
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 04 January 2022
Sec. Cancer Genetics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.778461
This article is part of the Research Topic Epigenetics and Molecular Genetics of Rare Tumors View all 9 articles
 Maria A. Pantaleo1,2
Maria A. Pantaleo1,2 Milena Urbini3*
Milena Urbini3* Angela Schipani2Margherita Nannini1
Angela Schipani2Margherita Nannini1 Valentina Indio3
Valentina Indio3 Antonio De Leo2,4Bruno Vincenzi5Antonella Brunello6Giovanni Grignani7Mariaelena Casagrande8Elena Fumagalli9Elena Conca10Maristella Saponara11Elisa Gruppioni12Annalisa Altimari12
Antonio De Leo2,4Bruno Vincenzi5Antonella Brunello6Giovanni Grignani7Mariaelena Casagrande8Elena Fumagalli9Elena Conca10Maristella Saponara11Elisa Gruppioni12Annalisa Altimari12 Dario De Biase4,13Giovanni Tallini2,4Gloria Ravegnini13Daniela Turchetti14Marco Seri14Andrea Ardizzoni1Paola Secchiero15
Dario De Biase4,13Giovanni Tallini2,4Gloria Ravegnini13Daniela Turchetti14Marco Seri14Andrea Ardizzoni1Paola Secchiero15 Annalisa Astolfi15
Annalisa Astolfi15Background: SDH-deficient gastrointestinal stromal tumors (GIST) account for 20–40% of all KIT/PDGFRA-negative GIST and are due to mutations in one of the four SDH-complex subunits, with SDHA mutations as the most frequent. Here we sought to evaluate the presence and prevalence of SDHA variants in the germline lineage in a population of SDHA-deficient GIST.
Methods: Germline SDHA status was assessed by Sanger sequencing on a series of 14 patients with gastric SDHA-deficient GIST.
Results: All patients carried a germline SDHA pathogenic variant, ranging from truncating, missense, or splicing variants. The second hit was the loss of the wild-type allele or an additional somatic mutation. One-third of the patients were over 50 years old. GIST was the only disease presentation in all cases except one, with no personal or familial cancer history. Seven metastatic cases received a multimodal treatment integrating surgery, loco-regional and medical therapy. The mean follow-up time was of 10 years, confirming the indolent clinical course of the disease.
Conclusion: SDHA germline variants are highly frequent in SDHA-deficient GIST, and the disease may occur also in older adulthood. Genetic testing and surveillance of SDHA-mutation carriers and relatives should be performed.
SDH-deficient gastrointestinal stromal tumors (GIST) account for 20–40% of all KIT/PDGFRA-negative GIST (1). Several evidences have suggested that SDH-deficient GIST exclusively arise from the stomach with mainly multifocal primary localization, frequently present lymph node involvement, generally affect younger population, and above all, have an indolent behavior even with metastatic disease (2). SDH deficiency in GIST is defined by the loss of expression of SDHB protein at immunohistochemistry and is mainly due to mutations in the four SDH mitochondrial complex subunits: SDHA, SDHB, SDHC, and SDHD (3). SDHA mutations are the most frequent among SDH alterations in GIST, accounting approximately for half of the cases. Other rare epigenetic events involve the recurrent aberrant DNA methylation of SDHC seen in GIST associated to the Carney triad (CT), which is a rare condition with synchronous or metachronous occurrence of GIST, paragangliomas, and pulmonary chondromas (4–6).
Most of SDH mutations in GIST are germline, in particular germline mutations in SDHB, SDHC, and SDHD occur in about 20–30% of SDH-deficient disease and may be referred to as a Carney-Stratakis syndrome (CSS) (4). This syndrome was firstly described in 2002 as a hereditary condition characterized by the occurrence of GIST and paraganglioma. Germline SDHA pathogenic variants have been rarely described in apparently sporadic cases (7, 8). Currently, germline testing is recommended for all SDH-deficient GIST, but no clear guidelines for genetic counseling and follow-up of SDH mutation carriers and relatives have yet been released (9). Moreover, regarding SDHA mutations, many issues are still unclear such as the germline status and the clinical implications that are not yet linked to well-defined hereditary syndrome. The aim of this work is to evaluate the presence and the prevalence of SDHA variants in the germline lineage in an SDH-deficient GIST population harboring SDHA somatic mutations.
Sixteen patients with a gastric SDHA-mutant GIST were studied. All cases were negative for SDHB immunohistochemistry, and SDHA mutations were assessed by Sanger sequencing of coding exons and exon-flanking regions as previously reported (10).
The tumor and patients’ characteristics are reported in Table 1. The study was performed in accordance with the Declaration of Helsinki protocols. The study was reviewed and approved by the local Institutional Ethical Committee of Azienda Ospedaliero-Universitaria Policlinico S. Orsola-Malpighi, Bologna, Italy (approval number 113/2008/U/Tess), and informed consent was provided by all living patients.
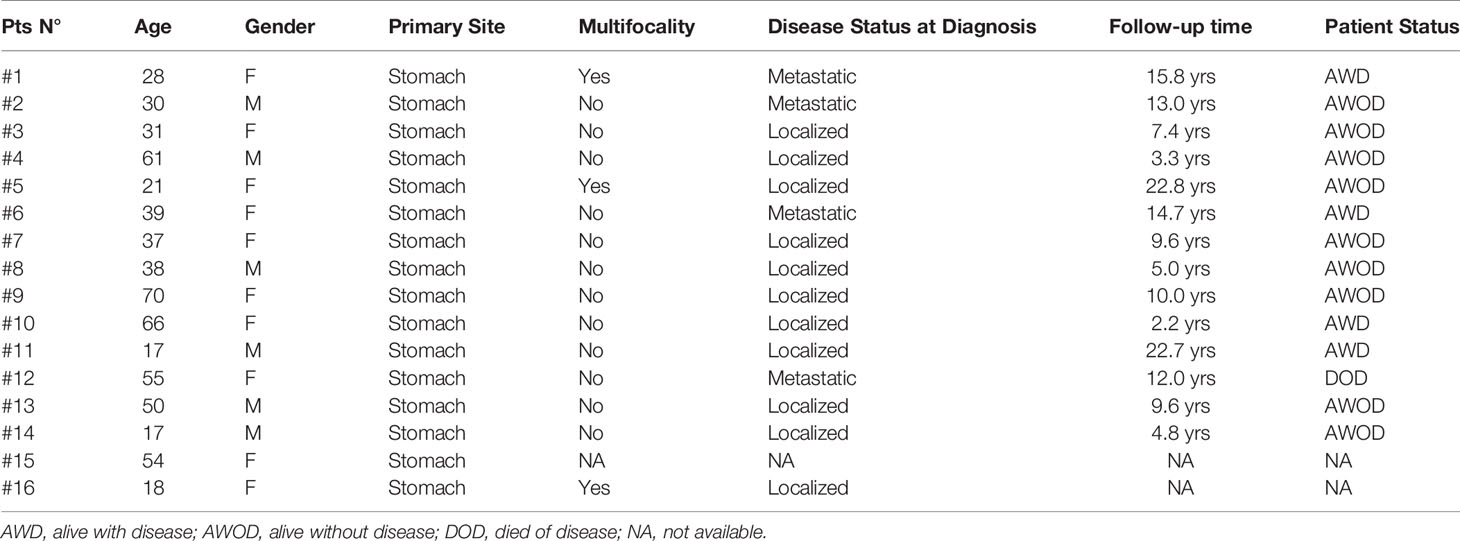
Table 1 Patients and tumor characteristics.
For germline analysis, DNA was extracted from peripheral blood or FFPE normal tissue with the QiaAmp mini or micro kit (Qiagen). For somatic analysis, manual macrodissection of the tumor area was performed using a scalpel on areas selected by an expert pathologist on FFPE slides. At least 70% tumor enrichment was required for sample inclusion, and DNA was extracted using QiaAmp micro kit. SDHA variants were identified by Sanger sequencing. Exonic and flanking intronic regions of SDHA were amplified with FastStart TAQ polymerase (Roche) and sequenced on both strands using the Big Dye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems) on ABI 3730 Genetic Analyzer (Applied Biosystems). Primer pairs were designed with Primer Express 3.0 Software (Applied Biosystems) to specifically amplify SDHA exons and not the related pseudogenes (10). Germline mutational analysis was performed on peripheral blood in nine cases and on matched normal tissue extracted from FFPE in five cases. Unfortunately, in two cases the matched normal counterpart was not available. Allele frequency in the general population was reported from the Genome Aggregation Database (gnomAD) v2.1.1, reporting data from 141,456 individuals. Variant classification was performed following ACMG recommendations using the VarSome shared data resource (https://varsome.com/).
Germline variants were identified in all 14 patients for which the normal counterpart was available (Table 2). Five cases harbored truncating non-sense variants (Ser384*, Arg31*, Trp119*, Arg210*), seven other cases carried missense variants (Gly233Val, Arg171His, Arg589Gln, Gly257Ala, Arg600Gln, Arg585Gln), and two harbored exon-flanking intronic variants predicted to affect splicing. In particular, the prediction with the tool Alternative Splice Site Predictor (ASSP) revealed that the c.457-2_457del is likely to generate an alternative acceptor splice site of exon 5, while the c.1663+3 G>C probably leads to the loss of donor splice site at exon 12 with consequent intron retention. All the two splice site alterations, as predicted, lead to a frameshift with a stop codon in the corresponding protein sequence. All the truncating variants, one splice-site, and two missense variants were classified as “Pathogenic” or “Likely Pathogenic” by the ACMG recommendations (Table 2). Conversely, the other five variants were classified as “Uncertain Significance” but were all predicted as damaging by the computational prediction algorithms implemented in Varsome. In eight cases, tumor DNA showed the loss of the corresponding wild-type allele, thus displaying homozygosity for the germline variant, while in the other six cases compound heterozygosity for an additional somatic mutation was detected (Arg589Trp, Arg451Cys, Arg171Cys, Arg585Gln, Thr308Met, and Gln176*) (Figure 1A). In both patients for which normal DNA was not available, tumors carried two mutational hits on SDHA. Germline SDHA variants identified in this study, along with those identified in previous reports on GIST patients, are summarized in Figure 1B. As expected, mutations are scattered along the whole coding sequence, with a peak frequency of the Arg31* variant, recurrently identified in previous studies (3, 7, 11–18) and present in one case in our series.
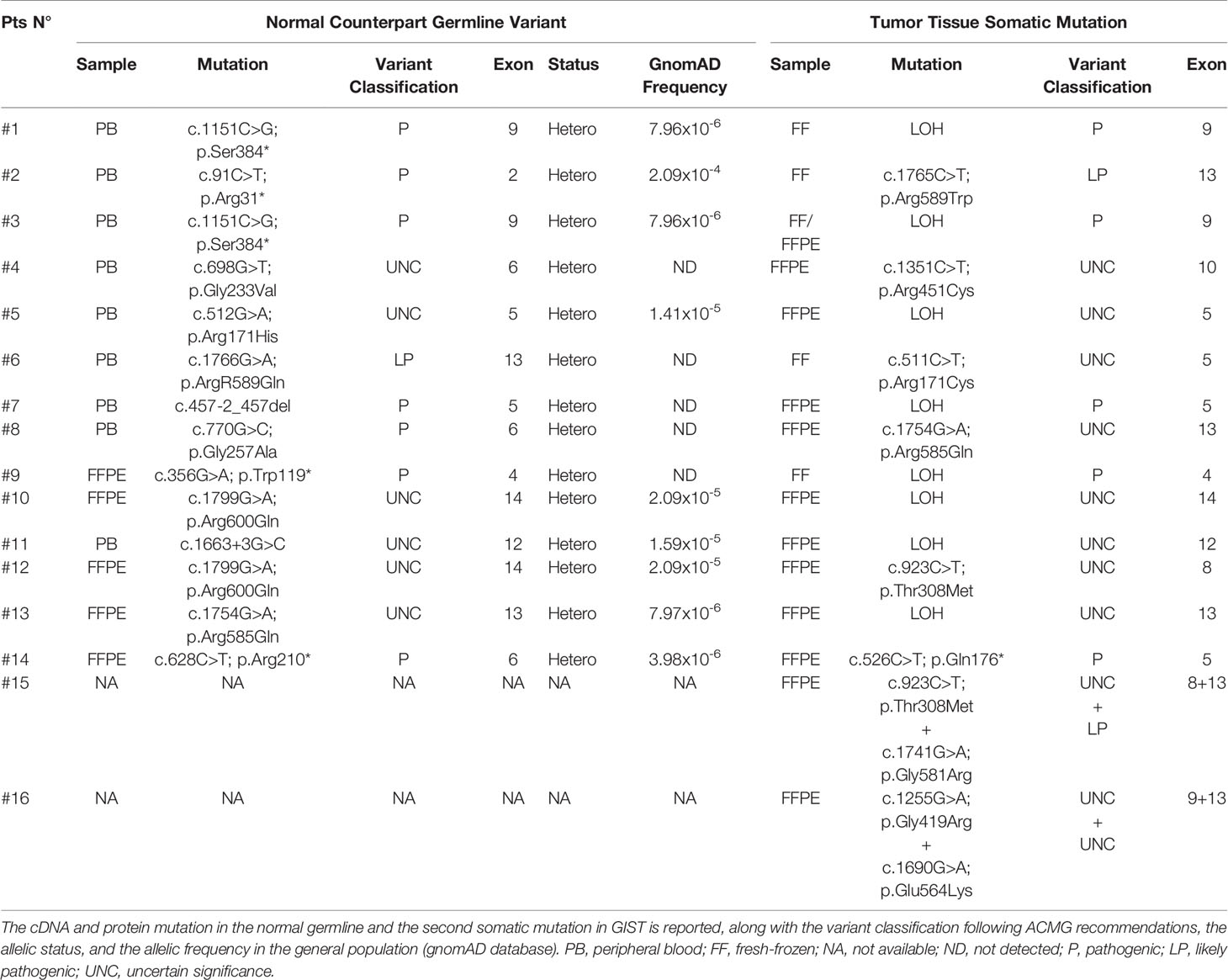
Table 2 Mutational analysis of germline and tumors in SDHA-deficient GIST patients.
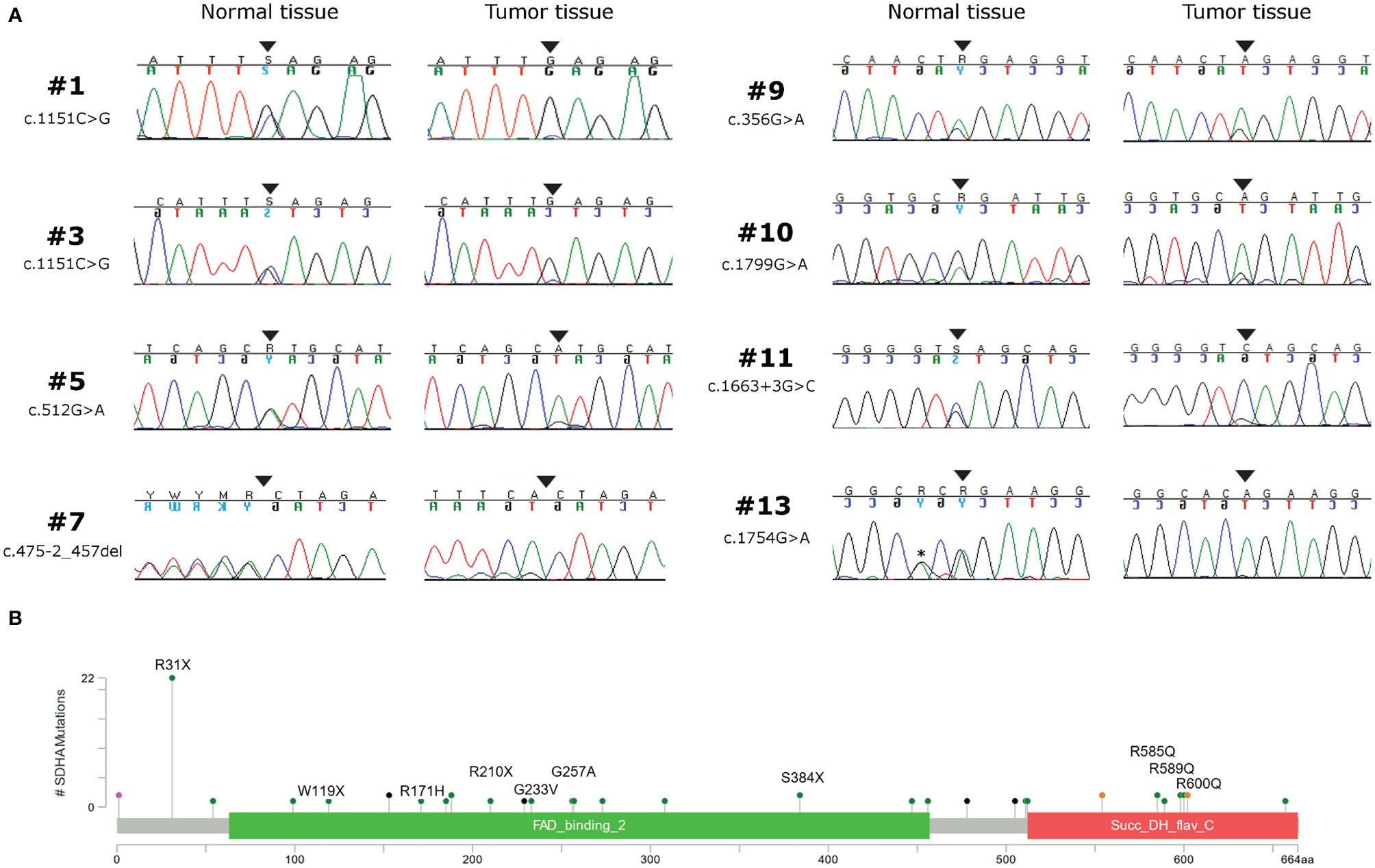
Figure 1 (A) Chromatograms showing the loss of heterozygosity of SDHA germline variants in tumor tissue of eight GIST patients. Black arrows indicate the position of the mutation; the asterisk indicates a silent single nucleotide polymorphism. (B) SDHA germline variant frequency of GIST patients coming from this series (written protein mutation) and from previous reports (3, 7, 8, 11–17), shown as lollipop plot.
Average age at diagnosis was 39.5 ± 4.5 years (range 17–70). Ten cases were young-adults (range: 17–39 years old) and 6 patients were older adults (>50 years old; range: 50–70). Combining our series with the previously published reports (3, 7, 8, 11–18), it emerges that GIST can arise at very different age ranges in SDHA-variant carriers, since mean age at diagnosis is 36.0 ± 2.3 years, but up to 22% of patients are being diagnosed at more than 50 years old (Figure 2). In all cases of adult patients, the GIST was unifocal. In the whole series, four patients displayed metastases already at the time of diagnosis, while all other harbored localized disease. In all cases except one, the GIST was the only disease presentation, and no personal or familial cancer history was revealed. Only one case of 61 years of age at diagnosis reported having been affected by paraganglioma in the past (more than 10 years before), but unfortunately the biological material was not available for a pathologic revision and genetic testing. Except for two cases lost at follow-up, the mean follow-up time was 10.9 years, ranging between 2.2 to 22.8 years. Among localized cases, three developed a recurrence with multiple metastases. Altogether, seven metastatic cases received a high-complexity treatment with standard medical therapy integrated with surgery and/or other loco-regional therapy (radiofrequency of liver metastasis and also chemoembolization in one case). Among these seven metastatic cases, one died of disease and one is currently free of disease through complete surgical removal of liver metastases.
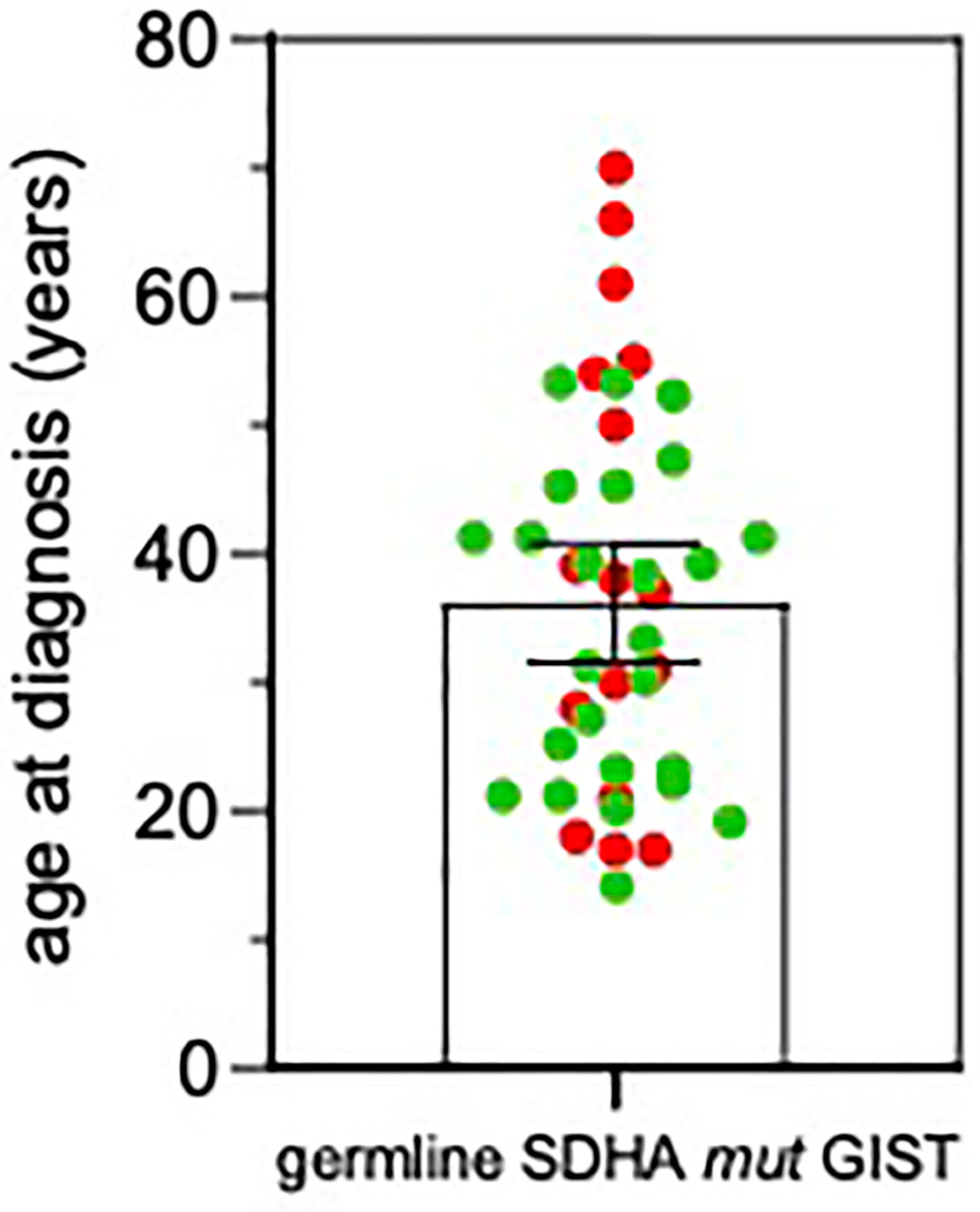
Figure 2 Age at GIST onset in SDHA-variant carriers. The mean and SEM are shown. Red, patients from this series; Green, patients from previously published reports (3, 7, 11–18).
In a population of SDH-deficient GIST harboring SDHA somatic mutations, we found germline SDHA variants in all cases for which normal DNA was available, and these findings underline that germline mutations in SDHA are highly frequent in SDHA-deficient GIST. Besides the non-sense and missense mutations, we also found two cases harboring damaging splice-site mutations (c.457-2_457 and c.1663+3 G>C) leading to a predicted premature protein sequence truncation. Overall, in our series we found seven truncating germline variants (five non-sense and two splice-site) present in all but one case in young patients. Conversely, four out of seven germline missense variants, whose effect on protein function and stability is harder to be assessed, were identified in older patients (more than 50 years old at diagnosis). Moreover, we did not find a high frequency of the Arg31* variant, whose recurrent identification in previous studies even led to the assumption of a putative founder effect (3, 7, 14, 16–18). Apart from the recurrent Arg31*, the high diversity of the variant type and position seen in our and in previous series supports a view in which SDHA can be inactivated by mutations scattered throughout its whole genomic sequence, suggesting that SDHA genetic analysis must be performed entirely and by qualified high-volume molecular diagnostic centers, as already suggested in other settings (19).
Lastly, all the described patients carried two mutational events at the SDHA locus, either the loss of the wild-type allele or a second somatic event in compound heterozygosity, in full agreement with the two-hit hypothesis of tumor suppressor genes inactivation. Therefore, this suggests the possibility of a germline first mutation also in the two cases of our series for which the matched normal DNA counterpart was not available. Interestingly, an in-depth biochemical study of the functional effects of SDHA variants of unknown significance supported the pathogenicity of two somatic mutations (p.Arg451Cys e p.Gly419Arg) classified as uncertain by ACMG recommendations, while it did not support the loss of function of the Arg171His variant (20).
Furthermore, our study definitely confirms the highly relevant association between germline SDHA pathogenic variants and GIST onset, which is supported by many previous studies reporting the development of GIST as the only cancer disease in SDHA germline-variant carrier population (3, 7, 13, 14, 18). In our series, only one case reported having been affected by paraganglioma more than 10 years before, but the diagnosis was not now revised. Recently, a large case-control study has estimated the penetrance of SDHx variants for PPGL, indicating a 1.7% lifetime disease penetrance for SDHA pathogenic variants (21). Up to now, the onset of other tumor types in the context of SDHA-mutant GIST appears to be very rare and limited to the concurrent description of paraganglioma in one case, of pulmonary chondroma in two cases, and of the full Carney triad in one patient (8, 14). Indeed, it was shown that mutations in SDHA account for around 30% of all KIT/PDGFRA-WT GIST (10), while being responsible for <1% of all pheochromocytoma and paraganglioma cases (PPGL), as opposed for example to SDHB that accounts for about 10% of all PPGL (22, 23). The only study that up to now suggesting the possible role of SDHA as a predisposing factor for other tumor types reported the development of neuroblastoma in one SDHA-mutation carrier showing the inactivation of the second allele (17). Therefore, our result, coupled with previous data and with the identification of a higher-than-expected frequency of SDHA germline pathogenic variants in a cohort of cancer patients with respect to healthy individuals from the Exome Aggregation Consortium (ExAC) database (17), further endorses the view of SDHA as a cancer gene mainly predisposing to GIST development. This aggregate result underscores the different tumor spectrum of SDHA-germline variant carriers and Carney Stratakis Syndrome patients, which are known to harbor SDHB, SDHC, or SDHD mutations, and to develop invariably the association of GIST and paraganglioma during their life course (24).
Actually, in literature and in our experience, a clear syndrome has not been clearly defined since all these cases appear to be sporadic without other neoplasms except from GIST, and therefore it still remains unclear if SDHA mutations should be accounted in the CSS. CSS by definition is due to SDHB, SDHC, and SDHD mutations and is characterized by the combination of GIST and paraganglioma inherited in an apparently autosomal dominant manner and with incomplete penetrance. Paragangliomas were described to be multicentric and GIST multifocal. Both these clinical presentations support the inherited nature of this tumor predisposition (4). Currently, germline testing is recommended for all SDH-deficient GIST including SDHA in some clinical practice guidelines (9, 25), but no clear protocol for genetic counseling and follow-up of SDHx variant carriers and relatives has been released, especially for those carrying a germline SDHA pathogenic variant not yet linked to well-defined hereditary syndrome.
Regarding the clinical findings in GIST patients with SDHA germline mutation, our series confirms the stomach as unique site of GIST onset but underlines a higher heterogeneity for the multifocality development and also for the metastatic presentation at diagnosis. Larger series and international efforts should be required to better define a phenotype/genotype correlation in this subset of disease between GIST features, other tumor development, and SDH genotyping in tumor/germline. No conclusive consideration can be made regarding the survival rates because of the heterogeneity of clinical presentations and the multiple treatments received in cases with metastatic disease. After all, our series confirms the indolent clinical course and long survival expectations since half of population presents a follow-up higher than 10 years, and four of these patients present a metastatic disease.
This study therefore shows that, differently from previously stated, the SDHA-mutant GIST patient is almost exclusively a germline SDHA-variant carrier that is prone to develop the tumor throughout his entire life and not just in his early adulthood (7, 10–12, 14). In fact, 38% of the patients from our series were older than 50 years, with peaks up to 70 years old. Actually, this result is supported by previous studies that occasionally reported age ranges up to 70 years old and subsets of SDHA-mutant GIST patients of more than 50 years old (3, 7, 13, 14, 16). This fact has very important implications supporting the need for genetic counseling, since even though complete pedigree analysis of SDHA mutation carrier families is still lacking and the clinical insights suggest a low penetrance of SDHA mutation in affected families (12, 26), the notion of a lifelong risk for GIST development urges the need for genetic testing and permanent clinical surveillance of SDHA-variant carriers.
As the process of referral to genetic counseling of the patients included in the study is still ongoing, conclusive information on documented history of cancer in relatives, segregation of the variant in the family, and clinical assessment of carriers, who will be included in management programs according to updated recommendations, are not yet available. Now, this may represent a formal limitation of the study, however not modifying its substantial conclusions.
In conclusion, germline pathogenic variants in SDHA are highly frequent in SDHA-deficient GIST in young and adult patients, and the disease may occur also in older adulthood. Genetic counseling of SDHA-variant carriers and relatives should be planned, and their clinical follow-up should be accurately defined.
The datasets presented in this study was submitted to ClinVar (SUB10460221). Variants identified are available under accession numbers from SCV002026125 to SCV002026144.
The studies involving human participants were reviewed and approved by Ethical Committe of Pol. Sant’Orsola Malpighi, University of Bologna. The patients/participants provided their written informed consent to participate in this study.
Conceptualization: MP, MU, AAs, and VI. Data curation: VI, AAs, MN, and MSa. Formal Analysis; MU, VI, AS, AAs, and GR. Funding acquisition: MP and MN. Investigation: MU, VI, AS, AAs, BV, AB, GG, MC, EF, EC, MSa, AL, and GT. Methodology: MU, VI, AS, AAs, EG, AAl, and GR. Project administration: MP. Software: VI, GR, and MU. Supervision: MP, AAs, AAr, and PS. Validation: MU, VI, AS, AA, GR, AL, EG, AAl, and DB. Visualization: DT, MSe, and GT. Writing—original draft: MP, AAs, and VI. Writing—review and editing: MP, AAs, MU, and VI. All authors contributed to the article and approved the submitted version.
This work was supported by Fondazione Carisbo, Bologna (Bando Ricerca medica traslazionale e clinica 2019), Italy, and by a research donation in memory of Alberto Arenghi, Caravaggio, BG, Italy. The funders have no role in the design of the study, the collection, analysis, and interpretation of data, the writing of the manuscript, or submission of the manuscript for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Special thanks to the contributing physicians, Bologna, Italy: Sabrina Angelini, Francesco Buia, Paolo Castellucci, Maurizio Cervellera, Matteo Cescon, Antonietta D’Errico, Massimo Del Gaudio, Stefano Fanti, Michelangelo Fiorentino, Fabio Niro, Maria Giulia Pirini, Nico Pagano, Donatella Santini, Valeria Tonini, and Valerio Di Scioscio.
GIST, gastrointestinal stromal tumor; CT, Carney triad; CSS, Carney-Stratakis syndrome; gnomAD, Genome Aggregation Database; PPGL, pheochromocytoma and paraganglioma; ExAC, Exome Aggregation Consortium.
1. Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, et al. Defects in Succinate Dehydrogenase in Gastrointestinal Stromal Tumors Lacking KIT and PDGFRA Mutations. Proc Natl Acad Sci USA (2011) 108(1):314–18. doi: 10.1073/pnas.1009199108
2. Pantaleo MA, Lolli C, Nannini M, Astolfi A, Indio V, Saponara M, et al. Good Survival Outcome of Metastatic SDH-Deficient Gastrointestinal Stromal Tumors Harboring SDHA Mutations. Genet Med (2015) 17(5):391–5. doi: 10.1038/gim.2014.115
3. Wagner AJ, Remillard SP, Zhang Y-X, Doyle LA, George S, Hornick JL. Loss of Expression of SDHA Predicts SDHA Mutations in Gastrointestinal Stromal Tumors. Mod Pathol (2013) 26(2):289–94. doi: 10.1038/modpathol.2012.153
4. Stratakis CA, Carney JA. The Triad of Parangangliomas, Gastric Stromal Tumors and Pulmonary Chondromas (Carney Triad), and the Dyad of Parangangliomas and Gastric Stromal Sarcomas (Carney-Stratakis Syndrome): A Molecular Genetics and Clinical Implications. J Int Med (2009) 266(1):43–52. doi: 10.1111/j.1365-2796.2009.02110.x
5. Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent Epimutation of SDHC in Gastrointestinal Stromal Tumors. Sci Transl Med (2014) 6(268):268ra177. doi: 10.1126/scitranslmed.3009961
6. Haller F, Moskalev EA, Faucz FR, Barthelmeß S, Wiemann S, Bieg M, et al. Aberrant DNA Hypermethylation of SDHC: A Novel Mechanism of Tumor Development in Carney Triad. Endocr Relat Cancer (2014) 21(4):567–77. doi: 10.1530/ERC-14-0254
7. Miettinen M, Killian JK, Wang ZF, Lasota J, Lau C, Jones L, et al. Immunohistochemical Loss of Succinate Dehydrogenase Subunit A (SDHA) in Gastrointestinal Stromal Tumors (GISTs) Signals SDHA Germline Mutation. Am J Surg Pathol (2013) 37(2):234–40. doi: 10.1097/PAS.0b013e3182671178
8. Carrera S, Beristain E, Sancho A, Iruarrizaga E, Rivero P, Mañe JM, et al. Germline C.1A>C Heterozygous Pathogenic Variant in SDHA Reported for the First Time in a Young Adult With a Gastric Gastrointestinal Stromal Tumour (GIST): A Case Report. Hered Cancer Clin Pract (2019) 17:23. doi: 10.1186/s13053-019-0124-6
9. Casali PG, Abecassis N, Aro HT, Bauer S, Biagini R, Bielack S, et al. ESMO Guidelines Committee and EURACAN. Gastrointestinal Stromal Tumours: ESMO-EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol (2018) 29(Suppl 4):iv68–78. doi: 10.1093/annonc/mdy095
10. Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, et al. Analysis of All Subunits, SDHA, SDHB, SDHC, SDHD, of the Succinate Dehydrogenase Complex in KIT/PDGFRA Wild-Type GIST. Eur J Hum Genet (2014) 22(1):32–9. GIST Study Group. doi: 10.1038/ejhg.2013.80
11. Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, et al. SDHA Loss of Function Mutations in a Subset of Young Adult Wild-Type Gastrointestinal Stromal Tumors. BMC Cancer (2012) 12:408. doi: 10.1186/1471-2407-12-408
12. Dwight T, Benn DE, Clarkson A, Vilain R, Lipton L, Robinson BJ, et al. Loss of SDHA Expression Identifies SDHA Mutations in Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Am J Surg Pathol (2013) 37(2):226–33. doi: 10.1097/PAS.0b013e3182671155
13. Belinsky MG, Rink L, Flieder DB, Jahromi MS, Schiffman JD, Godwin AK, et al. Overexpression of Insulin-Like Growth Factor 1 Receptor and Frequent Mutational Inactivation of SDHA in Wild-Type SDHB-Negative Gastrointestinal Stromal Tumors. Genes Chromosomes Cancer (2013) 52(2):214–24. doi: 10.1002/gcc.22023
14. Oudijk L, Gaal J, Korpershoek E, Van Nederveen FH, Kelly L, Schiavon G, et al. SDHA Mutations in Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors. Mod Pathol (2013) 26(3):456–63. doi: 10.1038/modpathol.2012.186
15. Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang JY, Li JL, et al. A Novel Germline Mutation in SDHA Identified in a Rare Case of Gastrointestinal Stromal Tumor Complicated With Renal Cell Carcinoma. Int J Clin Exp Pathol (2015) 8(10):12188–97.
16. Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol (2016) 2(7):922–8. doi: 10.1001/jamaoncol.2016.0256
17. Dubard Gault M, Mandelker D, DeLair D, Stewart CR, Kemel Y, Sheehan MR, et al. Germline SDHA Mutations in Children and Adults With Cancer. Cold Spring Harb Mol Case Stud (2018) 4(4):a002584. doi: 10.1101/mcs.a002584
18. Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, et al. SDHA Loss-Of-Function Mutations in KIT-PDGFRA Wild-Type Gastrointestinal Stromal Tumors Identified by Massively Parallel Sequencing. J Natl Cancer Inst (2011) 103(12):983–7. doi: 10.1093/jnci/djr130
19. Astolfi A, Indio V, Nannini M, Saponara M, Schipani A, De Leo A, et al. Targeted Deep Sequencing Uncovers Cryptic KIT Mutations in KIT/PDGFRA/SDH/RAS-P Wild-Type GIST. Front Oncol (2020) 10:504. doi: 10.3389/fonc.2020.00504
20. Bannon AE, Kent J, Forquer I, Town A, Klug LR, McCann K, et al. Biochemical, Molecular, and Clinical Characterization of Succinate Dehydrogenase Subunit A Variants of Unknown Significance. Clin Cancer Res (2017) 23(21):6733–43. doi: 10.1158/1078-0432.CCR-17-1397
21. Benn DE, Zhu Y, Andrews KA, Wilding M, Duncan EL, Dwight T, et al. Bayesian Approach to Determining Penetrance of Pathogenic SDH Variants. J Med Genet (2018) 55(11):729–34. doi: 10.1136/jmedgenet-2018-105427
22. Tufton N, Sahdev A, Drake WM, Akker SA. Can Subunit-Specific Phenotypes Guide Surveillance Imaging Decisions in Asymptomatic SDH Mutation Carriers? Clin Endocrinol (Oxf) (2019) 90(1):31–46. doi: 10.1111/cen.13877
23. Evenepoel L, Papathomas T, Krol N, Korpershoek E, De Krijger RR, Persu A, et al. Toward an Improved Definition of the Genetic and Tumor Spectrum Associated With SDH Germ-Line Mutations. Genet Med (2015) 17(8):610–20. doi: 10.1038/gim.2014.162
24. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and Molecular Genetics of Patients With the Carney-Stratakis Syndrome and Germline Mutations of the Genes Coding for the Succinate Dehydrogenase Subunits SDHB, SDHC, and SDHD. Eur J Hum Genet (2008) 16(1):79–88. doi: 10.1038/sj.ejhg.5201904
25. MacFarlane J, Seong KC, Bisambar C, Madhu B, Allinson K, Marker A, et al. A Review of the Tumour Spectrum of Germline Succinate Dehydrogenase Gene Mutations: Beyond Phaeochromocytoma and Paraganglioma. Clin Endocrinol (Oxf) (2020) 93(5):528–38. doi: 10.1111/cen.14289
Keywords: SDH-deficient GIST, SDHA, CT: Carney Triad, CSS: Carney-Stratakis syndrome, SDHA germinal mutations, gastrointestinal stromal tumors
Citation: Pantaleo MA, Urbini M, Schipani A, Nannini M, Indio V, De Leo A, Vincenzi B, Brunello A, Grignani G, Casagrande M, Fumagalli E, Conca E, Saponara M, Gruppioni E, Altimari A, De Biase D, Tallini G, Ravegnini G, Turchetti D, Seri M, Ardizzoni A, Secchiero P and Astolfi A (2022) SDHA Germline Variants in Adult Patients With SDHA-Mutant Gastrointestinal Stromal Tumor. Front. Oncol. 11:778461. doi: 10.3389/fonc.2021.778461
Received: 16 September 2021; Accepted: 30 November 2021;
Published: 04 January 2022.
Edited by:
Luis Del Valle, Louisiana State University, United StatesReviewed by:
Giovana Tardin Torrezan, A.C.Camargo Cancer Center, BrazilCopyright © 2022 Pantaleo, Urbini, Schipani, Nannini, Indio, De Leo, Vincenzi, Brunello, Grignani, Casagrande, Fumagalli, Conca, Saponara, Gruppioni, Altimari, De Biase, Tallini, Ravegnini, Turchetti, Seri, Ardizzoni, Secchiero and Astolfi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milena Urbini, bWlsZW5hLnVyYmluaUBpcnN0LmVtci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.