 Jasmeet Kaur
Jasmeet Kaur Shalmoli Bhattacharyya
Shalmoli Bhattacharyya- Department of Biophysics, Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, India
The subpopulation of cancer stem cells (CSCs) within tumor bulk are known for tumor recurrence and metastasis. CSCs show intrinsic resistance to conventional therapies and phenotypic plasticity within the tumor, which make these a difficult target for conventional therapies. CSCs have different metabolic phenotypes based on their needs as compared to the bulk cancer cells. CSCs show metabolic plasticity and constantly alter their metabolic state between glycolysis and oxidative metabolism (OXPHOS) to adapt to scarcity of nutrients and therapeutic stress. The metabolic characteristics of CSCs are distinct compared to non-CSCs and thus provide an opportunity to devise more effective strategies to target CSCs. Mechanism for metabolic switch in CSCs is still unravelled, however existing evidence suggests that tumor microenvironment affects the metabolic phenotype of cancer cells. Understanding CSCs metabolism may help in discovering new and effective clinical targets to prevent cancer relapse and metastasis. This review summarises the current knowledge of CSCs metabolism and highlights the potential targeted treatment strategies.
Introduction
Cancer causes significant deaths worldwide, despite major innovations in treatment therapy strategies, radiation- and chemo-therapy and drug delivery technologies. A major contributor to the cancer treatment-associated toxicities and resistance (1–3), is their inability to eradicate subset of cancer stem cells (CSCs) which drive tumour growth and heterogeneity. CSCs presence makes tumors resistant to conventional therapies (4). Density of CSCs is a proven prognostic marker in various cancers (5, 6), thus targeting CSCs is an effective way for treating cancer.
CSCs self-renewal and asymmetric division capacity help tumors to regenerate and propagate post-treatment. CSCs populations provide high radio- and chemo-resistance due to efficient DNA repair and cellular redox homeostasis, protective tumor microenvironment, escape from immune response and unique metabolic phenotype (7–10). CSCs use metabolic reprogramming to escape immune system (11) and grant them plasticity (12). Metabolic reprogramming induce M2 phenotype in tumor-associated macrophages (TAMs) (13, 14) and glycolysis induce IL-6 secretion in M2 macrophages (15). Secreted IL-6 promotes CSC phenotype in cancer cells (16) via activation of STAT3/NFκB signaling pathways (17). CSCs in turn induce M2 phenotype in TAMs to confer drug resistance and tumorigenicity in CSCs by blocking the anti-tumor CD8+ response during chemotherapy (18).
Till date the origin of CSCs remains elusive. Two models are postulated to explain the genetic and functional heterogeneity of cancer in a single patient: the clonal evolution model and the cancer stem cell (CSC) hypothesis (19). The clonal evolution model suggests that multiple stepwise oncogenic mutations in somatic cells leads to tumor formation and natural selection favors the tumor cells with aggressive phenotype (20, 21). The CSC hypothesis suggests that metabolic events occurring in cancer epithelial cells may generate CSCs (Figure 1). Altered metabolic events in cancer cells may affect chromatin organization and activate epigenetic program (22) which may further fuel metabolic-reprogramming of CSCs. Two proposed models explain how metabolic alterations could affect epigenetics (22). In the first model, metabolism reprogramming facilitates differentiation of one cell type to another by altering chromatin modifications without affecting the epigenomic landscape. The second model proposes that altered metabolism induces new potential cell types via creation of novel stable epigenetic states, thus reshaping the entire epigenomic landscape. In this model, altered metabolism remodels chromatin by either inducing gene expression or affecting availability of substrates and cofactors for chromatin-modifying enzymes. In either case, the end-result is a novel cell state that is irreversible as epigenomic landscape has changed.
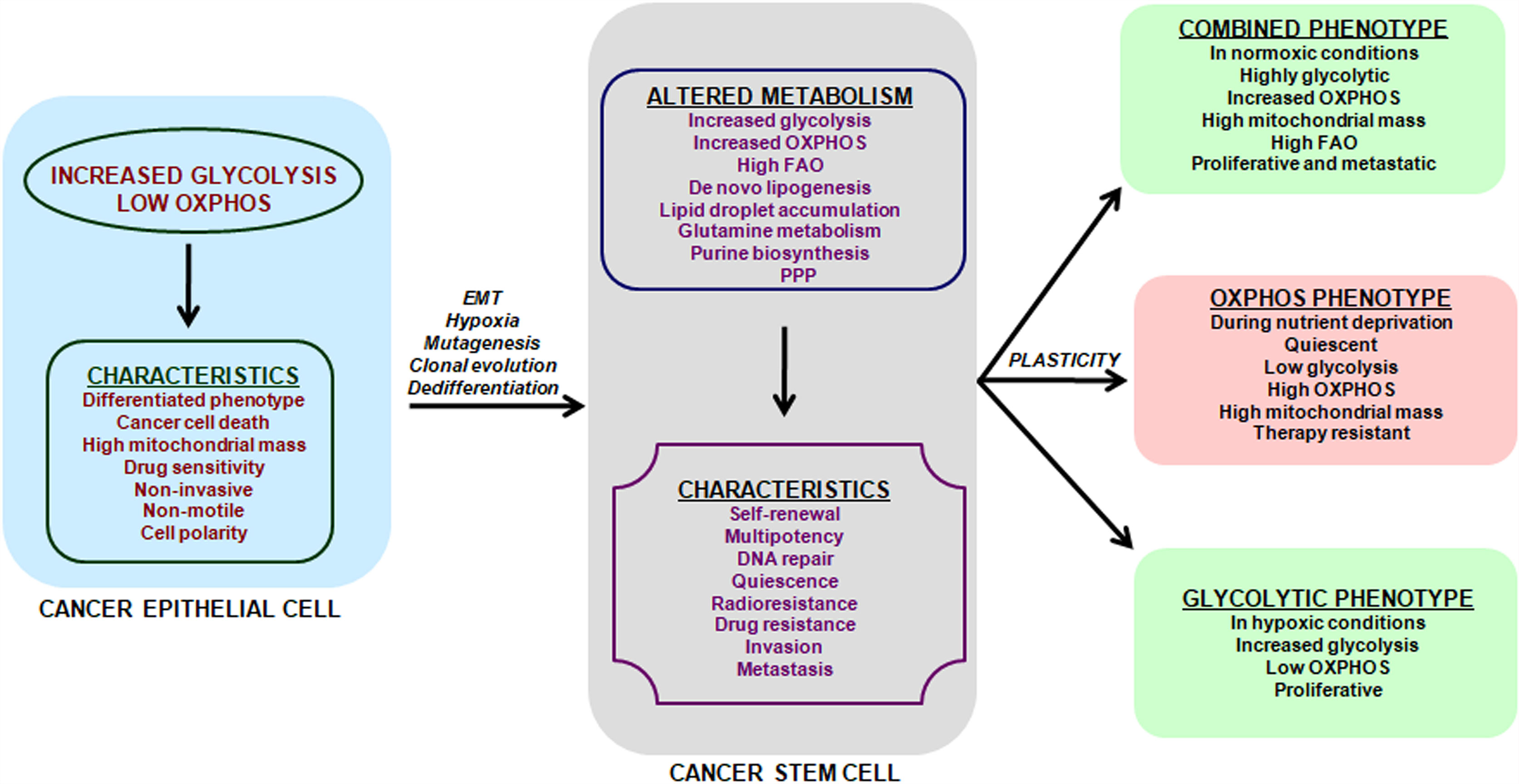
Figure 1 Metabolic features and plasticity of CSCs. Cancer is a heterogeneous disease with multiple sub-populations of cells and CSCs form the self-renewing and tumorigenic core in a tumor. Cancer cells have a predominant glycolytic phenotype and use aerobic glycolysis for tumor growth. This altered metabolism in cancer cells may trigger EMT, hypoxia, cell de-differentiation, mutagenesis and clonal evolution for acquisition of CSCs phenotype. The metabolic alterations in CSCs promote cell-renewal, immune system escape and invasive and metastatic potential. CSCs unlike cancer cells can use glycolysis, OXPHOS or both, depending on their oncogenic background and bio-energetic needs. This freedom of metabolic choice makes CSCs metabolically plastic and they easily shuffle between metabolic phenotypes, based on their state i.e. proliferative or quiescent. EMT, epithelial to mesenchymal transition; FAO, fatty acid oxidation; PPP, Pentose phosphate pathway.
Metabolic characterization of CSCs has been a challenging task, as CSCs lack a common metabolic phenotype across cancer types. CSCs metabolic pattern differ from adult stem cells (SCs) and use either glycolysis or OXPHOS (Figure 1) triggering cellular plasticity in CSCs (23). Thus, an understanding of CSCs metabolic features will help target CSCs specifically and prevent cancer progression. Current review summarizes the various metabolic features of CSCs along with therapeutic interventions that can be adopted to target key energy processes in CSCs. Targeting the metabolic flexibility in CSCs can emerge as an effective strategy for preventing or minimizing disease progression and recurrence.
Metabolic Features of CSCs
Growth factors, nutrients and oxygen in the tumor microenvironment provide necessary energy sources and growth signals for CSCs generation and proliferation. Recently, metabolism has been identified as a major component in CSCs biology, as oncogenic alterations has been observed to cause metabolite-driven dissemination of CSCs (19). Multipotent SCs use glycolysis (24, 25), have fewer mitochondria and produce less reactive oxygen species (ROS) (26, 27). Higher ROS levels cause SCs dysfunction (28–30) and shift to OXPHOS with increased ROS production leads to differentiated SCs progeny.
Glycolysis
CSCs were hypothesized to be glycolytic (31), as SCs rely primarily on glycolysis to generate energy (32). However, CSCs are more glycolytic than SCs in various cancers (33–35). Upregulation of glycolytic genes precede pluripotency markers expression, thus switching from OXPHOS to glycolysis promotes stemness in CSCs and is not an outcome of attaining pluripotency (36). CSCs’ glucose uptake and hence lactate and ATP production is higher (37) and glycolysis inhibition or glucose starvation cause CSCs’ death (19, 38). Glycolytic CSCs are shown in CD133+ liver carcinoma cells (39), osteosarcoma-initiating cells (40), breast cells (41) and glioblastoma cells (35).
Glycolysis is preferred in breast CD44+CD24lowEPCAM+ CSCs, sphere-forming radio-resistant nasopharyngeal carcinoma cells (42) and CD133+CD49f+ tumor initiating cells (TICs) in hepatocellular carcinoma (43). Elevated expression of oncogenic MYC drove stemness in these cancer types (44) and MYC-driven glycolytic program determined tumorigenic potential (45), thus making MYC a likely candidate linking glycolysis and stemness.
Lactate supports stemness by upregulation of transcription factor SP1 and increases aggressiveness, invasiveness and immune-suppression through sterol regulatory element-binding protein 1 (SREBP1) (46–51). Hypoxia-inducible factor-1 (HIF-1) promotes glycolysis in CSCs and declines OXPHOS and TCA cycle (52). HIF-1 reduces ROS production and upregulates glucose transporters (GLUT) and hexokinase (HK2) expression, pyruvate kinase (PK) activity and LDHA levels and downregulates pyruvate dehydrogenase (PDH) levels (52). HIF-1 promotes self-renewal and pluripotency in various cancers making them treatment resistant (19, 53, 54).
NANOG-expressing hepatocellular CSCs have higher glycolysis and fatty acid oxidation (FAO) rates, and lower OXPHOS and ROS generation (43). CSCs secretome have enriched levels of glycolytic and antioxidant pathways proteins and secreted high levels of ALDH than differentiated cells from colorectal tumors (24). ALDH detoxifies anticancer drugs such as maphosphamide and CSCs secreting ALDH promoted self-preservation and protected nearby differentiated mature cancer cells, leading to therapy resistance (24). Ovarian CSCs with glycolysis enrichment, de novo fatty acid synthesis, and decreased mitochondrial respiration and anaplerotic flux, led to aggressive tumors with therapy resistance to cisplatin in comparison to mature cancer cells (34).
Mitochondrial Respiration
As an energy source, OXPHOS is more efficient than glycolysis, but has a slower rate to produce energy. Quiescent or slow-cycling tumor-initiating CSCs prefer OXPHOS metabolism over glycolysis (Figure 1), consume less glucose, have lower lactate and higher ATP levels (55–57). OXPHOS-dependent CSCs with low glycolytic reserves are shown in acute myeloid leukemia, CD133+ glioblastoma, melanoma, pancreatic and ovarian cancer (58–63). In breast CSCs, elevated OXPHOS levels trigger chemotherapeutic resistance through synergistic action of MYC and MCL1 (64).
CSCs using OXPHOS have higher mitochondrial mass with increase in membrane potential and rates of oxygen consumption (62, 65, 66). Mitochondrial mass is a vital metabolic biomarker of CSCs (65, 67). Tumor cells without mitochondrial DNA (mtDNA) grew slowly and acquisition of mtDNA from host cells led to tumor-initiation and drug resistance in these tumor cells (68), suggesting mitochondrial function as a target for CSCs treatment. Master mitochondrial biogenesis regulator, peroxisome proliferator-activator 1 alpha (PGC1α) maintained stemness characteristics (69) in breast cancer (70) and pancreatic CD133+ CSCs (66) and increased chemoresistance in CSCs (64, 71–73). NANOG is a pluripotency gene that supports tumorigenesis through OXPHOS and fatty acid metabolism (43). Some breast CSCs show elevated glucose consumption and ATP production, higher mitochondrial activity but lower lactate levels, suggesting that OXPHOS and glycolysis may not be mutually exclusive to CSCs (62).
Glutamine Metabolism
Glycolysis and OXPHOS may not completely support CSCs metabolism, thus glutamine compensates for glucose shortage (74, 75). Although a non-essential amino acid, glutamine becomes essential for cancer cells (76) and CSCs from lung, pancreatic and ovarian cancer have shown glutamine dependence (77, 78). CSCs rely on glutamine for carbon and amino-nitrogen for protein, nucleotide and lipids biosynthesis (79). Glutamine metabolism is rewired by mutations in mitochondrial DNA (mtDNA) (80) and oncogenic alterations in KRAS (81, 82) and c-Myc (83) in tumor cells. Glutamine metabolism in c-Myc-over-expressing cells suggests a pluripotency gene profile dependence on glutamine (84). In pancreatic CSCs, glutamine unavailability reduced stemness characteristics and increased radiation therapy sensitivity (77). L-DON (a glutamine analog) inhibited glucose metabolism and prevented systemic metastasis to liver, lung and kidney in mice (85).
Lipid Metabolism
Cells use an anabolic process of fatty acid synthesis (FAS) to derive energy from fatty acid metabolism for cell growth and proliferation, and a catabolic process of fatty acid oxidation (FAO) for NADH and ATP production (86). CSCs are extremely reliant on de novo lipid biosynthesis, lipid oxidation and lipid metabolizing enzymes (87, 88).
Lipid accumulation correlates with tumor stage in mice with prostate cancer (89). De novo lipid synthesis associated transcription factor, SREBP-2 activated c-Myc transcription in prostate cancer, enhancing CSCs properties (90). Increased lipid droplet content in colorectal CSCs (91), upregulated lipogenesis in glioma (92) and pancreatic cancer CSCs (93), and increased fatty acid oxidation (FAO) in breast cancer (94) and leukemic cells (95) maintained stemness. High levels of unsaturated lipids in ovarian CSCs promotes cancer stemness and tumor initiation capacity (96).
CSCs use mitochondrial FAO for ATP and NADPH generation to survive loss of matrix attachment (97, 98). Pluripotency factor NANOG-induced FAO genes expression promoted chemoresistance in TICs in hepatocellular carcinoma (43). Hematopoietic stem cells (HSCs) and leukemia-initiating cells depend on FAO for self-renewal (95, 99) and thus FAO inhibition is a potential pharmacological opportunity to target CSCs (98). Lipid metabolism enzymes, ACSVL3 (acyl-CoA synthetase very-long-chain 3) and ALOX5 (arachidonic acid 5-lipoxygenase) promoted glioblastoma CSCs self-renewal and tumorigenicity (100, 101).
Other Metabolic Features
Mutations in isocitrate dehydrogenase (IDH1 and 2) promote stem-ness in leukemia by aberrant conversion of α-ketoglutarate (αKG) to an analogue named 2-hydroxyglutarate (2-HG). Intracellular accumulation of 2-HG promoted a pro-leukemic phenotype by inhibiting tet methylcytosine dioxygenase 2 (TET2) function, increased self-renewal and impaired differentiation of hematopoietic SCs (102–104).
Elevated purine synthesis promoted stemness in brain tumor initiating cells (BTICs) and correlated with significantly poorer overall survival in glioblastoma patients (105). MYC regulates purine synthesis enzymes and its liaison with de novo purine synthesis mediated selective dependence of BTICs on glucose-sustained anabolic metabolism. Inhibition of purine synthesis prevented BTICs growth by inhibiting their self-renewal capacity, but differentiated glioma cells remained unaffected (105). Thus frailty of purine synthesis in CSCs makes it a potential therapeutic target,
Lysine catabolism promoted self-renewal of CD110+ colorectal cancer tumor-initiating cells (TICs) by generating acetyl-CoA. Acetyl-CoA triggered LDL receptor-related protein 6 (LRP6) acetylation and phosphorylation, and finally activation of WNT signaling (106). Lysine catabolism promoted drug-resistance and metastasis to liver in CD110+ TICs by glutamate and glutathione synthesis, which modulated the redox status (106). Collectively, CSCs use an array of metabolism alterations to fuel their self-renewal, thus making these metabolic dependencies open to targeted therapies.
Clinical Implications
CSCs have both distinct and flexible metabolic phenotypes between glycolysis and OXPHOS-dependent. Despite limited clinical evidence, targeting CSCs through selective metabolic modulation is an effective and promising avenue for cancer treatment. In our view, synergistic treatments using a standard cytotoxic agent and a metabolic-based therapy will improve eradication of CSCs. Table 1 lists the available metabolic targeting agents undergoing clinical trials in various cancers.
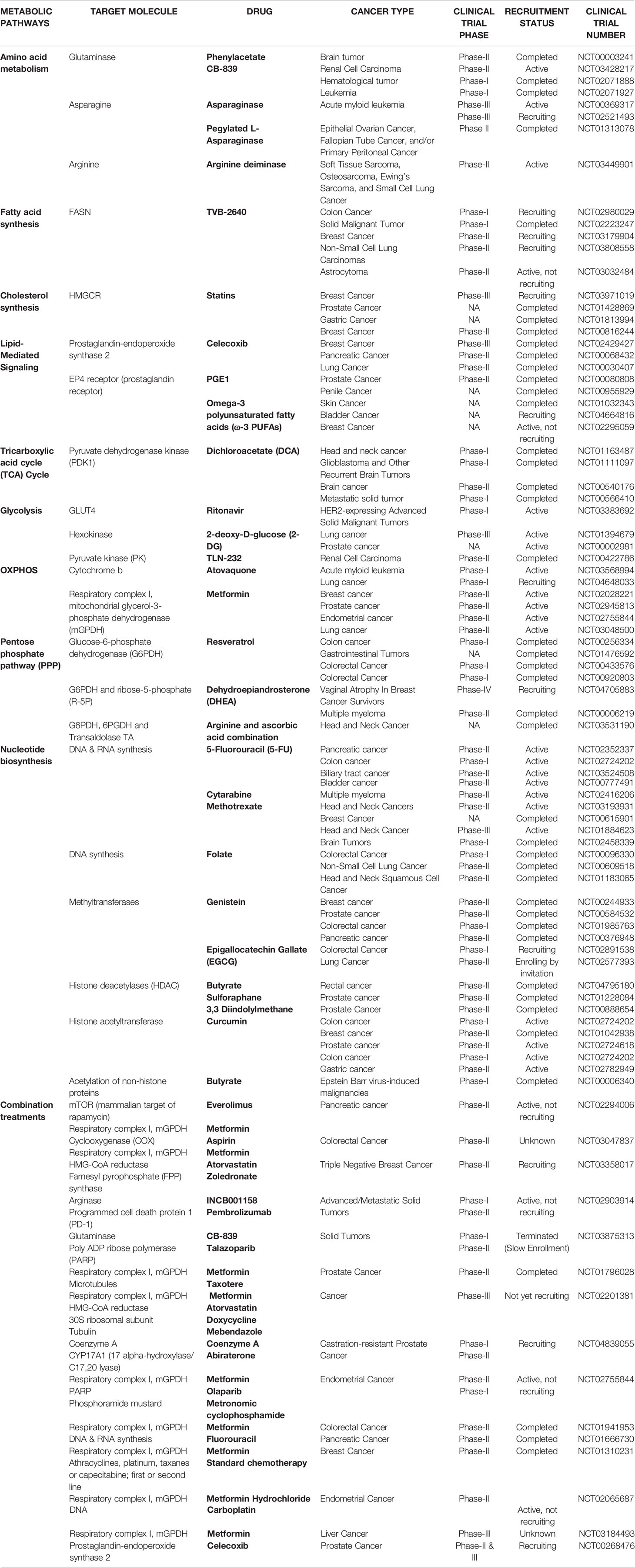
Table 1 Clinical trial status of drugs targeting metabolic pathways.
Targeting Glycolysis
Glycolytic CSCs can be targeted for glycolytic enzymes (hexokinase (HK), phosphoglycerate kinase, pyruvate kinase) and glucose transporters (GLUT1-4). Direct inhibition of GLUTs results in a total disruption of glucose uptake and hence energy metabolism, and GLUT inhibitors such as phloretin, fasentin and WZB117 have shown anticancer effects in preclinical models (107–110). However, ubiquitous expression of GLUTs even in normal cells challenges the explicit inhibition of CSCs glucose uptake and leads to side-effects.
HK enzymes catalyze the first step of glycolysis and their inhibition via 2-deoxy-D-glucose (2-DG), benserazide, lonidamine (LN) and genistein-27 (GEN-27) are being used for cancer treatment (111–114). 2-DG is a synthetic analog of glucose that competitively inhibits glucose transport (115) and can be used in combination with cisplatin/docetaxel as an anti-cancer agent (116, 117). 2-DG inhibited glycolysis and CSCs phenotype in triple-negative breast cancer cells (118) and 2-DG with biguanides (such as 3-bromopyruvate, 3-BP) prevented colon cancer cell proliferation (119).
Pyruvate is converted into mitochondrial acetyl-CoA in the cytosol and is negatively regulated by pyruvate dehydrogenase kinase (PDK) enzyme. This shifts cellular metabolism from OXPHOS to glycolysis and thus targeting PDK can inhibit cellular proliferation of CSCs. Dichloroacetate (DCA) activates mitochondrial pyruvate dehydrogenase (PDH) by inhibiting PDK (120), is fairly well-tolerated with fewer side effects and is being tested in several anticancer clinical trials (121, 122).
CSCs can oscillate between metabolic phenotypes during oxygen deprivation and glucose starvation, and thus targeting mechanisms underlying these metabolic adaptations can effectively eliminate CSCs. Hypoxia-inducible factors (HIFs) promote tumor progression in response to localized hypoxia by switching to glycolysis from OXPHOS, activating Notch pathway and expression of Oct4 transcription factor (123, 124). This suggests HIF-1α’s role in self-renewal and multipotency and targeting HIFs can be a prospective treatment for CSCs. Metformin, although an antidiabetic drug, attenuated glycolysis flux in hepatocellular carcinoma cells (125) and improved radiotherapy response in prostate and colon cancer tumor xenograft models (126). Epigallocatechin gallate (EGCG) is an inhibitor of glycolysis and its co-treatment with gemcitabine enhanced pancreatic cancer cell death both in vitro and in xenografts (127).
Targeting Mitochondrial Respiration
Several OXPHOS-targeting pharmacological agents are being explored in clinical trials for cancer treatment (Table 1) and have potential to target CSCs. OXPHOS inhibition overcame drug resistance in slow-cycling melanoma cells and mitochondria-targeted antibiotics prevented sphere formation and tumorigenesis in CSCs (61, 128). Metformin inhibited mitochondrial electron transport chain complex I and diminished OXPHOS (129). Metformin caused energy emergency and hence apoptosis in OXPHOS-dependent pancreatic cancer stem cells (CSCs), but spared their glycolytic differentiated progenies (66). Diabetic patients receiving metformin have a lower mortality rate from cancer and hence a better prognosis (130, 131). Phenformin, a biguanide formerly used in diabetes and a mitochondrial inhibitor induced non-small cell lung cancer (NSCLC) cells apoptosis (132).
CSCs mitochondrial mass and metabolism can be targeted using approved antibiotics like tetracyclines, salinomycin and erythromycins. Antibiotic salinomycin inhibits OXPHOS (133) and salinomycin treatment reduced breast CSCs gene expression. Antibiotic tigecycline inhibited mitochondrial translation in mitochondrial associated ribosomes in OXPHOS-dependent leukemia cells (134).
CSCs using OXPHOS have a higher mitochondrial membrane potential (Δψm) and thus Δψm can be explored for selective accumulation of cytotoxic drugs. Triphenylphosphonium (TPP) accumulates in the mitochondrial matrix (135) and conjugation of TPP to doxorubicin prevented drug efflux by enhancing drug selectivity in cancer cells (136). Dual inhibition of glycolysis and OXPHOS in sarcoma cells, using 2-DG and oligomycin/metformin co-treatment (137), suggests that simultaneous inhibition of glycolytic and mitochondrial respiration is more effective to eradicate CSCs (138, 139).
Targeting Glutamine Metabolism
Although a non-essential amino acid, glutamine becomes essential as a favored respiratory fuel for cancer cells and thus depriving glutamine is a potential anti-cancer strategy. Glutamine metabolism can be blocked by inhibiting glutaminase 1 (GLS1), an enzyme that converts glutamine to glutamate. GLS1 inhibition disrupted redox balance in CSCs and sensitized lung and pancreatic cancers to radiotherapy (77, 140). GLS1 inhibitors, BPTES (141) and CB-839 reduce intracellular glutamate and 2-hydroxyglutarate (an oncometabolite) levels. Lower glutamate levels inhibited cell growth, induced apoptosis and differentiation in Acute Myeloid Leukemia (AML) cells (142). CB-839 is under clinical trials for various cancers including renal cell carcinoma, hematologic cancer and leukemia (Table 1).
Targeting Lipid Metabolism
Cancer cells predominately use glycolysis for ATP production instead of oxidizing energy-rich substrates. However, unlike non-cancerous cells dependence on dietary lipids, cancer cells use de novo lipogenesis. Thus targeting fatty acid synthase (FASN), a central enzyme to lipogenesis, is a promising strategy to eliminate CSCs. FASN inhibitor cerulenin reduced de novo lipogenesis and in turn proliferation, migration and stemness of glioma stem cells (GSCs), induced apoptosis in colon cancer cell lines (92, 143) and blocked proliferation of pancreatic spheres (93). C75 decreased HER2+ breast cancer cells self-renewal capacity at non-cytotoxic concentrations (144). However, due to toxicity issues in in-vivo studies owing to high selectivity of FASN inhibitors, only one FASN inhibitor (TVB-2640) is under clinical trials to date (Table 1).
Studies show that increased fatty acid production in cancer cells raises their dependence on desaturases (enzymes that add double bonds into acyl-CoA chains). Thus targeting desaturase enzyme activity may provide a novel approach to selectively interfere lipid metabolism in CSCs. Several stearoyl-CoA desaturase-1 (SCD-1) inhibitors have effectively targeted stemness in pre-clinical models of cancer. Inhibitors like CAY10556 and SC-26196 reduced stem-ness markers and inhibited in-vitro sphere formation and in-vivo tumorigenicity, by down-regulating Hedgehog and Notch expression in aldehyde dehydrogenase (ALDH)- and CD133-enriched ovarian cells and had no effect on differentiated cells (96). Similarly, SCD-1 inhibitors (SSI-4 or A939572) promoted differentiation in chemo-resistant hepatospheres with little toxicity in vivo (145). MF-438 reduced expression of self-renewal and pluripotency markers in lung ALDH1+ cells (146).
Along with de novo lipogenesis, cancer cells also take lipids from the extracellular milieu (147) using LDL receptor (LDLR) (148), CD36 fatty acid translocase, fatty acid transport proteins (FATPs) (149) or fatty acid-binding proteins (FABPs) (150). Inhibition of CD36 transporter with 2-methylthio-1,4-naphtoquinone reduced self-renewal and promoted apoptosis in CD133+ glioblastoma (151) and sulfosuccinimidyl oleate reduced chemo-resistant leukemic stem cells (152). CD36-neutralizing antibodies inhibited progression and metastasis of oral squamous cell carcinoma and had no reported toxicity in-vivo (153).
Highly proliferating cells also have a higher demand for components of cell membrane like cholesterol. Cholesterol is either taken up from exogenous sources or synthesized using FASN or mevalonate pathway (154). Statins inhibit cholesterol synthesis through the mevalonate pathway and their target enzyme is 3-hydroxy-3-methyl-glutharyl-coenzyme A reductase (HMGCR). Statins treatment decreased CSCs self-renewal capacity and number in breast (155), nasopharyngeal (156) carcinomas and CD133+ brain TICs (157). MYC controls over-expression of mevalonate pathway genes and thus anti-CSCs effects of statins could be due to MYC inhibition (157).
Synthesized or accumulated fatty acids are also converted to signaling lipids and energy via FAO, in addition to membrane incorporation or being stored. FAO is an essential energy source in non-glycolytic tumors (158, 159), as CSCs show higher FAO in nutrient-deprived conditions (63, 86, 160, 161). FAO promotes pluripotency and chemoresistance (94) by reducing ROS production (162, 163) and promoted metastatic capacity in sphere-derived cells (164). Etomoxir, an inhibitor of FAO, inhibited mammosphere formation in hypoxic breast CSCs (165) and eradicated half of quiescent leukemia SCs (99), suggesting that FAO inhibitors hinder CSCs survival. In hepatocellular carcinoma, etomoxir sensitized CSCs to sorafenib treatment (43). Soraphen A, cerulenin and resveratrol inhibited FAO and lowered stemness markers and spheroid formation in CSCs (92, 166, 167).
Lipids also support CSCs functionality by being second messengers in signal transduction pathways. Sphingolipids, eicosanoids (prostaglandin E2) and glycerophospholipids (lysophosphatidic acid (LPA)) boost CSCs number by activation of Notch, AKT and NF-kB pathways in breast, bladder, colorectal (CRC) and ovarian cancer (168–171). Lipid-mediated signaling in CSCs thus can be targeted using inhibitors and dietary supplements. Inhibition of autotoxin (ATX) (a lysophosphatidic acid (LPA)-producing enzyme) with S32826 or PF8380 reduced tumorigenicity and chemoresistance in-vivo (171). Inhibition of LPA production in cancer cells modulated the immune system by inducing monocytes differentiation to macrophages and launching cancer-associated fibroblasts (CAFs) phenotype (172, 173). Prostaglandins are major lipid mediator in CSCs and celecoxib treatment of ApcMin/þ mice reduced number of CD133+CD44+ cells and tumor burden (170). Celecoxib reduced patient-derived CSCs content and liver metastatic tumors number in NOD scid gamma (NSG) mice and weakened chemoresistance in bladder carcinomas, indicating its potential as an adjuvant therapy (169). In contrast, reduction of CD34+ cells in chronic myelogenous leukemia (CML) xenograft model by EP4 receptor (prostaglandin receptor) agonist misoprostol or PGE1 (FDA-approved), suggests a context-dependent role of prostaglandins in stem-ness (174). Further, dietary omega-3 polyunsaturated fatty acids (ω-3 PUFA) decreased CRC risk and reduced CD133+ content in CRC cell lines (175, 176). Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) supplementation decreased breast tumorspheres proliferation (177) and EPA with chemotherapy suppressed tumor growth in mice (178), suggesting an anti-CSCs properties of ω-3 PUFAs.
Combination Treatments
CSCs can also attain a combined metabolic phenotype where both glycolysis and OXPHOS are utilized (Figure 1). This phenotype can be attained by direct association of AMP-activated protein kinase (AMPK, master regulator of OXPHOS) and HIF-1 (master regulator of glycolysis) activities (179). High AMPK/HIF-1 activities leads to higher glycolysis and OXPHOS, and provide enhanced proliferation and clonogenicity compared to only glycolytic or OXPHOS phenotype (179). In addition, CSCs metabolize glutamine along with glucose for carbon and amino-nitrogen to synthesize amino acids, nucleotides and lipids (79). Additionally, CSCs also use de novo lipogenesis to increase their bioenergetic requirements and are linked in tumor metastasis (88). Also preclinical and clinical setting has shown that targeting a single metabolic pathway like glycolysis has low success rates and enhanced side effects as GLUT transporters are ubiquitous. Also, inhibition of hexokinase II with ionidamine showed no significant improvement in overall survival but led to elevated toxicity (114, 180–182). Thus combination treatments targeting two or more metabolic pathways will majorly erase CSCs, prevent tumor relapse and prevent side-effects of a single treatment.
Further, combining a standard cytotoxic therapy with a metabolic inhibitor will probably enhance CSCs eradication. Combinations of metformin and JQ-1 (bromodomain and extraterminal motif (BET) inhibitor) in pancreatic cancer (66) or PI3K inhibitor in ovarian cancer (183) blocked both OXPHOS and glycolysis. Apart from direct metabolic inhibition, targeting oncogenes regulating cellular metabolism will also eradicate CSCs effectively. KRAS mutation occurs in about 90% of pancreatic cancer cases (184) and KRAS drives glycolysis and nucleic acids synthesis (185, 186). c-MYC is essential for glycolysis in cancer (187, 188) and MYC suppression prevents mitochondrial inhibitors resistance (66, 75). Thus combination approaches can be extended to target CSCs as an anti-cancer strategy. Table 1 lists the clinical trials using combination treatments for various cancers.
Future Challenges
Figure 1 summarizes the known CSCs’ metabolic phenotypes and how these phenotypes switch with metabolic stressors like nutrient deprivation and hypoxia. However, melanoma cells attain a drug-tolerant “idling state” after enduring MAPK inhibition (MAPKi) and this state has a metabolically Low/Low (L/L) phenotype, where both AMPK/HIF-1 activity and OXPHOS/glycolysis are minimal (189). L/L phenotype does not favor tumorigenicity but supports cell division. These idle L/L drug-tolerant cells accumulate mutations to promote relapse post MAPKi melanoma treatment (189).
Further adding to the complexity of CSCs metabolism, Luo et al. (190) showed that breast cancer stem cells (BCSCs) have two states: quiescent mesenchymal-like (M) and proliferative epithelial-like (E). Proliferative E-BCSCs showed higher mitochondrial OXPHOS, whereas M-BCSCs have enrichment of glycolysis and gluconeogenesis pathways and hypoxia promotes M to E transition in BCSCs (190). Thus CSCs’ multiple metabolic phenotypes (glycolytic, OXPHOS, combined and L/L) explain the futility of current efforts to eradicate CSCs and a deeper understanding of CSCs metabolic plasticity would translate to better therapeutic strategies.
Concluding Remarks
CSCs provide treatment resistance and promote metastasis during tumor growth and targeting metabolism holds potential in overcoming cancer recurrence and metastasis by CSCs. Deciphering metabolic reprogramming in cancer showed differences between metabolic phenotypes of CSCs and their differentiated counterparts. CSCs metabolism shuffles between glycolysis and OXPHOS primarily, however the mechanisms of CSCs metabolic heterogeneity are still unknown. Current knowledge suggests that carefully designed metabolic therapies have potential to be more effective against CSCs. Further, co-targeting CSCs using metabolic drugs and traditional anticancer treatments could be more efficient. The ongoing clinical trials targeting CSCs show a promising future for cancer therapy and are worth exploring further. More preclinical and clinical studies are thus required to uncover novel metabolic targets in CSCs.
Author Contributions
SB conceptualized the article and reviewed the literature, JK did literature search and drafted the manuscript. Both SB and JK contributed to the final version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bristow RG, Alexander B, Baumann M, Bratman SV, Brown JM, Camphausen K, et al. Combining Precision Radiotherapy With Molecular Targeting and Immunomodulatory Agents: A Guideline by the American Society for Radiation Oncology. Lancet Oncol (2018) 19:e240–51. doi: 10.1016/S1470-2045(18)30096-2
2. Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of Combining Radiotherapy With Immune-Checkpoint Inhibition. Nat Rev Clin Oncol (2018) 15:477–94. doi: 10.1038/s41571-018-0046-7
3. Cleeland CS, Allen JD, Roberts SA, Brell JM, Giralt SA, Khakoo AY, et al. Reducing the Toxicity of Cancer Therapy: Recognizing Needs, Taking Action. Nat Rev Clin Oncol (2012) 9:471–8. doi: 10.1038/nrclinonc.2012.99
4. Lytle NK, Barber AG, Reya T. Stem Cell Fate in Cancer Growth, Progression and Therapy Resistance. Nat Rev Cancer (2018) 18:669–80. doi: 10.1038/s41568-018-0056-x
5. Krause M, Dubrovska A, Linge A, Baumann M. Cancer Stem Cells: Radioresistance, Prediction of Radiotherapy Outcome and Specific Targets for Combined Treatments. Adv Drug Delivery Rev (2017) 109:63–73. doi: 10.1016/j.addr.2016.02.002
6. Bütof R, Dubrovska A, Baumann M. Clinical Perspectives of Cancer Stem Cell Research in Radiation Oncology. Radiother Oncol (2013) 108:388–96. doi: 10.1016/j.radonc.2013.06.002
7. Linge A, Löck S, Gudziol V, Nowak A, Lohaus F, von Neubeck C, et al. Low Cancer Stem Cell Marker Expression and Low Hypoxia Identify Good Prognosis Subgroups in HPV(-) HNSCC After Postoperative Radiochemotherapy: A Multicenter Study of the DKTK-ROG. Clin Cancer Res (2016) 22:2639–49. doi: 10.1158/1078-0432.CCR-15-1990
8. Prasetyanti PR, Medema JP. Intra-Tumor Heterogeneity From a Cancer Stem Cell Perspective. Mol Cancer (2017) 16:41. doi: 10.1186/s12943-017-0600-4
9. Wang K-J, Wang C, Dai L-H, Yang J, Huang H, Ma X-J, et al. Targeting an Autocrine Regulatory Loop in Cancer Stem-Like Cells Impairs the Progression and Chemotherapy Resistance of Bladder Cancer. Clin Cancer Res (2019) 25:1070–86. doi: 10.1158/1078-0432.CCR-18-0586
10. Kreso A, Dick JE. Evolution of the Cancer Stem Cell Model. Cell Stem Cell (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
11. Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G. Tumour Microenvironment and Metabolic Plasticity in Cancer and Cancer Stem Cells: Perspectives on Metabolic and Immune Regulatory Signatures in Chemoresistant Ovarian Cancer Stem Cells. Semin Cancer Biol (2018) 53:265–81. doi: 10.1016/j.semcancer.2018.10.002
12. Mukha A, Dubrovska A. Metabolic Targeting of Cancer Stem Cells. Front Oncol (2020) 10:537930. doi: 10.3389/fonc.2020.537930
13. Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, et al. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J Immunol (2015) 194:6082–9. doi: 10.4049/jimmunol.1402469
14. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
15. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J Biol Chem (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
16. Chen Y, Tan W, Wang C. Tumor-Associated Macrophage-Derived Cytokines Enhance Cancer Stem-Like Characteristics Through Epithelial-Mesenchymal Transition. Onco Targets Ther (2018) 11:3817–26. doi: 10.2147/OTT.S168317
17. Korkaya H, Liu S, Wicha MS. Regulation of Cancer Stem Cells by Cytokine Networks: Attacking Cancer’s Inflammatory Roots. Clin Cancer Res (2011) 17:6125–9. doi: 10.1158/1078-0432.CCR-10-2743
18. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res (2013) 73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731
19. De Francesco EM, Sotgia F, Lisanti MP. Cancer Stem Cells (CSCs): Metabolic Strategies for Their Identification and Eradication. Biochem J (2018) 475:1611–34. doi: 10.1042/BCJ20170164
20. Meacham CE, Morrison SJ. Tumour Heterogeneity and Cancer Cell Plasticity. Nature (2013) 501:328–37. doi: 10.1038/nature12624
21. Nowell PC. The Clonal Evolution of Tumor Cell Populations. Science (1976) 194:23–8. doi: 10.1126/science.959840
22. Reid MA, Dai Z, Locasale JW. The Impact of Cellular Metabolism on Chromatin Dynamics and Epigenetics. Nat Cell Biol (2017) 19:1298–306. doi: 10.1038/ncb3629
23. Sancho P, Barneda D, Heeschen C. Hallmarks of Cancer Stem Cell Metabolism. Br J Cancer (2016) 114:1305–12. doi: 10.1038/bjc.2016.152
24. Emmink BL, Verheem A, Van Houdt WJ, Steller EJA, Govaert KM, Pham TV, et al. The Secretome of Colon Cancer Stem Cells Contains Drug-Metabolizing Enzymes. J Proteomics (2013) 91:84–96. doi: 10.1016/j.jprot.2013.06.027
25. Hammoudi N, Ahmed KBR, Garcia-Prieto C, Huang P. Metabolic Alterations in Cancer Cells and Therapeutic Implications. Chin J Cancer (2011) 30:508–25. doi: 10.5732/cjc.011.10267
26. Jang Y-Y, Sharkis SJ. A Low Level of Reactive Oxygen Species Selects for Primitive Hematopoietic Stem Cells That may Reside in the Low-Oxygenic Niche. Blood (2007) 110:3056–63. doi: 10.1182/blood-2007-05-087759
27. Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway Is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells (2010) 28:721–33. doi: 10.1002/stem.404
28. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell (2010) 7:380–90. doi: 10.1016/j.stem.2010.07.011
29. Suda T, Takubo K, Semenza GL. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell (2011) 9:298–310. doi: 10.1016/j.stem.2011.09.010
30. Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, et al. An MTCH2 Pathway Repressing Mitochondria Metabolism Regulates Haematopoietic Stem Cell Fate. Nat Commun (2015) 6:7901. doi: 10.1038/ncomms8901
31. Folmes CDL, Dzeja PP, Nelson TJ, Terzic A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell (2012) 11:596–606. doi: 10.1016/j.stem.2012.10.002
32. Jang H, Yang J, Lee E, Cheong J-H. Metabolism in Embryonic and Cancer Stemness. Arch Pharm Res (2015) 38:381–8. doi: 10.1007/s12272-015-0558-y
33. Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast Cancer Stem Cells Rely on Fermentative Glycolysis and Are Sensitive to 2-Deoxyglucose Treatment. Cell Death Dis (2014) 5:e1336. doi: 10.1038/cddis.2014.285
34. Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, et al. Ovarian Cancer Spheroid Cells With Stem Cell-Like Properties Contribute to Tumor Generation, Metastasis and Chemotherapy Resistance Through Hypoxia-Resistant Metabolism. PloS One (2014) 9:e84941. doi: 10.1371/journal.pone.0084941
35. Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, et al. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells: Preference for Hypoxia and High Dependency on Glycolysis. J Biol Chem (2011) 286:32843–53. doi: 10.1074/jbc.M111.260935
36. Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. Somatic Oxidative Bioenergetics Transitions Into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab (2011) 14:264–71. doi: 10.1016/j.cmet.2011.06.011
37. Liu P-P, Liao J, Tang Z-J, Wu W-J, Yang J, Zeng Z-L, et al. Metabolic Regulation of Cancer Cell Side Population by Glucose Through Activation of the Akt Pathway. Cell Death Differ (2014) 21:124–35. doi: 10.1038/cdd.2013.131
38. Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, et al. Energy Metabolism Characterization of a Novel Cancer Stem Cell-Like Line 3AB-OS. J Cell Biochem (2014) 115:368–79. doi: 10.1002/jcb.24671
39. Song K, Kwon H, Han C, Zhang J, Dash S, Lim K, et al. Active Glycolytic Metabolism in CD133(+) Hepatocellular Cancer Stem Cells: Regulation by MIR-122. Oncotarget (2015) 6:40822–35. doi: 10.18632/oncotarget.5812
40. Mizushima E, Tsukahara T, Emori M, Murata K, Akamatsu A, Shibayama Y, et al. Osteosarcoma-Initiating Cells Show High Aerobic Glycolysis and Attenuation of Oxidative Phosphorylation Mediated by LIN28B. Cancer Sci (2020) 111:36–46. doi: 10.1111/cas.14229
41. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, et al. Targeting Unique Metabolic Properties of Breast Tumor Initiating Cells. Stem Cells (2014) 32:1734–45. doi: 10.1002/stem.1662
42. Shen Y-A, Wang C-Y, Hsieh Y-T, Chen Y-J, Wei Y-H. Metabolic Reprogramming Orchestrates Cancer Stem Cell Properties in Nasopharyngeal Carcinoma. Cell Cycle (2015) 14:86–98. doi: 10.4161/15384101.2014.974419
43. Chen C-L, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-Like Cells Through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab (2016) 23:206–19. doi: 10.1016/j.cmet.2015.12.004
44. Gabay M, Li Y, Felsher DW. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb Perspect Med (2014) 4:a014241. doi: 10.1101/cshperspect.a014241
45. Folmes CDL, Martinez-Fernandez A, Faustino RS, Yamada S, Perez-Terzic C, Nelson TJ, et al. Nuclear Reprogramming With C-Myc Potentiates Glycolytic Capacity of Derived Induced Pluripotent Stem Cells. J Cardiovasc Transl Res (2013) 6:10–21. doi: 10.1007/s12265-012-9431-2
46. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
47. Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and Lactate Increase Cancer Cell “Stemness,” Driving Recurrence, Metastasis and Poor Clinical Outcome in Breast Cancer: Achieving Personalized Medicine via Metabolo-Genomics. Cell Cycle (2011) 10:1271–86. doi: 10.4161/cc.10.8.15330
48. Lin S, Sun L, Lyu X, Ai X, Du D, Su N, et al. Lactate-Activated Macrophages Induced Aerobic Glycolysis and Epithelial-Mesenchymal Transition in Breast Cancer by Regulation of CCL5-CCR5 Axis: A Positive Metabolic Feedback Loop. Oncotarget (2017) 8:110426–43. doi: 10.18632/oncotarget.22786
49. Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, et al. Metabolic Heterogeneity Confers Differences in Melanoma Metastatic Potential. Nature (2020) 577:115–20. doi: 10.1038/s41586-019-1847-2
50. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
51. Daneshmandi S, Wegiel B, Seth P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers (Basel) (2019) 11:450. doi: 10.3390/cancers11040450
52. Yuen CA, Asuthkar S, Guda MR, Tsung AJ, Velpula KK. Cancer Stem Cell Molecular Reprogramming of the Warburg Effect in Glioblastomas: A New Target Gleaned From an Old Concept. CNS Oncol (2016) 5:101–8. doi: 10.2217/cns-2015-0006
53. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer (2016) 2:758–70. doi: 10.1016/j.trecan.2016.10.016
54. Tong W-W, Tong G-H, Liu Y. Cancer Stem Cells and Hypoxia-Inducible Factors (Review). Int J Oncol (2018) 53:469–76. doi: 10.3892/ijo.2018.4417
55. Cuyàs E, Corominas-Faja B, Menendez JA. The Nutritional Phenome of EMT-Induced Cancer Stem-Like Cells. Oncotarget (2014) 5:3970–82. doi: 10.18632/oncotarget.2147
56. LaBarge MA. The Difficulty of Targeting Cancer Stem Cell Niches. Clin Cancer Res (2010) 16:3121–9. doi: 10.1158/1078-0432.CCR-09-2933
57. Moore KA, Lemischka IR. Stem Cells and Their Niches. Science (2006) 311:1880–5. doi: 10.1126/science.1110542
58. Adams JM, Strasser A. Is Tumor Growth Sustained by Rare Cancer Stem Cells or Dominant Clones? Cancer Res (2008) 68:4018–21. doi: 10.1158/0008-5472.CAN-07-6334
59. Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, et al. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preserving Glioblastoma Cancer Stem Cells. Genes Dev (2012) 26:1926–44. doi: 10.1101/gad.188292.112
60. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
61. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B(high) Cells. Cancer Cell (2013) 23:811–25. doi: 10.1016/j.ccr.2013.05.003
62. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc Natl Acad Sci USA (2011) 108:16062–7. doi: 10.1073/pnas.1106704108
63. Pastò A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer Stem Cells From Epithelial Ovarian Cancer Patients Privilege Oxidative Phosphorylation, and Resist Glucose Deprivation. Oncotarget (2014) 5:4305–19. doi: 10.18632/oncotarget.2010
64. Lee K-M, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab (2017) 26:633–647.e7. doi: 10.1016/j.cmet.2017.09.009
65. Lamb R, Bonuccelli G, Ozsvári B, Peiris-Pagès M, Fiorillo M, Smith DL, et al. Mitochondrial Mass, a New Metabolic Biomarker for Stem-Like Cancer Cells: Understanding WNT/FGF-Driven Anabolic Signaling. Oncotarget (2015) 6:30453–71. doi: 10.18632/oncotarget.5852
66. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
67. Farnie G, Sotgia F, Lisanti MP. High Mitochondrial Mass Identifies a Sub-Population of Stem-Like Cancer Cells That Are Chemo-Resistant. Oncotarget (2015) 6:30472–86. doi: 10.18632/oncotarget.5401
68. Tan J, Ong CK, Lim WK, Ng CCY, Thike AA, Ng LM, et al. Genomic Landscapes of Breast Fibroepithelial Tumors. Nat Genet (2015) 47:1341–5. doi: 10.1038/ng.3409
69. Lamb R, Ozsvari B, Bonuccelli G, Smith DL, Pestell RG, Martinez-Outschoorn UE, et al. Dissecting Tumor Metabolic Heterogeneity: Telomerase and Large Cell Size Metabolically Define a Sub-Population of Stem-Like, Mitochondrial-Rich, Cancer Cells. Oncotarget (2015) 6:21892–905. doi: 10.18632/oncotarget.5260
70. De Luca A, Fiorillo M, Peiris-Pagès M, Ozsvari B, Smith DL, Sanchez-Alvarez R, et al. Mitochondrial Biogenesis is Required for the Anchorage-Independent Survival and Propagation of Stem-Like Cancer Cells. Oncotarget (2015) 6:14777–95. doi: 10.18632/oncotarget.4401
71. Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. Pgc1α Expression Defines a Subset of Human Melanoma Tumors With Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell (2013) 23:287–301. doi: 10.1016/j.ccr.2012.11.020
72. Yajima T, Ochiai H, Uchiyama T, Takano N, Shibahara T, Azuma T. Resistance to Cytotoxic Chemotherapy-Induced Apoptosis in Side Population Cells of Human Oral Squamous Cell Carcinoma Cell Line Ho-1-N-1. Int J Oncol (2009) 35:273–80.
73. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting Mitochondrial Biogenesis to Overcome Drug Resistance to MAPK Inhibitors. J Clin Invest (2016) 126:1834–56. doi: 10.1172/JCI82661
74. Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell (2014) 15:169–84. doi: 10.1016/j.stem.2014.06.002
75. Kim JH, Lee KJ, Seo Y, Kwon J-H, Yoon JP, Kang JY, et al. Effects of Metformin on Colorectal Cancer Stem Cells Depend on Alterations in Glutamine Metabolism. Sci Rep (2018) 8:409. doi: 10.1038/s41598-017-18762-4
76. Choi Y-K, Park K-G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol Ther (Seoul) (2018) 26:19–28. doi: 10.4062/biomolther.2017.178
77. Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of Glutamine Metabolism Counteracts Pancreatic Cancer Stem Cell Features and Sensitizes Cells to Radiotherapy. Oncotarget (2015) 6:31151–63. doi: 10.18632/oncotarget.5150
78. Sato M, Kawana K, Adachi K, Fujimoto A, Yoshida M, Nakamura H, et al. Spheroid Cancer Stem Cells Display Reprogrammed Metabolism and Obtain Energy by Actively Running the Tricarboxylic Acid (TCA) Cycle. Oncotarget (2016) 7:33297–305. doi: 10.18632/oncotarget.8947
79. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
80. Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells With Mitochondrial DNA Mutations. Cell Metab (2018) 27:1007–1025.e5. doi: 10.1016/j.cmet.2018.03.002
81. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res (2020) 80:1630–43. doi: 10.1158/0008-5472.CAN-19-1363
82. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat Med (2017) 23:1362–8. doi: 10.1038/nm.4407
83. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc Natl Acad Sci USA (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
84. Lu W, Pelicano H, Huang P. Cancer Metabolism: Is Glutamine Sweeter Than Glucose? Cancer Cell (2010) 18:199–200. doi: 10.1016/j.ccr.2010.08.017
85. Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine Targeting Inhibits Systemic Metastasis in the VM-M3 Murine Tumor Model. Int J Cancer (2010) 127:2478–85. doi: 10.1002/ijc.25431
86. Carracedo A, Cantley LC, Pandolfi PP. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat Rev Cancer (2013) 13:227–32. doi: 10.1038/nrc3483
87. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Emerging Role of Lipid Metabolism Alterations in Cancer Stem Cells. J Exp Clin Cancer Res (2018) 37:118. doi: 10.1186/s13046-018-0784-5
88. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Saccà M, Ciliberto G. Metabolic Features of Cancer Stem Cells: The Emerging Role of Lipid Metabolism. Oncogene (2018) 37:2367–78. doi: 10.1038/s41388-018-0141-3
89. O’Malley J, Kumar R, Kuzmin AN, Pliss A, Yadav N, Balachandar S, et al. Lipid Quantification by Raman Microspectroscopy as a Potential Biomarker in Prostate Cancer. Cancer Lett (2017) 397:52–60. doi: 10.1016/j.canlet.2017.03.025
90. Li X, Wu JB, Li Q, Shigemura K, Chung LWK, Huang W-C. SREBP-2 Promotes Stem Cell-Like Properties and Metastasis by Transcriptional Activation of C-Myc in Prostate Cancer. Oncotarget (2016) 7:12869–84. doi: 10.18632/oncotarget.7331
91. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells (2015) 33:35–44. doi: 10.1002/stem.1837
92. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PloS One (2016) 11:e0147717. doi: 10.1371/journal.pone.0147717
93. Brandi J, Dando I, Pozza ED, Biondani G, Jenkins R, Elliott V, et al. Proteomic Analysis of Pancreatic Cancer Stem Cells: Functional Role of Fatty Acid Synthesis and Mevalonate Pathways. J Proteomics (2017) 150:310–22. doi: 10.1016/j.jprot.2016.10.002
94. Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27:1357. doi: 10.1016/j.cmet.2018.04.018
95. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ Pathway for Fatty Acid Oxidation Regulates Hematopoietic Stem Cell Maintenance. Nat Med (2012) 18:1350–8. doi: 10.1038/nm.2882
96. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20:303–314.e5. doi: 10.1016/j.stem.2016.11.004
97. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and Oncogene Rescue of Metabolic Defects Caused by Loss of Matrix Attachment. Nature (2009) 461:109–13. doi: 10.1038/nature08268
98. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VCJ, Laurent G, et al. A Metabolic Prosurvival Role for PML in Breast Cancer. J Clin Invest (2012) 122:3088–100. doi: 10.1172/JCI62129
99. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic Inhibition of Fatty Acid Oxidation Sensitizes Human Leukemia Cells to Apoptosis Induction. J Clin Invest (2010) 120:142–56. doi: 10.1172/JCI38942
100. Wang B, Yu S, Jiang J, Porter GW, Zhao L, Wang Z, et al. An Inhibitor of Arachidonate 5-Lipoxygenase, Nordy, Induces Differentiation and Inhibits Self-Renewal of Glioma Stem-Like Cells. Stem Cell Rev Rep (2011) 7:458–70. doi: 10.1007/s12015-010-9175-9
101. Sun P, Xia S, Lal B, Shi X, Yang KS, Watkins PA, et al. Lipid Metabolism Enzyme ACSVL3 Supports Glioblastoma Stem Cell Maintenance and Tumorigenicity. BMC Cancer (2014) 14:401. doi: 10.1186/1471-2407-14-401
102. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
103. Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET Family Proteins and Their Role in Stem Cell Differentiation and Transformation. Cell Stem Cell (2011) 9:193–204. doi: 10.1016/j.stem.2011.08.007
104. Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-Oncogenic Role of Mutant IDH2 in Leukemia Initiation and Maintenance. Cell Stem Cell (2014) 14:329–41. doi: 10.1016/j.stem.2013.12.016
105. Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y, et al. Purine Synthesis Promotes Maintenance of Brain Tumor Initiating Cells in Glioma. Nat Neurosci (2017) 20:661–73. doi: 10.1038/nn.4537
106. Wu Z, Wei D, Gao W, Xu Y, Hu Z, Ma Z, et al. TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110+ Tumor-Initiating Cells. Cell Stem Cell (2015) 17:47–59. doi: 10.1016/j.stem.2015.05.016
107. Lin S-T, Tu S-H, Yang P-S, Hsu S-P, Lee W-H, Ho C-T, et al. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of P53-Mediated Signaling. J Agric Food Chem (2016) 64:6826–37. doi: 10.1021/acs.jafc.6b02861
108. Wu K-H, Ho C-T, Chen Z-F, Chen L-C, Whang-Peng J, Lin T-N, et al. The Apple Polyphenol Phloretin Inhibits Breast Cancer Cell Migration and Proliferation via Inhibition of Signals by Type 2 Glucose Transporter. J Food Drug Anal (2018) 26:221–31. doi: 10.1016/j.jfda.2017.03.009
109. Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, et al. A Novel Inhibitor of Glucose Uptake Sensitizes Cells to FAS-Induced Cell Death. Mol Cancer Ther (2008) 7:3546–55. doi: 10.1158/1535-7163.MCT-08-0569
110. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, et al. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In Vivo. Mol Cancer Ther (2012) 11:1672–82. doi: 10.1158/1535-7163.MCT-12-0131
111. Tao L, Wei L, Liu Y, Ding Y, Liu X, Zhang X, et al. Gen-27, a Newly Synthesized Flavonoid, Inhibits Glycolysis and Induces Cell Apoptosis via Suppression of Hexokinase II in Human Breast Cancer Cells. Biochem Pharmacol (2017) 125:12–25. doi: 10.1016/j.bcp.2016.11.001
112. Li W, Zheng M, Wu S, Gao S, Yang M, Li Z, et al. Benserazide, a Dopadecarboxylase Inhibitor, Suppresses Tumor Growth by Targeting Hexokinase 2. J Exp Clin Cancer Res (2017) 36:58. doi: 10.1186/s13046-017-0530-4
113. Coleman MC, Asbury CR, Daniels D, Du J, Aykin-Burns N, Smith BJ, et al. 2-Deoxy-D-Glucose Causes Cytotoxicity, Oxidative Stress, and Radiosensitization in Pancreatic Cancer. Free Radic Biol Med (2008) 44:322–31. doi: 10.1016/j.freeradbiomed.2007.08.032
114. Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, De Matteis A, et al. Time to Progression in Metastatic Breast Cancer Patients Treated With Epirubicin Is Not Improved by the Addition of Either Cisplatin or Lonidamine: Final Results of a Phase III Study With a Factorial Design. J Clin Oncol (2002) 20:4150–9. doi: 10.1200/JCO.2002.08.012
115. Dwarakanath B, Jain V. Targeting Glucose Metabolism With 2-Deoxy-D-Glucose for Improving Cancer Therapy. Future Oncol (2009) 5:581–5. doi: 10.2217/fon.09.44
116. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A Phase I Dose-Escalation Trial of 2-Deoxy-D-Glucose Alone or Combined With Docetaxel in Patients With Advanced Solid Tumors. Cancer Chemother Pharmacol (2013) 71:523–30. doi: 10.1007/s00280-012-2045-1
117. Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. 2-Deoxy-D-Glucose Combined With Cisplatin Enhances Cytotoxicity via Metabolic Oxidative Stress in Human Head and Neck Cancer Cells. Cancer Res (2007) 67:3364–70. doi: 10.1158/0008-5472.CAN-06-3717
118. O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose Inhibits Aggressive Triple-Negative Breast Cancer Cells by Targeting Glycolysis and the Cancer Stem Cell Phenotype. Sci Rep (2019) 9:3788. doi: 10.1038/s41598-019-39789-9
119. Lea MA, Qureshi MS, Buxhoeveden M, Gengel N, Kleinschmit J, Desbordes C. Regulation of the Proliferation of Colon Cancer Cells by Compounds That Affect Glycolysis, Including 3-Bromopyruvate, 2-Deoxyglucose and Biguanides. Anticancer Res (2013) 33:401–7.
120. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic Modulation of Glioblastoma With Dichloroacetate. Sci Transl Med (2010) 2:31ra34. doi: 10.1126/scitranslmed.3000677
121. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, et al. Phase 1 Trial of Dichloroacetate (DCA) in Adults With Recurrent Malignant Brain Tumors. Invest New Drugs (2014) 32:452–64. doi: 10.1007/s10637-013-0047-4
122. Chu QS-C, Sangha R, Spratlin J, Vos LJ, Mackey JR, McEwan AJB, et al. A Phase I Open-Labeled, Single-Arm, Dose-Escalation, Study of Dichloroacetate (DCA) in Patients With Advanced Solid Tumors. Invest New Drugs (2015) 33:603–10. doi: 10.1007/s10637-015-0221-y
123. Peng F, Wang J-H, Fan W-J, Meng Y-T, Li M-M, Li T-T, et al. Glycolysis Gatekeeper PDK1 Reprograms Breast Cancer Stem Cells Under Hypoxia. Oncogene (2018) 37:1119. doi: 10.1038/onc.2017.407
124. Keith B, Simon MC. Hypoxia-Inducible Factors, Stem Cells, and Cancer. Cell (2007) 129:465–72. doi: 10.1016/j.cell.2007.04.019
125. Hu L, Zeng Z, Xia Q, Liu Z, Feng X, Chen J, et al. Metformin Attenuates Hepatoma Cell Proliferation by Decreasing Glycolytic Flux Through the HIF-1α/PFKFB3/PFK1 Pathway. Life Sci (2019) 239:116966. doi: 10.1016/j.lfs.2019.116966
126. Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming Metabolism With Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin Cancer Res (2013) 19:6741–50. doi: 10.1158/1078-0432.CCR-13-1787
127. Wei R, Hackman RM, Wang Y, Mackenzie GG. Targeting Glycolysis With Epigallocatechin-3-Gallate Enhances the Efficacy of Chemotherapeutics in Pancreatic Cancer Cells and Xenografts. Cancers (Basel) (2019) 11:1496. doi: 10.3390/cancers11101496
128. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, et al. Antibiotics That Target Mitochondria Effectively Eradicate Cancer Stem Cells, Across Multiple Tumor Types: Treating Cancer Like an Infectious Disease. Oncotarget (2015) 6:4569–84. doi: 10.18632/oncotarget.3174
129. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife (2014) 3:e02242. doi: 10.7554/eLife.02242
130. Bowker SL, Yasui Y, Veugelers P, Johnson JA. Glucose-Lowering Agents and Cancer Mortality Rates in Type 2 Diabetes: Assessing Effects of Time-Varying Exposure. Diabetologia (2010) 53:1631–7. doi: 10.1007/s00125-010-1750-8
131. Kheirandish M, Mahboobi H, Yazdanparast M, Kamal W, Kamal MA. Anti-Cancer Effects of Metformin: Recent Evidences for Its Role in Prevention and Treatment of Cancer. Curr Drug Metab (2018) 19:793–7. doi: 10.2174/1389200219666180416161846
132. Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 Inactivation Dictates Therapeutic Response of non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell (2013) 23:143–58. doi: 10.1016/j.ccr.2012.12.008
133. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell (2009) 138:645–59. doi: 10.1016/j.cell.2009.06.034
134. Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell (2011) 20:674–88. doi: 10.1016/j.ccr.2011.10.015
135. Murphy MP. Targeting Lipophilic Cations to Mitochondria. Biochim Biophys Acta (2008) 1777:1028–31. doi: 10.1016/j.bbabio.2008.03.029
136. Chamberlain GR, Tulumello DV, Kelley SO. Targeted Delivery of Doxorubicin to Mitochondria. ACS Chem Biol (2013) 8:1389–95. doi: 10.1021/cb400095v
137. Issaq SH, Teicher BA, Monks A. Bioenergetic Properties of Human Sarcoma Cells Help Define Sensitivity to Metabolic Inhibitors. Cell Cycle (2014) 13:1152–61. doi: 10.4161/cc.28010
138. Cheong J-H, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual Inhibition of Tumor Energy Pathway by 2-Deoxyglucose and Metformin Is Effective Against a Broad Spectrum of Preclinical Cancer Models. Mol Cancer Ther (2011) 10:2350–62. doi: 10.1158/1535-7163.MCT-11-0497
139. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer Stem Cell Metabolism. Breast Cancer Res (2016) 18:55. doi: 10.1186/s13058-016-0712-6
140. Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Simecka CM, Kore RA, et al. Glutaminase Inhibitor CB-839 Increases Radiation Sensitivity of Lung Tumor Cells and Human Lung Tumor Xenografts in Mice. Int J Radiat Biol (2019) 95:436–42. doi: 10.1080/09553002.2018.1558299
141. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel Mechanism of Inhibition of Rat Kidney-Type Glutaminase by Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide (BPTES). Biochem J (2007) 406:407–14. doi: 10.1042/BJ20070039
142. Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, et al. Inhibiting Glutaminase in Acute Myeloid Leukemia: Metabolic Dependency of Selected AML Subtypes. Oncotarget (2016) 7:79722–35. doi: 10.18632/oncotarget.12944
143. Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, et al. Enhanced Antitumor Activity of Cerulenin Combined With Oxaliplatin in Human Colon Cancer Cells. Int J Oncol (2013) 43:431–8. doi: 10.3892/ijo.2013.1978
144. Corominas-Faja B, Vellon L, Cuyàs E, Buxó M, Martin-Castillo B, Serra D, et al. Clinical and Therapeutic Relevance of the Metabolic Oncogene Fatty Acid Synthase in HER2+ Breast Cancer. Histol Histopathol (2017) 32:687–98. doi: 10.14670/HH-11-830
145. Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, et al. Stearoyl-CoA Desaturase Regulates Sorafenib Resistance via Modulation of ER Stress-Induced Differentiation. J Hepatol (2017) 67:979–90. doi: 10.1016/j.jhep.2017.06.015
146. Pisanu ME, Noto A, De Vitis C, Morrone S, Scognamiglio G, Botti G, et al. Blockade of Stearoyl-CoA-Desaturase 1 Activity Reverts Resistance to Cisplatin in Lung Cancer Stem Cells. Cancer Lett (2017) 406:93–104. doi: 10.1016/j.canlet.2017.07.027
147. Medes G, Thomas A, Weinhouse S. Metabolism of Neoplastic Tissue. IV. A Study of Lipid Synthesis in Neoplastic Tissue Slices In Vitro. Cancer Res (1953) 13:27–9.
148. Brown MS, Goldstein JL. A Receptor-Mediated Pathway for Cholesterol Homeostasis. Science (1986) 232:34–47. doi: 10.1126/science.3513311
149. Kazantzis M, Stahl A. Fatty Acid Transport Proteins, Implications in Physiology and Disease. Biochim Biophys Acta (BBA) - Mol Cell Biol Lipids (2012) 1821:852–7. doi: 10.1016/j.bbalip.2011.09.010
150. Furuhashi M, Hotamisligil GS. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat Rev Drug Discovery (2008) 7:489–503. doi: 10.1038/nrd2589
151. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem Cells (2014) 32:1746–58. doi: 10.1002/stem.1716
152. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
153. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting Metastasis-Initiating Cells Through the Fatty Acid Receptor CD36. Nature (2017) 541:41–5. doi: 10.1038/nature20791
154. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis (2016) 5:e189. doi: 10.1038/oncsis.2015.49
155. Ginestier C, Monville F, Wicinski J, Cabaud O, Cervera N, Josselin E, et al. Mevalonate Metabolism Regulates Basal Breast Cancer Stem Cells and Is a Potential Therapeutic Target. Stem Cells (2012) 30:1327–37. doi: 10.1002/stem.1122
156. Peng Y, He G, Tang D, Xiong L, Wen Y, Miao X, et al. Lovastatin Inhibits Cancer Stem Cells and Sensitizes to Chemo- and Photodynamic Therapy in Nasopharyngeal Carcinoma. J Cancer (2017) 8:1655–64. doi: 10.7150/jca.19100
157. Wang X, Huang Z, Wu Q, Prager BC, Mack SC, Yang K, et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res (2017) 77:4947–60. doi: 10.1158/0008-5472.CAN-17-0114
158. Liu Y, Zuckier LS, Ghesani NV. Dominant Uptake of Fatty Acid Over Glucose by Prostate Cells: A Potential New Diagnostic and Therapeutic Approach. Anticancer Res (2010) 30:369–74.
159. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell (2012) 22:547–60. doi: 10.1016/j.ccr.2012.08.014
160. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-Transformed Cells Support Growth by Scavenging Unsaturated Fatty Acids From Lysophospholipids. Proc Natl Acad Sci USA (2013) 110:8882–7. doi: 10.1073/pnas.1307237110
161. Daniëls VW, Smans K, Royaux I, Chypre M, Swinnen JV, Zaidi N. Cancer Cells Differentially Activate and Thrive on De Novo Lipid Synthesis Pathways in a Low-Lipid Environment. PloS One (2014) 9:e106913. doi: 10.1371/journal.pone.0106913
162. Lee EA, Angka L, Rota S-G, Hanlon T, Mitchell A, Hurren R, et al. Targeting Mitochondria With Avocatin B Induces Selective Leukemia Cell Death. Cancer Res (2015) 75:2478–88. doi: 10.1158/0008-5472.CAN-14-2676
163. Chen X, Song M, Zhang B, Zhang Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev (2016) 2016:1580967. doi: 10.1155/2016/1580967
164. Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V, et al. Metabolic Reprogramming and Dependencies Associated With Epithelial Cancer Stem Cells Independent of the Epithelial-Mesenchymal Transition Program. Stem Cells (2016) 34:1163–76. doi: 10.1002/stem.2286
165. De Francesco EM, Maggiolini M, Tanowitz HB, Sotgia F, Lisanti MP. Targeting Hypoxic Cancer Stem Cells (CSCs) With Doxycycline: Implications for Optimizing Anti-Angiogenic Therapy. Oncotarget (2017) 8:56126–42. doi: 10.18632/oncotarget.18445
166. Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG, et al. Chemical Inhibition of Acetyl-CoA Carboxylase Suppresses Self-Renewal Growth of Cancer Stem Cells. Oncotarget (2014) 5:8306–16. doi: 10.18632/oncotarget.2059
167. Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, et al. Resveratrol Suppresses Growth of Cancer Stem-Like Cells by Inhibiting Fatty Acid Synthase. Breast Cancer Res Treat (2011) 130:387–98. doi: 10.1007/s10549-010-1300-6
168. Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-Phosphate Promotes Expansion of Cancer Stem Cells via S1PR3 by a Ligand-Independent Notch Activation. Nat Commun (2014) 5:4806. doi: 10.1038/ncomms5806
169. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-Induced Tumour Repopulation Abrogates Bladder Cancer Chemoresistance. Nature (2015) 517:209–13. doi: 10.1038/nature14034
170. Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology (2015) 149:1884–1895.e4. doi: 10.1053/j.gastro.2015.07.064
171. Seo EJ, Kwon YW, Jang IH, Kim DK, Lee SI, Choi EJ, et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells Through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells (2016) 34:551–64. doi: 10.1002/stem.2279
172. Ray R, Rai V. Lysophosphatidic Acid Converts Monocytes Into Macrophages in Both Mice and Humans. Blood (2017) 129:1177–83. doi: 10.1182/blood-2016-10-743757
173. Radhakrishnan R, Ha JH, Jayaraman M, Liu J, Moxley KM, Isidoro C, et al. Ovarian Cancer Cell-Derived Lysophosphatidic Acid Induces Glycolytic Shift and Cancer-Associated Fibroblast-Phenotype in Normal and Peritumoral Fibroblasts. Cancer Lett (2019) 442:464–74. doi: 10.1016/j.canlet.2018.11.023
174. Li F, He B, Ma X, Yu S, Bhave RR, Lentz SR, et al. Prostaglandin E1 and Its Analog Misoprostol Inhibit Human CML Stem Cell Self-Renewal via EP4 Receptor Activation and Repression of AP-1. Cell Stem Cell (2017) 21:359–373.e5. doi: 10.1016/j.stem.2017.08.001
175. De Carlo F, Witte TR, Hardman WE, Claudio PP. Omega-3 Eicosapentaenoic Acid Decreases CD133 Colon Cancer Stem-Like Cell Marker Expression While Increasing Sensitivity to Chemotherapy. PloS One (2013) 8:e69760. doi: 10.1371/journal.pone.0069760
176. Yang T, Fang S, Zhang H-X, Xu L-X, Zhang Z-Q, Yuan K-T, et al. N-3 PUFAs Have Antiproliferative and Apoptotic Effects on Human Colorectal Cancer Stem-Like Cells In Vitro. J Nutr Biochem (2013) 24:744–53. doi: 10.1016/j.jnutbio.2012.03.023
177. Erickson KL, Hubbard NE. Fatty Acids and Breast Cancer: The Role of Stem Cells. Prostaglandins Leukot Essent Fatty Acids (2010) 82:237–41. doi: 10.1016/j.plefa.2010.02.019
178. Vasudevan A, Yu Y, Banerjee S, Woods J, Farhana L, Rajendra SG, et al. Omega-3 Fatty Acid Is a Potential Preventive Agent for Recurrent Colon Cancer. Cancer Prev Res (Phila) (2014) 7:1138–48. doi: 10.1158/1940-6207.CAPR-14-0177
179. Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating Cancer Metabolic Plasticity by Coupling Gene Regulation With Metabolic Pathways. Proc Natl Acad Sci USA (2019) 116:3909–18. doi: 10.1073/pnas.1816391116
180. Gadducci A, Brunetti I, Muttini MP, Fanucchi A, Dargenio F, Giannessi PG, et al. Epidoxorubicin and Lonidamine in Refractory or Recurrent Epithelial Ovarian Cancer. Eur J Cancer (1994) 30A:1432–5. doi: 10.1016/0959-8049(94)00231-s
181. De Lena M, Lorusso V, Bottalico C, Brandi M, De Mitrio A, Catino A, et al. Revertant and Potentiating Activity of Lonidamine in Patients With Ovarian Cancer Previously Treated With Platinum. J Clin Oncol (1997) 15:3208–13. doi: 10.1200/JCO.1997.15.10.3208
182. De Marinis F, Rinaldi M, Ardizzoni A, Bruzzi P, Pennucci MC, Portalone L, et al. The Role of Vindesine and Lonidamine in the Treatment of Elderly Patients With Advanced non-Small Cell Lung Cancer: A Phase III Randomized FONICAP Trial. Italian Lung Cancer Task Force. Tumori (1999) 85:177–82. doi: 10.1177/030089169908500306
183. Li C, Liu VWS, Chan DW, Yao KM, Ngan HYS. LY294002 and Metformin Cooperatively Enhance the Inhibition of Growth and the Induction of Apoptosis of Ovarian Cancer Cells. Int J Gynecol Cancer (2012) 22:15–22. doi: 10.1097/IGC.0b013e3182322834
184. Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature (2016) 531:47–52. doi: 10.1038/nature16965
185. Blum R, Kloog Y. Metabolism Addiction in Pancreatic Cancer. Cell Death Dis (2014) 5:e1065. doi: 10.1038/cddis.2014.38
186. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras Maintains Pancreatic Tumors Through Regulation of Anabolic Glucose Metabolism. Cell (2012) 149:656–70. doi: 10.1016/j.cell.2012.01.058
187. Lin W, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, et al. Dormant Cancer Cells Contribute to Residual Disease in a Model of Reversible Pancreatic Cancer. Cancer Res (2013) 73:1821–30. doi: 10.1158/0008-5472.CAN-12-2067
188. He T-L, Zhang Y-J, Jiang H, Li X-H, Zhu H, Zheng K-L. The C-Myc-LDHA Axis Positively Regulates Aerobic Glycolysis and Promotes Tumor Progression in Pancreatic Cancer. Med Oncol (2015) 32:187. doi: 10.1007/s12032-015-0633-8
189. Jia D, Paudel BB, Hayford CE, Hardeman KN, Levine H, Onuchic JN, et al. Drug-Tolerant Idling Melanoma Cells Exhibit Theory-Predicted Metabolic Low-Low Phenotype. Front Oncol (2020) 10:1426. doi: 10.3389/fonc.2020.01426
Keywords: metabolism, cancer stem cell, glucose, glutamine, OxPhos
Citation: Kaur J and Bhattacharyya S (2021) Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 11:756888. doi: 10.3389/fonc.2021.756888
Received: 11 August 2021; Accepted: 18 October 2021;
Published: 05 November 2021.
Edited by:
Michael P. Lisanti, University of Salford Manchester, United KingdomReviewed by:
Pritam Sadhukhan, Johns Hopkins University, United StatesHerbert Levine, Rice University, United States
Copyright © 2021 Kaur and Bhattacharyya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shalmoli Bhattacharyya, c2hhbG1vbGkyMDA3QHlhaG9vLmNvLmlu; c2hhbG1vbGliQGdtYWlsLmNvbQ==