 Till Braun
Till Braun Annika Dechow1
Annika Dechow1 Gregor Friedrich
Gregor Friedrich Johanna Stachelscheid
Johanna Stachelscheid Marco Herling
Marco Herling
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 19 November 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.775363
This article is part of the Research Topic Pathogenesis, Treatment, and Future Directions for Rare T-Cell Leukemias View all 12 articles
T-cell prolymphocytic leukemia (T-PLL) is the most common mature T-cell leukemia. It is a typically aggressively growing and chemotherapy-resistant malignancy with a poor prognosis. T-PLL cells resemble activated, post-thymic T-lymphocytes with memory-type effector functions. Constitutive transcriptional activation of genes of the T-cell leukemia 1 (TCL1) family based on genomic inversions/translocations is recognized as a key event in T-PLL’s pathogenesis. TCL1’s multiple effector pathways include the enhancement of T-cell receptor (TCR) signals. New molecular dependencies around responses to DNA damage, including repair and apoptosis regulation, as well as alterations of cytokine and non-TCR activation signaling were identified as perturbed hallmark pathways within the past years. We currently witness these vulnerabilities to be interrogated in first pre-clinical concepts and initial clinical testing in relapsed/refractory T-PLL patients. We summarize here the current knowledge on the molecular understanding of T-PLL’s pathobiology and critically assess the true translational progress around this to help appraisal by caregivers and patients. Overall, the contemporary concepts on T-PLL’s pathobiology are condensed in a comprehensive mechanistic disease model and promising interventional strategies derived from it are highlighted.
T-cell prolymphocytic leukemia (T-PLL) is an aggressive peripheral T-cell malignancy (1) and represents the most common mature T-cell leukemia in Western countries (incidence ≈ 2.0/million/year) (2). Patients suffering from T-PLL typically present with exponentially rising white blood cell counts, (hepato-) splenomegaly, and small-node lymphadenopathy. CNS involvement has been described as a severe clinical manifestation in a minority of T-PLL (<5% of cases) (3, 4). The rapidly expanding and chemotherapy-refractory course is reflected by a median overall survival from diagnosis of less than 3 years (5, 6). Up to now, the humanized CD52-antibody alemtuzumab is the only substance that induces acceptably high response rates, (in >80% of patients at first line). Notably, nearly all patients relapse within 2 years after alemtuzumab, with very limited options to salvage (4, 7).
First described in 1973 (8), the diagnosis of T-PLL was mainly based on cytomorphological characteristics (6). In the following decades, the pathogenetic concept of T-PLL was centered around cytogenetic abnormalities. Inversions or translocations of the TCL1A locus are the most common chromosomal aberrations and are central in establishing the diagnosis of T-PLL (9). Within the last 5-7 years, genomic and epigenomic studies have remarkably expanded our pathogenetic understanding of T-PLL. More recently, molecular hallmarks around perturbed responses to DNA damage, including repair and apoptosis, as well as alterations of cytokine signaling and epigenetic deregulations, were identified as exploitable dependencies. Here, we condense these novel advances in a comprehensive mechanistic disease concept and highlight promising interventional strategies that are being derived from it.
In >95% of T-PLL, aberrant constitutive expression of the proto-oncogenes TCL1A or MTCP1 by inversions or translocations are observed that juxtapose the TCL1A (at 14q32.1) or MTCP1 (at Xq28) loci to the 14q11.2 locus and by that under control of highly active TRA gene enhancer elements. This prevents physiological downregulation of TCL1A or MTCP1 and is considered the initial event of T-PLL’s leukemogenesis (10). Both oncogenes have shown their oncogenic potential in transgenic mouse models (11–13). Under physiological conditions, expression of the TCL1A oncogene is silenced in CD4/CD8 double-positive (dp) thymocytes (14, 15). At this stage, rearrangements of the TRA locus, encoding for the T-cell receptor (TCR) α-chain, take place (16). Whole-genome sequencing and breakpoint analyses identified that all T-PLL had a breakpoint involving recombination signal sequences (RSS) of the J region of the TRA locus. On the opposite side of the inversion/translocation, breakpoints were more variable, but also involved classical or cryptic RSS (17). In accordance with the finding that virtually all T-PLL express the surface TCR complex (18), the other allele of the analyzed T-PLL cases showed legitimate TRA rearrangements, leading to the expression of a functional TCR (17). Together, these findings suggest, that the aberrant TRA-TCL1A/MTCP1 rearrangements occur during the opening of the TRA locus at the CD4/CD8 dp thymocyte stage in a RAG1/2 dependent manner (17), followed by legitimate recombination of the locus on the other allele. High TCL1A expression is associated with genomic instability (19), thereby forming the basis for additional genomic hits driving oncogenesis (9, 10). However, whether the illegitimate rearrangement is the first hit in the pathogenesis of T-PLL is uncertain. A preceding mono-allelic deletion or mutation of ATM, which are highly recurrent in T-PLL cells, is possible as well. This is supported by a high incidence of T-PLL in patients with germline ATM defects as well as its involvement in the regulation of monoallelic cleavage and genomic stability during TRA recombination (20).
Complex karyotypes (≥3 structural or numerical cytogenetic aberrations) are seen in ~70% of T-PLL and were associated with a poorer prognosis (21). T-PLL genomes usually show complex somatic DNA copy number alterations (CNA) in array-based profiling (10, 21, 22). Generally, losses of chromosomal regions are more frequent than gains. These somatic CNA usually affect hundreds of genes in a patient and are not closely associated with altered expression of the respective genes, indicating additional modes of transcriptional dysregulation beyond CNA. Besides the above-described aberrations affecting genes of the TCL1 family, genomic losses of chromosome 11q and gains of chromosome 8q are most recurrently observed. Losses affecting chromosome 11 involve the tumor suppressor ATM (11q22.3) as the minimally deleted region (6, 10, 19, 21–30). This is implicated in T-PLL development by dysregulation of proper DNA damage repair as highlighted by more complex karyotypes in ATM deleted cases (10). The genomic region encoding for the downstream effector of ATM, p53, is only disrupted in a minority of T-PLL (10). Gains of chromosome 8q can mainly be attributed to a trisomy of 8q, resulting from isochromosomes (8)(q10) (29). Overexpression of the proto-oncogene MYC (8q24.21) is not strictly associated with the presence of 8q gains and vice versa. Other genes like AGO2 at 8q24.3 are more frequently involved in these 8q amplifications. Overexpression of AGO2, which centrally regulates RNA interference, may additionally contribute to T-PLL development (10).
At lower frequencies, genomic losses of chromosomes 6q, 8p, 12p, 13q, and 22q as well as genomic amplifications of 6p and 22q are observed in T-PLL cells (10, 21–23, 27). Up to now, the underlying target genes of these structural aberrations and their functional contributions have not been fully revealed. First promising concepts could derive from a systems biology approach (31). Genome-wide gene expression and copy number profiles of T-PLL patients could be utilized to learn a T-PLL specific gene regulatory network (32). Such a network would allow to predict potential impacts of individual CNA on known cellular signaling pathways or treatment response signatures by network propagation (32), as demonstrated for oligodendrogliomas (33) and prostate carcinomas (34). Thus, more intensified efforts on integrating available genome-wide data could help to identify new potential driver candidates and their downstream targets in T-PLL.
Besides the highly prevalent structural lesions involving the oncogenes TCL1A, AGO2, and MYC, as well as in the tumor suppressor ATM, various single-nucleotide variants (SNVs) were linked to the molecular pathogenesis of T-PLL cells (10, 26, 35, 36). Generally, SNVs occur at similar rates in T-PLL as in other hematologic and solid tumors (10). Most of these primarily somatic SNVs seem to accumulate during T-PLL’s leukemogenesis in the context of high levels of oxidative damage and in the absence of efficient repair mechanisms to counteract these hazards (10). Fittingly, ATM, the central apical regulator of DNA integrity, shows high rates of damaging SNVs, in addition to the above-described partial inactivation by mono-allelic losses (10, 24, 26, 35–38). These missense, nonsense, or frameshift mutations of ATM mainly cluster within its FAT or PI3K domains (10).
Other frequently mutated genes in T-PLL are CHEK2, SAMHD1, and MSH, which are also involved in DNA damage repair mechanisms, which further supports a concept of T-PLL’s incompetence in safeguarding mechanisms of repair or cell death execution (10, 26, 35, 36). Remarkably, SAMHD1 and ATM belong to the small fraction of genes, whose mutations show variant allele fractions (VAFs) of more than 80% (10, 35), suggesting acquisition of these lesions early in leukemogenesis.
Within the last decade, genomic aberrations affecting the JAK/STAT signaling pathway emerged as an additional hallmark of T-PLL (10, 26, 35, 36, 38–42). The JAK3 gene shows the highest frequency of such gain-of-function mutations, followed by STAT5 and JAK1 (43). These primarily missense mutations target the conserved pseudokinase (JAK1, JAK3) or SH2 domains (STAT5) in most T-PLL cases. Notably, SNVs affecting components of the JAK/STAT signaling pathway occur at relatively low VAFs, indicating their rather sub-clonal character (10). However, the central role of deregulated JAK/STAT signaling is substantiated by genomic losses of genes that encode for negative regulators of this pathway (e.g. DUSP4, SOCS genes) (43). Together with the high frequency of JAK/STAT gene mutations, basal phosphorylation of distal STAT5 is observed in virtually every T-PLL case (10, 43). In addition, the WNT as well as the Notch signaling pathways, are disturbed by SNVs in a minority of T-PLL cases (10, 26). Rare mutations further involve cell cycle regulation (e.g. CDC27) and apoptosis regulation (e.g. BCLAF1) (10).
Analyses of the transcriptome of T-PLL cells have been performed intensively in bulk RNA samples, either by gene expression arrays or by RNA sequencing (RNA-seq). In line with rearrangements of the chromosome 14q, TCL1A was the most upregulated gene in virtually every cohort (10, 35, 42, 44). The other TCL1 family members, TCL1B and MTCP1, showed additional overexpression, although to a lower extent (10). In agreement with the gains at chromosome 8q, the proto-oncogene MYC as well as the miR-processing regulator AGO2 showed overexpression on mRNA level (10, 42). Highlighting the importance of deregulated JAK/STAT signaling in T-PLL, downstream targets of this pathway (e.g. BCL2L1) showed a significant upregulation (42).
Among the genes with the most significantly altered expression were those involved in TCR/cytokine signaling. Prominent examples are downregulated CTLA4 and SLAMF6. They are central mediators of immune signal transduction and regulation of lymphocyte activation and we implicate their loss in the activated T-cell phenotype of the T-PLL cell (10, 18, 22). Moreover, potential underlying causes for the inability of T-PLL cells to undergo cell death upon DNA damage were identified in their altered transcriptome: Pro-apoptotic genes (e.g. GIMAP5, various Caspases) were significantly downregulated (10, 22). Transcriptome studies can also be utilized to identify individualized treatment options for T-PLL patients. In a first case study, RNA-seq data were integrated with exome-seq and ex vivo single-drug sensitivities, establishing a customized platform on individual predictions of responses to drug combinations (39).
Recently, the miR-ome of T-PLL cells was analyzed by small RNA-seq in two independent cohorts (44, 45). T-PLL cells showed a global miR expression signature of ~35 significantly deregulated miRs, resembling the miR expression profile of TCR-activated healthy T-cells (45). By combining the small RNA-seq with transcriptome sequencing data, regulatory networks involving cell survival signaling and DNA repair pathways were uncovered. In both cohorts, the miR-141/200c cluster showed the strongest upregulation among all miRs and separated T-PLL cases into two major subgroups with normal vs. upregulated expression. Preliminary data revealed a role of this cluster in TGF-β signaling (44) as well as in cell cycle regulation (45). Further perturbations of miR expression include overexpression of miR-223-3p and miR-181a/miR-181 as well as downregulation of the miR-21 and the miR-29 cluster. The functional consequences of these deregulations have yet to be demonstrated in T-PLL. Nevertheless, based on the expression of miR-200a-3p, miR-223-3p, and miR-424-5p, a first overall survival score for T-PLL (miROS-TPLL) was established and might improve clinical stratifications (45).
Gene set enrichment analyses of T-PLL transcriptomes identified pathways of epigenetic regulation as significantly altered (10). These findings were additionally highlighted by a high incidence of mutations in epigenetic modifiers (e.g. EZH2, TET2, KMTs) (10, 26, 35, 36). However, systematic analyses of DNA-methylation, profiles of histone modifications, and states of chromatin accessibility have not yet been published. First data in a small cohort of T-PLL implicate massive epigenetic reprogramming, as shown by genome-wide alterations of chromatin states at promoters and active enhancers identified via H3K4me3 and H3K27ac ChIP-seq (46). These alterations correlated with changes in expression of frequently deregulated genes (e.g. TCL1A, MYC, EZH2, AGO2), presenting additional ways of their deregulation beyond the described genomic aberrations. Vice versa, a role of TCL1A/MTCP1 activation and/or ATM inactivation in epigenetic disturbances is also conceivable (47, 48).
Besides (epi)genetic changes, the dependence of leukemic cells on signals from microenvironmental sources for proliferation and survival has been shown for various entities, including T-cell neoplasms (49). Such interactions are mediated by adhesion molecules, cell surface ligands, chemokines, cytokines, and their respective receptors (50). So far, little is known about the (specific) micromilieu of T-PLL cells and how they shape it. Upregulation of cytokines (e.g. TNF, IL-8), cytokine receptors (e.g. CD25 (IL-2Rα), CD122 (IL-2Rβ), CD124, or CD127), as well as of chemokine receptors (e.g. CCR3 and CCR4) provide first hints of a deregulated crosstalk between T-PLL and bystander cells (18). Furthermore, mutations of chemokine receptors (e.g. CXCR3) are described (10). The potential proactive role of the micromilieu in T-PLL’s leukemogenesis is further implicated by the secretion of the Th1-associated cytokines IFN-γ, IL-2, IL-10, TNF-α/β, and IL-8 of T-PLL cells upon TCR stimulation (18). Mechanistic proof for an involvement of CCR7 in the sustenance of T-PLL cell survival derives from studies with CCR7-blocking antibodies. They impaired survival signaling pathways in T-PLL cells in vitro and increased the survival of mice transplanted with the T-PLL-like cell line SUP-T11 (51). More work is required to study the composition of T-PLL’s microenvironment (i.e. cell types and humoral factors) and the involved molecular interactions.
TCR signaling is the major growth regulatory system of T-cells. It shapes their maturation, differentiation, and activation, hence their effector and tolerogenic capacity (52, 53). Amplification of TCR signaling represents a feature of many T-cell malignancies, although generated by distinct mechanisms (54): (i) decreased input thresholds for continuous exogeneous TCR activation, (ii) autonomous activation of TCR-signaling intermediates, (iii) downregulation of inhibitory coregulators, or (iv) stand-ins for TCR signals, such as strong cytokine-inputs or their mimics, e.g. via the ALK oncogene. T-PLL cells usually express at least one surface component of the TCR/coreceptor complex and show robust TCR-signal competence when stimulated ex vivo (9, 18). Their gene expression profiles show prominent signatures of TCR activation (10). Notably, TCL1A acts as a physically engaging coactivator of TCR-kinases such as AKT, ZAP70, or ERK, and by that is a TCR-signal enhancer, hence, a sensitizer towards low-abundance signals. That places T-PLL into model (i) of the TCR-centric pathogenetic view of T-cell neoplasms (18, 54).
Enhanced TCR signaling is further established in T-PLL cells by impaired control mechanisms [model (iii)], e.g. by downregulation of negative coregulators such as SLAMFs or checkpoint molecules such as CTLA4 (10). The resulting activated phenotype of T-PLL cells is additionally accompanied by a TCL1A-mediated inability to execute FAS-mediated and activation-induced cell death (18).
In line with their TCR signaling competence, T-PLL cells reveal a phenotype of mature, antigen-experienced, non-conventional memory T-cells (18). As an underlying principle, it is tempting to speculate that through enhanced TCR signaling, the transition of naïve T-cells into an expanding pool of memory T-cells is accelerated. The lack of a common TCR clonotype across cases would indicate that not a specific antigen drives TCR-mediated outgrowth in T-PLL (18, 55). More likely is an MHC-dependent TCR activation through various low-avidity (auto)antigens or antigen-independent tonic signals at place, either MHC-driven or via TCR self-activation in enabled memory T-cells. Although treatment strategies that target TCR signaling intermediates have shown promising potential (56), the TCR dependence of T-PLL cells at the overt leukemic stage is not conclusively clarified.
Recent advances in omics technologies over the last decade have elevated the molecular understanding of T-PLL to another level (Figure 1, Supplementary Table 1). Translocations and inversions of chromosome 14q at the dp thymocyte stage are perceived to initiate T-PLL’s leukemogenesis (10, 17). These genomic aberrations lead to overexpression of the proto-oncogenes TCL1A and MTCP1 and result in apoptotic resistance and genomic instability (19). TCL1 family-activating lesions form a functionally perturbing cooperation with (preceding or subsequent) lesions that impair the tumor suppressor ATM, which further incapacitate the T-PLL cell to execute safeguarding responses (10). Likely, additional perturbations are operational for this TCL1up/ATMdef leukemic precursor to finally escape T-cell homeostatic control. These are acquired by lesions that activate JAK/STAT signaling (43), by miR (processing) deregulations (44, 45), by MYC amplification (6, 10), and by deregulated epigenetic mechanisms (10, 36). To a lesser degree we understand, on which central functional levels, such as TCR- or cytokine signaling or autocrine forward-feeding loops, these (epi)genetic events have a direct or less immediate impact.
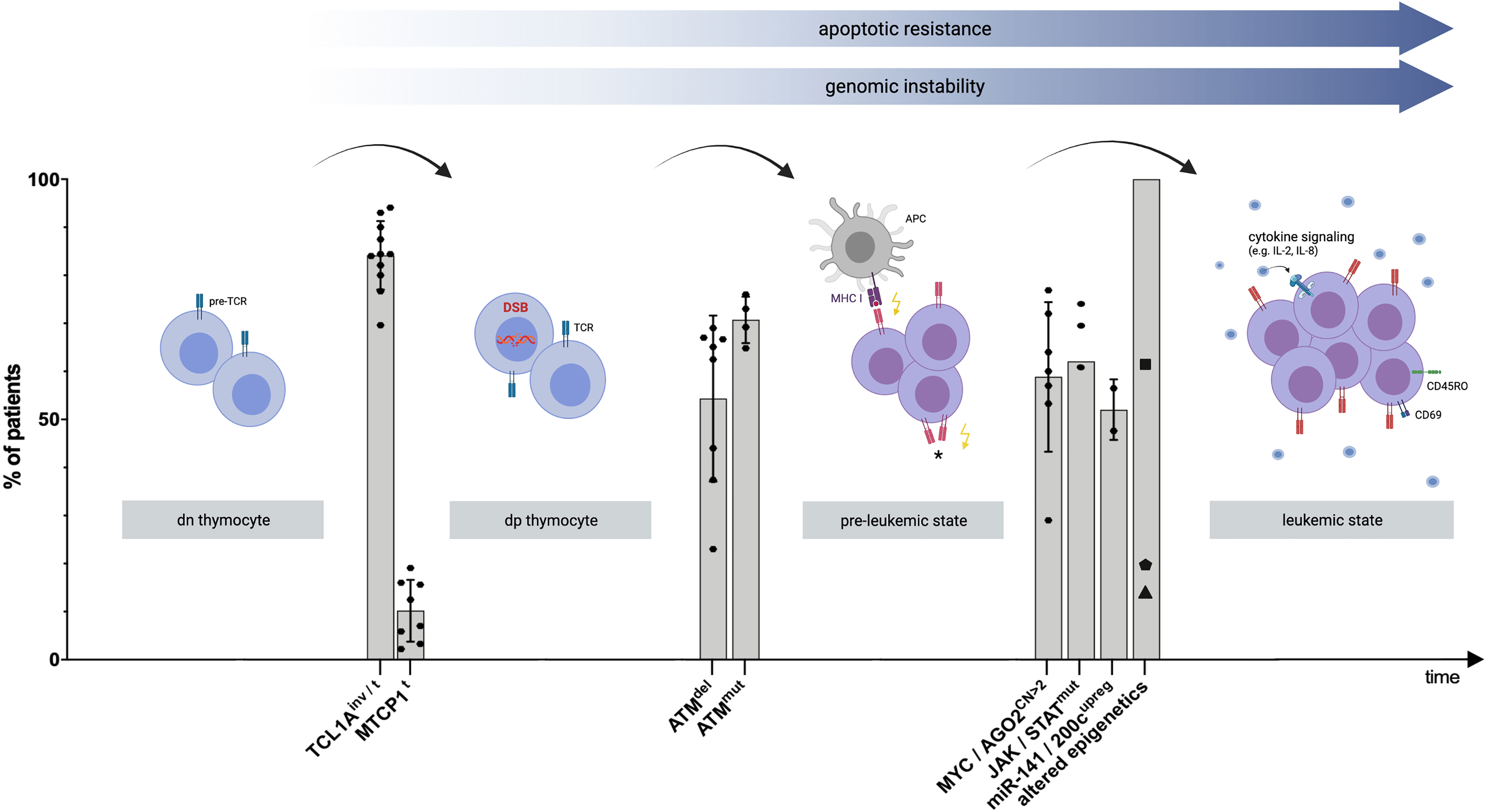
Figure 1 Proposed model of clonal evolution of T-PLL cells. Schematic visualization explaining T-PLL’s leukemogenesis, based on recent genomic profiling series and corresponding functional assessments. Timeline: Chronology of genomic events leading to the progression to an advanced state of (pre)malignant T-cell development. Y-Axis: Percentage of all analyzed T-PLL patients presenting the respective genomic aberration. Each dot represents a prevalence, derived from selected publications (Supplementary Table 1). The median, as well as standard deviation, out of these publications was calculated for each genomic event. The variability between the studies can be attributed to the different methods and cohort sizes (for more information refer to Supplementary Table 1). The first ‘stage’ involves the double-negative (dn) thymocyte, carrying the pre-T cell receptor (pre-TCR) complex. Translocations (t) and inversions (inv) of chromosome 14q at the dp thymocyte stage result in constitutive expression of the proto-oncogenes TCL1A or MTCP1 in a vast majority of T-PLL cases (9, 17). These hits impair the genomic stability of the affected T-cell by reduced DNA repair capacities of DNA double-stranded breaks (DSB) or other (oxidative) insults (10). Deletions (del) and mutations (mut) involving ATM lead to a functionally hypomorphic apical regulator of repair, cell fate, and cell cycle control of the T-PLL precursor. This pre-leukemic cell becomes unable to execute such safeguarding mechanisms upon genotoxic stress (10). Among subsequent perturbations, TCL1A overexpression lowers TCR-signaling thresholds (18), enabling the cell to sustain on low-level input, either by major histocompatibility complex (MHC)-dependent (auto) antigen-presenting cells (APC), or by self-MHC drive only, or by autonomous TCR activation (*, not proven). A central distal node is the JAK/STAT transcriptional machinery. Besides major growth pathways such as the TCR and cytokine-mediated cascades feeding into it, there also is a high prevalence of hyperactivating mutations that target JAK1, JAK3, or STAT5B (18) and a high incidence of losses of JAK/STAT negative regulators (43). Further leukemic outgrowth and progression to an exponentially proliferating T-PLL cell are likely mediated by additional aberrations, including copy number (CN) gains on chromosome 8q, leading to MYC amplification and AGO2 overexpression (10). Furthermore, deregulations of T-PLL’s miR-ome, exemplarily represented by the upregulation (upreg) of the miR-141/200c family (45), and of T-PLL’s epigenome in virtually all patients as shown by altered chromatin states at promoters and active enhancers (46), potentially mediated by frequent mutations in KMTs(▪), TET2 (⬟), and EZH2 (▲) (10), contribute to the final leukemic outgrowth of a transformed and activated T-cell (as shown by the T-cell activation marker CD69) with memory-type effector functions (as shown by CD45RO surface expression) (18). The figure was created by the authors using Biorender.com.
Overall, many questions of T-PLL’s pathogenesis remain unresolved, like (i) the role of pro-survival signals of T-PLL’s bystander cells, (ii) the dependence of T-PLL cells on their TCR in clonal sustenance, (iii) the nature of T-PLL’s epigenome, and (iv) the mechanisms of disease progression and treatment resistance. Especially the latter aspect calls for single-cell resolved analyses to illustrate clonal oscillations.
The identification of key drivers of the molecular pathogenesis of T-PLL offers the possibility for the development of new drugs that target its crucial pathways. Here, central pathogenetic relevance is likely not equivalent to a major vulnerability, which requires more thorough interrogations. However, there is sound reason to be optimistic that we will soon see novel strategies against T-PLL cells to become the basis for future combinatorial therapies. Exemplarily, agents targeting TCR signaling or the JAK/STAT pathway (18, 56) show encouraging results, preclinically and/or in first case reports (57, 58). In addition, the inability of T-PLL cells to induce adequate responses to DNA insults was translated into therapeutic strategies to reactivate p53 via MDM2/MDMx inhibitors or targeting BCL2 family members (e.g. Venetoclax) (10, 59, 60). There are ongoing activities in the search for efficacious combinations of the, as single agent clinically only moderately active Venetoclax, with other classes of inhibitors in relapsed/refractory (r/r) T-PLL (59–62). In addition, epigenetic disturbances of T-PLL cells further emphasize hypomethylating agents (e.g. Cladribine) as well as inhibitors of deacetylating enzymes (e.g. Romidepsin) as options (10, 63, 64). Combining these drugs, which target molecular vulnerabilities of T-PLL cells, with the current standard therapy of alemtuzumab represents another promising approach. Another challenge to be addressed is the ‘purposing’ of the innate or adaptive immune system to specifically attack T-PLL cells (65).
TB, AD, GF, MS, JS, and MH contributed to initial and subsequent drafts of the manuscript. TB, AD, and MH designed and drew the figure and corresponding table. All authors contributed to the article and approved the submitted version.
This research was funded by the DFG Research Unit FOR1961 (Control-T; HE3553/4-2), the Köln Fortune Program, and the Fritz Thyssen Foundation (10.15.2.034MN). This work was also funded by the EU Transcan-2 consortium ‘ERANET-PLL’ (01KT1906A/B) and by the ERAPerMed consortium ‘JAKSTAT-TARGET’ (ERAPERMED2018-066).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.775363/full#supplementary-material
1. Staber PB, Herling M, Bellido M, Jacobsen ED, Davids MS, Kadia TM, et al. Consensus Criteria for Diagnosis, Staging, and Treatment Response Assessment of T-Cell Prolymphocytic Leukemia. Blood (2019) 134:1132–43. doi: 10.1182/blood.2019000402
2. Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A Systematic Approach to Diagnosis of Mature T-Cell Leukemias Reveals Heterogeneity Among WHO Categories. Blood (2004) 104:328–35. doi: 10.1182/blood-2004-01-0002
3. Mori J, Oshima K, Kimura S, Ikezoe T. Treatment of T-Cell Prolymphocytic Leukemia With Central Nervous System Involvement Using Intrathecal Alemtuzumab Administration. Case Rep Hematol (2020) 2020:8822172. doi: 10.1155/2020/8822172
4. Dearden C. How I Treat Prolymphocytic Leukemia. Blood (2012) 120:538–51. doi: 10.1182/blood-2012-01-380139
5. Pflug N, Cramer P, Robrecht S, Bahlo J, Westermann A, Fink A-M, et al. New Lessons Learned in T-PLL: Results From a Prospective Phase-II Trial With Fludarabine–Mitoxantrone–Cyclophosphamide–Alemtuzumab Induction Followed by Alemtuzumab Maintenance. Leuk Lymphoma (2019) 60:649–57. doi: 10.1080/10428194.2018.1488253
6. Matutes E, Brito-Babapulle V, Swansbury J, Ellis J, Morilla R, Dearden C, et al. Clinical and Laboratory Features of 78 Cases of T-Prolymphocytic Leukemia. Blood (1991) 78:3269–74. doi: 10.1182/blood.v78.12.3269.3269
7. Dearden CE, Matutes E, Cazin B, Tjønnfjord GE, Parreira A, Nomdedeu B, et al. High Remission Rate in T-Cell Prolymphocytic Leukemia With CAMPATH-1h. Blood (2001) 98:1721–6. doi: 10.1182/blood.V98.6.1721
8. Catovsky D, Galetto J, Okos A, Galton DA, Wiltshaw E, Stathopoulos G. Prolymphocytic Leukaemia of B and T Cell Type. Lancet (London England) (1973) 2:232–4. doi: 10.1016/s0140-6736(73)93135-8
9. Herling M, Patel KA, Teitell MA, Konopleva M, Ravandi F, Kobayashi R, et al. High TCL1 Expression and Intact T-Cell Receptor Signaling Define a Hyperproliferative Subset of T-Cell Prolymphocytic Leukemia. Blood (2008) 111:328–37. doi: 10.1182/blood-2007-07-101519
10. Schrader A, Crispatzu G, Oberbeck S, Mayer P, Pützer S, von Jan J, et al. Actionable Perturbations of Damage Responses by TCL1/ATM and Epigenetic Lesions Form the Basis of T-PLL. Nat Commun (2018) 9:697. doi: 10.1038/s41467-017-02688-6
11. Virgilio L, Lazzeri C, Bichi R, Nibu K, Narducci MG, Russo G, et al. Deregulated Expression of TCL1 Causes T Cell Leukemia in Mice. Proc Natl Acad Sci U S A (1998) 95:3885–9. doi: 10.1073/pnas.95.7.3885
12. Gritti C, Dastot H, Soulier J, Janin A, Daniel MT, Madani A, et al. Transgenic Mice for MTCP1 Develop T-Cell Prolymphocytic Leukemia. Blood (1998) 92:368–73. doi: 10.1182/blood.V92.2.368
13. Joiner M, Le Toriellec E, Despouy G, Stern MH. The MTCP1 Oncogene Modifies T-Cell Homeostasis Before Leukemogenesis in Transgenic Mice. Leukemia (2007) 21:362–6. doi: 10.1038/sj.leu.2404476
14. Virgilio L, Narducci MG, Isobe M, Billips LG, Cooper MD, Croce CM, et al. Identification of the TCL1 Gene Involved in T-Cell Malignancies. Proc Natl Acad Sci U S A (1994) 91:12530–4. doi: 10.1073/pnas.91.26.12530
15. Hoyer KK, Herling M, Bagrintseva K, Dawson DW, French SW, Renard M, et al. T Cell Leukemia-1 Modulates TCR Signal Strength and IFN-γ Levels Through Phosphatidylinositol 3-Kinase and Protein Kinase C Pathway Activation. J Immunol (2005) 175:864–73. doi: 10.4049/jimmunol.175.2.864
16. Roth DB. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol Spectr (2014) 2:10. doi: 10.1128/microbiolspec.MDNA3-0041-2014
17. Patil P, Cieslak A, Bernhart SH, Toprak UH, Wagener R, López C, et al. Reconstruction of Rearranged T-Cell Receptor Loci by Whole Genome and Transcriptome Sequencing Gives Insights Into the Initial Steps of T-Cell Prolymphocytic Leukemia. Genes Chromosomes Cancer (2020) 59:261–7. doi: 10.1002/gcc.22821
18. Oberbeck S, Schrader A, Warner K, Jungherz D, Crispatzu G, von JJ, et al. Non-Canonical Effector Functions of the T-Memory-Like T-PLL Cell Are Shaped by Cooperative TCL1A and TCR Signaling. Blood (2020) 136:2786–802. doi: 10.1182/blood.2019003348
19. Petrinelli P, Elli R, Marcucci L, Tabolacci E, Barbieri C, Antonelli A. Telomeric Associations and Chromosome Instability in Ataxia Telangiectasia T Cells Characterized by TCL1 Expression. Cancer Genet Cytogenet (2001) 125:46–51. doi: 10.1016/S0165-4608(00)00358-7
20. Chaumeil J, Micsinai M, Ntziachristos P, Deriano L, Wang JM-H, Ji Y, et al. Higher-Order Looping and Nuclear Organization of Tcra Facilitate Targeted Rag Cleavage and Regulated Rearrangement in Recombination Centers. Cell Rep (2013) 3:359–70. doi: 10.1016/j.celrep.2013.01.024
21. Hu Z, Medeiros LJ, Fang L, Sun Y, Tang Z, Tang G, et al. Prognostic Significance of Cytogenetic Abnormalities in T-Cell Prolymphocytic Leukemia. Am J Hematol (2017) 92:441–7. doi: 10.1002/ajh.24679
22. Dürig J, Bug S, Klein-Hitpass L, Boes T, Jöns T, Martin-Subero JI, et al. Combined Single Nucleotide Polymorphism-Based Genomic Mapping and Global Gene Expression Profiling Identifies Novel Chromosomal Imbalances, Mechanisms and Candidate Genes Important in the Pathogenesis of T-Cell Prolymphocytic Leukemia With Inv(14)(Q11q32. Leukemia (2007) 21:2153–63. doi: 10.1038/sj.leu.2404877
23. Costa D, Queralt R, Aymerich M, Carrió A, Rozman M, Vallespí T, et al. High Levels of Chromosomal Imbalances in Typical and Small-Cell Variants of T-Cell Prolymphocytic Leukemia. Cancer Genet Cytogenet (2003) 147:36–43. doi: 10.1016/s0165-4608(03)00161-4
24. Stoppa-Lyonnet D, Soulier J, Laugé A, Dastot H, Garand R, Sigaux F, et al. Inactivation of the ATM Gene in T-Cell Prolymphocytic Leukemias. Blood (1998) 91:3920–6. doi: 10.1182/blood.V91.10.3920
25. Yamaguchi M, Yamamoto K, Miki T, Mizutani S, Miura O. T-Cell Prolymphocytic Leukemia With Der(11)T(1;11)(Q21;Q23) and ATM Deficiency. Cancer Genet Cytogenet (2003) 146:22–6. doi: 10.1016/S0165-4608(03)00104-3
26. Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, et al. Integrated Genomic Sequencing Reveals Mutational Landscape of T-Cell Prolymphocytic Leukemia. Blood (2014) 124:1460–72. doi: 10.1182/blood-2014-03-559542
27. Soulier J, Pierron G, Vecchione D, Garand R, Brizard F, Sigaux F, et al. A Complex Pattern of Recurrent Chromosomal Losses and Gains in T-Cell Prolymphocytic Leukemia. Genes Chromosom Cancer (2001) 31:248–54. doi: 10.1002/gcc.1141
28. Mossafa H, Brizard A, Huret J-L, Brizard F, Lessard M, Guilhot F, et al. Trisomy 8q Due to I(8q) or Der(8) T(8;8) Is a Frequent Lesion in T-Prolymphocytic Leukaemia: Four New Cases and a Review of the Literature. Br J Haematol (1994) 86:780–5. doi: 10.1111/j.1365-2141.1994.tb04829.x
29. Maljaei SH, Brito-Babapulle V, Hiorns LR, Catovsky D. Abnormalities of Chromosomes 8, 11, 14, and X in T-Prolymphocytic Leukemia Studied by Fluorescence In Situ Hybridization. Cancer Genet Cytogenet (1998) 103:110–6. doi: 10.1016/S0165-4608(97)00410-X
30. Tirado CA, Starshak P, Delgado P, Rao N. T-Cell Prolymphocytic Leukemia (T-PLL), a Heterogeneous Disease Exemplified by Two Cases and the Important Role of Cytogenetics: A Multidisciplinary Approach. Exp Hematol Oncol (2012) 1:21. doi: 10.1186/2162-3619-1-21
31. Seifert M, Friedrich B, Beyer A. Importance of Rare Gene Copy Number Alterations for Personalized Tumor Characterization and Survival Analysis. Genome Biol (2016) 17:204. doi: 10.1186/s13059-016-1058-1
32. Seifert M, Beyer A. Regnet: An R Package for Network-Based Propagation of Gene Expression Alterations. Bioinformatics (2018) 34:308–11. doi: 10.1093/bioinformatics/btx544
33. Gladitz J, Klink B, Seifert M. Network-Based Analysis of Oligodendrogliomas Predicts Novel Cancer Gene Candidates Within the Region of the 1p/19q Co-Deletion. Acta Neuropathol Commun (2018) 6:49. doi: 10.1186/s40478-018-0544-y
34. Seifert M, Peitzsch C, Gorodetska I, Börner C, Klink B, Dubrovska A. Network-Based Analysis of Prostate Cancer Cell Lines Reveals Novel Marker Gene Candidates Associated With Radioresistance and Patient Relapse. PloS Comput Biol (2019) 15:e1007460. doi: 10.1371/journal.pcbi.1007460
35. Johansson P, Klein-Hitpass L, Choidas A, Habenberger P, Mahboubi B, Kim B, et al. SAMHD1 Is Recurrently Mutated in T-Cell Prolymphocytic Leukemia. Blood Cancer J (2018) 8:11. doi: 10.1038/s41408-017-0036-5
36. López C, Bergmann AK, Paul U, Murga Penas EM, Nagel I, Betts MJ, et al. Genes Encoding Members of the JAK-STAT Pathway or Epigenetic Regulators Are Recurrently Mutated in T-Cell Prolymphocytic Leukaemia. Br J Haematol (2016) 173:265–73. doi: 10.1111/bjh.13952
37. Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, et al. Biallelic Mutations in the ATM Gene in T-Prolymphocytic Leukemia. Nat Med (1997) 3:1155–9. doi: 10.1038/nm1097-1155
38. Stengel A, Kern W, Zenger M, Perglerová K, Schnittger S, Haferlach T, et al. Genetic Characterization of T-PLL Reveals Two Major Biologic Subgroups and JAK3 Mutations as Prognostic Marker. Genes Chromosom Cancer (2016) 55:82–94. doi: 10.1002/gcc.22313
39. He L, Tang J, Andersson EI, Timonen S, Koschmieder S, Wennerberg K, et al. Patient-Customized Drug Combination Prediction and Testing for T-Cell Prolymphocytic Leukemia Patients. Cancer Res (2018) 78:2407–18. doi: 10.1158/0008-5472.CAN-17-3644
40. Greenplate A, Wang K, Tripathi RM, Palma N, Ali SM, Stephens PJ, et al. Genomic Profiling of T-Cell Neoplasms Reveals Frequent JAK1 and JAK3 Mutations With Clonal Evasion From Targeted Therapies. JCO Precis Oncol (2018) 2:1–16. doi: 10.1200/PO.17.00019
41. Bellanger D, Jacquemin V, Chopin M, Pierron G, Bernard OA, Ghysdael J, et al. Recurrent JAK1 and JAK3 Somatic Mutations in T-Cell Prolymphocytic Leukemia. Leukemia (2013) 28:417. doi: 10.1038/leu.2013.271
42. Andersson EI, Pützer S, Yadav B, Dufva O, Khan S, He L, et al. Discovery of Novel Drug Sensitivities in T-PLL by High-Throughput Ex Vivo Drug Testing and Mutation Profiling. Leukemia (2017) 32:774. doi: 10.0.4.14/leu.2017.252
43. Wahnschaffe L, Braun T, Timonen S, Giri AK, Schrader A, Wagle P, et al. JAK/STAT-Activating Genomic Alterations Are a Hallmark of T-PLL. Cancers (Basel) (2019) 11:1833. doi: 10.3390/cancers11121833
44. Erkeland SJ, Stavast CJ, Schilperoord-Vermeulen J, Dal Collo G, Van de Werken HJG, Leon LG, et al. The miR-200c/141-ZEB2-Tgfβ Axis Is Aberrant in Human T-Cell Prolymphocytic Leukemia. Haematologica (2021). doi: 10.3324/haematol.2020.263756
45. Braun T, Glass M, Wahnschaffe L, Otte M, Mayer P, Franitza M, et al. Micro-RNA Networks in T-Cell Prolymphocytic Leukemia Reflect T-Cell Activation and Shape DNA Damage Response and Survival Pathways. Haematologica (2020). doi: 10.3324/haematol.2020.267500
46. Tian S, Zhang H, Zhang P, Kalmbach M, Lee J-H, Ordog T, et al. Epigenetic Alteration Contributes to the Transcriptional Reprogramming in T-Cell Prolymphocytic Leukemia. Sci Rep (2021) 11:8318. doi: 10.1038/s41598-021-87890-9
47. Zhang P, Zhang M. Epigenetics in the Pathogenesis and Treatment of Cutaneous T-Cell Lymphoma. Front Oncol (2021) 11:663961. doi: 10.3389/fonc.2021.663961
48. Palamarchuk A, Yan PS, Zanesi N, Wang L, Rodrigues B, Murphy M, et al. Tcl1 Protein Functions as an Inhibitor of De Novo DNA Methylation in B-Cell Chronic Lymphocytic Leukemia (CLL). Proc Natl Acad Sci U S A (2012) 109:2555–60. doi: 10.1073/pnas.1200003109
49. Bennani NN, Ansell SM. Tumor Microenvironment in T-Cell Lymphomas BT - T-Cell and NK-Cell Lymphomas: From Biology to Novel Therapies. Querfeld C, Zain J, Rosen ST, editors. Cham: Springer International Publishing (2019) p. 69–82. doi: 10.1007/978-3-319-99716-2_3
50. Jin M-Z, Jin W-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct Target Ther (2020) 5:166. doi: 10.1038/s41392-020-00280-x
51. Cuesta-Mateos C, Fuentes P, Schrader A, Juárez-Sánchez R, Loscertales J, Mateu-Albero T, et al. CCR7 as a Novel Therapeutic Target in T-Cell PROLYMPHOCYTIC Leukemia. biomark Res (2020) 8:54. doi: 10.1186/s40364-020-00234-z
52. Pollizzi KN, Powell JD. Integrating Canonical and Metabolic Signalling Programmes in the Regulation of T Cell Responses. Nat Rev Immunol (2014) 14:435–46. doi: 10.1038/nri3701
53. Warner K, Weit N, Crispatzu G, Admirand J, Jones D, Herling M. T-Cell Receptor Signaling in Peripheral T-Cell Lymphoma - a Review of Patterns of Alterations in a Central Growth Regulatory Pathway. Curr Hematol Malig Rep (2013) 8:163–72. doi: 10.1007/s11899-013-0165-2
54. Hwang J-R, Byeon Y, Kim D, Park S-G. Recent Insights of T Cell Receptor-Mediated Signaling Pathways for T Cell Activation and Development. Exp Mol Med (2020) 52:750–61. doi: 10.1038/s12276-020-0435-8
55. Kotrova M, Novakova M, Oberbeck S, Mayer P, Schrader A, Knecht H, et al. Next-Generation Amplicon TRB Locus Sequencing can Overcome Limitations of Flow-Cytometric Vβ Expression Analysis and Confirms Clonality in All T-Cell Prolymphocytic Leukemia Cases. Cytom Part A (2018) 93:1118–24. doi: 10.1002/cyto.a.23604
56. Dondorf S, Schrader A, Herling M. Interleukin-2-Inducible T-Cell Kinase (ITK) Targeting by BMS-509744 Does Not Affect Cell Viability in T-Cell Prolymphocytic Leukemia (T-PLL). J Biol Chem (2015) 290:10568–9. doi: 10.1074/jbc.L115.644641
57. Gomez-Arteaga A, Margolskee E, Wei MT, van Besien K, Inghirami G, Horwitz S. Combined Use of Tofacitinib (Pan-JAK Inhibitor) and Ruxolitinib (a JAK1/2 Inhibitor) for Refractory T-Cell Prolymphocytic Leukemia (T-PLL) With a JAK3 Mutation. Leuk Lymphoma (2019) 60:1626–31. doi: 10.1080/10428194.2019.1594220
58. Wei M, Koshy N, van Besien K, Inghirami G, Horwitz SM. Refractory T-Cell Prolymphocytic Leukemia With JAK3 Mutation: In Vitro and Clinical Synergy of Tofacitinib and Ruxolitinib. Blood (2015) 126. doi: 10.1182/blood.V126.23.5486.5486
59. Herbaux C, Kornauth C, Poulain S, Chong SJF, Collins MC, Valentin R, et al. BH3 Profiling Identifies Ruxolitinib as a Promising Partner for Venetoclax to Treat T-Cell Prolymphocytic Leukemia. Blood (2021) 137:3495–506. doi: 10.1182/blood.2020007303
60. Kornauth CF, Herbaux C, Boidol B, Guillemette C, Mayerhöfer ME, Jäger U, et al. The Combination of Venetoclax and Ibrutinib Is Effective in Relapsed/Refractory T-Prolymphocytic Leukemia and Influences BCL-2-Family Member Dependencies. Hematol Oncol (2019) 37:482–4. doi: 10.1002/hon.161_2631
61. Hampel PJ, Parikh SA, Call TG, Shah MV, Bennani NN, Al-Kali A, et al. Venetoclax Treatment of Patients With Relapsed T-Cell Prolymphocytic Leukemia. Blood Cancer J (2021) 11:47. doi: 10.1038/s41408-021-00443-1
62. Kornauth C, Herbaux C, Boidol B, Guillemette C, Caron P, Mayerhöfer ME, et al. Rationale for the Combination of Venetoclax and Ibrutinib in T-Prolymphocytic Leukemia. Haematologica (2021) 106:2251–6. doi: 10.3324/haematol.2020.271304
63. Hasanali ZS, Saroya BS, Stuart A, Shimko S, Evans J, Vinod Shah M, et al. Epigenetic Therapy Overcomes Treatment Resistance in T Cell Prolymphocytic Leukemia. Sci Transl Med (2015) 7:293ra102. doi: 10.1126/scitranslmed.aaa5079
64. Toutah K, Nawar N, Timonen S, Sorger H, Raouf YS, Bukhari S, et al. Development of HDAC Inhibitors Exhibiting Therapeutic Potential in T-Cell Prolymphocytic Leukemia. J Med Chem (2021) 64:8486–509. doi: 10.1021/acs.jmedchem.1c00420
Keywords: T-PLL, clonal evolution, pathogenesis, TCL1A, ATM
Citation: Braun T, Dechow A, Friedrich G, Seifert M, Stachelscheid J and Herling M (2021) Advanced Pathogenetic Concepts in T-Cell Prolymphocytic Leukemia and Their Translational Impact. Front. Oncol. 11:775363. doi: 10.3389/fonc.2021.775363
Received: 13 September 2021; Accepted: 18 October 2021;
Published: 19 November 2021.
Edited by:
Swami P. Iyer, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Marta Sonia González Pérez, University Clinical Hospital of Santiago, SpainCopyright © 2021 Braun, Dechow, Friedrich, Seifert, Stachelscheid and Herling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Herling, bWFyY28uaGVybGluZ0BtZWRpemluLnVuaS1sZWlwemlnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.