 Haiying Fu1
Haiying Fu1 Huarong Zhou2
Huarong Zhou2 Yanyan Qiu2
Yanyan Qiu2 Jianfei Wang3
Jianfei Wang3 Zhiming Ma3
Zhiming Ma3 Hongping Li3
Hongping Li3 Feng Zhang2
Feng Zhang2 Chenxi Qiu1
Chenxi Qiu1 Jianzhen Shen2*
Jianzhen Shen2* Tingbo Liu2*
Tingbo Liu2*- 1Department of Hematology, The Third Affiliated People’s Hospital of Fujian University of Traditional Chinese Medicine, The Third People’s Hospital of Fujian Province, Fuzhou, China
- 2Department of Hematology, Fujian Medical University Union Hospital, Fujian Institute of Hematology, Fujian Provincial Key Laboratory on Hematology, Fuzhou, China
- 3Research and Development Division, Oriomics Biotech Inc, Hangzhou, China
Background: Mounting studies have sought to identify novel mutation biomarkers having diagnostic and prognostic potentials. Nevertheless, the understanding of the mutated pathways related to development and prognosis of B-cell lymphoma is still lacking. We aimed to comprehensively analyze the mutation alterations in genes of canonical signaling pathways and their impacts on the clinic outcomes of patients with B-cell lymphoma.
Methods: Circulating cell-free DNA (cfDNA) samples from 79 patients with B-cell lymphomas were used for targeted sequencing with a 560-gene panel for depicting mutation landscapes and identifying gene fusion events. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses of mutated genes were performed. The associations of mutation status of genes and seven canonical oncogenic pathways with progression-free survival (PFS) were assessed using Kaplan-Meier test and multivariate Cox analysis. The variant allele frequencies (VAFs) of genes in TP53 and Hippo pathways in paired baseline and post-treatment samples from 18 B-cell lymphoma patients were compared. Finally, the associations of identified fusion genes, mutated genes, and pathways with treatment response were evaluated based on objective response rates (ORRs) comparisons of groups.
Results: We identified 666 mutations from 262 genes in baseline cfDNAs from 79 B-cell lymphoma patients, and found some genes were preferentially mutated in our cohort such as GNAQ, GNAS, H3F3A, DNMT3A, HLA-A, and HLA-B. These frequently mutated genes were significantly associated with negative “regulation of gene expression, epigenetic” and virus infections such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus 1 infections. We detected five fusion genes in at least two patients with B-cell lymphoma, and among them, TCF7L2_WT1 gene fusion was most frequently detected in 30.4% of patients (24 of 79 cases). SEPT6_TRIM33 gene fusion, mutated TP53 and Hippo pathways were significantly associated with poor PFS, and SEPT6_TRIM33 fusion gene and mutated TP53 pathway were independent prognostic factors for B-cell lymphoma. A decreased VAF of TP53 p.Y88C and LATS2 p.F972L was detected in patients with complete response to treatments. Moreover, a significant difference in ORR was observed in patients with NPM1_NR4A3 and SEPT6_TRIM33 fusions.
Conclusions: SEPT6_TRIM33 gene fusion and mutated TP53 and Hippo pathways may serve as prognostic makers for B-cell lymphoma patients.
Introduction
B-cell lymphomas are heterogeneous malignancies that vary in clinical presentations and molecular phenotypes (1, 2), ranging from highly aggressive to very indolent. The complex pathogeneses of lymphomas result into various types of therapies, different treatment responses, and extremely variable clinical outcomes (3). B-cell lymphomas represent the most common subgroup of Non-Hodgkin lymphoma (NHL), which is the 13th most common cancer and the 12th most common cause of cancer death worldwide with over 544,352 new cases estimated for 2020 (4), thus posing a significant public health concern. In clinics, it is of great importance to assess the response to treatments and monitor clinical outcomes, which may affect the decision making in the wide range of therapeutic selection. Although imaging scans, including computed tomography (CT) and positron emission tomography (PET), are the traditional standards for initial diagnosis and early indication of treatment response of lymphoma, they are limited to use after onset of symptoms (5). With the development of next-generation sequencing, genomic analysis of circulating tumor DNA (ctDNA) emerges as an effective non-invasive method for identifying prognostic and predictive molecular makers in a broad spectrum of B-cell lymphomas using targeted sequencing panels (6).
Recently, mounting evidence has sought to identify novel molecular biomarkers with prognostic value and therapeutic response potentials in B-cell lymphomas. For example, the presence of MYC-IG rearrangement was identified as a predictor for treatment failure in patients with diffuse large B-cell lymphoma (DLBCL) who received immunochemotherapy (7). Novel evidence showed that DLBCL patients with MYC rearrangement, especially MYC double-hit or triple-hit constellation, had inferior outcomes (8). In addition, the occurrence of gene mutations like CREBBP and EP300 mutations during disease course may predict worse overall survival (OS) and progression-free survival (PFS) for germinal center B cell-like DLBCL (9). NFKBIE functions as a NF-κB inhibitor, and the deletion of which was associated with inferior survival in primary mediastinal B-cell lymphoma (10). KMD2D mutation was associated with poor outcomes as well in patients with mantle cell lymphoma (MCL) (11). Nevertheless, these findings are only to assess the prognostic significance from the perspective of genomic alterations, and the comprehensive knowledge on the deregulated pathways caused by multi-mutations in B-cell lymphoma has rarely been addressed.
B-cell development and activation are accompanied by a series of genetic events, and abnormalities in these genetic events are responsible for the alterations of genes associated with B-cell proliferation or apoptosis, and consequently contribute to B lymphomagenesis (12). Usually, hallmarks of cancers include genetic alterations of gene fusions caused by chromosomal rearrangements or instability and genetic alterations in a group of genes in canonical signaling pathways including cell cycle, Hippo, Notch, PI3-Kinase, RTK-RAS, p53, and Wnt pathways (13, 14). However, the clinical implications of gene fusion events and mutated canonical pathways in B-cell lymphoma prognosis have not yet been fully elucidated. In this study, we sought to gain a global view of somatic mutations and gene fusions events, identify mutated pathways, as well as evaluate their impacts on the clinic outcomes of patients with B-cell lymphomas.
Materials and Methods
Study Population
Seventy-nine patients who were diagnosed as having B-cell lymphomas in the Union Hospital Affiliated to Fujian Medical University between December 2017 and April 2021 were included in this study. Pathological diagnosis was performed by pathological reviews to confirm subtypes of lymphomas following the criteria of WHO classification of Tumors of Hematopoietic and Lymphoid tissue criteria (15). Demographic and clinicopathologic characteristics and outcomes were collected. Peripheral blood (10 ml) was drawn from each patient for circulating cell-free DNA (cfDNA) profiling assays. The study design was approved by the Ethics Committee of the Union Hospital Affiliated to Fujian Medical University in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Targeted Panel Sequencing
Total DNAs from blood samples were extracted using the Magnetic Serum/Plasma DNA Maxi Kit (DP710, TIANGEN Biotechnology, China), following the manufacturer’s instructions. The extracted cfDNA samples were quantified using Qubit dsDNA HS Assay Kit (Invitrogen) and sheared into 150 to 200 bp fragments, and cfDNA with input concentrations (≥5 ng) and without overly genomic DNA contamination was subjected to library construction prior to fragment length analysis using Qsep100 (Bioptic, Taiwan, China). Fragmented DNA libraries were constructed using nano DNA library prep kit (for Illumina®) (Nanodigmbio, Nanjing, China) for end-repairing, adding A-tailing, and adapter ligation. DNA libraries were then subjected to PCR amplification and purification. The fragment quantification was performed using Qsep100 automatic nucleic acid analysis system (BiOptic, Taiwan, China). Subsequently, the lymphoma panel libraries were captured hybridly with a designed panel of 560 cancer-related genes (Pancare®, Oriomics Biotech Inc, Zhejiang, China) using xGen® Hybridization and Wash Kit (IDT). The barcoded duplex adapters were eventually blocked by NadPrep NanoBlockers (for illumine) (Nanodigmbio, Nanjing, China). The quality control of final libraries was assessed by Qubit 3.0 fluorimeter (Invitrogen) and Qsep100 (Bioptic, Taiwan, China). The qualified libraries with minimum 10 ng/μl concentration were sequenced on the Illumina NextSeq 550 Dx platform with a mean coverage depth of 9,960× for captured regions.
Bioinformatics Analysis
After obtaining the raw sequencing data in the Fastq format, the fastp 0.20.0 software was utilized to perform quality analysis by removing adapter sequence, reads containing N base calls, and low-quality reads (quality reading below 20) (16). All clean paired-end reads were aligned to the human reference genome (17) using Burrows-Wheeler Aligner (BWA). Variant calling was performed using Mutect2 from Genome Analysis Toolkit (GATK 3.4.0, https://software.broadinstitute.org/gatk/; Broad Institute, Cambridge, MA, USA). Then, the detected mutations were further selected following established criteria: (1) variant allele frequency (VAF) more than 0.5% with at least three minimum variant supporting reads; (2) dual UMI ≥1; (3) strand specificity ranging from 0.1 to 0.9; (4) EXAC_EAS or genomAD_EAS less than 0.05. Final mutations were annotated using ANNOVAR (18), and only functional mutations, e.g., non-synonmous, frameshift deletion and insertion were selected for subsequent bioinformatics analysis. DNAs from leukocytes of the same patients were sequenced as germline controls. Maftools package (https://bioconductor.org/packages/release/bioc/vignettes/maftools/) (19), an efficient tool for comprehensive analysis of somatic variant for cancers, was applied to analyze the number of variants in each sample and draw oncoplots for visualizing the mutation landscape. For fusion gene analysis, GeneFuse software (version 0.6.1) was used to detect fusions from fusion file including cancer-related fusions genes from COSMIC (17). In order to investigate the potential biological roles of mutated genes, we used ClusterProfiler in R software to conduct the Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional analyses (12). The significant enrichment terms were identified by the threshold value of false discovery rate (FDR) <0.05. For mutated pathway analysis, one pathway was defined as mutated signaling pathway when patients harbored one or more mutated gene members of this pathway.
Statistical Analysis
Statistical analyses were conducted using R package (version 3.61). PFS was defined as time elapsed between first sampling to disease progression, or death, or last follow-up, whichever came first. Kaplan-Meier test in survival package (version 3.1-11) was applied to assess associations of mutation status of genes and pathways with PFS. Factors significantly associated with PFS in the univariate analysis were subjected to multivariable Cox proportional hazards regression analysis. Objective response rate (ORR) was defined as the percentage of patients who received complete response (CR) or partial response (PR) during the treatment using response evaluation criteria in lymphoma (RECIL) (20). ORRs between groups were compared to explore the associations of fusion genes, mutated genes, and pathways with treatment response using Chi-square test. A P value of < 0.05 was considered statistically significant.
Results
Clinical Characteristics
The demographics and baseline characteristics of 79 B-cell lymphoma patients are presented in Table 1. The median age of patients at blood drawn was 56 years, and the numbers of males and females were comparable (40/39). Histologically, 55 (69.6%) patients were diagnosed with DLBCL, six (7.6%) with follicular lymphoma (FL), four (5.1%) with marginal zone lymphoma (MCL), and two (2.5%) with high-grade B-cell lymphoma (HBL). In addition to three cases without detailed subtype information, the other 76 patients comprised 85.5% aggressive B-cell lymphoma (N=65) and 14.5% indolent B-cell lymphoma (N=11). Among 72 cases with definite staging, 5 (6.3%), 14 (17.7%), 10 (12.7%), and 43 (54.4%) patients had stage I, II, III, IV lymphomas, respectively. Majorities of the patients received R-CHOP (34.2%) or R-COEP (27.8%) as their first-line therapy. Fifty-seven percentage of patients achieved CR (N=26) or PR (N=19) to treatments they received, while 8.9% of patient had a stable (SD, N=2) or progressive disease (PD, N=5), during a median follow-up of 7.08 months.
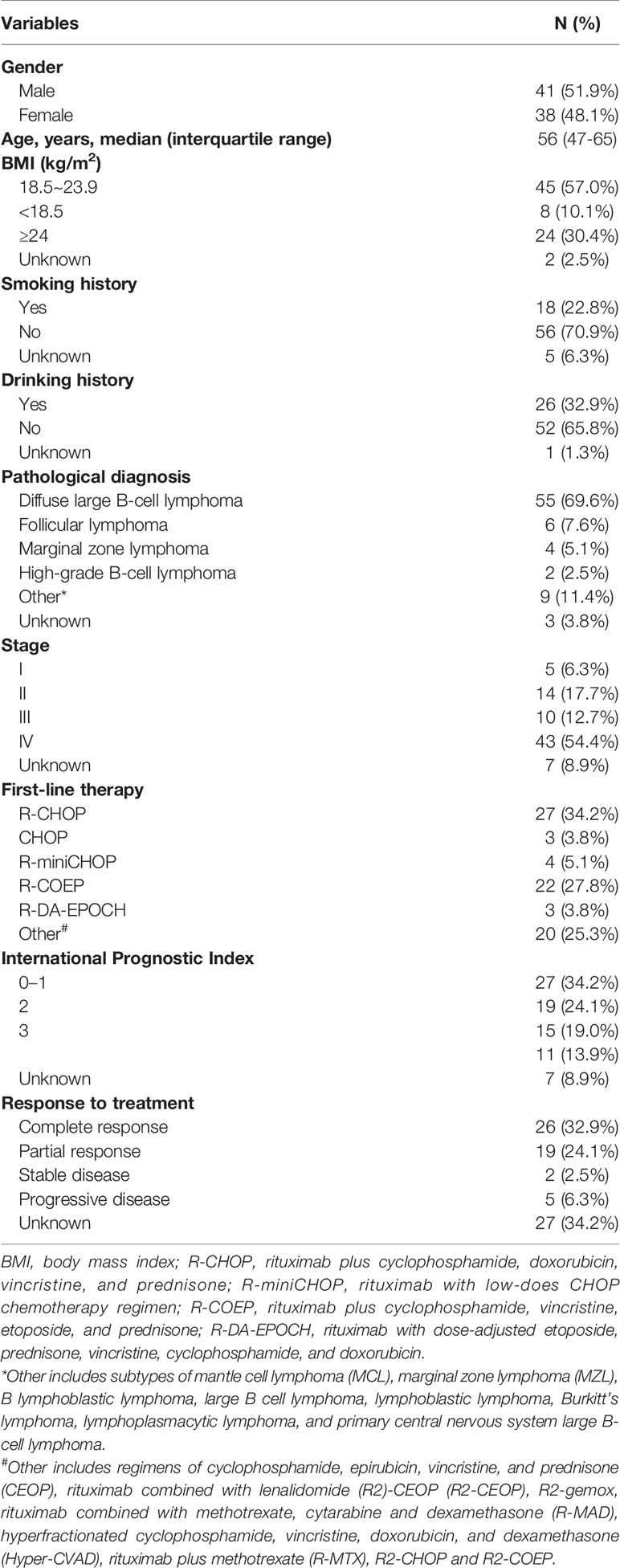
Table 1 Patient demographic and clinical characteristics (N=79).
Genomic Alterations in Patients With B-Cell Lymphomas
In order to gain a global view of the genomic variations of B-cell lymphomas, the somatic mutations and gene fusion events in 79 patients with B-cell lymphomas were identified based on targeted panel sequencing. In total, we identified 666 mutations, in which the major mutation types included missense mutation (79.0%), non-sense mutation (10.4%), frameshift deletion (5.1%), in-frameshift deletion (2.5%), frameshift insertion (2.4%), and non-stop mutation (0.6%). In addition, mutations from 262 of 560 cancer-related genes were detected in 59 patients, and among these mutated genes, 46 genes had a mutation frequency of more than 5%. The top 30 commonly mutated genes are shown in Figure 1, and the top 11 preference genes were TP53 (20%), MST1 (19%), KMT2D (18%), GNAQ (16%), MYD88 (15%), DNMT3A (11%), GNAS (11%), PIM1 (11%), ARID1A (10%), CREBBP (10%), and H3F3A (10%). Of note, there were nine preference mutated genes (e.g., TP53, KMT2D, CREBBP, BCL2, ATM, and PIM1) that were overlapped between the top 30 frequently mutated genes of our results, and the top 20 frequently mutated genes detected in lymphoid tissues of 413 DLBCL patients from COSMIC database. It also showed that the frequencies of these overlapped genes were comparable (Supplementary Table 1), demonstrating the reliability of our sequencing assays.
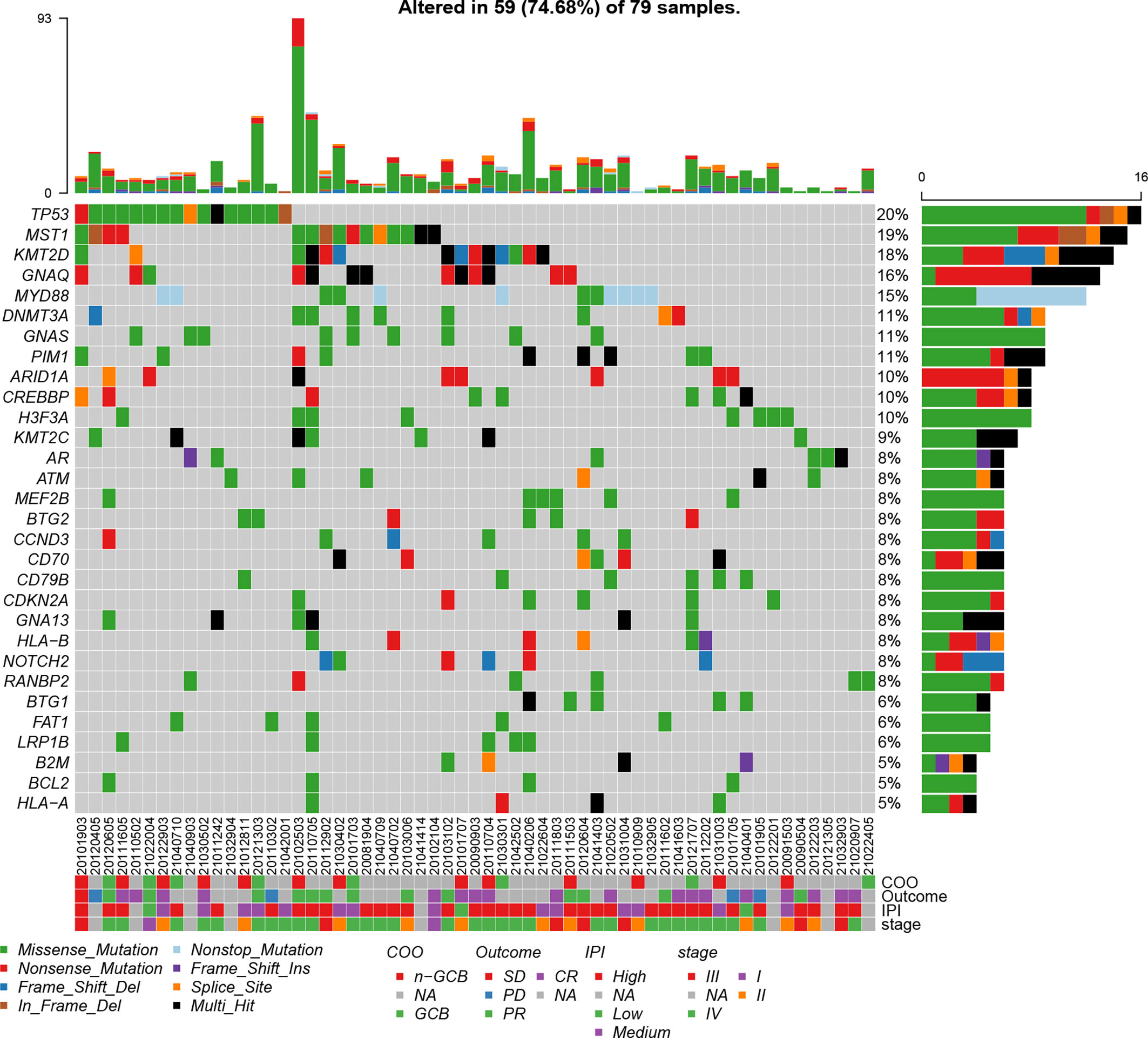
Figure 1 Mutation plot showing top 30 frequently mutated genes in 79 B-cell lymphoma patients. Color-coded is indicated by type of mutation. Y axis shows the percentages of patients with at least one mutation in the specified gene.
Gene fusions are an important class of somatic alterations in hematological malignancies (21). Thus, we examined the gene fusion events in our cohort and found that five fusion genes were detected in at least of two cases, comprising of FAM65B_NTRK3, NPM1_NR4A3, RANBP2_MCPH1, SEPT6_TRIM33, and TCF7L2_WT1. There were four patients having SEPT6_TRIM33 gene fusion, while nine patients harbored RANBP2_MCPH1 gene fusion. Notably, TCF7L2_WT1 gene fusion was most frequently detected in 29.1% patients (23 of 79 cases) (Table 2).
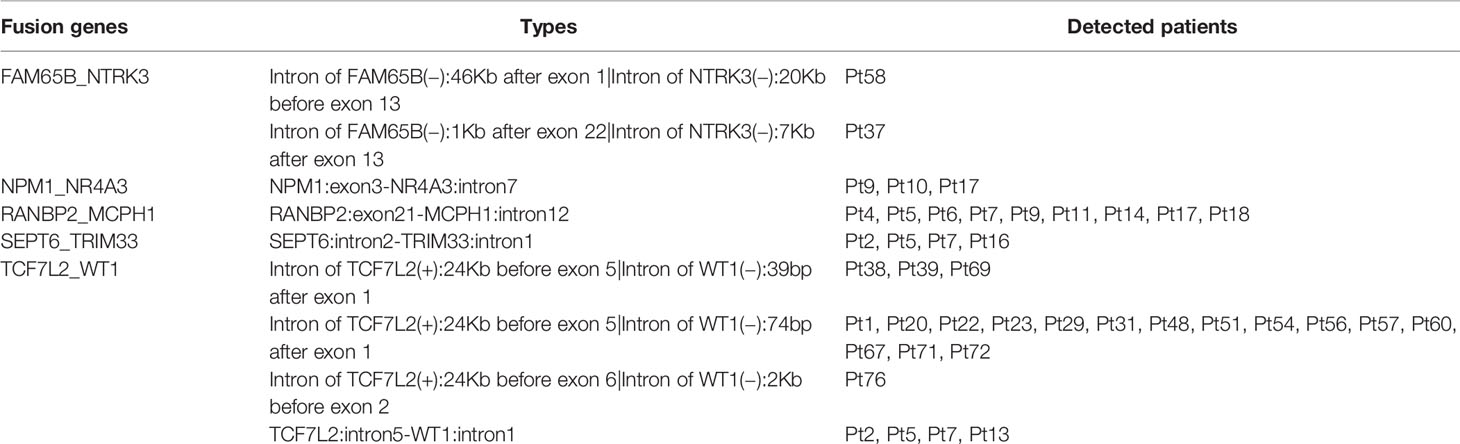
Table 2 Fusion genes identified from79 B-cell lymphomas.
Functional Enrichment Analysis of Somatic Mutated Genes
To better understand the biological roles of mutated genes, GO and KEGG enrichment analysis were performed. GO enrichment analysis revealed that the mutated genes were significantly enriched in regulation of G1/S transition of mitotic cell cycle (biological process [BP], e.g., CDKN2A, ATM, TP53 and BCL2), gene silencing (BP, e.g., DNMT3A, RANBP2, and KMT2D), negative regulation of gene expression, epigenetic (BP, e.g., H3F3A and DNMT3A), MHC protein complex (cellular component [CC], HLA-A, HLA-B, and B2M), and transcription factor binding (molecular function [MF], e.g., ARID1A, PIM1, AR, and CREBBP). KEGG pathway analysis showed that the mutated genes were significantly associated with virus infections involved pathways such as human T-cell leukemia virus 1 infection (e.g., HLA-A and HLA-B), cytomegalovirus infection (e.g., HLA-A, HLA-B, GNAQ, GNAS, and GNA13), Epstein-Barr virus (EBV) infection (e.g., HLA-A and HLA-B), human immunodeficiency virus 1 (HIV) infection (e.g., HLA-A, HLA-B, and GNAQ), and papillomavirus infection (e.g., HLA-A, HLA-B, and GNAS) (Figure 2).
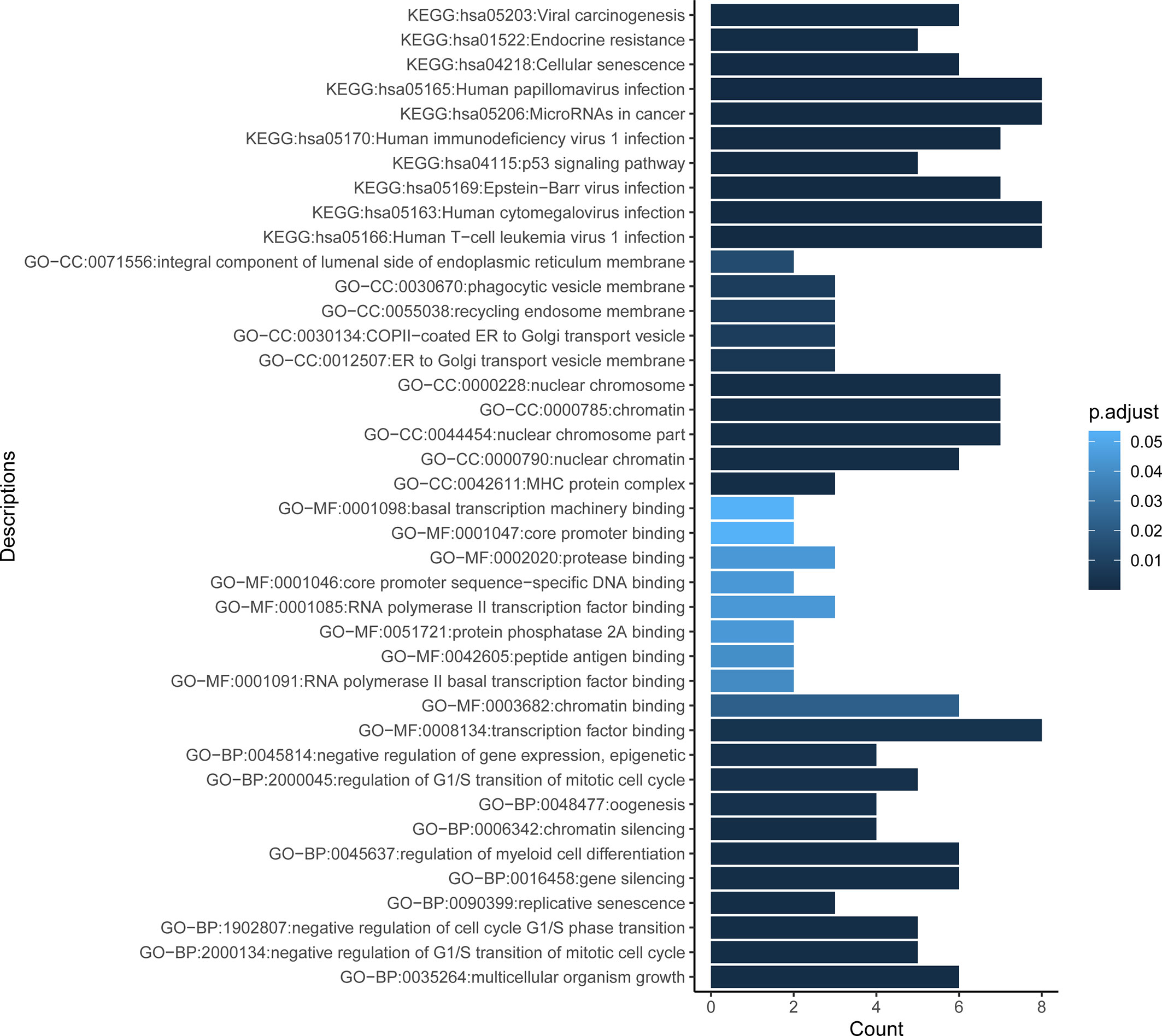
Figure 2 Functional enrichment result reveals the top 10 function terms for GO and KEGG analysis. Gene Ontology (GO) comprises three categories: molecular function (MF), biological process (BP), and cellular component (CC). X axis represents the count of genes enriched in corresponding terms. Color depth of columns is positively related with the P value of term. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Mutations in Seven Canonical Oncogenic Signaling Pathways
With the aim of identifying preferentially mutated pathways in B-cell lymphoma, we evaluated the mutational events of component genes involved in seven canonical oncogenic pathways. The mutation profiles of these pathways are shown in Figure 3. TP53 mutational profile was featured primarily by TP53 mutations, while CDKN2A, CCND3, FAT1, CREBBP, and PTEN were the most frequently mutated genes in cell cycle, Hippo, NOTCH, and PI3K mutational profiles, respectively. Strikingly, TP53 and NOTCH pathways were both most commonly mutated oncogenic pathways in B-cell lymphoma in our cohort. Almost one-fifth of patients harbored mutations in component genes of each of these two pathways (24.05%, 19 of 79 cases). In addition, we found that 7 (8.86%), 6 (7.59%), and 6 (7.59%) of 79 patients had mutations in Hippo, WNT, and cell cycle pathways, respectively (Supplementary Figure 1). There were 19 and 21 patients harboring mutations in component genes of at least two mutated pathways and only one mutated pathway, respectively.
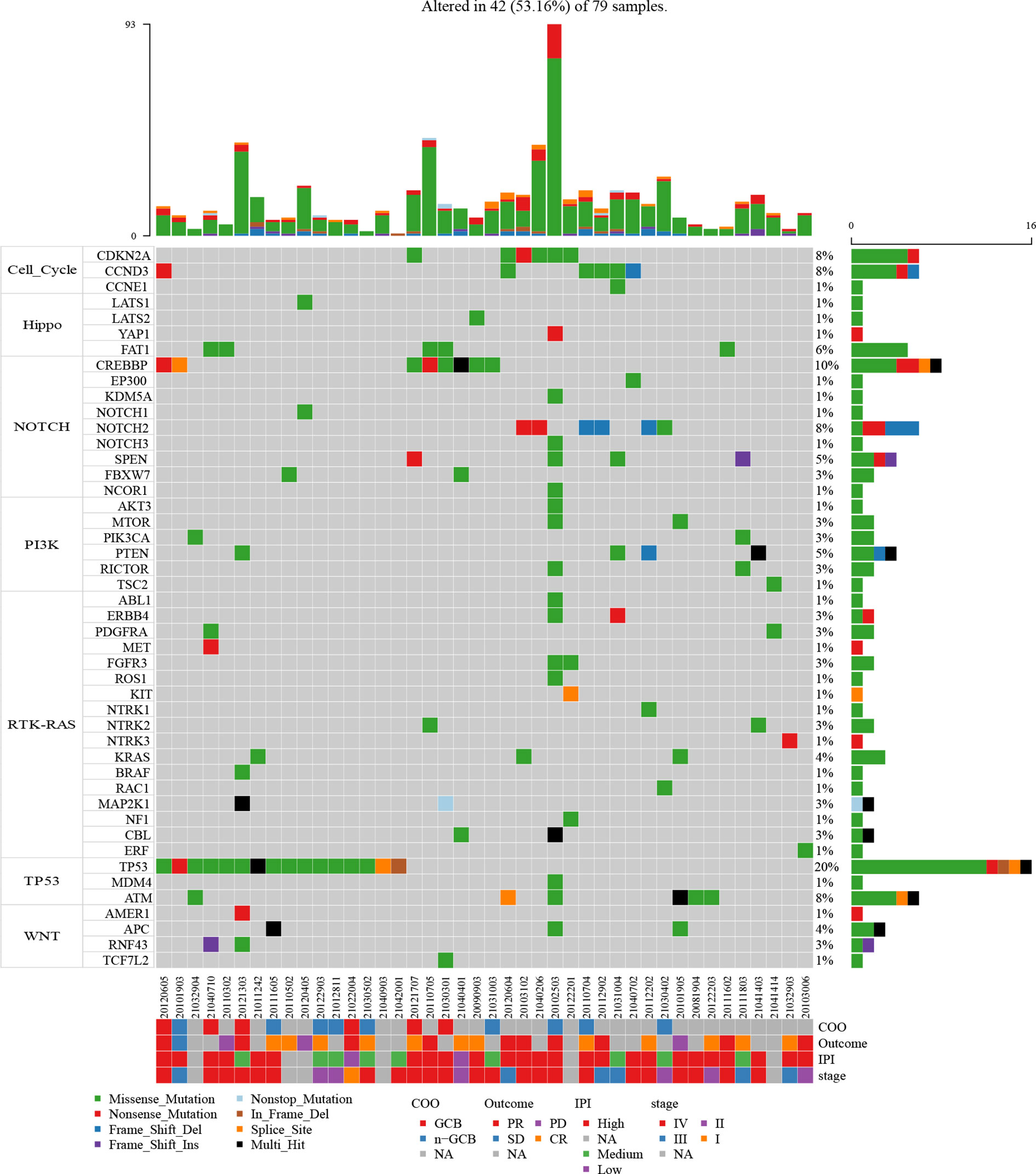
Figure 3 Mutation plot of seven canonical oncogenic signaling pathways (cell cycle, Hippo, Notch, PI3-Kinase, RTK-RAS, p53, and Wnt pathways) in 79 B-cell lymphoma patients. Color-coded is indicated by type of mutation. Y axis shows the percentages of patients with at least one mutation in the specified gene.
Prognostic Roles of Fusion Genes, Mutated Genes, and Pathways
To further investigate the clinical influence of genomic aberrations in groups of fusion genes, the top 30 commonly mutated genes, and seven canonical oncogenic pathways, we assessed the associations between the status of these genomic alterations and PFS in 52 patients with complete data. The baseline characteristics of these 52 patients are shown in Supplementary Table 2. The results revealed that among the five fusion genes, only patients with SEPT6_TRIM33 fusion gene had significantly shorter PFS (P<0.0001, Figure 4A). In addition, patients harboring any of these five fusion genes showed a trend towards worse prognosis (P=0.034, Figure 4B). Unfavorable outcome was also observed in B-cell lymphoma patients (N=52) with mutated TP53 gene (P=0.0086, Figure 4C), mutated TP53 pathway (P=0.013, Figure 4D), and mutated Hippo pathway (P=0.0038, Figure 4E). There was no significant difference in PFS between patients with and without mutations in other commonly mutated genes or pathways. We further conducted subgroup analysis in DLBCL patients (N=38), and the results showed that TP53-mutated DLBCL patients had a significantly unfavorable prognosis compared to TP53 wild-type DLBCL patients (P=0.0047, Supplementary Figure 2A). Similarly, DLBCL patients without mutated TP53 pathway tended to have prolonged PFS (P=0.061, Supplementary Figure 2B). However, we could not compare prognosis between DLBCL patients with and without mutated SEPT6_TRIM33 fusion gene or Hippo pathway, due to the small number of patients having this genomic alteration.
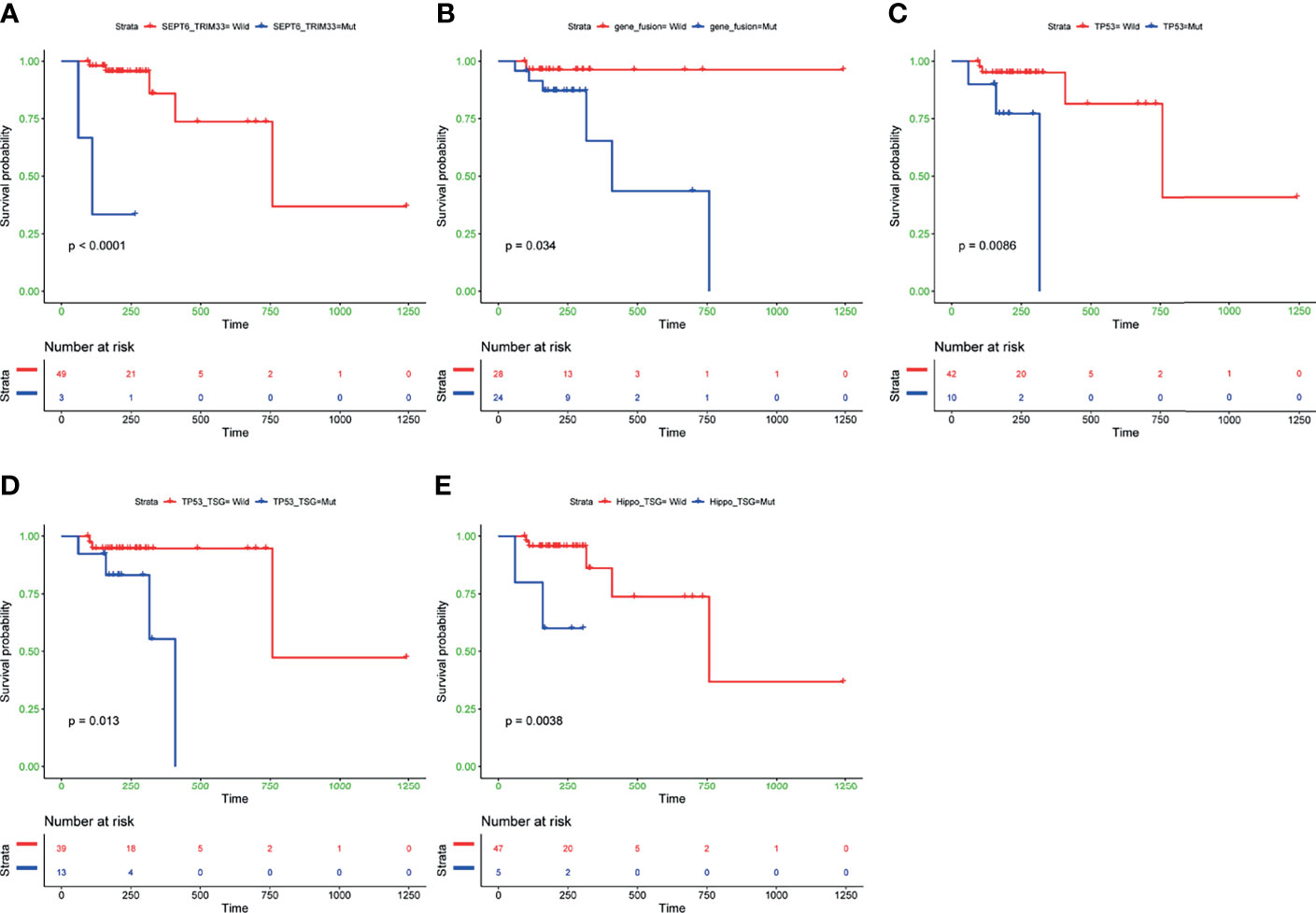
Figure 4 Kaplan-Meier curves of PFS differences between subgroups with and without SEPT6_TRIM33 gene fusion (A), any of five fusion genes detected in at least of two cases (B), TP53 gene mutation (C), mutated TP53 pathway (D), mutated Hippo pathway (E). PFS, progression-free survival. P values were calculated by the log-rank test.
To explore whether co-mutations of TP53 and Hippo pathways could have more significant impact than single TP53 or Hippo pathways mutations on the patients’ survival, we compared survivals across subgroups with co-mutations, with any mutations, and without a mutation in these two pathways. The results revealed that B-cell lymphoma patients with both wild-type TP53 and Hippo pathways had the best prognosis, while the patients with the presence of co-mutated TP53 and Hippo pathways had the worst PFS (P<0.0001, Supplementary Figure 3).
After identification of SEPT6_TRIM33 fusion genes, mutated TP53 and Hippo pathways as factors significantly associated with prognosis of B-cell lymphomas, multivariable Cox analyses adjusted by clinic covariates [age, gender, BMI, smoking history, drinking status and treatment (R-CHOP or R-CHOP-like regimes vs others)] were conducted. The result suggested that SEPT6_TRIM33 fusion gene [hazard ratio (HR): 242.69, P=0.046] and mutated TP53 pathway (HR: 14.06, P=0.03) were independent prognostic factors for B-cell lymphoma patients (Figure 5).

Figure 5 Prognostic significances of fusion gene, mutated pathways, and clinic factors by multivariable Cox proportional hazards regression analysis.
VAFs Changes of Mutated Genes in TP53 and Hippo Pathways
Owing to our above findings that mutated TP53 and Hippo pathways were significantly associated with patient prognosis, we wondered whether the status of the genes in these two pathways may be altered after chemotherapy, which may be useful for guiding subsequent therapies. To that end, we analyzed the VAFs of genes in the TP53 and Hippo pathways in paired baseline and post-treatment samples from 18 B-cell lymphoma patients. The characteristics of these 18 patients are presented in Supplementary Table 3. As shown in Figure 6, Pt18 (DLBCL subtype), who achieved PR after four cycles of R-DA-EDOCH treatment, presented an increased VAF of TP53 p.Y87N mutation (TP53 pathway; 0 vs 17.86%), but a decreased VAF of FAT1 p.E1292K mutation (Hippo pathway; 44.74 vs 0%), which might be responsible for partial sensitivity or acquired resistance to R-DA-EDOCH. Interestingly, two CR patients detected a decreased VAF of TP53 p.Y88C (Pt42, DLBCL subtype, five cycles of R-miniCHOP) and LATS2 p.F972L (Pt5, FL subtype, four cycles of R-COEP), respectively. These results suggested that VAF changes of mutated genes in TP53 and Hippo pathways might be an indicator for treatment responses.
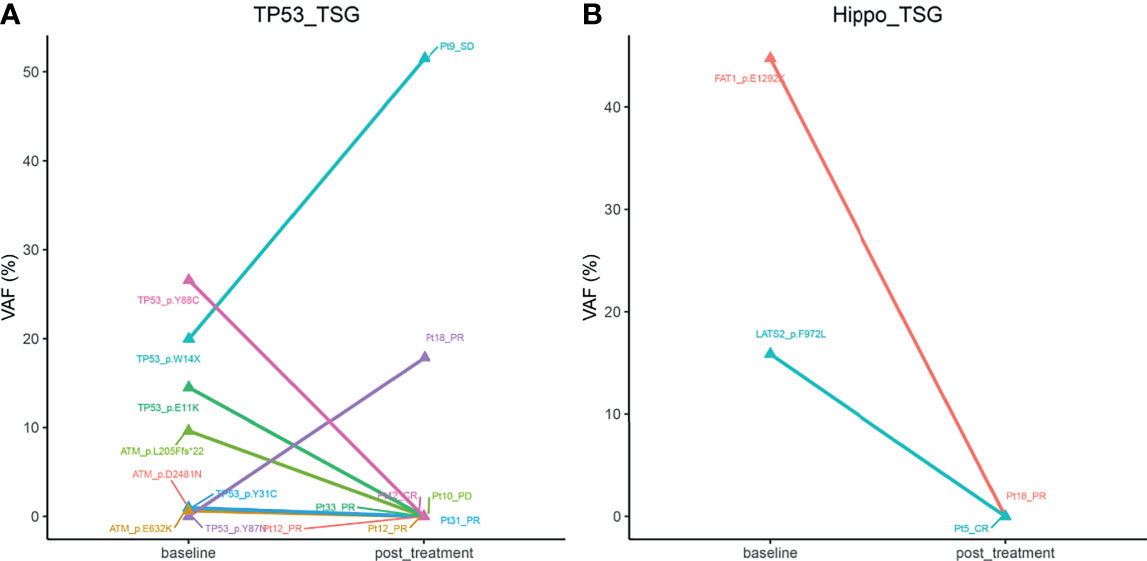
Figure 6 The alterations of mean VAFs of mutated genes in Tp53 (A) and hippo pathways (B) in paired baseline and post-treatment samples from 18 B-cell lymphoma patients. VAF, variant allele frequency.
Genomic Alterations Correlate to Treatment Response
In order to investigate the relationship between fusion genes, mutated genes and pathways, and response to treatment, ORRs were compared between patients with and without a specific genomic alteration. As a result, significant differences in ORR were observed for patients with NPM1_NR4A3 (33.33 vs. 88%, P=0.01) and SEPT6_TRIM33 (33.33 vs. 88%, P=0.01) fusions versus patients without corresponding fusion (Table 3). Although patients with other fusion genes, TP53 mutation (70 vs. 88.37%, P=0.14), mutated TP53 (69 vs. 90%, P=0.07), and Hippo pathways (60 vs. 87.5%, P=0.33) trended towards a lower ORR, the difference was not statistically significant (Table 3).
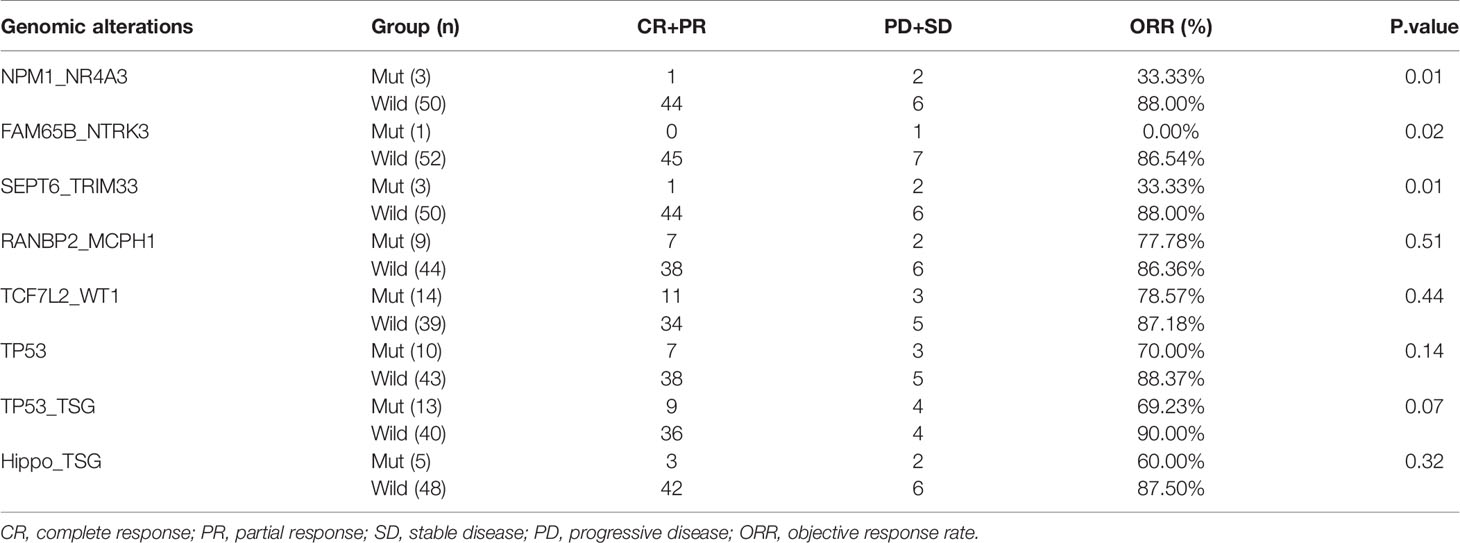
Table 3 Relationship between genomic alterations and clinical response to treatment.
Discussion
A handful of somatic mutations in B-cell lymphomas has been identified to participate in cancer development and progression (22, 23). Notably, the lymphomagenesis and progression caused by the coordination of multiple mutated genes at the pathway level are considered more reasonable than at the single gene level (24, 25). It is known that genomic alterations and their affected pathways may have diagnostic, prognostic, and therapeutic implications for B-cell lymphomas (2). However, the understanding of mutated pathways related to B-cell lymphoma development and patient prognosis is still inadequate, especially when genomic biomarkers are detected using the liquid biopsy approach. To that end, we detected the mutation landscapes and gene fusion events in cfDNA samples of 79 B-cell lymphoma patients based on targeted panel sequence to screen novel biomarkers having prognosis values. In this study, we found five gene fusions (e.g., TCF7L2_WT1) were frequently detected in B-cell lymphomas. The SEPT6_TRIM33 gene fusion, mutated TP53, and Hippo pathways conferred unfavorable prognosis. Moreover, SEPT6_TRIM33 fusion gene and mutated TP53 pathway were prognostic factors independent of clinical confounders. These findings may provide novel molecular biomarkers for prognostic stratification and potential therapeutic targets of B-cell lymphomas.
In this work, we totally identified 666 mutations from 262 genes in cfDNA samples from 79 B-cell lymphomas patients. Through comparing preferentially mutated genes between our cohort and 413 DLBCL tissues samples from COSMIC database, we found some genes were especially mutated with high frequencies in our cohort such as GNAQ, GNAS, H3F3A, DNMT3A, HLA-A, and HLA-B, in addition to nine overlapped genes (e.g., TP53, KMT2D, MYD88, ATM, and BCL2). The mutations identified in these overlapped genes are responsible for regulation of G1/S transition of mitotic cell cycle (e.g., ATM and TP53). Tumor suppressor-associated mutations in TP53 and ATM may not induce cell cycle arrest and DNA repair (26). Notably, these frequently mutated genes in our cohort were significantly associated with functional categories involved negative regulation of gene expression, epigenetic (H3F3A and DNMT3A), and virus infections such as cytomegalovirus, EBV, HIV, and papillomavirus infections (e.g., GNAS, GNAQ, HLA-A, and HLA-B). GNAS and GNAQ encode G-protein alpha subunit, and the viruses like cytomegalovirus and EBV have been identified to encode G proteins and their coupled receptors in their genomes to induce cancer initiation (27). The variations of antigen-presentation molecules HLA may alter epitope recognition by the T-cell receptor, which enables HIV to escape cytotoxic T lymphocytes responses (28). Of note, NHL is one of the most common cancers in HIV-positive patients (29). Collectively, the present work gives insight into the biological processes and pathways involved in lymphomagenesis.
Fusion genes have been extensively used in cancer diagnosis and determining prognostic impact (30). Recently, adopting capture-based panel sequencing approach has enabled the identification of novel gene fusions in cancer patients (31). In this study, we identified novel TCF7L2_WT1 gene fusion as one of the most frequently detected genomic aberrations in B-cell lymphomas. Transcription Factor 7-Like 2 (TCF7L2), also known as TCF4 transcription factor, is a key effector for the activation of Wnt signaling pathway in adult T cell leukemia cells (32). Wilms’ tumor 1 (WT1), a tumor-suppressor gene, can interact with lymphoid enhancer-binding factor 1 (LEF1) to mediate Wnt signaling activation (33). TCF4/LEF1 DNA binding effectors can interact with β-catenin to increase cyclin D1 and c-Myc expression, demonstrating the activation of Wnt signaling pathway (34). In line with the evidence that canonical Wnt signaling plays an important role in the development and progression of B-cell lymphomas (35), in our study population, we inferred the novel TCF7L2_WT1 gene fusion may associate with B-cell lymphomas progression through triggering the dysregulation of Wnt signaling. Although TCF7L2_WT1 gene fusion was not identified as a validated genomic alteration in tumors, an identified partner gene of EWSR1 for WT1 (WT1-EWSR1) reported in COSMIC database was also found as a partner gene of TCF7L2 in colon adenocarcinom (36). In addition, our result revealed the prognostic value of SEPT6_TRIM33 gene fusion in B-cell lymphomas. Septin 6 (SEPT6), a member of the septin family of GTPases, which preserves breakpoint at chromosomes Xq24, was found as a fusion partner with Leukemia gene (MLL1 or KMT2A) related to leukemia (37). Tripartite Motif Containing 33 (TRIM33), acting as an E3 ubiquitin-protein ligase, can prevent apoptosis in B lymphoblastic leukemia though mediating Bim activation (38). In addition, TRIM33 located at 1p13 region was identified as a tumor suppressor chronic myelomonocytic leukemia and associated with the survival of all B cell neoplasms (39). Combined with our survival results, we suggested that SEPT6-TRIM33 gene fusion conferred poor prognosis in B cell neoplasms, which was valuable for prognostication and therapeutic decision making.
In this study, we analyzed the mutation profiles of seven canonical oncogenic pathways in B-cell lymphoma and found TP53 pathway was one of the most frequently mutated oncogenic pathways in B-cell lymphoma cohort. The regulation of TP53 governs a multitude of cellular processes, including cell cycle arrest, apoptosis, and changes in metabolism (40). Mutations in this pathway may cause genomic instability and deregulated transcription of genes involved in cell cycle, DNA repair, and apoptosis (41). Reportedly, somatic mutations in abnormal TP53 pathway and TP53 itself are responsible for lymphoma generation, progression, and invasion (41). We detected mutations in TP53 pathway genes such as MDM4 and ATM, especially TP53, which acts as a vital player in the cancer arena, was the most frequently mutated genes in B-cell lymphomas in our cohort (20%, 16/79). Mutations in p53 or its modulators such as MDM4 and ATM may affect p53 tumor suppressor function (42). In addition, we observed the mutations of TP53 gene and mutated TP53 pathways were implicated with unfavorable patient prognosis. This was consistent with a previous study, in which TP53 was mutated in 23.8% of aggressive B-cell lymphoma, and mutated TP53 could serve as an adverse independent predictor for PFS (43).
Hippo signaling is another important tumor suppressor pathway, in which the core of module comprises of the tumor-suppressive MST-LATS kinases and downstream oncogenic effectors YAP and TAZ (44). So far, the mutations of genes in Hippo pathway have been rarely researched, possibly due to their low frequencies. Especially, DLBCL patients were reported to have few somatic alterations in Hippo pathway than other cancer types (45). In this study in B-cell lymphomas, we also found the low mutation frequencies of the major components of Hippo pathway like LATS1 (1%), LATS2 (1%), YAP1 (1%), and FAT1 (6%). Nonetheless, our result suggested the poor prognosis of mutated Hippo pathway for B-cell lymphoma. In addition, our result revealed that B-cell lymphoma patients with the presence of co-mutated TP53 and Hippo pathways had the worst prognosis, compared to patients with mutations in either pathway or without any mutation in both pathways. This result can be explained by that there may be a direct cross-talk between TP53 and Hippo tumor-suppressor pathways in multiple molecular interfaces such as p53-LATS axis (40) and p53-PTPN14-YAP axis (46).
Interestingly, a significantly lower ORR was observed in patients with SEPT6-TRIM33 fusion, which was consistent with shorter PFS in these patients. Although patients with TP53 mutation, mutated TP53 and Hippo pathways trended towards a lower ORR, the difference was not statistically significant. Notably, PFS was significantly shorter in these patients. Aberrant activation of YAP/TAZ or loss-of-function of tumor suppressors in the Hippo pathway enhances tumor cell resistance against anticancer therapeutic drugs (47). TP53 mutations are related to poor or no responses to chemotherapy such as cytarabine, rituximab, and autologous stem-cell transplant for mantle cell lymphoma (47). In our cohort, we found TP53 p.Y87N mutation in Pt18 (DLBCL subtype) might induce resistance against DA-EDOCH. Integrating, these findings demonstrated that patients with specific mutation alterations, particularly in tumor-suppressor genes or pathways, might associate with poor treatment response and PFS. It has revealed that Verteporfin can reverse 5-Fu resistance in colorectal cancer through targeting the Hippo pathway in a YAP-dependent manner (48). Thus, identification of alterations in TP53 and Hippo pathways or novel gene fusions that are implicated in drug resistance and poor prognosis may guide pharmacological interventions by targeting the alterations and thus improve survival of B-cell lymphoma patients.
Despite this study has identified novel genomic biomarkers associated with prognosis of B-cell lymphoma patients, a major limitation of this study was a relatively small sample size, especially the small number of patients with identified fusion genes or mutated Hippo pathways, which makes subgroup analyses infeasible. Moreover, although we performed in silico analyses (GO and KEGG) to indicate the potential functional roles of mutated genes identified in this study, future in vivo and in vitro studies may further reveal the biological and molecular mechanisms underlying the associations between these genomic biomarkers and outcomes of B-cell lymphomas, especially DLBCL. Other limitations included heterogeneous patients with various therapeutic regimens, relatively short follow-up time, and the lack of important covariates such as education level, performance status. Therefore, larger prospective studies are needed to further verify our present findings.
In summary, we comprehensively analyzed the molecular profiling of B-cell lymphoma and found that SEPT6_TRIM33 fusion gene, mutations in single TP53 gene, mutated TP53 and Hippo pathways would together predict inferior prognosis for B-cell lymphomas. The present work also gives insight into the potential biological processes and pathways involved in lymphomagenesis. The study may provide novel genomic biomarkers of prognostic significance and potential therapeutic targets of B-cell lymphomas.
Data Availability Statement
Data from this study, including the detected gene mutation points and fusion genes of each sample, are available in the main text, supplementary materials, or have been deposited in Figshare in the repository DOI: 10.6084/m9.figshare.16902844. Raw genetic sequencing data in this article are not readily available due to restrictions by national legislation, specifically the Administrative Regulations of the People’s Republic of China on Human Genetic Resources. Requests to acquire the sequencing data can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Union Hospital Affiliated to Fujian Medical University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
HF, JS, and TL conceived and designed the project. HZ, YQ, and FZ organized sample collection. ZM and CQ analyzed and interpreted the data. HF, JS, TL, and JW discussed the results. HF wrote the manuscript. TL and HL edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the financial contribution by a grant from the Joints Funds for the Innovation of Science and Technology, Fujian Province (2018Y9010 and 2019Y9069), Construction project of Fujian medical center of hematology (Min201704), and National and Fujian Provincial Key Clinical Specialty Discipline Construction Program, P.R.C. [(2011)1018 and (2012)149].
Conflict of Interest
JW, ZM, and HL are employees of Oriomics Biotech Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.765544/full#supplementary-material
Supplementary Table 1 | The overlapped mutated genes between the top 20 commonly mutated genes from COSMIC database and top 30 mutated genes our cohort.
Supplementary Table 2 | Clinical characteristics of 52 patients for prognosis analysis.
Supplementary Table 3 | Characteristics of subgroup patients with paired baseline and post-treatment samples (N=18).
Supplementary Figure 1 | Pie chart shows the mutation rates of seven canonical oncogenic signaling pathways (cell cycle, Hippo, Notch, PI3-Kinase, RTK-RAS, p53, and Wnt pathways) in 79 patients.
Supplementary Figure 2 | Kaplan-Meier curves of PFS differences between the TP53-mutated and TP53 wild-type DLBCL patients (A) and between DLBCL patients with or without mutated TP53 pathway (B).
Supplementary Figure 3 | Kaplan-Meier curves of PFS differences across subgroups with co-mutations, any mutations, and without any mutation in TP53 and Hippo pathways.
References
1. Solé C, Larrea E, Di Pinto G, Tellaetxe M, Lawrie CH. MiRNAs in B-Cell Lymphoma: Molecular Mechanisms and Biomarker Potential. Cancer Lett (2017) 405:79–89. doi: 10.1016/j.canlet.2017.07.020
2. Onaindia A, Medeiros LJ, Patel KP. Clinical Utility of Recently Identified Diagnostic, Prognostic, and Predictive Molecular Biomarkers in Mature B-Cell Neoplasms. Mod Pathol (2017) 30(10):1338–66. doi: 10.1038/modpathol.2017.58
3. Rosenquist R, Beà S, Du MQ, Nadel B, Pan-Hammarström Q. Genetic Landscape and Deregulated Pathways in B-Cell Lymphoid Malignancies. J Intern Med (2017) 282(5):371–94. doi: 10.1111/joim.12633
4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
5. Melani C, Wilson WH, Roschewski M. Monitoring Clinical Outcomes in Aggressive B-Cell Lymphoma: From Imaging Studies to Circulating Tumor DNA. Best Pract Res Clin Haematol (2018) 31(3):285–92. doi: 10.1016/j.beha.2018.07.004
6. Darrah JM, Herrera AF. Updates on Circulating Tumor DNA Assessment in Lymphoma. Curr Hematol Malig Rep (2018) 13(5):348–55. doi: 10.1007/s11899-018-0468-4
7. Copie-Bergman C, Cuillière-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, et al. MYC-IG Rearrangements Are Negative Predictors of Survival in DLBCL Patients Treated With Immunochemotherapy: A GELA/LYSA Study. Blood (2015) 126(22):2466–74. doi: 10.1182/blood-2015-05-647602
8. Rosenwald A, Bens S, Advani R, Barrans S, Copie-Bergman C, Elsensohn MH, et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol (2019) 37(35):3359–68. doi: 10.1200/jco.19.00743
9. Juskevicius D, Jucker D, Klingbiel D, Mamot C, Dirnhofer S, Tzankov. a. Mutations of CREBBP and SOCS1 Are Independent Prognostic Factors in Diffuse Large B Cell Lymphoma: Mutational Analysis of the SAKK 38/07 Prospective Clinical Trial Cohort. J Hematol Oncol (2017) 10(1):1–10. doi: 10.1186/s13045-017-0438-7
10. Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M, et al. Frequent NFKBIE Deletions Are Associated With Poor Outcome in Primary Mediastinal B-Cell Lymphoma. Blood (2016) 128(23):2666–70. doi: 10.1182/blood-2016-03-704528
11. Ferrero S, Rossi D, Rinaldi A, Bruscaggin A, Spina V, Eskelund CW, et al. KMT2D Mutations and TP53 Disruptions Are Poor Prognostic Biomarkers in Mantle Cell Lymphoma Receiving High-Dose Therapy: A FIL Study. Haematologica (2020) 105(6):1604–12. doi: 10.3324/haematol.2018.214056
12. Meng X, Min Q, Wang JY. B Cell Lymphoma. Adv Exp Med Biol (2020) 1254:161–81. doi: 10.1007/978-981-15-3532-1_12
13. López-Nieva P, Fernández-Navarro P, Graña-Castro O, Andrés-León E, Santos J, Villa-Morales M, et al. Detection of Novel Fusion-Transcripts by RNA-Seq in T-Cell Lymphoblastic Lymphoma. Sci Rep (2019) 9(1):5179. doi: 10.1038/s41598-019-41675-3
14. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell (2018) 173(2):321–37.e310. doi: 10.1016/j.cell.2018.03.035
15. Bruneau J, Molina TJ. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. In: Molina TJ, editor. Hematopathology. (2020). (Cham: Springer International Publishing (2020). p. 501–5.
16. Chen S, Zhou Y, Chen Y, Gu J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics (2018) 34(17):i884–90. doi: 10.1093/bioinformatics/bty560
17. Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. Genefuse: Detection and Visualization of Target Gene Fusions From DNA Sequencing Data. Int J Biol Sci (2018) 14(8):843–8. doi: 10.7150/ijbs.24626
18. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38(16):e164–4. doi: 10.1093/nar/gkq603
19. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: Efficient and Comprehensive Analysis of Somatic Variants in Cancer. Genome Res (2018) 28(11):1747–56. doi: 10.1101/gr.239244.118
20. Younes A, Hilden P, Coiffier B, Hagenbeek A, Salles G, Wilson W, et al. International Working Group Consensus Response Evaluation Criteria in Lymphoma (RECIL 2017). Ann Oncol (2017) 28(7):1436–47. doi: 10.1093/annonc/mdx097
21. Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, et al. The Mutational Landscape in Pediatric Acute Lymphoblastic Leukemia Deciphered by Whole Genome Sequencing. Hum Mutat (2015) 36(1):118–28. doi: 10.1002/humu.22719
22. Mareschal S, Pham-Ledard A, Viailly PJ, Dubois S, Bertrand P, Maingonnat C, et al. Identification of Somatic Mutations in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type by Massive Parallel Sequencing. J Invest Dermatol (2017) 137(9):1984–94. doi: 10.1016/j.jid.2017.04.010
23. Bolen CR, Klanova M, Trneny M, Sehn LH, He J, Tong J, et al. Prognostic Impact of Somatic Mutations in Diffuse Large B-Cell Lymphoma and Relationship to Cell-of-Origin: Data From the Phase III GOYA Study. Haematologica (2020) 105(9):2298–307. doi: 10.3324/haematol.2019.227892
24. Zhang W, Wang SL. An Integrated Framework for Identifying Mutated Driver Pathway and Cancer Progression. IEEE/ACM Trans Comput Biol Bioinform (2019) 16(2):455–64. doi: 10.1109/tcbb.2017.2788016
25. Vandin F, Upfal E, Raphael BJ. De Novo Discovery of Mutated Driver Pathways in Cancer. Genome Res (2012) 22(2):375–85. doi: 10.1101/gr.120477.111
26. Moia R, Boggione P, Mahmoud AM, Kodipad AA, Adhinaveni R, Sagiraju S, et al. Targeting P53 in Chronic Lymphocytic Leukemia. Expert Opin Ther Targets (2020) 24(12):1239–50. doi: 10.1080/14728222.2020.1832465
27. Arang N, Gutkind JS. G Protein-Coupled Receptors and Heterotrimeric G Proteins as Cancer Drivers. FEBS Lett (2020) 594(24):4201–32. doi: 10.1002/1873-3468.14017
28. Zimbwa P, Milicic A, Frater J, Scriba TJ, Willis A, Goulder PJR, et al. Precise Identification of a Human Immunodeficiency Virus Type 1 Antigen Processing Mutant. J Virol (2007) 81(4):2031–8. doi: 10.1128/JVI.00968-06
29. Zhang YX, Gui XE, Zhong YH, Rong YP, Yan YJ. Cancer in Cohort of HIV-Infected Population: Prevalence and Clinical Characteristics. J Cancer Res Clin Oncol (2011) 137(4):609–14. doi: 10.1007/s00432-010-0911-y
30. Saleem M, Yusoff NM. Fusion Genes in Malignant Neoplastic Disorders of Haematopoietic System. Hematology (2016) 21(9):501–12. doi: 10.1080/10245332.2015.1106816
31. Wang D, Ma K, Deng W, Li J, Xiang S, Zhang Y, et al. Development and Analytical Validation of a Targeted Next-Generation Sequencing Panel to Detect Actionable Mutations for Targeted Therapy. Onco Targets Ther (2021) 14:2423–31. doi: 10.2147/OTT.S299381
32. Kurashina R, Ohyashiki JH, Kobayashi C, Hamamura R, Zhang Y, Hirano T, et al. Anti-Proliferative Activity of Heat Shock Protein (Hsp) 90 Inhibitors via β-catenin/TCF7L2 Pathway in Adult T Cell Leukemia Cells. Cancer Lett (2009) 284(1):62–70. doi: 10.1016/j.canlet.2009.04.012
33. Tan H-Y, Wang N, Li S, Hong M, Guo W, Man K, et al. Repression of WT1-Mediated LEF1 Transcription by Mangiferin Governs β-Catenin-Independent Wnt Signalling Inactivation in Hepatocellular Carcinoma. Cell Physiol Biochem (2018) 47(5):1819–34. doi: 10.1159/000491063
34. Mao CD, Byers SW. Cell-Context Dependent TCF/LEF Expression and Function: Alternative Tales of Repression, De-Repression and Activation Potentials. Crit Rev Eukaryot Gene Expr (2011) 21(3):207–36. doi: 10.1615/critreveukargeneexpr.v21.i3.10
35. Janovská P, Bryja V. Wnt Signalling Pathways in Chronic Lymphocytic Leukaemia and B-Cell Lymphomas. Br J Pharmacol (2017) 174(24):4701–15. doi: 10.1111/bph.13949
36. Krystel-Whittemore M, Taylor MS, Rivera M, Lennerz JK, Le LP, Dias-Santagata D, et al. Novel and Established EWSR1 Gene Fusions and Associations Identified by Next-Generation Sequencing and Fluorescence in-Situ Hybridization. Hum Pathol (2019) 93:65–73. doi: 10.1016/j.humpath.2019.08.006
37. Peterson EA, Petty EM. Conquering the Complex World of Human Septins: Implications for Health and Disease. Clin Genet (2010) 77(6):511–24. doi: 10.1111/j.1399-0004.2010.01392.x
38. Wang E, Kawaoka S, Roe J-S, Shi J, Hohmann AF, Xu Y, et al. The Transcriptional Cofactor TRIM33 Prevents Apoptosis in B Lymphoblastic Leukemia by Deactivating a Single Enhancer. Elife (2015) 4:e06377. doi: 10.7554/eLife.06377
39. Crawford LJ, Johnston CK, Irvine AE. TRIM Proteins in Blood Cancers. J Cell Communication Signaling (2018) 12(1):21–9. doi: 10.1007/s12079-017-0423-5
40. Furth N, Aylon Y, Oren M. P53 Shades of Hippo. Cell Death Differ (2018) 25(1):81–92. doi: 10.1038/cdd.2017.163
41. Lu TX, Young KH, Xu W, Li JY. TP53 Dysfunction in Diffuse Large B-Cell Lymphoma. Crit Rev Oncol Hematol (2016) 97:47–55. doi: 10.1016/j.critrevonc.2015.08.006
42. Post SM, Pant V, Abbas H, Quintás-Cardama A. Prognostic Impact of the MDM2SNP309 Allele in Leukemia and Lymphoma. Oncotarget (2010) 1(3):168. doi: 10.18632/oncotarget.123
43. Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, et al. TP53 Mutation and Survival in Aggressive B Cell Lymphoma. Int J Cancer (2017) 141(7):1381–8. doi: 10.1002/ijc.30838
44. Gomez M, Gomez V, Hergovich A. The Hippo Pathway in Disease and Therapy: Cancer and Beyond. Clin Transl Med (2014) 3:22. doi: 10.1186/2001-1326-3-22
45. Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK-S, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep (2018) 25(5):1304–17. doi: 10.1016/j.celrep.2018.10.001
46. Aylon Y, Oren M. Tumor Suppression by P53: Bring in the Hippo! Cancer Cell (2017) 32(4):397–9. doi: 10.1016/j.ccell.2017.09.010
47. Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 Mutations Identify Younger Mantle Cell Lymphoma Patients Who do Not Benefit From Intensive Chemoimmunotherapy. Blood (2017) 130(17):1903–10. doi: 10.1182/blood-2017-04-779736
Keywords: B-cell lymphoma, gene fusion, mutated pathway, TP53 signaling pathway, Hippo pathway, prognosis
Citation: Fu H, Zhou H, Qiu Y, Wang J, Ma Z, Li H, Zhang F, Qiu C, Shen J and Liu T (2021) SEPT6_TRIM33 Gene Fusion and Mutated TP53 Pathway Associate With Unfavorable Prognosis in Patients With B-Cell Lymphomas. Front. Oncol. 11:765544. doi: 10.3389/fonc.2021.765544
Received: 08 September 2021; Accepted: 01 November 2021;
Published: 01 December 2021.
Edited by:
Rengyun Liu, The First Affiliated Hospital of Sun Yat-sen University, ChinaCopyright © 2021 Fu, Zhou, Qiu, Wang, Ma, Li, Zhang, Qiu, Shen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianzhen Shen, ZG9jdG9yc2p6QDE2My5jb20=; Tingbo Liu, bGl1dGI2NjhAMTM5LmNvbQ==