 Marina Bataller1
Marina Bataller1 Almudena Sánchez-García1
Almudena Sánchez-García1 Yoelsis Garcia-Mayea1
Yoelsis Garcia-Mayea1 Cristina Mir1
Cristina Mir1 Isabel Rodriguez2
Isabel Rodriguez2 Matilde Esther LLeonart1,3*
Matilde Esther LLeonart1,3*- 1Biomedical Research in Cancer Stem Cells Group, Vall d´Hebron Research Institute (VHIR), Barcelona, Spain
- 2Assistant Director of Nursing, Nursing Management Service Hospital Vall d’Hebron, Barcelona, Spain
- 3Spanish Biomedical Research Network Centre in Oncology, CIBERONC, Madrid, Spain
Drug resistance continues to be one of the major challenges to cure cancer. As research in this field evolves, it has been proposed that numerous bioactive molecules might be involved in the resistance of cancer cells to certain chemotherapeutics. One well-known group of lipids that play a major role in drug resistance are the sphingolipids. Sphingolipids are essential components of the lipid raft domains of the plasma membrane and this structural function is important for apoptosis and/or cell proliferation. Dysregulation of sphingolipids, including ceramide, sphingomyelin or sphingosine 1-phosphate, has been linked to drug resistance in different types of cancer, including breast, melanoma or colon cancer. Sphingolipid metabolism is complex, involving several lipid catabolism with the participation of key enzymes such as glucosylceramide synthase (GCS) and sphingosine kinase 1 (SPHK1). With an overview of the latest available data on this topic and its implications in cancer therapy, this review focuses on the main enzymes implicated in sphingolipids metabolism and their intermediate metabolites involved in cancer drug resistance.
1 Introduction
Cancer incidence and mortality are growing fast worldwide, with a higher frequency in countries with higher socioeconomic development, as life expectancy continues to rise. However, in countries with stronger health care systems, cancer mortality is decreasing due to early detection and treatment. For several decades, cancer has been the second leading cause of death globally where lung cancer is at the top of the list as the leading cause of cancer deaths. Nevertheless, the most commonly diagnosed types of cancer vary among countries depending on the degree of socioeconomic development and life style factors (1).
Despite the rapid advance of cancer therapies, treatment-resistant relapse remains a major challenge in cancer treatment. Treatment resistance can be classified as intrinsic or acquired resistance, depending on its origin. Intrinsic resistance arises from the administration of chemotherapy treatment, and it is due to preexisting factors of the tumor, so the tumor cannot respond to the initial treatment. On the other hand, acquired resistance appears during or after the administration of treatment and is usually the main contributing factor for relapse. Some theories explain this resistance as sporadic genetic mutations maintained by Darwinian selection through the exposure to the chemotherapeutic agent (2, 3). There are several mechanisms by which tumoral cells acquire this resistance to treatment: inactivation of the drug, multi-drug resistance (MDR) mechanisms, cell death inhibition increasing resistance to apoptosis, changes in cell metabolism, epigenetics modulation, increased DNA repair and gene mutation or amplification that cause the resistance to the chemotherapy (4).
Sphingolipids were named by Thudichum JL in 1884 because of their enigmatic nature (5). It is now known that sphingolipids are a family of bioactive membrane lipids that contribute to the regulation of the fluidity of the plasmatic membrane. The sub-domain structure of the lipid bilayers forms lipid rafts, which act as first and/or second messengers in different pathways as they function as bio-effector molecules (6). The enzymes involved in the sphingolipids’ metabolism have been studied during the last decade and have been directly linked to the control of cell growth, proliferation and apoptosis, among other cellular functions. Table 1 summarizes the functions and characteristics of the main enzymes of sphingolipids’ metabolism involved in cancer. This review focuses on the importance of these enzymes with a specific focus on their response to drug therapy.
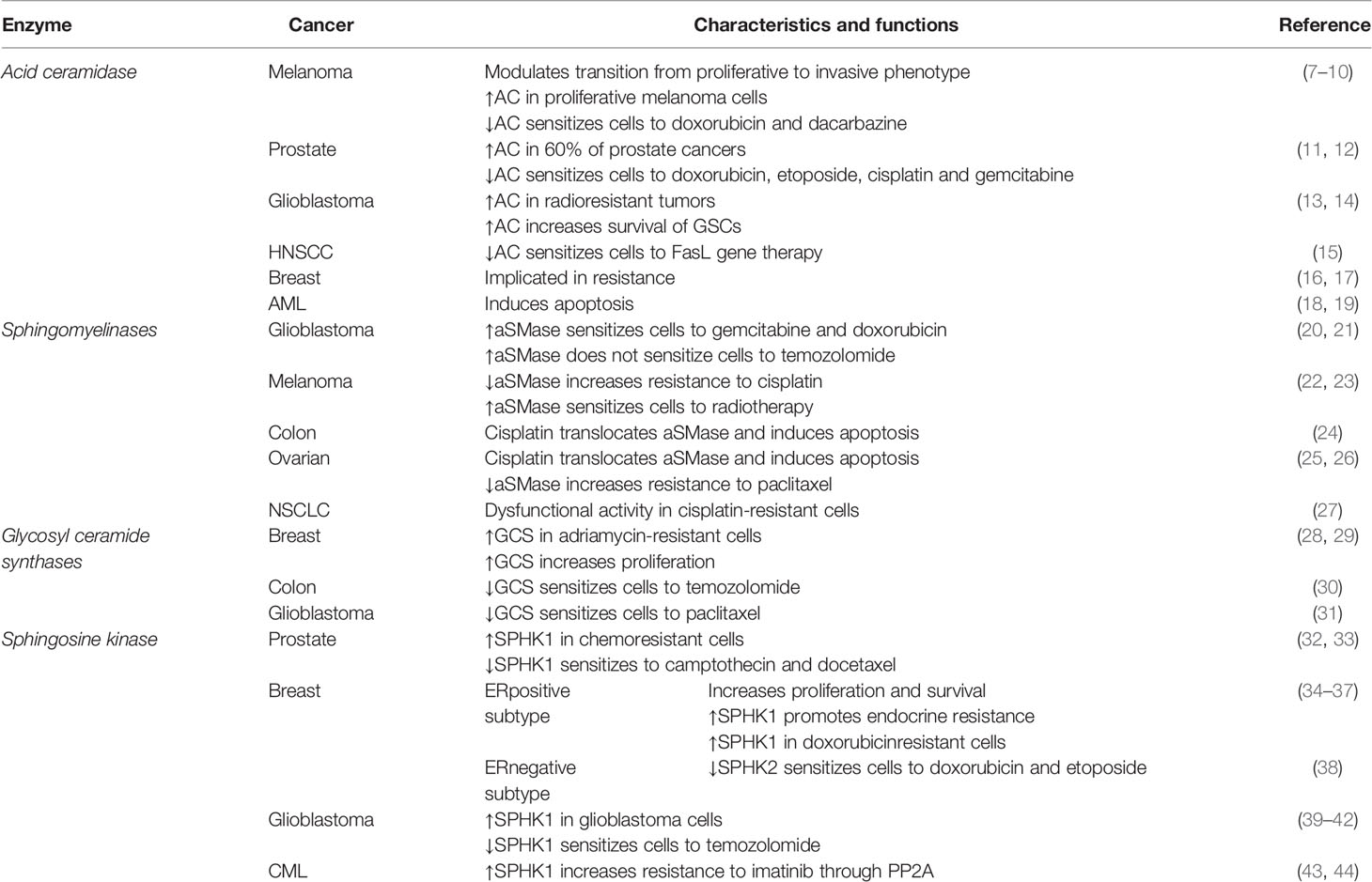
Table 1 Functions and characteristics of the main enzymes in sphingolipids’ metabolism involved in cancer.
2 Cellular Functions of Sphingolipids
The first sphingolipid that was identified was sphingosine, whose involvement has been described in the cytoskeleton, endocytosis, cell cycle and apoptosis regulation (45). However, the sphingolipids that have been most frequently implicated in cancer are ceramide and sphingosine 1-phosphate (S1P). Ceramide functions in the cell differ depending on the subcellular location where ceramide is accumulated. For example, when ceramide is generated in the plasma membrane, it is involved in growth inhibition, oxidative stress-mediated cell death, and lipid raft functions (46, 47); whereas when it is located in the lysosomes, ceramide mediates cell-stress responses such as cell senescence and apoptosis (48–50). In contrast, S1P plays a role in cell survival and proliferation, cell migration and invasion, autophagy and inflammation (51, 52). Furthermore, glucosylceramide, a derivative from ceramide, regulates the post-Golgi trafficking, as the enzyme that catalyzes the step from ceramide to glucosylceramide located in the Golgi apparatus (53). Figure 1 represents the structure of these sphingolipids and some of the main cellular processes in which they are involved in cancer.
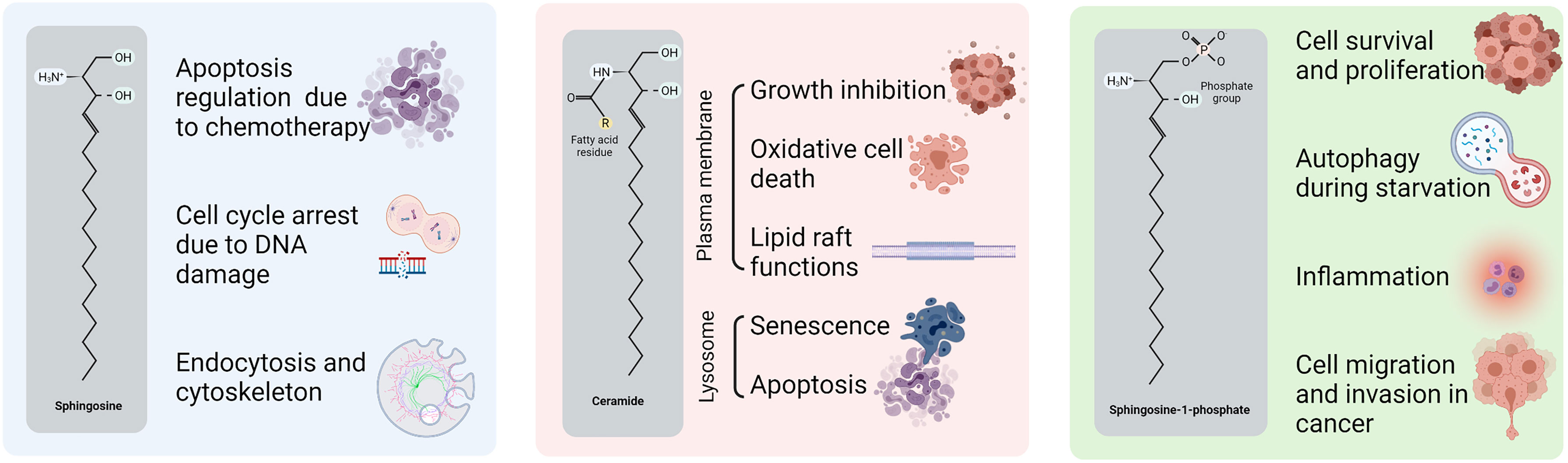
Figure 1 Structure and functions of sphingolipids in cancer. Sphingosine, ceramide and S1P structures with the principal carbon chain and the main chemist groups highlighted. Ceramide functions are divided by the location in the cell.
3 Sphingolipids’ Metabolism
It has been reported that sphingolipids metabolism has a unique metabolic entry point and a unique metabolic exit point. The first one is through the enzyme SPT (serine palmitoyl transferase), which forms the sphingolipid ceramide by the condensation of serine and palmitate in the synthesis pathway. The exit point is mediated by S1P lyase (SGPL), an enzyme that breaks down S1P into different non-sphingolipid molecules. Ceramide has a central position in both the catabolism and anabolism of sphingolipids. For this reason, it is considered as a metabolic hub in the sphingolipids’ metabolism pathway.
Specifically, the synthesis of ceramide starts with the condensation of serine and palmitoyl CoA by SPT, resulting in 3-keto-dihydrosphingosine, which then results in dihydrosphingosine by the action of 3-ketosphinganine reductase (KDSR) and it is later acetylated into dihydroceramide by ceramide synthases (CerS 1-6). Finally, the desaturation of dihydroceramide by dihydroceramide desaturase (DES) results in ceramide, which can be glycosylated by glucosylceramide synthase (GCS) to form glucosylceramide or, alternatively, it can form galactosylceramide by the action of galactosyltransferase (CGT) or it can acquire a phosphocholine headgroup by sphingomyelin synthases (SMS) to eventually form sphingomyelin. The phosphorylation of ceramide by ceramide kinase (CK) results in ceramide 1-phosphate (C1P) (54–56).
The catalysis of ceramide is performed by specific hydrolases. Ceramidase (CDase) breaks down ceramide and generates sphingosine, which can be recycled back into ceramide by CerS 1-6 or phosphorylated by a sphingosine kinase (SPHK1/2) to form S1P. Moreover, S1P can generate sphingosine by sphingosine phosphatase 1/2 (SGPP1/2) or it can exit the sphingolipids metabolic pathway through S1P cleavage by SGPL, obtaining ethanolamine-1-phosphate and hexadecenal. In addition, other hydrolases can produce ceramide, such as sphingomyelinase (SMase) which breaks down sphingomyelin (54). Figure 2 illustrates the main metabolic pathways of sphingolipids metabolism.
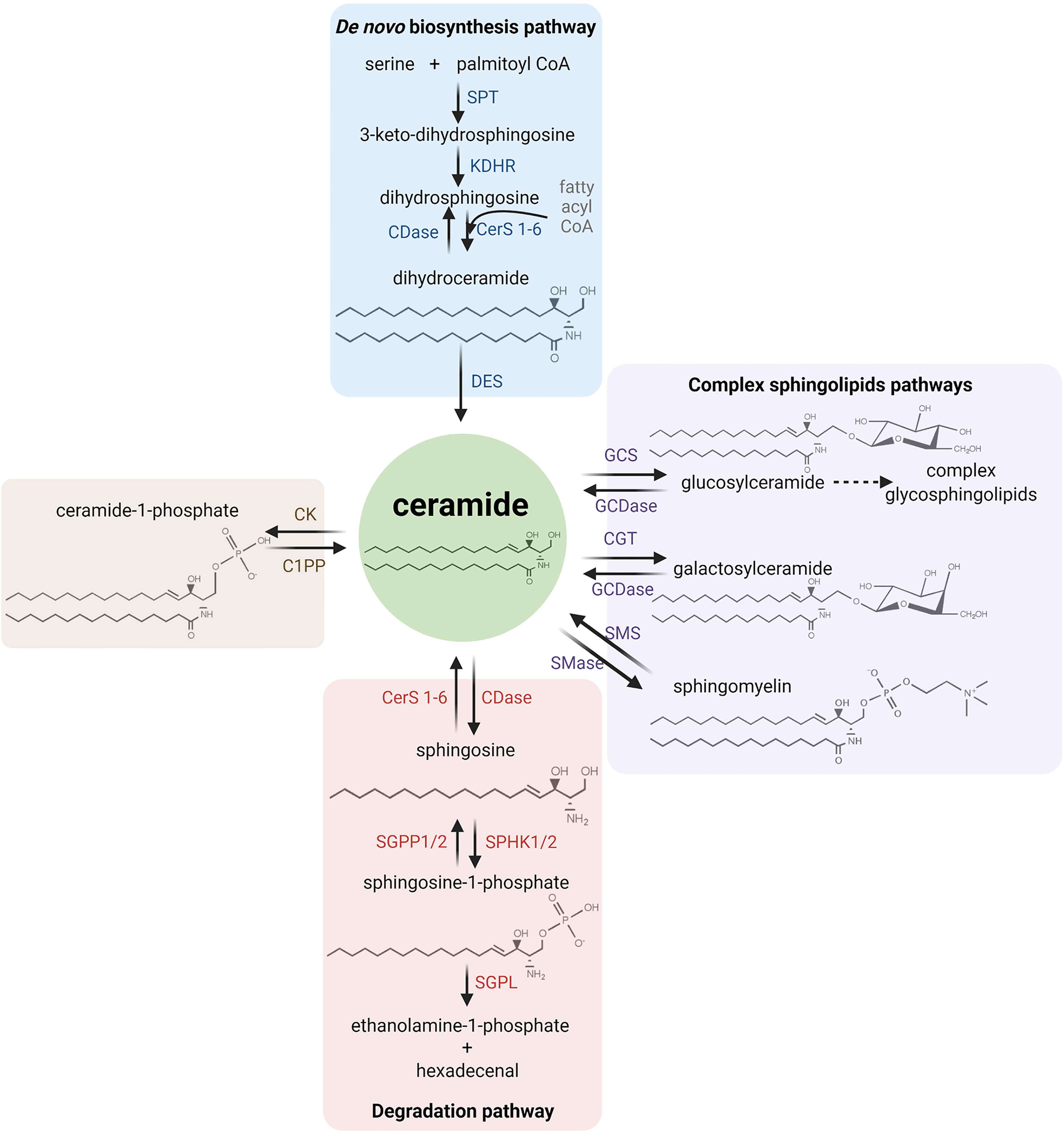
Figure 2 Sphingolipid metabolism. The synthesis of ceramide starts with the condensation of serine and palmitoyl CoA (the active form of palmitate) by serine palmitoyltransferase (SPT), resulting in 3-keto-dihydrosphingosine (also called 3-ketosphinganine). Subsequently, dihydrosphingosine (or sphinganine) is formed after the reduction of 3-keto-dihydrosphingosine by 3-ketosphinganine reductase (KDSR). Next, dihydrosphingosine is acetylated by a (dihydro-)ceramide synthase (referred to simply as ceramide synthase, CerS 1-6) to form dihydroceramide. Finally, desaturation of dihydroceramide by dihydroceramide desaturase (DES) results in ceramide. Ceramide can then be glycosylated by glucosylceramide synthase (GCS) to form glucosylceramide, or it can also form galactosylceramide by galactosyltransferase (CGT). On the other hand, ceramide can also acquire a phosphocholine headgroup due to the action of the sphingomyelin synthases (SMS) to eventually form sphingomyelin. Alternatively, ceramide kinase (CK) catalyzes the phosphorylation of ceramide to form ceramide 1-phosphate (C1P). Ceramidase (CDase) breaks down ceramide and generates sphingosine, which can be recycled back into ceramide by CerS 1-6 or phosphorylated by a sphingosine kinase (SPHK1/2) to form sphingosine-1-phosphate (S1P). In addition, S1P can be dephosphorylated by sphingosine phosphatase (SGPP1/2) to generate sphingosine again or it is cleaved by S1P lyase (SGPL) to obtain ethanolamine-1-phosphate and hexadecenal. On the other hand, the breakdown of sphingomyelin by sphingomyelinase (SMase), the breakdown of glucosyl/galactosylceramide by glucosyl/galactosylceramidase (GCDase) and the dephosphorylation of C1P by ceramide 1-phosphatase (C1PP) generate ceramide.
4 Enzymes Involved in Sphingolipids’ Metabolism and Drug Resistance
4.1 Glucosylceramide Synthase (GCS)
GCS, encoded by the UGCG (UDP-glucose ceramide glucosyltransferase) gene, is the enzyme that transfers an UDP-glucose molecule to ceramide thus generating glucosylceramide, the precursor for all complex glycosphingolipids (54). Glycosylated sphingolipids cluster in the plasma membrane forming glycosphingolipid-enriched microdomains (GEMs), which are functional clusters that membrane proteins use as signaling platforms (57). Besides the plasma membrane, these GEMs can be found in the membranes of some subcellular organelles, such as mitochondria and are involved in the regulation of diverse cell functions including apoptosis (58).
Overexpression of GCS was reported in various cancers, such as breast and colon cancer (7). Moreover, drug resistant cancer cells from ovarian cancer, cervical cancer, melanoma, colon cancer and leukemia have shown GCS overexpression (11, 13, 59). Likewise, while doxorubicin exerts its action as an intercalating DNA agent and generates free radicals to damage growing cells, it also upregulates GCS expression and leads to drug resistance in cells through the modulation of the Sp1 transcription factor (15). Hence, efforts are being made to find a GCS inhibitor to downregulate GCS expression or inhibit its catalytic function.
4.1.1 Breast Cancer
GCS overexpression is related to increased cellular proliferation (18) and poor prognosis in breast cancer patients (12). The most interesting feature of this enzyme is its functional connection with the ATP binding cassette subfamily B member 1 (ABCB1) gene, which encodes the ABCB1 protein, also called P-glycoprotein 1 (P-gp). Silencing of the UGCG gene by the promoter CpG island methylation shows an inverse correlation with drug resistance in ductal breast cancer cells (60). This means that the demethylation of the CpG island in the UGCG promoter could increase the generation of multidrug-resistant clones.
Liu et al. demonstrated that GCS upregulates ABCB1 expression and modulates cancer drug resistance (59). In addition, studies from Zhang et al. revealed that in turn, ABCB1 can regulate GCS expression (61). Liu et al. also showed that silencing of GCS downregulates ABCB1 expression and sensitizes multidrug-resistant cells to chemotherapy through Src and β-catenin signaling (59). There is also evidence that GCS overexpression in breast cancer leads to AKT activation (p-AKT), which induces ABCB1 expression. At present, p-AKT is known to phosphorylate GSK-3β, the enzyme that phosphorylates β-catenin so it can exit the nucleus, hence this finding could validate the previous theory (18). Morad and Cabot suggested that ABCB1 is also located in the Golgi membrane acting as a flippase, and it is responsible for the transfer of glucosylceramide from the cytosol to the Golgi lumen, therefore promoting ceramide clearance (62). However, a different study revealed that flippase activity of ABCB1 is only needed for neutral, and not acidic, glycosphingolipids generation. There are also other studies which postulate that glucosylceramides regulate their own entrance to the Golgi apparatus, depending on the length of their ceramides’ chains, hence the flippase activity of ABCB1 would not be necessary or it would be an alternative mechanism to ceramide regulation (63). Further studies in this field are required to decipher the role of ABCB1 in the Golgi membrane and its connection with GCS (8) (Figure 3).
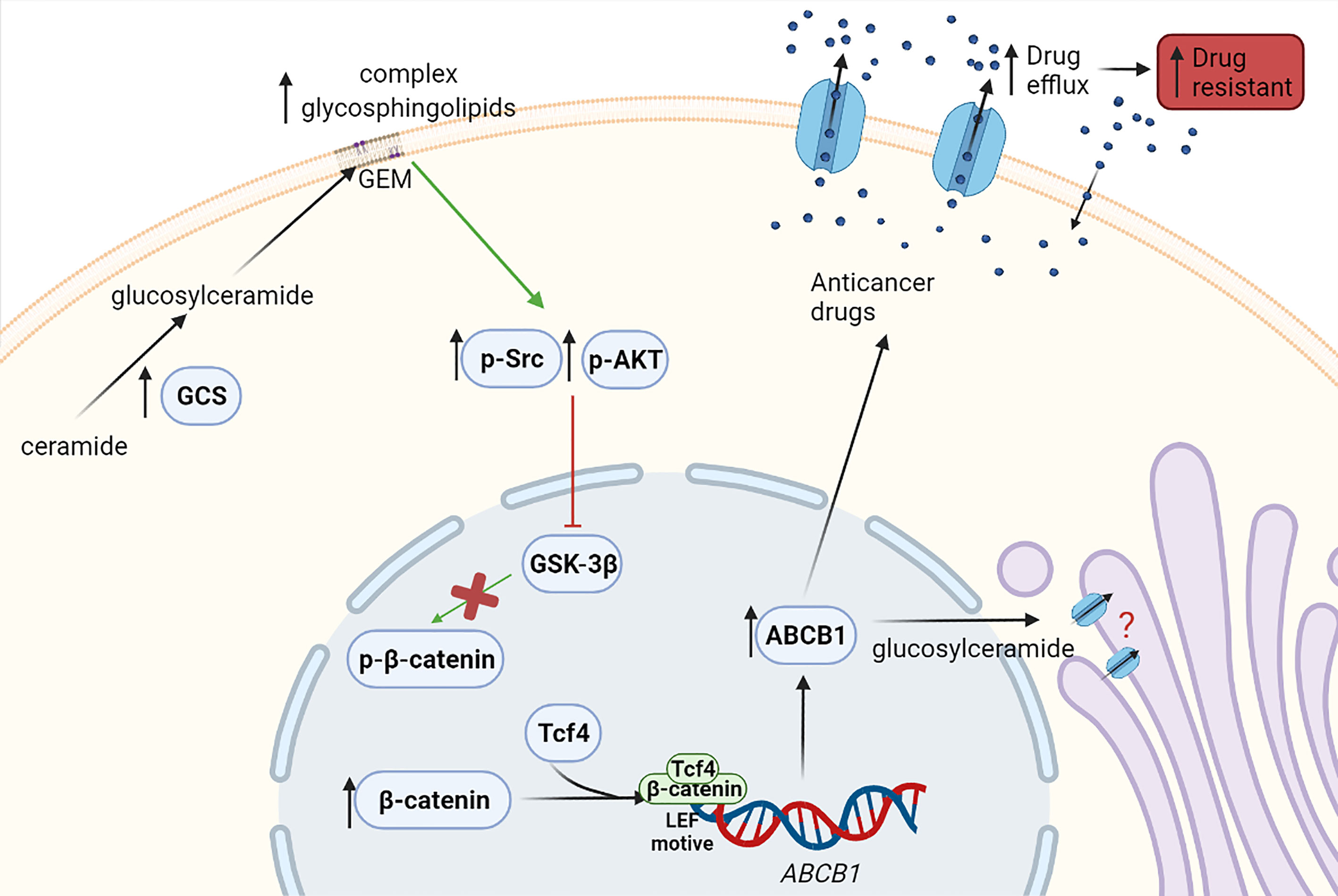
Figure 3 Effect of the enzyme GCS in drug resistance. Overexpressed GCS leads to increased concentrations of glycosphingolipids in the membrane, which may mediate the phosphorylation of Src (p-Src) and AKT (p-AKT) and their activation. Then, GSK-3β phosphorylated by p-Src or p-AKT becomes inactive and cannot phosphorylate β-catenin. Consequently, β-catenin cannot exit the nucleus and thus, β-catenin levels in the nucleus increase. β-catenin within the nucleus might bind in a transcriptional complex with Tcf4 (T-cell factor 4). This β-catenin/Tcf4 complex would then be able to bind a LEF (lymphoid enhancer factor) motive in ABCB1 (ATP-binding cassette subfamily B member 1) gene promoter, resulting in its transcription and increasing ABCB1 expression, thus leading to anticancer drug efflux and multidrug resistance. Likewise, it has been described that ABCB1 is located in the Golgi membrane and promotes the transfer of glucosylceramide from the cytosol to the Golgi, helping with the ceramide clearance from cytosol. However, further studies are required to validate this ABCB1 function. Green arrows show induction of expression and red arrows show inhibition.
Although, it has been demonstrated that the GCS role in drug resistance is usually through ABCB1 overexpression, Liu et al. described that GCS is overexpressed in adriamycin-resistant MCF-7 breast cancer cells and that the multidrug resistance in these cells is independent from ABCB1 (59). Thus, it is possible that multidrug resistance in some cancer models is also derived from other unidentified proteins that are able to interact with GCS (9).
4.1.2 GCS in Other Cancer Types
Inhibition of GCS in doxorubicin-resistant cells and temozolomide/paclitaxel-resistant cells is sensitized to treatment in colon cancer cells (64) and glioblastoma cells (10), respectively. In chronic myeloid leukemia (CML) cells, GCS can increase the expression of ABCB1 through NF-κβ signaling, a different mechanism than the one described in breast cancer (65). Moreover, in a murine melanoma model, inhibition of GCS in intrinsically chemoresistant cancer stem cells sensitized them to some genotoxic drugs (66).
4.2 Sphingosine Kinase (SPHK)
SPHK is a conserved family of lipid kinases and the enzymes responsible for the phosphorylation of sphingosine to generate S1P, maintaining the ceramide-S1P rheostat (67). There are two mammalian SPHK isoenzymes: SPHK1 and SPHK2; which have different locations in the cell and have different substrate specificities, kinetic properties and tissue expression (14, 16, 17). Regarding their structure, both isoenzymes have five conserved domains of ~50% identity (19). Accordingly, it is thought that some of the differences in the physiological effects between the two enzymes are due to the fact that SPHK2 has an extended NH2 terminus where a putative BH3 binding domain is located (68). Upon the first apoptosis stimulus, the BH3 domain is responsible to orchestrate apoptosis. The subcellular location of these isoenzymes is variable since it depends on the pathological state and tissue type. Usually, SPHK1 is found in the cell cytoplasm, whereas SPHK2 can be found either in the nucleus or in the cytoplasm (67, 69). Although only these two isoforms have been identified in human tissues, some studies suggest the presence of additional uncharacterized isoforms (67, 70). SPHK plays a pivotal role by regulating cell growth and acting as an oncogene in tumorigenesis (71–74). Furthermore, it has been shown that both isoenzymes exert important functions in angiogenesis, a crucial step in the metastatic spread process (16).
SPHK mRNA is overexpressed in some cancer tissues, including breast, colon, lung, ovary, stomach, uterus and kidney (75). In addition, the SPHK protein is overexpressed in prostate cancer and glioblastoma (76, 77). A study in the model organism Dictyostelium discoideum (a eukaryote species that belongs to the phylum Amoebozoa) showed that SPHK activity regulated the sensitivity to cisplatin, but not doxorubicin, etoposide and 5-fluoruracil, indicating the anti-cancer drug specificity. Overexpression of SPHKs sgkA and sgkB (homologous to human SPHK1, SPHK2, respectively) was observed to result in increased resistance to cisplatin (78). Similarly, it is expected that both SPHK1 and SPHK2 would provide cancer resistance in humans. Some examples based on cancer types are shown in the following sections.
4.2.1 Prostate Cancer
Chemoresistant prostate cancer cells have high levels of SPHK1 (76). Pchejetski et al. proposed that SPHK1 activity is a chemotherapy sensor, since prostate cancer cells sensitive to the chemotherapeutic drug camptothecin reduced SPHK1 levels after treatment (20). In contrast, cells resistant to camptothecin did not significantly change SPHK1 levels after the treatment (20). Supporting this finding, Akao et al. demonstrated that SPHK1 activity in chemoresistant prostate cancer cells was significantly increased by treatment with camptothecin in a concentration-dependent manner and that this increment of the activity was due to increased protein and mRNA levels (76). Likewise, Pchejetski et al. also revealed that the inhibition of SPHK1 sensitized prostate cells not only to camptothecin but also to docetaxel (20). Since then, other studies have shown the sensitization to docetaxel by SPHK1 inhibition in docetaxel-resistant prostate cancer cells (22, 24). For example, Alshaker et al. showed that the SPHK inhibitor RAD001 sensitizes docetaxel-resistant cells to the docetaxel treatment both in vitro and in vivo (22). Additional in vivo studies are needed to demonstrate the potential of SPHK inhibitors in prostate cancer.
4.2.2 Breast Cancer
It has been demonstrated that SPHK increases proliferation and survival in estrogen receptor (ER)-positive breast cancers, and it is associated with poor prognosis in the ER-negative subtype (25, 27). Moreover, overexpression of SPHK in MCF-7 cell line (ER-positive) promotes resistance to hormone therapy. A remarkable finding is that SPHK1 and SPHK2 expression among the different breast cancer subtypes is highly variable, making it difficult to generalize about the implication of these enzymes in breast cancer (69).
On the other hand, doxorubicin-resistant breast cancer cell lines showed a high expression of SPHK1 and its inhibition with fingolimod (immunosuppressive drug) caused a decrease in proliferation (21). Antoon et al. disclosed that ER-negative resistant breast cancer cells overexpressed SPHK; and specific inhibition of SPHK2 by ABC294640 decreased the growth of chemoresistant breast cancer cells in vivo enhancing doxorubicin and etoposide-induced apoptosis when using combined therapy (79). Some of these inhibitors are currently under clinical trials as described below.
4.2.3 Glioblastoma
SPHK has been found overexpressed in glioblastoma cells and its overexpression has been correlated with poor prognosis (77). Several studies illustrated that the inhibition of SPHK1 results in a decrease in cell viability following the temozolomide treatment in temozolomide-resistant glioblastoma cells (23, 80, 81). Furthermore, there is also data that supports the therapeutic value of SPHK inhibitors as radiosensitizers (26).
4.2.4 Chronic Myeloid Leukemia
Interestingly, resistance to tyrosine kinase inhibitors (such as imatinib) used for the treatment of chronic myeloid leukemia (CML) has been linked to alterations of sphingolipid metabolism and signaling. Overexpression of SPHK1 has been described to mediate imatinib resistance in CML patient-derived cells (82). The mechanism by which SPHK1 mediates imatinib resistance in CML cells is thought to take place through the modulation of the protein phosphatase 2 (PP2A). Inhibition of PP2A by SPHK1 might attenuate ubiquitination and proteasomal degradation of BCR-ABL, the mutated tyrosine kinase, enhancing its stability and resulting in subsequent drug resistance (83). Other studies revealed that SPHK1 overexpression in CML imatinib-resistant cells is regulated by signaling through PI3K, AKT2 and mTOR, with AKT playing a major role in the modulation of this resistance (84). Recently, Sun and Wang showed that a combined therapy of SPHK inhibitors and all-trans retinoic acid (ATRA), which has been described as limited to a treatment for CML, exert synergistic effects to inhibit proliferation in CML cells (85). Thus, the study of SPHK inhibitors might be important for CML treatment.
4.3 Acid Ceramidase (AC)
Ceramidases (CDases) are the enzymes responsible for the breakdown of ceramide to generate sphingosine. Human CDases can be classified into 5 different types, depending on their cellular location, primary structure and optimal pH needed for their catalytic activity: the 3 main ones are alkaline, neutral and acid CDases (86, 87). In particular, acid CDase (AC), encoded by the ASAH1 gene, is a 50 kDa enzyme which belongs to the N-terminal nucleophile superfamily of hydrolases. AC is the only CDase that requires an additional protein to reach its optimal activity, saposin-D, a lysosomal protein which helps to present ceramide as a substrate to AC (88).
AC is overexpressed in some human cancers. For example, overexpression of AC has been demonstrated in melanoma (89) and prostate cancer (28) cell lines and biopsies as well as in head and neck squamous cell carcinoma (HNSCC) (90, 91), glioblastoma (92) and acute myeloid leukemia (AML) (93). Since AC upregulation has been linked to apoptosis resistance (29, 94), it is expected that the drugs able to decrease AC would be an efficient tool to sensitize resistant cells. The AC characterization is accordingly shown for the following cancer models.
4.3.1 Melanoma
The most abundant skin cells (fibroblasts and keratinocytes) express low levels of AC. However, the number of cells with high levels of AC increase during cancer progression, as melanocytes start growing uncontrollably (89). The phenotype-switching model is a model of tumor progression that describes cancer development, resistance to therapy and metastasis; it is considered as one of the origins of intratumoral heterogeneity, a feature highly associated with therapy resistance. It states the existence of two different phenotypes: the proliferative phenotype, that is less invasive, and the invasive phenotype, which is less proliferative (30, 31, 95). Studies in melanoma cells revealed that lysosomal AC and the sphingolipid metabolism drive the transition between the proliferative and the invasive phenotype (96). AC expression is higher in proliferative melanoma cells compared with other skin cells (89). Interestingly, in metastatic melanoma, it has been described that downregulation of AC, but not neutral or alkaline CDases, increases ceramide levels in the cell and confers the cells sensitization to dacarbazine (97), the main chemotherapeutic drug (DNA-alkylating agent) used for this type of cancer before immune checkpoint inhibitors or BRAF inhibitors became available for clinical use (98). Recently, it has been reported that AC ablation restores melanoma sensitivity to doxorubicin, a different chemotherapeutic agent used for melanoma treatment that affects sphingolipids’ metabolism, since it forces cells treated with doxorubicin to undergo apoptosis. It is thought that this restoration of doxorubicin sensitivity is due to the increase of ceramide accumulation subsequent to AC inhibition (99). Overall, AC inhibition or downregulation could represent an interesting approach to sensitize melanoma cells to some cancer drugs.
4.3.2 Prostate Cancer
It has been shown that AC is overexpressed in 60% of prostate cancer tumors (28). This overexpression is linked to prostate cancer progression, and it modulates sphingolipid levels in prostate cancer cells, resulting in higher levels of very long chain ceramides. In addition, higher levels of AC made the cells more resistant to apoptosis following treatment with doxorubicin, etoposide, cisplatin or gemcitabine, known chemotherapeutics used to treat prostate cancer. Accordingly, downregulation of AC reversed the resistance to these therapies. For this reason, the combination of current chemotherapy and AC inhibitors is proposed as an efficient way to improve prostate cancer treatment (94). In this sense, Kus et al. demonstrated the induction of apoptosis in prostate cancer cells by using a ceramidase inhibitor, ceranib-2 (100). Besides its role in chemotherapy resistance, AC overexpression has also been linked to resistance to radiotherapy in prostate cancer (101, 102). This is important because resistance to radiotherapy is an undeveloped field of exploration and in the case of prostate cancer - in which it is relatively easy for cells to acquire resistance to androgens – the treatment with radiotherapy acquires an essential role.
4.3.3 AC in Other Cancer Types
The implication of AC in treatment resistance in breast cancer, AML, HNSCC and glioblastoma has also been explored. In glioblastoma, it has been demonstrated that AC levels are higher in radioresistant tumors, suggesting that AC may confer radio resistance (103). In this particular type of cancer, AC has been directly linked to the increase in the survival of glioblastoma stem-like cells (GSCs), that are usually more resistant to anticancer therapies (92). In HNSCC, it has been shown in both in vitro and in vivo techniques that the use of an AC inhibitor (LCL 204) sensitizes the tumor to FasL gene therapy. For this reason, the combination of FasL gene therapy with LCL 204 may become an effective new treatment for HNSCC tumors (90). With a proteomics/bioinformatics approach, Yang et al. found that AC, among other proteins, was associated with breast cancer drug resistance (32, 39). Finally, inhibition of AC in AML has been shown to increase ceramide levels and induce apoptosis (93, 104). Hence, AC can be considered as a potential target for several cancer models of a particularly aggressive nature such as HNSCC, melanoma or glioblastoma therapy.
4.4 Sphingomyelinases (SMases)
SMases are the enzymes that carry out the hydrolysis of sphingomyelin (SM) to generate ceramide. In 1999, Samet and Barenholz (33) proposed a classification of eukaryote SMases into 5 categories, which differed based on cation dependency, pH cation optima and intracellular location: acid sphingomyelinase (aSMase), secretory sphingomyelinase (sSMase), -dependent neutral sphingomyelinases (nSMase), -independent neutral sphingomyelinases and alkaline sphingomyelinases (bSMases) (33, 105, 106). At present, they are usually labelled by their optimal pH (54): aSMases, located in the lysosome and lipid rafts; nSMases, located in the plasma membrane; and bSMases, located in the endoplasmic reticulum (34). Most SMase studies focus on aSMase, encoded by the SMPD1 (sphingomyelin phosphodiesterase 1) gene (35), which produces a protein with different molecular weights (from 58kDa to 75kDa), depending on the tissue of origin (36, 105). It has been estimated that around 70% of all cellular aSMase are located in lipid rafts, which constitute an important group of the plasma membranes’ structures involved in the regulation of various cellular processes. Specifically, aSMase could be involved in the increase of membrane fluidity because its activity results in cholesterol release from the membranes (37).
The action of SMases has been found to be an essential step for the efficacy of chemotherapy and radiotherapy (38, 40, 41). For example, Santana et al. discovered that aSMase-deficient human lymphoblasts and mice specimens cannot induce apoptosis after ionizing radiation treatment (42). Moreover, many studies have linked this enzyme with drug resistance in glioblastoma (107), melanoma (43), colon cancer (44), ovarian cancer (108) and non-small cell lung cancer (NSCLC) (109). Thus, the potential role of this enzyme not only in chemo- but radio resistance might situate aSMase as a central axis in therapy resistance. These studies are summarized in the next sections. The role of SMases in different cancer models is shown below.
4.4.1 Glioblastoma
In glioblastoma, some studies showed that aSMase overexpression sensitized glioma cells to gemcitabine and doxorubicin, two chemotherapeutics used for glioblastoma treatment (107). Paradoxically, Gramatzki et al. detected opposite results several years later with a different chemotherapeutic, demonstrating that the overexpression of aSMase did not sensitize glioblastoma cells to radiation or chemotherapy with temozolomide, the current chemotherapeutic considered as standard (110). Nevertheless, Gramatzki et al. also found in the same study that increased levels of ceramide by aSMase-independent pathways decreased the survival of temozolomide-resistant glioma cell lines (110). A different study showed that aSMase, but not nSMase, hydrolyzed sphingomyelin to generate ceramide and induced apoptosis in p53-deficient glioblastoma cells; while in p53 wild-type glioblastoma cells, p53 expression upregulated AC and blocked the ceramide response, thus allowing the cells to evade apoptosis. Therefore, p53 status might be important for the response to treatment in glioma cells (111). The exact mechanism of p53-ceramide interaction is still not fully understood, but it is known that SMases can regulate apoptosis in glioma cells with differential responses, somehow related to p53 expression and p53 downstream targets (112). Figure 4 represents how the SMase expression modulates the response to chemotherapy.
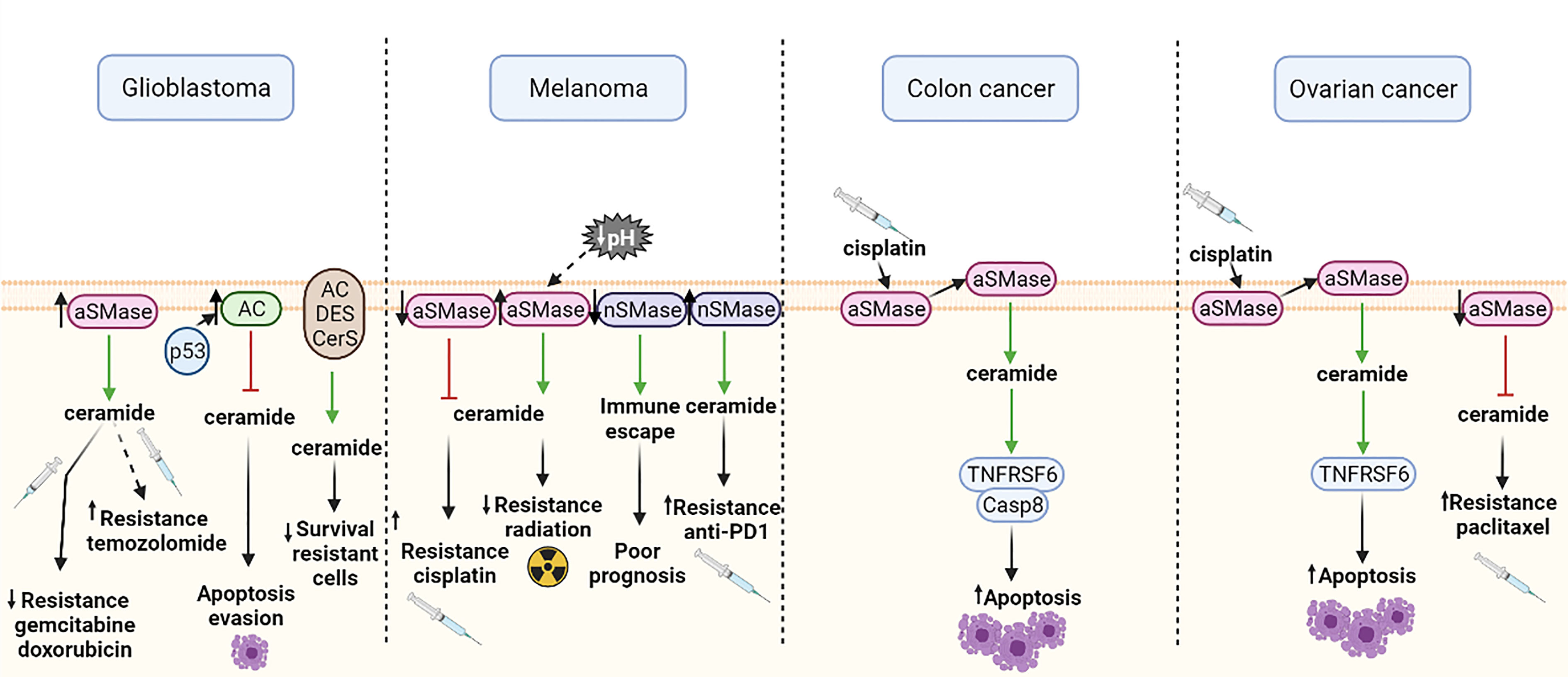
Figure 4 Effects of different SMase expressions in chemotherapy response or cellular functions in glioblastoma, melanoma, colon cancer and ovarian cancer. Green arrows show induction of expression and red arrows show inhibition. aSMase, acid sphingomyelinase; nSMase, neutral sphingomyelinase; AC, acid ceramidase; DES, dihydroceramide desaturase; CerS, ceramide synthase; Casp8, caspase 8; TNFRSF6, tumor necrosis factor receptor superfamily member 6. All the figures were created with BioRender.
4.4.2 Melanoma
Studies of the role of SMase in melanoma cells show that cells with low aSMase expression exhibit higher resistance to cisplatin, probably because of their lower levels of ceramide (43). Furthermore, aSMase expression is also linked to radiotherapy resistance, since overexpression of aSMase in mice with melanoma sensitized the tumors to irradiation. Regarding this matter, some studies suggest that a lower pH of some solid tumors may increase the activity of aSMase and, consequently, radiosensitivity (113). Likewise, it has been demonstrated that downregulation of nSMase2 contributes to immune escape and is associated with poor prognosis in human melanoma. Recently, it has been suggested that nSMase2 overexpression might be useful to overcome resistance to anti-PD-1 (114) (Figure 4). This has clinical relevance since immunotherapy has been revealed as an effective alternative therapy in approximately 20% of metastatic melanoma patients.
4.4.3 Colon Cancer
As mentioned earlier, aSMase is mainly located in the plasma membrane lipid rafts. Lacour et al. proposed a molecular mechanism of cisplatin-induced cytotoxicity involving aSMase in human colon cancer cells (44). This study suggested that treatment with cisplatin induces a translocation of aSMase to the extracellular surface of the plasma membrane. Subsequently, ceramide production is activated and the TNFRSF6 (tumor necrosis factor receptor superfamily member 6) proapoptotic protein and caspase 8 (Casp8) are redistributed into membrane fractions enriched in cholesterol and sphingolipids (44) (Figure 4). However, the detailed molecular mechanism of cisplatin-induced cytotoxicity and how aSMase is involved still remains unclear. Further investigation in this field is required to unveil the specific molecular mechanism in order to design a personalized therapy.
4.4.4 Ovarian Cancer
Maurmann et al. postulate that aSMase activation and increased TNFRSF6 levels in cisplatin-resistant ovarian cells may suggest a similar mechanism as the one described by Lacour et al. for cisplatin-induced cytotoxicity (44, 108). In this study, they also demonstrated that in cisplatin-resistant cells, the aSMase activation of TNFRSF6 is dependent on cisplatin concentration (108). Furthermore, besides the cisplatin resistance, it has also been described that aSMase inhibition in ovarian cancer cells increase paclitaxel treatment resistance. It has been reported that aSMases and nSMases are activated by paclitaxel treatment in drug-sensitive cells, but are not affected in resistant cells (115) (Figure 4). Hence for this cancer model, alternative therapies should cover the targeting of resistant cells.
4.4.5 SMase in Other Cancer Types
There is also evidence of the importance of aSMase in other types of cancer, such as NSCLC, but further studies are needed to decipher the role of this enzyme in chemoresistance (109).
5 Clinical Implications
Sphingolipids act both as structural components of membranes and as bio-effector molecules. In the last decade, the study of sphingolipids has been important for the development of innovative therapeutic strategies for drug-resistant tumors. The central axis of sphingolipids´ metabolism is ceramide, which has been the focus of numerous studies. Ceramide can be generated or catabolized by many enzymes, leading to different sphingolipids. AC breaks down ceramide to produce sphingosine, and its inhibition usually sensitizes drug-resistant cells to treatment. Similarly, the inhibition of SPHK, that phosphorylates sphingosine into S1P, and GCS, that generates glucosylceramide from ceramide, also sensitizes drug-resistant cells. All these enzymes are involved in the elimination of ceramide by the cell; hence, lower levels of ceramide are linked to cancer drug resistance. In contrast, the inhibition of SMases, which generate ceramide from the sphingomyelin breakdown, leads to increased drug-resistance in cancer. Taking all of these factors into account, it seems clear that the regulation of these enzymes affects the levels of ceramide in the cell, subsequently providing resistance or sensitization to some chemotherapeutics (15). Many authors support the idea that, independently of which enzyme is deregulated, drug resistance is acquired when there are low levels of ceramide in the cell. For example, doxorubicin enhances ceramide production inside the cell by activation of SMases or enzymes of ceramide synthesis, which should drive the cell to apoptosis (15, 116). However, it has been demonstrated that ceramide upregulates GCS expression, resulting in a resistant phenotype (15). Unlike the other three enzymes (AC, SMase and SPHK), the drug resistance produced by GCS may not only be related to ceramide generation, since GCS products (glycosphingolipids) are involved in the increase of multi-drug resistance through ABCB1 expression. Moreover. GCS expression which is epigenetically regulated correlates with drug resistance in breast cancer cells (60). Nevertheless, to our knowledge the mechanism has not been identified by which SMase, SPHK or AC modulate their expression levels.
In general, although targeting sphingolipids such as ceramide or glycosphingolipids is a major approach to deal with drug resistance in cancer, targeting crucial enzymes involved in these pathways seems to be a promising strategy in this field. These enzymes could be considered as potential targets to modulate ceramide levels in the cell and, therefore, resistance to treatment, particularly in drug-resistant tumors. Further research about the protein levels of these enzymes should be extended to all cancer types to determine if the use of the modulators of these enzymes (inhibitors or enhancers) would be effective either to prevent or to revert resistance to current conventional treatments. Overall, inhibitors of AC, GCS and SPHK and enhancers of SMases could be potential targets for the treatment of drug-resistant tumors in the indicated cancer types. Some modulators for these enzymes have already been studied and are currently used for clinical purposes to manage treatment resistance. Unfortunately, none of these modulators has been approved for cancer treatment to date, although some of them are currently under study. For instance, tamoxifen, a classical drug to treat several cancer types with a complex action mechanism, inhibits AC (117). Therefore, for those cancers that are not treated with tamoxifen, administering this drug in combination with the standard treatment may prevent resistance to that standard treatment (62, 117). Likewise, treatment with doxorubicin or etoposide leads to a significant downregulation of SPHK1 and, consequently, an accumulation of ceramide within the cell which confers therapy sensitization to resistant cells (118).
In addition, some sphingolipid-targeting drugs are under study because of their potential use in cancer treatment in chemoresistant tumors. This is the case of the SPHK1 inhibitor PF-543 since it has been demonstrated that its dansylated form (DPF-543) provides high cytotoxicity. This compound also triggers aSMase activation, leading to a higher accumulation of ceramides within the cell (119). Other SPHK inhibitors, such as F-12509, SKI-I and FTY720 (Fingolimod), specifically inhibit SPHK1; ABC294640 (Opaganib) and K145 specifically inhibit SPHK2; and SKI-II which inhibits both SPHK1 and SPHK2. The inhibitor ABC294640 has recently reached phase 2 of clinical trials as a drug for refractory multiple myeloma patients (NCT02757326, www.clinicaltrials.gov). FTY720 is also being tested in clinical trials (www.clinicaltrials.gov) for breast cancer (NCT02490930) and glioma patients (NCT03941743). Likewise, several AC inhibitors, such as LCL521 and ceranib-2, have been described to improve chemotherapy effects (120, 121). Additional research on these molecules is required to evaluate their potential effects as a cancer therapy. Similarly, some GCS inhibitors, such as PPMP (D,L-Threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol) and PDMP (D,L-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol), have been tested in vitro with promising results. For instance, PPMP increased sensitivity of chemoresistant HNSCC to cisplatin and PDMP increased sensitivity of pancreatic cells (122, 123).
Although these compounds reached clinical trials, most of them did not show sufficient efficacy in patients to continue the study. The lack of efficacy of these compounds in patients could be explained by several reasons. First, different enzymes of sphingolipid metabolism are thought to have redundant functions, meaning that even if one enzyme is inhibited by one drug, a different enzyme could perform its function. For example, some studies suggest that SPHK1 and SPHK2 may have redundant functions and that one may compensate for the deficiency of the other (16, 17). A second explanation could be that these compounds that reached clinical trials present non-specific effects or that the compound is effective at high doses that are not clinically tolerable in patients (liver toxicity). A third reason could be the adverse effects that such compounds cause in patients under study, leading to discontinuation and definitive transfer to the clinic. Adverse effects may include hematuria, vomiting, hyperglycemia, personality disorders, fever, and dizziness. Overall, these compounds remain a promising strategy to treat chemoresistant tumors once these problems are overcome.
6 Conclusions
1. Reliable markers based upon sphingolipid enzymes able to stratify patients (i.e., responders to cancer therapy versus non-responders) into current therapies including immunotherapy are expected.
2. AC is a promising target and several AC inhibitors are already under study, although no clinical trials have been performed with these inhibitors to date.
3. Since SMases need to be overexpressed to potentially reduce drug resistance, such overexpression has only been reached by the addition of SMases or genetic modulation, but not pharmacologically. Additional studies of this enzyme are needed to test if the overexpression of this enzyme can be achieved pharmacologically.
4. Research on the characterization of SPHK inhibitors is growing due to the relation between GCS and multidrug resistance. However, none of those research projects has reached clinical trials yet. Further research on GCS and multidrug resistance relation in different cancer types is required, since the majority of the studies published to date have been performed in breast cancer.
5. The most promising drugs to modulate sphingolipids are SPHK inhibitors, which have been subject to in-depth study. Currently, some of these inhibitors, such as ABC294640 and fingolimod are in clinical trials with good results.
6. In the next few years, animal models should reveal whether the therapeutic exploitation of sphingolipids metabolism modulation will determine future treatments for oncology patients.
Author Contributions
MB: writing the manuscript. AS-G: design of figures. YG-M: reading and improving the scientific quality. CM: reading and improving the scientific quality. IR: reading and improving the scientific quality. ML: supervision and correction of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Teresa Moline and Rosa Somoza from the VHIR. This work was supported by grants from the Instituto de Salud Carlos III (ISCIII; PI20/00556 and CP03/00101 [ML]) and CIBERONC (ML). This work was also co-financed by the European Regional Fund (ERDF) and AECC (Spanish Association of Cancer Research) (Founding Ref. GC16173720CARR [ML]). YG-M, CM, and AS-G were supported by the VHIR, iP-FIS (ISCIII) and VHIR fellowships, respectively.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Yardley DA. Drug Resistance and the Role of Combination Chemotherapy in Improving Patient Outcomes. Int J Breast Cancer (2013) 2013:1–15. doi: 10.1155/2013/137414
3. Schmidt F, Efferth T. Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals (2016) 9:33–44. doi: 10.3390/ph9020033
4. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull (2017) 7:339–48. doi: 10.15171/apb.2017.041
5. Thudichum JLW. A Treatise on the Chemical Constitution of the Brain Based Throughout Upon Original Researches. London: Bailliere, Tindall and Cox (1884).
6. Futerman AH, Hannun YA. The Complex Life of Simple Sphingolipids. EMBO Rep (2004) 5:777–82. doi: 10.1038/sj.embor.7400208
7. Gouazé V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, et al. Overexpression of Glucosylceramide Synthase and P-Glycoprotein in Cancer Cells Selected for Resistance to Natural Product Chemotherapy. Mol Cancer Ther (2004) 3:633–9.
8. Wegner MS, Gruber L, Mattjus P, Geisslinger G, Grösch S. The UDP-Glucose Ceramide Glycosyltransferase (UGCG) and the Link to Multidrug Resistance Protein 1 (MDR1). BMC Cancer (2018) 18:153–63. doi: 10.1186/s12885-018-4084-4
9. Liu YY, Han TY, Giuliano AE, Cabot MC. Expression of Glucosylceramide Synthase, Converting Ceramide to Glucosylceramide, Confers Adriamycin Resistance in Human Breast Cancer Cells. J Biol Chem (1999) 274:1140–6. doi: 10.1074/jbc.274.2.1140
10. Giussani P, Bassi R, Anelli V, Brioschi L, De Zen F, Riccitelli E, et al. Glucosylceramide Synthase Protects Glioblastoma Cells Against Autophagic and Apoptotic Death Induced by Temozolomide and Paclitaxel. Cancer Invest (2012) 30:27–37. doi: 10.3109/07357907.2011.629379
11. Liu Y-Y, Han T-Y, Giuliano AE, Cabot MC. Ceramide Glycosylation Potentiates Cellular Multidrug Resistance. FASEB J (2001) 15:719–30. doi: 10.1096/fj.00-0223com
12. Liu YY, Patwardhan GA, Xie P, Gu X, Giuliano AE, Cabot MC. Glucosylceramide Synthase, a Factor in Modulating Drug Resistance, Is Overexpressed in Metastatic Breast Carcinoma. Int J Oncol (2011) 39:425–31. doi: 10.3892/ijo.2011.1052
13. Xie P, Shen YF, Shi YP, Ge SM, Gu ZH, Wang J, et al. Overexpression of Glucosylceramide Synthase in Associated With Multidrug Resistance of Leukemia Cells. Leuk Res (2008) 32:475–80. doi: 10.1016/J.LEUKRES.2007.07.006
14. Alshaker H, Srivats S, Monteil D, Wang Q, Low CMR, Pchejetski D. Field Template-Based Design and Biological Evaluation of New Sphingosine Kinase 1 Inhibitors. Breast Cancer Res Treat (2018) 172:33–43. doi: 10.1007/s10549-018-4900-1
15. Liu Y-Y, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, et al. A Role for Ceramide in Driving Cancer Cell Resistance to Doxorubicin. FASEB J (2008) 22:2541–51. doi: 10.1096/FJ.07-092981
16. Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential Role for Sphingosine Kinases in Neural and Vascular Development. Mol Cell Biol (2005) 25:11113. doi: 10.1128/MCB.25.24.11113-11121.2005
17. Hait NC, Sarkar S, Le Stunff H, Mikami A, Maceyka M, Milstien S, et al. Role of Sphingosine Kinase 2 in Cell Migration Toward Epidermal Growth Factor. J Biol Chem (2005) 280:29462–9. doi: 10.1074/jbc.M502922200
18. Wegner MS, Schömel N, Gruber L, Örtel SB, Kjellberg MA, Mattjus P, et al. UDP-Glucose Ceramide Glucosyltransferase Activates AKT, Promoted Proliferation, and Doxorubicin Resistance in Breast Cancer Cells. Cell Mol Life Sci (2018) 75:3393–410. doi: 10.1007/s00018-018-2799-7
19. Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, et al. Molecular Cloning and Functional Characterization of a Novel Mammalian Sphingosine Kinase Type 2 Isoform. J Biol Chem (2000) 275:19513–20. doi: 10.1074/jbc.M002759200
20. Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, et al. Sphingosine Kinase-1 as a Chemotherapy Sensor in Prostate Adenocarcinoma Cell and Mouse Models. Cancer Res (2005) 65:11667–75. doi: 10.1158/0008-5472.CAN-05-2702
21. Katsuta E, Yan L, Nagahashi M, Raza A, Sturgill JL, Lyon DE, et al. Doxorubicin Effect Is Enhanced by Sphingosine-1-Phosphate Signaling Antagonist in Breast Cancer. J Surg Res (2017) 219:202. doi: 10.1016/J.JSS.2017.05.101
22. Alshaker H, Wang Q, Kawano Y, Arafat T, Böhler T, Winkler M, et al. Everolimus (RAD001) Sensitizes Prostate Cancer Cells to Docetaxel by Down-Regulation of HIF-1α and Sphingosine Kinase 1. Oncotarget (2016) 7:80943–56. doi: 10.18632/oncotarget.13115
23. Bassi R, Brambilla S, Tringali C, Giussani P. Extracellular Sphingosine-1-Phosphate Downstream of EGFR Increases Human Glioblastoma Cell Survival. Int J Mol Sci (2021) 22:6824–38. doi: 10.3390/IJMS22136824
24. Sauer L, Nunes J, Salunkhe V, Skalska L, Kohama T, Cuvillier O, et al. Sphingosine Kinase 1 Inhibition Sensitizes Hormone-Resistant Prostate Cancer to Docetaxel. Int J Cancer (2009) 125:2728–36. doi: 10.1002/IJC.24640
25. Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P. Restoring Endocrine Response in Breast Cancer Cells by Inhibition of the Sphingosine Kinase-1 Signaling Pathway. Endocrinology (2009) 150:4484–92. doi: 10.1210/EN.2009-0391
26. Oancea-Castillo LR, Klein C, Abdollahi A, Weber K-J, Régnier-Vigouroux A, Dokic I. Comparative Analysis of the Effects of a Sphingosine Kinase Inhibitor to Temozolomide and Radiation Treatment on Glioblastoma Cell Lines. Cancer Biol Ther (2017) 18:400. doi: 10.1080/15384047.2017.1323583
27. Ruckhäberle E, Rody A, Engels K, Gaetje R, Von Minckwitz G, Schiffmann S, et al. Microarray Analysis of Altered Sphingolipid Metabolism Reveals Prognostic Significance of Sphingosine Kinase 1 in Breast Cancer. Breast Cancer Res Treat (2008) 112:41–52. doi: 10.1007/s10549-007-9836-9
28. Seelan RS, Qian C, Yokomizo A, Bostwick DG, Smith DI, Liu W. Human Acid Ceramidase Is Overexpressed But Not Mutated in Prostate Cancer. Genes Chromosom Cancer (2000) 29:137–46. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1018>3.0.CO;2-E
29. Liu X, Elojeimy S, Turner LS, Mahdy AEM, Zeidan YH, Bielawska A, et al. Acid Ceramidase Inhibition: A Novel Target for Cancer Therapy. Front Biosci (2008) 13:2293–8. doi: 10.2741/2843
30. Hoek KS, Goding CR. Cancer Stem Cells Versus Phenotype-Switching in Melanoma. Pigment Cell Melanoma Res (2010) 23:746–59. doi: 10.1111/J.1755-148X.2010.00757.X
31. Kemper K, de Goeje PL, Peeper DS, Amerongen Rv. Phenotype Switching: Tumor Cell Plasticity as a Resistance Mechanism and Target for Therapy. Cancer Res (2014) 74:5937–41. doi: 10.1158/0008-5472.CAN-14-1174
32. Yang WS, Moon H-G, Kim HS, Choi E-J, Yu M-H, Noh D-Y, et al. Proteomic Approach Reveals FKBP4 and S100A9 as Potential Prediction Markers of Therapeutic Response to Neoadjuvant Chemotherapy in Patients With Breast Cancer. J Proteome Res (2011) 11:1078–88. doi: 10.1021/PR2008187
33. Samet D, Barenholz Y. Characterization of Acidic and Neutral Sphingomyelinase Activities in Crude Extracts of HL-60 Cells. Chem Phys Lipids (1999) 102:65–77. doi: 10.1016/S0009-3084(99)00076-6
34. Hannun YA, Luberto C, Argraves KM. Enzymes of Sphingolipid Metabolism: From Modular to Integrative Signaling. Biochemistry (2001) 40:4893–903. doi: 10.1021/bi002836k
35. Marchesini N, Hannun YA. Acid and Neutral Sphingomyelinases: Roles and Mechanisms of Regulation. Biochem Cell Biol (2011) 82:27–44. doi: 10.1139/O03-091
36. Kusuda S, Chang-Yi C, Takahashi M, Tezuka T. Localization of Sphingomyelinase in Lesional Skin of Atopic Dermatitis Patients. J Invest Dermatol (1998) 111:733–8. doi: 10.1046/J.1523-1747.1998.00370.X
37. Ridgway ND, Lagace TA, Cook HW, Byers DM. Differential Effects of Sphingomyelin Hydrolysis and Cholesterol Transport on Oxysterol-Binding Protein Phosphorylation and Golgi Localization. J Biol Chem (1998) 273:31621–8. doi: 10.1074/JBC.273.47.31621
38. Aureli M, Murdica V, Loberto N, Samarani M, Prinetti A, Bassi R, et al. Exploring the Link Between Ceramide and Ionizing Radiation. Glycoconj J (2014) 31:449–59. doi: 10.1007/s10719-014-9541-y
39. Li Yh, Liu Ht, Xu J, Xing Ay, Zhang J, Wang Yw, et al. The Value of Detection of S100A8 and ASAH1 in Predicting the Chemotherapy Response for Breast Cancer Patients. Hum Pathol (2018) 74:156–63. doi: 10.1016/J.HUMPATH.2018.01.004
40. Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, Mc loughlin M, Fuks Z, et al. Ionizing Radiation Acts on Cellular Membranes to Generate Ceramide and Initiate Apoptosis. J Exp Med (1994) 180:525–35. doi: 10.1084/jem.180.2.525
41. Senchenkov A, Litvak DA, Cabot MC. Targeting Ceramide Metabolism - A Strategy for Overcoming Drug Resistance. J Natl Cancer Inst (2001) 93:347–57. doi: 10.1093/jnci/93.5.347
42. Santana P, Peña LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, et al. Acid Sphingomyelinase–Deficient Human Lymphoblasts and Mice Are Defective in Radiation-Induced Apoptosis. Cell (1996) 86:189–99. doi: 10.1016/S0092-8674(00)80091-4
43. Cervia D, Assi E, De Palma C, Giovarelli M, Bizzozero L, Pambianco S, et al. Essential Role for Acid Sphingomyelinase-Inhibited Autophagy in Melanoma Response to Cisplatin. Oncotarget (2016) 7:24995–5009. doi: 10.18632/oncotarget.8735
44. Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O, et al. Cisplatin-Induced CD95 Redistribution Into Membrane Lipid Rafts of HT29 Human Colon Cancer Cells. Cancer Res (2004) 64:3593–8. doi: 10.1158/0008-5472.CAN-03-2787
45. Smith ER, Merrill J, Obeid LM, Hannun YA. Effects of Sphingosine and Other Sphingolipids on Protein Kinase C. Methods Enzymol (2000) 312:361–73. doi: 10.1016/s0076-6879(00)12921-0
46. Testai FD, Landek MA, Dawson G. Regulation of Sphingomyelinases in Cells of the Oligodendrocyte Lineage. J Neurosci Res (2004) 75:66–74. doi: 10.1002/jnr.10816
47. Ségui B, Cuvillier O, Adam-Klages S, Garcia V, Malagarie-Cazenave S, Lévêque S, et al. Involvement of FAN in TNF-Induced Apoptosis. J Clin Invest (2001) 108:143. doi: 10.1172/JCI11498
48. Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. Role of Ceramide in Cellular Senescence. J Biol Chem (1995) 270:30701–8. doi: 10.1074/jbc.270.51.30701
49. Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed Cell Death Induced by Ceramide. Science (80- ) (1993) 259:1769–71. doi: 10.1126/science.8456305
50. Hannun YA, Obeid LM. Sphingolipids and Their Metabolism in Physiology and Disease. Nat Rev Mol Cell Biol (2018) 19:175–91. doi: 10.1038/nrm.2017.107
51. Hla T. Physiological and Pathological Actions of Sphingosine 1-Phosphate. Semin Cell Dev Biol (2004) 15:513–20. doi: 10.1016/j.semcdb.2004.05.002
52. Spiegel S, Milstien S. Sphingosine-1-Phosphate: An Enigmatic Signalling Lipid. Nat Rev Mol Cell Biol (2003) 4:397–407. doi: 10.1038/nrm1103
53. Radin NS, Shayman JA, Inokuchi J-I. Metabolic Effects of Inhibiting Glucosylceramide Synthesis With PDMP and Other Substances. Adv Lipid Res (1993) 26:183–211.
54. Hannun YA, Obeid LM. Principles of Bioactive Lipid Signalling: Lessons From Sphingolipids. Nat Rev Mol Cell Biol (2008) 9:139–50. doi: 10.1038/nrm2329
55. Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, et al. Involvement Of The Dihydroceramide Desaturase In Cell Cycle Progression In Human Neuroblastoma Cells. J Biol Chem (2007) 282:16718. doi: 10.1074/JBC.M700647200
56. Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (Longevity Assurance Genes) Become CerS (Ceramide Synthases)? Insights Into the Regulation of Ceramide Synthesis. J Biol Chem (2006) 281:25001–5. doi: 10.1074/jbc.R600010200
57. Hakomori SI. Cell Adhesion/Recognition and Signal Transduction Through Glycosphingolipid Microdomain. Glycoconj J (2000) 17:143–51. doi: 10.1023/A:1026524820177
58. Garofalo T, Manganelli V, Grasso M, Mattei V, Ferri A, Misasi R, et al. Role of Mitochondrial Raft-Like Microdomains in the Regulation of Cell Apoptosis. Apoptosis (2015) 20:621–34. doi: 10.1007/S10495-015-1100-X
59. Liu YY, Gupta V, Patwardhan GA, Bhinge K, Zhao Y, Bao J, et al. Glucosylceramide Synthase Upregulates MDR1 Expression in the Regulation of Cancer Drug Resistance Through Csrc and β-Catenin Signaling. Mol Cancer (2010) 9:145. doi: 10.1186/1476-4598-9-145
60. Liu J, Zhang X, Liu A, Zhang D, Su Y, Liu Y, et al. Altered Methylation of Glucosylceramide Synthase Promoter Regulates Its Expression and Associates With Acquired Multidrug Resistance in Invasive Ductal Breast Cancer. Oncotarget (2016) 7:36755. doi: 10.18632/ONCOTARGET.9337
61. Zhang X, Wu X, Li J, Sun Y, Gao P, Zhang C, et al. MDR1 (Multidrug Resistence 1) can Regulate GCS (Glucosylceramide Synthase) in Breast Cancer Cells. J Surg Oncol (2011) 104:466–71. doi: 10.1002/JSO.21958
62. Morad SAF, Cabot MC. Tamoxifen Regulation of Sphingolipid Metabolism—Therapeutic Implications. Biochim Biophys Acta (2015) 1851:1134. doi: 10.1016/J.BBALIP.2015.05.001
63. De Rosa MF, Sillence D, Ackerley C, Lingwood C. Role of Multiple Drug Resistance Protein 1 in Neutral But Not Acidic Glycosphingolipid Biosynthesis. J Biol Chem (2004) 279:7867–76. doi: 10.1074/jbc.M305645200
64. Hosain SB, Khiste SK, Uddin MB, Vorubindi V, Ingram C, Zhang S, et al. Inhibition of Glucosylceramide Synthase Eliminates the Oncogenic Function of P53 R273H Mutant in the Epithelial-Mesenchymal Transition and Induced Pluripotency of Colon Cancer Cells. Oncotarget (2016) 7:60575–92. doi: 10.18632/oncotarget.11169
65. Zhang X, Xie K, Zou J, Li Y, Mu H, Zhang B, et al. NF-κb Mediates the Effect of Glucosylceramide Synthase on P-Glycoprotein Modulation in a Drug-Resistance Leukemia Cell Line. Chin J Med Genet (2014) 31:34–8. doi: 10.3760/CMA.J.ISSN.1003-9406.2014.01.008
66. Ghosh S, Juin SK, Bhattacharyya Majumdar S, Majumdar S. Crucial Role of Glucosylceramide Synthase in the Regulation of Stem Cell-Like Cancer Cells in B16F10 Murine Melanoma. Mol Carcinog (2021) 60(12):840–58. doi: 10.1002/mc.23347
67. Antoon JW, Beckman BS. Sphingosine Kinase: A Promising Cancer Therapeutic Target. Cancer Biol Ther (2011) 11:647–50. doi: 10.4161/cbt.11.7.14921
68. Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, et al. SphK1 and SphK2, Sphingosine Kinase Isoenzymes With Opposing Functions in Sphingolipid Metabolism. J Biol Chem (2005) 280:37118–29. doi: 10.1074/jbc.M502207200
69. Antoon JW, White MD, Driver JL, Burow ME, Beckman BS. Sphingosine Kinase Isoforms as a Therapeutic Target in Endocrine Therapy Resistant Luminal and Basal-A Breast Cancer. Exp Biol Med (2012) 237:832–44. doi: 10.1258/ebm.2012.012028
70. Banno Y, Kato M, Hara A, Nozawa Y. Evidence for the Presence of Multiple Forms of Sph Kinase in Human Platelets. Biochem J (1998) 335:301–4. doi: 10.1042/bj3350301
71. Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S. Sphingosine Kinase Type 1 Promotes Estrogen-Dependent Tumorigenesis of Breast Cancer MCF-7 Cells. Exp Cell Res (2002) 281:115–27. doi: 10.1006/EXCR.2002.5658
72. Edsall LC, Cuvillier O, Twitty S, Spiegel S, Milstien S. Sphingosine Kinase Expression Regulates Apoptosis and Caspase Activation in PC12 Cells. J Neurochem (2001) 76:1573–84. doi: 10.1046/J.1471-4159.2001.00164.X
73. Xia P, Gamble JR, Wang L, Pitson SM, Moretti PAB, Wattenberg BW, et al. An Oncogenic Role of Sphingosine Kinase. Curr Biol (2000) 10:1527–30. doi: 10.1016/S0960-9822(00)00834-4
74. Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine Kinase, Sphingosine-1-Phosphate, and Apoptosis. Biochim Biophys Acta - Mol Cell Biol Lipids (2002) 1585:193–201. doi: 10.1016/S1388-1981(02)00341-4
75. French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, et al. Discovery and Evaluation of Inhibitors of Human Sphingosine Kinase. Cancer Res (2003) 63:5962–9.
76. Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, et al. High Expression of Sphingosine Kinase 1 and S1P Receptors in Chemotherapy-Resistant Prostate Cancer PC3 Cells and Their Camptothecin-Induced Up-Regulation. Biochem Biophys Res Commun (2006) 342:1284–90. doi: 10.1016/j.bbrc.2006.02.070
77. Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine Kinase-1 Expression Correlates With Poor Survival of Patients With Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J Neuropathol Exp Neurol (2005) 64:695–705. doi: 10.1097/01.JNEN.0000175329.59092.2C
78. Min J, Traynor D, Stegner AL, Zhang L, Hanigan MH, Alexander H, et al. Sphingosine Kinase Regulates the Sensitivity of Dictyostelium Discoideum Cells to the Anticancer Drug Cisplatin. Eukaryot Cell (2005) 4:178. doi: 10.1128/EC.4.1.178-189.2005
79. Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, et al. Targeting Nfκb Mediated Breast Cancer Chemoresistance Through Selective Inhibition of Sphingosine Kinase-2. Cancer Biol Ther (2011) 11:678–89. doi: 10.4161/cbt.11.7.14903
80. Bektas M, Johnson SP, Poe WE, Bigner DD, Friedman HS. A Sphingosine Kinase Inhibitor Induces Cell Death in Temozolomide Resistant Glioblastoma Cells. Cancer Chemother Pharmacol (2009) 64:1053–8. doi: 10.1007/s00280-009-1063-0
81. Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, et al. Targeting Sphingosine Kinase 1 Inhibits Akt Signaling, Induces Apoptosis, and Suppresses Growth of Human Glioblastoma Cells and Xenografts. Cancer Res (2009) 69:6915–23. doi: 10.1158/0008-5472.CAN-09-0664
82. Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, et al. Alterations of Ceramide/Sphingosine 1-Phosphate Rheostat Involved in the Regulation of Resistance to Imatinib-Induced Apoptosis in K562 Human Chronic Myeloid Leukemia Cells. J Biol Chem (2007) 282:10922–34. doi: 10.1074/jbc.M610157200
83. Salas A, Ponnusamy S, Senkal CE, Meyers-Needham M, Selvam SP, Saddoughi SA, et al. Sphingosine Kinase-1 and Sphingosine 1-Phosphate Receptor 2 Mediate Bcr-Abl1 Stability and Drug Resistance by Modulation of Protein Phosphatase 2A. Blood (2011) 117:5941–52. doi: 10.1182/blood-2010-08-300772
84. Marfe G, Di Stefano C, Gambacurta A, Ottone T, Martini V, Abruzzese E, et al. Sphingosine Kinase 1 Overexpression Is Regulated by Signaling Through PI3K, AKT2, and mTOR in Imatinib-Resistant Chronic Myeloid Leukemia Cells. Exp Hematol (2011) 39:653–65. doi: 10.1016/j.exphem.2011.02.013
85. Sun D, Wang S. Sphingosine Kinases Are Involved in the Regulation of All-Trans Retinoic Acid Sensitivity of K562 Chronic Myeloid Leukemia Cells. Oncol Lett (2021) 22:581–90. doi: 10.3892/ol.2021.12842
86. Xu R, Antwi Boasiako P, Mao C. Alkaline Ceramidase Family: The First Two Decades. Cell Signal (2021) 78:109860–72. doi: 10.1016/j.cellsig.2020.109860
87. Mao C, Obeid LM. Ceramidases: Regulators of Cellular Responses Mediated by Ceramide, Sphingosine, and Sphingosine-1-Phosphate. Biochim Biophys Acta (2008) 1781:424. doi: 10.1016/J.BBALIP.2008.06.002
88. Azuma N, O’brien JS, Moser HW, Kishimoto Y. Stimulation of Acid Ceramidase Activity by Saposin D. Arch Biochem Biophys (1994) 311:354–7. doi: 10.1006/ABBI.1994.1248
89. Realini N, Palese F, Pizzirani D, Pontis S, Basit A, Bach A, et al. Acid Ceramidase in Melanoma: Expression, Localization, and Effects of Pharmacological Inhibition. J Biol Chem (2016) 291:2422–34. doi: 10.1074/jbc.M115.666909
90. Elojeimy S, Liu X, Mckillop JC, El-Zawahry AM, Holman DH, Cheng JY, et al. Role of Acid Ceramidase in Resistance to FasL: Therapeutic Approaches Based on Acid Ceramidase Inhibitors and FasL Gene Therapy. Mol Ther (2007) 15:1259–63. doi: 10.1038/sj.mt.6300167
91. Norris JS, Bielawska A, Day T, El-Zawahri A, ElOjeimy S, Hannun Y, et al. Combined Therapeutic Use of AdGFPFasL and Small Molecule Inhibitors of Ceramide Metabolism in Prostate and Head and Neck Cancers: A Status Report. Cancer Gene Ther 2006 1312 (2006) 13:1045–51. doi: 10.1038/sj.cgt.7700965
92. Doan NB, Alhajala H, Al-Gizawiy MM, Mueller WM, Rand SD, Connelly JM, et al. Acid Ceramidase and Its Inhibitors: A De Novo Drug Target and a New Class of Drugs for Killing Glioblastoma Cancer Stem Cells With High Efficiency. Oncotarget (2017) 8:112662. doi: 10.18632/ONCOTARGET.22637
93. Tan SF, Liu X, Fox TE, Barth BM, Sharma A, Turner SD, et al. Acid Ceramidase Is Upregulated in AML and Represents a Novel Therapeutic Target. Oncotarget (2016) 7:83208–22. doi: 10.18632/oncotarget.13079
94. Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AEM, et al. The Functional Effects of Acid Ceramidase Overexpression in Prostate Cancer Progression and Resistance to Chemotherapy. Cancer Biol Ther (2007) 6:1455–60. doi: 10.4161/cbt.6.9.4623
95. Li FZ, Dhillon AS, Anderson RL, McArthur G, Ferrao PT. Phenotype Switching in Melanoma: Implications for Progression and Therapy. Front Oncol (2015) 5:31. doi: 10.3389/FONC.2015.00031
96. Leclerc J, Garandeau D, Pandiani C, Gaudel C, Bille K, Nottet N, et al. Lysosomal Acid Ceramidase ASAH1 Controls the Transition Between Invasive and Proliferative Phenotype in Melanoma Cells. Oncogene (2019) 38:1282–95. doi: 10.1038/S41388-018-0500-0
97. Bedia C, Casas J, Andrieu-Abadie N, Fabriàs G, Levade T. Acid Ceramidase Expression Modulates the Sensitivity of A375 Melanoma Cells to Dacarbazine. J Biol Chem (2011) 286:28200–9. doi: 10.1074/jbc.M110.216382
98. Fujimura T, Kambayashi Y, Ohuchi K, Muto Y, Aiba S. Treatment of Advanced Melanoma: Past, Present and Future. Life (2020) 10:1–15. doi: 10.3390/LIFE10090208
99. Lai M, Amato R, La Rocca V, Bilgin M, Freer G, Spezia P, et al. Acid Ceramidase Controls Apoptosis and Increases Autophagy in Human Melanoma Cells Treated With Doxorubicin. Sci Rep (2021) 11:1–14. doi: 10.1038/s41598-021-90219-1
100. Kus G, Kabadere S, Uyar R, Kutlu HM. Induction of Apoptosis in Prostate Cancer Cells by the Novel Ceramidase Inhibitor Ceranib-2. Vitr Cell Dev Biol - Anim (2015) 51:1056–63. doi: 10.1007/s11626-015-9932-9
101. Mahdy AEM, Cheng JC, Li J, Elojeimy S, Meacham WD, Turner LS, et al. Acid Ceramidase Upregulation in Prostate Cancer Cells Confers Resistance to Radiation: AC Inhibition, a Potential Radiosensitizer. Mol Ther (2009) 17:430–8. doi: 10.1038/mt.2008.281
102. Cheng JC, Bai A, Beckham TH, Marrison ST, Yount CL, Young K, et al. Radiation-Induced Acid Ceramidase Confers Prostate Cancer Resistance and Tumor Relapse. J Clin Invest (2013) 123:4344. doi: 10.1172/JCI64791
103. Doan NB, Nguyen HS, Montoure A, Al-Gizawiy MM, Mueller WM, Kurpad S, et al. Acid Ceramidase Is a Novel Drug Target for Pediatric Brain Tumors. Oncotarget (2017) 8:24753–61. doi: 10.18632/oncotarget.15800
104. Tan SF, Pearson JM, Feith DJ, Loughran TP. The Emergence of Acid Ceramidase as a Therapeutic Target for Acute Myeloid Leukemia. Expert Opin Ther Targets (2017) 21:583–90. doi: 10.1080/14728222.2017.1322065
105. Goñi FM, Alonso A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett (2002) 531:38–46. doi: 10.1016/S0014-5793(02)03482-8
106. Modrak DE, Gold DV, Goldenberg DM. Sphingolipid Targets in Cancer Therapy. Mol Cancer Ther (2006) 5:200–8. doi: 10.1158/1535-7163.MCT-05-0420
107. Grammatikos G, Teichgräber V, Carpinteiro A, Trarbach T, Weller M, Hengge UR, et al. Overexpression of Acid Sphingomyelinase Sensitizes Glioma Cells to Chemotherapy. Antioxidants Redox Signal (2007) 9:1449–56. doi: 10.1089/ars.2007.1673
108. Maurmann L, Belkacemi L, Adams NR, Majmudar PM, Moghaddas S, Bose RN. A Novel Cisplatin Mediated Apoptosis Pathway Is Associated With Acid Sphingomyelinase and FAS Proapoptotic Protein Activation in Ovarian Cancer. Apoptosis (2015) 20:960–74. doi: 10.1007/s10495-015-1124-2
109. Paul I, Chacko AD, Stasik I, Busacca S, Crawford N, McCoy F, et al. Acquired Differential Regulation of Caspase-8 in Cisplatin-Resistant Non-Small-Cell Lung Cancer. Cell Death Dis (2012) 3:e449. doi: 10.1038/CDDIS.2012.186
110. Gramatzki D, Herrmann C, Happold C, Becker KA, Gulbins E, Weller M, et al. Glioma Cell Death Induced by Irradiation or Alkylating Agent Chemotherapy Is Independent of the Intrinsic Ceramide Pathway. PLoS One (2013) 8:e63527–43. doi: 10.1371/journal.pone.0063527
111. Hara S, Nakashima S, Kiyono T, Sawada M, Yoshimura S, Iwama T, et al. P53-Independent Ceramide Formation in Human Glioma Cells During γ-Radiation-Induced Apoptosis. Cell Death Differ (2004) 11:853–61. doi: 10.1038/sj.cdd.4401428
112. Hawkins CC, Ali T, Ramanadham S, Hjelmeland AB. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules (2020) 10:1–23. doi: 10.3390/biom10101357
113. Smith EL, Schuchman EH. Acid Sphingomyelinase Overexpression Enhances the Antineoplastic Effects of Irradiation In Vitro and In Vivo. Mol Ther (2008) 16:1565–71. doi: 10.1038/mt.2008.145
114. Montfort A, Bertrand F, Rochotte J, Gilhodes J, Filleron T, Milhès J, et al. Neutral Sphingomyelinase 2 Heightens Anti-Melanoma Immune Responses and Anti–PD-1 Therapy Efficacy. Cancer Immunol Res (2021) 9:568–82. doi: 10.1158/2326-6066.CIR-20-0342
115. Prinetti A, Millimaggi D, D’Ascenzo S, Clarkson M, Bettiga A, Chigorno V, et al. Lack of Ceramide Generation and Altered Sphingolipid Composition Are Associated With Drug Resistance in Human Ovarian Carcinoma Cells. Biochem J (2006) 395:311. doi: 10.1042/BJ20051184
116. Murate T, Suzuki M, Hattori M, Takagi A, Kojima T, Tanizawa T, et al. Up-Regulation of Acid Sphingomyelinase During Retinoic Acid-Induced Myeloid Differentiation of NB4, a Human Acute Promyelocytic Leukemia Cell Line. J Biol Chem (2002) 277:9936–43. doi: 10.1074/jbc.M111594200
117. White-Gilbertson S, Lu P, Jones CM, Chiodini S, Hurley D, Das A, et al. Tamoxifen Is a Candidate First-in-Class Inhibitor of Acid Ceramidase That Reduces Amitotic Division in Polyploid Giant Cancer Cells—Unrecognized Players in Tumorigenesis. Cancer Med (2020) 9:3142. doi: 10.1002/CAM4.2960
118. Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, et al. Overcoming MDR-Associated Chemoresistance in HL-60 Acute Myeloid Leukemia Cells by Targeting Shingosine Kinase-1. Leukemia (2006) 20:95–102. doi: 10.1038/sj.leu.2404023
119. Shamshiddinova M, Gulyamov S, Kim H-J, Jung S-H, Baek D-J. Lee Y-M. A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation. Int J Mol Sci (2021) 22:9190–208. doi: 10.3390/IJMS22179190
120. Vethakanraj HS, Sesurajan BP, Padmanaban VP, Jayaprakasam M, Murali S, Sekar AK. Anticancer Effect of Acid Ceramidase Inhibitor Ceranib-2 in Human Breast Cancer Cell Lines MCF-7, MDA MB-231 by the Activation of SAPK/JNK, P38 MAPK Apoptotic Pathways, Inhibition of the Akt Pathway, Downregulation of Erα. Anticancer Drugs (2018) 29:50–60. doi: 10.1097/CAD.0000000000000566
121. Bai A, Mao C, Jenkins RW, Szulc ZM, Bielawska A, Hannun YA. Anticancer Actions of Lysosomally Targeted Inhibitor, LCL521, of Acid Ceramidase. PLoS One (2017) 12:e0177805–15. doi: 10.1371/JOURNAL.PONE.0177805
122. Wang T, Wei J, Wang N, Ma JL, Hui PP. The Glucosylceramide Synthase Inhibitor PDMP Sensitizes Pancreatic Cancer Cells to MEK/ERK Inhibitor AZD-6244. Biochem Biophys Res Commun (2015) 456:821–6. doi: 10.1016/j.bbrc.2014.12.019
Keywords: glucosylceramide synthase (GCS), sphingosine kinase 1 (SPHK1), sphingomyelinase (SMase), acid ceramidase (AC), cancer, shingolipids
Citation: Bataller M, Sánchez-García A, Garcia-Mayea Y, Mir C, Rodriguez I and LLeonart ME (2021) The Role of Sphingolipids Metabolism in Cancer Drug Resistance. Front. Oncol. 11:807636. doi: 10.3389/fonc.2021.807636
Received: 02 November 2021; Accepted: 07 December 2021;
Published: 23 December 2021.
Edited by:
Stefano Tiziani, University of Texas at Austin, United StatesReviewed by:
Min Hee Kang, Texas Tech University Health Sciences Center, United StatesAnna Bianchi-Smiraglia, University at Buffalo, United States
Copyright © 2021 Bataller, Sánchez-García, Garcia-Mayea, Mir, Rodriguez and LLeonart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matilde Esther LLeonart, bWF0aWxkZS5sbGVvbmFydEB2aGlyLm9yZw==