 Elena Ionica1*
Elena Ionica1* Gisela Gaina2
Gisela Gaina2 Mihaela Tica3
Mihaela Tica3 Mariana-Carmen Chifiriuc1,4
Mariana-Carmen Chifiriuc1,4 Gratiela Gradisteanu-Pircalabioru5
Gratiela Gradisteanu-Pircalabioru5- 1Department of Biochemistry and Molecular Biology, Faculty of Biology, University of Bucharest, Bucharest, Romania
- 2Laboratory of Cell Biology, Neuroscience and Experimental Miology, Victor Babes National Institute of Pathology, Bucharest, Romania
- 3Bucharest Emergency University Hospital, Bucharest, Romania
- 4Biological Science Division, Romanian Academy of Sciences, Bucharest, Romania
- 5Research Institute of the University of Bucharest, University of Bucharest, Bucharest, Romania
In order to ensure that primary endpoints of clinical studies are attained, the patients’ stratification is an important aspect. Selection criteria include age, gender, and also specific biomarkers, such as inflammation scores. These criteria are not sufficient to achieve a straightforward selection, however, in case of multifactorial diseases, with unknown or partially identified mechanisms, occasionally including host factors, and the microbiome. In these cases, the efficacy of interventions is difficult to predict, and as a result, the selection of subjects is often random. Colorectal cancer (CRC) is a highly heterogeneous disease, with variable clinical features, outcomes, and response to therapy; the CRC onset and progress involves multiple sequential steps with accumulation of genetic alterations, namely, mutations, gene amplification, and epigenetic changes. The gut microbes, either eubiotic or dysbiotic, could influence the CRC evolution through a complex and versatile crosstalk with the intestinal and immune cells, permanently changing the tumor microenvironment. There have been significant advances in the development of personalized approaches for CRC screening, treatment, and potential prevention. Advances in molecular techniques bring new criteria for patients’ stratification—mutational analysis at the time of diagnosis to guide treatment, for example. Gut microbiome has emerged as the main trigger of gut mucosal homeostasis. This may impact cancer susceptibility through maintenance of the epithelial/mucus barrier and production of protective metabolites, such as short-chain fatty acids (SCFAs) via interactions with the hosts’ diet and metabolism. Microbiome dysbiosis leads to the enrichment of cancer-promoting bacterial populations, loss of protective populations or maintaining an inflammatory chronic state, all of which contribute to the development and progression of CRC. Meanwhile, variations in patient responses to anti-cancer immuno- and chemotherapies were also linked to inter-individual differences in intestine microbiomes. The authors aim to highlight the contribution of epithelial and gut microbiome inflammatory biomarkers in the improvement of CRC patients’ stratification towards a personalized approach of early diagnosis and treatment.
1 Introduction
Noncommunicable diseases (NCDs) are now responsible for most of the global deaths, among which cancer is a serious health problem in all countries, regardless of socio-economic status. According to data provided by the World Health Organization (WHO) in 2020, at a global level one in five people faces a cancer diagnosis during their lifetime. The most important actions to increase life expectancy should be increasing prevention, diagnosis, treatment and management, palliative care, and surveillance. According to the Agency for Research on Cancer, the disease is the first or second leading cause of death in 2019 in the USA, while ranking third or fourth in an additional 22 countries. An estimated 18.1 million people around the world (excluding nonmelanoma skin cancer) developed cancer, out of which 9.9 million died from the disease before age 70 in 112 of 183 countries (1, 2). Lung cancer (11.6% of all cases), followed by female breast (11.6%) and colorectal (CRC) cancers (10.2%) are the most frequently diagnosed cancers (3).
Although developed countries have the highest CRC incidence and mortality rates worldwide, CRC incidence has recently started increasing in low-income and middle-income nations (2, 4). This reflects changes in lifestyle factors and diet, an increased intake of animal-source foods, a more sedentary lifestyle, leading to decreased physical activity and increased prevalence of excess body weight (1). However, complex causes reflect both aging and global population growth, and also changes in the prevalence and distribution of the main risk factors for cancer. In some cases, these factors were associated with socioeconomic development (5). It has been observed that in countries undergoing major transitions, such as countries from Eastern Europe, South Europe, South Central Asia, and South America, the incidence rate tends to increase uniformly with the Human Development Index (HDI) (1). Therefore, primary prevention remains the most important strategy to reduce the global prevalence of colorectal cancer.
In 2020, CRC ranks third in terms of incidence and second in terms of mortality (1, 5). Even in countries where national CRC screening programs exist the mortality is around 30%, and only a small proportion of CRCs are diagnosed through population-based screening programs (1, 5).
Before the year 2000, the changes in pattern behavioral risk factors (smoking reduction, change in dietary pattern) and also increasing number of CRC screening programs around the world (SUA, UK, Switzerland, Austria) was associated with a decreased rate of CRC incidence. However, there is still a gap in diagnosing individuals at a localized stage, probably because screening programs include only patients older than 50 years and only track behavioral changes. The screening is based on colonoscopy, a method that predominantly prevents tumor metastasis by removing premalignant polyps (6).
After 2000, due to rapid technological improvement and affordable costs of molecular techniques, scientists were able to define criteria for CRC patients’ stratification based on sex, age, family history, genetic susceptibility, and endoscopy and colonoscopy examinations. Consequently, the individualized treatment has been achieved more rapidly. In 2015, in a joint effort to understand the heterogeneous clinical and drug outcomes observed in CRC patients, a consortium of researchers defined a new criterion for CRC patients’ stratification based on Consensus Molecular Subtyping (CMS). This highlighted the interconnection between six classification systems that were finally grouped into four molecular subtypes. The first subtype (CMS1) is based on microsatellite instability (MSI), significance of immune activation and hypermethylation, specific processes for the early-stage sporadic colorectal cancer. For early diagnosis and treatment of CRC patients, the MSI model (CMS1) has become a key biomarker due to advances in the understanding of the immune microenvironment of colorectal cancer. CRC patients with different microsatellite statuses exhibited different compositions and distributions of immune cells and cytokines within their tumor microenvironments (TMEs). A complex crosstalk between different regulatory pathways was observed in the TME, with a key role in the occurrence, progression, and treatment of tumours, representing an important source of potential therapeutic targets. Intestinal microbiome (IM) has emerged as the main environmental trigger of intestinal mucosal homeostasis that may also influence cancer susceptibility through maintenance of the epithelial/mucus barrier and production of protective metabolites via interactions with host diet and metabolism. Dysbiosis leads to the enrichment of cancer-promoting bacterial populations, loss of protective populations, development, and progression of CRC by maintaining an inflammatory chronic state, etc., while variations in patient responses to cancer immuno- and chemotherapies have been linked to inter-individual differences in intestine microbiomes (7–9). Accumulating evidence suggests that chronic inflammation and the metabolites accumulated ensuing inflammation contribute to tumor initiation and tumor progression (10–12).
In this article, we aim to focus on the complex network signaling pathway crosstalk between colonocytes, microbiome, and tumor cells at the earlier-stage sporadic colorectal cancer, and to reveal the role of inflammatory biomarkers in CRC screening programs.
2 Advances in the Molecular Stratification of Colorectal Cancer
CRC is a highly heterogeneous disease, with varying clinical outcomes, morphological features, and genetic and gene regulatory levels which contribute to differences in individual response to therapy. The histopathological parameters typically used for diagnosis are not enough to recognize high risk patients, while molecular features are still sparsely used in current clinical practice.
Explosion in knowledge regarding genetic, epigenetic, and biochemical alterations, associated with the evolution of CRC, correlated with advances in molecular biology, sequencing, computer science and modern bioinformatics have facilitated the development of different patients’ stratification strategies. A systematic review of literature on developed stratification models on CRC indicates that current screening guidelines for CRC stratify patients based on different criteria as presented in Table 1.

Table 1 CRC patients’ stratification criteria.
As shown in Table 1, selection criteria for the enrolment of prospective subjects include age, gender, molecular signature, inflammation scores, or Dietary Inflammatory Index (DII®). The DII, a literature-derived population-based dietary index, was analyzed by Shivappa in nine studies that examined the association between the diets’ inflammatory potential and CRC. In their meta-analysis they demonstrate that limiting the consumption of pro-inflammatory foods, such as red meat, and increasing consumption of anti-inflammatory nutrients may play an important role in reducing the risk of CRC. Anti-inflammatory foods include fiber, monounsaturated fatty acids, polyunsaturated fatty acids, omega 3, omega 6, vitamins and minerals, all associated with a lower DII score. While pro-inflammatory foods include carbohydrates, proteins, trans fat, total fat, cholesterol, saturated fatty acids, iron, etc., associated with a higher DII score (32, 33). Evidence suggests that modifiable lifestyle factors, namely, excess adiposity, poor diet, and physical inactivity play an important role in the occurrence and progression of this disease (34). While for diseases with known underlying mechanism or targeted therapy, the selection of subjects can be straightforward to obtain high efficacy of a given intervention, this cannot be achieved, unfortunately, in the case of CRC. It is a multifactorial disease, with unknown or partially elucidated mechanisms, sometimes including host factors, local environmental factors, lifestyle factors (35), microbiome, and host diets that have been extensively studied. All of these play a causal and protective role in the development of CRC (36). Based on 19 factors, Jeon and col. created a risk score that summarizes an individuals’ overall lifestyle and environment CRC risk profile by using data from 14 population-based studies. They demonstrate that both lifestyle, environmental factors, and common genome-wide association study (GWAS) variants are independent risk predictors for CRC (16).
Advances in molecular techniques bring new criteria for patients’ stratification, e.g., mutational analysis at the time of diagnosis to guide treatment. By reviewing the literature, Coleman and co. found that the caloric content and composition of diets are linked to the development of obesity and were associated with persistent and transmissible alterations in the IM. It was suggested that differences in diet and in the IM may be responsible for variations in CRC prevalence between two similar human populations (37).
The CRC onset and progress involves multiple sequential steps with accumulation of genetic alterations including mutations, gene amplification, and epigenetic changes. Although inherited genetic susceptibility has a key role in a subset of CRC cases, in general, CRC cases are sporadic and non-inherited. Traditionally, CRC can be divided into familial CRC (hereditary CRC, HCRC) and sporadic CRC (non-hereditary CRC, NHCRC) (38).
Other than HCRC which accounts for approximately 20–25% of all types of CRC, as determined by non-modifiable risk factors such as familial history of CRC, adenomatous polyps, specific mutations occurring in genes involved in the tumorigenic pathway, an approximate of 75% of CRC tumors have some genetic defects that occur throughout life, stemming from a multitude of factors.
Even if new molecular pathways, critical for tumor development, are continuously discovered and their molecular targeting is a success in the experimental models, the number of effective therapies for CRC patients is still very limited.
While in the early 1990s, researchers were able to link the mutations of some genes to the susceptibility of CRC and postulated the idea that CRC progression is correlated with the accumulation of genetic changes, more recently there has been an increased emphasis on the heterogeneity of colon cancer, and the involvement of the intratumoral environment (39–43).
The two main pathways used to classify CRC and guide the personalized therapeutic strategy are the chromosomal instability pathway (CIN) that accounts for 85% of cases, and the microsatellite instability pathway (MSI) that represents 15% of total CRC cases (44).
Initially, this classification was used for dichotomization into good and poor prognosis patients, whereas current research reveals that separation into strictly two groups does not reflect the biological diversity specific to colon cancer (45).
Most of the sporadic CRC cases are explained using CIN model, which suggests a predictable progression with sequential accumulation of mutations in specific genes like adenomatous polyposis coli (APC), WNT, etc. The model provides signs that can be used for risk assessment, early detection, prognosis, and treatment of the disease. According to this model, the sequential accumulation of mutations that eventually leads to CRC provides a level of opportunity to prevent CRC before these mutations reach a threshold level.
The MSI model represents a class of colon cancer characterised by high mutational load caused by defective DNA mismatch repair (MMR). The microsatellites, representing repetitive DNA sequences, are especially sensitive to mutation due to dysfunctional MMR and a high abundance of microsatellite length alteration. The detection of MSI is identified via a PCR-based assay categorizing tumours as either MSI-high (MSI-H), MSI-low (MSI-L) or microsatellite stable (MSS), based on the number of microsatellite markers demonstrating instability. In sporadic MSI tumors, defective DNA MMR usually result from CpG (5’-CG-3’) island promoter methylation and therefore the inactivation of the MLH1 gene which encodes a key gene in MMR pathway. This promoter methylation is accompanied by other methylation processes of promoters throughout the genome. This phenomenon is known as CpG island methylator phenotype (CIMP). Testing for CIMP is performed via PCR for hypermethylation in CACNA1G, MLH1, NEUROG1, RUNX3, and SOCS1 (46). Based on MSI status, an international consortium consolidated separate findings into one overarching stratification system, the Consensus Molecular Subtypes (CMS) of colorectal cancer (22).
As mentioned, a consortium of researchers highlighted the interconnection between six classification systems that were finally grouped into four molecular subtypes (CMS): CMS1 (MSI Immune) (14% of classified molecular clusters), microsatellite instability, significance of immune activation and hypermethylation; CMS2 (Canonical) (37%), shows changes in the WNT and MYC signalling pathways in epithelial cells; CMS3 (Metabolic) (13%), presents disorders in the signalling pathways in epithelial cells and metabolic changes; and the CMS4 subtype (Mesenchymal) (23%) shows changes in mesenchymal–epithelial cells, prominent stromal invasion and angiogenesis, changes that are accompanied by activation of growth factor β (TGF-β) (22, 47). The remaining 13% of the analyzed molecular clusters could not be included in any subtype because the characteristics are specific to several subtypes; it is considered to have a phenotype of transition from one cluster to another. This classification is the most comprehensive, preserving the relation between specific molecular changes, biological classification, and clinical classification.
3 Tumor Microenvironment Heterogeneity as a Potential Source of CRC Biomarkers and Therapeutic Targets
Over the last 10–15 years, the CRC research field has moved from mainly assessing mutations to measuring gene expression patterns, in order to describe a broader spectrum of CRC heterogeneity (48). The molecular heterogeneity involves several molecular pathways and molecular changes unique to a tumor of an individual and, although the existence of heterogeneity in CRC has been recognized for a longer period, it is sparingly incorporated as a determining factor in current clinical practice.
Development of TME is driven by genetic instability of cancer cells and by epigenetic factors in response to endogenous stress stimuli or exogenous factors like pH changes, aberrant angiogenesis, hypoxia, glucose deprivation, acidosis, and oxidative stress. Endogenous stressors are associated with imbalanced cell growth, increased mutation rate, errors in lipid and glycoprotein biosynthesis and decreased amino acid supplies (49, 50).
The tumor stromal-inflammatory interface represents a dynamic space which includes growth factors (hepatocyte growth factor (HGF), insulin-like growth factors (IGF), nerve growth factor (NGF), wingless-type MMTV integration site family member 1 (WNT1), epidermal growth factor (EGF), fibroblast growth factor 2 (FGF2), vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF).), cytokines (IL-1, IL-6, IL-8), chemokines (CXC chemokines CXCL1 to CXCL12, CC chemokines CCL2 and CCL20), enzymes (LOX family oxidases and LOX-like proteins 1–4, COX2), matrix metalloproteinases (MMP-1, MMP-2, MMP-7, MMP-9, MMP-13, and MMP-14), ECM proteins (fibronectin, collagen I and III, EDA-fibronectin, tenascin C, and SPARC), and metabolic intermediates that are secreted during the exchanges (between various molecular information) associated with stromal and cancer cells transitions. Recruitment, activation, reprogramming and persistence of inflammatory and stromal cells in the extracellular space are the consequences of a reciprocal relationship between TME and cancer cells (51). Studies have shown that infiltration of immune cells into TME is an important factor affecting tumor heterogeneity and prognosis, and infiltrated immune cells and cytokines secreted by the inflammatory process may play dual roles by inhibiting or promoting tumors.
In light of all these, the above mentioned MSI model (CMS1) is of paramount importance for the early diagnosis and treatment of CRC patients due to advances in the understanding of the immune microenvironment of colorectal cancer. CRC patients with different microsatellite statuses present different compositions and distributions of immune cells and cytokines within their TMEs (52).
A complex crosstalk between different regulatory pathways was observed in the TME, which played a key role in the occurrence, progression, and treatment of tumours, representing an important potential therapeutic target (53). As so, the TME is permanently remodeling and reprogramming according to the modification of numerous physical, biochemical, and stromal cells functions like pH, oxidative stress, ECM stiffness, metabolism, inflammation, and immunity. Therefore, inhibition of one specific target leads to different answers, at different levels in different regulatory pathways. In addition, the cell is able to avoid the path that is blocked so that the entire signaling network functions; cancer cells develop new adaptative strategies to adjust their phenotype to the variation of TME conditions (50). New developed heterogenous cancer cells subpopulations appear and show different characteristics in terms of plasticity, stem-like phenotype, metabolic reprograming, invasion, immunosuppression, and therapeutic resistance (51). According to Alsibai, the most widely known plasticity processes are the epithelial–mesenchymal transition (EMT) and the reversible mesenchymal–epithelial transition (MET). Cancer cells follow similar EMT processes to establish metastases. EMT facilitates epithelial cancer cells to enter a mesenchymal-like state by endowing the migratory and invasive properties, which enables a primary tumor to move and colonize distant organs and form metastases. This is a critical step in early phase of cancer metastasis and is closely linked to carcinogenesis, invasion, recurrence, and therapy resistance (50). One of the goals of the researchers for stratifying patients according to the specific crosstalk patterns is to analyze how they communicate with the main signaling pathways involved in carcinogenesis, as well as to show the nature of the relationship between different subtypes of tumor cells and protein expression.
4 Contribution of Gut Microbiota Derived Markers to CRC Patients’ Stratification
Nearly all the digestion and absorption of nutrients is carried out in the gastrointestinal tract (GI). Abundant gut microbes in the GI, estimated to total about 1013–1014 CFU (colony forming units)/ml, are involved in the process. Bacterial products together with food residue make up most of the contents in the intestine and are directly or indirectly in contact with the mucus layer separating them from the epithelial cells. The gut microbiome, the largest micro-ecosystem in the human body, is symbiotic with the host and maintains normal physiological processes in a dynamic equilibrium state (54). It establishes a complex interaction with host cells and even a small disturbance may lead to the breakdown of intestinal homeostasis just like the ‘butterfly effect’ (55). The maintenance of this precise balance requires the control of epithelial cells via different immune mechanisms and significantly contributes to the constitution of the intestinal immune barrier (56). It is widely acknowledged that the gut microbiome has an important role in the development of a properly functioning mucosal and systemic immune system. The intestinal microbes, either eubiotic or dysbiotic, could influence the CRC evolution, through a complex and versatile crosstalk with the intestinal and immune cells, permanently changing the tumor microenvironment.
With the rapid development of molecular biology, genomics, bioinformatic analysis technology, researchers linked CRC to intestinal dysbiosis (57, 58), and to specific microbial composition, structure, and function signatures (59).
Intestinal dysbiosis leads to the enrichment of cancer-promoting bacterial populations, loss of protective populations, development, and progression of CRC by maintaining an inflammatory chronic state etc., while variations in patient responses to cancer immuno- and chemotherapies were linked to inter-individual differences in gut microbiome.
However, despite long-standing associations between diet, the microbiome, and CRC (60, 61), the specific mechanisms by which the gut microbiome may influence not only the starting events of carcinogenesis, but also its progression, have only been highlighted recently (8, 9, 54). Several studies investigating the CRC microbiota have shown that the dysbiosis associated with CRC is characterized by a relatively decreased abundance of obligate anaerobes, an increase in potential pathogenic bacteria (pathobionts), and a reduction of butyrate-producing bacteria (62), such as Bifidobacteria, Lactobacillus, and Bacteroides. Molecular fingerprinting and clone sequencing methods revealed that the microbiota of subjects with adenomas harboured a higher proportion of Proteobacteria and lower abundance of Bacteroidetes when compared to that of control subjects (63). These initial findings were later confirmed in a study that used 16S rRNA gene amplicon 454 pyrosequencing methods to characterize the microbiome. Sanapareddy et al. (13) found an enrichment of potential pathogens like Helicobacter, Bacteroides fragilis, Fusobacterium nucleatum, Acinetobacter, Pseudomonas, Escherichia coli, and other Proteobacteria in mucosal biopsies of adenoma patients compared to non-adenoma controls (64).
Brim et al. (14) analyzed the faecal microbiota of African American patients with colorectal adenomas and reported altered microbial changes between adenoma patients and healthy controls (65). Using experimental models of CRC, Wei et al. (15) showed that precancerous lesions had dysbiosis associated with an increased abundance of Allobaculum spp. and Ruminococcus obeum (66). These data suggest that changes in the adherent microbial community composition may have a role in the development of adenomas. Recently, Yachida et al. (67) conducted the largest metagenomics (n = 616) and metabolomics (n = 406) analysis on human CRC. Specific shifts in microbiome composition, metabolome and bacterial gene abundance were correlated with different stages of CRC progression. Members of the Firmicutes, Fusobacteria, and Bacteroidetes phyla were increased in carcinoma patients compared to healthy controls and adenomas. Moreover, early stages of disease were characterised by an enrichment of taxa such as Actinomyces odontolyticus, Atopobium parvulum, Phascolarctobacterium succinatutens, and Desulfovibrio longreachensis. Lactic acid-producing bacteria such as Bifidobacterium animalis, Streptococcus thermophiles, and Streptococcus mutans were prevalent only in the healthy control group. Progression from healthy to advanced adenoma was characterized by a significant increase of Bacteroides species such as B. dorei and B. massiliensis whereas an increase in B. massiliensis, B. ovatus, B. vulgatus, and E. coli was reported for the progression from advanced adenoma to carcinoma (68).
The pathogenic bacteria prevalent in the CRC patients’ microbiome can secrete toxic metabolites that affect intestinal epithelial cells and cause a chronic inflammatory response, which contributes to the development of CRC (69, 70).
Recent mounting evidence suggests that gut microbes can sense certain features of cancer cells which they use for their own advantage. Fusobacterium nucleatum interacts with tumor cells via its adhesion molecule FadA which binds eukaryotic Annexin A1 in a process mediated by E-cadherin, forming a complex with β-catenin, a central effector of the Wnt pathway (Figure 1) (71). Peptostreptococcus anaerobius, a microbe enriched in colon tumors can selectively adhere to the CRC mucosa in vivo. Due to its surface protein PCWBR2 (putative cell wall binding repeat 2), P. anaerobius promotes tumor growth due to activation of the PI3K–AKT pathway and NF-κB-driven inflammation (72).
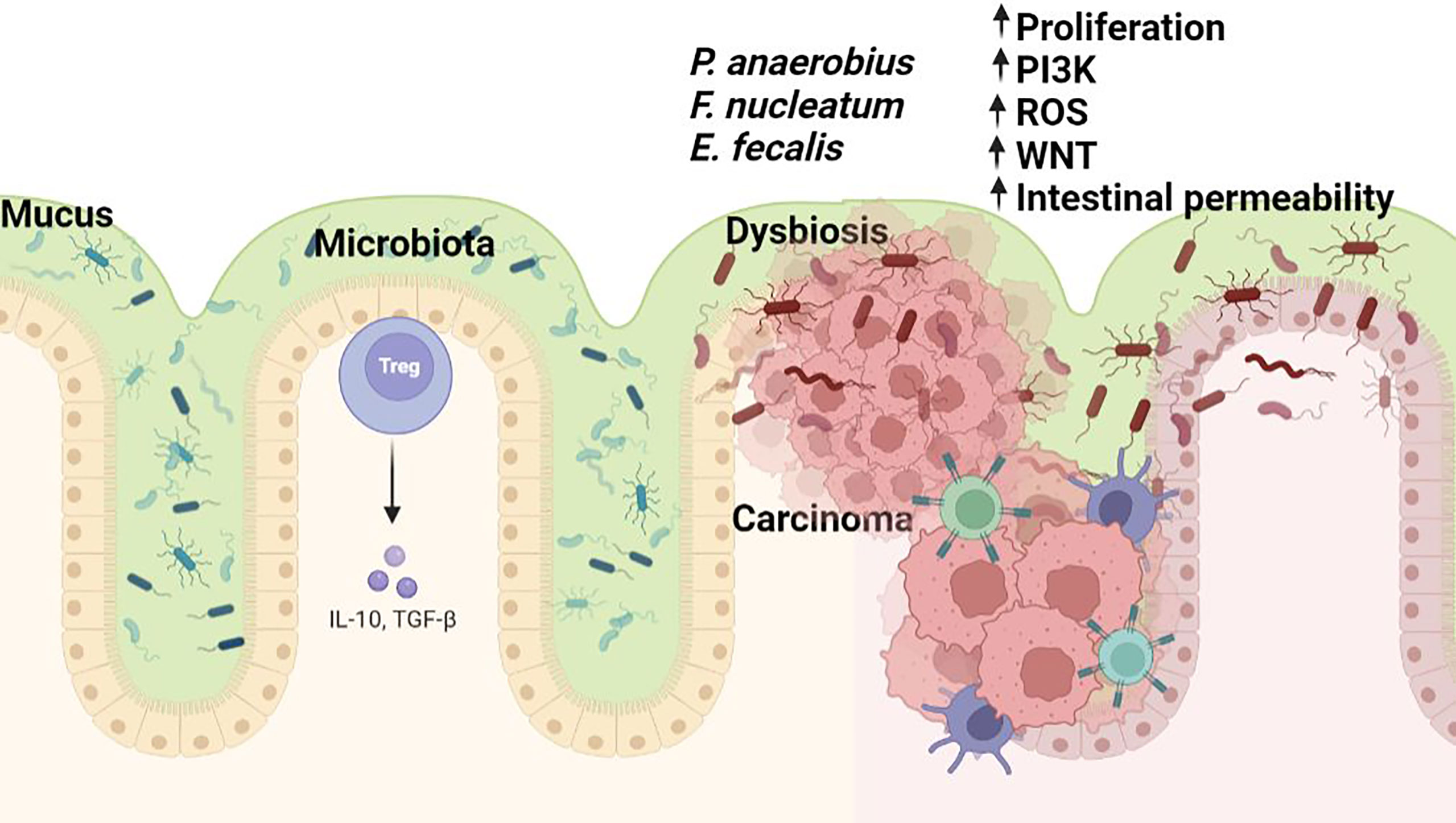
Figure 1 The microbiome in colorectal carcinogenesis. Several bacterial taxa including Fusobacterium sp., Enterococcus sp., and P.anaerobius are commonly associated with colorectal cancer. Dysbiosis hinders the gut barrier function of epithelial tight junctions and the mucus layer favouring exposure of the intestinal epithelium to bacteria and their metabolites (some of which may harbour carcinogenic potential). Bacterial translocation causes enhanced inflammation associated with the production of toxic chemicals or procarcinogen molecules such as reactive oxygen species (ROS) by inflammatory cells (i.e., macrophages). All these changes lead to oxidative and subsequent DNA damage. Figure created in Biorender.com.
Importantly, the general conclusion of various studies is that there is not a unique specific microbe that is responsible for CRC, but a group of bacteria whose detrimental actions surpass those of the beneficial microorganisms.
Therefore, to restore the gut eubiosis and achieve a beneficial modulation of the gut microbiome composition and metabolic activities, probiotics might be used to reduce the risk of CRC development. However, although probiotics and prebiotics have shown success in attenuating CRC and its concomitant effects (73), the intimate mechanisms of action in this type of interventional treatments are unknown (74).
5 Inflammation as an Earlier Driver of Colorectal Tumorigenesis
Chronic inflammation is one of the hallmarks of CRC and was found to be present from the earliest stages of tumor onset.
5.1 Protection Mechanisms Against Gut Inflammation—The Role of Molecular Signaling Crosstalk
Generally, the GI tract has a very good signaling pathway for the rapid activation of anti-inflammatory mechanisms, and the intestinal epithelium is dynamically renewed within a week. This is due to the crypt-progenitor stem cells which can proliferate, differentiate, and are shed at the luminal surface. The intestinal epithelial stem cells generate multiple cell lineages, namely, absorptive enterocytes (80% of the cells), mucus-producing goblet cells (single-cell glands that produce and secrete mucin), enteroendocrine cells, and antimicrobial peptide-producing Paneth cells (75).
The intestinal epithelium provides a dynamic physical barrier that separates the mucosal tissue from pathogens, dietary antigens, and commensal bacteria, and facilitates selective absorption of nutrients, water, and electrolytes. The intestinal epithelial cells (IECs) have specific properties that allow them to manage the complex interaction between the host and the microenvironment, and to maintain the tissue homeostasis. Beside IECs, other types of cells, including immune cells, and the overlaying mucus layer contribute to the protection of the intestinal layer. Damage to the GI barrier leads to homeostasis disruptions, pathological inflammatory responses, and tumorigenesis.
The organization of the mucus layer varies along the length of the colon. Multiple studies examining mucus properties carried out in both mice and humans describe two mucus layers in the colon that include a firm mucus layer adjacent to the epithelium that is devoid of bacteria, which serves as the primary innate defense barrier, with some bacteria colonizing the thin outer layer, and an inner layer which is largely sterile (76, 77). Membrane mucins (hydrophilic branched glycoproteins) provide a safe epithelial cell barrier while playing an important role in signal transduction (78). The distribution of types of mucin differs from one part of gastrointestinal tract to another, and even if MUC5AC is a component of stomach mucus layer (79), it was observed in the distal colon along with MUC2, the major intestinal mucus layer component, during inflammation associated with ulcerative colitis and adenocarcinomas (80, 81).
The mucous layer serves as the outer-most colonic barrier exposed to pathogens and contains mainly mucin2 (MUC2) secreted largely by the goblet cells. Recent research has shown that in some goblet cells (localized at the colonic crypt entrance) there is an endocytosis of LPS and P3CSK4 TRL-ligands that target TRL-MyD88 signaling, which induce ROS, causing the activation of caspase 1 and 11. Subsequently, this process leads to the Ca2+-dependent exocytosis of MUC2 and intercellular signaling connections, prompting the secretion of MUC2 by the adjacent responsive goblet cells (82). It has been shown that MUC2 imprint anti-inflammatory gene markers required for oral tolerance on antigen-presenting cells (APCs). Interfering with the APC and epithelial cell interaction reduces the transfer of goblet cell products to APCs, reducing the induction of mucosal reactions. The terminal differentiation of goblet cells is directly controlled by the transcription factor SAM pointed domain-containing ETS transcription factor (Spdef) (83) and also via a network of transcriptional factors regulated by the Notch, Wnt/β-catenin, PI3-kinase/Akt and bone morphogenetic protein (BMP) signaling pathways known to influence developmental and inflammation pathways (84). Studies have demonstrated that the IL-6 mediated Jak/STAT3 pathway may drive goblet cells differentiation via its downstream PI3-kinase/Akt signal peptide corroborating the previous finding of visible damage of mucosa in IL-6−/− mice (85, 86).
According to Miller, consensus molecular subtype 3 (CMS3) CRC tumors and cell lines are enriched for the expression of goblet cell marker genes. Furthermore, these CMS subtype tumors are mutually exclusive from mucinous adenocarcinoma pathologies (87).
Paneth cells are fully differentiated cells that maintain asepsis of intestinal crypts (88) by secreting the several anti-microbial molecules like defensin-5 (DEFA5) and defensin-6 (DEFA6) and different enzymes, namely, lysozymes and phospholipase A2. Paneth cells are present in chronic non-neoplastic conditions such as inflammatory bowel diseases, and also in neoplastic conditions such as adenoma or carcinoma. The prevalence of Paneth cell differentiation in adenomas varies from 0.2 to 70% (89). In the case of an inflammatory process, Paneth cells occur and tend to linger on after the inflammation was resolved and the crypt structure improved. In the GT, expression of sPLA2-IIA has been localized in Paneth cells of the small intestine, metaplastic Paneth cells of gastric and colonic mucosa, and also columnar epithelial cells of inflammated colonic mucosa (90).
Another defensive barrier is represented by the junctions between epithelial cells, namely, tight junction (TJ), adhesion junction (AJ), desmosomes connection, and gap junction from top to basement, (86, 91–93). The TJ present in the cardinal position is composed of occludin, claudins, junctional adhesion molecule (JAM), and zonula occludens (ZO) and limits the passage of macromolecules and microorganisms (91). Early mutagenic events, including loss of adenomatous polyposis coli (APC) and/or activation of β-catenin signaling, may also alter MUC2 and tight junctional proteins and allow for infiltration of pro-tumorigenic microbial products (76). The AJ is an ancient junctional complex that initiates and maintains epithelial cell–cell contacts and has E-cadherin (CDH1) as key transmembrane protein, which mediates calcium-dependent homotypic intercellular adhesions. On the cytoplasmic face of the AJ, E-cadherin associates with p120 (CTNND1), β-catenin (CTNNB1), and α-catenin (CTNNA1), forming a complex that is anchored to cortical actin filaments (75, 94) (Figure 2). The assembly and stability of TJ and AJ are maintained by a common regulatory mechanism that involves interactions with the cortical actin cytoskeleton. The cytosolic plaque of TJ and AJ contain actin-binding proteins like α-actin, vinculin, ZO family, afadin, and cingulin that anchor the junctional complexes to F-actin bundles. β-Catenin is a member of the cadherin-based cell adhesive complex, which also acts as a transcription factor if the protein is translocated to the nucleus. When it is not bound to E-cadherin and participating in cell-to-cell adhesion, a cytoplasmic degradation complex (consisting of APC, Axin, GSK-3β, and β-catenin) leads to β-catenin phosphorylation and degradation (75). The extracellular region of E-cadherin from the cell surface binds to cadherins present on adjacent cells, whereas its intracellular region contains binding sites which interact with catenin’s and other regulatory proteins. Regulation of the cytoplasmic level of β-catenin is achieved by the opposite action of Wnt and adenomatous polyposis coli (APC)/E-cadherin (Figure 2). The APC protein binds β-catenin to the “Arm” repetitive sequences serving as a negative regulator, while Wnt is a positive regulator of the cytoplasmic level of β-catenin (94, 95).
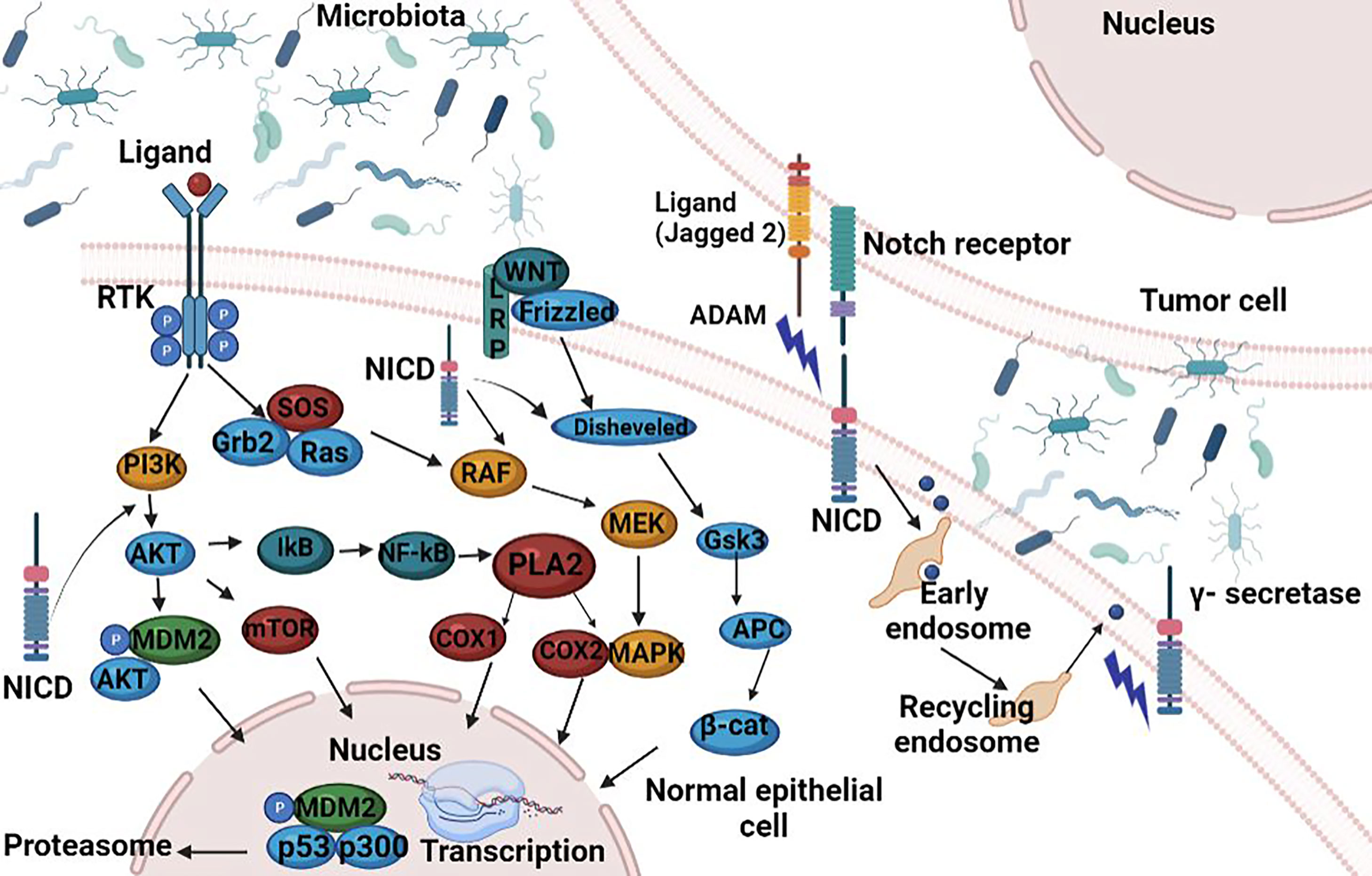
Figure 2 Functional activities of β-catenin. (1) activation of the APC; (2) involvement of the APC in the E-catenin unit (ECCU); (3) activation of the β-catenin degradation site at the C-terminal end of the APC; (4) binding of the APC to the filaments of tubulin; (5) binding of APC to DLG/EB1 proteins. When β-catenin accumulates in the cytosol, the Wnt signalling pathway is inhibited and GSK3β signalling pathway is activated. Active GSK3β simultaneously phosphorylates β-catenin and APC, which it also activates. The APC phosphorylation is possible only if Axin protein is included in the β-catenin-GSK3β-APC assembly also. Once activated, the APC protein is capable to bind free cytoplasmatic β-catenin. APC, “adenomatous polyposis coli”; GSH3β, glycogen synthase kinase 3β; DLG and EB1, tumor suppressor proteins; CRD, cysteine-rich domain; Frzb, Frizzby protein; Frizzled, Wnt protein receptor; Dsh, Dishevelled protein; DLG, protein “human large disc”; EB1, protein EB1. Figure created in Biorender.com.
In target cells, the Wnt cellular signaling pathway has the effect of increasing the cytoplasmic level of β-catenin. The Wnt signaling pathway, known as canonical, is one of the major pathways involved in embryogenesis and homeostasis of colorectal tissue, and therefore in carcinogenesis in cases of abnormal activation (96). The role of canonical Wnt signaling in CRC development has been well documented and is presented in Figure 3. In the absence of Wnt signals, a multiprotein destruction complex including Axin and the APC facilitates phosphorylation of serine residues in the N terminus of cytosolic β-catenin, which leads to its ubiquitination and proteosomal destruction (95).
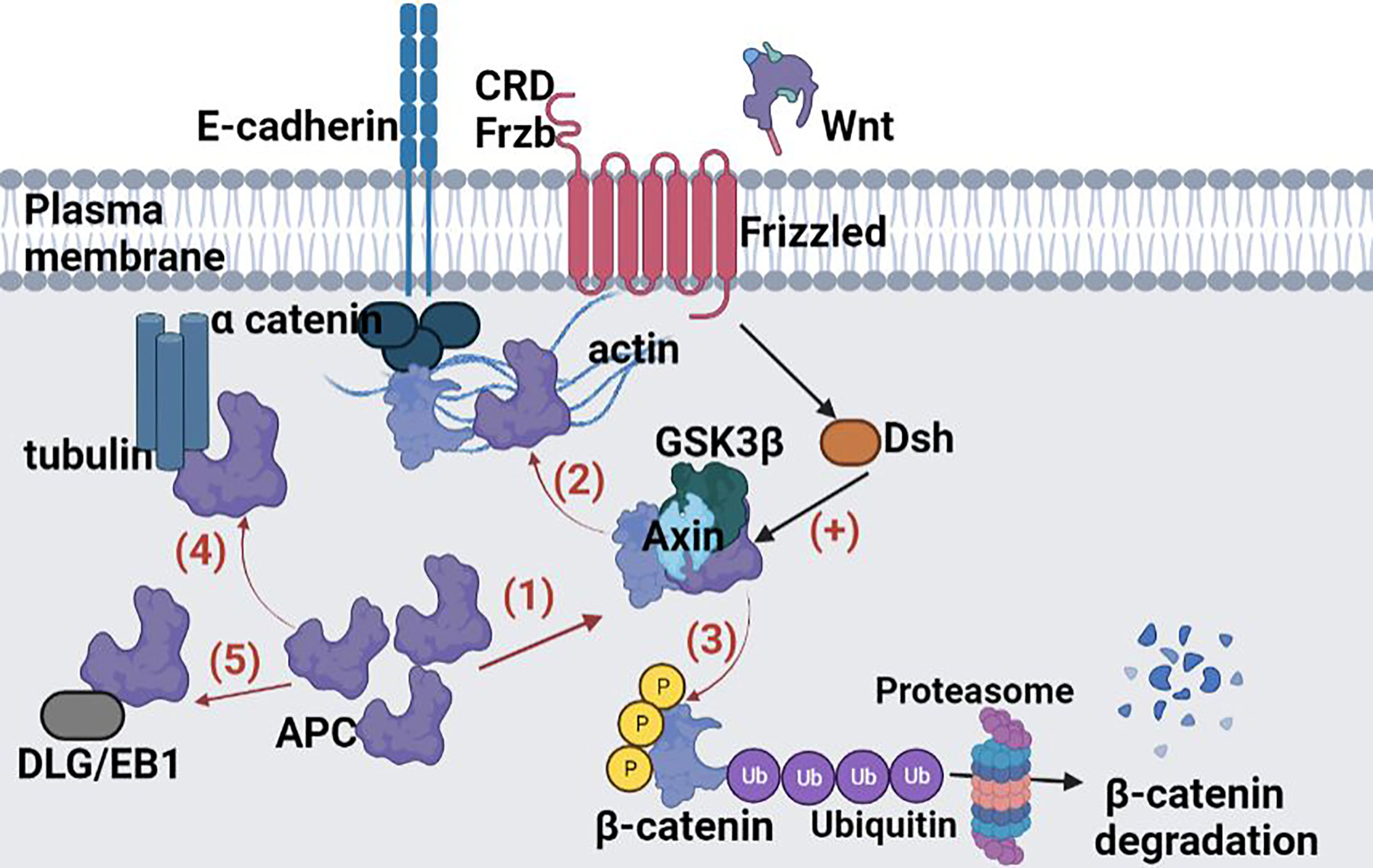
Figure 3 Crosstalk between Notch, Wnt, MAPK and AA signalling pathways. A small increase in cytoplasmic β-catenin levels leads to activation of Wnt gene transcription. The Wnt glycoprotein is synthesized and will be activated by binding to one of the members of the Frizzled family (Frzb)–the Wnt receptor. From the receptor, the signal is transmitted inside the cell to casein kinase II (CKII); it activates and phosphorylates the Disheveled protein (Dsh). The active Disheveled protein is translocated from the cytosol to the cell membrane where, through a signalling process, protein kinase C is phosphorylated, and in turn phosphorylate GSK3β to a N-terminal Ser residue. The effect is the inhibition of GSK3β and finally the β-catenin accumulation in the cytosol. From the cytosol β-catenin is translocated into the nucleus, independent from Lef/Tcf transcription factors translocation. At this level the protein binds Lef/Tcf architectural transcription factors and Wnt gene transcription is induced. Following the interaction with β-catenin, the transcription factors Lef and Tcf can no longer bind to the corresponding sites in the promoter region of some genes and thus the transcriptional process of those genes can take place. In this way Wnt promotes post-transcriptional stabilization and accumulation of β-catenin in the cytosol. Figure created in Biorender.com.
The APC signaling pathway has the opposite effect to the Wnt signaling pathway, leading to decreased cytoplasmic β-catenin levels (Figure 2).
The deregulation of Wnt signaling is the most frequent molecular aberration in CRC, with inactivating mutations in the APC gene occurring in ∼75% of all tumors (22). The two important partners of Wnt family involved in colorectal carcinogenesis are Wnt5a and Wnt11. On the canonical pathway, after binding to its receptor, extracellular Ca2+ initiates the transcription and translation of the canonical Wnt5a in intestinal epithelial cells (97), which upregulates the expression of tumor suppressor 15-PGDH, and further promotes the differentiation of colorectal cancer cells (98). On the noncanonical pathway, Wnt11 enhances the activation of protein kinase C and Ca2+/calmodulin-dependent protein-kinase II (CaMKII), which reduces the E-cadherin-mediated cell–cell interaction contact, and further stimulates proliferation and promotes morphological transformation in the intestinal epithelial cells (99). APC encodes a large scaffolding protein that is part of the AXIN destruction complex, which is necessary for the phosphorylation and degradation of β-catenin (100).
Activating mutations in CTNNB1 (β- catenin), or in other Wnt signaling activators (101) can also hyperactivate Wnt signalling in CRC, as can inactivating mutations in Wnt repressors (102). Alternatively, miRNAs can also modulate Wnt signaling through the repression of pathway components. For example, miRNAs correlate with microsatellite instability (MSI) status (103, 104), tumor location (63, 105), BRAF and KRAS mutation (106), (107) and tumor stage (108).
5.1.1 The Antitumorigenic Activity of Phospholipase A2 Type IIA in CRC
Phospholipase A2 type IIA (sPLA2-IIA) is a 14-kDa enzyme found in several tissues and secretory products and is often referred to as an “inflammatory sPLA”. Its expression is induced by pro-inflammatory cytokines and lipopolysaccharides (LPS), and its activity is associated with inflammation, host defence against bacteria, blood coagulation, and atherosclerosis. Many human cells can secrete PLA2G2A, namely, smooth muscle cells, endothelial cells, macrophages, and Paneth cells. Among others, PLA2G2A is involved in arachidonic acid metabolism, antimicrobial activity, exocytosis of endocrine cells, the release of pro-inflammatory mediators, cell proliferation, and cancer. These functions are mediated by both enzyme catalytic and non-catalytic activities of PLA2-IIA. Functional defects in sPLA2-IIA in tumor cells may interfere with the regulatory mechanisms of tumor growth (94). The plasma concentration of sPLA2-IIA increases dramatically in the plasma of CRC patients’ severe infections and other diseases involving generalized inflammation (109), and approximately 55% of CRC patients exhibit high expression of sPLA2-IIA in their epithelial cytoplasm (110). By our studies we indicate that sPLA2-IIA is over-expressed in Paneth cells in adenomas comparative to malignant colorectal tumours, and its PLA2G2A gene is upregulated in human colon adenomas and is frequently subject of loss-of-heterozygosity (LOH) (111, 112). Recently, our results were confirmed by Schewe et al. (17) who provided a potential mechanism by which sPLA2-IIA, expressed by Paneth cells in the small intestine, supresses colon cancer (113). In their studies, Avoranta et al. observed an overexpression of sPLA2-IIA in most colorectal adenomas, where the expression is disturbed, and sPLA2-IIA protein level is dramatically reduced in malignant colorectal tumours as compared to adenomas. In addition, peritumoral mucosa shows increased expression and content of sPLA2-IIA (114). Elevated expression of sPLA2-IIA inhibited colorectal cancer invasion and metastasis through the Wnt/β-catenin signaling pathway (115) and was found to be associated with improved patient survival (116).
The sPLA2-IIA is quickly released (seconds or minutes) from secretory granules following the stimulation of the cell with Ca2+ ions or with stimulating agents such as thrombin or ionophores. In contrast to this rapid stage, sPLA2-IIA can also be released after a long period of time (from a few hours to a few days) following the action of some cytokines. The role of these cytokines is to modulate gene expression for sPLA2-IIA, which is why the secretion is delayed.
The most important cytokines that induce the expression of sPLA2-IIA are IL-1α, IL-1β, and TNF-α. As presented in Figure 4, these cytokines induce transcription and secretion of sPLA2-IIA through a mechanism involving the MAP-kinase pathway (MAPK).
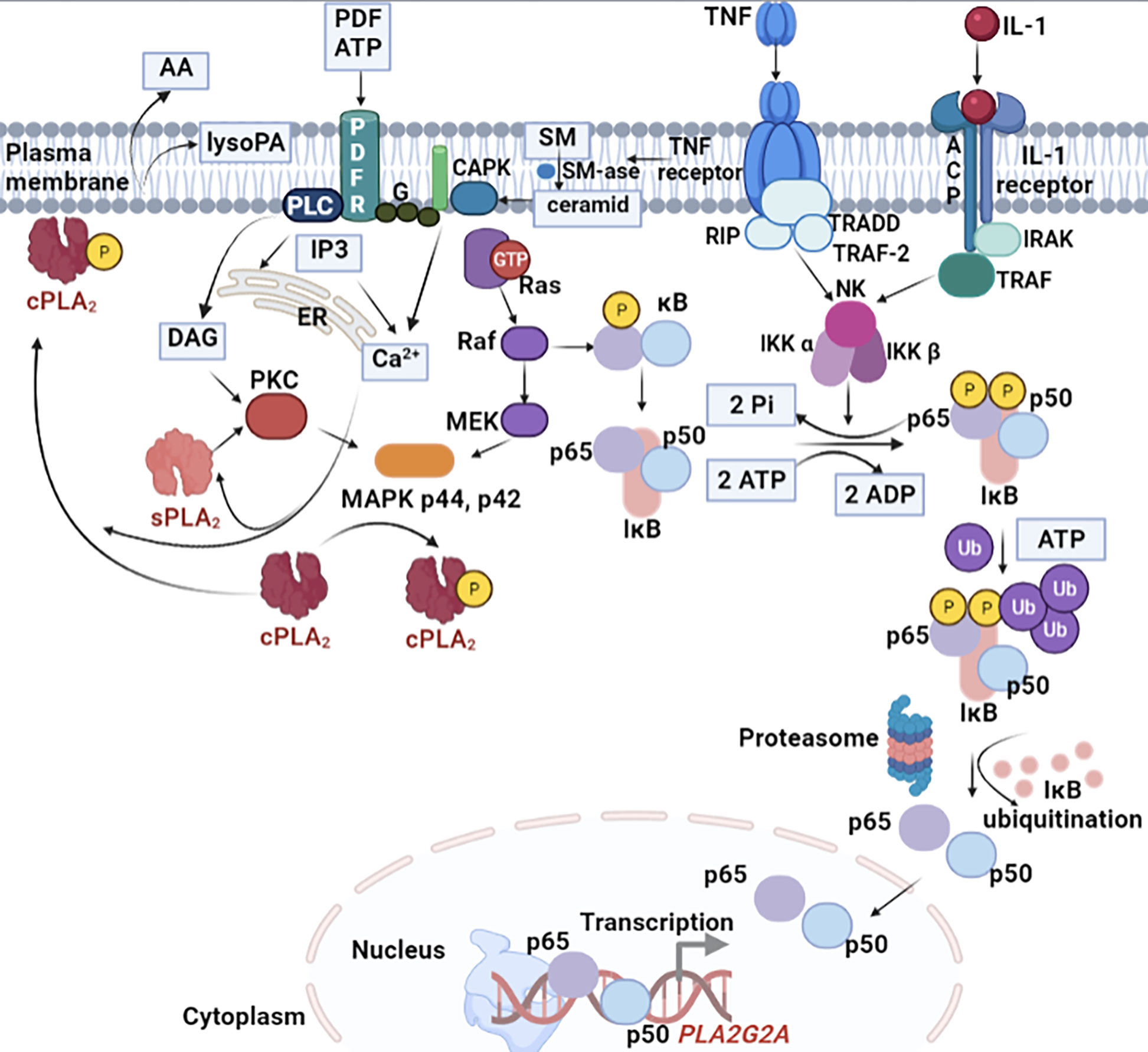
Figure 4 Cellular signalling pathway crosstalk induced by IL-1 for sPLA2-IIA expression. IL-1 induce transcription and secretion of sPLA2-IIA through a mechanism involving the MAPK. A few hours after the enzyme secretion, the signalling pathway is followed by the generation of prostanoids. The induction process is time-dependent and generally occurs after an initial lag period of about 8 h. Both IL-1 and TNF-α stimulate gene transcription and stabilize its RNA. In certain cells, such as for example macrophages, the expression sPLA2-IIA is strongly induced by the two cytokines stimulation under lipopolysaccharides (LPS) action. The MAPK cascade is activated in response to several cytokines, in particular TNF-α and IL-1, and is associated with the activation of nuclear transcriptional factor kB (NF-kB) and several serine/treonin kinases. TNF-α and IL-1 initiates the hydrolysis of sphingomyelin from the plasma membrane. The formed ceramide activates a specific protein kinase (CAPK) on which the MAPK cascade initiation relies on. While the MAPK pathway occurs in the cytosol, the processes that initiate it take place inside the plasma membrane.
Interleukin-6 (IL-6) is a cytokine that recognizes two response elements located in the promoter region of the PLA2G2A gene and induces the expression of the enzyme. The study on hepatomas from the HepG2 cell line revealed that IL-6 induces the expression of both sPLA2-IIA and other acute phase proteins. These observations led the researchers to suggest that sPLA2-IIA is one of the acute phase proteins whose plasma levels increase during the inflammatory response. Il-6 may also induce the expression of sPLA2 type II in human cell lines with invasive gastric cancer.
The most important role of sPLA2-IIA is the release of arahidonic acid (AA) from the structure of membrane phospholipids. The AA concentration inside the cell depends both on the released amount because of the PLA2 action, and on the re-incorporated amount into the structure of the phospholipid following the action of acyltransferase.
5.1.2 The Pro-Inflammatory Activity of Cyclooxygenase-2 in CRC
Cyclooxygenase (COX) enzymes catalyze the conversion of AA, derived from membrane phospholipids by PLA2, into prostanoids which modulate the immune response, blood clotting, and have a role in different pathological conditions, including inflammation.
COX regulates colon carcinoma-induced angiogenesis by two mechanisms: COX-1 regulates angiogenesis in endothelial cells, while COX-2 can modulate production of angiogenic factors by colon cancer cells. While COX-1 is constitutively expressed in many tissues and cell types but in some cases is increased during differentiation, the expression of COX-2 is not usually detectable in normal tissue but is induced by numerous growth factors, hormones, cytokines, and tumor promoters.
COX inhibitors exhibit dramatic antineoplastic activity in several tumor model systems. More than 80% of CRC patients have increased COX-2 levels when compared to adjacent normal tissue. Researchers suggested that COX-2 is expressed in the first steps of colorectal tumorigenesis suggesting an important role of this enzyme in tumor progression. The COX activity is coupled to several terminal synthases that produce five different prostanoids: prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2a (PGF2a), prostaglandin I2/prostacyclin (PGI2), and thromboxane A2 (TXA2), some of them also being involved in colon cancer. They have a pro-inflammatory effect, which stimulates cell proliferation, angiogenesis, and resistance to apoptosis. Among the prostanoids, PGE2 has been proposed as the principal prostanoid promoting tumor growth and survival in CRC. PGE2 is present in the healthy colon but its levels are elevated in CRC and correlate with tumour size and progression. More than that, production of PGE2 is induced by COX-2 which in turn increase further expression of COX-2 in colon cancer cells (117, 118). It was observed that COX-2 is stimulated by pro-inflammatory cytokines (TNF, IL1, IL6) produced by the inflammatory cells (119).
5.1.3 NOTCH1 Signaling—A Key in CRC Progression
The Notch signaling pathway significantly regulates the intestinal enterocyte lineage, activating the hairy and enhancer of split 1 (Hes1) transcription factor, and repressing the basic helix-loop-helix (bHLH) transcription factor mouse atonal homolog 1 (Math1), also known as Atonal homologue 1 (Atoh1). Notch signaling pathway activation disrupts the differentiation of secretory cells with the villi coated primarily with absorptive enterocytes associated with Hes1 activation (120). The NOTCH1 cell signaling mechanism is conserved in most multicellular organisms and functions as a receptor for membrane-bound ligands Jagged1, Jagged2, and Delta1 in the regulation of cell-fate determination. According to Schmalhofer et al. (18), the Jagged1-induced Notch1 signalling activation leads to the inhibition of E-cadherin expression, affecting cell–cell adhesion and the simultaneous increase of N-cadherin and vimentin expressions, and nuclear localization of β-catenin (Figure 2), which in the end induces an invasive and mesenchymal phenotype (121). Notch signaling has been considered as an oncogene involved in the pathogenesis of CRC (122). In a research published in 2019, Lloyd-Lewis postulates the idea that Notch acts as a biological kapellmeister (orchestra conductor), coordinating spatial cues generated by cell flows during morphogenesis, to dictate cell fate decisions at specific developmental times (123). Earlier studies revealed deregulated Notch signaling in several solid human tumors including CRC. The Notch pathway, is a short-range communication system in which contact between a cell expressing a membrane-bound ligand and a cell expressing a transmembrane receptor initiate a regulatory signal which is sent through a cascade of transcriptional regulatory events that affects the expression of a wide number of genes, resulting in remarkable cell-context dependent pleiotropic effects. Notch proteolysis is required for downstream signaling (124).
Notch also acts as a molecular bridge between stem cells and their non-cellular microenvironment. There is a lot of evidence indicating a bidirectional intercellular communication involving Notch signals between tumor cells and stromal cells in some malignancies (125). Alterations of several signaling pathways, which are correlated with Notch canonical pathway, were described in solid tumors associated with growth activation, resistance to apoptosis, angiogenesis, and invasion/metastatic behavior, but nothing was described related to Notch non-canonical pathway.
In macrophages, the Notch pathway can be activated by Notch ligands that are expressed by macrophages and by Notch ligands expressed on epithelial cells, stromal cells at inflammatory sites or intestinal stem cells. Inflammatory cytokines like TNF and IL1β serve as Notch activators: TNF induces expression of Notch1, Notch4, and Jagged2 and also, the NICD nuclear translocation (126), while IL1β, another important proinflammatory cytokine, induce Notch target genes Hes 1 and Hey1 (127) expression.
At the same time, it was shown that NF-kB signaling interacts with the Notch pathway in many pathologies, including cancer (128). Another group of signaling molecules involved in mediating Notch activation are mitogen-activated protein kinases (MAPKs), a family of serin/threonine protein kinases that are key regulators in inflammation (129).
6 Conclusions
An improved approach to cancer biology is expected to explain the reasons for therapeutic failures in the group of patients with early-stage colorectal carcinomas. Colorectal cancer develops because of multiple sequential steps due to the accumulation of genetic alterations including mutations, gene amplification, and epigenetic changes.
To allow survival, evolution has created a robust system of cell-fate regulation that is highly resistant to the loss of one or even a few components. Unfortunately, uncontrolled endogenous and exogenous factors may disturb the regulation system and abnormal cells with abnormal growth mechanisms can develop and modify the normal homeostasis of the body. In CRC specifically, the cellular regulation from the epithelial layer is even more complex due to the presence of microbiota and metabolites resulting from the degradation of nutrients in the daily diet. Therefore, a deeper understanding of the functional roles of the gut microbiome and its interactions with the human host is needed to enable the application of microbiome knowledge to the clinics. As suggested by several authors, imbalances in the normal content of the gut microbiome led to colonization of driver bacteria that induce a chronic inflammation of the colonocytes. This inflammation changes the microenvironment and allows the colonization by passenger bacteria, which may contribute to carcinogenesis process from adenomatosis to tumor formation. So far, there is no universal specific microbiome signature associated with CRC, but the present findings suggest that microbiome could add important information for improving patients’ stratification, providing future leads for preventing CRC tumorigenesis and optimizing CRC therapies through personalized treatment decisions. Analyzing how they communicate with the main signalling pathways involved in carcinogenesis and which is the relationship between different subtypes of tumor cells, microbial signatures and hot response is one of the researchers’ goals for stratifying patients according to the specific crosstalk patterns.
Generally, the GI tract has a very good signalling pathway for a rapid activation of anti-inflammatory mechanisms, and the intestinal epithelium is dynamically renewed within a week. In inflammation processes, the tumor stromal-inflammatory interface represents a dynamic space where growth factors, cytokines, chemokines, enzymes, matrix metalloproteinases, ECM proteins, and metabolic intermediates communicate and facilitate the transition of normal epithelial cells to cancer cells. Further, the cell growth is disrupted, the mutation rate increases, more errors are accumulating in the lipid and glycoprotein biosynthesis, while amino acid supplies decrease. The complex crosstalk between different regulatory pathways allows the tumoral cell to avoid the blocked path and to reroute the signalling network. Moreover, the cell-fate control systems are not only interconnected but also highly redundant, such that if a gene or protein is disabled, another can perform a similar function. Therefore, the system can reset itself to a new status and a new population of cancer cells with new adaptative strategies to the variation of TME conditions is developed. The MSI model (CMS1) became a key biomarker for the early diagnosis and treatment CRC patients, unveiling different microsatellite statuses, compositions and distributions of immune cells and cytokines within TMEs. Unfortunately, the TME versatility hijacked by cancer cells turns the search for a specific drug into a difficult task. Moreover, inhibition of one of specific targets leads to different answers, at different levels, and in different regulatory pathways.
The variability in clinical presentation, aggressiveness, and patterns of treatment failure suggest the necessity of establishing distinct phenotypes-genotypes correlations that can help future treatment strategies. To predict response to therapy, the molecular tests which are currently in use include microsatellite instability (MSI) testing to detect inheritable disease, APC, KRAS, BRAF and other mutational analyses (Table 2). In this respect, for the prevention and early diagnosis of CRC, among other currently explored CRC biomarkers, diagnosis must also focus on specific inflammatory markers such as sPLA2-IIA, COX-2, PGE2, β-catenin, or NOTCH1, presented in Table 3.
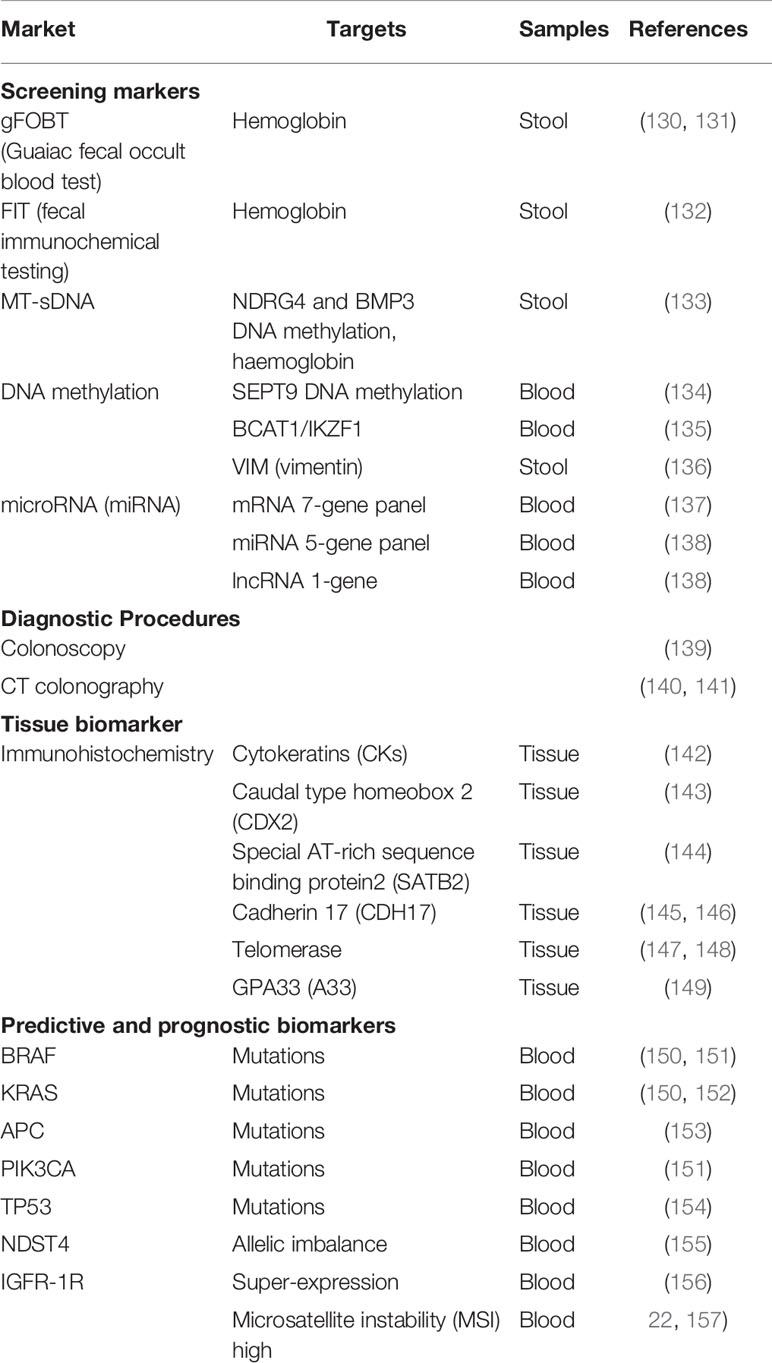
Table 2 Biochemical markers used for colorectal cancer screening, diagnosis, and assessment of treatment efficacy.
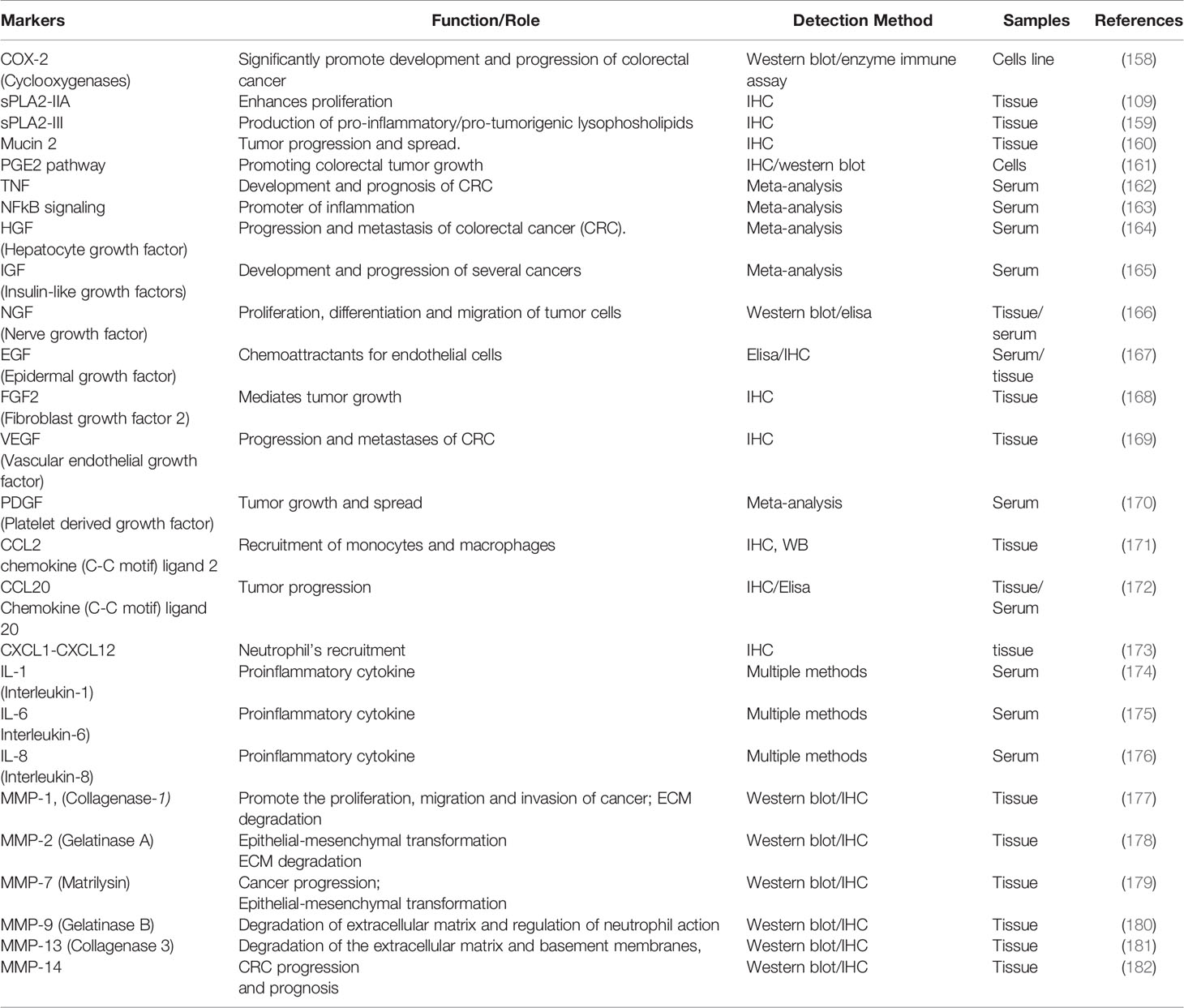
Table 3 Specific inflammatory markers that can be used in early-stage colorectal cancer diagnosis.
In the future, the complex network signaling pathway crosstalk between colonocytes, microbiome, and tumor cells at the earlier-stage sporadic colorectal cancer may be of interest for pharmaceutical and bio-pharmaceutical companies in developing target plasma biomarkers, that can be used in screening programs and for establishing phenotype/genotype signatures. Further on, these could help define combinatory strategies to enhance and improve therapies.
Author Contributions
EI designed the paper structure, performed the literature search, wrote the first draft of the manuscript, and prepared one figure. EI, GG-P, MT, and C-MC wrote sections of the manuscripts. GG analyzed the literature data and prepare the tables. C-MC and GG-P contributed to the design of the manuscript structure. GG-P prepared three pictures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferley J, Siegel R, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Arnold M, Sierra MS, Laversannea M, Soerjomataram I, Jemal A, Bray F. Global Patterns and Trends in Colorectal Cancer Incidence and Mortality. 4. Gut (2017) 66(4):683–91. doi: 10.1136/gutjnl-2015-310912
3. World Health Organization.. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All. Geneva: World Health Organization (2020).
4. Siegel RL, Miller KD, Fedawa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal Cancer Statistics. Cancer J Clin (2017) 67(3):177–93. doi: 10.3322/caac.21395
5. Bray F, Ferlay J, Sorerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
6. Lieberman DA, Weiss DG, Bond JH, Garewal H, Chejfec G. Use of Colonoscopy to Screen Asymptomatic Adults for Colorectal Cancer. Veterans Affairs Cooperative Study Group. N Engl J Med (2000) 343:162–8. doi: 10.1056/NEJM200007203430301
7. Chen Y, Chen Z, Tang Y, Xiao Q. The Involvement of Noncanonical Wnt Signaling in Cancers. Biomed Pharmacother (2021) 133:110946. doi: 10.1016/j.biopha.2020.110946
8. Liu Z, de Vries B, Gerritsen J, Smidt H, Zoetendal EG. Microbiome-Based Stratification to Guide Dietary Interventions to Improve Human Health. Nutr Res (2020) 82:1–10. doi: 10.1016/j.nutres.2020.07.004
9. Sanchez-Alcoholando L, Ramos-Molia B, Otero A, Laborda-Ilanes A, Ordonez R, Medina JA. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy. Cancers (2020) 12(6):1406–34. doi: 10.3390/cancers12061406
10. Chen J, Pitmon E Wang K. Microbiome, Inflammation and Colorectal Cancer. Semin Immunol (2017) 32:43–53. doi: 10.1016/j.smim.2017.09.006
13. Prabhalaran S, Leong J, Petrelli N, Khatri VJ. Precision Medicine in Colorectal Surgery. Surg Oncol Clin N Am (2020) 29:23–4. doi: 10.1016/j.soc.2019.09.001
14. Hsu L, Joen J, Brenner H, Gruber S, Schoen R, Berndt S, et al. A Model to Determine Colorectal Risk Using Common Genetic Susceptibility Loci. Gastroenterology (2015) 148(7):1330–9(e)14. doi: 10.1053/j.gastro.2015.02.010
15. Northcutt M, Shi Z, Ziljstra M, Shah A, Zheng S, Yen E, et al. Polygenic Risk Score is a Predictor of Adenomatous Polyps at Screening Colonoscopy. BMC Gastroenterol (2021) 21:65. doi: 10.1186/s12876-021-01645-4
16. Jeon J, Du M, Schoen RE, Hoffmeister M, Newcomb PA, Berndt SI. Determining Risk of Colorectal Cancer and Strating Age of Screening Based on Lifeltyle, Environmental and Genetic Factors. Gastroenterology (2018) 154(8):2159–64.e19. doi: 10.1053/j.gastro.2018.02.021
17. Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizii T, Sboner A, et al. Personalized In Vitro and In Vivo Cancer Models for Precision Medicine. Cancer Discovery (2017) 7(5):462–77. doi: 10.1158/2159-8290.CD-16-1154
18. Clevers H. Modeling Development and Disease With Organoids. Cell (2016) 165(7):1586–97. doi: 10.1016/j.cell.2016.05.082
19. Zhang Y, Xu H, Frisman D. Genomic Determinants of Somatic Copy Number Alterations Across Human Cancers. Hum Mol Genet (2016) 25(5):1019–30. doi: 10.1093/hmg/ddv623
20. Zack T, Schumacher S, Carter S, Cherniak A, Saksena G, Tabak B, et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat Genet (2013) 45(10):1134–40. doi: 10.1038/ng.2760
21. Zhang L, Shi J, Ouyang J, Zhang R, Tao Y, Yuan D, et al. X-CNV: Genome-Wide Prediction of the Pathogenicity of Copy Number Variations. Genome Med (2021) 13(1):132. doi: 10.1186/s13073-021-00945-4
22. Guineey J, Dienstmann R, Wang X, de Reynies A, Schilicker A, Bot BM, et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat Med (2015) 21(11):1350–6. doi: 10.1038/nm.3967
23. Buikhuisen JY, Torang A, Medema JP. Exploring and Modelling Colon Cancer Inter-Tumour Heterogeneity: Opportunities and Challenges. Oncogenesis (2020) 9:66. doi: 10.1038/s41389-020-00250-6
24. Bijlsma MF, Sadanandam A, Tan P, Vermeulen L. Molecular Subtypes in Cancers of the Gastrointestinal Tract. Nat Rev Gastroenterol Hepatol 14(6):333–42. doi: 10.1038/nrgastro.2017.33
25. Menter DG, Davis JS, Broom BM, Overman MJ, Morris J, Kopetz S. Back to the Colorectal Cancer Consensus Molecular Subtype. Curr Gastroenterol Rep (2019) 21(2):5. doi: 10.1007/s11894-019-0674-9
26. American Cancer Society. Cancer Facts & Figures 2020. Atlanta: American Cancer Society (2020). Available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf.
27. Galon J, Mleenik B, Berger A, Lagoree C, Angell HK, Bindea G, et al. Towards the Introduction of the ”Immunoscore” in the Classification of Malignant Tumours. Clin Cancer Res (2015) 21(7):1543–8. doi: 10.1158/1078-0432.CCR-14-0877
28. Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB. Molecular Profiling of Cancer Patients Enables Personalized Contribution Therapy: The PREDICT Study. Nat Med (2019) 25(5):744–50. doi: 10.1038/s41591-019-0407-5
29. Rodon J, Soria J-C, Berger R, Miller WH, Rubin E, Kugel A, et al. Genomic and Transcriptomic Profiling Expands Precision Cancer Medicine: The WHINTER Trial. Nat Med (2019) 25(5):751–8. doi: 10.1038/s41591-019-0424-4
30. Harris L, Chen A, O'Dwyer P, Flaherty K, Hamilton S, McShane L, et al. Update on the NCI-Molecular Analysis for Therapy Choice. (NCI-MATCH/EAY131) Precision Medicine Trial. Mol Cancer Ther (2017) 17(1):Abstract B080. doi: 10.1158/1535-7163.TARG-17-B080
31. Markowitz SD, Bertagnolli MM. Molecular Origin of Cancer: Molecular Basis of Colorectal Cancer. N Engl J Med (2009) 361(25):2449–60. doi: 10.1056/NEJMra0804588
32. Thanikachalam K, Khan G. Colorectal Cancer and Nutrition. Nutrients (2019) 11(1):164. doi: 10.3390/nu11010164
33. Shivappa N, Godos J, Hebert JR, Wirth MD, Piuri G, Speciani AF, et al. Dietary Inflammatory Index and Colorectal Cancer Risk - A Meta-Analysis. Nutrients (2017) 9(9):1043. doi: 10.3390/nu9091043
34. Veettil SK, Wong TY, Loo YS, Playdom MC, Lai NM, Giovanucci EL, et al. Role of Diet in Colorectal Cancer Incidence Mbrella Review of Meta-Analyses of Prospective Observational Studies. JAMA Netw Open (2021) 4(2):e2037341. doi: 10.1001/jamanetworkopen.2020.37341
35. Ahmad R, Singh JK, Wunnava A, Al-Obeed O, Abdulla M, Srivastana SK, et al. Emerging Trends in Colorectal Cancer: Dysregulated Signaling Pathways. (Review). Int J Mol Med (2021) 47(14):1–25. doi: 10.3892/ijmm.2021.4847
36. Research, World Cancer Research Fund/ American Institute for Cancer. Diet, Nutrition, Physical Activity and Colorectal Cancer.” Dietandcancerreport.Org (2018) (Accessed August 22, 2021). httpp://www.dietcancerreport.org.
37. Coleman OI, Haller D. Dysbiosis of the Intestinal Microbiota and Colorectal Cancer. Colorectal Neoplasia and the Colorectal Microbiome. Chap. 7. In: Colorectal Neoplasia and the Colorectal Microbiome. Amsterdam: Elsvier Inc (2020). p. 135–55. doi: 10.1016/B978-0-12-819672-4.00007-6
38. Wen J, Xu Q, Yuan Y. Single Nucleotide Polymorphisms and Sporadic Colorectal Cancer Susceptibility: A Field Synopsis and Meta-Analysis. Cancer Cell Int (2018) 18:155. doi: 10.1186/s12935-018-0656-2
39. Peltomaki P, Sistonen P, Mecklin JP, Pylkkanen L, Jarvinen H, Simons JW. Evidence Supporting Exclusion of the DCC Gene and a Portion of Chromosome 18q as the Locus for Susceptibility to Hereditary Nonpolyposis Colorectal Carcinoma in Five Kindreds. Cancer Res 51(16):4135–40.
40. Hollstein M, Sidransky D, Vogelstein B, Harris CC. P53 Mutations in Human Cancers. Science (2015) 253(5015):49–53. doi: 10.1126/science.1905840
41. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM. Chromosome 17 Deletions and P53 Gene Mutations in Colorectal Carcinomas. Science (1989) 244(4901):217–21. doi: 10.1126/science.2649981
42. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC Mutations Occur Early During Colorectal Tumorigenesis. Nature (1992) 359(6392):235–7. doi: 10.1038/359235a0
43. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, et al. Identification of FAP Locus Genes From Chromosome 5q21. Science (1991) 25(5020):661–5. doi: 10.1126/science.1651562
44. Aziz MA, Yousef Z, Saleh AM, Mohammad S, Knawy BA, et al. Towards Personalized Medicine of Colorectal Cancer. Crit Rev Oncology/Hematology. (2017) 118:70–8. doi: 10.1016/j.critrevonc.2017.08.007
45. Buikhuisen JY, Torang A, Medema JP. Exploring and Modelling Colon Cancer Inter-Tumour Heterogeneity: Opportunities and Challenges. Oncogenesis (2020) 9:66. doi: 10.1038/s41389-020-00250-6
46. Koulis C, Yap R, Engel R, Jarde T, Wilkins S, Solon G, et al. Personalized Medicine-Current and Emerging Predictive and Prognostic Biomarkers in Colorectal Cancer. Cancer (2020) 12(4):812. doi: 10.3390/cancers12040812
47. Wang W, Kandimalla R, Huang H, Zhu L, Li Y, Gao F, et al. Molecular Subtyping of Colorectal Cancer: Recent Progress, New Challenges and Emerging Opportunities. Semin Cancer Biol (2018) 55:1–16. doi: 10.1016/j.semcancer.2018.05.002
48. Ogino S, Fuchs CS, Giovannucci E. How Many Molecular Subtypes? Implications of the Unique Tumor Principle in Personalized Medicine. Expert Rev Mol Diagn (2012) 12(6):621–8. doi: 10.1586/erm.12.46
49. Mahadevan NR, Zanetti M. Tumor Stress Inside Out: Cell-Extrinsic Effects of the Unfolded Protein Response in Tumor Cells Modulate the Immunological Landscape of the Tumor Microenvironment. J Immunol (2011) 187(9):4403–9. doi: 10.4049/jimmunol.1101531
50. Drak AK, Didier M. Tumor Microenvironment and Noncoding RNAs as Co-Drivers of Epithelial–Mesenchymal Transition and Cancer Metastasis. Dev Dyn (2017) 247(3):2–68. doi: 10.1002/dvdy.24548
51. Meseur D, Vacher S, Alsibai KD, Nicolas A. Pivotal Role of Pervasive Neoplastic and Stromal Cells Reprogramming in Circulating Tumor Cells Dissemination and Metastatic Colonization. Cancer Microenviron (2014) 7(3):95–115. doi: 10.1007/s12307$+14$+158$2
52. Junge B, Hongsheng C, Xuefeng B. Relationship Between Microsatellite Status and Immune Microenvironment of Colorectal Cancer and its Application to Diagnosis and Treatment. J Clin Lab Anal (2021) 35(6):e23810. doi: 10.1002/jcla.23810
53. Schulz M, Salamero-Boix A, Niesel K, Alekseeva T, Sevenich L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front Immunol (2019) 10:1713. doi: 10.3389/fimmu.2019.01713
54. Yinwei C, Zhou J, Wang L. Role and Mechanism of Gut Microbiota in Human Disease. Front Cell Infect Microbiol (2021) 11:625913. doi: 10.3389/fcimb.2021.625913
55. Abhari FM, Pirestani M, Dalimi A. Anti-Amoebic Activity of a Cecropin-Melittin Hybrid Peptide. (CM11) Against Trophozoites of Entamoeba Histolytica. Wien Klin Wochenschr (2019) 131(17-18):427–34. doi: 10.1007/s00508-019-01540-9
56. Guo Y, Wang B, Wang T, Gao L, Yang Z-J, Wang F-F. Biological Characteristics of IL-6 and Related Intestinal Diseases. Int J Biol Sci (2021) 17(1):204–19. doi: 10.7150/ijbs.51362
57. McCullough ML, Patel AV, Kushi LH, Patel R, Willett WC, Doyle C, et al. Following Cancer Prevention Guidelines Reduces Risk of Cancer, Cardiovascular Disease, and All-Cause Mortality. Cancer Epidemiol Biomarkers Prev (2011) 20(7):1089–109. doi: 10.1158/1055-9965.EPI-10-1173
58. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science (2015) 350(6264):1084–9. doi: 10.1126/science.aac4255
59. Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, et al. The Gut Microbiome Modulates Colon Tumorigenesis. mBio (2013) 4(6):e00692–13. doi: 10.1128/mBio.00692-13
60. Aries V, Crowter JS, Drasar BS, Hill MJ, Williams RE. Bacteria and the Aetiology of Cancer of the Large Bowel. Gut (1969) 10(5):334–35. doi: 10.1136/gut.10.5.334
61. Hill MJ, Drasar BS, Hawksworth G, Aries V, Crowther JS, Williams RE. Bacteria and Aetiology of Cancer of Large Bowel. Lancet (1971) 1(7690):95–100. doi: 10.1016/s0140-6736(71)90837-3
62. Chen D, Jin D, Huang S, Wu J, Xu M, Liu T, et al. Clostridium Butyricum, a Butyrate-Producing Probiotic, Inhibits Intestinal Tumor Development Through Modulating Wnt Signaling and Gut Microbiota. Cancer Lett (2020) 469:456–67. doi: 10.1016/j.canlet.2019.11.019
63. Schee K, Lorenz S, Molton Worren M, Gunther CC, Holden M, Hovig E, et al. Deep Sequencing the microRNA Transcriptome in Colorectal Cancer. PloS One (2013) 8(6):e66165. doi: 10.1371/journal.pone.0066165
64. Sanapareddy N, Legee RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, et al. Increased Rectal Microbial Richness is Associated With the Presence of Colorectal Adenomas in Humans. ISME (2012) 6(10):1858–68. doi: 10.1038/ismej.2012.43
65. Brim H, Yooseph S, Zoetendal EG, Lee E, Tarralbo M, Laiyemo AO, et al. Microbiome Analysis of Stool Samples From African Americans With Colon Polyps. PloS One (2013) 8(12):e81352. doi: 10.1371/journal.pone.0081352
66. Wei H, Dong L, Wang T, Zhang M, Hua W, Zhang C, et al. Structural Shifts of Gut Microbiota as Surrogate Endpoints for Monitoring Host Health Changes Induced by Carcinogen Exposure. FEMS Microbiol Ecol (2010) 73(3):577–86. doi: 10.1111/j.1574-6941.2010.00924.x
67. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat Med (2019) 25(6):96876. doi: 10.1038/s41591-019-0458-7
68. Yakida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat Med (2019) 25(6):968–76. doi: 10.1038/s41591-019-0458-7
69. Cheng Y, Ling Z, Li L. The Intestinal Microbiota and Colorectal Cancer. Front Immunol (2020) 11:615056. doi: 10.3389/fimmu.2020.615056
70. Si H, Yang Q, Hu H, Ding C, Wang H, Lin X. Colorectal Cancer Occurrence and Treatment Basedon Changes in Intestinal flora. Semin Cancer Biol (2021) 70:3–10. doi: 10.1016/j.semcancer.2020.05.004
71. Rubinstein MR, Baik KE, Lagana SM, Han RP, Raab WJ, Sahoo D, et al. Fusobacterium Nucleatum Promotes Colorectal Cancer by Inducing Wnt/β-Catenin Modulator Annexin A1. EMBO Rep (2021) 20(4):e47683. doi: 10.15252/embr.201847638
72. Silva M, Brunner V, Tshurtschenthaler M. Microbiota and Colorectal Cancer: From Gut to Bedside. (2021) 12:. doi: 10.3389/fphar.2021.760280
73. Compare D, Nardone G. The Bacteria-Hypothesis of Colorectal Cancer: Pathogenetic and Therapeutic Implications. Transl Gastrointest Cancer (2013) 3(1):44–53. doi: 10.3978/j.issn.2224-4778.2013.05.37
74. Kuugbee ED, Shang X, Ganallat Y, Bamba D, Awadasseid A, Suliman MA, et al. Structural Change in Microbiota by a Probiotic Cocktail Enhances the Gut Barrier and Reduces Cancer via TLR2 Signaling in a Rat Model of Colon. Cancer Dig Dis Sci (2016) 61(10):2908–20. doi: 10.1007/s10620-016-4238-7
75. Luissint A-C, Parkos CA, Nusrat A. Inflammation and the Intestinal Barrier: Leukocyte–Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology (2016) 151(4):616–32. doi: 10.1053/j.gastro.2016.07.008
76. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Juach D, et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature (2012) 8(491):254–8. doi: 10.1038/nature11465
77. Zhang M, Chenchen W. The Relationship Between Intestinal Goblet Cells and the Immune Response. Biosci Rep (2020) 40(10):BSR20201471. doi: 10.1042/BSR20201471
78. Bergestrom KS, Xia L. Mucin Type O-Glycans and Their Roles in Intestinal Homeostasis. Glycobiology (2013) 3(9):1026–37. doi: 10.1093/glycob/cwt045
79. Nordman H, Davies JR, Lindell G, de Bolos C, Real F, Carlstedt I. Gastric MUC5AC and MUC6 are Large Oligomeric Mucins That Differ in Size, Glycosylation and Tissue Distribution. Biochem J (2002) 364:191–200. doi: 10.1042/bj3640191
80. Forgue-Lafitte ME, Fabiani B, Levy PP, Maurin N, Flejou JF, Bara J. Abnormal Expression of M1/MUC5AC Mucin in Distal Colon of Patients With Diverticulitis, Ulcerative Colitis and Cancer. Int J Cancer (2007) 121(7):1543–49. doi: 10.1002/ijc.22865
81. Herath M, Hosie S, Bornstein JC, Franks AE, Hill-Yardin EL. The Role of the Gastrointestinal Mucus System in Intestinal Homeostasis: Implications for Neurological Disorders. Front Cell Infect Microbiol (2020) 10:248(248):248. doi: 10.3389/fcimb.2020.00248
82. Songwei Y, Yu M. Role of Goblet Cells in Intestinal Barrier and Mucosal Immunity. J Inflammation Res (2021) 14:3171–83. doi: 10.2147/JIR.S318327
83. Noah TK, Kazanjian A, Whitesett J, Shroyer NF. SAM Pointed Domain ETS Factor. (SPDEF) Regulates Terminal Differentiation and Maturation of Intestinal Goblet Cells. Exp Cell Res (2010) 316(3):452–65. doi: 10.1016/j.yexcr.2009.09.020
84. van de Flier LG, Clevers H. Stem Cells, Self-Renewal, and Differentiation in the Intestinal Epithelium. Annu Rev Physiol (2009) 71(1):241–60. doi: 10.1146/annurev.physiol.010908
85. Kuhn KA, Schulz HM, Regner EH, Severs EL, Hendrickson JD, Mehta G, et al. Bacteroidales Recruit IL-6-Producing Intraepithelial Lymphocytes in the Colon to Promote Barrier Integrity. Mucosal Immunol (2018) 11(2):357–68. doi: 10.1038/mi.2017.55
86. Swierczynski M, Szymaszkiewicz A, Fichna J, Zielinska M. New Insights Into Molecular Pathways in Colorectal Cancer: Adiponectin, Interleukin-6 and Opioid Signaling. BBA - Rev Cancer (2021) 1875:188460. doi: 10.1016/j.bbcan.2020.188460
87. Millers S, Ghobashi A, O'Hagan HM. Consensus Molecular Subtyping of Colorectal Cancers is Influenced by Goblet Cell Content. Cancer Genet (2021) 254-255:34–9. doi: 10.1016/j.cancergen.2021.01.009
88. Bevins C, Salzman NH. Paneth Cells, Antimicrobial Peptides and Maintenance of Intestinal Homeostasis. Nat Rev Microbiol (2011) 9(5):356–68.
89. Nihed A, Soumaya M, Atika B, Ilhem BJ, Atef BA, Fehmi H, et al. Paneth Cell Adenocarcinoma of the Colon: A Rare Entity. Int J Surg Case Rep (2019) 65:313–6. doi: 10.1016/j.ijscr.2019.10.071
90. Peng Z, Chang Y, Fan J, Ji W, Su C. Phospholipase A2 Superfamily in Cancer. Cancer Lett (2021) 497:165–77. doi: 10.1016/j.canlet.2020.10.021
91. Coyne CB, Bergelson JM. CAR: A Virus Receptor Within the Tight Junction. Adv Drug Delivery Rev (2005) 57(6):869–82. doi: 10.1016/j.addr.2005.01.007
92. Nagai M, Yaoita E, Kuwano R, Nameta M, Ohshiro K, Isome M, et al. Coxsackievirus and Adenovirus Receptor, a Tight Junction Membrane Protein, is Expressed in Glomerular Podocytes in the Kidney. Lab Invest (2003) 83(6):901–11. doi: 10.1097/01.lab.0000073307.82991.cc
93. Xu Z, Waeckerlin R, Urbanowski MD, van Marle G, Hobman TC. West Nile Virus Infection Causes Endocytosis of a Specific Subset of Tight Junction Membrane Proteins. PloS One (2012) 7(5):e37886. doi: 10.1371/journal.pone.0037886
94. Ionica E, Tica M, Tica V, Naumescu A, Uta M, Vlaicu O. Genotype-Phenotype Disturbances of Some Biomarkers in Colorectal Cancer. In: Cooper DN, Chen J-M, editors. Mutations in Human Genetic Disease. London: InTech (2012). p. 1–28. doi: 10.5772/WILL-BE-SET-BY-INTECH
95. Tian X, Liu Z, Niu B, Zhang J, Tan TK, Lee SR, et al. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J BioMed Biotechnol (2011) 2011:567305. doi: 10.1155/2011/567305
96. Gala MK, Chan AT. Molecular Pathways: Aspirin and Wnt Signaling—A Molecularly Targeted Approach to Cancer Prevention and Treatment. Clin Cancer Res (2015) 21(7):1543–8. doi: 10.1158/1078-0432.CCR-14-0877
97. MacLeod RJ, Hayes M, Pachevo I. Wnt5a Secretion Stimulated by the Extracellular Calcium-Sensing Receptor Inhibits Defective Wnt Signaling in Colon Cancer Cells. Am J Physiol Gastrointest Liver Physiol (2007) 293(1):G403–11. doi: 10.1152/ajpgi.00119.2007
98. Mehdawi LN, Prasad CP, Ehrnstrom R, Andresson T, Sjolander A. Non-Canonical WNT5A Signaling Up-Regulates the Expression of the Tumor Suppressor 15-PGDH and Induces Differentiation of Colon Cancer Cells. Mol Oncol (2016) 10(9):1415–29. doi: 10.1016/j.molonc.2016.07.011
99. Ouko L, Ziegel TR, Yang VW, Eisenberg LM, Gu LH. Wnt11 Signaling Promotes Proliferation, Transformation, and Migration of IEC6 Intestinal Epithelial Cells. J Biol Chem (2004) 279(5):26707–15. doi: 10.1074/jbc.M402877200
100. MacDonald BT, Tamai K, He X. Wnt/beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev Cell (2009) 17(1):9–26. doi: 10.1016/j.devcel.2009.06.016
101. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature (2012) 488:660–4. doi: 10.1038/nature11282
102. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour Suppressor RNF43 is a Stem-Cell E3 Ligase That Induces Endocytosis of Wnt Receptors. Nature (2012) 488:665–9. doi: 10.1038/nature11308
103. Yamamoto H, Imai K. Microsatellite Instability: An Update. Arch Toxicol (2015) 89(6):899–921. doi: 10.1007/s00204-015-1474-0
104. Slattery ML, Wolff E, Hoffman MD, Pellatt DF, Milash B, Wolff RK. MicroRNAs and Colon and Rectal Cancer: Differential Expression by Tumor Location and Subtype. Genes Chromosom Cancer (2011) 50(3):196–206. doi: 10.1002/gcc.20844
105. Shao S, Neely BA, Kao TC, Eckhaus J, Bourgeois J, Brooks J. Proteomic Profiling of Serial Prediagnostic Serum Samples for Early Detection of Colon Cancer in the U.S Military. Cancer Epidemiol Biomarkers Prev (2017) 26(5):711–8. doi: 10.1158/1055-9965.EPI-16-0732
106. Slattery ML, Herrick JS, Mullany LE, Wolf E, Hoffman MD, Pellatt DF, et al. Colorectal Tumor Molecular Phenotype and miRNA: Expression Profiles and Prognosis. Mod Pathol (2016) 29(8):915–27. doi: 10.1038/modpathol.2016.73
107. Nosho K, Igarashi H, Nojima M, Ito M, Maruyama R, Yashii S, et al. Association of microRNA-31 With BRAF Mutation, Colorectal Cancer Survival and Serrated Pathway. Carcinogenesis (2014) 35(4):776–83. doi: 10.1093/carcin/bgt374
108. Rohr C, Kerick M, Kuhn A, Kashofer K, Timmermann B, Daskalaki A, et al. High- Throughput miRNA and mRNA Sequencing of Paired Colorectal Normal, Tumor and Metastasis Tissues and Bioinformatic Modeling of miRNA-1 Therapeutic Applications. PloS One (2013) 8(7):e67461. doi: 10.1371/journal.pone.0067461
109. Yamashita S, Ogawa M, Sakamato K, Abe T, Arakawa H, Yamashita J. Elevation of Serum Group II Phospholipase A2 Levels in Patients With Advanced Cancer. Clin Chim Acta (1994) 228(2):91–9. doi: 10.1016/0009-8981(94)90280-1
110. Edhemovic`s I, Snoi M, Kljun A, Golouh R. Immunohistochemical Localization of Group II Phospholipase A2 in the Tumours and Mucosa of the Colon and Rectum. Eur J Surg Oncol (2001) 27(6):545–8. doi: 10.1053/ejso.2001.1134
111. Mihalcea A, Tica V, Georgescu SE, Tesio C, Dinischiotu A, Condac E, et al. Allelic Imbalance on Chromosomes 1 and 5 in Colorectal Carcinoma. In: Gruev B, Nikolova și M, Donev A, editors. Balkan Scientific Conference of Biology. Plovdiv: Plovdiv University Press (2005). p. 568–75.
112. Mihalcea A, Condac E, Tica V, Gerogescu SE, Vrabie CD, Copaescu C, et al. Analyse of Allelic Modification on Patients With Colorectal Cancer at the Chromosome 1 Level. Bull Anal Soc Nat Biol Cell (2005) 10(5):341–44.
113. Schewe M, Franken PF, Sacchetti A, Schmitt M, Joosten R, Bottcher R, et al. Secreted Phospholipases A2 are Intestinal Stem Cell Niche Factors With Distinct Roles in Homeostasis, Inflammation, and Cancer. Cell Stem Cell (2016) 19:38–51. doi: 10.1016/j.stem.2016.05.023
114. Avoranta T, Sundstrom J, Korkeila E, Syrjanen K, Pyrhonen S, Laine J. The Expression and Distribution of Group IIA Phospholipase A2 in Human Colorectal Tumours. Virchows Arch (2010) 457(6):665–7. doi: 10.1007/s00428-010-0992-7
115. Ganesan K, Ivanova T, Wu Y, Rajasegaran V, Wu J, Lee MH, et al. Inhibition of Gastric Cancer Invasion and Metastasis by PLA2G2A, a Novel Beta-Catenin/TCF Target Gene. Cancer Res (2008) 68(11):4277–86. doi: 10.1158/0008-5472.CAN-07-6517
116. Wang X, Huang CJ, Yu GZ, Wang JJ, Wang R, Li YM, et al. Expression of Group IIA Phospholipase A2 is an Independent Predictor of Favorable Outcome for Patients With Gastric Cancer. Hum Pathol (2013) 44(10):2020–27. doi: 10.1016/j.humpath.2013.01.027
117. Hidalgo-Estevez AM, Stamatakis K, Jimenez-Martinez M, Lopez-Perez R, Fresno M. Cyclooxygenase 2-Regulated Genes an Alternative Avenue to the Development of New Therapeutic Drug for Colorectal Cancer. Front Oncol (2020) 11:533:533. doi: 10.3389/fphar.2020.00533
118. Karpisheh V, Nikkhoo A, Hojihat-Farsangi M, Namdar A, Azizi G, Ghalamfarsa G. Prostaglandin E2 as a Potent Therapeutic Target for Treatment of Colon Cancer. Prostaglandins Other Lipid Mediat. (2019) 144:106338. doi: 10.1016/j.prostaglandins.2019.106338
119. Garcia-Albeniz X, Chan AT. Aspirin for the Prevention of Colorectal Cancer. Best Pract Res Clin Gastroenterol (2011) 25(4-5):461–72. doi: 10.1016/j.bpg.2011.10.015
120. Tyagi A, Sharma AK, Damodaran C. A Review on Notch Signaling and Colorectal Cancer. Cells (2020) 9:1549. doi: 10.3390/cells9061549
121. Schmalhofer O, Simone B. Thomas B. E-Cadherin, β-Catenin, and ZEB1 in Malignant Progression of Cancer. Cancer Metastasis Rev (2009) 28(1-2):151–66. doi: 10.1007/s10555-008-9179-y
122. Shang Y, Smith S, Hu X. Role of Notch Signaling in Regulating Innate Immunity and Inflammation in Healthand Disease. Protein Cell (2016) 7(3):159–74. doi: 10.1007/s13238-016-0250-0
123. Lloyd-Lewis B, Mourikis P, Fre S. Notch Signalling: Sensor and Instructor of the Microenvironment to Coordinate Cell Fate and Organ Morphogenesis. Curr Opin Cell Biol (2019) 61:16–23. doi: 10.1016/j.ceb.2019.06.003
124. Zhang P, Yang Y, Nolo R, Zweidler-McKay PA, Hughes DPM. Regulation of NOTCH Signaling by Reciprocal Inhibition of HES1 and Deltex 1 and its Role in Osteosarcoma Invasiveness. Oncogene (2010) 29(20):2916–26. doi: 10.1038/onc.2010.62
125. Dotto PG. Crosstalk of Notch With P53 and P63 in Cancer Growth Control. Nature (2009) 9(8):587–95. doi: 10.1038/nrc2675
126. Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T, et al. Induction of Notch Signaling by Tumor Necrosis Factor in Rheumatoid Synovial fibroblasts. Oncogene (2003) 22:7796–803. doi: 10.1038/sj.onc.1206965
127. Maniati E, Bossard M, Cook N, Candido JB, Emma-Shahri N, Nedospasov S, et al. Crosstalk Between the Canonical NF-kappaB and Notch Signaling Pathways Inhibits Paragamma Expression and Promotes Pancreatic Cancer Progression in Mice. J Clin Invest (2011) 121(12):4685–99. doi: 10.1172/JCI45797
128. Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 Pathway Sustains NF-kappaB Activation Through CYLD Repression in T Cell Leukemia. Cancer Cell (2010) 18(3):268–81. doi: 10.1016/j.ccr.2010.08.006
129. Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, et al. Integrated Regulation of Toll-Like Receptor Responses by Notch and Interferon-Gamma Pathways. Immunity (2008) 29(5):691–703. doi: 10.1016/j.immuni.2008.08.016
130. Zhu MM, Xu XT, Nie F, Tong JL, Xiao SD, Ran ZH. Comparison of Immunochemical and Guaiac-Based Fecal Occult Blood Test in Screening and Surveillance for Advanced Colorectal Neoplasms: A Meta-Analysis. J Dig Dis (2010) 11(3):148–60. doi: 10.1111/j.1751-2980.2010.00430.x
131. Guittet L, Bouvier V, Mariotte N, Valee JP, Levillain R, Tichet J, et al. Comparison of a Guaiac and an Immunochemical Faecal Occult Blood Test for the Detection of Colonic Lesions According to Lesion Type and Location. Br J Cancer (2009) 100(8):1230–35. doi: 10.1038/sj.bjc.6604996
132. Lee JK, Liles EG, Bent S, Levin TR, Corley DA. Accuracy of Fecal Immunochemical Tests for Colorectal Cancer: Systematic Review and Meta-Analysis. Ann Intern Med (2014) 160(3):171–206. doi: 10.7326/M13-1484
133. Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N Engl J Med (2014) 370(14):1287–97. doi: 10.1056/NEJMoa1311194
134. Payne SR. From Discovery to the Clinic: The Novel DNA Methylation Biomarker Msept9 for the Detection of Colorectal Cancer in Blood. Epigenomics (2010) 2(4):575–85. doi: 10.2217/epi.10.35
135. Pedersen SK, Baker RT, McEvoy A, Murray DH, Thomas M, Molloy PL, et al. A Two-Gene Blood Test for Methylated DNA Sensitive for Colorectal Cancer. PloS One (2015) 10(4):e0125041. doi: 10.1371/journal.pone.0125041
136. Ned RM, Melillo S, Marrone M. Fecal DNA Testing for Colorectal Cancer Screening: The ColoSure Test. PloS Curr (2011) 3:RRN1220. doi: 10.1371/currents.RRN1220
137. Marshall KW, Mohr S, Khettabi FE, Nossovo N, Chao S, Bao W, et al. A Blood-Based Biomarker Panel for Stratifying Current Risk for Colorectal Cancer. Int J Cancer (2010) 126(5):1177–86. doi: 10.1002/ijc.24910
138. Ganepola GA, Nizin J, Rutledge JR, Chang DH. Use of Blood-Based Biomarkers for Early Diagnosis and Surveillance of Colorectal Cancer. World J Gastrointest Oncol (2014) 6:83–97. doi: 10.4251/wjgo.v6.i4.83
139. Nguyen MT, Weinberg DS. The Impact of Colonoscopy on Colorectal Cancer Incidence and Mortality. In: de Miskovitz P, editor. Colonoscopy. London (2011). p. 1–21. doi: 10.5772/19254
140. Johnson CD, Chen MH, Toledano AY, Heiken JP, Dachman A, Kuo MD, et al. Accuracy of CT Colonography for Detection of Large Adenomas and Cancers. N Engl J Med (2008) 359(12):1207–17. doi: 10.1056/NEJMoa08009961207-17
141. Pickhardt PJ, Choi JR, Hwang I, Butler JA, Puckett ML, Hildebrandt HA, et al. Computed Tomographic Virtual Colonoscopy to Screen for Colorectal Neoplasia in Asymptomatic Adults. N Engl J Med (2013) 349(23):2191–200. doi: 10.1056/NEJMoa031618
142. Bayrak R, Yenidünya S, Haltas H. Cytokeratin 7 and Cytokeratin 20 Expression in Colorectal Adenocarcinomas. Pathol Res Pract (2011) 207(3):156–60. doi: 10.1016/j.prp.2010.12.005
143. Werling RW, Yaziji H, Bacchi CE, Gown AM. CDX2, a Highly Sensitive and Specific Marker of Adenocarcinomas of Intestinal Origin: An Immunohistochemical Survey of 476 Primary and Metastatic Carcinomas. Am J Surg Pathol (2003) 27(3):303–10. doi: 10.1097/00000478-200303000-00003
144. Dragomir A, de Wit M, Johansson C, Uhlen M, Pontén F. The Role of SATB2 as a Diagnostic Marker for Tumors of Colorectal Origin: Results of a Pathology-Based Clinical Prospective Study. Am J Clin Pathol (2014) 141(5):630–8. doi: 10.1309/AJCPWW2URZ9JKQJU
145. Su MC, Yuan RH, Lin CY, Jeng YM. Cadherin-17 is a Useful Diagnostic Marker for Adenocarcinomas of the Digestive System. Mod Pathol (2008) 21(11):1379–86. doi: 10.1038/modpathol.2008.107
146. Lin F, Shi J, Zhu S, Li A, Chen T, Wang HL, et al. Cadherin-17 and SATB2 are Sensitive and Specific Immunomarkers for Medullary Carcinoma of the Large Intestine. Arch Pathol Lab Med (2014) 138(8):1015–26. doi: 10.5858/arpa.2013-0452-OA
147. Shay JW, Zou Y, Hiyama E, Wright WE. Telomerase and Cancer. Hum Mol Genet (2001) 10(7):677–85. doi: 10.1093/hmg/10.7.677
148. Roig AI, Wright WE, Shay JW. Is Telomerase a Novel Target for Metastatic Colon Cancer? Curr Colorectal Cancer Rep (2009) 54:203–8. doi: 10.1007/s11888-009-0028-3
149. Wong NACS, Adamczyk LA, Evans S, Cullen J, Oniscu A, Oien KA. A33 Shows Similar Sensitivity to But is More Specific Than CDX2 as an Immunomarker of Colorectal Carcinoma. Histopathology (2017) 71(1):34–41. doi: 10.1111/his.13194
150. Febbo PG, Ladanyi M, Aldape KD, De Marzo AM, Hammond ME, Hayes DF, et al. NCCN Task Force Report: Evaluating the Clinical Utility of Tumor Markers in Oncology. J Natl Compr Cancer Netw (2011) 9(5):S1–32. doi: 10.6004/jnccn.2011.0137
151. Schmoll HJ, Cutsem EV, Stein A, Valentini V, Glimelius B, Haustermans K, et al. ESMO Consensus Guidelines for Management of Patients With Colon and Rectal Cancer. A Personalized Approach to Clinical Decision Making. Ann Oncol (2012) 23(10):2479–516. doi: 10.1093/annonc/mds236
152. Locker GY, Hamilton S, Harris J, Jessup JM, Kemeny N, Macdonald JS, et al. ASCO 2006 Update of Recommendations for the Use of Tumor Markers in Gastrointestinal Cancer. J Clin Oncol (2006) 24(33):5313–27. doi: 10.1200/JCO.2006.08.2644
153. Liang TJ, Wang HX, Zheng YY, Cao YQ, Wu X, Zhou X, et al. APC Hypermethylation for Early Diagnosis of Colorectal Cancer: A Meta-Analysis and Literature Review. Oncotarget (2017) 8(28):46468–79. doi: 10.18632/oncotarget.17576
154. Borresen-Dale AL, Lothe RA, Meling GI, Hainaut P, Rognum OT, Skovlund E. TP53 and Long-Term Prognosis in Colorectal Cancer. Clin Cancer Res (1998) 4(1):203–10.
155. Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, et al. Mad-Related Genes in the Human. Nat Genet (1996) 13(3):347–9. doi: 10.1038/ng0796-347
156. Codony-Servat J, Cuatrecaracas M, Asensio E, Montironi C, Martinez-Cardus A, Marin-Anguilera M, et al. Nuclear IGF-1R Predicts Chemotherapy and Targeted Therapy Resistance in Metastatic Colorectal Cancer. Br J Cancer (2017) 117(12):1777–86. doi: 10.1038/bjc.2017.279
157. Alwers E, Blaker H, Walter V, Jansen L, Kloor M, Arnold A, et al. External Validation of Molecular Subtype Classifications of Colorectal Cancer Based on Microsatellite Instability, CIMP, BRAF and KRAS. Cancer (2019) 19:681–90. doi: 10.1186/s12885-019-5842-7
158. Fujino H, Seira N, Kurata N, Araki Y, Nakamura H, Regan JW, et al. Prostaglandin E2-Stimulated Prostanoid EP4 Receptors Induce Prolonged De Novo Prostaglandin E2 Synthesis Through Biphasic Phosphorylation of Extracellular Signal-Regulated Kinases Mediated by Activation of Protein Kinase A in HCA-7 Human Colon Cancer Cell. Eur J Pharmacol (2015) 768:149–59. doi: 10.1016/j.ejphar.2015.10.044
159. Murase R, Taketomi Y, Miki Y, Nishito Y, Saito M, Fukami K, et al. Group III Phospholipase A2 Promotes Colitis and Colorectal Cancer. Sci Rep (2017) 7:12261–73.
160. Betge J, Schneider NI, Pollheimer MJ, Lindtner RA, Kornpart P, Ebert MP, et al. MUC1, MUC2, MUC5AC, and MUC6 in Colorectal Cancer: Expression Profiles and Clinical Significance. Virchows Arch (2016) 469(3):255–65. doi: 10.1007/s00428-016-1970-5
161. Rigas B, Goldman IS, Levine L. Altered Eicosanoid Levels in Human Colon Cancer. J Lab Clin Med (1993) 122(5):518–23.
162. Tojek K, Anaszewicz M, Szukay B, Czerniak B, Socha E, Lis K, et al. Circulating Leptin, Adiponectin, and Tumor Necrosis Factor-Alpha in Patients Undergoing Surgery Due to Colorectal Cancer. Digestion (2021) 102(2):246–55. doi: 10.1159/000504507
163. Scartozzi M, Bearzi I, Pierantoni C, Mandolesi A, Loupakis F, Zaniboni A, et al. Nuclear factor-kB Tumor Expression Predicts Response and Survival in Irinotecan-Refractory Metastatic Colorectal Cancer Treated With Cetuximab-Irinotecan Therapy. J Clin Oncol (2007) 25(25):3930–5. doi: 10.1200/JCO.2007.11.5022
164. Huang C, Zhou Q, Hu Y, Wen Y, Qiu Z, Liang M, et al. Hepatocyte Growth Factor is a Prognostic Marker in Patients With Colorectal Cancer: A Meta-Analysis. Oncotarget (2017) 8(14):23459–68. doi: 10.18632/oncotarget.15589
165. Murphy N, Carreras-Torres R, Song M, Chan AT, Martin RM, Papdimitriou N, et al. Circulating Levels of Insulin-Like Growth Factor 1 and Insulin-Like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology (2020) 158(5):1300–13e20. doi: 10.1053/j.gastro.2019.12.020
166. Emoto S, Ishigami H, Yamashita H, Yamaguchi H, Kaisaki S, Kitayama J. Clinical Significance of CA125 and CA72-4 in Gastric Cancer With Peritoneal Dissemination. Gastric Cancer (2012) 152(2):154–61. doi: 10.1007/s10120-011-0091-8
167. de Jong JS, van Diest PJ, van der Valk P, Baak JP. Expression of Growth Factors, Growth-Inhibiting Factors, and Their Receptors in Invasive Breast Cancer. II: Correlations With Proliferation and Angiogenesis. J Pathol (1998) 184(1):53–7. doi: 10.1002/(SICI)1096-9896(199801)184:1<53::AID-PATH6>3.0.CO;2-7
168. Im JH, Buzzelli JN, Jones K, Frankini F, Gordon-Weeks A, Markels B, et al. Muschel RJFGF2 Alters Macrophage Polarization, Tumour Immunity and Growth and can be Targeted During Radiotherapy. Nat Commun (2020) 11:4064. doi: 10.1038/s41467-020-17914-x
169. Bendardaf R, El-Serafi IA, Syrjänen K, Collan Y, Pyrhönen S. The Effect of Vascular Endothelial Growth Factor-1 Expression on Survival of Advanced Colorectal Cancer Patients. Libyan J Med (2017) 12(1):1290741. doi: 10.1080/19932820.2017.1290741
170. Yu J, Ustach C, Kim HR. Platelet-Derived Growth Factor Signaling and Human Cancer. J Biochem Mol Biol (2003) 36(1):49–59. doi: 10.5483/bmbrep.2003.36.1.049
171. Hu H, Sun L, Guo C, Liu Q, Zhou Z, Peng L, et al. Tumor Cell-Microenvironment Interaction Models Coupled With Clinical Validation Reveal CCL2 and SNCG as Two Predictors of Colorectal Cancer Hepatic Metastasis. Clin Cancer Res (2009) 15(17):5485–93. doi: 10.1158/1078-0432
172. Wang D, Yang L, Yu W, Wu Q, Lian J, Li F, et al. Colorectal Cancer Cell-Derived CCL20 Recruits Regulatory T Cells to Promote Chemoresistance via FOXO1/CEBPB/NF-κb Signaling. J Immunother Cancer (2019) 7:215. doi: 10.1186/s40425-019-0701-2
173. Oladipo O, Conlon S, O'Grady A, Purcell C, Wilson C, Maxwell PJ, et al. The Expression and Prognostic Impact of CXC-Chemokines in Stage II and III Colorectal Cancer Epithelial and Stromal Tissue. Br J Cancer (2011) 104(3):480–7. doi: 10.1038/sj.bjc.6606055
174. Voronov E, Carmi Y, Apte RN. The Role IL-1 in Tumor-Mediated Angiogenesis. Front Physiol (2014) 5:114. doi: 10.3389/fphys.2014.00114
175. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 Released by Colon Cancer-Associated Fibroblasts is Critical for Tumour Angiogenesis: Anti-Interleukin-6 Receptor Antibody Suppressed Angiogenesis and Inhibited Tumour-Stroma Interaction. Br J Cancer (2014) 1102(2):469–78. doi: 10.1038/bjc.2013.748
176. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis (2019) 10:273. doi: 10.1038/s41419-019-1435-2
177. Weiss MB, Abel EV, Mayberry MM, Basile KJ, Berger AC, Aplin AE. TWIST1 is an ERK1/2 Effector That Promotes Invasion and Regulates MMP-1 Expression in Human Melanoma Cells. Cancer Res (2012) 72(24):6382–92. doi: 10.1158/0008-5472.CAN-12-1033
178. Chatterjee K, Jana S, DasMahapatra P, Swarnakar S. EGFR-Mediated Matrix Metalloproteinase-7 Up-Regulation Promotes Epithelial-Mesenchymal Transition via ERK1-AP1 Axis During Ovarian Endometriosis Progression. FASEB J (2018) 32(8):4560–72. doi: 10.1096/fj.201701382RR
179. Maurel J, Nadal C, Garcia-Albeniz X, Gallego R, Carcereny E, Almendro V, et al. Serum Matrix Metalloproteinase 7 Levels Identifies Poor Prognosis Advanced Colorectal Cancer Patients. Int J Cancer (2007) 121(5):1066–71. doi: 10.1002/ijc.22799
180. Wilson S, Damery S, Stocken DD, Dowswell G, Holder R, Ward ST, et al. Serum Matrix Metalloproteinase 9 and Colorectal Neoplasia: A Community-Based Evaluation of a Potential Diagnostic Test. Br J Cancer (2012) 2014:1431–8. doi: 10.1155/2014/897523
181. Vincent-Chong VK, Salahsourifar I, Karen-Ng LP, Siow MY, Kallarakkal TG, Ramanathan A, et al. Overexpression of MMP13 is Associated With Clinical Outcomes and Poor Prognosis in Oral Squamous Cell Carcinoma. ScientificWorldJournal (2014) 2014):897523. doi: 10.1155/2014/897523
Keywords: colorectal cancer, inflammation, gut microbiota, biomarkers, patients’ stratification
Citation: Ionica E, Gaina G, Tica M, Chifiriuc M-C and Gradisteanu-Pircalabioru G (2022) Contribution of Epithelial and Gut Microbiome Inflammatory Biomarkers to the Improvement of Colorectal Cancer Patients’ Stratification. Front. Oncol. 11:811486. doi: 10.3389/fonc.2021.811486
Received: 12 November 2021; Accepted: 20 December 2021;
Published: 07 February 2022.
Edited by:
Octav Ginghina, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Woojung Shin, Harvard Medical School, United StatesJian Tan, The University of Sydney, Australia
Copyright © 2022 Ionica, Gaina, Tica, Chifiriuc and Gradisteanu-Pircalabioru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Ionica, ZWxlbmEuaW9uaWNhQHVuaWJ1Yy5ybw==