 Tycho de Bakker1,2Fabrice Journe2,3Géraldine Descamps3Sven Saussez3Tatiana Dragan1Ghanem Ghanem2Mohammad Krayem1,2*Dirk Van Gestel1
Tycho de Bakker1,2Fabrice Journe2,3Géraldine Descamps3Sven Saussez3Tatiana Dragan1Ghanem Ghanem2Mohammad Krayem1,2*Dirk Van Gestel1- 1Department of Radiation Oncology, Institut Jules Bordet, Université Libre de Bruxelles, Brussels, Belgium
- 2Laboratory of Clinical and Experimental Oncology (LOCE), Institut Jules Bordet, Université Libre de Bruxelles, Brussels, Belgium
- 3Laboratory of Human Anatomy and Experimental Oncology, Faculty of Medicine and Pharmacy, University of Mons, Mons, Belgium
TP53 mutation is one of the most frequent genetic alterations in head and neck squamous cell carcinoma (HNSCC) and results in an accumulation of p53 protein in tumor cells. This makes p53 an attractive target to improve HNSCC therapy by restoring the tumor suppressor activity of this protein. Therapeutic strategies targeting p53 in HNSCC can be divided into three categories related to three subtypes encompassing WT p53, mutated p53 and HPV-positive HNSCC. First, compounds targeting degradation or direct inhibition of WT p53, such as PM2, RITA, nutlin-3 and CH1iB, achieve p53 reactivation by affecting p53 inhibitors such as MDM2 and MDMX/4 or by preventing the breakdown of p53 by inhibiting the proteasomal complex. Second, compounds that directly affect mutated p53 by binding it and restoring the WT conformation and transcriptional activity (PRIMA-1, APR-246, COTI-2, CP-31398). Third, treatments that specifically affect HPV+ cancer cells by targeting the viral enzymes E6/E7 which are responsible for the breakdown of p53 such as Ad-E6/E7-As and bortezomib. In this review, we describe and discuss p53 regulation and its targeting in combination with existing therapies for HNSCC through a new classification of such cancers based on p53 mutation status and HPV infection.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is diagnosed in 890.000 patients each year worldwide, ranking it as the sixth most common cancer in the world (1). HNSCC is a collection of cancers encompassing the oral cavity, nasopharynx, oropharynx, hypopharynx and larynx (2). The most common genetic aberrations are the overexpression of the epidermal growth factor receptor (EGFR) and inhibiting mutations of p53 in 95% and 75-85% of non-HPV-related cases respectively (3). Additionally, the PI3K/AKT/mTOR signalling pathway is the most commonly mutated signalling pathway with up to 62% of HNSCC patients showing activating mutations. The hyperactivation of this pathway contributes to increased cell growth, proliferation and cellsurvival as well as regulation of apoptosis and DNA damage repair (4).
Due to the prevalence of p53 mutation, p53 is a frequent topic of many HNSCC studies. A large portion of those reports involves the reactivation of p53 which has become a promising treatment approach in combination with targeted therapies for HNSCC (5). In addition, p53 is responsible for the transcription activation of many genes involved in cell cycle arrest, apoptosis, senescence, DNA repair and metabolism (6). Indeed, activation of p53 (Figure 1A) occurs when either the cell accumulates too much DNA damage, an oncogene is activated, or in case of stressors such as nutrient deprivation or hypoxia (7, 8). Depending on the severity of these activators, the cell will undergo different responses such as induction of cell cycle arrest, apoptosis, senescence, induction of protective antioxidant activities, DNA repair as well as the alteration of the mitochondrial respiration (9). In cancer cells without functional p53, the cell has a deletion in one of the TP53 alleles and a mutated, non-functional form of TP53 in the other allele. Consequently, the cell can accumulate additional mutations, without resulting in apoptosis or cell cycle arrest for repair, and further activate oncogenes that result in oncogenesis (10) (Figure 1B).
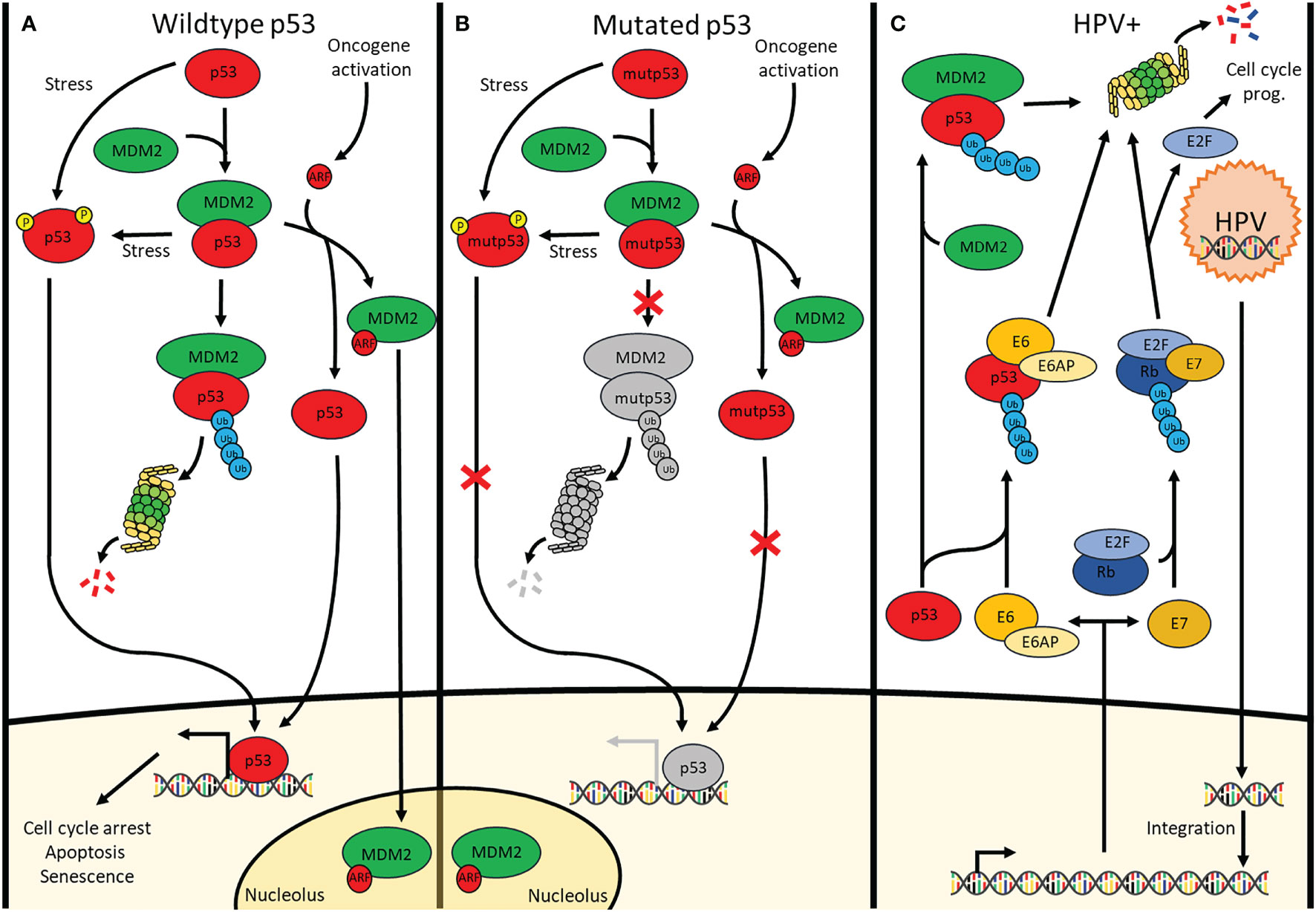
Figure 1 Signalling pathways of p53 in cells containing (A) wildtype p53 cells, (B) mutated p53 cells and (C) HPV-positive cells. (A) p53 is continuously polyubiquitinylated by MDM2 under physiological conditions. Additionally, there is MDM4 (Not shown) which inhibits p53 through direct interaction. Stressors cause p53 phosphorylation preventing or removing the binding of MDM2 & MDM4 to p53 allowing downstream transcription activation. Upon oncogene activation, the INK4A locus is transcribed, one of its products being ARF which sequesters MDM2 in the nucleolus thus releasing p53 allowing it to transcribe its target genes involved in cell cycle arrest, apoptosis and senescence. (B) Although mutated p53 undergoes the same pathways as wildtype p53, the mutation causes a lack of downstream transcription activation. (C) Upon infection by HPV, the viral DNA integrates into the genome where it is transcribed for the production of new viral particles. Viral E6 and E7 enzymes are transcribed as well causing the breakdown of p53 and Rb, respectively. This respectively results in loss of apoptosis induction and uncontrolled cell cycle progression.
In cells with wild-type p53 (WT p53), double- and single-stranded breaks in the DNA activate kinases such as ataxia-telangiectasia mutated (ATM) and ATM and Rad3 related (ATR), respectively (11), which in turn directly phosphorylate p53 at Ser-15 and will activate checkpoint kinase 2 (CHK2) and checkpoint kinase 1 (CHK1), respectively. The latter will subsequently phosphorylate Ser-20 of p53, inducing a change in its conformation and therefore removing inhibition of p53 by mouse double minute 2 (MDM2) which dissociates from p53 (12, 13). Additionally, ATM phosphorylates MDM2 and MDM4/X lead to the degradation of both of these p53 inhibitors, once again activating p53 (14). Under normal conditions, MDM2 continually causes polyubiquitination resulting in the proteasomal breakdown of p53. By contrast, MDM4/X binds p53 to inhibit its function as a transcription factor albeit without inducing the degradation of p53 (15). Upon activation of an oncogene, the Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) locus gets activated. This occurs through the inhibition of the histone modifier Enhancer of zeste homolog 2 (EZH2) causing derepression of the CDKN2A locus. Activation of this locus results in the expression of both p16, also known as INK4A, as well as p14, also known as ADP-ribosylation factor(ARF). ARF, in turn, binds MDM2 causing its dissociation from p53 and translocation and sequestration into the nucleolus, thereby activating p53. At the same time, p16 binds and inhibits Cyclin-dependent kinase 4/6 (CDK4/6) which prevents retinoblastoma protein (Rb) phosphorylation and thus also prevents the release of E2 factor (E2F), preventing cell cycle progression (16). One of p53’s target genes is p21, a protein with a similar function to p16, also causing inhibition of E2F and preventing cell cycle progression (17). The combination of p16 & p53 activation causes irreversible cell cycle arrest also known as senescence (18). In about 50% of cancers, p53 is mutated and in most of the other cancers, other parts of the p53 pathway are defective resulting in reduced or no p53 signalling (19, 20). This occurs through mechanisms such as overexpression of MDM2 or MDM4/X, downregulation of ARF or deregulation of microRNAs (21). In HNSCC, there are other ways in which p53 is inactivated besides inactivating mutations of p53. Based on data acquired from a 243-patientcohort collected by the cancer genome atlas (22) the highest percentage of mutations causing p53 inactivation in human papilloma virus (HPV) negative HNSCC occurs in TP53 itself (84%) followed by CDKN2A (57%) which either contains a homozygous deletion or a mutation (22). A recent computational analysis of the TCGA database revealed that, although more or less severe depending on the types of mutations and localization of the mutation within the gene, patients with mutated TP53 HNSCC were shown to have reduced overall survival (23). The 8 most common p53 mutations are all located in the DNA binding region which can be found between residues 103-292 (3) Among these mutants are 2 groups, a group that has mutated the specific residues responsible for binding the DNA and a second group that changes the conformation of the DNA binding element thereby also preventing DNA binding (24). Some of the treatments in this review specifically target these mutated conformations to induce the refolding into their original wild-type conformation. Other treatments target cofactors associated with p53 in order to result in reactivation of p53 (Table 1). Evidently, the mutational status of p53 in cancer that is to be treated is important for the choice of treatment that is to be used. While many different mechanisms for treatments have been targeted, not all treatment modalities have the same efficacy. The 84% mutation rate of p53 as well as the worse prognosis in HNSCC connected to these mutations warrant research towards a treatment capable of reactivating p53.
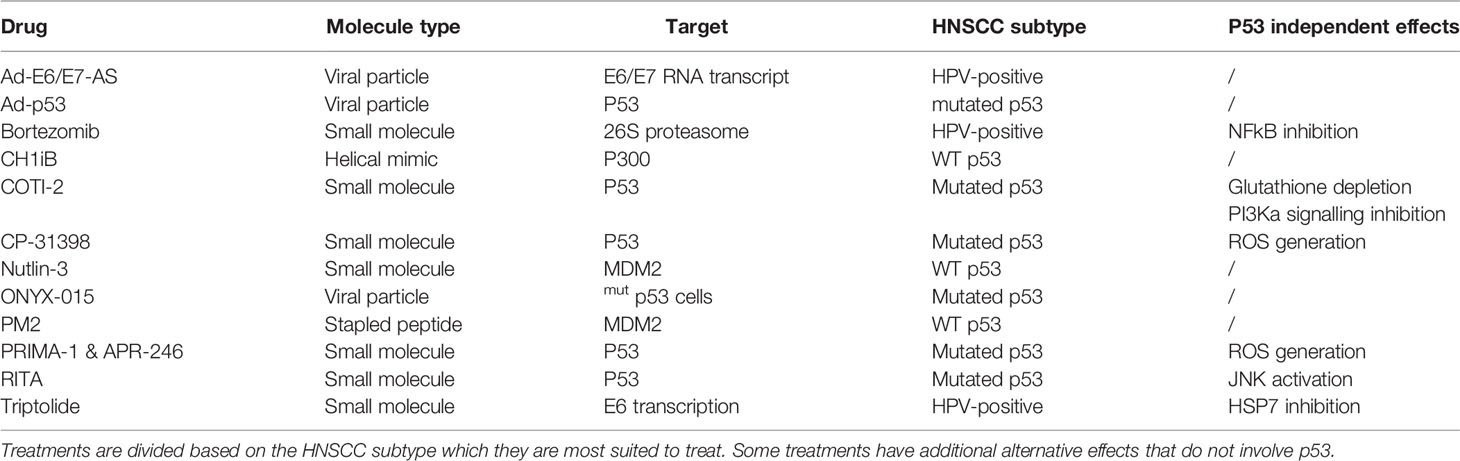
Table 1 The different types of molecules and mechanisms used to reactivate p53 by targeting either p53 directly or another protein preventing p53 activity.
HPV is the cause of a portion of HNSCC, particularly in the oropharynx. In recent years, HPV infection has become more and more prevalent (25) and might soon take the place of the most common cause of HNSCC from chronic exposure to tobacco and alcohol. Additionally, the average age of HPV-related head and neck cancer is significantly lower than that of non-HPV-related HNSCC (26). However, HPV-related cases of HNSCC have a more favorable prognosis than non-HPV-related HNSCC (27). Besides the difference in prognosis, the fundamental mechanism causing HNSCC is also different. The human papillomavirus contains 6 early proteins and 2 late ones. These late proteins are responsible for the capsid synthesis which is needed for the envelopment of the virus upon its duplication. The early proteins are involved in the initial steps of the infection, and include E1, E2 and E4 which are responsible for viral genome replication, viral DNA replication and transcription, and packing of the viral genome by late protein L1 and L2. However, some early proteins are known to be oncoproteins. Cellular transformation is mainly caused by the two viral proteins E6 and E7 (Figure 1C). In this context, E6 interacts with E6 associated protein (E6AP) and functions as a ubiquitin ligase specifically for p53 degradation. Nevertheless, the p53-E6AP-E6 complex does not always lead to degradation as p53 has 2 domains with which it interacts within the complex. There is a high-risk HPV-E6 which binds both a C-terminal binding domain not leading to degradation as well as a core-binding domain which does lead to degradation. By contrast, the low-risk HPV-E6 only binds the C-terminal domain and thus does not induce the degradation of p53. With high-risk HPV, E6 can lead to accumulation of mutations, eventually leading to the activation of oncogenes and the transformation into cancer cells (28). Simultaneously, E7 causes the breakdown of Rb which leads to a lack of cell cycle arrest and thus continued cell cycle progression once again contributing to oncogenesis. Of note, E7 contributes to accumulating p16 in the cytoplasm and nucleus of cancer cells, such accumulation being viewed as a master predictive marker of a transcriptionally active HPV infection.
Since HNSCC encompasses tumors in many different sites, treatment may be slightly different depending on the site. In general, the treatment schemes by stage are as follows. Treatment for early-stage disease (stage I-II) consists of either surgery or RT (29). Depending on adverse features in the resection, adjuvant radiotherapy +/- chemotherapy is advised (29). Advanced disease (stage III-IVa/b) is treated with the combination of surgery and postoperative RT or by post-operative chemoradiotherapy (CRT) in case of high risk factors (extracapsular extension and/or R1 resection) (29). Concomitant chemoradiotherapy (CCRT) is indicated when the patient is inoperable, in case of organ preservation or when surgery is considered to be too mutilating. Stage IVc cancers are highly influenced by the performance score and comorbidities which can call for palliative local or systemic treatment (30). The advent of T-cell-based immunotherapies in recent years now allows us to improve overall survival of recurrent HNSCC in approximately 20% of these patients (31, 32). Pembrolizumab and Nivolumab are immune checkpoint inhibitors whose mode of action is to reactivate cytotoxic T cells so that they can kill tumor cells. However, resistance to these treatments which is related to the immunosuppressive tumor microenvironment (TME) led to clinical trials combining immunotherapeutic treatment with targeting of the TME (33). Over the past decade, in young patients affected by an HPV+ oropharyngeal carcinoma, a treatment de-escalation program is under research to avoid the harmful side effects of cisplatin and radiotherapy in this population. This concept mainly consists of reduced radiotherapy doses, using less toxic chemotherapy agents such as Cetuximab or even removing chemotherapy administration (34). So far, this approach has been disappointing.
In this review, we will summarize the p53 reactivating treatments reported to get an anti-HNSCC activity in vitro, in vivo and/or in clinical settings. In addition, based on p53 mutational status and HPV infection, we will propose a new classification for HNSCC encompassing 1°) wildtype p53, 2°) mutated p53, and 3°) HPV-positive head and neck carcinomas, related to specific treatment strategies targeting p53 in order to restore its activity.
Molecular Compounds that Primarily Target Mutated p53 HNSCC
From large screening analyses, several small molecules were identified to directly interact with p53 and restore its activity. In many cases, such compounds had also p53-independent effects that may contribute to the antitumor effect in many cancers including HNSCC.
COTI-2
In 2016, a compound screening known as CHEMSAS discovered a new compound, COTI-2 (35), which is a third-generation thiosemicarbazone and has both a p53 dependent as well as a p53 independent mechanism to affect cancer cells. Although the exact mechanism for both modes of action is not well known, the independent mechanism involves downregulation of the PI3K/AKT/mTOR signalling pathway (5, 36). When comparing the IC50 of cell lines containing WT p53 versus mutated p53, it was observed that cell lines with mutated p53 were more sensitive to COTI-2 than cells with WT p53. Additionally, by using conformation-dependent antibodies, it was determined that COTI-2 could cause refolding of unfolded or misfolded p53 into a wild-type conformation (37). Using surface plasmon resonance, Synnot et al. evaluated the binding affinity of immobilized p53 with different concentrations of COTI-2. There it was seen that COTI-2 interacts with both the full-length p53 in addition to the DNA binding domain of p53 (37). The inherent properties of a thiosemicarbazone is its ability to form complexes with copper ions. These copper complexes subsequently interact with glutathione (GSH) which then allows the complex to be exported by the ABCC1, conferring resistance of the cell to COTI-2 (38).
COTI-2 shows both synergistic effects with cisplatin and radiotherapy in the treatment of HNSCC in both in vitro and in vivo setting (5). Currently, COTI-2 is being evaluated in a clinical trial encompassing gynaecological cancers, colorectal cancer, lung cancer and head and neck squamous cell carcinoma (NCT02433626) (39). However, at the time of writing, no results have been posted yet.
CP-31398
In 1999, a compound screening for protection of p53 from thermal denaturation was performed for a collection of 100.000 small molecules (40). Using two antibodies, mAb240 which binds an epitope only accessible when p53 is in an inactive conformation and mAb1620 which binds an epitope that becomes accessible upon p53 taking on the active conformation (34),Foster et al (40) showed that addition of CP-31398 (N′-[2-[2-(4-methoxyphenyl)ethenyl]-4-quinazolinyl]-N,N-dimethyl-1,3-propanediamine dihydrochloride), a prototype compound, increased the mAb1620 steady state and therefore the active conformation of p53. CP-31398 also shows elongated reporter expression in a p53 activated luciferase reporter constructs transfected into H1299 cells containing mutated p53, reflecting the restoration of the WT p53 function (40). Besides just activating p53, CP-31398 also causes p53 translocation to the mitochondria where it induces a release of cytochrome C which will subsequently initiate apoptosis (41). Further research indicates that the treatment of p53 mutant cells with CP-31398 increases the DNA binding capacity of the DNA binding domain of p53 (42). Other papers published results that indicated that CP-31398 has p53 independent growth-suppressive effects (43). This independent growth suppression is related to elevated ROS generation (44). CP-31398 was also tested for growth inhibition and induction of apoptosis of several HNSCC cell lines (45). While not being tested on HNSCC in vivo, CP-31398 has been shown to reduce growth or inhibit progression of different types of cancers such as skin (41), liver (46) and colorectal cancers (47) in animals.
RITA
RITA “reactivation of p53 and induction of apoptosis”, like nutlin-3, also interrupts the interaction between MDM2 and p53. RITA acts by binding p53 instead of MDM2 like nutlin-3 does (48). To identify the means of interaction between RITA and p53 in the cells, Issaeva et al. used the inherent fluorescent properties of RITA in a fluorescence correlation spectroscopy study. Based on the change in diffusion time (G(τ)) they showed that RITA interacts directly with p53 and does not interact with either MDM2 or GST. It was shown that RITA blocks the interaction of p53 with MDM2 and thus prevents degradation of p53 (48). However, at this time, RITA was not found to actively promote p53 activation through its binding (48). In vivo experiments seemed to show that the antitumor effect of RITA is WT p53 dependent (48). On the contrary, in 2011, Roh et al. showed that despite p53 mutational status, RITA induces an antitumor effect in FaDu, an HNSCC cell line containing a mutated form of p53 (45). This was also supported by an earlier paper in which the p53 activity of several different cell lines containing a diverse collection of mutations was tested for their response to RITA (49). This means that RITA can restore function to mutated p53 cells as well as WT p53 cells, and indicates that, in some way, RITA induces a conformational change in p53 as well as inhibits MDM2 from binding and causing degradation of p53 (49). Additionally, while not only causing apoptosis, RITA also seems to induce senescence in the HNSCC cell lines HN30 and HN1 (50). However, when p53 null PCI13 cells were treated with RITA, their clonogenic ability seemed to decrease. This observation points towards a p53-independent effect of RITA that was not described previously (50). Further investigation has also shown that RITA has a p53-independent activation of JNK signalling contributing to apoptosis (51). RITA has been shown to synergize with 3-MA, a PI3K inhibitor, both in vivo and in vitro in chemotherapy-resistant cells (52).
PRIMA-1 and APR-246
In 2002, Bykov et al. screened a library of low molecular weight compounds that suppress the proliferation of human cancer cells that contain mutated p53 (53). They discovered the compound 2,2-bis(hydroxymethyl)-1-azabicyclo[2,2,2]octan-3-one which was named “p53 reactivation and induction of massive apoptosis” or PRIMA-1. Three years later, they reported a structural analog APR-246 which is the methylated form of PRIMA-1. This modification allows for better diffusion across the cell membrane (54). When either compound enters the cell, the compound gets converted into methylene quinuclidinone (MQ).
MQ binds Cys124 of p53 located in the pocket between loop L2 and sheet S3 in the core domain of p53. This will occur in both WT as well as mutated p53, resulting in a WT conformation of p53 to induce transcription of p53 target genes (55). In addition to Cys124, another important cysteine residue is Cys277 (located in p53 core domain). According to substitution experiments, Cys277 showed to be essential for conserving the thermostabilising function of MQ. This was followed by testing the response of cells with p53 containing the same C277A and C124A substituted residues to APR-246 over time. Apoptotic markers such as Annexin V, p21, Bax and Fas were measured using FACS analysis. This showed that the cells containing either of the two substitutes had markedly less staining of these markers compared to the non-substituted control. These experiments indicate that both residues are essential for the proper reactivation of p53 in cells treated with APR-246 (56). Besides the direct reactivation of p53, APR-246 also induces several p53 independent effects. During the assessment of the antitumor effect of PRIMA-1, a change in the redox balance of the cell was observed. Reactive oxygen species (ROS) levels of the cell were increased while glutathione levels were decreased (55). Due to its sulfhydryl binding properties, MQ binds sulfhydryl groups other than those of p53, such as those in glutathione and antioxidant enzymes such as TrxR1, Prx3 or GPx-1. This in turn causes the accumulation of massive amounts of ROS in the cell synergizing with the reactivation of p53 to induce apoptosis. Furthermore, APR-246 dysregulates the NFE2L2/HMOX axis. This causes the activation of HMOX1 by NFE2L2 resulting in a general protective response that tries to balance the cellular redox. Therefore, knocking down the NFE2L2 gene causes increased sensitivity to PRIMA-1 (57). To achieve this effect in practice, PI3K or mTOR inhibitors can be used to break down NFE2L2 (57).
In recent years, treatments combined with PRIMA-1 or APR-246 have shown synergistic effects both in in vitro and in vivo settings. The combination of APR-246 and chemotherapeutics showed a synergistic effect. It is thought that reinstating the WT conformation of p53 may sensitize the cell to chemotherapeutics (54). Additionally, although not shown in head and neck squamous cell carcinoma, PRIMA-1 synergizes with radiotherapy in melanoma (58). Besides combinations with conventional treatments, it has been shown that piperlongumine (59) and PARP-1 (60) inhibitors respectively synergize with and sensitize HNSCC cells to PRIMA-1 and APR-246. Piperlongumine is a compound extracted from the piper longum L. plant which induces cell death both in vitro and in vivo in pancreatic cancer, breast cancer, leukemia (61). However, this effect is not observed in non-transformed cells. Piperlongumine directly binds to glutathione S transferase Pi 1 causing an increase in intracellular ROS and subsequent apoptotic cancer cell death. In addition to apoptosis, piperlongumine also causes cell cycle arrest, autophagy and inhibits migration and invasion (61). Besides targeting glutathione S transferase Pi 1 directly, docking of piperlongumine to AKT causes a downregulation of AKT phosphorylation both in vitro and in vivo (62). On the other hand, PARP-1 inhibitor affects the PARP protein primarily implicated in DNA damage repair, and also seem to inhibit TrxR1. This causes a decrease in available glutathione for a breakdown of ROS and therefore results in increased intracellular ROS levels as well (60). Both piperlongumine and PARP-1 inhibitors have functions that influence the redox state of the cells and this is the most likely mechanism in which these compounds synergize with PRIMA-1 and APR-246. Nowadays, several clinical trials have been performed with PRIMA-1 and APR-246, however, none with HNSCC patients.
Ad-p53
In recent years, viral particles which do not harm normal human cells have been investigated as a form of treatment. These viruses are used to lyse the oncogenic cells and are therefore called oncolytic viruses. Among the viruses being investigated are the herpes simplex virus (63), maraba virus (64) and adenoviral particles. For those types of treatments in this review, we focus on the viral treatments that specifically influence p53 but that is not the only mechanism by which these viral treatments exert antitumoral effects. One treatment termed Ad-p53, also known under the name gendicine, uses an adenoviral particle that can selectively infect both dividing and quiescent cancer cells. The adenoviral vector remains in the cell as an episomal vector which is capable of producing high amounts of the recombinant TP53 gene transcript. The WT p53, in turn, causes cell death in head and neck cancer cell lines in vitro (65). Additionally, treatment with Ad-p53 sensitizes the cells to radiotherapy (66). Finally, in xenograft mice, intratumoral injection of the adenoviral particle causes a significant reduction in tumour size (67).
ONYX-015
A similar adenovirus called ONYX-015 specifically kills p53 deficient cells. The viral protein E1b55k would normally prevent the activation of p53 allowing the replication of the adenovirus. In ONYX-015, there is an 827bp deletion in the E1B gene region that causes a premature stop codon in E1B55k, this allows p53 sufficient cells to induce apoptosis upon viral infection, preventing full replication of the virus. However, p53 deficient cells, which is often the cases with cancer cells, do not induce apoptosis regardless of the absence of E1B55k. This means that the virus can replicate and causes apoptosis upon accumulation of newly synthesized viral particles. This mechanism allows the virus to specifically infect and kill cancer cells (68, 69). However, later on, it was discovered that ONYX-015 also replicated in WT p53 cells. This seemed to be caused by a loss of p14ARF in those cells. It is thought that this is due to the lack of inhibition of MDM2, the p53 inhibitor. In the absence of p14ARF, MDM2 takes over the role of E1b55k and causes inhibition of p53. This in turn allows the replication of ONYX-015 (70).
Molecules That Target WTp53 HNSCC
Although many HNSCCs carry a mutated form of p53, 15-25% have WT p53 that is inhibited by overexpressed regulators such as MDM2 and MDMX/4 (3, 71). Different treatments can target this interaction in one of two ways. Either by preventing the inhibitor from binding p53 by occupying the binding site on p53 itself or by removing the inhibitor from p53 by occupying the p53 binding pocket on the inhibitor. Among these treatments are different peptides (72) and small molecules.
PM2
Although many stapled peptides such as SAH-p53-8 (73) and PM2 (72, 74) are developed and tested on many different cancers, only PM2 has been tested in HNSCC. A stapled peptide differs from standard peptides due to a crosslinking bond between different R- groups from several amino acids forming the peptide. This crosslink allows for a specific helical conformation and improves the stability and resistance of the peptide to protease degradation (75). A stapled peptide will interfere with the protein-protein interaction, which in this case is between p53 and MDM2 and/or MDMX/4.
PM2 is an inhibitor of both MDM2 and MDMX/4 which prevents their binding to p53. PM2 binds 3 residues in the N-terminal hydrophobic cleft of MDM2 thereby preventing its interaction with p53 (75). Since MDM2 is the E3 ubiquitin ligase for p53, its inhibition, therefore, causes accumulation of p53 in the cell (75). Although only tested in a limited amount of head and neck cancers, PM2 synergizes with radiotherapy (76). Additionally, PM2 sensitizes cells to targeted radionuclide therapy (72). Combining PM2 and 177Lu-labeled anti CD44v6 antibody (177Lu-AbN44v6) increases the antitumor effect when compared to monotherapies. However, this combination requires the expression of CD44v6 which is a cancer stem cell marker in many tumors, and is one of the CD44 isoforms (72).
Nutlin-3
Nutlins are a group of cis-imidazoline analogs that were discovered in 2004 during a screening for compounds that could inhibit the p53-MDM2 interaction. After testing three nutlins for their IC50 values, Nutlin-3 showed the most potent inhibitory effect. Additionally, it was the arbitrarily assigned enantiomer A that was about 150 times more potent than enantiomer B (IC50: 0.09µM vs. 13.6µM). The interaction of the nutlins with MDM2 was determined using crystallography. This shows that the nutlins bind MDM2 in the p53 binding pocket, thereby preventing its binding to p53 itself (77). Using a pan-caspase inhibitor before administration of the active enantiomer caused a reduction in apoptosis, showing that nutlin-3 causes cell death in a caspase-dependent manner involving p53. In vivo, Nutlin-3 also showed a high percentage of tumor growth inhibition (77).
In 2010, nutlin-3 was shown to radio sensitize laryngeal carcinoma cells (78). By using several different cell lines with differing p53 mutational status, researchers showed that treating cells with the combination of radiotherapy and nutlin-3 decreased clonogenicity more than with radiotherapy alone. Additionally, cell lines containing WT p53 had increased senescence compared to mutant cells when treated with nutlin-3 combined with radiotherapy compared to radiotherapy alone. This indicates that the radiosensitizing effect of nutlin-3 requires WT p53 (78). In addition to radiosensitizing, Nutlin-3 also enhances the cytotoxicity of chemotherapeutic agents such as cisplatin. Once again, this effect is more pronounced in cells that express WT p53 (45).
Generally, p53 is highly mutated in HNSCC, however, this is not the case for nasopharyngeal cancer where the mutation of p53 is rarer (13%) (79). Therefore, nutlin-3, which requires WT p53 to efficiently exert its function may help improve the outcome for patients with nasopharyngeal cancer. Then, as expected, when testing in nasopharyngeal cancer cell lines, nutlin-3 sensitizes these cells to cisplatin (80, 81).
In recent years, a combination of nutlin-3 and immunotherapy has been explored extensively as well. CD4+ T cells that are reactive against a selected peptide from MDM2 have been developed and showed promising effects against cancer progression. When tested in combination with nutlin-3, the expression of MDM2 in cancer cells was increased which in turn increases the anti-tumour response of the CD4+ T cells (82).
Finally, while not proven in HNSCC, Nutlin-3 has been found to activate anti-apoptotic signalling pathways such as the JNK-pathway which gets activated by p53 and subsequently activates heme oxidase -1 (HO-1). HO-1 may subsequently metabolize heme groups whose metabolites possess anti-apoptotic properties (83). Additionally, p53 may also indirectly phosphorylate MEK and ERK from the RAS/RAF/MEK/ERK pathway which is known to confer anti-apoptotic signals (84). Activation of both pathways involves translocation of activated p53 to mitochondria where it may induce the release of ROS and activation of both JNK as well as MEK and ERK. Finally, Nutlin-3 also interacts with members of the BCL-2 family. However, in which way this affects their function is not well known (85).
CH1iB
Some drugs target the cellular mechanisms that are specific to HPV-positive HNSCC. The incidence of HPV+-HNSCC increases year over year making this type of treatment more and more relevant. One of these treatments targets the CH1 domain (amino acids 350 to 450) of p300, a co-activator of transcription factor like p53. Besides degrading p53, HPV E6 also associated with p300 preventing the acetylation of p53 (86). Acetylated p53 is more stable and has increased transcriptional activity. Xie et al. (86) found that blocking the interaction between HPV E6 and p300 through an exogenic expression of the CH1 domain could prevent the breakdown of p53 due to competitive inhibition of HPV E6 by the exogenic CH1 (86).
Xie et al. transfected HPV+ cells (UMSCC47 and UPCI : SCC090) with an exogenous CH1 polypeptide (86). The evaluation of p53 in these cell lines showed that the transfected cells had more acetylated p53 than the non-transfect cells. The transformation of cells with exogenous CH1 showed a significant decrease in clonogenic survival compared to the control. Ectopic expression of the CH1 domain of p300 in HPV-positive HNSCC also sensitized the cells to cisplatin (86). In HPV- cells, the same results were obtained as p300 also binds MDM2. Therefore, exogenous CH1 also blocks the interaction between p300 and MDM2 which subsequently increases the p53 activity (86). To implement this principle in the clinic, a helical mimic named CH1iB inhibiting the p300-HPV E6 enzyme interaction has been developed. However, it is important to note that only inhibition of binding site B of CH1 with the mimic CH1iB showed to be efficacious. CH1iB was reported to reduce the interaction between p300 and HPV E6 thus allowing p300 to acetylate p53 which resulted in an increase in p53 transcriptional activation (86, 87).
Molecules That Specifically Target HPV+ HNSCC
Ad-E6/E7-AS
Like Ad-p53 & ONYX-015 earlier in this paper, other adenovirus particles are used in combatting HNSCC. An adenoviral particle, first tested in cervical cancer cells, transfers the HPV16 E6/E7 antisense RNA (Ad-E6/E7-AS) (88). When Ad-E6/E7-AS infects the HPV+ cell, the antisense RNA will hybridize with the RNA for HPV proteins E6 and E7. The new dsRNA will be degraded by the cell preventing translation of the E6 and E7 transcript. Therefore, the p53 protein which is normally degraded is then expressed at a normal level due to the absence of the proteasomal breakdown originally caused by the HPV-derived E6 protein. When treated with Ad-E6/E7-AS, the growth of the head and neck cancer cell line SCC47 was stopped. Additionally, it was also tested in combination with cisplatin which showed a synergistic effect with Ad-E6/E7-AS (89).
Bortezomib
In 1995, Muogenics developed a molecule called Bortezomib. A compound that targets the proteasomal 26S complex and thereby also preventing the breakdown of p53 by either MDM2 or HPV E6. It increases apoptosis in HPV+ cell lines but does not affect sensitivity to chemo- or radiotherapy (90). In addition to the inhibition of p53 breakdown, bortezomib also inhibits the downregulation of IkB. The release of the p65-p50 complex is subsequently also inhibited and will prevent activation of the NF-kB target genes resulting in decreased proliferation and cell survival (91). In addition, phosphorylation of AKT is also decreased due to increased activity of the phosphatase PP2A which results in reduced signalling to mTOR contributing to bortezomib-induced apoptosis. Bortezomib has subsequently been shown to decrease proliferation both in vitro as well as in vivo (92).
Triptolide and Minnelide
Triptolide is a molecule that was originally isolated from the Chinese herb Tripterygium wilfordii. Ever since it has been extensively studied in several types of diseases and cancers (93), (94). Its more water-soluble derivative Minnelide, prevents E6 transcription thereby also preventing the breakdown of p53 and thus resulting in massive apoptosis (95). Minnelide has also been shown to induce apoptosis and helps overcome cisplatin resistance in PDXs in vivo (96). Additionally, Minnelide inhibits HSP70 and reduces the secretion of IFN-y resulting in decreased PD-L1 expression.
Targeting p53 Through Macrophage Polarization
Many studies have shown the involvement of immune cells in the progression of HNSCC, and tumor-associated macrophages (TAMs) are an important element of the TME of HNSCC promoting multiple aspects of cancer development. In HNSCC patients, it is now demonstrated that high density of TAMs correlates with poor prognosis (97). Furthermore, 80% of stained macrophages in the TME are macrophages of M2 phenotype, namely TAMs (98). It is increasingly clear that the polarization of macrophages into TAMs and their infiltration in the TME is a poor prognostic factor, making these TAMs a promising target for HNC patients. A first model of chronic liver damage has initially demonstrated that proliferating stellate cells deficient for p53 were able to stimulate polarization of macrophages into a tumor-promoting M2-state and enhance the proliferation of premalignant cells (99) Since then, many studies focused on the clinical and prognostic implication of p53 on macrophage infiltration in some cancers and revealed that patients with mutant TP53 had significantly higher macrophage infiltration than those with wild-type TP53 and this was correlated with poorer survival (100). Moreover, a recent study on bladder carcinoma highlighted the association between M2 macrophage polarization and p53 dysfunction (101). Interestingly, Fang et al. recently tested a pharmacological p53 activator in combination with PD-1 blockade and investigated the role of p53 on immune modulation. Their results were very encouraging as they demonstrated that p53 activation promotes antitumor immunity in the TME, with a decrease in M2 macrophages, both in TP53 wild-type and TP53 mutated mice tumors (102).
Targeting p53 in HNSCC in the Clinical Setting
Although many treatments have been tested in preclinical settings, only some have reached clinical trials in treating HNSCC patients. Adenoviral particles are most commonly used in clinical trials. The only small compound that is currently tested in clinical studies involving HNSCC is COTI-2, which is used in combination with cisplatin in a phase II trial. However, no results have been posted yet (NCT02433626).
The most researched aforementioned treatment is the Ad-p53. While it is not yet used in western clinics, Ad-p53 was approved in October of 2003 by the Chinese food and drug administration under the name gendicine (103). Between the years 2000 and 2010, multiple clinical trials using combinations of gendicine and radiotherapy or chemotherapy have been performed. Both the total response rate as well as the overall 1-, 2-, 3- and 5-year survival rates were significantly improved when gencidine was added to the treatment compared to the conventional treatment alone (104). One clinical trial shows that the complete response of the combination of gencidine and radiotherapy was increased 2.1 fold compared to radiotherapy alone (62.5% versus 29.6%). Additionally, the 3-year overall survival was 14.4% higher in the combination group. Currently, for HNSCC, 4 clinical trials currently evaluate the efficacy of Ad-p53 either alone or in combination with other treatments such as immune checkpoint inhibitors or the standard of care, concomitant chemoradiotherapy (NCT02842125, NCT00003257, NCT00017173, NCT03544723).
Similarly, ONYX-015 has also been used in a clinical setting in HNSCC patients. A 2016 Phase II clinical trial showed ONYX-015 to be safe for use. In the same study, it is suggested that the treatment also shows modest antitumor activity (105). The last Phase III trial using ONYX-015 in combination with cisplatin and fluorouracil was withdrawn in 2013 without posting any results (NCT00006106).
Conclusion
This review discusses the current status of pharmacological manipulation of p53 for the treatment of HNSCC. Due to the need for new treatment combinations in HNSCC, many studies have looked at different p53 reactivators for an answer. The three described strategies to reactivate p53 in HNSCC are proposed regarding to the mutational status of p53 and the infection by HPV, which lead to a new classification of HNSCC encompassing WT p53, mutated p53 and HPV positive cancer cells. Such subtypes may be associated with effective and specific treatment strategies that must be optimized before clinical application. While some of these drugs (COTI-2 and adenoviral particles) have been tested in clinical settings, more research is needed to find more specific, safe and effective p53 reactivators to improve the treatment of HNSCC.
Author Contributions
TB and MK collected the data and drafted the manuscript. DG, FJ, and GD revised the manuscript. TD, SS, and GG provided additional revisions. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by “Les Amis de l’Institut Jules Bordet”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Laura QM, Chow MD. Head and Neck Cancer. New Engl J Med (2020) 382:60–72. doi: 10.3109/02841869609101661
2. Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clinic Proc (2016) 91:386–96. doi: 10.1016/j.mayocp.2015.12.017
3. Zhou G, Liu Z, Myers JN. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J Cell Biochem (2016) 117:2682–92. doi: 10.1002/jcb.25592.TP53
4. Schötz U, Balzer V, Brandt F-W, Ziemann F, Subtil FSB, Rieckmann T, et al. Dual PI3K/mTOR Inhibitor NVP-BEZ235 Enhances Radiosensitivity of Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines Due to Suppressed Double-Strand Break (DSB) Repair by Non-Homologous End Joining. Cancers (2020) 12:467. doi: 10.3390/cancers12020467
5. Lindemann A, Novel Thiosemicarbazone Derivative A. Exhibits Antitumor Activity in HNSCC Through P53-Dependent and -Independent Mechanisms. Clin Cancer Res (2019) 25:5650–62. doi: 10.1158/1078-0432.CCR-19-0096
6. Levine AJ, Momand J, Finlay CA. The P53 Tumour Suppressor Gene. Nature (1991) 351:453–6. doi: 10.1038/351453a0
8. Levine AJ, Hu W, Feng Z. The P53 Pathway: What Questions Remain to be Explored? Cell Death Differ (2006) 13:1027–36. doi: 10.1038/sj.cdd.4401910
9. Horn HF, Vousden KH. Coping With Stress: Multiple Ways to Activate P53. Oncogene (2007) 26:1306–16. doi: 10.1038/sj.onc.1210263
10. Chin D, Boyle GM, Theile DR, Parsons PG, Coman WB. Molecular Introduction to Head and Neck Cancer (HNSCC) Carcinogenesis. Br J Plast Surg (2004) 57:595–602. doi: 10.1016/j.bjps.2004.06.010
11. Reaper PM, Griffiths MR, Long JM, Charrier JD, MacCormick S, Charlton PA, et al. Selective Killing of ATM- or P53-Deficient Cancer Cells Through Inhibition of ATR. Nat Chem Biol (2011) 7:428–30. doi: 10.1038/nchembio.573
12. Ou YH, Chung PH, Sun TP, Shieh SY. P53 C-Terminal Phosphorylation by CHK1 and CHK2 Participates in the Regulation of DNA-Damage-Induced C-Terminal Acetylation. Mol Biol Cell (2005) 16:1684–95. doi: 10.1091/mbc.e04-08-0689
13. Sun B, Ross SM, Rowley S, Adeleye Y, Clewell RA. Contribution of ATM and ATR Kinase Pathways to P53-Mediated Response in Etoposide and Methyl Methanesulfonate Induced DNA Damage. Environ Mol Mutagen (2017) 58:72–83. doi: 10.1002/em.22070
14. Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM Activates P53 by Regulating MDM2 Oligomerization and E3 Processivity. EMBO J (2009) 28:3857–67. doi: 10.1038/emboj.2009.294
15. Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ (2018) 25:133–43. doi: 10.1038/cdd.2017.174
16. Rayess H, Wang MB, Srivatsan ES. Cellular Senescence and Tumor Suppressor Gene P16. Int J Cancer (2012) 130:1715–25. doi: 10.1002/ijc.27316
17. Dimri GP, Nakanishi M, Desprez PY, Smith JR, Campisi J. Inhibition of E2F Activity by the Cyclin-Dependent Protein Kinase Inhibitor P21 in Cells Expressing or Lacking a Functional Retinoblastoma Protein. Mol Cell Biol (1996) 16:2987–97. doi: 10.1128/MCB.16.6.2987
18. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A Senescence Program Controlled by P53 and P16ink4a Contributes to the Outcome of Cancer Therapy. Cell (2002) 109:335–46. doi: 10.1016/S0092-8674(02)00734-1
19. Levine AJ. Reviewing the Future of the P53 Field. Cell Death Differ (2018) 25:1–2. doi: 10.1038/cdd.2017.181
20. Duffy MJ, Synnott NC, McGowan PM, Crown J, O’Connor D, Gallagher WM. P53 as a Target for the Treatment of Cancer. Cancer Treat Rev (2014) 40:1153–60. doi: 10.1016/j.ctrv.2014.10.004
21. Prokocimer M, Molchadsky A, Rotter V. Dysfunctional Diversity of P53 Proteins in Adult Acute Myeloid Leukemia: Projections on Diagnostic Workup and Therapy. Blood (2017) 130:699–712. doi: 10.1182/blood-2017-02-763086
22. The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
23. Caponio VCA, Troiano G, Adipietro I, Zhurakivska K, Arena C, Mangieri D, et al. Computational Analysis of TP53 Mutational Landscape Unveils Key Prognostic Signatures and Distinct Pathobiological Pathways in Head and Neck Squamous Cell Cancer. Br J Cancer (2020) 123:1302–14. doi: 10.1038/s41416-020-0984-6
24. Chen JM, Rosal R, Smith S, Pincus MR, Brandt-Rauf PW. Common Conformational Effects of P53 Mutations. J Protein Chem (2001) 20:101–5. doi: 10.1023/a:1011065022283
25. Castellsagué X, Alemany L, Quer M, Halec G, Quirós B, Tous S, et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. J Natl Cancer Inst (2016) 108:djv403. doi: 10.1093/jnci/djv403
26. Chaturvedi A, Engels E, Anderson W, Gillison M. Incidence Trends for Human Papillomavirus-Related (HPV-R) and Unrelated (HPV-U) Head and Neck Squamous Cell Carcinomas (HNSCC) in the United States (Us). J Clin Oncol (2007) 25:6001. doi: 10.1200/jco.2007.25.18_suppl.6001
27. Koneva LA, Zhang Y, Virani S, Hall PB, McHugh JB, Chepeha DB, et al. HPV Integration in HNSCC Correlates With Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol Cancer research: MCR (2018) 16:90–102. doi: 10.1158/1541-7786.MCR-17-0153
28. Rampias T, Sasaki C, Psyrri A. Molecular Mechanisms of HPV Induced Carcinogenesis in Head and Neck. Oral Oncol (2014) 50:356–63. doi: 10.1016/j.oraloncology.2013.07.011
29. Lee YG, Kang EJ, Keam B, Choi JH, Kim JS, Park KU, et al. Treatment Strategy and Outcomes in Locally Advanced Head and Neck Squamous Cell Carcinoma: A Nationwide Retrospective Cohort Study (KCSG HN13-01). BMC Cancer (2020) 20:1–9. doi: 10.1186/s12885-020-07297-z
30. Oosting SF, Haddad RI. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front Oncoll (2019) 9:815. doi: 10.3389/fonc.2019.00815
31. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. New Engl J Med (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
32. Cohen EEW, Soulières D, le Tourneau C, Dinis J, Licitra L, Ahn M-J, et al. Pembrolizumab Versus Methotrexate, Docetaxel, or Cetuximab for Recurrent or Metastatic Head-and-Neck Squamous Cell Carcinoma (KEYNOTE-040): A Randomised, Open-Label, Phase 3 Study. Lancet (London England) (2019) 393:156–67. doi: 10.1016/S0140-6736(18)31999-8
33. Economopoulou P, Kotsantis I, Psyrri A. Tumor Microenvironment and Immunotherapy Response in Head and Neck Cancer. Cancers (2020) 12:3377. doi: 10.3390/cancers12113377
34. Mehanna H, Rischin D, Wong SJ, Gregoire V, Ferris R, Waldron J, et al. De-Escalation After DE-ESCALATE and RTOG 1016: A Head and Neck Cancer InterGroup Framework for Future De-Escalation Studies. J Clin Oncol: Off J Am Soc Clin Oncol (2020) 38:2552–7. doi: 10.1200/JCO.20.00056
35. Salim KY, Maleki Vareki S, Danter WR, Koropatnick J. COTI-2, a Novel Small Molecule That is Active Against Multiple Human Cancer Cell Lines In Vitro and In Vivo. Oncotarget (2016) 7:41363–79. doi: 10.18632/oncotarget.9133
36. Salim KY, Maleki Vareki S, Danter WR, Koropatnick J. 38 - COTI-2, a New Anticancer Drug Currently Under Clinical Investigation, Targets Mutant P53 and Negatively Modulates the PI3K/AKT/mTOR Pathway. Eur J Cancer (2016) 69:S19. doi: 10.1016/S0959-8049(16)32638-7
37. Synnott NC, O’Connell D, Crown J, Duffy MJ. COTI-2 Reactivates Mutant P53 and Inhibits Growth of Triple-Negative Breast Cancer Cells. Breast Cancer Res Treat (2020) 179:47–56. doi: 10.1007/s10549-019-05435-1
38. Bormio Nunes JH, Hager S, Mathuber M, Pósa V, Roller A, Enyedy ÉA, et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J Med Chem (2020) 63:13719–32. doi: 10.1021/acs.jmedchem.0c01277
39. Alsahafi E, Begg K, Amelio I, Raulf N, Lucarelli P, Sauter T, et al. Clinical Update on Head and Neck Cancer: Molecular Biology and Ongoing Challenges. Cell Death Dis (2019) 10:540–57. doi: 10.1038/s41419-019-1769-9
40. Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological Rescue of Mutant P53 Conformation and Function. Science (1999) 286:2507–10. doi: 10.1126/science.286.5449.2507
41. Tang X, Zhu Y, Han L, Kim AL, Kopelovich L, Bickers DR, et al. CP-31398 Restores Mutant P53 Tumor Suppressor Function and Inhibits UVB-Induced Skin Carcinogenesis in Mice. J Clin Invest (2007) 117:3753–64. doi: 10.1172/JCI32481
42. Demma MJ, Wong S, Maxwell E, Dasmahapatra B. CP-31398 Restores DNA-Binding Activity to Mutant P53 In Vitro But Does Not Affect P53 Homologs P63 and P73. J Biol Chem (2004) 279:45887–96. doi: 10.1074/jbc.M401854200
43. Rippin TM, Bykov VJN, Freund SMV, Selivanova G, Wiman KG, Fersht AR. Characterization of the P53-Rescue Drug CP-31398 In Vitro and in Living Cells. Oncogene (2002) 21:2119–29. doi: 10.1038/sj.onc.1205362
44. Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a Novel P53-Stabilizing Agent, Induces P53-Dependent and P53-Independent Glioma Cell Death. Oncogene (2003) 22:8233–45. doi: 10.1038/sj.onc.1207198
45. Roh J-L, Kang SK, Minn I, Califano JA, Sidransky D, Koch WM. P53-Reactivating Small Molecules Induce Apoptosis and Enhance Chemotherapeutic Cytotoxicity in Head and Neck Squamous Cell Carcinoma. Oral Oncol (2011) 47:8–15. doi: 10.1016/j.oraloncology.2010.10.011
46. He XX, Zhang YN, Yan JW, Yan JJ, Wu Q, Song YH. CP-31398 Inhibits the Growth of P53-Mutated Liver Cancer Cells In Vitro and In Vivo. Tumor Biol (2016) 37:807–15. doi: 10.1007/s13277-015-3857-5
47. He X, Kong X, Yan J, Yan J, Zhang Y, Wu Q, et al. CP-31398 Prevents the Growth of P53-Mutated Colorectal Cancer Cells In Vitro and In Vivo. Tumor Biol (2015) 36:1437–44. doi: 10.1007/s13277-014-2389-8
48. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LGGC, Masucci M, et al. Small Molecule RITA Binds to P53, Blocks P53-HDM-2 Interaction and Activates P53 Function in Tumors. Nat Med (2004) 10:1321–8. doi: 10.1038/nm1146
49. Zhao CY, Grinkevich VV, Nikulenkov F, Bao W, Selivanova G. Rescue of the Apoptotic-Inducing Function of Mutant P53 by Small Molecule RITA. Cell Cycle (2010) 9:1847–55. doi: 10.4161/cc.9.9.11545
50. Chuang H-C, Yang LP, Fitsgerald AL, Osman A, Woo SH. The P53-Reactivating Small Molecule RITA Induces Senescence in Head and Neck Cancer Cells. PloS One (2014) 9:104821. doi: 10.1371/journal.pone.0104821
51. Weilbacher A, Gutekunst M, Oren M, Aulitzky WE. & Kuip, H. Van Der. RITA can Induce Cell Death in P53-Defective Cells Independently of P53 Function via Activation of JNK/SAPK and P38. Cell Death Dis (2014) 5:e1318. doi: 10.1038/cddis.2014.284
52. Shin D, Kim EH, Lee J, Roh JL. RITA Plus 3-MA Overcomes Chemoresistance of Head and Neck Cancer Cells via Dual Inhibition of Autophagy and Antioxidant Systems. Redox Biol (2017) 13:219–27. doi: 10.1016/j.redox.2017.05.025
53. Bykov VJN, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the Tumor Suppressor Function to Mutant P53 by a Low-Molecular-Weight Compound. Nat Med (2002) 8:282–8. doi: 10.1038/nm0302-282
54. Bykov VJN, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1MET Synergizes With Cisplatin to Induce Tumor Cell Apoptosis. Oncogene (2005) 24:3484–91. doi: 10.1038/sj.onc.1208419
55. Wassman CD, Baronio R, Demir Ö, Wallentine BD, Chen C-K, Hall L v, et al. Computational Identification of a Transiently Open L1/S3 Pocket for Reactivation of Mutant P53. Nat Commun (2013) 4:1407. doi: 10.1038/ncomms2361
56. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 Reactivates Mutant P53 by Targeting Cysteines 124 and 277. Cell Death Dis (2018) 9:1–12. doi: 10.1038/s41419-018-0463-7
57. Ali D, Mohammad DK, Mujahed H, Jonson-Videsäter K, Nore B, Paul C, et al. Anti-Leukaemic Effects Induced by APR-246 are Dependent on Induction of Oxidative Stress and the NFE2L2/HMOX1 Axis That can be Targeted by PI3K and mTOR Inhibitors in Acute Myeloid Leukaemia Cells. Br J Haematol (2016) 174:117–26. doi: 10.1111/bjh.14036
58. Krayem M, Sabbah M, Najem A, Wouters A, Lardon F, Simon S, et al. The Benefit of Reactivating P53 Under MAPK Inhibition on the Efficacy of Radiotherapy in Melanoma. Cancers (2019) 11:11. doi: 10.3390/cancers11081093
59. Hang W, Yin Z-XX, Liu G, Zeng Q, Shen X-FF, Sun Q-HH, et al. Piperlongumine and P53-Reactivator APR-246 Selectively Induce Cell Death in HNSCC by Targeting GSTP1. Oncogene (2018) 37:3384–98. doi: 10.1038/s41388-017-0110-2
60. Yin Z-X, Hang W, Liu G, Wang Y-SS, Shen X-FF, Sun Q-HH, et al. PARP-1 Inhibitors Sensitize HNSCC Cells to APR-246 by Inactivation of Thioredoxin Reductase 1 (TrxR1) and Promotion of ROS Accumulation. Oncotarget (2018) 9:1885–97. doi: 10.18632/oncotarget.21277
61. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine Rapidly Induces the Death of Human Pancreatic Cancer Cells Mainly Through the Induction of Ferroptosis. Int J Oncol (2018) 52:1011–22. doi: 10.3892/ijo.2018.4259
62. Lv F, Deng M, Bai J, Zou D, Wang J, Li H, et al. Piperlongumine Inhibits Head and Neck Squamous Cell Carcinoma Proliferation by Docking to Akt. Phytother Res (2020) 34:3345–58. doi: 10.1002/ptr.6788
63. Yura Y. Presage of Oncolytic Virotherapy for Oral Cancer With Herpes Simplex Virus. Japanese Dental Sci Rev (2017) 53:53–60. doi: 10.1016/j.jdsr.2016.10.001
64. Atherton MJ, Stephenson KB, Nikota JK, Hu QN, Nguyen A, Wan Y, et al. Preclinical Development of Peptide Vaccination Combined With Oncolytic MG1-E6E7 for HPV-Associated Cancer. Vaccine (2018) 36:2181–92. doi: 10.1016/j.vaccine.2018.02.070
65. Liu TJ, El-Naggar AK, McDonnell TJ, Steck KD, Wang M, Taylor DL, et al. Apoptosis Induction Mediated by Wild-Type P53 Adenoviral Gene Transfer in Squamous Cell Carcinoma of the Head and Neck. Cancer Res (1995) 55:3117–22.
66. Pirollo KF, Hao Z, Rait A, Jang Y-J, Fee WE Jr, Ryan P, et al. P53 Mediated Sensitization of Squamous Cell Carcinoma of the Head and Neck to Radiotherapy. Oncogene (1997) 14:1735–46. doi: 10.1038/sj.onc.1201116
67. Clayman GL, El-Naggar AK, Roth JA, Zhang W-W, Goepfert H, Taylor DL, et al. In Vivo Molecular Therapy With P53 Adenovirus for Microscopic Residual Head and Neck Squamous Carcinoma. Cancer Res (1995) 55:1–6.
68. Dix BR, Edwards SJ, Braithwaite AW. Does the Antitumor Adenovirus ONYX-015/Dl1520 Selectively Target Cells Defective in the P53 Pathway? J Virol (2001) 75:5443–7. doi: 10.1128/JVI.75.12.5443-5447.2001
69. Ries S, Korn WM. ONYX-015: Mechanisms of Action and Clinical Potential of a Replication-Selective Adenovirus. Br J Cancer (2002) 86:5–11. doi: 10.1038/sj.bjc.6600006
70. Ries SJ, Brandts CH, Chung AS, Biederer CH, Hann BC, Lipner EM, et al. Loss of P14(ARF) in Tumor Cells Facilitates Replication of the Adenovirus Mutant Dl1520 (ONYX-015). Nat Med (2000) 6:1128–33. doi: 10.1038/80466
71. Hoffmann TK, Sonkoly E, Hauser U, van Lierop A, Whiteside TL, Klussmann JP, et al. Alterations in the P53 Pathway and Their Association With Radio- and Chemosensitivity in Head and Neck Squamous Cell Carcinoma. Oral Oncol (2008) 44:1100–9. doi: 10.1016/j.oraloncology.2008.02.006
72. Mortensen ACL, Morin E, Brown CJ, Lane DP, Nestor M. Enhancing the Therapeutic Effects of In Vitro Targeted Radionuclide Therapy of 3D Multicellular Tumor Spheroids Using the Novel Stapled MDM2/X-P53 Antagonist PM2. EJNMMI Res (2020) 10. doi: 10.1186/s13550-020-0613-7
73. Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, et al. A Stapled P53 Helix Overcomes HDMX-Mediated Suppression of P53. Cancer Cell (2010) 18:411–22. doi: 10.1016/j.ccr.2010.10.024
74. Mortensen ACL, Spiegelberg D, Brown CJ, Lane DP, Nestor M. The Stapled Peptide PM2 Stabilizes P53 Levels and Radiosensitizes Wild-Type P53 Cancer Cells. Front Oncol (2019) 9:923. doi: 10.3389/fonc.2019.00923
75. Wei SJ, Chee S, Yurlova L, Lane D, Verma C, Brown C, et al. Avoiding Drug Resistance Through Extended Drug Target Interfaces: A Case for Stapled Peptides. Oncotarget (2016) 7:32232–46. doi: 10.18632/oncotarget.8572
76. Spiegelberg D, Mortensen AC, Lundsten S, Brown CJ, Lane DP, Nestor M. The MDM2/MDMX-P53 Antagonist PM2 Radiosensitizes Wild-Type P53 Tumors. Cancer Res (2018) 78:5084–93. doi: 10.1158/0008-5472.CAN-18-0440
77. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science (2004) 303:844–8. doi: 10.1126/science.1092472
78. Arya AK, El-Fert A, Devling T, Eccles RM, Aslam MA, Rubbi CP, et al. Nutlin-3, the Small-Molecule Inhibitor of MDM2, Promotes Senescence and Radiosensitises Laryngeal Carcinoma Cells Harbouring Wild-Type P53. Br J Cancer (2010) 103:186–95. doi: 10.1038/sj.bjc.6605739
79. Effert P, McCoy R, Abdel-Hamid M, Flynn K, Zhang Q, Busson P, et al. Alterations of the P53 Gene in Nasopharyngeal Carcinoma. J Virol (1992) 66:3768–75. doi: 10.1128/jvi.66.6.3768-3775.1992
80. Yee-Lin V, Pooi-Fong W, Soo-Beng AK. Nutlin-3, A P53-Mdm2 Antagonist for Nasopharyngeal Carcinoma Treatment. Mini-Reviews Med Chem (2017) 18. doi: 10.2174/1389557517666170717125821.
81. Voon YL, Ahmad M, Wong PF, Husaini R, Ng WTW, Leong CO, et al. Nutlin-3 Sensitizes Nasopharyngeal Carcinoma Cells to Cisplatin-Induced Cytotoxicity. Oncol Rep (2015) 34:1692–700. doi: 10.3892/or.2015.4177
82. Kono M, Kumai T, Hayashi R, Yamaki H, Komatsuda H, Wakisaka R, et al. Interruption of MDM2 Signaling Augments MDM2-Targeted T Cell-Based Antitumor Immunotherapy Through Antigen-Presenting Machinery. Cancer Immunol Immunother (2021) 1–14. doi: 10.1007/s00262-021-02940-5
83. Choe YJ, Lee SY, Ko KW, Shin SJ, Kim HS. Nutlin-3 Induces HO-1 Expression by Activating JNK in a Transcription-Independent Manner of P53. Int J Oncol (2014) 44:761–8. doi: 10.3892/ijo.2013.2227
84. Lee SY, Shin SJ, Kim HS. ERK1/2 Activation Mediated by the Nutlin-3-Induced Mitochondrial Translocation of P53. Int J Oncol (2013) 42:1027–35. doi: 10.3892/ijo.2013.1764
85. Shin JS, Ha JH, He F, Muto Y, Ryu KS, Yoon HS, et al. Structural Insights Into the Dual-Targeting Mechanism of Nutlin-3. Biochem Biophys Res Commun (2012) 420:48–53. doi: 10.1016/j.bbrc.2012.02.113
86. Xie X, Piao L, Bullock BN, Smith A, Su T, Zhang M, et al. Targeting HPV16 E6-P300 Interaction Reactivates P53 and Inhibits the Tumorigenicity of HPV-Positive Head and Neck Squamous Cell Carcinoma. Oncogene (2014) 33:1037–46. doi: 10.1038/onc.2013.25
87. Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of Hypoxia Inducible Factor 1-Transcription Coactivator Interaction by a Hydrogen Bond Surrogate α-Helix. J Am Chem Soc (2010) 132:941–3. doi: 10.1021/ja9082864
88. Hamada K, Sakaue M, Alemany R, Zhang WW, Horio Y, Roth JA, et al. Adenovirus-Mediated Transfer of HPV 16 E6/E7 Antisense RNA to Human Cervical Cancer Cells. Gynecologic Oncol (1996) 63:219–27. doi: 10.1006/gyno.1996.0310
89. Kojima Y, Otsuki N, Kubo M, Kitamoto J, Takata E, Saito H, et al. Adenovirus-Mediated Transfer of HPV 16 E6/E7 Antisense RNA Combined With Cisplatin Inhibits Cellular Growth and Induces Apoptosis in HPV-Positive Head and Neck Cancer Cells. Cancer Gene Ther (2018) 25:274–83. doi: 10.1038/s41417-018-0024-3
90. Seltzsam S, Ziemann F, Dreffke K, Preising S, Arenz A, Schötz U, et al. In HPV-Positive HNSCC Cells, Functional Restoration of the P53/P21 Pathway by Proteasome Inhibitor Bortezomib Does Not Affect Radio- or Chemosensitivity. Trans Oncol (2019) 12:417–25. doi: 10.1016/j.tranon.2018.11.013
91. van Waes C, Chang AA, Lebowitz PF, Druzgal CH, Chen Z, Elsayed YA, et al. Inhibition of Nuclear Factor-κb and Target Genes During Combined Therapy With Proteasome Inhibitor Bortezomib and Reirradiation in Patients With Recurrent Head-and-Neck Squamous Cell Carcinoma. Int J Radiat Oncol Biol Phys (2005) 63:1400–12. doi: 10.1016/j.ijrobp.2005.05.007
92. Lin YC, Chen KC, Chen CC, Cheng AL, Chen KF. CIP2A-Mediated Akt Activation Plays a Role in Bortezomib-Induced Apoptosis in Head and Neck Squamous Cell Carcinoma Cells. Oral Oncol (2012) 48:585–93. doi: 10.1016/j.oraloncology.2012.01.012
93. Brinker AM, Ma J, Lipsky PE, Raskin I. Medicinal Chemistry and Pharmacology of Genus Tripterygium (Celastraceae). Phytochemistry (2007) 68:732–66. doi: 10.1016/j.phytochem.2006.11.029
94. Kuo CS, Yang CY, Lin CK, Lin GJ, Sytwu HK, Chen YW. Triptolide Suppresses Oral Cancer Cell PD-L1 Expression in the Interferon-γ-Modulated Microenvironment In Vitro, In Vivo, and in Clinical Patients. Biomed Pharmacother (2021) 133:111057. doi: 10.1016/j.biopha.2020.111057
95. Caicedo-Granados E, Lin R, Fujisawa C, Yueh B, Sangwan V, Saluja A. Wild-Type P53 Reactivation by Small-Molecule MinnelideTM in Human Papillomavirus (HPV)-Positive Head and Neck Squamous Cell Carcinoma. Oral Oncol (2014) 50:1149–56. doi: 10.1016/j.oraloncology.2014.09.013
96. Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, et al. A Preclinical Evaluation of Minnelide as a Therapeutic Agent Against Pancreatic Cancer. Sci Trans Med (2012) 4. doi: 10.1126/scitranslmed.3004334
97. Troiano G, Caponio VCA, Adipietro I, Tepedino M, Santoro R, Laino L, et al. Prognostic Significance of CD68 + and CD163 + Tumor Associated Macrophages in Head and Neck Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. Oral Oncol (2019) 93:66–75. doi: 10.1016/j.oraloncology.2019.04.019
98. Ou D, Adam J, Garberis I, Blanchard P, Nguyen F, Levy A, et al. Influence of Tumor-Associated Macrophages and HLA Class I Expression According to HPV Status in Head and Neck Cancer Patients Receiving Chemo/Bioradiotherapy. Radiotherapy Oncol (2019) 130:89–96. doi: 10.1016/j.radonc.2018.08.013
99. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-Cell-Autonomous Tumor Suppression by P53. Cell (2013) 153:449–60. doi: 10.1016/j.cell.2013.03.020
100. El-Arabey AA, Abdalla M, Abd-Allah AR. SnapShot: TP53 Status and Macrophages Infiltration in TCGA-Analyzed Tumors. Int Immunopharmacol (2020) 86:106758. doi: 10.1016/j.intimp.2020.106758
101. Kobatake K, Ikeda K, Nakata Y, Yamasaki N, Ueda T, Kanai A, et al. Kdm6a Deficiency Activates Inflammatory Pathways, Promotes M2 Macrophage Polarization, and Causes Bladder Cancer in Cooperation With P53 Dysfunction. Clin Cancer Res: an Off J Am Assoc Cancer Res (2020) 26:2065–79. doi: 10.1158/1078-0432.CCR-19-2230
102. Fang DD, Tang Q, Kong Y, Wang Q, Gu J, Fang X, et al. MDM2 Inhibitor APG-115 Synergizes With PD-1 Blockade Through Enhancing Antitumor Immunity in the Tumor Microenvironment. J Immunother Cancer (2019) 7:327. doi: 10.1186/s40425-019-0750-6
103. Peng Z. Current Status of Gendicine in China: Recombinant Human Ad-P53 Agent for Treatment of Cancers. Hum Gene Ther (2005) 16:1016–27. doi: 10.1089/hum.2005.16.1016
104. Zhang WW, et al. The First Approved Gene Therapy Product for Cancer Ad-P53 (Gendicine): 12 Years in the Clinic. Hum Gene Ther (2018) 29:160–79. doi: 10.1089/hum.2017.218
Keywords: p53, mutation, HPV, HNSCC, targeted therapy
Citation: de Bakker T, Journe F, Descamps G, Saussez S, Dragan T, Ghanem G, Krayem M and Van Gestel D (2022) Restoring p53 Function in Head and Neck Squamous Cell Carcinoma to Improve Treatments. Front. Oncol. 11:799993. doi: 10.3389/fonc.2021.799993
Received: 22 October 2021; Accepted: 15 December 2021;
Published: 06 January 2022.
Edited by:
Kevin Joseph Harrington, Institute of Cancer Research (ICR), United KingdomReviewed by:
Gabriella D’Orazi, G. D’Annunzio University of Chieti-Pescara, ItalyMarco Mascitti, Marche Polytechnic University, Italy
Copyright © 2022 de Bakker, Journe, Descamps, Saussez, Dragan, Ghanem, Krayem and Van Gestel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Krayem, Mohammad.krayem@bordet.be