project (grant numbers: 212102310675 and 212102310738), and major public welfare projects in Henan Province (grant number: 201300310400).
Author ZL is currently employed by Amoy Diagnostics Co., Ltd., Xiamen, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We appreciate all the other colleagues in the Department of Molecular Pathology of Henan Cancer Hospital for their generous contributions to this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.812656/full#supplementary-material
1. Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary Breast and Ovarian Cancer: New Genes in Confined Pathways. Nat Rev Cancer (2016) 16:599–612. doi: 10.1038/nrc.2016.72
2. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
3. Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, et al. ENIGMA–Evidence-Based Network for the Interpretation of Germline Mutant Alleles: An International Initiative to Evaluate Risk and Clinical Significance Associated With Sequence Variation in BRCA1 and BRCA2 Genes. Hum Mutat (2012) 33:2–7. doi: 10.1002/humu.21628
4. Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence Variant Classification and Reporting: Recommendations for Improving the Interpretation of Cancer Susceptibility Genetic Test Results. Hum Mutat (2008) 29:1282–91. doi: 10.1002/humu.20880
5. Federici G, Soddu S. Variants of Uncertain Significance in the Era of High-Throughput Genome Sequencing: A Lesson From Breast and Ovary Cancers. J Exp Clin Cancer Res (2020) 39:46. doi: 10.1186/s13046-020-01554-6
6. Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, et al. Functional and Mutational Landscapes of BRCA1 for Homology-Directed Repair and Therapy Resistance. eLife (2017) 6:e21350. doi: 10.7554/eLife.21350
7. Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate Classification of BRCA1 Variants With Saturation Genome Editing. Nature (2018) 562:217–22. doi: 10.1038/s41586-018-0461-z
8. Fraile-Bethencourt E, Diez-Gomez B, Velasquez-Zapata V, Acedo A, Sanz DJ, Velasco EA. Functional Classification of DNA Variants by Hybrid Minigenes: Identification of 30 Spliceogenic Variants of BRCA2 Exons 17 and 18. PloS Genet (2017) 13:e1006691. doi: 10.1371/journal.pgen.1006691
9. Jian X, Boerwinkle E, Liu X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic Acids Res (2014) 42:13534–44. doi: 10.1093/nar/gku1206
10. Parsons MT, Tudini E, Li H, Hahnen E, Wappenschmidt B, Feliubadaló L, et al. Large Scale Multifactorial Likelihood Quantitative Analysis of BRCA1 and BRCA2 Variants: An ENIGMA Resource to Support Clinical Variant Classification. Hum Mutat (2019) 40:1557–78. doi: 10.1002/humu.23818
11. Wallace AJ. New Challenges for BRCA Testing: A View From the Diagnostic Laboratory. Eur J Hum Genet (2016) 24(Suppl 1):S10–8. doi: 10.1038/ejhg.2016.94
12. Lopez-Urrutia E, Salazar-Rojas V, Brito-Elias L, Coca-Gonzalez M, Silva-Garcia J, Sanchez-Marin D, et al. BRCA Mutations: Is Everything Said? Breast Cancer Res Treat (2019) 173:49–54. doi: 10.1007/s10549-018-4986-5
13. Jimenez-Sainz J, Jensen RB. Imprecise Medicine: BRCA2 Variants of Uncertain Significance (VUS), the Challenges and Benefits to Integrate a Functional Assay Workflow With Clinical Decision Rules. Genes (2021) 12(5):780. doi: 10.3390/genes12050780
14. Lee JS, Oh S, Park SK, Lee MH, Lee JW, Kim SW, et al. Reclassification of BRCA1 and BRCA2 Variants of Uncertain Significance: A Multifactorial Analysis of Multicentre Prospective Cohort. J Med Genet (2018) 55:794–802. doi: 10.1136/jmedgenet-2018-105565
15. So MK, Jeong TD, Lim W, Moon BI, Paik NS, Kim SC, et al. Reinterpretation of BRCA1 and BRCA2 Variants of Uncertain Significance in Patients With Hereditary Breast/Ovarian Cancer Using the ACMG/AMP 2015 Guidelines. Breast Cancer (2019) 26:510–9. doi: 10.1007/s12282-019-00951-w
16. Fanale D, Fiorino A, Incorvaia L, Dimino A, Filorizzo C, Bono M, et al. Prevalence and Spectrum of Germline BRCA1 and BRCA2 Variants of Uncertain Significance in Breast/Ovarian Cancer: Mysterious Signals From the Genome. Front Oncol (2021) 11:682445. doi: 10.3389/fonc.2021.682445
17. Steffensen AY, Dandanell M, Jonson L, Ejlertsen B, Gerdes AM, Nielsen FC, et al. Functional Characterization of BRCA1 Gene Variants by Mini-Gene Splicing Assay. Eur J Hum Genet (2014) 22:1362–8. doi: 10.1038/ejhg.2014.40
18. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
19. Robles-Diaz L, Goldfrank DJ, Kauff ND, Robson M, Offit K. Hereditary Ovarian Cancer in Ashkenazi Jews. Fam Cancer (2004) 3:259–64. doi: 10.1007/s10689-004-9552-0
20. Li J, Han S, Zhang C, Luo Y, Wang L, Wang P, et al. Identification of BRCA1:c.5470_5477del as a Founder Mutation in Chinese Ovarian Cancer Patients. Front Oncol (2021) 11:655709. doi: 10.3389/fonc.2021.655709
21. Li A, Xie R, Zhi Q, Deng Y, Wu Y, Li W, et al. BRCA Germline Mutations in an Unselected Nationwide Cohort of Chinese Patients With Ovarian Cancer and Healthy Controls. Gynecol Oncol (2018) 151:145–52. doi: 10.1016/j.ygyno.2018.07.024
22. You Y, Li L, Lu J, Wu H, Wang J, Gao J, et al. Germline and Somatic BRCA1/2 Mutations in 172 Chinese Women With Epithelial Ovarian Cancer. Front Oncol (2020) 10:295. doi: 10.3389/fonc.2020.00295
23. Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J Clin Oncol (2020) 38:1222–45. doi: 10.1200/JCO.19.02960
24. Eccles BK, Copson E, Maishman T, Abraham JE, Eccles DM. Understanding of BRCA VUS Genetic Results by Breast Cancer Specialists. BMC cancer (2015) 15:936. doi: 10.1186/s12885-015-1934-1
25. Baralle D, Baralle M. Splicing in Action: Assessing Disease Causing Sequence Changes. J Med Genet (2005) 42:737–48. doi: 10.1136/jmg.2004.029538
26. Houdayer C, Caux-Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, et al. Guidelines for Splicing Analysis in Molecular Diagnosis Derived From a Set of 327 Combined In Silico/In Vitro Studies on BRCA1 and BRCA2 Variants. Hum Mutat (2012) 33:1228–38. doi: 10.1002/humu.22101
27. Tang X, Zhang T, Cheng N, Wang H, Zheng CH, Xia J, et al. usDSM: A Novel Method for Deleterious Synonymous Mutation Prediction Using Undersampling Scheme. Brief Bioinform (2021) 22(5):bbab123. doi: 10.1093/bib/bbab123
28. Gaildrat P, Killian A, Martins A, Tournier I, Frebourg T, Tosi M. Use of Splicing Reporter Minigene Assay to Evaluate the Effect on Splicing of Unclassified Genetic Variants. Methods Mol Biol (2010) 653:249–57. doi: 10.1007/978-1-60761-759-4_15
29. Gelli E, Colombo M, Pinto AM, De Vecchi G, Foglia C, Amitrano S, et al. Usefulness and Limitations of Comprehensive Characterization of mRNA Splicing Profiles in the Definition of the Clinical Relevance of BRCA1/2 Variants of Uncertain Significance. Cancers (2019) 11(3):295. doi: 10.3390/cancers11030295
30. Sanz DJ, Acedo A, Infante M, Durán M, Pérez-Cabornero L, Esteban-Cardeñosa E, et al. A High Proportion of DNA Variants of BRCA1 and BRCA2 Is Associated With Aberrant Splicing in Breast/Ovarian Cancer Patients. Clin Cancer Res (2010) 16:1957–67. doi: 10.1158/1078-0432.CCR-09-2564
31. Thery JC, Krieger S, Gaildrat P, Révillion F, Buisine MP, Killian A, et al. Contribution of Bioinformatics Predictions and Functional Splicing Assays to the Interpretation of Unclassified Variants of the BRCA Genes. Eur J Hum Genet (2011) 19:1052–8. doi: 10.1038/ejhg.2011.100
32. Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A Systematic Genetic Assessment of 1,433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer-Predisposition Genes. Am J Hum Genet (2007) 81:873–83. doi: 10.1086/521032
33. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting Splicing From Primary Sequence With Deep Learning. Cell (2019) 176:535–48 e24. doi: 10.1016/j.cell.2018.12.015
34. Pirim D, Kaya N, Yildirim EU, Sag SO, Temel SG. Characterization and In Silico Analyses of the BRCA1/2 Variants Identified in Individuals With Personal and/or Family History of BRCA-Related Cancers. Int J Biol Macromol (2020) 162:1166–77. doi: 10.1016/j.ijbiomac.2020.06.222
35. Quiroz-Zarate A, Harshfield BJ, Hu R, Knoblauch N, Beck AH, Hankinson SE, et al. Expression Quantitative Trait Loci (QTL) in Tumor Adjacent Normal Breast Tissue and Breast Tumor Tissue. PloS One (2017) 12:e0170181. doi: 10.1371/journal.pone.0170181
36. Choi J, Topouza DG, Tarnouskaya A, Nesdoly S, Koti M, Duan QL. Gene Networks and Expression Quantitative Trait Loci Associated With Adjuvant Chemotherapy Response in High-Grade Serous Ovarian Cancer. BMC cancer (2020) 20:413. doi: 10.1186/s12885-020-06922-1
37. Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, et al. Large-Scale Cis- and Trans-eQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat Genet (2021) 53:1300–10. doi: 10.1038/s41588-021-00913-z
38. Gong J, Mei S, Liu C, Xiang Y, Ye Y, Zhang Z, et al. PancanQTL: Systematic Identification of cis-eQTLs and trans-eQTLs in 33 Cancer Types. Nucleic Acids Res (2018) 46:D971–D6. doi: 10.1093/nar/gkx861
39. Muranen TA, Khan S, Fagerholm R, Aittomäki K, Cunningham JM, Dennis J, et al. Association of Germline Variation With the Survival of Women With BRCA1/2 Pathogenic Variants and Breast Cancer. NPJ Breast Cancer (2020) 6:44. doi: 10.1038/s41523-020-00185-6
40. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat Rev Cancer (2011) 12:68–78. doi: 10.1038/nrc3181
41. Yeo G, Burge CB. Maximum Entropy Modeling of Short Sequence Motifs With Applications to RNA Splicing Signals. J Comput Biol (2004) 11:377–94. doi: 10.1089/1066527041410418
42. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: An Online Bioinformatics Tool to Predict Splicing Signals. Nucleic Acids Res (2009) 37:e67. doi: 10.1093/nar/gkp215
43. Shapiro MB, Senapathy P. RNA Splice Junctions of Different Classes of Eukaryotes: Sequence Statistics and Functional Implications in Gene Expression. Nucleic Acids Res (1987) 15:7155–74. doi: 10.1093/nar/15.17.7155
44. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved Splice Site Detection in Genie. J Comput Biol (1997) 4:311–23. doi: 10.1089/cmb.1997.4.311
45. Pertea M, Lin X, Salzberg SL. GeneSplicer: A New Computational Method for Splice Site Prediction. Nucleic Acids Res (2001) 29:1185–90. doi: 10.1093/nar/29.5.1185
46. Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. Using Positional Distribution to Identify Splicing Elements and Predict Pre-mRNA Processing Defects in Human Genes. Proc Natl Acad Sci USA (2011) 108:11093–8. doi: 10.1073/pnas.1101135108
47. Woolfe A, Mullikin JC, Elnitski L. Genomic Features Defining Exonic Variants That Modulate Splicing. Genome Biol (2010) 11:R20. doi: 10.1186/gb-2010-11-2-r20
Keywords: BRCA1/2, splicing variants, HBOC, variants classification, synonymous variants
Citation: Li J, Wang P, Zhang C, Han S, Xiao H, Liu Z, Wang X, Liu W, Wei B, Ma J, Li H and Guo Y (2022) Characterization of Synonymous BRCA1:c.132C>T as a Pathogenic Variant. Front. Oncol. 11:812656. doi: 10.3389/fonc.2021.812656
Received: 10 November 2021; Accepted: 08 December 2021;
Published: 11 January 2022.
Edited by:
Florentia Fostira, National Centre of Scientific Research Demokritos, GreeceReviewed by:
Irene Konstantopoulou, National Centre of Scientific Research Demokritos, GreeceCopyright © 2022 Li, Wang, Zhang, Han, Xiao, Liu, Wang, Liu, Wei, Ma, Li and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjun Guo, Z3VveW9uZ2p1bkB6enUuZWR1LmNu; Hongle Li, bGxobDczQDE2My5jb20=; Jun Li, c2VyYXBoLmxlZWp1bkBvdXRsb29rLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.