 Xuan Cheng
Xuan Cheng Jian-Xiong Zhao
Jian-Xiong Zhao Feng Dong
Feng Dong Xu-Chen Cao
Xu-Chen Cao- 1The First Department of Breast Cancer, Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin, China
- 2Key Laboratory of Cancer Prevention and Therapy, Tianjin, China
- 3Tianjin’s Clinical Research Center for Cancer, Tianjin, China
- 4Key Laboratory of Breast Cancer Prevention and Therapy, Tianjin Medical University, Ministry of Education, Tianjin, China
- 5Department of Neurosurgery, Tianjin Medical University General Hospital and Laboratory of Neuro-Oncology, Tianjin Neurological Institute, Tianjin, China
- 6State Key Laboratory of Experimental Hematology, The Province and Ministry Co-sponsored Collaborative Innovation Center for Medical Epigenetics, Tianjin Key Laboratory of Cellular Homeostasis and Human Diseases, Department of Cell Biology, Tianjin Medical University, Tianjin, China
Distant metastasis is the principal cause of mortality for breast cancer patients. Targeting specific mutations that have been acquired during the evolution process of advanced breast cancer is a potential means of enhancing the clinical efficacy of treatment strategies. In metastatic breast cancer, ARID1A is the most prevalent mutation of the SWI/SNF complex, which regulates DNA repair, recombination, and gene transcription. The low expression of ARID1A is associated with poor disease-free survival and overall survival of patients with luminal A or HER2-rich breast cancer. In addition, ARID1A plays a prominent role in maintaining luminal characteristics and has an advantage for identifying responses to treatment, including endocrine therapies, HDAC inhibitors and CDK4/6 inhibitors. The therapeutic vulnerabilities initiated by ARID1A alterations encourage us to explore new approaches to cope with ARID1A mutant-related drug resistance or metastasis. In this review, we describe the mutation profiles of ARID1A in metastatic breast cancer and the structure and function of ARID1A and the SWI/SNF complex as well as discuss the potential mechanisms of ARID1A-mediated endocrine resistance and therapeutic potential.
Background
Breast cancer has become the most frequently occurring malignancy worldwide, with 2.3 million women diagnosed with breast cancer in 2020 (1). Despite advances in early diagnosis and comprehensive therapeutic regimens, 20–30% of breast cancer patients who are diagnosed with new or recurrent advanced-stage or metastatic breast cancers contribute to 90% of cancer-related deaths (2, 3). Because of their high heterogeneity, aggressive metastatic breast cancers have variable responses to treatment and different patient prognoses. The common distant metastatic organs are bone, lung, liver, and brain, with 5-year overall survival rates of 22.8, 16.8, and 8.5% and extremely short survival rates which are lower than 80%, the 5-year overall survival rate of breast cancer patients without metastasis (4).
Genetic alterations with clinical significance in advanced breast cancer are more abundant and more complicated than those in early-stage breast cancer (5). Massive parallel sequencing of 8,654 advanced breast cancers showed that 80.4% of the tumors harbored a genetic variation in at least one pathway with therapeutic implications (6). Although some driver genes, such as PIK3CA, TP53, ERBB2, ESR1, AKT1, and BRCA1, are detected in some early breast cancers, these genes are more frequently altered in patients with distant metastasis (7). Moreover, several studies have further demonstrated that the accumulation of genetic alterations during the process of cancer evolution between primary tumors and metastases and the exposure to systemic therapy itself lead to endocrine/chemotherapy resistance of tumor cells, resulting in treatment failure or disease progression (4, 8, 9). Thus, it is important and urgent to develop and research targeted agents aimed at newly discovered driver aberrations to guide personalized therapy against metastatic breast cancer.
The SWItch mating type/Sucrose Non-Fermenting (SWI/SNF) family, a member of chromatin remodeling complexes which include four distinct families in eukaryotes, was initially identified in Saccharomyces cerevisiae and is evolutionarily conserved, particularly among the ATPase subunits (10). SWI/SNF complexes utilize the energy of ATP hydrolysis to remodel chromatin accessibility by means of nucleosome sliding or nucleosome ejection and insertion (11). The bromodomain of ATPase subunits assists in targeting acetylated histones for SWI/SNF complexes (12). The AT-rich interactive domain (ARID) subunits are crucial for SWI/SNF-mediated chromatin remodeling and recruit SWI/SNF complexes to chromatin via nonselective DNA binding activity (13–15) or through interacting with other transcription factors. These complexes modulate multiple cellular processes and are associated with gene transcription and DNA repair. It is worth noting that mammalian SWI/SNF complexes contribute to not only activation but also repression of transcription. Brahma-related gene 1 (BRG1) and homologous Brahma participate in both transcriptional activation and repression of ER-driven genes in ligand- and context-dependent reprogramming (16–18).
A high prevalence of mutations in SWI/SNF chromatin remodeling complexes has been found in 20–25% of all human cancers (19, 20). Most of the mutations result in decreased protein expression of the SWI/SNF subunit, which influences subsequent chromatin changes and further transcription outcomes (10). The tumor suppressor AT-rich interactive domain protein 1A (ARID1A) is the most commonly mutated gene in SWI/SNF complexes. ARID1A is located on chromosome 1p35.11 and encodes a protein of approximately 250 kD. ARID1A is localized in the nucleus and has a broad tissue distribution. ARID1A is characterized by a 100-amino acid ARID domain conserved in eukaryotic organisms. The ARID domain is important for mediating ARID1A-associated SWI/SNF family DNA binding. It has been reported that the ARID domain does not preferentially interact with the AT-rich sequence, but it can recognize the pyrimidine-rich sequence of the β-globin locus (21). In addition, some LXXLL motifs in the C-terminal region of ARID1A are critical for ARID1A–glucocorticoid receptor (GR) interaction and subsequent GR-mediated transcription (21). Specific vulnerabilities attributed to the loss of tumor suppressors in tumor cells often create potential targets for therapy. ARID1A is one of the few genes that are mutated in pre-existing genomically unstable cancer cells, and the importance of ARID1A mutations is underestimated.
ARID1A is now known to have the highest mutation incidence, up to 46–57%, in ovarian clear cell carcinoma (22, 23). Several reviews have summarized the synthetic lethal strategies for ARID1A mutation (24, 25), but few review articles are available specifically in the context of metastatic breast cancer. Here we review the current understanding of ARID1A, including mutation profiles, prognostic values, and biological functions in metastatic breast cancer, discuss how ARID1A mutation mediates drug resistance, and summarize the potential therapeutic strategies for ARID1A-mutated metastatic breast cancer to curb this dreaded disease.
ARID1A Mutation and Prognostic Values in Breast Cancer
In breast cancer, ARID1A is regarded as a tumor suppressor (26) and cooperates with CEBPα to suppress cell proliferation and migration (27). Inactivating ARID1A mutations were only present in 2.5% of all breast cancers. Nevertheless, the aberration frequency increased up to 12% in metastatic breast cancers with treatment resistance and 10% in inflammatory breast cancer (IBC), which has a higher tumor mutation burden than non-IBC (19, 28). Recently, in the search for newly emerging driver genes in metastatic breast cancer via whole-genome sequencing, Yates et al. found that genes related to the SWI/SNF family, especially ARID1A, were commonly wild type in primary breast cancer but inactive in recurrent breast cancer (29). Corresponding ARID1A alterations are shown in Figure 1.
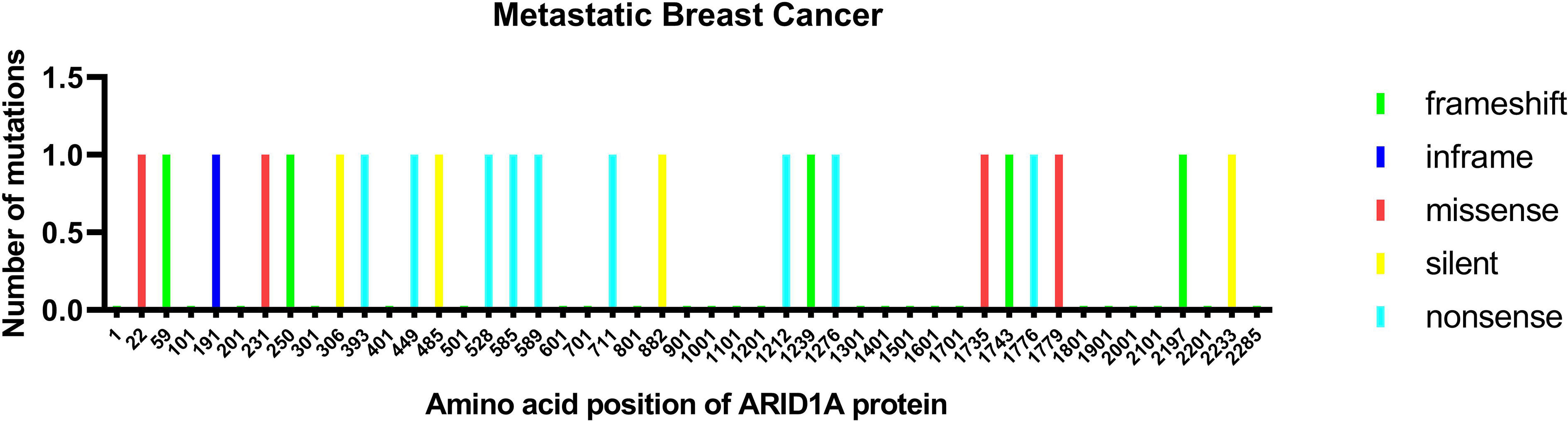
Figure 1 ARID1A protein mutation in metastatic breast cancer. The mutations are colored by the type of mutations: missense, nonsense, silent, and inframe.
Previous studies support ARID1A deletion as an independent prognostic factor for invasive breast cancer (30–32). The low mRNA expression of ARID1A is related to shorter overall survival in luminal A and human epidermal growth factor receptor 2 (HER2)-rich breast cancer (31). Takao et al. analyzed the immunohistochemical staining to evaluate the relationship between the downregulation of ARID1A and poor disease-free survival (33). Moreover, ARID1A may have a high prognostic value for drug sensitivity. Xu et al. verified a vital function of ARID1A in breast luminal lineage maintenance, which further revealed that the expression of ARID1A in the luminal type was higher than that in the nonluminal type (31, 32). The loss of ARID1A was more general in post-endocrine therapy metastatic cancer and regarded as the top candidate whose loss indicated fulvestrant resistance (32, 34). In addition, ARID1A loss or downregulation drives paclitaxel resistance and HER2/PI3K/mTOR-targeting drug resistance in breast cancer (35, 36). Therefore, ARIDIA is closely related to tumor metastasis or drug resistance-mediated tumor recurrence.
Mechanisms Underlying the Therapeutic Responses of ER+ Breast Cancer
Estrogen receptor-positive (ER+) breast cancer is the most common breast cancer subtype. Endocrine therapy, as the mainstay of ER+ breast cancer treatment, can disrupt the interaction of estrogen with the ER ligand binding domain and inhibit the ER signaling pathway either by selective ER modulators/degraders (SERMs/SERDs) antagonizing ER or by aromatase inhibitors (AIs), which decreases the level of systemic estrogen by blocking the conversion of androgens to estrogens. While these treatments markedly reduce the risk of recurrence and death in ER+ breast cancer patients (37–39), resistance to endocrine therapy remains a major challenge (40).
Razavi et al. found that ARID1A loss-of-function mutations were acquired following treatment with endocrine therapy (41). A recent study has shown that ARID1A is indispensable for the activity of both tamoxifen (SERM) and fulvestrant (SERD), and the subsequent repression of ER target gene transcription is dependent on specific enhancers. Nagarajan et al. found that ARID1A relies on the FOXA1 protein to bind to the regulatory elements of chromatin (28). Tamoxifen-bound ER is enriched and binds to the FOXA1-dependent ARID1A binding site with an ARID1A-containing SWI/SNF complex to suppress gene expression under tamoxifen treatment. This is not associated with chromatin accessibility but with histone deacetylase 1 (HDAC1) recruitment. ARID1A loss promotes bromodomain-containing protein-4 (BRD4)-mediated transcription on account of reduced HDAC1 binding and increased histone H4 acetylation (H4ac) levels. Indeed ARID1A deletion makes breast cancer cells sensitive to bromodomain and extraterminal domain (BET) inhibitors and HDAC inhibitors. This provides rational treatment strategies for inactivated ARID1A-mediated endocrine resistance in breast cancer (28). Chidamide (an oral HDAC inhibitor with subtype specificity for the inhibition of HDAC1, HDAC2, HDAC3, and HDAC10) combined with exemestane (AI) improved the progression-free survival in advanced HR+ HER2- breast cancer patients who progressed after a previous endocrine therapy compared with placebo plus exemestane (42).
Furthermore, in ER+ breast cancer, ARID1A was found to participate in the maintenance of luminal identity by modulating cellular plasticity (32). ARID1A loss mainly results in the loss of chromatin accessibility, which tends to be in intergenic or intragenic regions. In addition, the recruitment of master luminal transcription factors such as ER, forkhead box protein A1 (FOXA1), and GATA-binding factor 3 (GATA3) and histone modification H3K27ac levels at correspondingly accessible sites is clearly decreased, impacting enhancer activity and ER-dependent transcription. Meanwhile, TEA domain transcription factor 4, which is enriched in basal-like cells, is recruited to accessible sites, indicating the switch of phenotypes from the luminal type to the basal-like type in ARID1A-loss breast cancer. The lack of ER expression in ER-positive breast cancer is related to endocrine-resistant therapy and ER-negative metastatic relapse (43). Hence, it is plausible that inactive ARID1A mutants are found in endocrine-resistant breast cancer.
Dysregulated cellular proliferation is a key hallmark of cancer, and targeting cell proliferative signaling is a widely used strategy for tumor treatment (44). In the cell division cycle, cyclin-dependent kinase-4 (CDK4)- and CDK6-mediated retinoblastoma phosphorylation is necessary to drive cells from G1 to S phase (45). Hence, drugs inhibiting enzymatic activities to induce cell cycle arrest have been used and proven to have significant efficacy in tumor therapy, including metastatic breast cancer (46, 47). As early as 2005, it was reported that ARID1A participates in cell cycle regulation and that its deletion induces impaired cell cycle arrest, indicating that ARID1A depletion may be associated with carcinogenesis (48). As mentioned above, inactivated ARID1A is related to endocrine resistance (28). An important mechanism of endocrine resistance is cell cycle dysregulation, which inhibits antiproliferative effects (49, 50). Based on the underlying mechanisms mentioned above and the remarkable outcomes of clinical trials, palbociclib or ribociclib (CDK4/6 inhibitor) combined with standard endocrine therapy is regarded as the first-line therapy for recurrent unresectable or metastatic ER+ breast cancer according to the NCCN guidelines (51–53). These findings indicate that CDK4/6 inhibitors are promising therapeutic strategies for ARID1A-mutated metastatic breast cancer.
Promising Treatment Targets
Genetic mutations and altered molecular pathways serve as promising targets in personalized therapeutic approaches. Previous studies have revealed that ARID1A mutations can pre-exist in primary cancers and can be enriched during metastasis. Loss-of-function mutations in ARID1A often lead to the disruption of mammalian SWI/SNF complex integrity and subsequent transcriptional dysfunction. Therefore, ARID1A mutations simultaneously create an altered regulation of distinct target gene sets and generate a series of cellular dependencies or synthetic lethality. Here we summarize newly discovered potential therapeutic targets for metastasis- or endocrine-resistant breast cancer driven by mutated ARID1A (Figure 2) and the treatment approaches being tested in clinical trials involving patients with ARID1A-mutated cancer (Table 1).
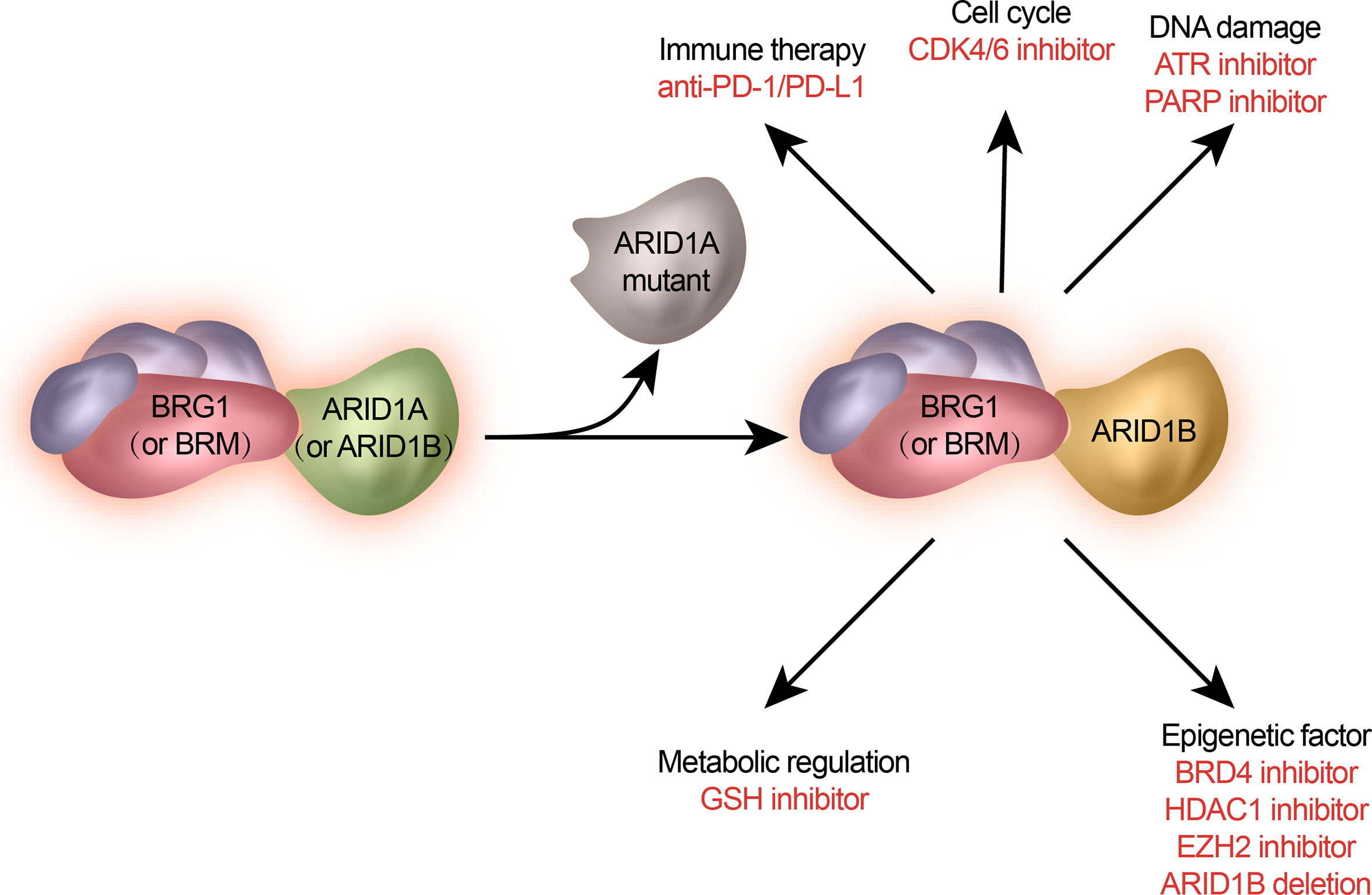
Figure 2 Potential treatments of ARID1A-mutated metastatic breast cancer. ARID1A mutation gives rise to specific signaling pathways and cellular functions. They are selected as therapeutic targets against ARID1A mutations.
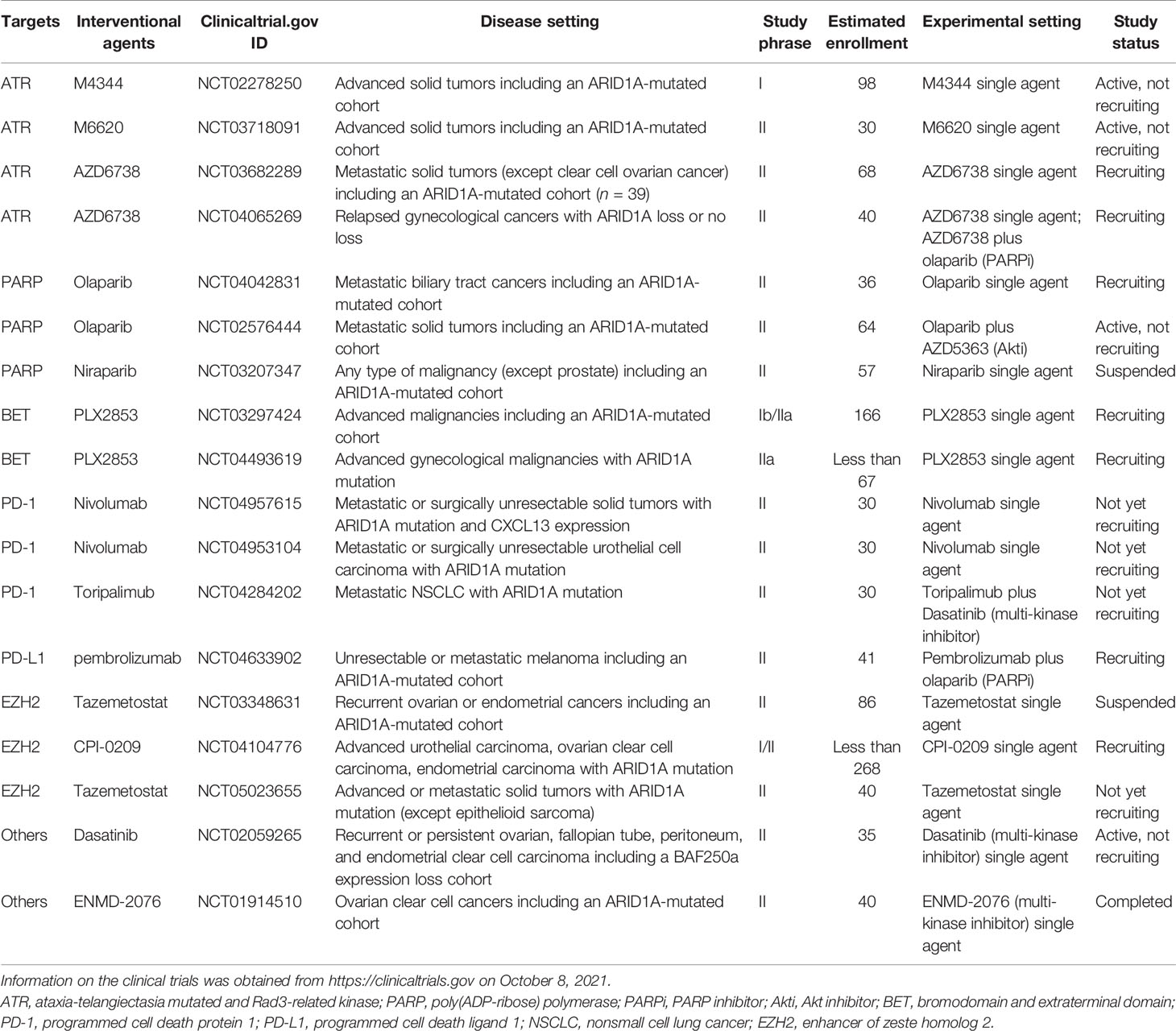
Table 1 Clinical trials involving patients with ARID1A-mutated cancer.
DNA Damage Repair—PARP and ATR
DNA damage response networks are important for the maintenance of genomic integrity especially in cancer cells. Cancer cells often undergo various exogenous and endogenous events, such as oxidative stress and replication stress, causing genomic instability. Oncogene-induced DNA damage, as one of the cancer-intrinsic features of damage, offers a possible basis for conditional synthetic lethality. Nuclear enzyme poly (ADP-ribose) polymerase 1 (PARP1) is best known for detecting single-stranded DNA breaks and affecting the process of DNA repair and transcription regulation. PARP1 is activated by zinc finger motif-mediated nicked DNA recognition. The ADP-ribosyltransferase catalytic domain of PARP then actuates the PARylation of PARP1 substrate proteins, mediating the recruitment of single-strand break repair (SSBR) machinery for DNA repair. Finally, PARP1 autoPARylation causes the release of PARP1 from DNA to another location for the next cycle of the SSBR process (54, 55). PARP inhibitors (PARPis), primarily designed for the catalytic site to interfere with DNA repair, cooperate with the pre-existing defects in homologous recombination (HR) repair to induce tumor cell death (56, 57). The oral PARPi olaparib is approved for the treatment of patients with metastatic breast cancer and a germline (g) BRCA mutation (58, 59). Recently, the last clinical trial of HER2-negative early breast cancer with (g) BRCA mutation indicated the significant efficacy of prolonging invasive and distant disease-free survival (60). Furthermore, the TBCRC 048 trial has expanded the clinical indications of PARPis for metastatic breast cancer patients with somatic (s) BRCA mutations or g/s mutations in HR-related genes such as gPALB2 other than BRCA1/2 (61). In 2015, it was reported that ARID1A deficiency makes cancer cells more sensitive to PARPis, providing an innovative treatment strategy for patients with ARID1A-mutated breast tumors (62). Conversely, the team of Sourav found that ARID1A loss makes breast cancer insensitive to PARP inhibitors (63). This is probably related to the use of different breast cancer cell lines; therefore, primary cancer cells from patients will be used to further resolve this paradoxical phenomenon.
Ataxia-telangiectasia-mutated and Rad3-related protein kinase (ATR), a member of the phosphatidylinositol 3-kinase-like kinase family, is a central regulator of the cellular DNA damage response (64–67). In response to DNA damage, ATR can recruit ARID1A to DNA double-strand breaks for processing to single-strand ends and maintaining DNA damage signaling. A preclinical study indicated that the combination of ATR inhibitors and Wee1 inhibitors can increase DNA damage and suppress breast cancer metastasis (68). Meanwhile, ARID1A was identified as an ATR synthetic lethal partner. The inhibition of ATR disrupts the cell cycle, genomic stability, and cell survival in ARID1A-deficient tumor cells. Consequently, ATR inhibitors are also deemed to be a potential single-agent therapy for ARID1A-mutated cancers, including breast cancer (69).
Epigenetic Regulation—BET and HDAC
Bromodomain and extraterminal domain (BET) proteins are chromatin readers that recognize acetylated proteins by a conserved bromodomain to regulate gene transcription (70, 71). BET proteins essentially contain two tandem bromodomains (BD1 and BD2), an extraterminal domain (ET), and a C-terminal domain (CTD). The ET domain recruits effector proteins for its regulatory function (72). The CTD interacts with positive transcription elongation factor b and promotes the phosphorylation of serine residues of the RNA Pol II C-terminal motif for transcriptional activation (73, 74). BET proteins consist of the ubiquitously expressed BRD2, BRD3, and BRD4 and the testis-restricted BRDT (75).
Previous studies have revealed that BRD4 plays key roles in embryogenesis and metastasis in breast cancer. BRD4 can regulate the self-renewal of mouse embryonic stem cells by promoting Nanog expression (76), and its loss causes early post-implantation defects in mice (77). In breast cancer, the long isoform BRD4 is a negative predictor of cancer metastasis and survival (78, 79), but unexpectedly, the short isoform BRD4 can induce EMT transition, CSC-like properties (79), migration, and metastasis through Engrailed-1-mediated enhancer regulation (80). BRD4 is considered a therapeutic target of tamoxifen-resistant breast cancer (81) because it participates in the regulation of ERα function (82, 83). In basal-like breast cancer, BRD4 links nonhistone Twist diacetylation to tumorigenesis (84). Moreover, there is a bromodomain-independent effect of BRD4 in BET-resistant TNBC cells (85). Given the importance of BRD4 in breast cancer, BRD4 is regarded as a therapeutic target by interacting with the bromodomain (86, 87) or proteolysis targeting chimeric (PROTAC) molecule (88–90). More importantly, ARID1A deficiency makes ER+ breast cancer cells sensitive to BET inhibitors (28). Although BET inhibitor resistance has become a challenging issue, synergy with multiple epigenetic agents, particularly histone deacetylase inhibitors, is expected to overcome this problem (91, 92).
HDAC1 protein is associated with the breast cancer stem cell phenotype and distant metastasis (93, 94). Reduced levels of BRMS1L (breast cancer metastasis suppressor 1 like) activates Wnt signaling receptor FZD10 expression by regulating HDAC1 recruitment and histone H3K9ac levels to promote breast cancer cell invasion and migration (94). In addition, it has been reported that HDAC1 participates in ARID1A-mediated gene expression. ARID1A loss inhibits HDAC1 recruitment and promotes H4ac-mediated BRD4 binding for gene transcription and growth (28). Because ARID1A mutations are mainly present in metastatic tumors or tumors progressing under endocrine therapy, a synthetic lethality-based endocrine treatment strategy with BET and/or HDAC1 inhibitors is expected to be used for ARID1A-mutated breast cancer (32).
Residual SWI/SNF Activity—ARID1B
Mammalian SWI/SNF can be mainly composed of canonical Brahma-related gene 1 (Brg1)-associated factor (BAF) complexes and polybromo Brg1-associated factor (PBAF) complexes (95, 96). Brg1 and homologous Brahma (Brm) are the central ATPase subunits in SWI/SNF complexes (97). In addition to the shared ATPase Brg1/Brm, the SWI/SNF family also contains variant subunits and complex-specific subunits. ARID1A (BAF250A) and ARID1B (BAF250B) subunits are distinct from BAF complexes in a mutually exclusive fashion (15) replaced by ARID2 (BAF200) in PBAF complexes (98).
Considering that ARID1A mutations such as nonsense and frameshift usually result in loss-of-function traits, this defective protein is not suitable for targeting. ARID1A loss may make cancer cells more dependent on ARID1B-associated BAFs. Depletion of ARID1B destabilizes the SWI/SNF complex and suppresses the proliferation of ARID1A-deficient breast cancer (99). Meanwhile, the synthetic lethality between ARID1A and ARID1B has also been found to be a conserved function in colorectal cancer (100), ovarian clear cell carcinomas (101), and ovarian cancer (102) with an ARID1A-mutant background. Hence, ARID1B may be a promising target for the potential treatment of ARID1A-mutated cancers.
Previous studies have revealed that ARID1A, ARID1B, and ARID2 contain some mutations and are inactivated in recurrent breast cancer (29). In TNBC, ARID1B expression was upregulated and related to worse clinical prognoses (103, 104). In view of the importance of ARID1B for facilitating BAF ATPase module binding to chromatin, approaches with small stabilized peptides can be considered for disrupting the association of ARID1B with the BAF core module (105), particularly SMARCC and SMARCD (99). A homologous approach has been used to interfere with the enhancer of zeste homologue 2 (EZH2)/EED complex in EZH2-dependent cancer (106). BET-PROTACs were used to target and degrade BRD4 protein to revert BETi resistance (90). These strategies can be employed for ARID1B treatment. Hence, strategies of synthetic lethality between ARID1A and ARID1B or ARID2 are worthy of further exploration.
Tumor Immunological Microenvironment
Previous preclinical and clinical studies have reported that ARID1A mutation can enhance the immunogenicity of tumor cells in breast cancer (107) and improve tumor sensitivity to immune checkpoint inhibitors in various cancers, including metastatic urothelial cancer (108), advanced nonsmall cell lung cancer (109), ovarian clear cell cancer (110), colorectal cancer (111, 112), gastrointestinal cancer (113), and colon cancer (114), which can provide insights into ARID1A-mutated breast cancer. Meanwhile, ATM inhibitors and HDAC6 inhibitors can further potentiate the efficacy of antitumor immunity in ARID1A-mutated ovarian cancer (115, 116). The efficacy of immune checkpoint inhibitors in ARID1A-mutated metastatic breast cancer is expected to be good.
Glutathione Metabolic Pathway
A recent study showed that ARID1A-deficient cancer cells are specifically vulnerable to inhibitors of the glutathione (GSH) metabolic pathway due to lower GSH levels (117). Furthermore, downregulation of GSH was associated with poor prognosis in breast cancer (118). GSH is also considered a potential therapeutic target for metastatic breast cancer, including metastatic brain colonization and breast cancer bone metastasis (119–121). Consequently, targeting the GSH metabolic pathway may offer hope for breakthroughs in the fight against ARID1A-deficient metastatic breast cancer.
EZH2 Inhibition
Furthermore, EZH2 is also regarded to be specifically vulnerable in ARID1A-mutated cancers (122). EZH2 is a core member of the polycomb repressive complex 2 (PRC2) and is responsible for catalyzing H3K27me3, which is associated with transcriptional repression (123, 124). EZH2 cooperates with ARID1A to regulate PIK3IP1 expression and the PI3K/AKT signaling pathway, and EZH2 inhibitors can reduce the proliferation of ARID1A-mutated tumors. Given that ARID1A-deficient tumors are sensitive to both tumor immunotherapy and PARP inhibitors, the combination of EZH2 inhibitors with PARP inhibitors or tumor immunotherapy with ARID1A-deficient tumors may be potential treatment strategies (62, 125, 126). EZH2 expression levels or protein modifications are involved in breast cancer initiation (127–129), progression or metastasis (130–133), breast cancer stem cell regulation (134), and drug resistance (135–141). The high expression level of EZH2 is also considered an indicator of aggressive breast cancer (128). The MEK–ERK pathway (142) and lncRNA ANCR (143) can regulate EZH2 levels to impact breast cancer metastasis. Meanwhile, EZH2 undergoes post-translational modifications, including arginine methylation, lysine methylation (144), and serine/threonine phosphorylation (145), regulating protein methyltransferase activity (146), protein stability (147), or cellular localization (148), which ultimately affect cancer metastasis. Considering the crucial role of EZH2 in the process of breast cancer metastasis and endocrine resistance (138), EZH2 inhibition represents a novel treatment option for ARID1A-mutated metastatic breast cancer.
Conclusion and Discussion
Cancer metastasis, as the paramount factor in cancer-related death, places both an economic and psychological burden on breast cancer patients. Previous studies on genomic alterations acquired during metastatic evolution highlight the value of molecular features for therapeutic strategies and have contributed to the substantial improvement in cancer outcomes. Recently, ARID1A, a subunit of the SWI/SNF complex, was found to be commonly mutated in recurrent breast cancer and is considered a promising but challenging drug target. Inactivated ARID1A mutations are important to maintain the luminal identity of breast cancer and are associated with endocrine resistance. Based on synthetic lethality and cellular dependence, therapeutic strategies targeting ARID1A-mutated breast cancer have been reported, such as some inhibitors of PARP, EZH2, and BRD4. Moving forward, further mechanistic studies and clinical trials are needed to determine the efficacy of cancer targets driven by ARID1A mutations.
There is still much confusion regarding ARID1A mutations and metastatic breast cancers. Although we know that ARID1A is the most commonly mutated gene in SWI/SNF complexes and has a higher incidence in metastatic breast cancer, the functions and mutation frequencies of ARID1A in different subtypes of breast cancer are not clear. Whether the occurrence of ARID1A-mutated metastatic breast cancer has an organ tendency needs more statistical analysis. There are more potential therapeutic targets that have been discovered and proven effective in preclinical studies, but the clinical trial outcomes are limited, and the underlying mechanisms are not fully understood. Indeed the responses of BET inhibitors in TNBC are exactly the opposite of those in ER+ breast cancer (149). In addition, the indications and the combination strategies of different therapeutic targets remain key questions and active areas of research. CDK4/6 inhibitors (150) or BRD4 inhibitors (151, 152), for example, drive antitumor immunity and provide a reasonable combination strategy with immunotherapies, while CDK4/6 may interfere with the activity of ATR-associated drugs that rely on replicative stress (153). Consequently, more investigations of ARID1A-mutated breast cancer are necessary and expected.
Author Contributions
XuaC, J-XZ, X-CC, and FD prepared and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. World Cancer Day 2021: Spotlight on IARC Research Related to Breast Cancer. Available at: https://www.iarc.who.int/featured-news/world-cancer-day-2021/.
2. Cancer Genome Atlas N. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
3. Tosello G, Torloni MR, Mota BS, Neeman T, Riera R. Breast Surgery for Metastatic Breast Cancer. Cochrane Database Syst Rev (2018) 3:CD011276. doi: 10.1002/14651858.CD011276.pub2
4. Liang Y, Zhang H, Song X, Yang Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin Cancer Biol (2020) 60:14–27. doi: 10.1016/j.semcancer.2019.08.012
5. Kimbung S, Loman N, Hedenfalk I. Clinical and Molecular Complexity of Breast Cancer Metastases. Semin Cancer Biol (2015) 35:85–95. doi: 10.1016/j.semcancer.2015.08.009
6. Ross JS, Gay LM. Comprehensive Genomic Sequencing and the Molecular Profiles of Clinically Advanced Breast Cancer. Pathology (2017) 49(2):120–32. doi: 10.1016/j.pathol.2016.11.005
7. Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic Characterization of Metastatic Breast Cancers. Nature (2019) 569(7757):560–4. doi: 10.1038/s41586-019-1056-z
8. Arnedos M, Vicier C, Loi S, Lefebvre C, Michiels S, Bonnefoi H, et al. Precision Medicine for Metastatic Breast Cancer–Limitations and Solutions. Nat Rev Clin Oncol (2015) 12(12):693–704. doi: 10.1038/nrclinonc.2015.123
9. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 Mutations in Hormone-Resistant Metastatic Breast Cancer. Nat Genet (2013) 45(12):1446–51. doi: 10.1038/ng.2823
10. St Pierre R, Kadoch C. Mammalian SWI/SNF Complexes in Cancer: Emerging Therapeutic Opportunities. Curr Opin Genet Dev (2017) 42:56–67. doi: 10.1016/j.gde.2017.02.004
11. Saha A, Wittmeyer J, Cairns BR. Chromatin Remodelling: The Industrial Revolution of DNA Around Histones. Nat Rev Mol Cell Biol (2006) 7(6):437–47. doi: 10.1038/nrm1945
12. Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a Core Chromatin Remodeling Complex From SWI/SNF Subunits. Mol Cell (1999) 3(2):247–53. doi: 10.1016/S1097-2765(00)80315-9
13. Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The Human SWI-SNF Complex Protein P270 Is an ARID Family Member With Non-Sequence-Specific DNA Binding Activity. Mol Cell Biol (2000) 20(9):3137. doi: 10.1128/MCB.20.9.3137-3146.2000
14. Patsialou A, Wilsker D, Moran E. DNA-Binding Properties of ARID Family Proteins. Nucleic Acids Res (2005) 33(1):66–80. doi: 10.1093/nar/gki145
15. Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, et al. Two Related ARID Family Proteins Are Alternative Subunits of Human SWI/SNF Complexes. Biochem J (2004) 383(Pt 2):319–25. doi: 10.1042/BJ20040524
16. Direnzo J, Shang Y, Phelan M, Sif S, Myers M, Kingston R, et al. BRG-1 Is Recruited to Estrogen-Responsive Promoters and Cooperates With Factors Involved in Histone Acetylation. Mol Cell Biol (2000) 20(20):7541. doi: 10.1128/MCB.20.20.7541-7549.2000
17. Belandia B, Orford RL, Hurst HC, Parker MG. Targeting of SWI/SNF Chromatin Remodelling Complexes to Estrogen-Responsive Genes. EMBO J (2002) 21(15):4094–103. doi: 10.1093/emboj/cdf412
18. Zhang B, Chambers KJ, Faller DV, Wang S. Reprogramming of the SWI/SNF Complex for Co-Activation or Co-Repression in Prohibitin-Mediated Estrogen Receptor Regulation. Oncogene (2007) 26(50):7153–7. doi: 10.1038/sj.onc.1210509
19. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat Genet (2013) 45(6):592–601. doi: 10.1038/ng.2628
20. Shain AH, Pollack JR. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PloS One (2013) 8(1):e55119. doi: 10.1371/journal.pone.0055119
21. Nie Z, Xue Y, Yang D, Zhou S, Deroo BJ, Archer TK, et al. A Specificity and Targeting Subunit of a Human SWI/SNF Family-Related Chromatin-Remodeling Complex. Mol Cell Biol (2000) 20(23):8879–88. doi: 10.1128/MCB.20.23.8879-8888.2000
22. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N Engl J Med (2010) 363(16):1532–43. doi: 10.1056/NEJMoa1008433
23. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science (2010) 330(6001):228–31. doi: 10.1126/science.1196333
24. Caumanns JJ, Wisman GBA, Berns K, van der Zee AGJ, de Jong S. ARID1A Mutant Ovarian Clear Cell Carcinoma: A Clear Target for Synthetic Lethal Strategies. Biochim Biophys Acta Rev Cancer (2018) 1870(2):176–84. doi: 10.1016/j.bbcan.2018.07.005
25. Mathur R. ARID1A Loss in Cancer: Towards a Mechanistic Understanding. Pharmacol Ther (2018) 190:15–23. doi: 10.1016/j.pharmthera.2018.05.001
26. Huang J, Zhao YL, Li Y, Fletcher JA, Xiao S. Genomic and Functional Evidence for an ARID1A Tumor Suppressor Role. Genes Chromosomes Cancer (2007) 46(8):745–50. doi: 10.1002/gcc.20459
27. Guo X, Zhang Y, Mayakonda A, Madan V, Ding LW, Lin LH, et al. ARID1A and CEBPalpha Cooperatively Inhibit UCA1 Transcription in Breast Cancer. Oncogene (2018) 37(45):5939–51. doi: 10.1038/s41388-018-0371-4
28. Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, et al. ARID1A Influences HDAC1/BRD4 Activity, Intrinsic Proliferative Capacity and Breast Cancer Treatment Response. Nat Genet (2020) 52(2):187–97. doi: 10.1038/s41588-019-0541-5
29. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell (2017) 32(2):169–84 e7. doi: 10.1016/j.ccell.2017.07.005
30. Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, et al. An Integrated Genomic Approach Identifies ARID1A as a Candidate Tumor-Suppressor Gene in Breast Cancer. Oncogene (2012) 31(16):2090–100. doi: 10.1038/onc.2011.386
31. Zhang J, Hou S, You Z, Li G, Xu S, Li X, et al. Expression and Prognostic Values of ARID Family Members in Breast Cancer. Aging (2021) 13(4):5621–37. doi: 10.18632/aging.202489
32. Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, et al. ARID1A Determines Luminal Identity and Therapeutic Response in Estrogen-Receptor-Positive Breast Cancer. Nat Genet (2020) 52(2):198–207. doi: 10.1038/s41588-019-0554-0
33. Takao C, Morikawa A, Ohkubo H, Kito Y, Saigo C, Sakuratani T, et al. Downregulation of ARID1A, A Component of the SWI/SNF Chromatin Remodeling Complex, in Breast Cancer. J Cancer (2017) 8(1):1–8. doi: 10.7150/jca.16602
34. Priedigkeit N, Ding K, Horne W, Kolls JK, Du T, Lucas PC, et al. Acquired Mutations and Transcriptional Remodeling in Long-Term Estrogen-Deprived Locoregional Breast Cancer Recurrences. Breast Cancer Res BCR (2021) 23(1):1. doi: 10.1186/s13058-020-01379-3
35. Lin YF, Tseng IJ, Kuo CJ, Lin HY, Chiu IJ, Chiu HW. High-Level Expression of ARID1A Predicts a Favourable Outcome in Triple-Negative Breast Cancer Patients Receiving Paclitaxel-Based Chemotherapy. J Cell Mol Med (2018) 22(4):2458–68. doi: 10.1111/jcmm.13551
36. Berns K, Sonnenblick A, Gennissen A, Brohée S, Hijmans EM, Evers B, et al. Loss of ARID1A Activates ANXA1, Which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22(21):5238. doi: 10.1158/1078-0432.CCR-15-2996
37. Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, et al. Effect of Anastrozole and Tamoxifen as Adjuvant Treatment for Early-Stage Breast Cancer: 10-Year Analysis of the ATAC Trial. Lancet Oncol (2010) 11(12):1135–41. doi: 10.1016/S1470-2045(10)70257-6
38. Regan MM, Neven P, Giobbie-Hurder A, Goldhirsch A, Ejlertsen B, Mauriac L, et al. Assessment of Letrozole and Tamoxifen Alone and in Sequence for Postmenopausal Women With Steroid Hormone Receptor-Positive Breast Cancer: The BIG 1-98 Randomised Clinical Trial at 8·1 Years Median Follow-Up. Lancet Oncol (2011) 12(12):1101–8. doi: 10.1016/S1470-2045(11)70270-4
39. Robertson JFR, Bondarenko IM, Trishkina E, Dvorkin M, Panasci L, Manikhas A, et al. Fulvestrant 500 Mg Versus Anastrozole 1 Mg for Hormone Receptor-Positive Advanced Breast Cancer (FALCON): An International, Randomised, Double-Blind, Phase 3 Trial. Lancet (2016) 388(10063):2997–3005. doi: 10.1016/S0140-6736(16)32389-3
40. Strasser-Weippl K. Advances in Adjuvant Hormonal Therapy for Postmenopausal Women. J Clin Oncol (2005) 23(8):1751–9. doi: 10.1200/JCO.2005.11.038
41. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell (2018) 34(3):427–38.e6. doi: 10.1016/j.ccell.2018.08.008
42. Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, et al. Tucidinostat Plus Exemestane for Postmenopausal Patients With Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2019) 20(6):806–15. doi: 10.1016/S1470-2045(19)30164-0
43. Kuukasjärvi T, Kononen J, Helin H, Holli K, Isola J. Loss of Estrogen Receptor in Recurrent Breast Cancer Is Associated With Poor Response to Endocrine Therapy. J Clin Oncol Off J Am Soc Clin Oncol (1996) 14(9):2584–9. doi: 10.1200/JCO.1996.14.9.2584
44. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646. doi: 10.1016/j.cell.2011.02.013
45. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct Binding of Cyclin D to the Retinoblastoma Gene Product (pRb) and pRb Phosphorylation by the Cyclin D-Dependent Kinase CDK4. Genes Dev (1993) 7(3):331–42. None. doi: 10.1101/gad.7.3.331
46. Dickler MN, Tolaney SM, Rugo HS, Cortes J, Dieras V, Patt D, et al. MONARCH 1, A Phase II Study of Abemaciclib, A CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients With Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(17):5218–24. doi: 10.1158/1078-0432.CCR-17-0754
47. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination With Letrozole Versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol (2015) 16(1):25–35. doi: 10.1016/S1470-2045(14)71159-3
48. Nagl NG Jr., Patsialou A, Haines DS, Dallas PB, Beck GR Jr., Moran E. The P270 (ARID1A/SMARCF1) Subunit of Mammalian SWI/SNF-Related Complexes Is Essential for Normal Cell Cycle Arrest. Cancer Res (2005) 65(20):9236–44. doi: 10.1158/0008-5472.CAN-05-1225
49. Span PN, Tjan-Heijnen VC, Manders P, Beex LV, Sweep CG. Cyclin-E Is a Strong Predictor of Endocrine Therapy Failure in Human Breast Cancer. Oncogene (2003) 22(31):4898–904. doi: 10.1038/sj.onc.1206818
50. Pérez-Tenorio G, Berglund F, Merca AE, B, Rutqvist L, Skoog L, et al. Cytoplasmic P21waf1/CIP1 Correlates With Akt Activation and Poor Response to Tamoxifen in Breast Cancer. Int J Oncol (2006) 28(5):1031–42. doi: 10.3892/ijo.28.5.1031
51. Gao JJ, Cheng J, Bloomquist E, Sanchez J, Wedam SB, Singh H, et al. CDK4/6 Inhibitor Treatment for Patients With Hormone Receptor-Positive, HER2-Negative, Advanced or Metastatic Breast Cancer: A US Food and Drug Administration Pooled Analysis. Lancet Oncol (2020) 21(2):250–60. doi: 10.1016/S1470-2045(19)30804-6
52. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant Plus Palbociclib Versus Fulvestrant Plus Placebo for Treatment of Hormone-Receptor-Positive, HER2-Negative Metastatic Breast Cancer That Progressed on Previous Endocrine Therapy (PALOMA-3): Final Analysis of the Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet Oncol (2016) 17(4):425–39. doi: 10.1016/S1470-2045(15)00613-0
53. Turner NC, Ro J, Andre F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med (2015) 373(3):209–19. doi: 10.1056/NEJMoa1505270
54. Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, et al. Structural Basis for Allosteric PARP-1 Retention on DNA Breaks. Science (2020) 368(6486):eaax6367. doi: 10.1126/science.aax6367
55. Curtin NJ, Szabo C. Poly(ADP-Ribose) Polymerase Inhibition: Past, Present and Future. Nat Rev Drug Discov (2020) 19(10):711–36. doi: 10.1038/s41573-020-0076-6
56. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res (2012) 72(21):5588–99. doi: 10.1158/0008-5472.CAN-12-2753
57. D'Andrea AD. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Repair (Amst) (2018) 71:172–6. doi: 10.1016/j.dnarep.2018.08.021
58. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients With BRCA1 or BRCA2 Mutations and Recurrent Ovarian Cancer: A Proof-of-Concept Trial. Lancet (2010) 376(9737):245–51. doi: 10.1016/S0140-6736(10)60893-8
59. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J Clin Oncol (2015) 33(3):244–50. doi: 10.1200/JCO.2014.56.2728
60. Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant Olaparib for Patients With BRCA1- or BRCA2-Mutated Breast Cancer. N Engl J Med (2021) 384(25):2394–405. doi: 10.1056/NEJMoa2105215
61. Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J Clin Oncol (2020) 38(36):4274–82. doi: 10.1200/JCO.20.02151
62. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov (2015) 5(7):752–67. doi: 10.1158/2159-8290.CD-14-0849
63. Hu HM, Zhao X, Kaushik S, Robillard L, Barthelet A, Lin KK, et al. A Quantitative Chemotherapy Genetic Interaction Map Reveals Factors Associated With PARP Inhibitor Resistance. Cell Rep (2018) 23(3):918–29. doi: 10.1016/j.celrep.2018.03.093
64. Keegan KS, Holtzman DA, Plug AW, Christenson ER, Hoekstra MF. The Atr and Atm Protein Kinases Associate With Different Sites Along Meiotically Pairing Chromosomes. Genes Dev (1996) 10(19):2423–37. doi: 10.1101/gad.10.19.2423
65. Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in Phosphorylation of Chk1 and Cell Cycle Regulation in Response to DNA Replication Blocks and UV-Damaged DNA in Xenopus Egg Extracts. Genes Dev (2000) 14(21):2745–56. doi: 10.1101/gad.842500
66. Liu Q, Guntuku S, Cui XS, Matsuoka S, Elledge SJ. Chk1 Is an Essential Kinase That Is Regulated by Atr and Required for the G2/M DNA Damage Checkpoint. Genes Dev (2000) 14(12):1448–59. doi: 10.1101/gad.14.12.1448
67. Shiloh Y, Ziv Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat Rev Mol Cell Biol (2013) 14(4):197–210. doi: 10.1038/nrm3546
68. Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J Clin Invest (2019) 129(3):1329–44. doi: 10.1172/JCI122622
69. Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat Commun (2016) 7:13837. doi: 10.1038/ncomms13837
70. Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int J Mol Sci (2016) 17(11):1849. doi: 10.3390/ijms17111849
71. Zeng L, Zhou MM. Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett (2002) 513(1):124–8. doi: 10.1016/S0014-5793(01)03309-9
72. Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol Cell Biol (2011) 31(13):2641–52. doi: 10.1128/MCB.01341-10
73. Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-Interacting Domain of BRD4 Inhibits HIV Transcription. Proc Natl Acad Sci USA (2007) 104(34):13690–5. doi: 10.1073/pnas.0705053104
74. Itzen F, Greifenberg AK, Bosken CA, Geyer M. Brd4 Activates P-TEFb for RNA Polymerase II CTD Phosphorylation. Nucleic Acids Res (2014) 42(12):7577–90. doi: 10.1093/nar/gku449
75. Shang E, Salazar G, Crowley TE, Xiang W, Lopez RA, Wang X, et al. Identification of Unique, Differentiation Stage-Specific Patterns of Expression of the Bromodomain-Containing Genes Brd2, Brd3, Brd4, and Brdt in the Mouse Testis. Gene Expression Patterns (2004) 4(5):513–9. doi: 10.1016/j.modgep.2004.03.002
76. Liu W, Stein P, Cheng X, Yang W, Shao NY, Morrisey EE, et al. BRD4 Regulates Nanog Expression in Mouse Embryonic Stem Cells and Preimplantation Embryos. Cell Death Differ (2014) 21(12):1950–60. doi: 10.1038/cdd.2014.124
77. Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and Early Postimplantation Defects in Mice Deficient for the Bromodomain-Containing Protein Brd4. Mol Cell Biol (2002) 22(11):3794–802. doi: 10.1128/MCB.22.11.3794-3802.2002
78. Crawford N, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, et al. Bromodomain 4 Activation Predicts Breast Cancer Survival. Proc Natl Acad Sci USA (2008) 105(17):6380–5. doi: 10.1073/pnas.0710331105
79. Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, et al. Deletion of the Proline-Rich Region of the Murine Metastasis Susceptibility Gene Brd4 Promotes Epithelial-to-Mesenchymal Transition- and Stem Cell-Like Conversion. Cancer Res (2011) 71(8):3121–31. doi: 10.1158/0008-5472.CAN-10-4417
80. Wu SY, Lee CF, Lai HT, Yu CT, Lee JE, Zuo H, et al. Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol Cell (2020) 78(6):1114–32.e10. doi: 10.1016/j.molcel.2020.04.034
81. Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. An Epigenomic Approach to Therapy for Tamoxifen-Resistant Breast Cancer. Cell Res (2014) 24(7):809–19. doi: 10.1038/cr.2014.71
82. Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep (2014) 8(2):460–9. doi: 10.1016/j.celrep.2014.06.016
83. Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell (2016) 164(1-2):293–309. doi: 10.1016/j.cell.2015.11.062
84. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, et al. Disrupting the Interaction of BRD4 With Diacetylated Twist Suppresses Tumorigenesis in Basal-Like Breast Cancer. Cancer Cell (2014) 25(2):210–25. doi: 10.1016/j.ccr.2014.01.028
85. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, et al. Response and Resistance to BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Nature (2016) 529(7586):413–7. doi: 10.1038/nature16508
86. Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 Regulates Breast Cancer Dissemination Through Jagged1/Notch1 Signaling. Cancer Res (2016) 76(22):6555–67. doi: 10.1158/0008-5472.CAN-16-0559
87. Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, et al. Spatially Constrained Tandem Bromodomain Inhibition Bolsters Sustained Repression of BRD4 Transcriptional Activity for TNBC Cell Growth. Proc Natl Acad Sci USA (2018) 115(31):7949–54. doi: 10.1073/pnas.1720000115
88. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for. Proc Natl Acad Sci USA (2001) 98(15):8554–9. doi: 10.1073/pnas.141230798
89. Maneiro MA, Forte N, Shchepinova MM, Kounde CS, Chudasama V, Baker JR, et al. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem Biol (2020) 15(6):1306–12. doi: 10.1021/acschembio.0c00285
90. Mio C, Gerratana L, Bolis M, Caponnetto F, Zanello A, Barbina M, et al. BET Proteins Regulate Homologous Recombination-Mediated DNA Repair: BRCAness and Implications for Cancer Therapy. Int J Cancer (2019) 144(4):755–66. doi: 10.1002/ijc.31898
91. Zhang B, Lyu J, Yang EJ, Liu Y, Wu C, Pardeshi L, et al. Class I Histone Deacetylase Inhibition Is Synthetic Lethal With BRCA1 Deficiency in Breast Cancer Cells. Acta Pharm Sin B (2020) 10(4):615–27. doi: 10.1016/j.apsb.2019.08.008
92. Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by Combining BET and HDAC Inhibitors in Breast Cancer Cells. Oncotarget (2015) 6(32):33623–35. doi: 10.18632/oncotarget.5601
93. Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, et al. Identification of a Cancer Stem Cell-Specific Function for the Histone Deacetylases, HDAC1 and HDAC7, in Breast and Ovarian Cancer. Oncogene (2017) 36(12):1707–20. doi: 10.1038/onc.2016.337
94. Gong C, Qu S, Lv XB, Liu B, Tan W, Nie Y, et al. BRMS1L Suppresses Breast Cancer Metastasis by Inducing Epigenetic Silence of FZD10. Nat Commun (2014) 5:5406. doi: 10.1038/ncomms6406
95. Muchardt C, Yaniv M. A Human Homologue of Saccharomyces Cerevisiae SNF2/SWI2 and Drosophila Brm Genes Potentiates Transcriptional Activation by the Glucocorticoid Receptor. EMBO J (1993) 12(11):4279–90. doi: 10.1002/j.1460-2075.1993.tb06112.x
96. Khavari PA, Peterson CL, Tamkun JW, Mendel DB, Crabtree GR. BRG1 Contains a Conserved Domain of the SWI2/SNF2 Family Necessary for Normal Mitotic Growth and Transcription. Nature (1993) 366(6451):170–4. doi: 10.1038/366170a0
97. Wang W, Côté J, Xue Y, Zhou S, Crabtree GR. Purification and Biochemical Heterogeneity of the Mammalian SWI-SNF Complex. EMBO J (1996) 15(19):5370. doi: 10.1002/j.1460-2075.1996.tb00921.x
98. Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, et al. PBAF Chromatin-Remodeling Complex Requires a Novel Specificity Subunit, BAF200, to Regulate Expression of Selective Interferon-Responsive Genes. Genes Dev (2005) 19(14):1662–7. doi: 10.1101/gad.1323805
99. Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, et al. ARID1B Is a Specific Vulnerability in ARID1A-Mutant Cancers. Nat Med (2014) 20(3):251–4. doi: 10.1038/nm.3480
100. Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, Stuschke M. Targeting ARID1A-Mutant Colorectal Cancer: Depletion of ARID1B Increases Radiosensitivity and Modulates DNA Damage Response. Sci Rep (2019) 9(1):18207. doi: 10.1038/s41598-019-54757-z
101. Sato E, Nakayama K, Razia S, Nakamura K, Ishikawa M, Minamoto T, et al. ARID1B as a Potential Therapeutic Target for ARID1A-Mutant Ovarian Clear Cell Carcinoma. Int J Mol Sci (2018) 19(6):1710. doi: 10.3390/ijms19061710
102. Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin Accessibility Underlies Synthetic Lethality of SWI/SNF Subunits in ARID1A-Mutant Cancers. Elife (2017) 6:e30506. doi: 10.7554/eLife.30506
103. Shao F, Guo T, Chua PJ, Tang L, Thike AA, Tan PH, et al. Clinicopathological Significance of ARID1B in Breast Invasive Ductal Carcinoma. Histopathology (2015) 67(5):709–18. doi: 10.1111/his.12701
104. Cui Y, Bai X, Niu M, Qin Y, Zhang X, Pang D. Upregulated Expression of AT-Rich Interactive Domain-Containing Protein 1B Predicts Poor Prognosis in Patients With Triple-Negative Breast Cancer. Oncol Lett (2019) 17(3):3289–95. doi: 10.3892/ol.2019.9961
105. Mashtalir N, D'Avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell (2018) 175(5):1272–88 e20. doi: 10.1016/j.cell.2018.09.032
106. Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted Disruption of the EZH2-EED Complex Inhibits EZH2-Dependent Cancer. Nat Chem Biol (2013) 9(10):643–50. doi: 10.1038/nchembio.1331
107. Okamura R, Kato S, Lee S, Jimenez RE, Sicklick JK, Kurzrock R. ARID1A Alterations Function as a Biomarker for Longer Progression-Free Survival After Anti-PD-1/PD-L1 Immunotherapy. J Immunother Cancer (2020) 8(1):e000438. doi: 10.1136/jitc-2019-000438
108. Goswami S, Chen Y, Anandhan S, Szabo PM, Sharma P. ARID1A Mutation Plus CXCL13 Expression Act as Combinatorial Biomarkers to Predict Responses to Immune Checkpoint Therapy in mUCC. Sci Trans Med (2020) 12(548):eabc4220. doi: 10.1126/scitranslmed.abc4220
109. Sun D, Tian L, Zhu Y, Wo Y, Liu Q, Liu S, et al. Subunits of ARID1 Serve as Novel Biomarkers for the Sensitivity to Immune Checkpoint Inhibitors and Prognosis of Advanced Non-Small Cell Lung Cancer. Mol Med (2020) 26(1):78. doi: 10.1186/s10020-020-00208-9
110. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat Med (2018) 24(5):556–62. doi: 10.1038/s41591-018-0012-z
111. Montal R, Sia D, Montironi C, Leow WQ, Esteban-Fabro R, Pinyol R, et al. Molecular Classification and Therapeutic Targets in Extrahepatic Cholangiocarcinoma. J Hepatol (2020) 73(2):315–27. doi: 10.1016/j.jhep.2020.03.008
112. Mehrvarz Sarshekeh A, Alshenaifi J, Roszik J, Manyam GC, Advani SM, Katkhuda R, et al. ARID1A Mutation May Define an Immunologically Active Subgroup in Patients With Microsatellite Stable Colorectal Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(6):1663–70. doi: 10.1158/1078-0432.CCR-20-2404
113. Li L, Li M, Jiang Z, Wang X. ARID1A Mutations Are Associated With Increased Immune Activity in Gastrointestinal Cancer. CellsV (2019) 8(7):678. doi: 10.3390/cells8070678
114. Bao X, Zhang H, Wu W, Cheng S, Dai X, Zhu X, et al. Analysis of the Molecular Nature Associated With Microsatellite Status in Colon Cancer Identifies Clinical Implications for Immunotherapy. J Immunother Cancer (2020) 8(2):e001437. doi: 10.1136/jitc-2020-001437
115. Wang L, Yang L, Wang C, Zhao W, Ju Z, Zhang W, et al. Inhibition of the ATM/Chk2 Axis Promotes cGAS/STING Signaling in ARID1A-Deficient Tumors. J Clin Invest (2020) 130(11):5951–66. doi: 10.1172/JCI130445
116. Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S, et al. HDAC6 Inhibition Synergizes With Anti-PD-L1 Therapy in ARID1A-Inactivated Ovarian Cancer. Cancer Res (2019) 79(21):5482–9. doi: 10.1158/0008-5472.CAN-19-1302
117. Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell (2019) 35(2):177–90 e8. doi: 10.1016/j.ccell.2018.12.009
118. Buser K, Joncourt F, Altermatt HJ, Bacchi M, Oberli A, Cerny T. Breast Cancer: Pretreatment Drug Resistance Parameters (GSH-System, ATase, P-Glycoprotein) in Tumor Tissue and Their Correlation With Clinical and Prognostic Characteristics. Ann Oncol Off J Eur Soc Med Oncol (1997) 8(4):335–41. doi: 10.1023/A:1008202723066
119. Wang F, Huang Q, Wang Y, Shi L, Shen Y, Guo S. NIR-Light and GSH Activated Cytosolic P65-shRNA Delivery for Precise Treatment of Metastatic Cancer. J Control Release (2018) 288:126–35. doi: 10.1016/j.jconrel.2018.09.002
120. Blazquez R, Rietkotter E, Wenske B, Wlochowitz D, Sparrer D, Vollmer E, et al. LEF1 Supports Metastatic Brain Colonization by Regulating Glutathione Metabolism and Increasing ROS Resistance in Breast Cancer. Int J Cancer (2020) 146(11):3170–83. doi: 10.1002/ijc.32742
121. Zhao YP, Ye WL, Liu DZ, Cui H, Cheng Y, Liu M, et al. Redox and pH Dual Sensitive Bone Targeting Nanoparticles to Treat Breast Cancer Bone Metastases and Inhibit Bone Resorption. Nanoscale (2017) 9(19):6264–77. doi: 10.1039/C7NR00962C
122. Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic Lethality by Targeting EZH2 Methyltransferase Activity in ARID1A-Mutated Cancers. Nat Med (2015) 21(3):231–8. doi: 10.1038/nm.3799
123. Czermin B, Melfi R, Mccabe D, Seitz V, Imhof A, Pirrotta V. Drosophila Enhancer of Zeste/ESC Complexes Have a Histone H3 Methyltransferase Activity That Marks Chromosomal Polycomb Sites. Cell (2002) 111(2):185–96. doi: 10.1016/S0092-8674(02)00975-3
124. Müller J, Hart CM, Francis NJ, Vargas ML, Simon JA. Histone Methyltransferase Activity of a Drosophila Polycomb Group Repressor Complex. Cell (2002) 111(2):197–208. doi: 10.1016/S0092-8674(02)00976-5
125. Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL, et al. EZH2 Contributes to the Response to PARP Inhibitors Through Its PARP-Mediated Poly-ADP Ribosylation in Breast Cancer. Oncogene (2018) 37(2):208–17. doi: 10.1038/onc.2017.311
126. Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. Epigenetic Driver Mutations in ARID1A Shape Cancer Immune Phenotype and Immunotherapy. J Clin Invest (2020) 130(5):2712–26. doi: 10.1172/JCI134402
127. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 Promotes Expansion of Breast Tumor Initiating Cells Through Activation of RAF1-Beta-Catenin Signaling. Cancer Cell (2011) 19(1):86–100. doi: 10.1016/j.ccr.2010.10.035
128. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 Is a Marker of Aggressive Breast Cancer and Promotes Neoplastic Transformation of Breast Epithelial Cells. Proc Natl Acad Sci (2003) 81(1):77–90. doi: 10.1073/pnas.1933744100
129. Jeong GY, Park MK, Choi HJ, An HW, Park YU, Choi HJ, et al. NSD3-Induced Methylation of H3K36 Activates NOTCH Signaling to Drive Breast Tumor Initiation and Metastatic Progression. Cancer Res (2021) 81(1):77–90. doi: 10.1158/0008-5472.CAN-20-0360
130. Shan L, Zhou X, Liu X, Wang Y, Su D, Hou Y, et al. FOXK2 Elicits Massive Transcription Repression and Suppresses the Hypoxic Response and Breast Cancer Carcinogenesis. Cancer Cell (2016) 30(5):708–22. doi: 10.1016/j.ccell.2016.09.010
131. Chu W, Zhang X, Qi L, Fu Y, Wang P, Zhao W, et al. The EZH2-PHACTR2-AS1-Ribosome Axis Induces Genomic Instability and Promotes Growth and Metastasis in Breast Cancer. Cancer Res (2020) 80(13):2737–50. doi: 10.1158/0008-5472.CAN-19-3326
132. Chang PH, Chen MC, Tsai YP, Tan GYT, Hsu PH, Jeng YM, et al. Interplay Between Desmoglein2 and Hypoxia Controls Metastasis in Breast Cancer. Proc Natl Acad Sci USA (2021) 118(3):e2014408118. doi: 10.1073/pnas.2014408118
133. Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, et al. Polycomb Protein EZH2 Regulates Tumor Invasion via the Transcriptional Repression of the Metastasis Suppressor RKIP in Breast and Prostate Cancer. Cancer Res (2012) 72(12):3091–104. doi: 10.1158/0008-5472.CAN-11-3546
134. Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, et al. EZH2 Expands Breast Stem Cells Through Activation of NOTCH1 Signaling. Proc Natl Acad Sci USA (2014) 111(8):3098–103. doi: 10.1073/pnas.1308953111
135. Bao Y, Oguz G, Lee WC, Lee PL, Ghosh K, Li J, et al. EZH2-Mediated PP2A Inactivation Confers Resistance to HER2-Targeted Breast Cancer Therapy. Nat Commun (2020) 11(1):5878. doi: 10.1038/s41467-020-19704-x
136. Deblois G, Tonekaboni SAM, Grillo G, Martinez C, Kao YI, Tai F, et al. Epigenetic Switch-Induced Viral Mimicry Evasion in Chemotherapy-Resistant Breast Cancer. Cancer Discov (2020) 10(9):1312–29. doi: 10.1158/2159-8290.CD-19-1493
137. Reijm EA, Timmermans AM, Look MP, Meijer-van Gelder ME, Stobbe CK, van Deurzen CHM, et al. High Protein Expression of EZH2 Is Related to Unfavorable Outcome to Tamoxifen in Metastatic Breast Cancer. Ann Oncol Off J Eur Soc Med Oncol (2014) 25(11):2185–90. doi: 10.1093/annonc/mdu391
138. Wu Y, Zhang Z, Cenciarini ME, Proietti CJ, Amasino M, Hong T, et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERalpha-GREB1 Transcriptional Axis. Cancer Res (2018) 78(3):671–84. doi: 10.1158/0008-5472.CAN-17-1327
139. Pathiraja TN, Nayak SR, Xi Y, Jiang S, Garee JP, Edwards DP, et al. Epigenetic Reprogramming of HOXC10 in Endocrine-Resistant Breast Cancer. Sci Trans Med (2014) 6(229):229ra41. doi: 10.1126/scitranslmed.3008326
140. Bado IL, Zhang W, Hu J, Xu Z, Wang H, Sarkar P, et al. The Bone Microenvironment Increases Phenotypic Plasticity of ER(+) Breast Cancer Cells. Dev Cell (2021) 56(8):1100–17.e9. doi: 10.1016/j.devcel.2021.03.008
141. Hirukawa A, Smith HW, Zuo D, Dufour CR, Savage P, Bertos N, et al. Targeting EZH2 Reactivates a Breast Cancer Subtype-Specific Anti-Metastatic Transcriptional Program. Nat Commun (2018) 9(1):2547. doi: 10.1038/s41467-018-04864-8
142. Fujii S, Tokita K, Wada N, Ito K, Yamauchi C, Ito Y, et al. MEK-ERK Pathway Regulates EZH2 Overexpression in Association With Aggressive Breast Cancer Subtypes. Oncogene (2011) 30(39):4118–28. doi: 10.1038/onc.2011.118
143. Li Z, Hou P, Fan D, Dong M, Ma M, Li H, et al. The Degradation of EZH2 Mediated by lncRNA ANCR Attenuated the Invasion and Metastasis of Breast Cancer. Cell Death Differ (2017) 24(1):59–71. doi: 10.1038/cdd.2016.95
144. Zeng Y, Qiu R, Yang Y, Gao T, Zheng Y, Huang W, et al. Regulation of EZH2 by SMYD2-Mediated Lysine Methylation Is Implicated in Tumorigenesis. Cell Rep (2019) 29(6):1482–98 e4. doi: 10.1016/j.celrep.2019.10.004
145. Nie L, Wei Y, Zhang F, Hsu YH, Chan LC, Xia W, et al. CDK2-Mediated Site-Specific Phosphorylation of EZH2 Drives and Maintains Triple-Negative Breast Cancer. Nat Commun (2019) 10(1):5114. doi: 10.1038/s41467-019-13105-5
146. Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol Cell (2018) 69(2):279–91.e5. doi: 10.1016/j.molcel.2017.12.024
147. Li Z, Wang D, Lu J, Huang B, Wang Y, Dong M, et al. Methylation of EZH2 by PRMT1 Regulates Its Stability and Promotes Breast Cancer Metastasis. Cell Death Differ (2020) 27(12):3226–42. doi: 10.1038/s41418-020-00615-9
148. Anwar T, Arellano-Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, et al. P38-Mediated Phosphorylation at T367 Induces EZH2 Cytoplasmic Localization to Promote Breast Cancer Metastasis. Nat Commun (2018) 9(1):2801. doi: 10.1038/s41467-018-05078-8
149. Shu S, Wu HJ, Ge JY, Zeid R, Harris IS, Jovanovic B, et al. Synthetic Lethal and Resistance Interactions With BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol Cell (2020) 78(6):1096–113.e8. doi: 10.1016/j.molcel.2020.04.027
150. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
151. Jing X, Shao S, Zhang Y, Luo A, Zhao L, Zhang L, et al. BRD4 Inhibition Suppresses PD-L1 Expression in Triple-Negative Breast Cancer. Exp Cell Res (2020) 392(2):112034. doi: 10.1016/j.yexcr.2020.112034
152. Xu Y, Wu Y, Zhang S, Ma P, Jin X, Wang Z, et al. A Tumor-Specific Super-Enhancer Drives Immune Evasion by Guiding Synchronous Expression of PD-L1 and PD-L2. Cell Rep (2019) 29(11):3435–47.e4. doi: 10.1016/j.celrep.2019.10.093
Keywords: ARID1A, metastatic breast cancer, SWI/SNF complex, endocrine resistance, synthetic lethality, therapeutic targets
Citation: Cheng X, Zhao J-X, Dong F and Cao X-C (2021) ARID1A Mutation in Metastatic Breast Cancer: A Potential Therapeutic Target. Front. Oncol. 11:759577. doi: 10.3389/fonc.2021.759577
Received: 16 August 2021; Accepted: 15 October 2021;
Published: 04 November 2021.
Edited by:
San-Gang Wu, First Affiliated Hospital of Xiamen University, ChinaReviewed by:
Svasti Haricharan, Sanford Burnham Prebys Medical Discovery Institute, United StatesRaksha Bhat, University of Houston, United States
Copyright © 2021 Cheng, Zhao, Dong and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Dong, ZG9uZ2ZlbmdAdG11LmVkdS5jbg==; Xu-Chen Cao, Y2FveHVjaGVuQHRtdS5lZHUuY24=
†These authors have contributed equally to this work